
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular Friedel–Crafts alkylation with a silylium-ion-activated cyclopropyl group: formation of tricyclic ring systems from benzyl-substituted vinylcyclopropanes and hydrosilanes†
Tao
He
 ,
Guoqiang
Wang
,
Peng-Wei
Long
,
Sebastian
Kemper
,
Elisabeth
Irran
,
Hendrik F. T.
Klare
* and
Martin
Oestreich
*
,
Guoqiang
Wang
,
Peng-Wei
Long
,
Sebastian
Kemper
,
Elisabeth
Irran
,
Hendrik F. T.
Klare
* and
Martin
Oestreich
*
Institut für Chemie, Technische Universität Berlin, Strasse des 17. Juni 115, 10623 Berlin, Germany. E-mail: hendrik.klare@tu-berlin.de; martin.oestreich@tu-berlin.de
First published on 29th October 2020
Abstract
A trityl-cation-initiated annulation of benzyl-substituted vinylcyclopropanes (VCPs) with hydrosilanes is reported. Two Si–C(sp3) bonds and one C(sp2)–C(sp3) bond are formed in this process where an intramolecular 6-endo-tet Friedel–Crafts alkylation of a silylium-ion-activated cyclopropane ring is the rate-determining key step. The reaction mechanism is proposed based on computations and is in agreement with experimental observations. The new reaction leads to an unprecedented silicon-containing 6/6/5-fused ring system. A phenethyl-substituted VCP derivative yields another unknown tricycle having 6/6/6 ring fusion by reacting in a related but different way involving a 6-exo-tet ring closure.
Introduction
We recently became interested in the reactivity of catalytically generated silicon electrophiles towards cyclopropane derivatives.1–3 Our investigations typically comprise B(C6F5)3/hydrosilane combinations4 and silylium-ion-like reactants5 emerging from hydrosilanes and the trityl salt Ph3C+[B(C6F5)4]− as an initiator. VCPs as substrates specifically caught our attention because these versatile building blocks can react in diverse ways.6 This is also true for their reactions with silicon electrophiles (Scheme 1, top).2,3 These reactions involve the intermediacy of β-silicon-stabilized7 cyclopropylcarbinyl cations8 with different counteranions (gray boxes). Depending on the hydride source, the outcomes could not be more different. For aryl-substituted VCPs, the B(C6F5)3-catalyzed hydrosilylation proceeds generally with little ring opening (top left).2 In contrast, treatment of these VCPs with Ph3C+[B(C6F5)4]− in the presence of various hydrosilanes affords silicon-containing six-membered rings as a result of a formal (5 + 1) cycloaddition accompanied by an aryl migration (top right).3 The situation changes again when replacing the aryl substituent by a benzyl group (Scheme 1, bottom). The additional methylene group makes a huge difference. The hydrosilylation under B(C6F5)3 catalysis is now plagued with ring opening, likely due to poorer stabilization of the carbocation intermediate (bottom left).2 Strikingly, the silylium-ion-promoted setup gives rise to yet another product where the aryl group becomes part of the product's ring system (bottom right). We report here a new trityl-cation-initiated cycloaddition of benzyl-substituted VCPs and hydrosilanes involving an intramolecular Friedel–Crafts alkylation. Our study includes a full experimental and computational mechanistic analysis.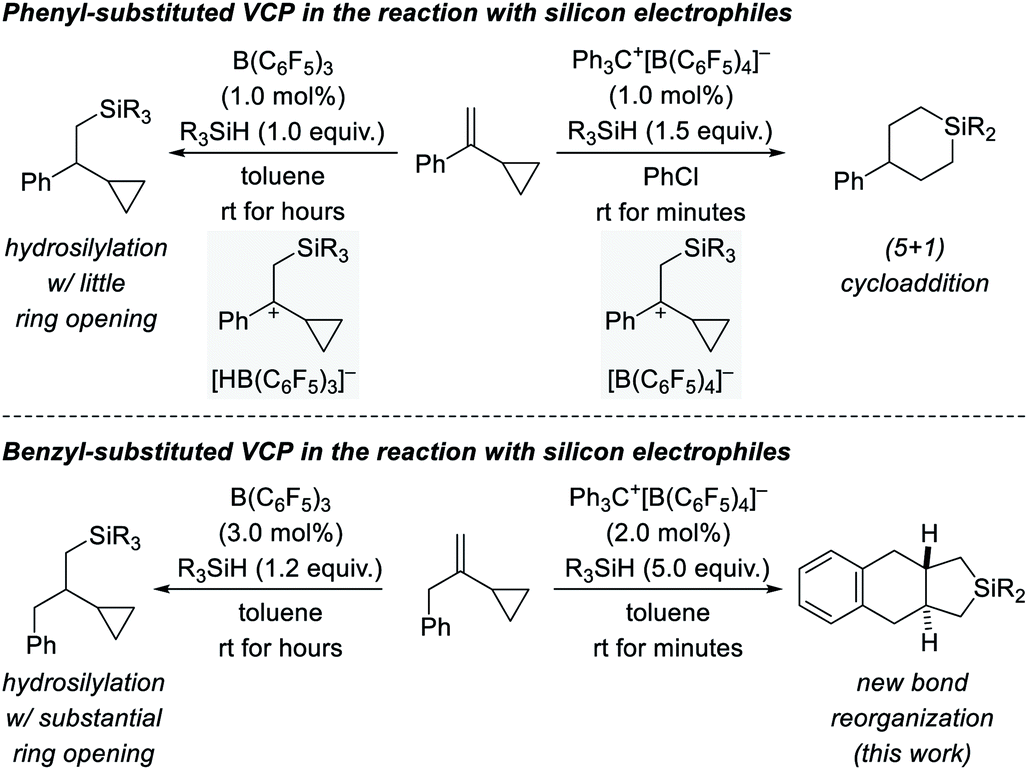 | ||
| Scheme 1 Diverse outcomes from the reaction of phenyl- and benzyl-substituted VCPs and hydrosilanes (R3SiH = 3°, 2°, and 1° hydrosilanes with R = alkyl and/or aryl). | ||
Results and discussion
Using 2 mol% of Ph3C+[B(C6F5)4]− as an initiator, we began investigating the reaction of VCP 1a and excess Et2SiH2 (2a) in various arene solvents at ambient temperature (Table 1). In benzene as solvent, 2,2-diethyl-2,3,3a,4,9,9a-hexahydro-1H-naphtho[2,3-c]silole (3aa) was found as the major product along with ring-opened 4-methyl-5-phenylpentyl-substituted silane 4ab and six-membered ring system 5aa (entry 1). As ring-opening hydrosilylation would yield an unsaturated compound,1 we speculated that the formation of saturated 4ab (through initially formed 4aa)9,10 involves an additional alkene hydrogenation by protonation of the VCP to form a cyclopropylcarbinyl cation followed by hydride transfer from the hydrosilane. The proton could be released from a Wheland intermediate in the course of an assumed Friedel–Crafts reaction. Addition of norbornene as a proton scavenger had no effect though (entry 2). In turn, the silicon-containing ring system 5aa could be the result of the aforementioned silylium-ion-promoted hydrosilylation of cyclopropanes1 coupled with an endo cyclization of an allylbenzene intermediate (see discussion of the mechanism).|
|
|||||
|---|---|---|---|---|---|
| Entry | Initiator (mol%) | Solvent | Yieldb (%) | ||
| 3aa | 4aa/4ab | 5aa | |||
| a All reactions were performed with VCP 1a (0.10 mmol) and the indicated amounts of the initiator and Et2SiH2 (2a) under argon atmosphere in the indicated arene solvent (0.5 mL, 0.2 M) at room temperature. Unless otherwise noted, conversion was greater than 95% for each entry as estimated by 1H NMR spectroscopy using CH2Br2 as an internal standard. b Yields were estimated by 1H NMR spectroscopy using CH2Br2 as an internal standard and tend to be too high because of the long relaxation time of CH2Br2. c 1.0 equiv. of norbornene used. d 2.0 equiv. of Et2SiH2 (2a) used. e 39% of VCP 1a recovered. f Performed at 0.1 M. | |||||
| 1 | Ph3C+[B(C6F5)4]− (2.0 mol%) | Benzene | 51 | 34 | 9 |
| 2c | Ph3C+[B(C6F5)4]− (2.0 mol%) | Toluene-d8 | 61 | 26 | 9 |
| 3d | Ph3C+[B(C6F5)4]− (2.0 mol%) | Benzene | 41 | 47 | 8 |
| 4e | Ph3C+[B(C6F5)4]− (1.0 mol%) | Benzene | 28 | 28 | <5 |
| 5 | Ph3C+[B(C6F5)4]− (5.0 mol%) | Benzene | 55 | 30 | 13 |
| 6f | Ph3C+[B(C6F5)4]− (2.0 mol%) | Benzene | 52 | 33 | 11 |
| 7 | Ph3C+[B(C6F5)4]− (2.0 mol%) | Chlorobenzene | 56 | 20 | 14 |
| 8 | Ph3C+[B(C6F5)4]− (2.0 mol%) | 1,2-C6H4Cl2 | 61 | 19 | 9 |
| 9 | Ph3C+[B(C6F5)4]− (2.0 mol%) | Toluene-d8 | 65 | 21 | 13 |
| 10 | Et3Si+[CHB11H5Br6]− (2.0 mol%) | Toluene-d8 | 50 | 23 | 25 |
| 11 | [(C6H6)·H]+[CHB11H5Br6]− (2.0 mol%) | Toluene-d8 | 48 | 26 | 23 |
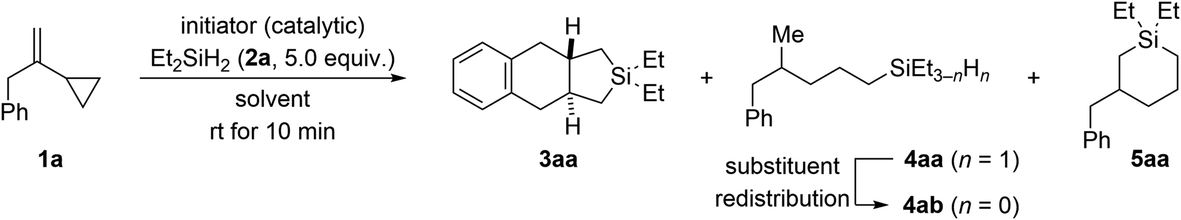
A diminished yield was observed at a lower loading of the dihydrosilane (2.0 instead of 5.0 equiv.; entry 3). Poor conversion was seen with less initiator; the product distribution was also affected unfavorably at 1.0 mol% catalyst loading (entry 4). More of Ph3C+[B(C6F5)4]− was without effect (entry 5). Likewise, the reaction outcome did not change at lower concentration (0.1 M instead of 0.2 M; entry 6). The solvent had a minor effect on the ratio of 3aa, 4ab, and 5aa (entries 7–9), and we eventually proceeded with toluene for the highest overall yield. In accordance with our mechanistic picture, both the counteranion-stabilized silylium ion Et3Si+[CHB11H5Br6]− (ref. 11) and the benzenium ion [(C6H6)·H]+[CHB11H5Br6]− (ref. 12) could also be used to initiate this cycloaddition, yet with more pronounced formation of undesired cyclic 5aa (entries 10 and 11).
With the optimized protocol in hand (Table 1, entry 9), we probed the substrate scope (Schemes 2–4). Electronic and steric effects of the substituent on the aryl group were examined with VCPs 1a–k (ortho and para, Scheme 2) and 1l–o (meta, Scheme 3). Parent VCP 1a afforded the cycloadduct 3aa in 54% isolated yield. Yields were moderate for VCPs with aryl rings bearing an electron-donating methyl or isopropyl group; the position of the substituent made no difference (1b–d → 3ba–da). VCP 1e decorated with a tert-butyl group did participate equally well but underwent predominant de-tert-butylation13 to mainly afford 3aa rather than 3ea. Similarly, Et3Si-substituted 1f suffered complete desilylation14 with none of 3fa being formed. Interestingly, the isolated yield of 80% for 3aa was significantly higher than that obtained for the parent system (54% for 1a → 3aa). We explain this with loss of a silylium ion instead of a proton, thus eliminating the aforementioned formation of the saturated byproducts 4aa and 4ab. This finding not only provides a solution of how to bypass unwanted alkene protonation but is also evidence for the intermediacy of Wheland complexes and as such a Friedel–Crafts-type mechanism. Halogen atoms were tolerated in this reaction but there was a clear difference between ortho- and para-substituted VCP derivatives (1g–k → 3ga–ka). Yields were moderate for the para- and low for the ortho-halogenated substrates (the preparation of the ortho-bromine-substituted VCP was unsuccessful). Both electronic and steric effects could be responsible for that. Besides, we also subjected VCP 6 with a thien-2-yl instead of the phenyl group to the procedure but did not obtain the cycloadduct; 6 was almost completely recovered (gray box, Scheme 2).
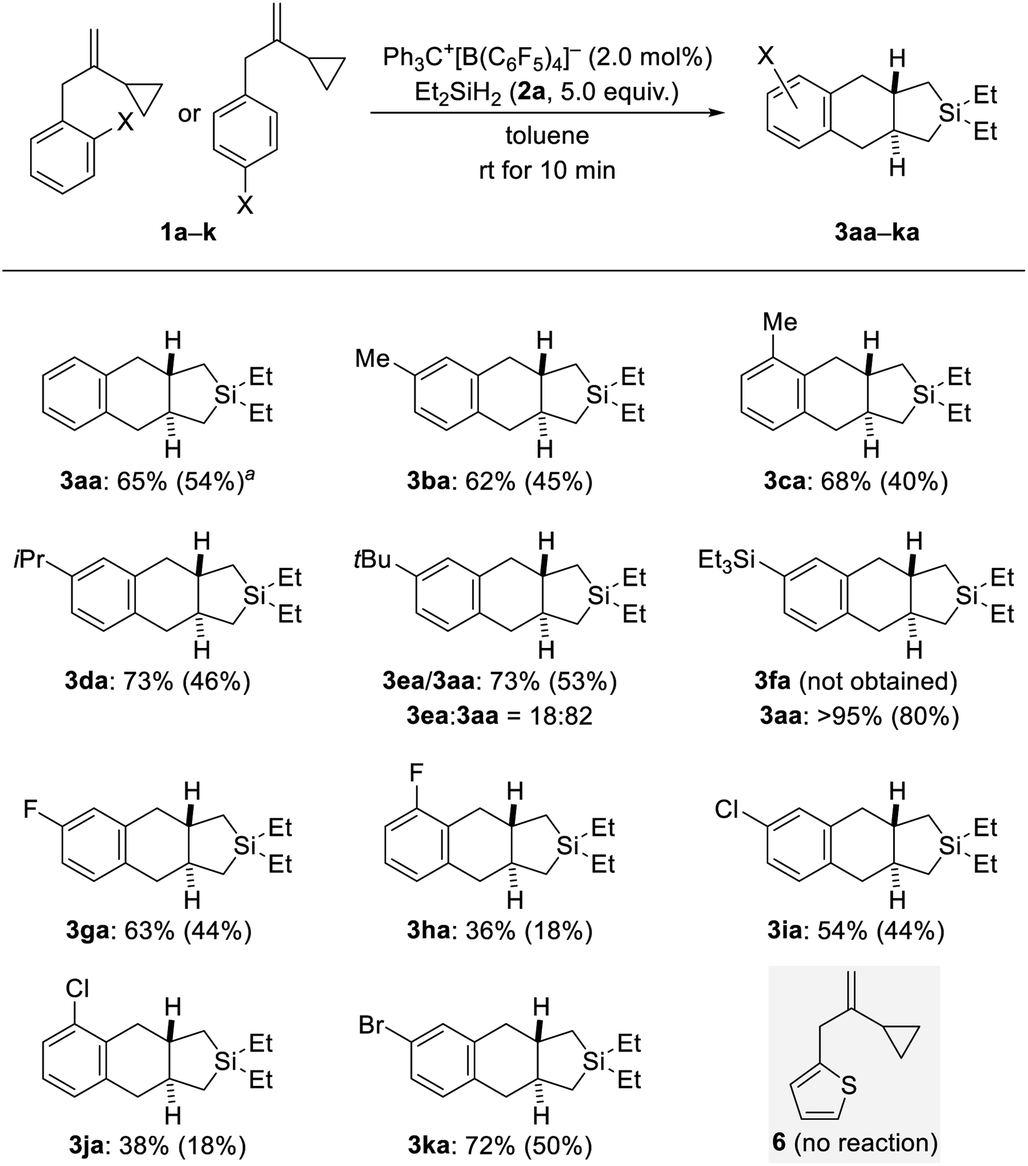 | ||
| Scheme 2 Scope I: variation of the ortho- and para-substituent of the benzyl group in VCPs 1. All reactions were performed on 0.30 mmol scale unless noted otherwise. Conversion was generally greater than 95% as estimated by 1H NMR spectroscopy using CH2Br2 as an internal standard. Yields were estimated by 1H NMR spectroscopy using CH2Br2 as an internal standard and tend to be too high because of the long relaxation time of CH2Br2; isolated yields in parentheses are of analytically pure material after flash chromatography on silica gel. a65% (48%) on a 1.2 mmol scale. | ||
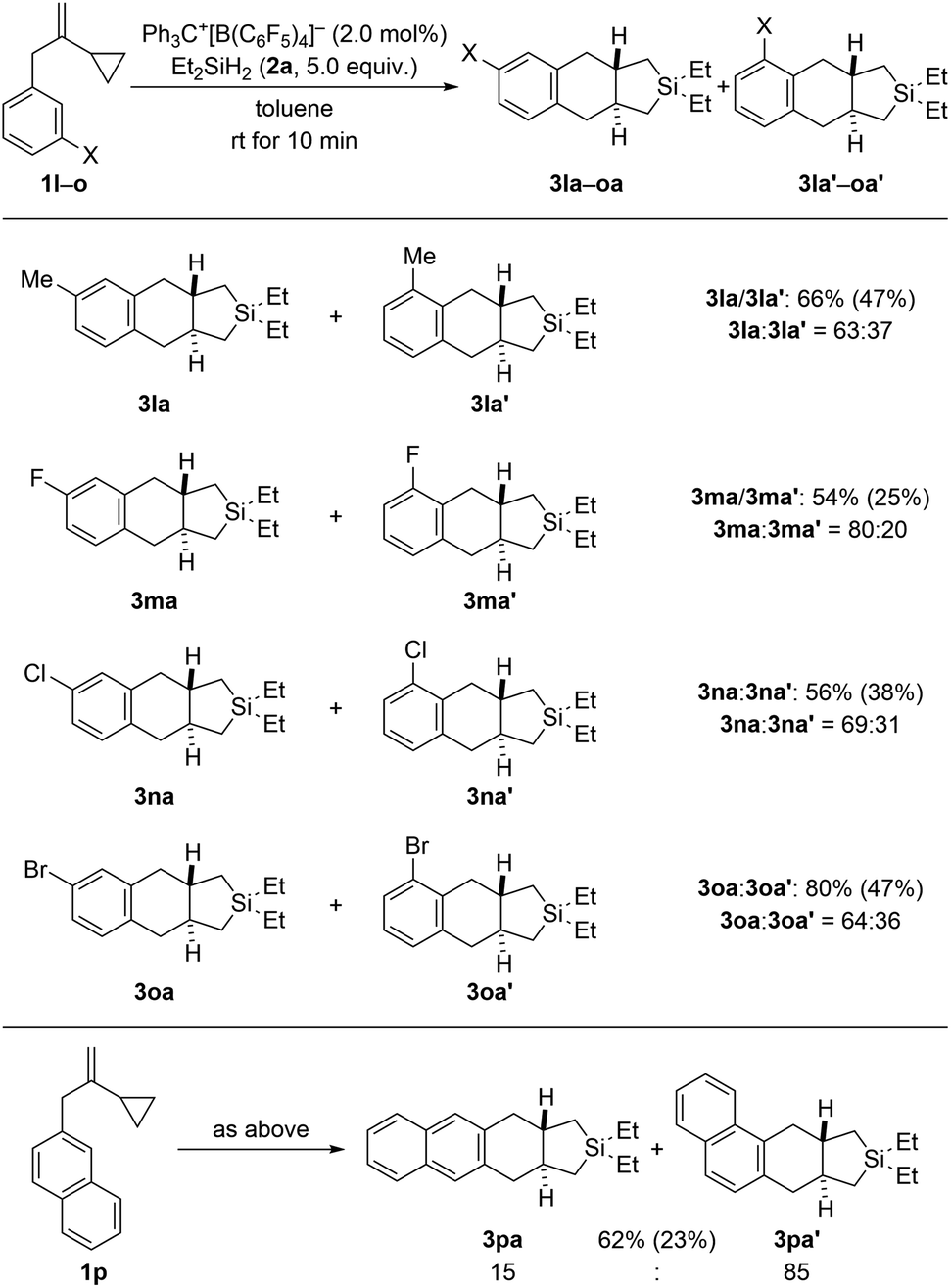 | ||
| Scheme 3 Scope II: variation of the meta-substituent of the benzyl group in VCPs 1. See caption of Scheme 2 for further details. Regio-isomeric ratios determined by 1H NMR spectroscopy after purification. | ||
Regioisomeric mixtures of the cycloadducts were generated when starting from representative VCPs with meta-substituted aryl groups (1l–o → 3la–oa/3la′–oa′, Scheme 3, top). Bond formation occurred preferentially in the less hindered ortho-position with regioisomeric ratios ranging from 63![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :37 to 80:20. Conversely, VCP 1p with a naphth-2-yl group yielded the kinetically favored, more hindered cycloadduct 3pa′ with good regiocontrol (Scheme 3, bottom). The cycloaddition of a bis(vinylcyclopropane) system was also tested (1q → 3qa/3qa′, Scheme 4, top); para-substituted 1q would first convert into a meta,para-disubstituted intermediate (not shown) which, in turn, would then give rise to a mixture of the regioisomers 3qa and 3qa′ (not shown) in the second cycloaddition event. A complex reaction mixture was experimentally found, and the separation by various chromatography methods failed. Part of the problem is that 3qa is formed as a mixture of racemic dl and meso diastereomers in approximately 50:50 ratio; the same likely applies to regioisomeric 3qa′ which we were not able to isolate (not shown). Slow evaporation of a solution of that regio- and diastereomeric mixture (four compounds; assuming only trans and no cis annulation) in ethyl acetate and cyclohexane (1:1) eventually led to the co-crystallization of bis-trans-isomers dl-3qa and meso-3qa. The meso compound is depicted in Scheme 4 (bottom; see the ESI† for details).15 The relative configuration of all annulation products was deduced from this molecular structure and assigned as trans, thereby confirming the result obtained from recorded and simulated 1H NMR spectra.
:37 to 80:20. Conversely, VCP 1p with a naphth-2-yl group yielded the kinetically favored, more hindered cycloadduct 3pa′ with good regiocontrol (Scheme 3, bottom). The cycloaddition of a bis(vinylcyclopropane) system was also tested (1q → 3qa/3qa′, Scheme 4, top); para-substituted 1q would first convert into a meta,para-disubstituted intermediate (not shown) which, in turn, would then give rise to a mixture of the regioisomers 3qa and 3qa′ (not shown) in the second cycloaddition event. A complex reaction mixture was experimentally found, and the separation by various chromatography methods failed. Part of the problem is that 3qa is formed as a mixture of racemic dl and meso diastereomers in approximately 50:50 ratio; the same likely applies to regioisomeric 3qa′ which we were not able to isolate (not shown). Slow evaporation of a solution of that regio- and diastereomeric mixture (four compounds; assuming only trans and no cis annulation) in ethyl acetate and cyclohexane (1:1) eventually led to the co-crystallization of bis-trans-isomers dl-3qa and meso-3qa. The meso compound is depicted in Scheme 4 (bottom; see the ESI† for details).15 The relative configuration of all annulation products was deduced from this molecular structure and assigned as trans, thereby confirming the result obtained from recorded and simulated 1H NMR spectra.
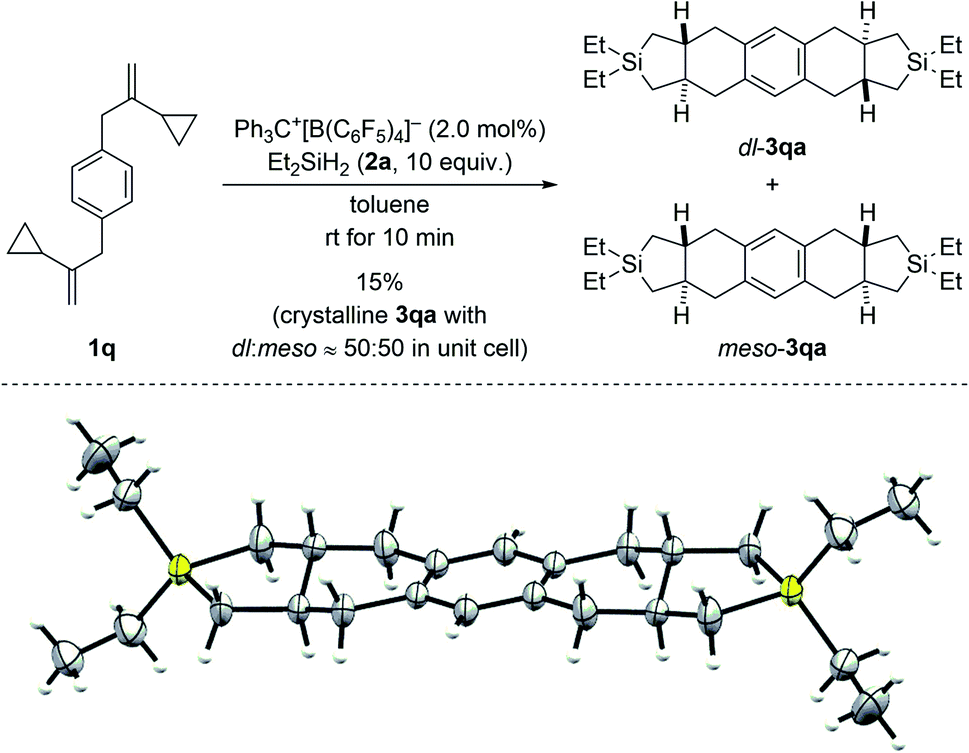 | ||
| Scheme 4 Scope III: reaction of a bis(VCP) system (top; see caption of Scheme 2 for further details) and molecular structure of the bis-trans-isomer meso-3qa (bottom; thermal ellipsoids are shown at the 50% probability level). | ||
In our previous study (see Scheme 1, top right),3 we had already shown that, apart from dihydrosilanes, monohydrosilanes can be used with similar success. The reason for this is the ability of silylium ions to cleave Si–C(sp3) as well as Si–C(sp2) bonds,16,17 corresponding to an exchange of alkyl or aryl group between two silicon centers.9 Hence, when there is no Si–H bond available (as in a quaternary silane) an alkyl or aryl group can be abstracted by another silylium ion (cf. the ESI† of ref. 3). We therefore tested a few tertiary hydrosilanes (2b–d) in the cycloaddition of model VCP 1a (Scheme 5). As expected, Et3SiH (2b) afforded the same product 3aa as obtained with Et2SiH2 (2a) albeit in lower yield. Demethylation was again preferred over cleavage of an ethyl group with EtMe2SiH (2c). Dearylation occurred when employing Me2PhSiH (2d). These observations are in line with those made earlier.3,16,17
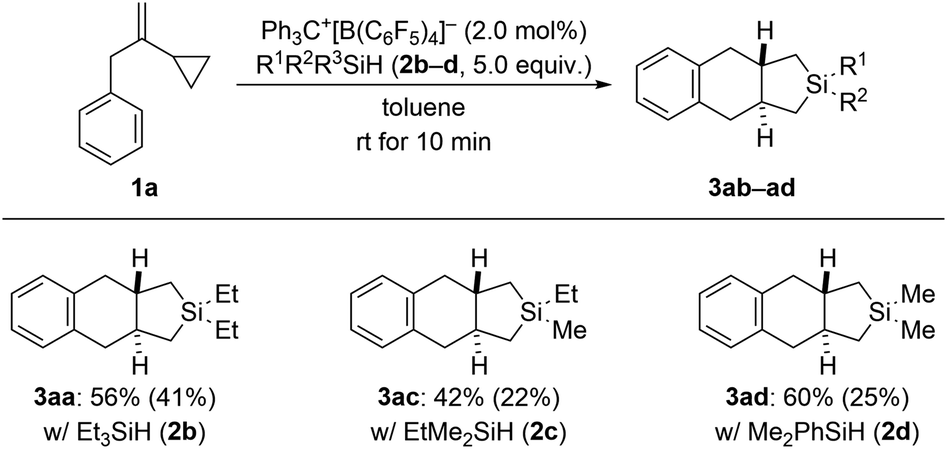 | ||
| Scheme 5 Scope IV: variation of the hydrosilane in the cycloaddition of VCP 1a. See caption of Scheme 2 for further details. | ||
The results with the phenyl-substituted VCPs3 and those with a benzyl substituent (see Scheme 1, bottom) underscore the structural richness that becomes accessible with this chemistry. We had already reported that the corresponding VCP with a cyclohexyl group led to a complex reaction mixture.3 However, going from a benzyl to a phenethyl group was successful, yielding yet another unknown silicon-containing ring system (7 → 8a, Scheme 6). The relative configuration was deduced from the 3JH,H coupling (10.6 Hz) in the 1H NMR spectrum of the highlighted H nuclei.
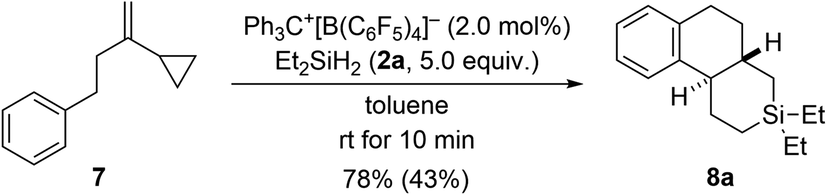 | ||
| Scheme 6 Scope V: going from benzyl to phenethyl in the VCP. See caption of Scheme 2 for further details. | ||
To gain insight into the reaction mechanism, we designed control experiments to distinguish between reaction pathways with the ring opening of the cyclopropyl group happening before or after manipulation of the alkene (Scheme 7). We showed before that silylium-ion-promoted ring-opening hydrosilylation of cyclopropanes is feasible1 but had excluded this possibility for phenyl-substituted VCPs3 in the (5 + 1) cycloaddition (see Scheme 1, top right). If hydrosilylation of the cyclopropyl group in VCPs 1 is the initial step, 2-substituted allylbenzene derivatives 9 are likely intermediates. When subjecting independently prepared 9ab to the standard procedure using Et3SiH (2b), an endo cyclization furnished product 5aa in high yield (top). With 5aa being formed as minor byproduct in the annulation reaction (cf.Table 1), we can state that ring opening preceding the functionalization of the alkene in the VCP is a competing pathway. In turn, potential intermediate 10aa2 arising from alkene hydrosilylation of VCP 1a and Et2SiH2 (2a) transformed cleanly into the cycloadduct 3aa upon treatment with trityl borate Ph3C+[B(C6F5)4]− in the absence of external hydrosilane (bottom). Hence, the opening of the cyclopropyl ring is interlinked with the intramolecular Friedel–Crafts-type bond formation at the aryl group18 and is downstream to the alkene hydrosilylation.
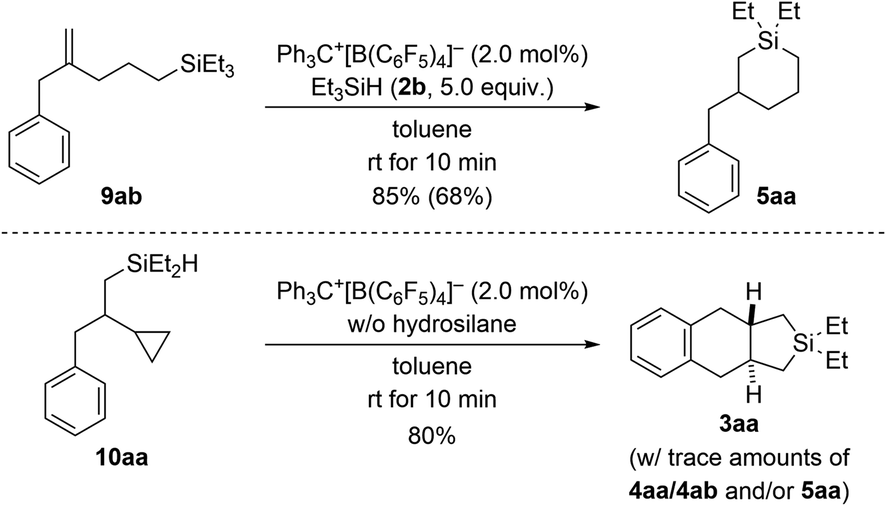 | ||
| Scheme 7 Control experiments I: verification of ring opening of the cyclopropyl group in VCPs prior to engagement of the alkene unit. See caption of Scheme 2 for further details. | ||
In another set of control experiments, we employed deuterium-labeled hydrosilane Et3SiD (2b-d1) and model VCP 1a-d1 with an ortho-C–D bond (Scheme 8). When 1a was reacted with 2b-d1, the deuterium label was exclusively incorporated into one of the angular positions of cycloadduct 3aa-d1 (top). To verify whether cleavage of the ortho-C–H bond is involved in the rate-determining step, 1a-d1 was subjected to the standard setup (bottom). However, H/D scrambling had occurred at all positions of the phenylene group in 3aa-d.19
 | ||
| Scheme 8 Control experiments II: deuterium-labeling of the hydrosilane or the VCP. See caption of Scheme 2 for further details. The deuteration grades were estimated by NMR spectroscopy. | ||
The rough mechanistic picture was refined by density functional theory (DFT) calculations at the M062X/cc-PVTZ//M062X/6-31G(d,p) level.20 Computations were performed on the model reaction of VCP 1a and Et2SiH2 (2a) with Ph3C+[B(C6F5)4]− as the initiator (Scheme 9; see the ESI for details and Fig. S85 and S86† for the free-energy profiles and the optimized structures). The solvent effect was taken into consideration using a polarizable continuum model (PCM)21 for both geometry optimizations and single-point energy calculations. Chlorobenzene was chosen to avoid the complexity of possible proton exchange between Wheland intermediates and toluene. The cycloadduct 3aa is obtained in moderate yield in chlorobenzene (Table 1, entry 7). We have previously shown that β-silicon-stabilized cyclopropylcarbinyl cation I, generated from the association of VCP 1a and the hydrogen-substituted silylium ion [Et2HSi(PhCl)]+, is much more stable than other donor-stabilized silylium ions such as the corresponding chlorobenzene-, hydrosilane-, or cyclopropane-stabilized systems.3 Unless otherwise noted, adduct I is considered the reference minimum for the estimation of the relative energy of intermediates or transition states calculated here.
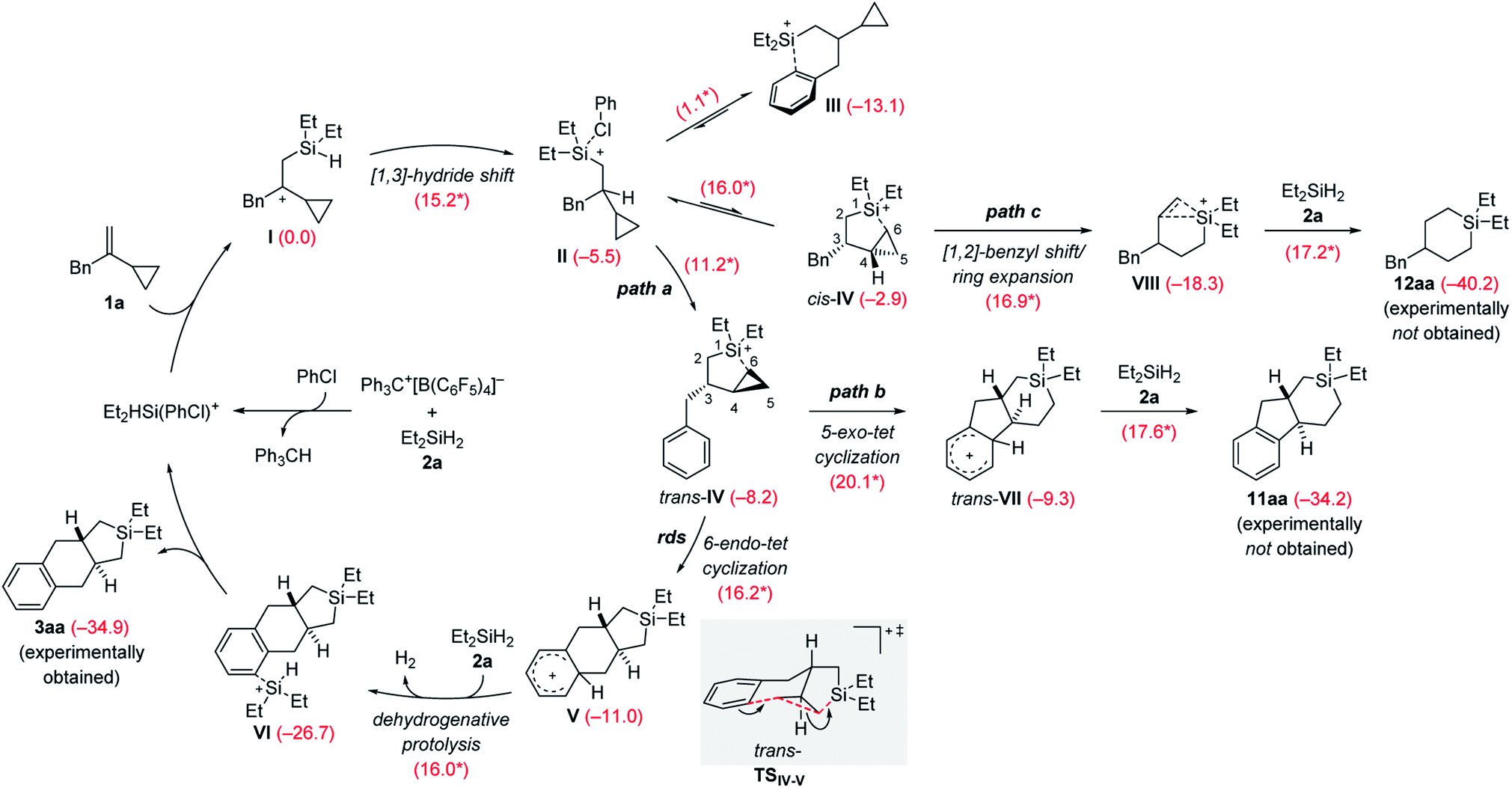 | ||
| Scheme 9 Catalytic cycle of the silylium-ion-promoted cycloaddition of VCP 1a and Et2SiH2 (2a). For each reaction step, the Gibbs free reaction energies and barriers (labeled with an asterisk) in kcal mol−1 were computed with the M06-2X functional (see the ESI† for details). rds = rate-determining step. | ||
The β-silylcarbenium ion I then undergoes an intramolecular [1,3]-hydride shift from the silicon atom to the benzylic carbon atom to arrive at the chlorobenzene-stabilized silylium ion II over a barrier of 15.2 kcal mol−1. We also calculated the potential intermolecular hydride transfer from Et2SiH2 (2a) to I but this pathway can be excluded because of an energetically higher transition state (19.0 versus 15.2 kcal mol−1, Fig. S84 in the ESI†). Subsequent reorganization can lead to several intramolecularly donor-stabilized silylium ions III (arene stabilization; see ref. 16b for a crystallographically characterized derivative) and cis-IV/trans-IV (cyclopropane stabilization with cis- or trans-configuration). In the case of benzyl-substituted VCPs, III is much more stable than either of the two cyclopropane-stabilized silylium ions IV. Conversion of III into cis-IV or trans-IV requires an activation barrier of 16.0 and 11.2 kcal mol−1, respectively. The trans-isomer is kinetically and thermodynamically more accessible than the cis-isomer. It is therefore trans-IV which engages in a subsequent intramolecular Friedel–Crafts alkylation reaction of a silylium-ion-activated cyclopropane ring.18
A 6-endo-tet ring closure by nucleophilic attack of the phenyl group at C5 with concomitant cleavage of the distal C5–C6 bond and formation of a bond between C6 and the silicon atom leads to tricyclic Wheland intermediate V (path a). This process through transition state trans-TSIV–V (gray box) is exergonic by −2.8 kcal mol−1 with an activation barrier of 16.2 kcal mol−1. It is also the rate-determining step, consistent with the rapid reaction rate at room temperature. The Brønsted acid V reacts with hydrosilane 2a by dehydrogenative protolysis17,22 to form the experimentally obtained product 3aaviaVI along with dihydrogen and the chlorobenzene-stabilized silylium-ion catalyst (ΔG‡ = 16.0 kcal mol−1). The overall Gibbs free energy change ΔG is −34.9 kcal mol−1 (calculated from the Gibbs energy difference between 3aa and 1a/2a). Based on transition state trans-TSIV–V, the predicted relative configuration of 3aa is trans, and is supported by 1H NMR spectroscopy (3JH,H = 11.7 Hz) and was eventually confirmed by X-ray diffraction (Scheme 4, bottom).
We considered other kinetically less favorable pathways such as a the 5-exo-tet cyclization (path b, cleavage of the proximal C4–C6 bond leading to 11aa) or [1,2]-benzyl shift/ring expansion (path c, leading to 12aa). Neither 11aa nor 12aa were observed experimentally. The route to 12aa corresponds to the previously reported (5 + 1) cycloaddition of phenyl-substituted VCPs3 (see Fig. S87–S90 in the ESI†). However, the annulation of a phenethyl-substituted VCP 7 to afford the 6/6/6-fused ring system 8a can be explained by a related 6-exo-tet cyclization corresponding to path b (see Schemes 6 and S12 in the ESI† for the computed pathway).
The Wheland intermediate V is the proton source in this reaction that can also be the starting point of side reactions. Instead of the above dehydrogenative protolysis of the hydrosilane over a barrier of 16.0 kcal mol−1 (V → VI, Scheme 9), this Brønsted acid can also transfer a proton to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of VCP 1a, releasing the product 3aa and the cyclopropylcarbinyl cation8IX (Scheme 10). The barrier of this protonation step is only 10.1 kcal mol−1 and is as such kinetically competitive. Subsequent hydride transfer from Et2SiH2 (2a) to IX gives a γ-silicon-stabilized carbenium ion X. This undergoes hydride abstraction from another molecule of 2a to yield open-chain product 4aa; the silylium ion released at the same time forms the β-silicon-stabilized cyclopropylcarbinyl cation I with VCP 1a. This ring-opening hydrosilylation of a cyclopropane promoted by a silylium ion is a known reaction and was recently studied by us in detail.1 With the low barriers involved, the formation of 4aa and eventually 4ab after rapid substituent redistribution9,10 cannot be avoided.
C double bond of VCP 1a, releasing the product 3aa and the cyclopropylcarbinyl cation8IX (Scheme 10). The barrier of this protonation step is only 10.1 kcal mol−1 and is as such kinetically competitive. Subsequent hydride transfer from Et2SiH2 (2a) to IX gives a γ-silicon-stabilized carbenium ion X. This undergoes hydride abstraction from another molecule of 2a to yield open-chain product 4aa; the silylium ion released at the same time forms the β-silicon-stabilized cyclopropylcarbinyl cation I with VCP 1a. This ring-opening hydrosilylation of a cyclopropane promoted by a silylium ion is a known reaction and was recently studied by us in detail.1 With the low barriers involved, the formation of 4aa and eventually 4ab after rapid substituent redistribution9,10 cannot be avoided.
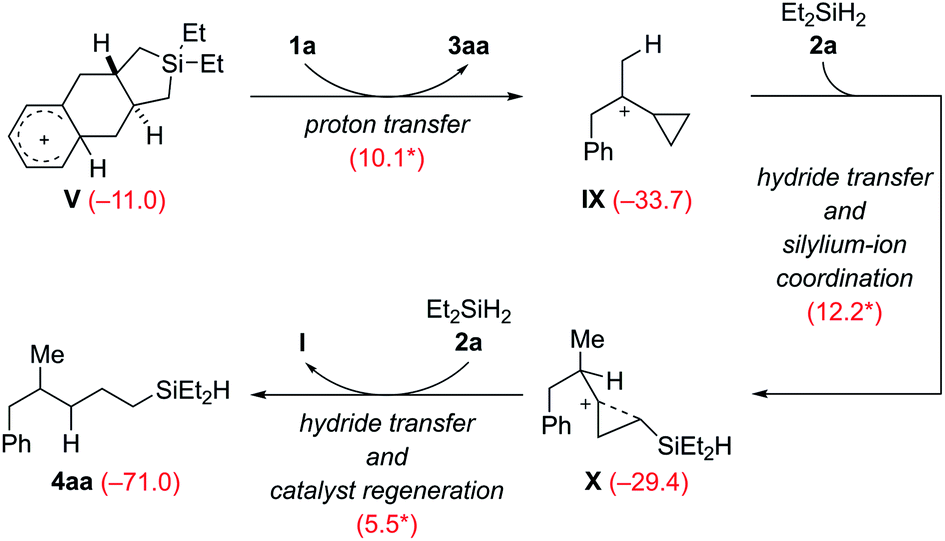 | ||
| Scheme 10 Computed pathway of the formation of byproduct 4aa, precursor of 4ab (see discussion of Table 1). | ||
Conclusion
The present work showcases that reactions of VCPs with different electrophilic silicon reagents do lead to drastically different outcomes (cf.Scheme 1). The substituent on the VCP and the choice of the silicon electrophile together with the counteranion of the β-silicon-stabilized cyclopropyl carbocation intermediates decide their fate. With silylium-ion-like reagents, benzyl- and phenethyl-substituted VCPs lead to silicon-containing 6/6/5- and 6/6/6-fused ring systems, respectively, both of which previously unknown motifs. The corresponding phenyl-substituted VCPs engage in a (5 + 1) cycloaddition under otherwise identical reaction conditions.3 The annulation sequence is initiated by the trityl cation and then maintained by self-regeneration of the silylium-ion reagent.5a Two Si–C(sp3) bonds and one C(sp2)–C(sp3) bond are formed with an intramolecular Friedel–Crafts alkylation of a silylium-ion-activated cyclopropane ring as the key step (6-endo-tet for 6/6/5 system starting from benzyl-substituted VCP and 6-exo-tet for 6/6/6 system starting from phenethyl-substituted VCP). Given the high number of possible reaction pathways, the chemoselectivity and preference for one product with any of the substrates is remarkable.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy (EXC 2008/1-390540038). G. W. and P.-W. L. thank the China Scholarship Council for a postdoctoral fellowship (2019–2020) and a predoctoral fellowship (2019–2023), respectively. All theoretical calculations were performed at the High Performance Computing Center (HPCC) of Nanjing University. M. O. is indebted to the Einstein Foundation Berlin for an endowed professorship.Notes and references
- A. Roy, V. Bonetti, G. Wang, Q. Wu, H. F. T. Klare and M. Oestreich, Org. Lett., 2020, 22, 1213–1216 CrossRef CAS.
- P.-W. Long, T. He and M. Oestreich, Org. Lett., 2020, 22, 7383–7386 CrossRef CAS.
- T. He, G. Wang, V. Bonetti, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2020, 59, 12186–12191 CrossRef CAS.
- (a) D. Weber and M. R. Gagné, in Organosilicon Chemistry: Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 33–85 Search PubMed; (b) M. Oestreich, J. Hermeke and J. Mohr, Chem. Soc. Rev., 2015, 44, 2202–2220 RSC.
- (a) J. C. L. Walker, H. F. T. Klare and M. Oestreich, Nat. Rev. Chem., 2020, 4, 54–62 CrossRef CAS; (b) P. Shaykhutdinova, S. Keess and M. Oestreich, in Organosilicon Chemistry: Novel Approaches and Reactions, ed. T. Hiyama and M. Oestreich, Wiley-VCH, Weinheim, 2019, pp. 131–170 Search PubMed; (c) V. Y. Lee and A. Sekiguchi, in Organosilicon Compounds, ed. V. Y. Lee, Academic Press, Oxford, 2017, vol. 1, pp. 197–230 Search PubMed.
- (a) J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Chem. Rev., DOI:10.1021/acs.chemrev.0c00160; (b) M. Meazza, H. Guo and R. Rios, Org. Biomol. Chem., 2017, 15, 2479–2490 RSC; (c) V. Ganesh and S. Chandrasekaran, Synthesis, 2016, 48, 4347–4380 CrossRef CAS; (d) L. Jiao and Z.-X. Yu, J. Org. Chem., 2013, 78, 6842–6848 CrossRef CAS.
- (a) J. B. Lambert, Y. Zhao, R. W. Emblidge, L. A. Salvador, X. Liu, J.-H. So and E. C. Chelius, Acc. Chem. Res., 1999, 32, 183–190; CrossRef CASfor a summary of the chemistry of silyl-substituted carbocations, see: (b) H.-U. Siehl and T. Müller, in The Chemistry of Organic Silicon Compounds, ed. Z. Rappoport and Y. Apeloig, Wiley, Chichester, 1989, part 2, pp. 595–701 Search PubMed.
- For a review, see: (a) Z. Goldschmidt and B. Crammer, Chem. Soc. Rev., 1988, 17, 229–267; RSCfor original work, see: (b) G. A. Olah, D. P. Kelly, C. L. Jeuell and R. D. Porter, J. Am. Chem. Soc., 1970, 92, 2544–2546 CrossRef CAS; (c) G. A. Olah, C. L. Jeuell, D. P. Kelly and R. D. Porter, J. Am. Chem. Soc., 1972, 94, 146–156 CrossRef CAS; (d) G. A. Olah and P. W. Westerman, J. Am. Chem. Soc., 1973, 95, 7530–7531 CrossRef CAS; (e) J. F. Wolf, P. G. Harch, R. W. Taft and W. J. Hehre, J. Am. Chem. Soc., 1975, 97, 2902–2904 CrossRef CAS; (f) H. Mayr and G. A. Olah, J. Am. Chem. Soc., 1977, 99, 510–513 CrossRef CAS; (g) K. B. Wiberg, D. Shobe and G. L. Nelson, J. Am. Chem. Soc., 1993, 115, 10645–10652 CrossRef CAS.
- (a) A. Schäfer, M. Reißmann, A. Schäfer, W. Saak, D. Haase and T. Müller, Angew. Chem., Int. Ed., 2011, 50, 12636–12638 CrossRef; (b) A. Schäfer, M. Reißmann, S. Jung, A. Schäfer, W. Saak, E. Brendler and T. Müller, Organometallics, 2013, 32, 4713–4722 CrossRef; (c) R. Labbow, F. Reiß, A. Schulz and A. Villinger, Organometallics, 2014, 33, 3223–3226 CrossRef CAS; (d) L. Omann, B. Pudasaini, E. Irran, H. F. T. Klare, M.-H. Baik and M. Oestreich, Chem. Sci., 2018, 9, 5600–5607 RSC.
- The formation of 4ab with an SiEt3 group rather than expected 4aa with an SiEt2H group can be explained by a known substituent redistribution.9 This was confirmed by treatment of an independently prepared sample of 4aa with 2.0 mol% of Ph3C+[B(C6F5)4]− at room temperature. Within ten minutes, silane 4ab (n = 0, 25%) was found together with monohydrosilane 4aa (n = 1, 54%) and the cognate dihydrosilane (n = 2, 21%; not shown). See the ESI† for details.
- Z. Xie, R. Bau, A. Benesi and C. A. Reed, Organometallics, 1995, 14, 3933–3941 CrossRef CAS.
- (a) C. A. Reed, N. L. P. Fackler, K.-C. Kim, D. Stasko, D. R. Evans, P. D. W. Boyd and C. E. F. Rickard, J. Am. Chem. Soc., 1999, 121, 6314–6315 CrossRef CAS; (b) D. Stasko and C. A. Reed, J. Am. Chem. Soc., 2002, 124, 1148–1149 CrossRef CAS; (c) C. A. Reed, K.-C. Kim, E. S. Stoyanov, D. Stasko, F. S. Tham, L. J. Mueller and P. D. W. Boyd, J. Am. Chem. Soc., 2003, 125, 1796–1804 CrossRef CAS.
- S. A. Saleh and H. L. Tashtoush, Tetrahedron, 1998, 54, 14157–14177 CrossRef CAS and cited references.
- (a) T. H. Chan and I. Fleming, Synthesis, 1979, 761–786 CrossRef CAS; (b) C. Eaborn, Pure Appl. Chem., 1969, 19, 375–388; Search PubMedsee also (c) S. Bähr and M. Oestreich, Angew. Chem., Int. Ed., 2017, 56, 52–59 CrossRef and cited references.
- CCDC 2034954 (3qa) contains the ESI crystallographic data for this paper.†.
- (a) Q. Wu, Z.-W. Qu, L. Omann, E. Irran, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2018, 57, 9176–9179 CrossRef CAS; (b) Q. Wu, A. Roy, E. Irran, Z.-W. Qu, S. Grimme, H. F. T. Klare and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 17307–17311 CrossRef CAS.
- Q. Wu, E. Irran, R. Muller, M. Kaupp, H. F. T. Klare and M. Oestreich, Science, 2019, 365, 168–172 CrossRef CAS.
- To the best of our knowledge, there is one remotely related example of a cyclopropane ring involved in an intramolecular Friedel–Crafts alkylation: V. Lanke, F.-G. Zhang, A. Kaushansky and I. Marek, Chem. Sci., 2019, 10, 9548–9554 RSC.
- F. Cacace, M. E. Crestoni and S. Fornarini, J. Am. Chem. Soc., 1992, 114, 6776–6784 CrossRef CAS.
- (a) Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed; (b) Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS.
- Q.-A. Chen, H. F. T. Klare and M. Oestreich, J. Am. Chem. Soc., 2016, 138, 7868–7871 CrossRef CAS and cited references.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization, crystallographic and computational data. CCDC 2034954. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05553k |
| This journal is © The Royal Society of Chemistry 2021 |