
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and enantioseparation of chiral Au13 nanoclusters protected by bis-N-heterocyclic carbene ligands†
Hong
Yi‡
 a,
Kimberly M.
Osten‡
a,
Tetyana I.
Levchenko
b,
Alex J.
Veinot
b,
Yoshitaka
Aramaki
*ac,
Takashi
Ooi
*ac,
Masakazu
Nambo
*a and
Cathleen M.
Crudden
*ab
a,
Kimberly M.
Osten‡
a,
Tetyana I.
Levchenko
b,
Alex J.
Veinot
b,
Yoshitaka
Aramaki
*ac,
Takashi
Ooi
*ac,
Masakazu
Nambo
*a and
Cathleen M.
Crudden
*ab
aInstitute of Transformative Bio-Molecules (WPI-ITbM), Nagoya University Furo, Chikusa, Nagoya 464-8602, Japan. E-mail: aramaki@chembio.nagoya-u.ac.jp; tooi@chembio.nagoya-u.ac.jp; mnambo@itbm.nagoya-u.ac.jp; cruddenc@chem.queensu.ca
bDepartment of Chemistry, Queen's University, Chernoff Hall, Kingston, Ontario K7L 3N6, Canada
cDepartment of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Nagoya 464-8601, Japan
First published on 2nd July 2021
Abstract
A series of chiral Au13 nanoclusters were synthesized via the direct reduction of achiral dinuclear Au(I) halide complexes ligated by ortho-xylyl-linked bis-N-heterocyclic carbene (NHC) ligands. A broad range of functional groups are tolerated as wingtip substituents, allowing for the synthesis of a variety of functionalized chiral Au13 nanoclusters. Single crystal X-ray crystallography confirmed the molecular formula to be [Au13(bisNHC)5Cl2]Cl3, with a chiral helical arrangement of the five bidentate NHC ligands around the icosahedral Au13 core. This Au13 nanocluster is highly luminescent, with a quantum yield of 23%. The two enantiomers of the Au13 clusters can be separated by chiral HPLC, and the isolated enantiomers were characterized by circular dichroism spectroscopy. The clusters show remarkable stability, including configurational stability, opening the door to further investigation of the effect of chirality on these clusters.
The intersection of chirality with nanomaterials has been called “one of the most dynamic areas in modern science”.1a Atomically precise nanoclusters are a particularly important class of nanomaterials in this area because of the ability to discern molecular structure with high precision, and the high structural purity obtainable.1 The unique properties of these nanomaterials opens up applications in chiral sensing,2 enantioselective nanocatalysis3 and chiroptics.4 Chirality in metal nanoclusters can be generated via an intrinsically chiral metal core,5 from an achiral core with chiral organic ligands6 or from an achiral core with achiral ligands that are arranged in an asymmetric fashion.7 Although impressive strides have been made in the synthesis of chiral metal clusters, considerable challenges remain with regards to designing chiral nanoclusters with stable chirality that is resistant to racemization and with optimized properties, for example photo-physical properties.
The Bürgi group have been pioneers in the creation of chiral thiolate-stabilized nanoclusters, highlighted by their work with Au38(SR)24, which is chiral by virtue of the arrangement of thiol staples around the core, and can be separated into its enantiomers by HPLC.8 Cluster Au38(SCH2CH2Ph)24 shows high stability at room temperature, with racemization occurring in approximately 30 minutes at 80 °C.9 Computational work from the Häkkinen group suggests that chiral inversions for Au38(SR)24 and Pd-doped Au38 can take place through a rotational reconstruction of the metal core without the need for ligand–metal bond cleavage.10
Smaller Au13 nanoclusters can also be prepared with achiral phosphine ligands depending on their orientation around the core. Shichibu and Konishi described this situation for [Au13(dppe)5Cl5]3+ (dppe = 1,2-bis(diphenylphosphino)ethane),11 in which the five dppe ligands take up a propeller-like arrangement.12 However, successful resolution of the individual enantiomers by chiral HPLC was not achieved, possibly due to fast racemization of these clusters in solution. The only examples of resolvable chiral Au clusters stabilized by phosphine ligands require the use of inherently chiral ligands.13
N-Heterocyclic carbenes (NHCs) have recently emerged as valuable alternatives to thiols or phosphines for the stabilization of metal surfaces, nanoparticles, and clusters.14 The neutral, electron-rich NHCs form a strong covalent bond with the metal,15 which is important in providing stability to metal nanoclusters.16 Our group recently described the utility of NHCs in protecting gold nanoclusters, reporting the first examples of NHC-containing Au11 and Au13 clusters.17,18 Zheng et al. have also described the synthesis of Au13,19 Au25 (ref. 20a) and Au44 (ref. 20b) nanoclusters, and Tsukuda has prepared Au23 clusters,21 illustrating the potential of NHCs as stabilizing ligands for a range of gold nanocluster architectures. Very recently, Zang et al. reported the synthesis of Au11 and Au13 nanoclusters protected by m-phenylene-bridged N-heterocyclic carbene ligands that result in different clusters based on subtle changes in N-substituents.22
Herein we report the synthesis of chiral Au13 nanoclusters prepared with achiral bidentate NHC ligands. The nanoclusters derive their chirality from a helical arrangement of the achiral ligand (Fig. 1). The stability introduced by the NHC ligand permits the separation of the nanocluster into its constituent enantiomers by chiral HPLC. Each enantiomer was characterized by circular dichroism. The clusters display impressive thermal stability and record-setting photoluminescent quantum yields.
 | ||
| Fig. 1 Synthesis of chiral Au13 clusters. | ||
Inspired by the work of Zheng, who showed that bisNHC ligands linked by a simple propyl chain can provide stable Au13 nanoclusters,19 we set out to examine bis-benzimidazolylidene ligands with aromatic linkers, designed to take advantage of the multitude of CH-π interactions observed in other Au13 clusters.18 Bis-benzimidazolium salts (1a–g) prepared by simple SN2 reactions were converted into the corresponding Au complexes (bisNHC)Au2X2 (2a–g) by reaction with Au(SMe2)Cl in the presence of K2CO3 (see ESI† for full details and characterization).23 Cluster synthesis was accomplished by reduction of complex 2a with NaBH4 followed by etching with HCl. This procedure afforded cluster [3a]Cl3 in 43% isolated yield based on Au, after purification by column chromatography (Fig. 2A).
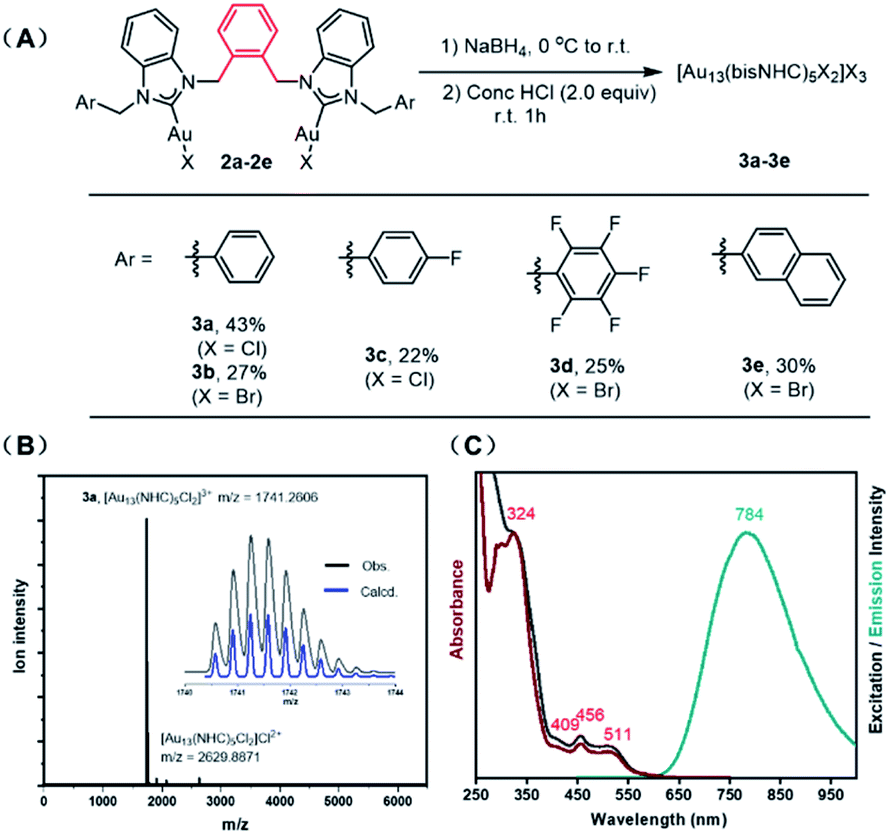 | ||
| Fig. 2 (A) Preparation of [Au13(bisNHC)5X2]X3 nanoclusters [3a–e]X3 (B) ESI-MS of [3a]Cl3; (C) UV-vis absorbance (red), photoluminescence excitation (black) and emission (green) spectra of [3a]Cl3 in dichloromethane. | ||
The effect of HCl on the synthetic protocol was examined by UV-vis spectroscopy and electrospray ionization mass spectrometry (ESI-MS). The absorbance spectra of the cluster were significantly sharper after HCl etching, consistent with the size-focusing effect observed in other systems (Fig. S1†).11,18 The molecular formula of the cluster [3a]Cl3 was confirmed by ESI-MS, which showed a dominant molecular ion peak at 1741.26 m/z corresponding to [Au13(bisNHCBn)5Cl2]3+ (Fig. 2B). Good agreement was observed between the experimental and theoretical isotopic distribution patterns. A minor peak at 2629.89 m/z was also observed, corresponding to the divalent ion [Au13(bisNHCBn)5Cl2]Cl2+ derived from incomplete ionization of the parent cluster.
The UV-vis spectrum of [3a]Cl3 in dichloromethane shows distinct absorption bands at 324, 409, 456, and 511 nm (Fig. 2C, red), which are in line with previously reported Au13 clusters.18 Solutions of [3a]Cl3 in dichloromethane show bright red-orange emission. Photoluminescence quantum yields measured by two independent methods is 23%, a value considerably higher than most superatom gold clusters, including our previous report of 16% for an NHC-protected Au13 cluster.18 The spectrum produced by excitation at 784 nm (Fig. 2C, black) is in good agreement with the UV-vis spectrum, supporting the conclusion that the measured quantum yield was not affected by contamination with other emissive species. Although the precise reason for the high quantum yield is currently under investigation, the rigid shell provided by the NHC with multiple π–π stacking interactions likely contributes.
Halide counterions had little effect on cluster formation, with bromide complex 2b affording 27% yield of [Au13(bisNHCBn)5Br2]Br3 ([3b]Br3) after purification. The nature of the linking unit was found to be critically important, with meta- and para-xylyl linked ligands (1f and 1g respectively) giving no observable Au13 clusters (see Fig. S2†). However, wingtip aromatic substituents were readily interchangeable, with complexes 2c–e all affording Au13 nanoclusters (see ESI† for full characterization).
Single crystals of [3a]Cl3 suitable for X-ray crystallography were grown from layering hexane onto a dichloromethane solution of the pure cluster. The structure features an icosahedral Au13 core with Au–Cl bonds capping the top and bottom of the core and the five bis-NHC ligands bound to the two twisted Au5 pentagons of the core (Fig. 3). The average Au–C bond length is 2.05(2) Å, similar to other NHC-stabilized nanoclusters.18 The average Au centre–Au peripheral bond distance of the icosahedral core is 2.7655(12) Å, and the average Au–Cl bond length of the terminal chloride ligands is 2.332(6) Å. A distinct structural characteristic is the face-to-face arrangement of one benzimidazole and the ortho-phenyl bridge, and the other benzimidazole and one benzyl phenyl ring on neighbouring ligands, with average distances of 3.3 Å and 3.4 Å, respectively, suggesting the presence of π–π stacking interactions (Fig. 3B).
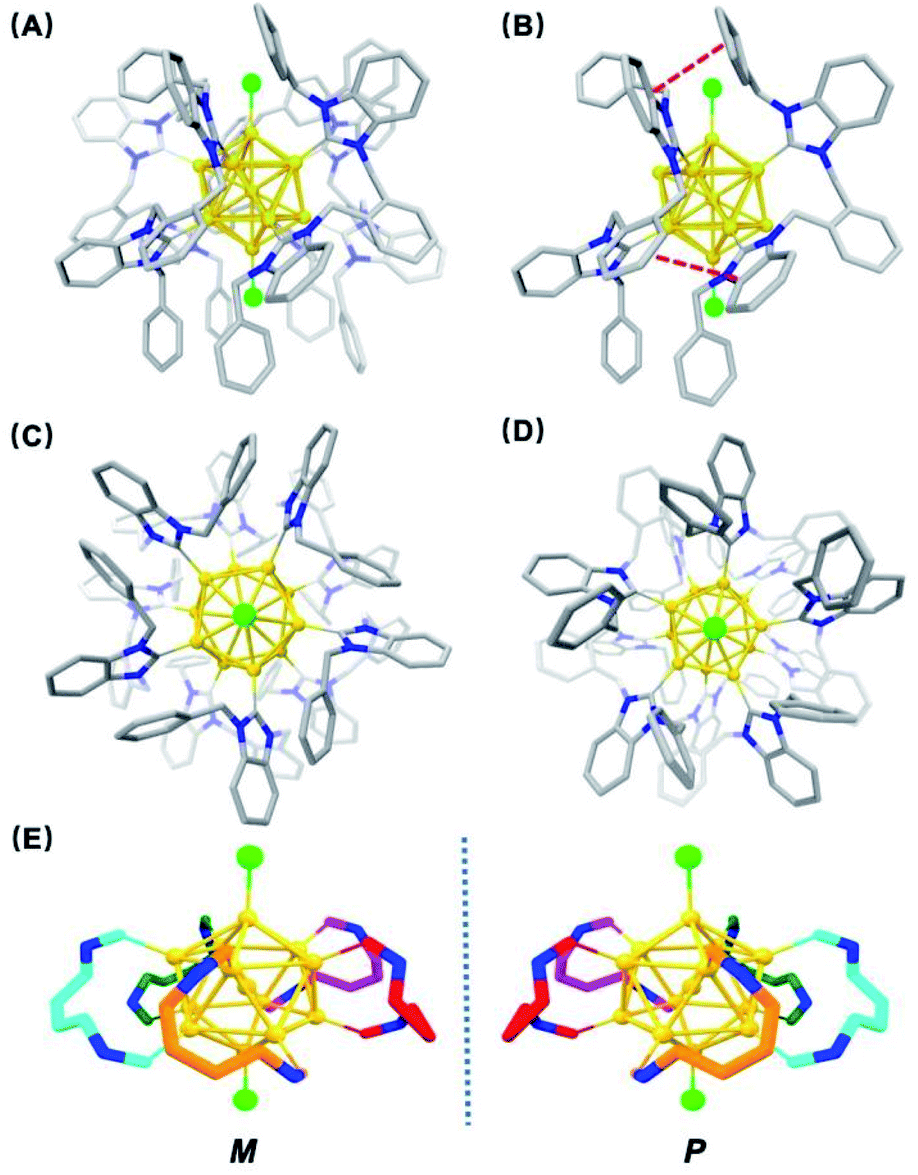 | ||
| Fig. 3 (A) Single-crystal X-ray structure of [Au13(bisNHCBn)5Cl2]Cl3 ([3a]Cl3). Anions and hydrogen atoms have been removed for clarity (color key: carbon, gray; nitrogen, blue; chlorine, green; gold, yellow); (B) side-view of structure showing interactions between benzimidazole/o-phenyl bridge and benzimidazole/benzyl arm phenyl rings on neighbouring ligands; (C) top-down and (D) bottom-up views of the structure. See ESI† for full ORTEP structures; (E) M/P descriptors of 3a. The chirality can be easily viewed from the left (counter-clockwise) and right (clockwise)-handed arrangement of the bisNHC ligand on the Au surface (dark blue: nitrogen of NHC). | ||
From the top view, the disposition of these mutually interacted components is unidirectional, rendering the top of 3a different from the bottom. The NHC ligands are oriented with pseudo C5 symmetry (Fig. 3C and D). From the bottom view, benzyl arms are oriented vertically without any possible interactions with the other components (Fig. 3D). Accordingly, 3a has a helical chirality with pseudo C5 symmetry (Fig. 3E). Cluster [3a]Cl3 was crystallized as a racemic mixture in Pna21 space group with mirror planes.
To investigate whether this structure is conserved in solution, the clusters were characterized by NMR spectroscopy. The 1H NMR spectrum of cluster [3a]Cl3 has four sets of diastereotopic methylene CH2 protons attributed to the benzyl and xylyl groups (Fig. 4A). This suggests a single ligand environment without C2 symmetry and with a low degree of rotational freedom, similar to what is observed in the solid-state (see ESI† for full spectral details of all clusters). The NMR shifts of some protons were also found to be anion dependent, with [3a][PF6]3 displaying significant changes in methylene resonance shifts compared to the chloride analogue (see ESI† for full details).
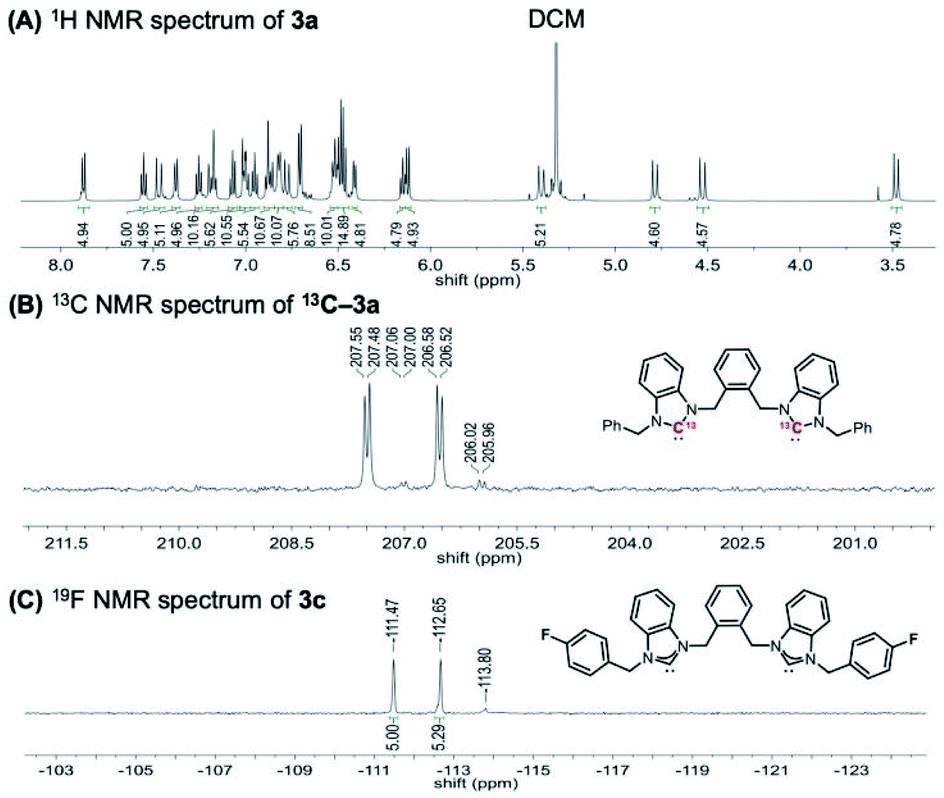 | ||
| Fig. 4 Solution structural characterization of [Au13(bisNHCAr)5Cl2]Cl3 clusters by NMR spectroscopy in CD2Cl2. (A) 1H NMR spectrum of [3a]Cl3. (B) Cluster region of the 13C{1H} NMR spectrum of 13C-[3a][PF6]3 bearing 13C(C2)-labelled bisNHC ligand; (C) 19F NMR spectrum of [3c]Cl3 (the signal at 113.80 ppm belongs to a small amount of [L2Au2]Cl2). | ||
The asymmetric ligand environment was also confirmed by synthesizing 13C-labeled cluster 13C-[3a][PF6]3 using 13C(C2)-labelled NHC precursor, which enables the direct observation of the M–C bond. The 13C{1H} NMR spectrum has two major carbene peaks, which appear as doublets at 207.5 and 206.6 ppm (Fig. 4B). A minor set of two doublets was also observed at 207.0 and 206.0 ppm, the origin of which is currently under further study in our group. Similarly, the 19F NMR spectrum of the fluorinated analogue [3c]Cl3 displays two signals at −111.5 and −112.7 ppm (Fig. 4C).
Thus, solution NMR spectroscopic results support the presence of ligand asymmetry in solution as well as solid state. The NMR studies are also consistent with conformational rigidity of the clusters, at least on the NMR timescale, as we have reported previously for other related NHC-functionalized clusters.18 These results suggested that the clusters might have high configurational stability, such that the separation of the two enantiomers via solution-based methods, such as chiral HPLC, might be feasible.
With solution NMR studies showing high structural rigidity of the cluster, we then proceeded to attempt to separate the two enantiomers. [3a]Cl3 was successfully separated into its constituent enantiomers using a chiral cellulose-based analytical HPLC column with methanol (MeOH) as the eluent, along with trifluoroacetic acid (TFA) and diethylamine (DEA) as additives. Two well-separated peaks were observed at 21.6 and 24.2 minutes in an approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio as expected for a racemic mixture (labelled as enantiomers 3a-en1 and 3a-en2 respectively, Fig. 5A). These conditions enabled the separation of a large batch (20 mg) of racemic cluster [3a]Cl3 by preparative chiral HPLC. The enantiomeric excess (ee) of the two collected fractions was determined by chiral HPLC and found to be 88% for 3a–en1 and 95% for 3a-en2. The UV-vis, NMR and mass spectra of the separated enantiomers were consistent with the starting racemic mixture, illustrating the stability of the Au13 structure under the separation conditions, although the ESI-MS and NMR spectra suggest that anion exchange with trifluoroacetate anion had occurred (Fig. S3–S5†).
:1 ratio as expected for a racemic mixture (labelled as enantiomers 3a-en1 and 3a-en2 respectively, Fig. 5A). These conditions enabled the separation of a large batch (20 mg) of racemic cluster [3a]Cl3 by preparative chiral HPLC. The enantiomeric excess (ee) of the two collected fractions was determined by chiral HPLC and found to be 88% for 3a–en1 and 95% for 3a-en2. The UV-vis, NMR and mass spectra of the separated enantiomers were consistent with the starting racemic mixture, illustrating the stability of the Au13 structure under the separation conditions, although the ESI-MS and NMR spectra suggest that anion exchange with trifluoroacetate anion had occurred (Fig. S3–S5†).
 | ||
| Fig. 5 Enantioseparation and characterization of chiral Au13 nanoclusters: (A) chromatogram showing the enantioseparation of rac-3a by chiral HPLC using UV-vis detection at 350 nm; (B) CD spectra of the two chiral enantiomers separated by HPLC and rac-3a for comparison. | ||
The as-separated enantiomers were characterized by circular dichroism (CD) spectroscopy (Fig. 5B). As expected, the CD spectra of the two enantiomers are mirror images and show distinct bands between 250 and 800 nm (main peaks: 259, 271, 296, 317, 333 nm, weak peaks at 376, 425 nm, and a broad peak at 500 nm). No CD signal was observed for the racemic mixture.
The thermal stability of cluster [3a]Cl3 was investigated by UV-vis spectroscopy. The UV-vis spectrum showed no discernible change after 3 days of heating at 60 °C in acetonitrile (Fig. 6A). Emission spectra were also identical within error after this time (Fig. S7†). The configurational stability of 3a was investigated by monitoring the change in enantiopurity upon heating to different temperatures. Samples of 3a-en1 (88% initial ee) were heated in MeOH at a variety of temperatures for 1 h. At 60 °C, there was no discernible change, but a slight drop to 81% ee was observed when the cluster was heated to 80 °C, and at 100 °C the ee further decreased to 72% (Fig. 6B). Changes in enantiomeric ratios were documented by HPLC (Fig. S8†) and CD (Fig. S9†). Work to clarify the mechanism of racemization is underway in our lab.
 | ||
| Fig. 6 (A) Thermal stability of rac-3a at 60 °C in acetonitrile monitored by UV-vis spectroscopy; (B) chiral stability of 3a-en-1 in methanol after treatment at different temperatures for 1 h. The enantiomeric purity after heating was examined by chiral HPLC. | ||
Conclusions
In conclusion, we have described the synthesis and structural characterization of a new chiral ortho-xylyl linked bisNHC-stabilized Au13 cluster [Au13(bisNHCBn)5Cl2]Cl3 ([3a]Cl3) and its bromide derivative [3b]Br3. These clusters have a similar icosahedral structure to previously reported Au13 clusters, including those ligated by monodentate,18 bis-NHC19 and bis-phosphine ligands.11 A variety of wingtip groups were compatible with nanocluster formation, allowing for the isolation of clusters incorporating diverse functionality (3c–e). The chirality of cluster 3a in the solid-state was investigated by X-ray crystallography, where it was discerned that chirality results from a helical arrangement of the surface ligands that is dictated by the choice of a bidentate ligand. The two enantiomers were separated by chiral HPLC and characterized by CD spectra. The clusters show high thermal stability and stability against racemization, which may facilitate their future applications in chiral catalysis and sensing.Data availability
The experimental data is included in the ESI.Author contributions
C. C. and M. N. conceived the project. H. Y. and K. O. carried out the synthesis, purification and characterization of the Au13 nanoclusters. T. L. performed the quantum yield of Au13 nanocluster. A. V., Y. A., and T. O. analysed the X-ray crystal of the cluster [3a]Cl3. All the authors wrote the paper, supplementary methods, and related materials.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by KAKENHI from JSPS (17H03030 to C. M. C.). H. Y. and K. M. O. thank JSPS for funding through the JSPS International Research Fellow program. M. N. thanks the support from The Mazda foundation. A. J. V. thanks NSERC for a Vanier Scholarship and the Walter C. Sumner foundation for additional scholarship support. T. I. L. thanks NSERC for a postdoctoral fellowship. We thank DAICEL corporation for the assistance of separation of enantiomers. JSPS and NU are acknowledged for funding of this research through The World Premier International Research Centre Initiative (WPI) program. NSERC and the Canada Foundation for Innovation (CFI) are thanked for financial support of this work in terms of operating and equipment grants to C. M. C. The authors thank Dr Kevin Stamplecoskie (Queen's University) for access to UV-vis absorbance and fluorescence spectrometers and for useful advice on fluorescence quantum yield measurements.References
- (a) S. Knoppe and T. Burgi, Acc. Chem. Res., 2014, 47, 1318–1326 CrossRef CAS; (b) Y. Zhu, J. Guo, X. Qiu, S. Zhao and Z. Tang, Acc. Mater. Res., 2020, 2, 21–35 CrossRef; (c) Y. Li, T. Higaki, X. Du and R. Jin, Adv. Mater., 2020, 32, e1905488 CrossRef PubMed; (d) C. Zeng and R. Jin, Chem.–Asian J., 2017, 12, 1839–1850 CrossRef CAS; (e) Q.-Y. Zhang and L. Zhao, Tetrahedron Lett., 2018, 59, 310–316 CrossRef CAS.
- D. Astruc, M. C. Daniel and J. Ruiz, Chem. Commun., 2004, 2637–2649 RSC.
- (a) T. Mallat, E. Orglmeister and A. Baiker, Chem. Rev., 2007, 107, 4863–4890 CrossRef CAS PubMed; (b) E. S. Andreiadis, M. R. Vitale, N. Mezailles, X. Le Goff, P. Le Floch, P. Y. Toullec and V. Michelet, Dalton Trans., 2010, 39, 10608–10616 RSC.
- (a) Y. Zhao, M. A. Belkin and A. Alu, Nat. Commun., 2012, 3, 870 CrossRef CAS PubMed; (b) L. Ohnoutek, N. H. Cho, A. W. Allen Murphy, H. Kim, D. M. Rasadean, G. D. Pantos, K. T. Nam and V. K. Valev, Nano Lett., 2020, 20, 5792–5798 CrossRef CAS.
- (a) A. Lechtken, D. Schooss, J. R. Stairs, M. N. Blom, F. Furche, N. Morgner, O. Kostko, B. von Issendorff and M. M. Kappes, Angew. Chem., Int. Ed., 2007, 46, 2944–2948 CrossRef CAS PubMed; (b) X. K. Wan, S. F. Yuan, Z. W. Lin and Q. M. Wang, Angew. Chem., Int. Ed., 2014, 53, 2923–2926 CrossRef CAS PubMed.
- (a) T. G. Schaaff and R. L. Whetten, J. Phys. Chem., 2000, 104, 2630–2641 CrossRef CAS; (b) H. Yao, K. Miki, N. Nishida, A. Sasaki and K. Kimura, J. Am. Chem. Soc., 2005, 127, 15536–15543 CrossRef CAS PubMed; (c) Y. Yanagimoto, Y. Negishi, H. Fujihara and T. Tsukuda, J. Phys. Chem. B, 2006, 110, 11611–11614 CrossRef CAS PubMed.
- (a) O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Hakkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210–8218 CrossRef CAS PubMed; (b) C. Zeng, T. Li, A. Das, N. L. Rosi and R. Jin, J. Am. Chem. Soc., 2013, 135, 10011–10013 CrossRef CAS PubMed.
- (a) I. Dolamic, S. Knoppe, A. Dass and T. Burgi, Nat. Commun., 2012, 3, 798 CrossRef; (b) S. Knoppe, I. Dolamic, A. Dass and T. Burgi, Angew. Chem., Int. Ed., 2012, 51, 7589–7591 CrossRef CAS PubMed.
- S. Knoppe, I. Dolamic and T. Burgi, J. Am. Chem. Soc., 2012, 134, 13114–13120 CrossRef CAS.
- S. Malola and H. Häkkinen, J. Am. Chem. Soc., 2019, 141, 6006–6012 CrossRef CAS.
- Y. Shichibu and K. Konishi, Small, 2010, 6, 1216–1220 CrossRef CAS PubMed.
- (a) J. Zhang, Y. Zhou, K. Zheng, H. Abroshan, D. R. Kauffman, J. Sun and G. Li, Nano Res., 2018, 11, 5787–5798 CrossRef CAS; (b) Y. Shichibu, Y. Ogawa, M. Sugiuchi and K. Konishi, Nanoscale Adv., 2021, 3, 1005–1011 RSC.
- Y. Yang, Q. Zhang, Z. J. Guan, Z. A. Nan, J. Q. Wang, T. Jia and W. W. Zhan, Inorg. Chem., 2019, 58, 3670–3675 CrossRef CAS PubMed.
- (a) C. M. Crudden, J. H. Horton, I. I. Ebralidze, O. V. Zenkina, A. B. McLean, B. Drevniok, Z. She, H. B. Kraatz, N. J. Mosey, T. Seki, E. C. Keske, J. D. Leake, A. Rousina-Webb and G. Wu, Nat. Chem., 2014, 6, 409–414 CrossRef CAS; (b) A. V. Zhukhovitskiy, M. J. MacLeod and J. A. Johnson, Chem. Rev., 2015, 115, 11503–11532 CrossRef CAS PubMed; (c) S. Engel, E. C. Fritz and B. J. Ravoo, Chem. Soc. Rev., 2017, 46, 2057–2075 RSC; (d) G. Wang, A. Ruhling, S. Amirjalayer, M. Knor, J. B. Ernst, C. Richter, H. J. Gao, A. Timmer, H. Y. Gao, N. L. Doltsinis, F. Glorius and H. Fuchs, Nat. Chem., 2017, 9, 152–156 CrossRef CAS PubMed; (e) C. A. Smith, M. R. Narouz, P. A. Lummis, I. Singh, A. Nazemi, C. H. Li and C. M. Crudden, Chem. Rev., 2019, 119, 4986–5056 CrossRef CAS PubMed.
- (a) C. M. Crudden and D. P. Allen, Coord. Chem. Rev., 2004, 248, 2247–2273 CrossRef CAS; (b) T. J. Robilotto, J. Bacsa, T. G. Gray and J. P. Sadighi, Angew. Chem., Int. Ed., 2012, 51, 12077–12080 CrossRef CAS PubMed; (c) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS; (d) L. Jin, D. S. Weinberger, M. Melaimi, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 9059–9063 CrossRef CAS PubMed.
- (a) T. H. Tsukuda and H. Häkkinen, Protected Metal Clusters: From Fundamentals to Applications, Elsevier, Amsterdam, 2015 Search PubMed; (b) Q. Tang, G. Hu, V. Fung and D. E. Jiang, Acc. Chem. Res., 2018, 51, 2793–2802 CrossRef CAS; (c) J. Yan, B. K. Teo and N. Zheng, Acc. Chem. Res., 2018, 51, 3084–3093 CrossRef CAS; (d) I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed; (e) R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed; (f) X. Kang, Y. Li, M. Zhu and R. Jin, Chem. Soc. Rev., 2020, 49, 6443–6514 RSC; (g) Z. Lei, X. L. Pei, H. Ube and M. Shionoya, Bull. Chem. Soc. Jpn., 2021, 94, 1324–1330 CrossRef CAS.
- M. R. Narouz, K. M. Osten, P. J. Unsworth, R. W. Y. Man, K. Salorinne, S. Takano, R. Tomihara, S. Kaappa, S. Malola, C. T. Dinh, J. D. Padmos, K. Ayoo, P. J. Garrett, M. Nambo, J. H. Horton, E. H. Sargent, H. Hakkinen, T. Tsukuda and C. M. Crudden, Nat. Chem., 2019, 11, 419–425 CrossRef CAS PubMed.
- M. R. Narouz, S. Takano, P. A. Lummis, T. I. Levchenko, A. Nazemi, S. Kaappa, S. Malola, G. Yousefalizadeh, L. A. Calhoun, K. G. Stamplecoskie, H. Hakkinen, T. Tsukuda and C. M. Crudden, J. Am. Chem. Soc., 2019, 141, 14997–15002 CrossRef CAS.
- H. Shen, S. Xiang, Z. Xu, C. Liu, X. Li, C. Sun, S. Lin, B. K. Teo and N. Zheng, Nano Res., 2020, 13, 1908–1911 CrossRef CAS.
- (a) H. Shen, G. Deng, S. Kaappa, T. Tan, Y. Z. Han, S. Malola, S. C. Lin, B. K. Teo, H. Hakkinen and N. Zheng, Angew. Chem., Int. Ed., 2019, 58, 17731–17735 CrossRef CAS PubMed; (b) H. Shen, Z. Xu, M. S. A. Hazer, Q. Wu, J. Peng, R. Qin, S. Malola, B. K. Teo, H. Hakkinen and N. Zheng, Angew. Chem., Int. Ed., 2021, 60, 3752–3758 CrossRef CAS PubMed.
- K. Hirano, S. Takano and T. Tsukuda, J. Phys. Chem. C, 2021, 125, 9930–9936 CrossRef CAS.
- P. Luo, S. Bai, X. Wang, J. Zhao, Z. N. Yan, Y. F. Han, S. Q. Zang and T. C. W. Mak, Adv. Opt. Mater., 2021, 2001936 CrossRef CAS.
- A. Collado, A. Gomez-Suarez, A. R. Martin, A. M. Slawin and S. P. Nolan, Chem. Commun., 2013, 49, 5541–5543 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2067324. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc03076k |
| ‡ Hong Yi and Kimberly M. Osten contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |