
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe development of biomass-derived carbon-based photocatalysts for the visible-light-driven photodegradation of pollutants: a comprehensive review
Bui Thanh Son ,
Nguyen Viet Long* and
Nguyen Thi Nhat Hang
,
Nguyen Viet Long* and
Nguyen Thi Nhat Hang
Nanotechnology, Thu Dau Mot University, Binh Duong Province, Vietnam. E-mail: nguyenvietlong@tdmu.edu.vn
First published on 14th September 2021
Abstract
Biomass-derived carbonaceous materials have recently attracted extensive interest on account of their exceptional physicochemical properties which make them promising candidates for various critical applications. Several achieved advances have been reported in the recent literature, mainly focusing on the areas of energy storage and conversion. There is no review dedicated specifically to the potential applications of biomass-derived carbon-based photocatalytic materials for environmental remediation using the visible spectral region. The excellent characteristics of carbon materials, such as good electronic conductivity, unique nanocrystal structures, inherent hydrophobicity, and the tunable surface characteristics, are fully compatible with diverse catalytic reactions including organic transformations and photocatalysis processes. Importantly, biomass-carbon-based materials are considered to be green and viable alternative photocatalysts due to their environmentally friendly and naturally abundant nature. This work aims to provide a comprehensive review of recent advances relating to the synthesis of biomass-derived carbon-based photocatalysts, focusing on their potential for the photodegradation of various pollutants. First, potential natural biomass sources, various synthetic routes, and the properties of carbon materials are systematically discussed. Recent advances in the production of biomass-carbon-based photocatalysts (including material design, mechanisms, and photocatalytic performance) are highlighted. Regarding ideas for the development of new biomass-derived photocatalysts, we outline research gaps that are worthy of further research in the future.
1. Introduction
Water quality is one of the huge challenges that the globe will face during the 21st century. Even though up to 70% of the Earth's surface is covered with water, only 2.5% of that is available for agricultural, industrial, and domestic use.1,2 In fact, the widespread disposal of untreated or insufficiently treated wastewater into streams and the presence of newly emerging pollutants, comprising pharmaceuticals, pesticides, and industrial chemicals, exacerbate the pollution of freshwater resources. For instance, the concentrations of ibuprofen and naproxen in natural water and wastewater are reported to range from hundreds of ng L−1 to tens of μg L−1.3 Consequently, the use of untreated or insufficiently treated water presents numerous serious risks to human beings, since soluble contaminants are able to enter the human body through food, where their accumulation can directly cause health problems such as diarrhea, renal failure, and cancer.4–6 Therefore, effectively eliminating contaminants from wastewater has become one of the most urgent issues in recent decades. Numerous attempts have been made to solve this matter, such as the use of biodegradation,7 coagulation and flocculation,8,9 ozonation,9 and membrane filtration10 techniques. However, the several obvious issues preventing the widespread use of these techniques include toxic by-product formation and high operational and maintenance costs.11,12First discovered by Fujishima and Honda in 1972, the photocatalytic splitting of water molecules can occur on the surface of a titanium dioxide (TiO2) electrode under ultraviolet (UV) light irradiation.13 This pioneering work truly inspired a new era of photocatalysis process. In recent decades, photocatalysis has emerged as a low-cost, efficient, and environmentally friendly technique to remove organic compounds from wastewater.6 Photocatalysis is defined as a photo-activated chemical reaction occurring on a semiconductor surface. The mechanism of the photocatalytic activities on semiconductor's surface under light irradiation is illustrated in Fig. 1.
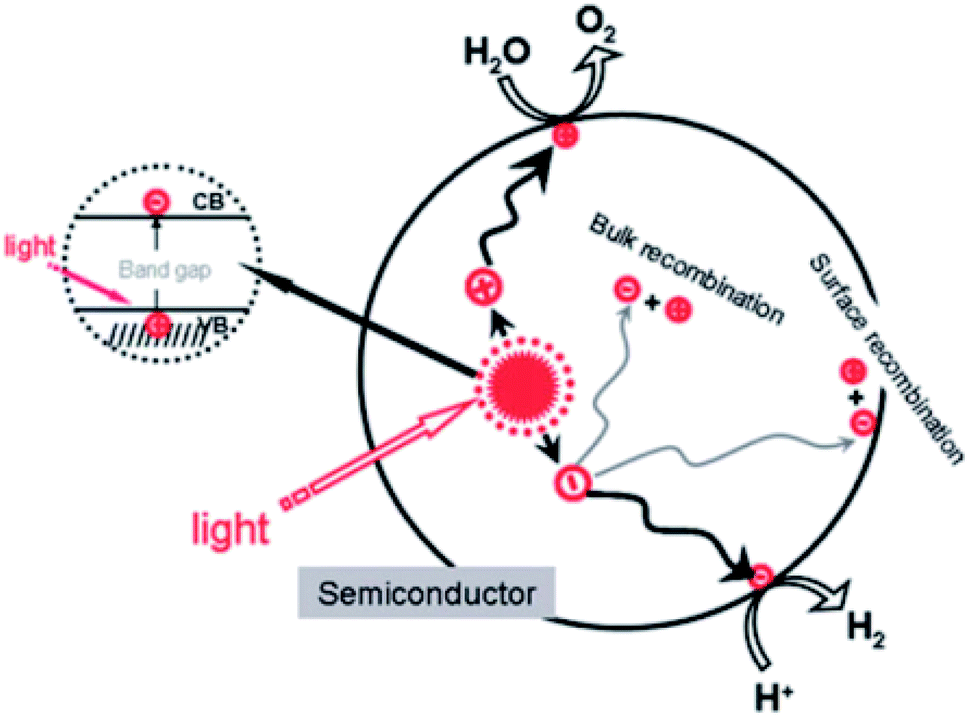 | ||
| Fig. 1 The mechanism for the photocatalysis. This figure has been reproduced from ref. 14 with permission from the American Chemical Society, copyright: 2010. | ||
When a semiconductor is illuminated by a suitable light source, this given extra energy allows electrons from the valance band of the semiconductor to easily become excited and migrate to the conduction band (e−CB), leaving free holes (h+VB) in the valance band. A “suitable” light source means one in which the energy of the light source is at least equal (or higher than) the band-gap energy of the semiconductor (see eqn (1)).15 At least two reactions will then occur simultaneously: the first oxidation reaction is derived from positive holes and the second reaction involves reduction caused by negative photogenerated electrons (see eqn (2) and (3)).16
 | (1) |
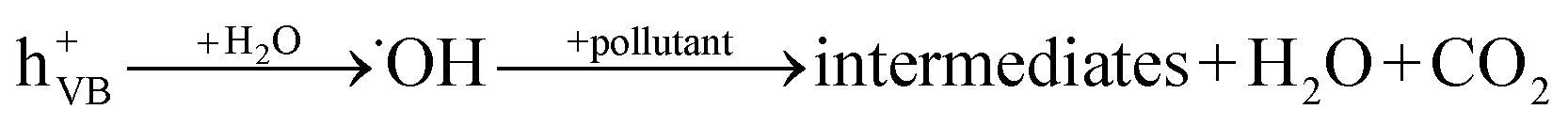 | (2) |
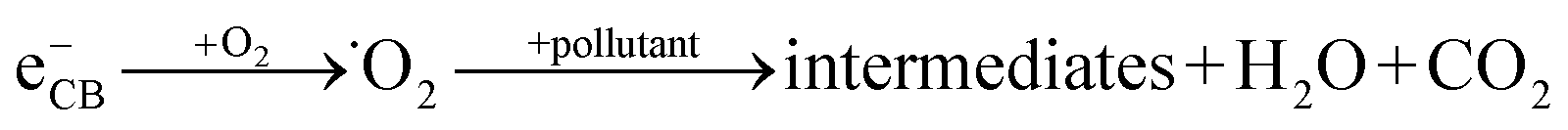 | (3) |
Several traditional semiconductors that have been widely applied include titanium dioxide (TiO2), zinc oxide (ZnO), iron oxide (Fe2O3), tungsten trioxide (WO3), vanadium oxide (V2O5), and zirconia (ZrO2).2,17–19 However, most semiconductors have a large band-gap energy (Eg), for example TiO2 (3.2 eV)19 and ZnO (3.37 eV),20 which means that they only work effectively under ultraviolet light. Additionally, the quick recombination of charge carriers can hinder the photocatalytic activities of these materials.
To address these drawbacks, one effective and innovative technique used recently is doping and constructing heterojunctions with biomass-derived carbon materials.21,22 Carbon-based nanostructured materials possess a high degree of porosity, tunable structures, large BET surface areas, high charge carrier mobility, good recycling characteristics, and high stability, making them appropriate support frames for catalysts.23 More importantly, the utilization of biomass waste as a carbon-rich precursor for producing activated carbon is becoming much more attractive, as it can be considered a “green” solution, overcoming serious issues related to waste disposal and lowering raw materials costs. Thus, biomass-carbon-based materials have been widely studied in recent years for energy storage and conversion purposes.24,25 Unfortunately, studies on synthesizing biomass-carbon-based photocatalysts for the photodegradation of pollutants are still in the early stage and examples are reported quite rarely.
This work has been aimed at providing a comprehensive overview of recent advances in the synthesis of biomass-derived carbon-based photocatalysts in light of their potential for the photodegradation of emerging pollutants. The specific objectives of this review article are as follows: (1) determining the chemical and elemental compositions of potential natural biomass sources; (2) comparing the various synthetic routes for biomass-based carbon materials; (3) discussing changes in the physiochemical properties of carbon materials in response to different synthesis conditions; and (4) summarizing and reviewing recent advances relating to the application of biomass-derived carbon-based photocatalysts for the removal of pollutants using the visible spectral region.
2. Natural-biomass-derived carbon materials: sources and classification
In nature, “biomass” refers to materials originating from animals, plants, and daily waste, which are proven to be rich in various elements such as carbon, hydrogen, oxygen, and nitrogen. Recently, natural-biomass-based materials have been widely studied and utilized as sustainable carbon-rich precursors for preparing carbon-based nanomaterials on account of possessing several typical attractive properties, such as abundant availability (low-cost), environmental friendliness, unique nanoporous structures (large surface areas), a wide range of morphologies, and mechanical stability.26,27 Biomass provides carbon-rich sources, and the microstructural characteristics and elemental compositions of as-synthesized materials vary depending on the natural biomass source used. Undoubtedly, the composition and structure of a carbon material depend on the elemental and chemical composition of the corresponding precursor. Therefore, natural biomass precursors play a key role in directly influencing the final yields and structural characteristics of carbon materials.In the field of environmental remediation, the development of carbon materials possessing unique properties, such as high surface areas, good stability/recyclability, and improved charge carrier propagation, has remained a hot topic in recent years. In that context, enhancing the production rates of aromatic carbon and enhancing carbon yields following thermal carbonization processes are key matters, and these can partly be achieved via selecting an ideal biomass precursor. It is imperative to note that a high oxygen content in the initial precursor can lead to more defects and lower crystallinity in the final structure, thus hindering the formation of aromatic carbon. Meanwhile, via releasing volatile compounds, high levels of non-crosslinked and aliphatic compounds can retard fusion and flow processes, thereby inhibiting the final carbon yields. Thus, potential biomass precursors have to possess simultaneously high carbon content levels, high content levels of highly crosslinked molecules with a unique nanoporous structure, and high elemental content levels of nitrogen, which are favorable for the in situ formation of nitrogen-modified carbon with remarkably improved conductivity.25 Natural biomass precursors can be classified into three groups: plant-derived precursors, animal-remains-derived precursors, and microorganism-derived precursors, as illustrated in Fig. 2.
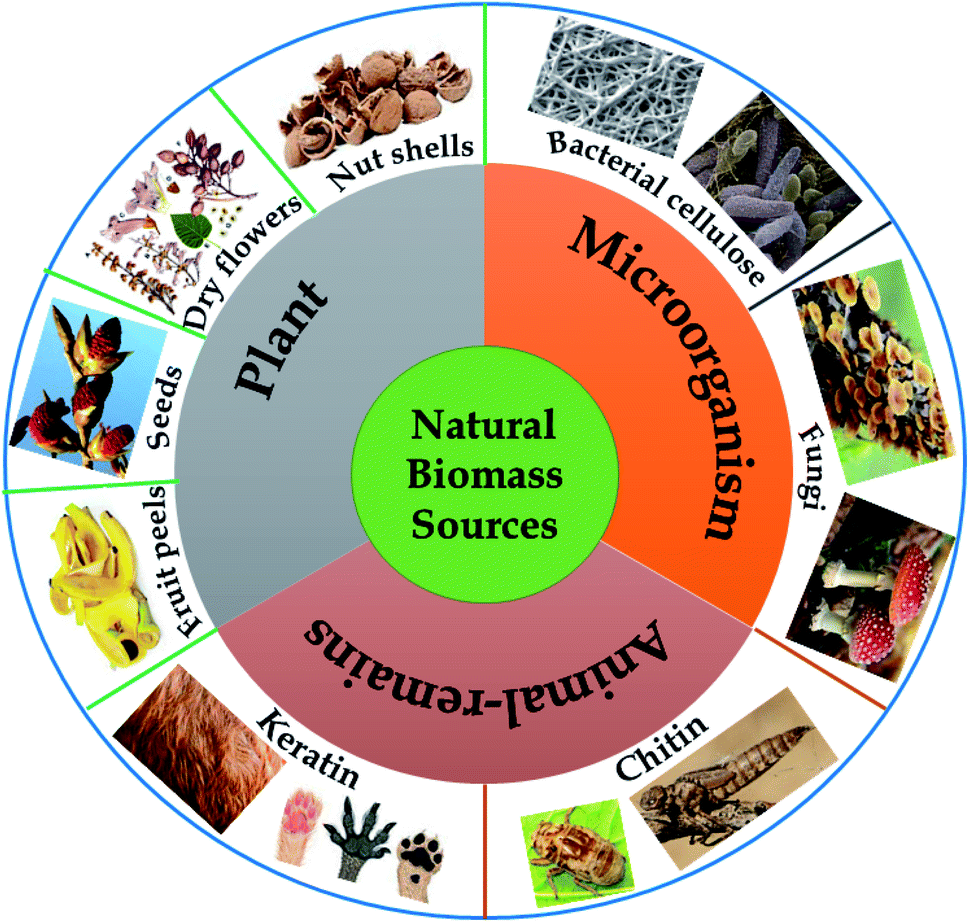 | ||
| Fig. 2 Natural biomass sources. | ||
2.1. Plant-derived precursors
The most common precursors come from the category of plant-derived biomass, and the quantitative chemical characteristics vary depending on the species and the part used. This group majorly involves lignin, hemicellulose, and cellulose. Accordingly, the chemical constituents and structures of lignin, hemicellulose, and cellulose are illustrated in Fig. 3. Bilba et al.28 found that 46.48% of the weight of dried coconut coir is lignin, while cellulose and hemicellulose only account for 21.46% and 12.36%, respectively. However, in dried hemp, cellulose makes up 67% of the weight, whilst lignin only makes up 3.3% of total weight.29 Several types of plant-derived biomass, such as Scots pine branches and Scots pine stumps (approximately 32% and 28%, respectively), show remarkable hemicellulose fractions.25,30,31 The main chemical compositions of various plant-based biomass sources are summarized in Table 1.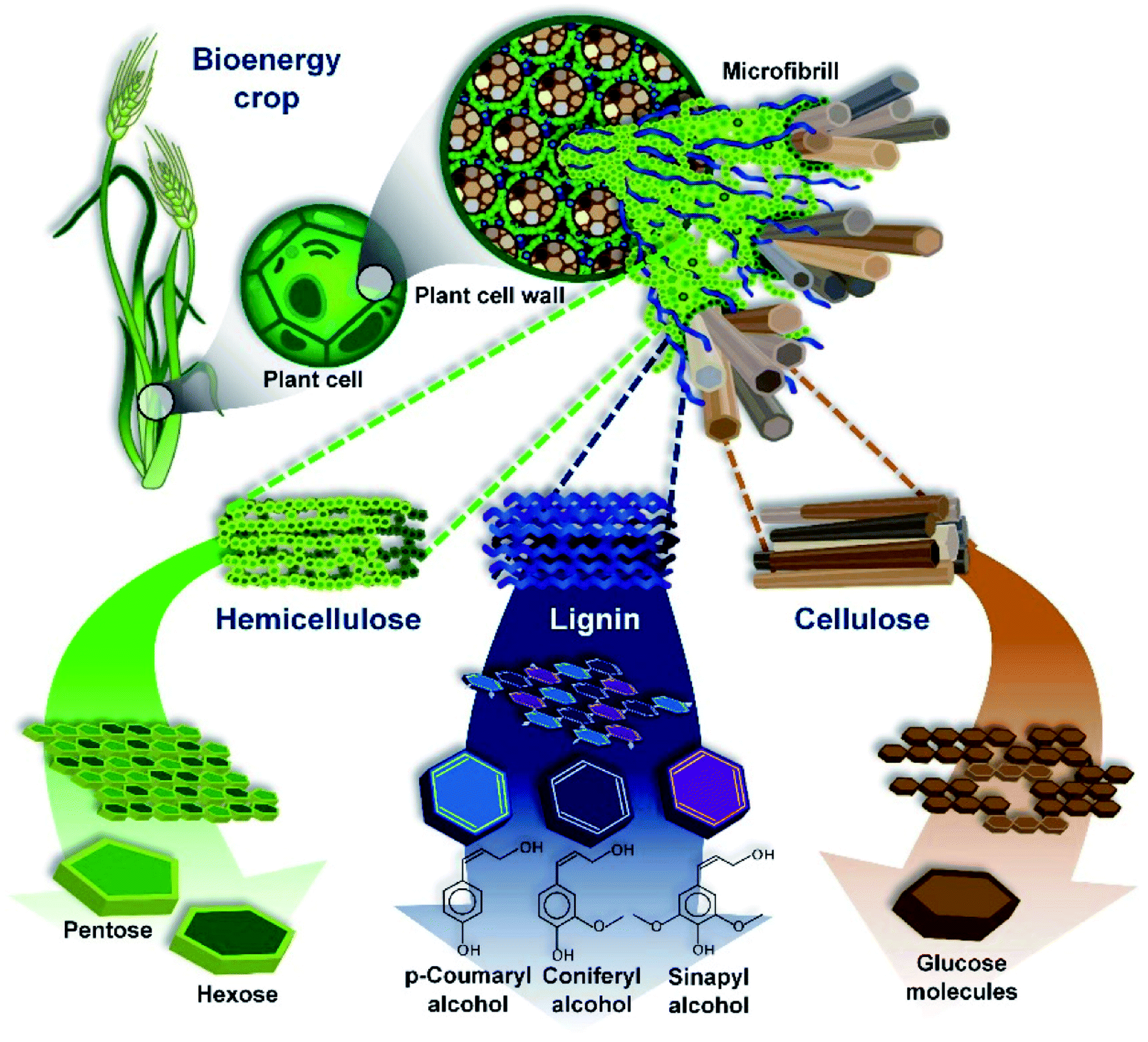 | ||
| Fig. 3 An overview of the structure of lignocellulosic biomass. This figure has been reproduced from ref. 38 with permission from MDPI, copyright: 2010. | ||
| Biomass | Cellulose (%) | Hemicellulose (%) | Lignin (%) | Ref. |
|---|---|---|---|---|
| Corn stalks | 35–37 | 24–26 | 18–20 | 32 |
| Corn cobs | 34–36 | 36–38 | 9–11 | 32 |
| Corn stover | 35–37 | 28–30 | 18–20 | 32 |
| Wheat straw | 34–36 | 28–30 | 15–17 | 32 |
| Rice straw | 34–36 | 25–27 | 7–9 | 32 |
| Barley straw | 33–40 | 20–35 | 8–17 | 33 |
| Switchgrass | 36–38 | 26–28 | 17–19 | 32 |
| Scots pine branches | 32.0 | 32.0 | 21.5 | 30 |
| Scots pine bark | 22.2 | 8.1 | 13.1 | 30 |
| Scots pine stem wood | 40.7 | 26.9 | 27.0 | 30 |
| Scots pine roots | 28.6 | 18.9 | 29.8 | 25 |
| Scots pine stumps | 36.4 | 28.2 | 19.5 | 25 |
| Sunflower seed hulls | 31.3 | 25.2 | 28.7 | 34 |
| Willow leaves | 18.5 | 14.7 | 20.0 | 35 |
| Poplar leaves | 22.3 | 12.8 | 23.2 | 35 |
| Coconut coir | 21.46 | 12.36 | 46.48 | 28 |
| Banana leaves | 25.65 | 17.04 | 24.84 | 28 |
| Hemp | 67.0 | — | 3.3 | 29 |
Among the aforementioned groups, lignin is the most stable during thermal decomposition and it consequently contributes considerably to the final carbon yield. In addition, several obvious interactions between lignin and cellulose at 550 °C have been observed.36,37 Thus, to be appropriate for environmental remediation applications, apart from having a high nitrogen content and low oxygen level, an ideal biomass precursor should contain significant amounts of lignin and an insignificant cellulose fraction to enhance the conductivity, controllable defects, and degree of graphitization.24
Dry flowers contain organic components such as saccharides, glucosides, vitamins, and proteins. Wei et al.39 utilized a mixture of camellia petals (as a carbon precursor) and ammonium persulfate (as a dopant precursor) to fabricate S/N co-doped porous carbon nanosheets with a remarkably improved BET surface area, higher than 1122 m2 g−1. In 2015, Chang et al.40 used paulownia flowers to successfully prepare nanostructured carbon materials. Paulownia is a deciduous tree commonly planted in many areas of China, and its biomass possesses a unique porous texture and high levels of various organic compounds. In comparison to the other organs, the leaves contain a great deal of saccharides and proteins, as well as intrinsic oxide- and nitrogen-based surface functional groups. Zhu et al.41 recently successfully synthesized three types of carbon materials, through pyrolysis and subsequent KOH-, ZnCl2-, and H3BO3-based activation, from dead ginkgo leaves from a typical Chinese ornamental plant. In 2019, bamboo leaves, as an abundant waste source, were converted into a carbon/copper oxide/cuprous oxide (CuOx@C) nanocomposite with unique hierarchical structure.42
Other typical plant-derived precursors that need to be mentioned here are seeds. As seeds possess the ideal morphology of hollow and thin-walled tubes, they can be utilized as a potential precursor to fabricate active carbon for environmental applications. Recently, Su et al.43 used poplar catkin biowaste as the starting material to fabricate a hollow activated carbon nanomesh with a hierarchical porous structure and a very high BET surface area (1893.0 m2 g−1) and total pore volume (1.495 cm3 g−1). Similar to other organs, fruit peel is an abundant waste source, and most of it is discarded or even burned, directly contributing to environmental issues. Fruit peel mainly consists of a great deal of fiber and saccharides, making it ideal for fabricating activated carbon materials. In 2018, Yang et al.44 utilized dried banana peel to synthesize MnO2/biomass-derived porous carbon (BPC) composites through a hydrothermal method. Interestingly, the authors found that the unique hierarchical naturally porous structure of dried banana peel can be maintained during the fabrication process, providing sufficient space for the growth of MnO2 and alleviating the agglomeration of MnO2 particles. In addition, Xiao et al.45 successfully synthesized hierarchical porous carbon materials from peanut shells with a large BET surface area (1549 m2 g−1).
2.2. Microorganism-derived precursors
In recent years, utilizing microorganism-derived biomass as a carbon-rich precursor source has opened up a new avenue for fabricating activated carbon materials. Microorganism-based precursors mainly consist of carbohydrates, proteins, fiber, and fat. For example, more than 95% of the weight of dried Lentinula edodes consists of carbohydrates (30.2%), crude protein (17.1%), crude fiber (39.4%), and fat (1.9%).46 Similarly, these components occupy more than 90% of the dry weights of numerous species, such as Agaricus bisporus (92%),47 B. aereus (91.5%),48 Tricholoma portentosum (90.1%),49 and Lactarius deliciosus (92.5%).50 Among these components, carbohydrates mainly contribute to the carbon yield as they naturally contain plenty of chitin, which can easily interact and then act as a main carbon source during the thermal carbonization process. In contrast, proteins, fibers, and fat provide insignificant carbon yields since these constituents decompose quickly during high-temperature carbonization and release volatile low-molecular-weight compounds.25Fungi are a potential candidate for fabricating carbon materials thanks to their accelerated growth and abundant nature. Mushrooms are a rich source of nitrogen. It is reported that the nitrogen levels in mushrooms are mainly in the range of 3–10% and therefore they can act as an available precursor for fabricating N-doped active carbon materials.25,51 Recently, Yao et al.23 utilized Bradyrhizobium japonicum (nitrogen content: 8–10 wt%) as a starting material to prepare 3D hierarchical porous carbon with a high BET surface area (1275 m2 g−1) and good electrical conductivity.
2.3. Animal-remains-derived precursors
Animal remains, such as seafood waste, arthropods, crustaceans, and mollusks, can be considered as a natural carbon-rich source; therefore, they can work as potential precursors for deriving carbon materials for environmental applications. This can be attributed to chitin, the primary and most crucial component of these aforementioned materials. For example, several typical species of crustacean contain very high levels (>50%) of chitin in their bodies, such as Cancer (crab) (72.1%), Nephrops (lobster) (69.8%), Lepas (goose barnacle) (58.3%), and Carcinus (crab) (64.2%) genera.52 The same trend was also reported for several typical kinds of insects, such as Diptera pupae (chitin content ∼ 54.8%) and Pieris pupae (∼64.0%).52Chitin is one of the most naturally abundant biopolymers, behind only cellulose. Chitin, a typical amino-polysaccharide, contains a high concentration of crucial bio-renewable nitrogen (∼7 wt%) and it is highly chemically/thermally stable.53–55 Therefore, chitin can provide a higher yield of carbon in comparison to cellulose. Notably, chitin is not only bio-crosslinked with catecholamine and glucan to form crosslinking networks, but it is also able to create strong intermolecular hydrogen bonds.56 This also explains why the chemical/thermal stability of chitin is much higher than that of cellulose, making it a much better option for preparing carbon materials.
In addition to chitin, keratin is another important component of animal remains. Chitin is normally the main constituent of sloughs and cuticles, while keratin is mainly found in the hair, claws, and horns of animals.24 Keratin, a form of fibrous structural proteins, possesses high chemical/thermal stability on account of strong intermolecular hydrogen bonds.57 Additionally, both strong covalent disulfide cystine crosslinking and non-covalent hydrogen bonds simultaneously exist within the molecular structure of keratin (see Fig. 4), resulting in remarkably improved stability during thermal carbonization processes, consequently providing high carbon yields.58 In 2014, Qian et al.59 successfully fabricated heteroatom-doped nanoporous carbon flakes through carbonization, using human hair fibers as the starting precursor. Interestingly, Rangel-Mendez et al.60 used shrimp heads (Litopenaeus sp.) as the primary source to extract chitin via lactic acid fermentation, followed by the formation of chitosan, which illustrated impressive efficiency (up to 68%) for removing cadmium(II) from aqueous environments. Recently, shrimp shells were also utilized as a chitin-rich starting material for synthesizing ultra-fine chitin nanofibers to eliminate arsenic metal ions from water.61 Owing to their superior properties, animal remains can become a primary source for chitin or keratin extraction, opening up new, economical, and green approaches for carbon material production.
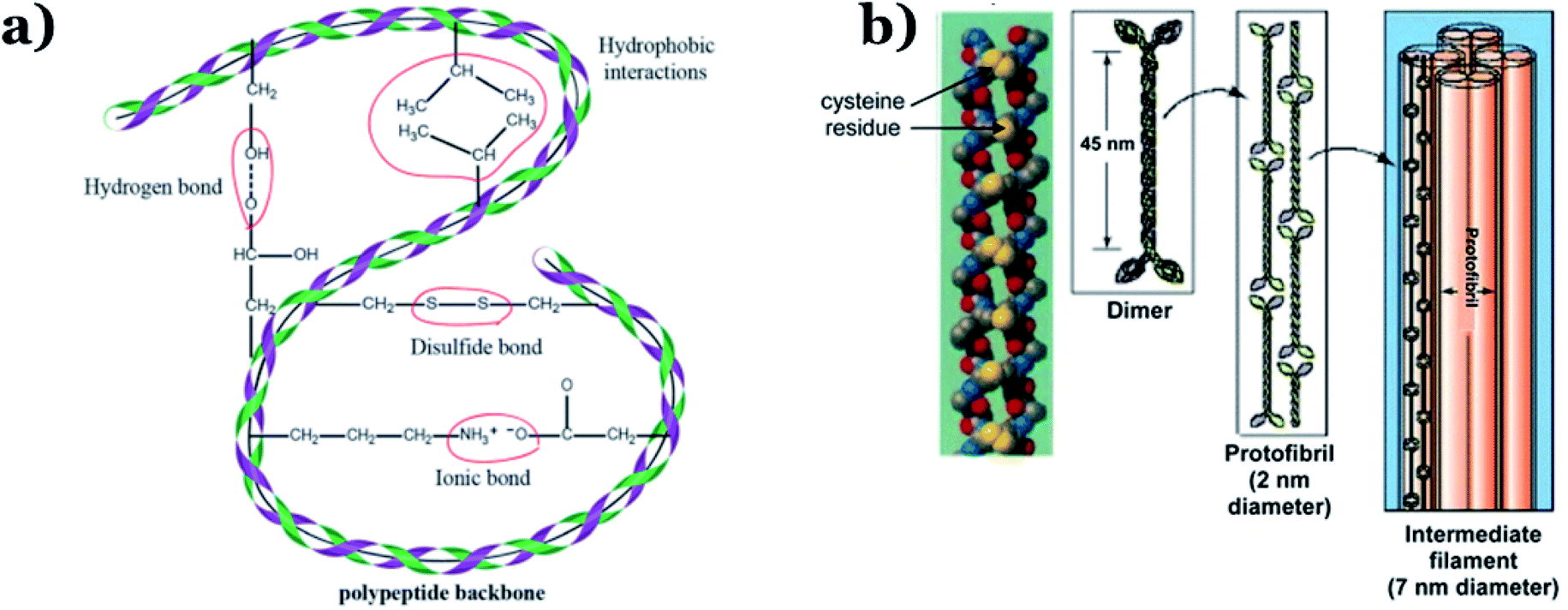 | ||
| Fig. 4 (a) Inter(intra)-molecular bonding in keratin. This figure has been reproduced from ref. 62 with permission from the Royal Society of Chemistry, copyright: 2017. (b) The structure of α keratin. This figure has been reproduced from ref. 63 with permission from Springer Nature, copyright: 2012. | ||
3. The synthesis and properties of biomass-derived carbon materials
Herein, various synthetic routes for obtaining natural-biomass-based carbon are discussed in detail (see Fig. 5). For each method, changes in the physiochemical properties of the as-synthesized carbon materials in response to different conditions are systematically investigated.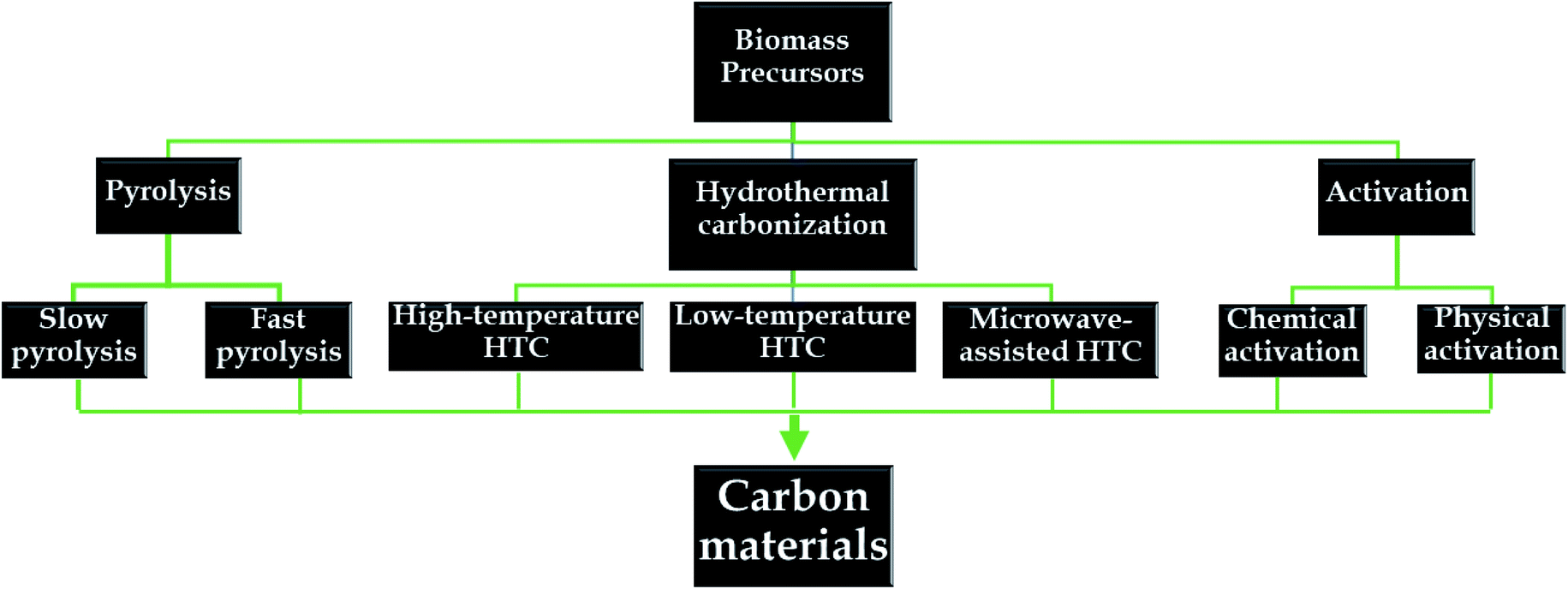 | ||
| Fig. 5 A summary of different routes for biomass-derived carbon preparation. | ||
3.1. Pyrolysis
Pyrolysis is a traditional fabrication method used with biomass, in which biomass is directly heated in the temperature range of 300 to 1200 °C in the absence of oxygen.64,65 The energy requirements for the pyrolysis of several agricultural-derived forms of biomass are reported to be in the range of 207 to 434 kJ kg−1.66 Since the entire process occurs in the absence of oxygen, there is no direct combustion of the precursors; instead, the main components of biomass, lignin, hemicellulose, and cellulose, will be thermally decomposed into gases and carbon materials (biochar).67 Yang et al.68 carried out separate tests on lignin, hemicellulose, and cellulose to understand their different characteristics during the pyrolysis process. The authors found that the decomposition of different components primarily occurred in obviously distinct temperature ranges. Accordingly, the decomposition of lignin was observed in a temperature range of 160–900 °C, whereas the other constituents decomposed obviously in a narrow range of 220–400 °C. Notably, of the above-mentioned components, the final carbon yield derived from lignin was the highest (40%). In pyrolysis processes, considerably distinct effects are obviously observed on the characteristics and yields of carbon upon using different heating rates. In terms of the processing conditions, pyrolysis can be classified as fast pyrolysis or slow pyrolysis. Accordingly, fast pyrolysis is a continuous process, aiming to inhibit the process of cracking products into non-condensable compounds. This process is carried out in a very high temperature range of 450 to 600 °C, with a high heating rate (above 200 °C min−1) and short residence time (<10 s).67 The driving force behind fast pyrolysis is the production of biomass-derived liquid fuel. Recent reports have indicated that the final yield of liquid fuel extracted from wood via fast pyrolysis can reach up to 75%.69,70On the contrary, the slow pyrolysis process is a batch process, carried out with a much longer residence time (>60 min) and at lower heating rates (in the range of 5–7 °C min−1).65 This process can provide maximum yields of carbon-based materials. In particular, moisture can obviously influence the biochar properties during slow pyrolysis. For instance, 15–20% is reported to be a suitable moisture range for preparing charcoal via pyrolysis.71,72 Aside from the heating rate and residence time, other conditions, such as temperature, particle size, pressure, etc., can directly influence the mechanism of action, characteristics, and final product yields.67,73 High temperatures and short residence times are beneficial for the formation of condensable products.74 It has been reported that carbon material, or biochar, produced at low temperatures during pyrolysis is less stable and possesses lower conductivity and a lower BET surface area.75 A report by Zanzi et al.76 described the effects of fast pyrolysis process conditions on agricultural-residue-based products. Wood, olive waste, and wheat straw were utilized as the initial carbon-rich precursors to fabricate gas and char via fast pyrolysis at high temperatures (800 to 1000 °C) in a free-fall reactor. The yield of olive-waste-derived char at 100 °C (14.6%) was much lower than that obtained at 800 °C (20.0%). Similar results were observed for the other precursors of wood and straw. Additionally, higher temperatures may lead to higher heating rates, which are not beneficial for biochar production. Interestingly, the authors realized that the utilization of small particles might result in an increase in the heating rate and, consequently, a reduction in the char yield. After rapid pyrolysis at 800 °C, when the particle size of both wood and olive waste was increased from 0.5 to 1.0 mm, the corresponding char yields increased from 5.6 to 7.2% and from 20 to 20.6%, respectively. Finally, the constituents of the biomass precursor also determine the distribution of products. Under the same conditions, with rapid pyrolysis at 800 °C, the char yield from olive waste was highest (20–27 wt%) in comparison to wood (7.2%) and straw (13%) precursors. This is due to the lignin content in olive waste (28 wt%) being higher than in wood (20 wt%) and wheat straw (∼21.7%). In summary, the characteristics of biomass-derived carbon materials obtained using different pyrolysis conditions are summarized in Table 2.
| Biomass precursor | Type of pyrolysis process | Process conditions (temperature, residence time) | Biochar yield (%) | Properties (SBET, m2 g−1; Vpore, cm3 g−1) | Ref. |
|---|---|---|---|---|---|
| Wood | Slow | 450 °C, 10 min | 29.2 | SBET: 4 | 77 |
| Wood | Slow | 750 °C, 10 min | 23.0 | SBET: 128 | |
| Wheat straw | Slow | 450 °C, 60 min | 27.5 | SBET: 16 | |
| Green garden waste | Slow | 450 °C, 60 min | 27.8 | SBET: 17 | |
| Dry algae | Slow | 600 °C, 60 min | 22.9 | SBET: 19 | |
| Sewage sludge | Fast | 500 °C, 20 min | 63.1 | SBET: 25 | 78 |
| Sewage sludge | Fast | 900 °C, 20 min | 53.3 | SBET: 67 | |
| Cattle manure | Slow | 300 °C, 2 h | 58.7 | SBET: 5.02; Vpore: 0.82 | 79 |
| Cattle manure | Slow | 400 °C, 2 h | 44.89 | SBET: 10.22; Vpore: 0.79 | |
| Cattle manure | Slow | 500 °C, 2 h | 39.84 | SBET: 2.38; Vpore: 0.85 | |
| Cattle manure | Slow | 700 °C, 2 h | 37.12 | SBET: 2.39: Vpore: 0.66 | |
| Oak wood | Fast | 400 °C and 450 °C | SBET: 8.8; Vpore: 0.86 | 80 | |
| Bark wood | Fast | 400 °C and 450 °C | SBET: 6.1 | ||
| Corn cobs | Fast | 500 °C | 18.9 | SBET: 0 | 81 |
| Corn stover | Fast | 500 °C | 17.0 | SBET: 3.10 | |
| Bagasse | Slow | 500 °C, 60 min | 43.7 | SBET: 202 | 82 |
| Cocopeat | Slow | 500 °C, 60 min | 62.9 | SBET: 13.7 | |
| Paddy straw | Slow | 500 °C, 60 min | 49.6 | SBET: 45.8 | |
| Wood stem | Slow | 500 °C, 60 min | 42.6 | SBET: 316 |
3.2. Hydrothermal carbonization
“Hydrothermal carbonization” (HTC) refers to a thermochemical process for the low-temperature pretreatment of biomass which contains a high content of moisture.83 The entire HTC process is carried out in an autoclave (a closed reactor), in which biochar with many chemical functional groups will be gradually formed from steam in a low temperature range of 180 to 250 °C over several hours.84 Hydrochar, or HTC-derived biochar, is produced as the desired product with a yield of 40–70%.85 Hydrochar is sequentially formed through hydrolysis, aromatization, dehydration, decarboxylation, and condensation. The main constituents of hydrochar are aliphatic compounds rather than aromatic compounds, with a considerable number of oxygen-containing groups.75 Notably, the presence of oxygen-containing groups endows hydrochar with hydrophilic properties and, as a result, its water-holding capacity is remarkably enhanced.86 In addition, the temperature, steam pressure, residence time, and biomass-to-water ratio are crucial parameters influencing the hydrochar properties. A higher temperature will enhance the BET surface area and total pore volume of hydrochar, thus improving its adsorption capacity, which is beneficial for pollutant elimination applications. However, at high temperatures, the secondary decomposition of solid residue can easily occur, which is favorable for the continuous conversion of condensable products into incondensable gas products.87 As a result, the final yield of hydrochar will decrease. Hoekman et al.88 concluded that a higher recovery efficiency of hydrochar from lignocellulose was obtained at lower temperatures. The authors found that the mass yield of hydrochar decreased from approximately 70% to nearly 50% when the temperature was increased from 215 to 255 °C. Recently, Sabio et al.87 utilized tomato-peel waste to prepare hydrochar via HTC. The final results pointed out that the treatment temperature is the key processing variable, having a major effect on the carbon yield. Under the same experimental conditions for 10 h, when the temperature was increased from 150 to 200 °C, the corresponding carbon yield also increased from 65% to approximately 61%. Similarly, a longer residence time leads to a decrease in the hydrochar yield. Via controlling the degree of decomposition of the initial biomass, the residence time can indirectly affect the texture formation of hydrochar and the size of its microspheres. Sevilla et al.89 utilized glucose, sucrose, and starch as separate precursors for HTC (at >170 °C) with residence times of 4.5 h and 15 h, resulting in the formation of uniform hydrochar microparticles with sizes ranging from 0.4 to 6.0 mm. Additionally, with an increase in the reaction time from 4.5 h to 15 h, the spherical particle diameter of glucose-derived hydrochar increased from 0.4 to 1.0. Romero-Anaya et al.90 pointed out that upon increasing the residence time or temperature, microspheres tend to aggregate on the surface of hydrochar. Accordingly, the morphology of hydrochar particles was obviously changed at longer residence times (12–48 h). Compared to pyrolysis, HTC requires much higher energy consumption on account of the longer residence time (up to several hours) and the effects of the high moisture levels in the precursors (or the biomass-to-water ratios).HTC can be classified into three groups: high-temperature HTC, low-temperature HTC, and microwave-assisted HTC. Generally, a low-temperature process offers a general platform for preparing uniform colloidal carbonaceous spheres with different functionalities. In a typical low-temperature HTC method, hydration, condensation, polymerization, and aromatization processes will sequentially take place. In the last stage, after polymerization reactions, recalcitrant substances are generated, and these then undergo further dehydration or pyrolysis to generate carbon materials. The high-temperature process is a promising way to prepare biomass-derived carbon materials with large surface areas and unique crystalline structures. Salvador et al.91 fabricated carbon materials from oak wood and anthracite at 650 °C and 700 °C, respectively, over 1 h via high-temperature HTC under gasification with supercritical water and steam. Unlike steam, which is only active at the surface of hydrochar, supercritical water not only carries out activation on the outside, but it can also enter the char interior and therefore enhance its porosity. A report by Calderon-Moreno et al.92 described the evolution of single-walled carbon nanotubes via high-temperature hydrothermal treatment at 800 °C and at a pressure of 100 MPa. Accordingly, after hydrothermal treatment at 600 °C for 48 h, the morphology of the sample became nanotube-like, in which crystalline clusters were inserted into an amorphous carbon-matrix layer (see Fig. 6a). Interestingly, when the temperature was increased to 800 °C, graphitic particle crystallization took place, and carbon nanotubes with multilayers obviously grew (Fig. 6b). There was no sign of amorphous carbon at this temperature.
 | ||
| Fig. 6 A TEM micrograph showing multiwalled nanotubes and crystalline graphitic clusters, covered by amorphous carbon, in a sample treated at 600 °C for 48 h (a) and a TEM micrograph showing SWNTs (single-walled carbon nanotubes) treated at 800 °C for 48 h (b). These figures have been reproduced from ref. 92 with permission from Elsevier, copyright: 2002. SEM micrographs of pine sawdust raw material (c), this material hydrothermally carbonized in a microwave oven (d), α-cellulose raw material (e), and this material hydrothermally carbonized in a microwave oven (f). These figures have been reproduced from ref. 95 with permission from Elsevier, copyright: 2009. | ||
In another approach, microwave irradiation is a potential alternative method utilized to replace carbohydrate hydrolysis for the carbonization of lignocellulosic materials.93 The several benefits of applying microwave irradiation towards biomass carbon preparation include shortened processing times and cost reduction.94 The structure of the final product also depends on the precursor material and it can be fine-tuned under certain conditions. In 2009, Guiotoku et al.95 reported a novel microwave-assisted hydrothermal carbonization method. In this study, pine sawdust and α-cellulose, as carbon-rich precursors, underwent microwave-based carbonization at 200 °C in acidic aqueous media for three different reaction times. The authors found that the pine-sawdust-derived carbon material basically maintained its micro-morphological features after hydrothermal carbonization, as illustrated in Fig. 6c and d. On the contrary, the morphology of α-cellulose showed noticeable alterations. The agglomeration of fibers (initial morphology) occurred, forming spherical particles with a size of around 1.2–2.0 μm in diameter (see Fig. 6e and f). The driving force behind this phenomenon was said to be interfacial energy minimization. Similar results were also obtained in the recent literature.96
3.3. Activation
Physical activation is a useful way to boost the adsorption capacity and physicochemical properties of carbon materials. In a physical activation process, a controlled atmosphere, consisting of carbon dioxide, air, or steam, is utilized as the ideal pyrolysis reaction environment, aiming to form internal pores in the carbon material.12 This also provide more active sites on the surface of biochar, which is beneficial for the pollutant adsorption capacity. More importantly, it has been proved that the characteristics of biochar, including the surface functional groups, BET surface area, and average pore size, can be controlled via adjusting process parameters such as the activation temperature, residence time, and atmosphere.97 A report by Ghouma et al.98 described that activated carbon material could be successfully synthesized from olive stones via a physical activation process using water vapor at 750 °C for 6 h. The as-synthesized carbon possessed a large surface area of 807 m2 g−1, a microporous volume of 0.30 cm3 g−1, and pore sizes between approximately 10 and 1 μm. Consequently, the adsorption capacity was improved to 131 mg g−1, demonstrating an obvious improvement in comparison to several types of lignocellulosic biomass-based carbon in the recent literature. Silvestre-Albero et al.99 described the impressive physicochemical properties of activated carbon material synthesized from peach stones with CO2 activation at a treatment temperature of 1123 K for 2 h, comprising a large BET surface area of nearly 1500 m2 g−1 and a mesopore volume of 0.63 cm3 g−1. In addition, the observed activated carbon owned a unique morphology containing curvy carbon multilayers mutually interconnected to create a 3D carbon matrix. Similarly, Cabal et al.100 used bean pods as a carbonaceous precursor to fabricate activated carbon at a carbonization temperature of 600 °C for 10 min at a heating rate of 60 °C min−1. Afterwards, the product was continuously activated at 700 °C with water vapor for one hour. The as-synthesized carbon possessed a high BET surface area of 258 m2 g−1, a pore volume of 0.206 cm3 g−1, and a naphthalene uptake capacity of 85 mg g−1. In addition, the high number of surface groups on the activated carbon are mainly basic groups, as the physical activation process based on steam is beneficial for the formation of oxygen-containing functional groups. In another research work, Zhao-qiang et al.101 synthesized high-quality activated carbon from Crofton weed through microwave carbonization with CO2 physical activation. Interestingly, the authors found that the activation duration, temperature, and CO2 flow rate are essential parameters influencing the adsorption capacity and activated carbon yield. Accordingly, when any of the above-mentioned parameters is increased, the production rate of carbon tends to decrease. The optimal conditions were determined to be a residence time of 90 min, an activation temperature of 980 °C, and a flow rate of 300 mL min−1, with the corresponding carbon yield being 18.03%. Under these conditions, the activated carbon possessed a large BET surface area of 1036 m2 g−1, a total pore volume of 0.71 mL g−1, and an average pore diameter of 2.75 nm.Chemical activation is an alternative solution for further improving the morphology of porous nanosheets and increasing the BET surface area of activated carbon using chemical agents, comprising acids, alkalis, or metal salts, during pyrolysis.102 In terms of the mechanism of action, the first step can be regarded as the chemical treatment of the biomass precursor. In this step, the biomass material will be mixed with activators (or chemical agents) at a certain ratio. The most popular activation agents used frequently include acids, such as H3PO4, H2SO4, and HNO3; alkalis, such as KOH and NaOH; and metal salts, such as ZnCl2 and CaCl2.103–105 Afterwards, mixtures of biomass and activation agents are pyrolyzed slowly in a temperature range of 400–1000 °C under a strictly controlled atmosphere in the presence of an inert gas.12 Finally, the activated carbon product is collected via washing and removing excess chemicals. Among the available chemical activation agents, alkalis are promising activators that have attracted high levels of attention as they can promote surface chemical group formation and enhance the porosity of biochar materials through dehydration and degradation processes. This can be explained based on the good penetration capacity of the activator into carbonized biomass skeletons, where it consequently interacts strongly with the carbon planes of the initial biomass material.12,102 Among the available alkaline agents, KOH is most frequently used in chemical activation processes. The reaction mechanism of KOH activation can be illustrated through the following reaction equations (see Fig. 7a):106,107
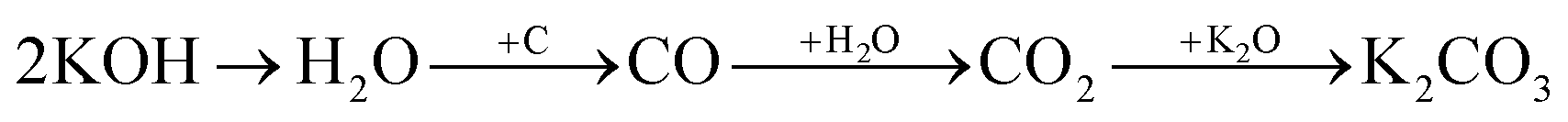 | (4) |
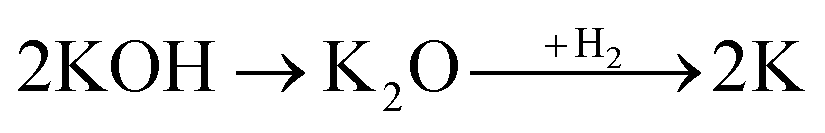 | (5) |
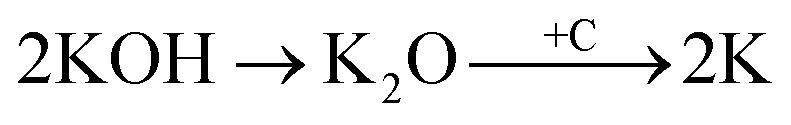 | (6) |
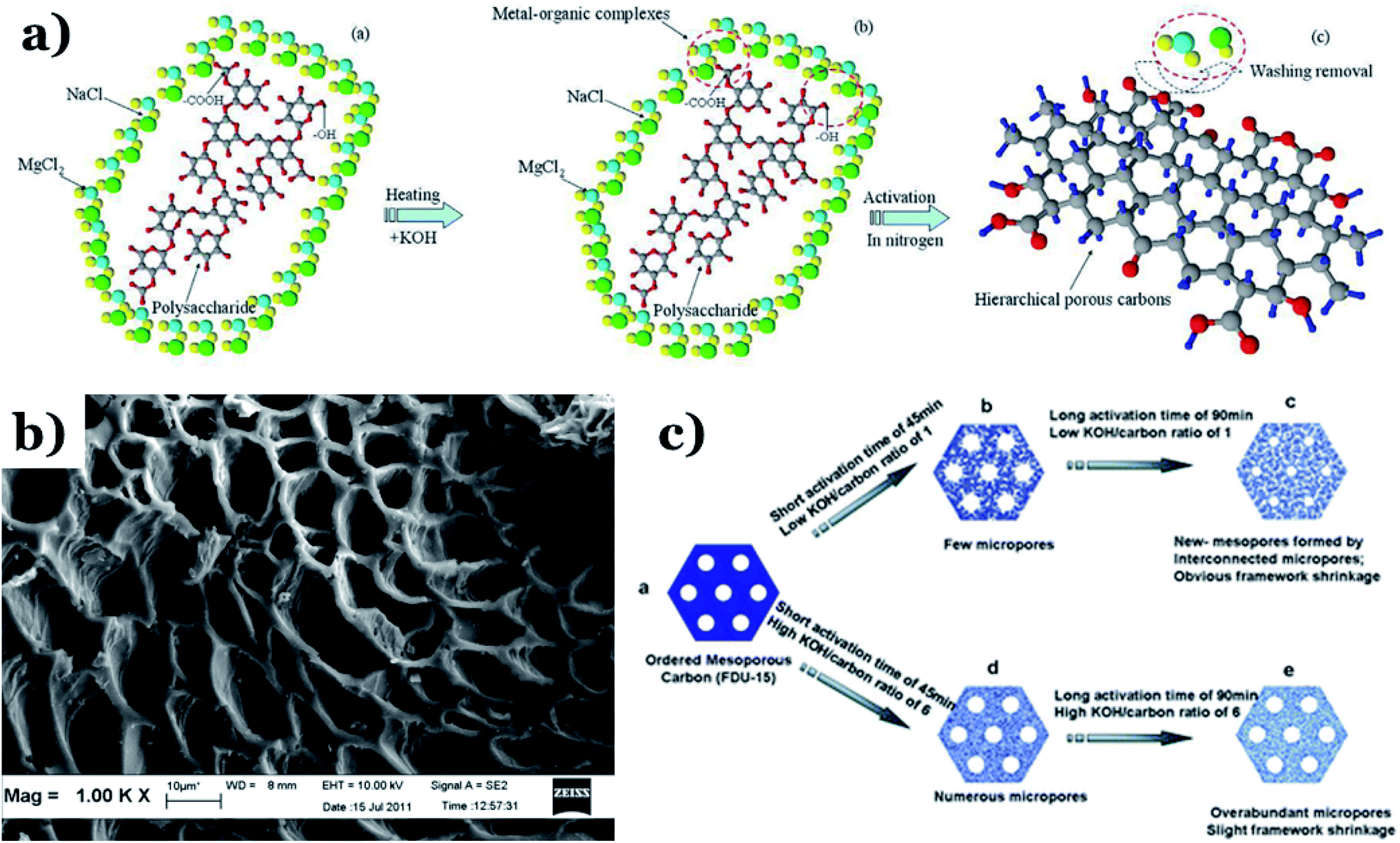 | ||
| Fig. 7 (a) A schematic illustration of hierarchical porous carbon framework formation. This figure has been reproduced from ref. 107 with permission from Elsevier, copyright: 2014. (b) An SEM image of sky-fruit-husk-derived carbon material. This figure has been reproduced from ref. 108 with permission from Elsevier, copyright: 2014. (c) The KOH-based activation of ordered mesoporous carbon. This figure has been reproduced from ref. 109 with permission from the Royal Society of Chemistry, copyright: 2012. | ||
It can be seen that both chemical and physical activation methods are systematically combined in this mechanism. The reaction chain in eqn (4) consists of several oxidation–reduction reactions with carbon (chemical activation) and reactions releasing H2O and CO2 (physical activation). This interesting combination has a positive impact on the porosity of the carbon frame. Notably, from eqn (5) and (6), potassium (K+) as a reaction product can enter the lattice of carbon, resulting in an extension of the lattice, consequently improving the porous structure of activated carbon.
The chemical activation process parameters, comprising the activation temperature, residence time, and ratio of activator to raw material (or the activator/C ratio), have considerable impacts on the structure and physicochemical properties of biochar. It has been proved that an increase in any one of the two factors of the activation temperature and activator/biomass ratio can promote the development of the carbon porosity, thus resulting in an improved level of porosity.12 Xing et al.110 pointed out that an increase in the activation temperature and the KOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :mesoporous-carbon ratio results in an improvement in the characteristics, such as the BET surface area and total pore volume, of activated samples. At a higher temperature of 100 °C, the hierarchical porous carbon possessed a large BET surface area and total pore volume of 2060 m2 g−1 and 1.91 cm3 g−1, respectively. The expansion and development of the pore texture can be enhanced via increasing the residence time and activation temperature, which is beneficial for the formation of microporous biochar (see Fig. 7b).108 This is a positive development for developing high-efficiency adsorption materials for environmental remediation, especially wastewater treatment. A report by Kv et al.109 described the effects of activation process conditions on the properties and morphologies of mesoporous carbon materials. The obtained results showed that adjusting only the KOH/C ratio and residence time can effectively enhance the BET surface area and the pore texture of activated carbon. It is imperative to mention that at low KOH/carbon ratios, KOH contained in micropores is easily released, and these micropores are then connected to form an interconnected mesoporous structure, as shown in Fig. 7c. This phenomenon leads to the extension and shrinkage of the carbon framework. Alternatively, when the KOH/C ratio is increased from 1 to 6 with the same activation time of 60 min, the corresponding BET surface area and pore volume of biochar particles increase from 1030 to 1400 m2 g−1 (BET) and from 0.52 to 0.69 cm3 g−1 (pore volume). Similarly, when the activation time is increased from 45 min to 90 min using the same KOH/C ratio of 1, the BET surface area and pore volume of activated samples are remarkably improved, from 930 to 1410 m2 g−1 (BET) and from 0.49 to 0.73 cm3 g−1 (pore volume). Similar results were obtained by Zahibi et al.111 In this study, walnut shell was used as a carbon-rich precursor, and it underwent chemical activation with ZnCl2 at the carbonization temperature under N2 flow at a rate of 5 °C min−1, forming the final porous carbon material. A comparison using two different walnut shell/ZnCl2 weight ratios, 1 and 2, was carried out. The obtained results pointed out that when this ratio was increased from 1 to 2, the specific surface area of the activated sample was reduced from 803 to 780 m2 g−1.
:mesoporous-carbon ratio results in an improvement in the characteristics, such as the BET surface area and total pore volume, of activated samples. At a higher temperature of 100 °C, the hierarchical porous carbon possessed a large BET surface area and total pore volume of 2060 m2 g−1 and 1.91 cm3 g−1, respectively. The expansion and development of the pore texture can be enhanced via increasing the residence time and activation temperature, which is beneficial for the formation of microporous biochar (see Fig. 7b).108 This is a positive development for developing high-efficiency adsorption materials for environmental remediation, especially wastewater treatment. A report by Kv et al.109 described the effects of activation process conditions on the properties and morphologies of mesoporous carbon materials. The obtained results showed that adjusting only the KOH/C ratio and residence time can effectively enhance the BET surface area and the pore texture of activated carbon. It is imperative to mention that at low KOH/carbon ratios, KOH contained in micropores is easily released, and these micropores are then connected to form an interconnected mesoporous structure, as shown in Fig. 7c. This phenomenon leads to the extension and shrinkage of the carbon framework. Alternatively, when the KOH/C ratio is increased from 1 to 6 with the same activation time of 60 min, the corresponding BET surface area and pore volume of biochar particles increase from 1030 to 1400 m2 g−1 (BET) and from 0.52 to 0.69 cm3 g−1 (pore volume). Similarly, when the activation time is increased from 45 min to 90 min using the same KOH/C ratio of 1, the BET surface area and pore volume of activated samples are remarkably improved, from 930 to 1410 m2 g−1 (BET) and from 0.49 to 0.73 cm3 g−1 (pore volume). Similar results were obtained by Zahibi et al.111 In this study, walnut shell was used as a carbon-rich precursor, and it underwent chemical activation with ZnCl2 at the carbonization temperature under N2 flow at a rate of 5 °C min−1, forming the final porous carbon material. A comparison using two different walnut shell/ZnCl2 weight ratios, 1 and 2, was carried out. The obtained results pointed out that when this ratio was increased from 1 to 2, the specific surface area of the activated sample was reduced from 803 to 780 m2 g−1.
4. Green photocatalysts for the photodegradation of various pollutants
The previous discussion revealed that biomass-activated carbon possesses potential physicochemical properties, including large specific surface areas, unique crystalline structures, long-term stability, and enriched surface functional groups, making it a promising candidate for energy and environmental applications.21 In terms of environmental remediation, biomass-derived activated carbon also shows strong light adsorption capacities, suggesting that it is suitable to be utilized as a photoactive species.112 Thus, when it comes to environmental remediation via photocatalysis, the potential applications of biomass-derived carbon materials are worthy of consideration. Table 3 summarizes relevant reports from the recent literature regarding the employment of biomass-carbon-based semiconductors for the photodegradation of various pollutants.| Photocatalyst | Synthesis method | Properties (SBET, m2 g−1, and Eg, eV) | Experimental conditions (light source and reaction time) | Pollutant and concentration | Overall efficiency (%) | Ref. |
|---|---|---|---|---|---|---|
| a CA: carbonaceous aerogel, DETA: diethylenetriamine, AC: activated carbon, BC: biomass carbon, YC: yeast-derived carbon, SAC: lotus-seedpod-derived activated carbon, LAC-T: lotus-leaf-derived activated carbon, Bio-CDs: biomass-derived carbon dots, CDs: carbon dots. | ||||||
| TiO2/CA | Hydrothermal | SBET: 113.33 | 500 W tungsten lamp (λ > 420 nm), 210 min | Ciprofloxacin, 40 ppm | 65 | 113 |
| TiO2/CA | Hydrothermal | SBET: 113.33 | 500 W tungsten lamp (λ > 420 nm), 210 min | Methylene blue, 40 ppm | 82 | 113 |
| TiO2/AC-DETA | Sol-hydrothermal | SBET: 173 | 300 W mercury lamp, 180 min | Cr(VI), 30 ppm | 90 | 114 |
| AC/Fe–TiO2 | Sol–gel | SBET: 644.66 | Visible light from a 20 W lamp, 120 min | NOx, ∼71% | 67.6 | 115 |
| C-Doped TiO2 | Microwave | SBET: 8.32, Eg: 3.14 | 25 W vis-LED light (≈450 nm, 46 W m−2), 120 min | Tetracycline hydrochloride, 5 ppm | 70 | 116 |
| rGO/TiO2 | Hydrothermal | — | 20 W UV lamp, 75 min | Methyl orange, 10 ppm | 100 | 117 |
| AC@ZnO/SnO2 | Ultra-sonication | SBET: 13.02, Eg: 2.76 | 400 W lamp (λ: 440–700 nm), 25 min | Linezolid antibiotic, 25 ppm | 94.6 | 118 |
| Lignin-derived carbon/ZnO | Carbonization | SBET: 139.53, Eg: 3.03 | Simulated solar light (500 W Xe lamp), 30 min | Rhodamine B, 15 ppm | 100 | 119 |
| ZnO/biochar from jute fibers | Pyrolysis | SBET: 62.2 | UV source (λ: 320–400 nm), 60 min | Methylene blue, 20 ppm | 99 | 120 |
| C/Fe3O4/Bi2O3 | Hydrothermal | SBET: 24.11 | Xe lamp with a UV filter, 90 min | Tetracycline, 20 ppm | 91 | 121 |
| g-C3N4@wood-derived carbon | Carbonization | SBET: 1064.39 | 300 W Xe lamp (λ > 400 nm), 60 min | Methylene blue, 20 ppm | 98 | 122 |
| Carbon/C3N4 | Thermal condensation | SBET: 152.6 | 300 W xenon lamp (λ > 420 nm), 90 min | 2-Mercaptobenzothiazole, 20 ppm | 91 | 123 |
| Chitin-derived carbon/g-C3N4 | Thermally induced polymerization | SBET: 49.1, Eg: 2.5 | 300 W xenon lamp (λ > 420 nm), 180 min | Rhodamine B, 5 ppm | 92.2 | 124 |
| BC/g-C3N4/Co3O4 | Hydrothermal and thermal condensation | SBET: 21.3038 | 300 W xenon lamp (λ > 420 nm), 60 min | Tetracycline | 70 | 125 |
| g-C3N4/YC/Cu2WS4 | Hydrothermal | SBET: 559.812 | 300 W xenon lamp (λ > 420 nm), 60 min | Cr(IV), 10 ppm | 98 | 126 |
| g-C3N4/YC/Cu2WS4 | Hydrothermal | SBET: 559.812 | 300 W xenon lamp (λ > 420 nm), 120 min | Tetracycline, 10 ppm | 78 | 126 |
| g-C3N4/C | Thermal condensation | SBET: 45.1 | 300 W xenon lamp (λ > 420 nm), 90 min | Tetracycline, 10 ppm | 80 | 127 |
| CdS@C | Pyrolysis carbonization and hydrothermal | Eg: 2.1 | Xenon lamp (CEL-HXF300, λ > 420 nm), 120 min | Rhodamine B, 40 ppm | 98.6 | 128 |
| CdS@C | Pyrolysis carbonization and hydrothermal | Eg: 2.1 | Xenon lamp (CEL-HXF300, λ > 420 nm), 120 min | Methylene blue | ∼85 | 128 |
| CdS@C | Pyrolysis carbonization and hydrothermal | Eg: 2.1 | Xenon lamp (CEL-HXF300, λ > 420 nm), 120 min | Acid red 11 | ∼95 | 128 |
| CdS@SAC | Carbonization | SBET: 943.2, Eg: 1.99 | 300 W Xe-arc lamp (λ > 420 nm), 120 min | Rhodamine B, 40 ppm | 94 | 129 |
| CdS@LAC-T | Carbonization and activation | SBET: 1245, Eg: 2.01 | 300 W Xe-arc lamp (λ > 420 nm), 150 min | Methylene blue, 40 ppm | 96.3 | 130 |
| CdS@LAC-T | Carbonization and activation | SBET: 1245, Eg: 2.01 | 300 W Xe-arc lamp (λ > 420 nm), 60 min | Methyl orange, 40 ppm | 97.8 | 130 |
| CdS@LAC-T | Carbonization and activation | SBET: 1245, Eg: 2.01 | 300 W Xe-arc lamp (λ > 420 nm), 60 min | Rhodamine B, 40 ppm | 95.9 | 130 |
| Carbon-supported CdS | Carbonization and hydrothermal | SBET: 169, Eg: 2.21 | Visible light, 90 min | Rhodamine B | 98 | 131 |
| N-Doped carbon | Hydrothermal and anaerobic pyrolysis | SBET: 3.4, Eg: 1.32 | 500 W Xe lamp (λ > 420 nm), 325 min | Cr(VI), 50 ppm | ∼78 (∼98.25 mg g−1) | 132 |
| Bio-CDs co-doped with S/Cl | Hydrothermal | — | Xe lamp (λ > 420 nm), 45 min | Rhodamine B, 30 ppm | 71.7 | 133 |
| Bio-CDs co-doped with S/Cl | Hydrothermal | — | Xe lamp (λ > 420 nm), 45 min | Methylene blue, 30 ppm | 94.2 | 133 |
| CDs co-doped with N/S | Hydrothermal | — | 300 W Xe lamp (λ > 420 nm), 60 min | Methylene blue, 20 ppm | kN/S-CDs = 2.34kCDs | 134 |
| CDs co-doped with N/S | Hydrothermal | — | 300 W Xe lamp (λ > 420 nm), 60 min | Methyl violet, 5 ppm | kN/S-CDs = 2.3kCDs | 134 |
| V-Doped carbon/Bi2O3 | Carbonization | SBET: 6.267 | Visible light, 90 min | Oxytetracycline hydrochloride, 5 ppm | 40 | 135 |
| Ce-Doped biomass-carbon-based g-C3N4 | Thermal condensation | SBET: 63.8 | 300 W Xe lamp (λ > 420 nm), 90 min | 2-Mercaptobenzothiazole, 10 ppm | 96 | 136 |
Aside from atom-doping modifications, activated carbon can also be modified via embedding photocatalyst nanoparticles onto its porous surface. The typical types of semiconductors that have been recently used to carry out this unique integration can be classified into three groups: metal oxides, g-C3N4, and CdS, as shown in Fig. 8. The interesting combination of biomass-derived carbon and semiconductors to form hybrid photocatalysts has been demonstrated to be a “wise form of cooperation” which simultaneously possesses synergistic effects relating to both adsorption and photodegradation processes in an advanced oxidation process system.2,22 This unique technique has recently demonstrated potential progress; however, the potential of these interesting hybrids has rarely been reported to date. Below, we summarize recent advances related to preparing biomass-derived carbon-based photocatalysts for environmental photocatalytic degradation applications using the visible spectrum range.
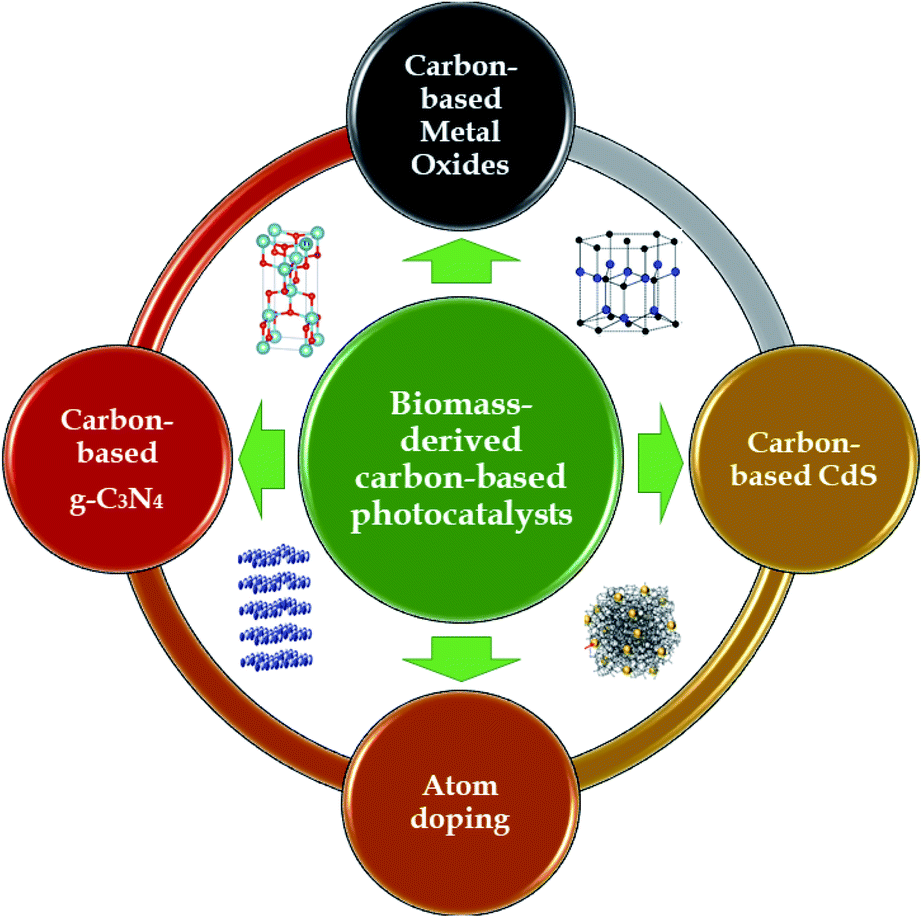 | ||
| Fig. 8 Biomass-derived carbon photocatalytic materials for environmental remediation. | ||
4.1. Carbon-based metal oxides
In hybrid systems integrating biomass-derived carbon and metal oxides, target pollutants molecules will very quickly adsorb on the surface of the carbon material, followed by prompt photodegradation in the presence of metal oxides.2 Among the available metal oxides, the first, and most common, candidate that needs to be mentioned is titanium dioxide (TiO2). TiO2 possesses high physical and chemical stability, nontoxicity, a suitable oxidation–reduction potential, and cost effectiveness.137,138 However, several serious disadvantages of TiO2 can be pointed out, including a high electron–hole recombination rate and a wide band-gap energy (up to 3.2 eV).139 Consequently, TiO2 only works efficiently under UV light, which makes up roughly 5% of the total solar spectrum.140 Numerous attempts have been made to enhance the photocatalytic performance of TiO2 under visible light. Among these, modification using non-metallic materials, especially carbon materials, has led to improved photocatalytic efficacy under visible light. Introducing carbon elements into the TiO2 structure has been proven to expand the absorption spectrum of TiO2 into the visible region. The driving forces behind this phenomenon are improvements in both the surface area characteristics and the charge-carrier separation efficiencies of the new hybrid systems.116Shi et al.113 fabricated a novel binary TiO2/carbonaceous aerogel (TiO2/CA) composite utilizing watermelon as a rich-carbon source via a cheap and facile hydrothermal process (Fig. 9a). An obtained SEM image of the 50 wt% TiO2/CA composite (as shown in Fig. 9c) demonstrated that the morphology of the hybrid consists of uniform-sized TiO2 nanoparticles dispersed on the surface of the porous architecture that was formed from carbonaceous nanofibers. The spacing distances between the lattice planes observed in the HRTEM image were determined to be 0.238 and 0.350 nm (Fig. 9f). The authors underlined that the unique porous structure and three-dimensional interconnected network of biomass-derived carbon and positive synergetic effects between the CA and TiO2 are the driving forces controlling the photocatalytic activity of hybrid TiO2/CA under visible-light irradiation. Compared with pure TiO2, the DRS absorption edge of the TiO2/CA composite showed remarkably enhanced absorption in the visible range (Fig. 9d). In addition, the large reduction in the PL intensities of the emission peaks of the TiO2/CA composites (see Fig. 9g) directly demonstrated the effectiveness of the synergetic effect at reducing the recombination of photogenerated electron–hole pairs when adding the CA into the host material. In the presence of only pure TiO2, the amount of ciprofloxacin (CIP, 40 ppm) slowly decreased over time to roughly 40% after 210 min under visible light irradiation, while TiO2/CA exhibited remarkably improved photoactivity, with more than 65% CIP degradation (see Fig. 9h).
 | ||
| Fig. 9 The synthetic pathway to a TiO2/CA composite (a); SEM (c) and HRTEM (f) images of the 50 wt% TiO2/CA composite; UV-vis DRS spectra of as-prepared samples (d); PL emission spectra of TiO2 and the TiO2/CA composites (g); and the relationship between Ct/C0 and the reaction time (t) during the photodegradation of CIP (h). These figures have been reproduced from ref. 113 with permission from the Royal Society of Chemistry, copyright: 2016. Possible linkages between Cr(VI) and TiO2/AC-DETA (b) and the adsorption–photoreduction of Cr(VI) on as-prepared catalysts (e). These figures have been reproduced from ref. 114 with permission from Elsevier, copyright: 2015. | ||
Fu et al.114 successfully synthesized diethylenetriamine-functionalized TiO2/activated carbon (TiO2/AC-DETA) via a facile sol-hydrothermal preparation. The results showed a significant enhancement of the photocatalytic activity of TiO2/AC-DETA compared to bare TiO2 or TiO2/AC. After 180 min, more than 90% of the original Cr(VI) (30 ppm) was removed completely in the presence of TiO2/AC-DETA, which is much higher than when using TiO2/AC (∼40%) or TiO2 alone (∼30%) (Fig. 9e). Additionally, considerable enhancements in the adsorption capacity were found when adding DETA to the surface of AC and TiO2/AC. The BET surface area of TiO2/AC-DETA was 173 m2 g−1, which is also higher than that of TiO2 (142 m2 g−1). More importantly, the authors found that functionalizing the carbon surface with diethylenetriamine plays a key role in improving the photocatalytic activity of the final composite. The rate of mass transfer of Cr(VI) from aqueous solution to the carbon surface will be quickly accelerated. In this case, several interactions between Cr(VI) and TiO2/AC-DETA were suggested, consisting of hydrogen bonding interactions, coordinative interactions, and electrostatic interactions (see Fig. 9b). The formation of chemical bonding in the form of C–O–Ti at the interfacial junction between AC and TiO2 promotes the migration of pollutant molecules from the carbon surface to the host material and the transport of photogenerated electrons from TiO2 to the carbon surface. In other research, nanostructured Fe-modified TiO2 was fabricated and loaded onto activated carbon, synthesized from coconut shell, to prepare a composite photocatalyst (AC/Fe–TiO2).115 Herein, vehicle exhaust (VE), comprising components such as CO, TiO2, and NOx, was employed as the target pollutant, and all photocatalytic tests were done under visible light. Compared to Fe–TiO2, the removal efficacy of VE using AC/Fe–TiO2 is obviously improved, and this is attributed to a synergistic effect between coconut-shell-derived activated carbon (adsorption process) and Fe–TiO2 (photocatalysis process). For instance, the photodegradation of NOx reached 44%, 60%, and the highest value of 67.6% when using AC alone, Fe–TiO2, and the hybrid of AC/Fe–TiO2 (after five loading rounds), respectively. Interestingly, loading an excessive amount of Fe-doped TiO2 onto AC can block the AC pores. This will lower the BET surface area and total pore volume, thereby reducing the photocatalytic activity. With an increase in the number of loading rounds from 1 to 5, the corresponding BET surface area of the final composite decreased from 777.13 to 644.66 m2 g−1. The corresponding photodegradation of VE, for example CO degradation (%), was also reduced from 5.8 to 4.8%. A recent report by Oseghe et al.116 showed the superior photocatalytic abilities of TiO2-doped pine-cone-based carbon synthesized via a microwave procedure. The presence of biomass-derived carbon retarded electron–hole recombination, thereby boosting the photocatalytic performance of TiO2. Interestingly, pine-cone-based carbon and microwave power can hinder the growth of the rutile phase and promote anatase phase formation. At an optimal microwave power of 800 W, the sample showed highest crystallite sizes for both the rutile (29.51 nm) and anatase (26.67 nm) phases. Also, the corresponding photodegradation efficacy toward tetracycline hydrochloride (5 ppm) was highest: 70% after 120 min of visible light exposure (∼450 nm). In a recent report by Faiz et al.,117 TiO2 nanoparticles were successfully incorporated onto the surface of reduced graphene oxide sheets (rGO), which were synthesized from tea-waste biomass. The stacking of graphene layers after the carbonization of tea waste can clearly be seen (Fig. 10a and b), followed by the exfoliation of the stacked graphite structure. Oxygen functional groups are inserted between planes, and the surface is greatly disturbed during the oxidation process (see Fig. 10c and d). Later, TiO2 particles were well-dispersed on the surface of the synthesized rGO sheets, as illustrated in Fig. 10e and f. Notably, the authors concluded that the addition of graphene could prevent the recombination of charge carriers, which was ascribed to π–π interactions and the 2D planar structure of graphene. However, an excessive amount of rGO has an adverse impact on the overall photocatalytic efficiency. The given reason is that active TiO2 sites will be blocked by the graphene sheets, thus hindering the ability of light to pass through the material. Even though biomass-derived graphene possesses numerous excellent properties, such as large BET surface areas and good electrical/thermal conductivities, reports detailing this material have still only mostly focused on its electrochemical energy storage and conversion applications to date.141,142
 | ||
| Fig. 10 FESEM images of (a and b) samples obtained after the carbonization of tea waste at 650 °C, (c and d) the synthesized rGO, and (e and f) the nanocomposite of GO/TiO2. These figures have been reproduced from ref. 117 with permission from IOPscience, copyright: 2020. | ||
Apart from TiO2, biomass-derived carbon is also able to enhance the photocatalytic activities of other conventional metal oxides, such as ZnO. Recently, a ternary ZnO/SnO2 framework in conjugation with date-seed-derived carbon (AC@ZnO/SnO2) was fabricated by Faisal et al.118 via an ultrasonication process. AC@ZnO/SnO2 possessed unique morphology, in which SnO2 nanorods were covered with nanosized ZnO particles and then tightly dispersed on the surface of date-seed-derived carbon nanosheets. The lattice fringes of the hexagonal structure of ZnO ((101) plane, d = 0.25 nm) and the tetragonal crystal structure of SnO2 ((110) plane, d = 0.33 nm) were observed (Fig. 11a). In addition, the optical properties of 10% AC@ZnO/SnO2 were remarkably enhanced, with an obtained band-gap energy of 2.76 eV, which is much lower than pure ZnO (2.97 eV), pure SnO2 (3.5 eV), and ZnO/SnO2 (2.91 eV) (Fig. 11b). Faisal and co-workers pointed out that carbon doping effectively improved the photocatalytic activity of ZnO/SnO2 heterostructures. The rate constant (k) of the newly designed photocatalyst reached 0.12 min−1 (as shown in Fig. 11c), the highest value among the synthesized samples. Corresponding to that, roughly 95% of the total linezolid content (initial concentration of 25 ppm) was removed completely under visible light irradiation (440–700 nm) when using 10% AC@ZnO/SnO2. The remarkable improvement found when using AC@ZnO/SnO2 can be attributed to a synergistic effect among the various components of the tri-system. Incorporating AC into the ZnO/SnO2 heterostructure leads to Sn–O–Zn bond formation; these bonds work as a “bridge”, extending the lifetimes of charge carriers (see Fig. 11e). In addition, the existence of more defects in the ZnO/SnO2 framework inhibited the recombination of charge carriers.
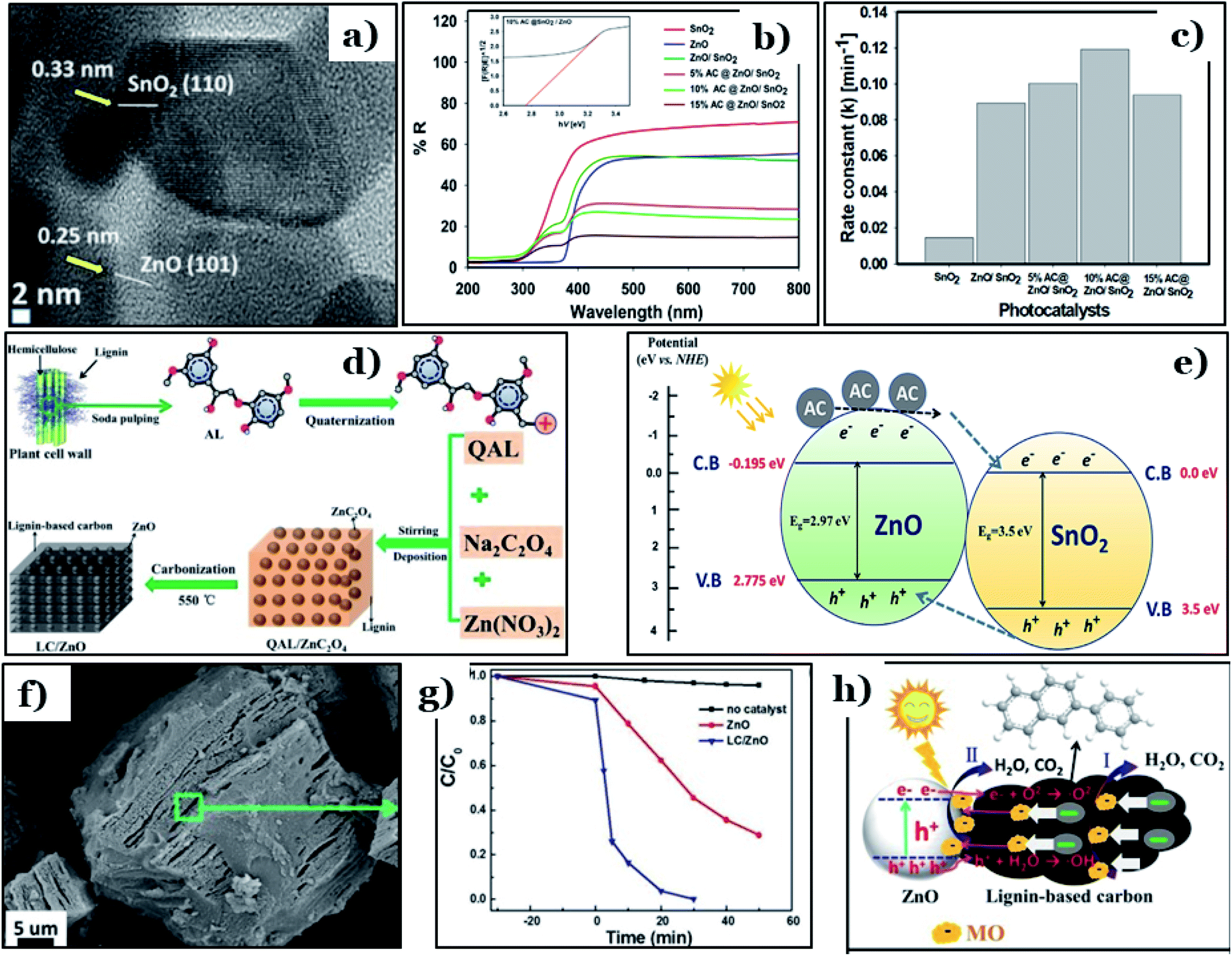 | ||
| Fig. 11 A high-magnification TEM image of 10% AC@ZnO/SnO2 (a); the UV-visible DRS spectra of pure SnO2, pure ZnO, ZnO/SnO2, and AC@ ZnO/SnO2 samples with different AC wt% levels (5.0–15.0%) and the plot of the transferred Kubelka–Munk function vs. the energy of the absorbed light for 10% AC@ZnO/SnO2 (inset) (b); the apparent rate constant (k) values for the photodegradation of the drug linezolid (c); and a diagram illustrating the charge transfer processes occurring within the heterostructured AC@ZnO/SnO2 photocatalyst (e). These figures have been reproduced from ref. 118 with permission from Elsevier, copyright: 2021. The synthetic route to the LC/ZnO hybrid composite (d); SEM images of the prepared LC/ZnO (f); the photocatalytic degradation of MO over pure ZnO nanoparticles and the LC/ZnO hybrid composite (g); and the photocatalytic mechanism for the degradation of MO over LC/ZnO (h). These figures have been reproduced from ref. 119 with permission from Elsevier, copyright: 2017. | ||
In another approach, Wang et al.119 successfully prepared a hybrid system of lignin-derived carbon/ZnO (LC/ZnO) via a green approach using industrial lignin as the carbon precursor, as shown in Fig. 11d. The microstructure of LC/ZnO was formed of tight bonds between LC sheets and ZnO particles, in which uniform nanosized ZnO nanoparticles were well-dispersed on carbon sheets (Fig. 11f). This unique morphology gave LC/ZnO a superior surface area (139.53 m2 g−1), nearly three times higher than pure ZnO (45.38 m2 g−1). Upon the addition of lignin-derived carbon, the calculated band-gap energy of LC/ZnO was 3.03 eV, an obvious reduction compared to ZnO (3.25 eV). In addition, rhodamine B (RhB, 15 ppm) was completely degraded under simulated solar light after only 30 min in the presence of LC/ZnO, while roughly 50% of RhB was removed using pure ZnO under the same conditions (Fig. 11g). The driving force behind the improved performance of the LC/ZnO composite was the synergistic interfacial interaction between the two components. The lignin-derived carbon sheets played an irreplaceable role in the hybrid system. Accordingly, excited electrons and holes from ZnO under photon irradiation migrated to the carbon nanosheets, improving the charge carrier separation efficacy. Then, on the surface of the carbon sheets, holes and electrons participate in oxidation–reduction reactions to produce active radicals, i.e., ˙OH and ˙O2− radicals. In the final step, thanks to the strong oxidative abilities, both radicals can decompose RhB molecules into by-products (Fig. 11h).
4.2. Carbon-based g-C3N4
In recent years, graphitic carbon nitride (g-C3N4) has been intensively studied as a potential photocatalyst for environmental remediation on account of its photochemical stability, facile fabrication, and suitable band-gap energy (∼2.7 eV).143 However, the fast charge-carrier recombination and small surface area of g-C3N4 are two large challenges that need to be addressed. In that context, many attempts have been made to solve this issue, such as elemental doping144 and heterojunction design.145 Among these strategies, combining g-C3N4 with nontoxic and abundant biomass-derived carbon materials is considered a “green” strategy, and it has recently showed great potential for improving the photocatalytic performance of g-C3N4.122,123In recent research, discarded natural basswood was employed to fabricate g-C3N4@wood-derived carbon (g-C3N4@WDC) via a facile two-stage method (see Fig. 12a).122 Interestingly, the morphology of g-C3N4@WDC possessed networked systems of internal channels in axial and radial directions. g-C3N4 nanoparticles were well dispersed on the insides and outsides of the carbon shells, suggesting that g-C3N4 is firmly combined with the ultrathin carbon layers (see Fig. 12c–f). The photocatalytic abilities of g-C3N4@WDC were proved through its remarkable elimination of MB (20 ppm), achieving up to 98% degradation after 60 min of visible light exposure (Fig. 12b). The addition of wood-derived ultrathin carbon layers played an irreplaceable role in overcoming the drawbacks of g-C3N4. Considerable numbers of illuminating photons will interact with and be transmitted through the highly ordered hierarchical porous structure originated from the carbon material, followed by being absorbed by g-C3N4 nanoparticles. More importantly, it is reported that wood-derived carbon possesses high electronic conductivity and an impressive electron storage capacity; therefore, excited electrons (e−) accumulated in the CB of g-C3N4 can easily be transported to the ultrathin carbon layers. These points remarkably promote the light-harvesting abilities and reduce e−/h+ recombination in the presence of g-C3N4@WDC.
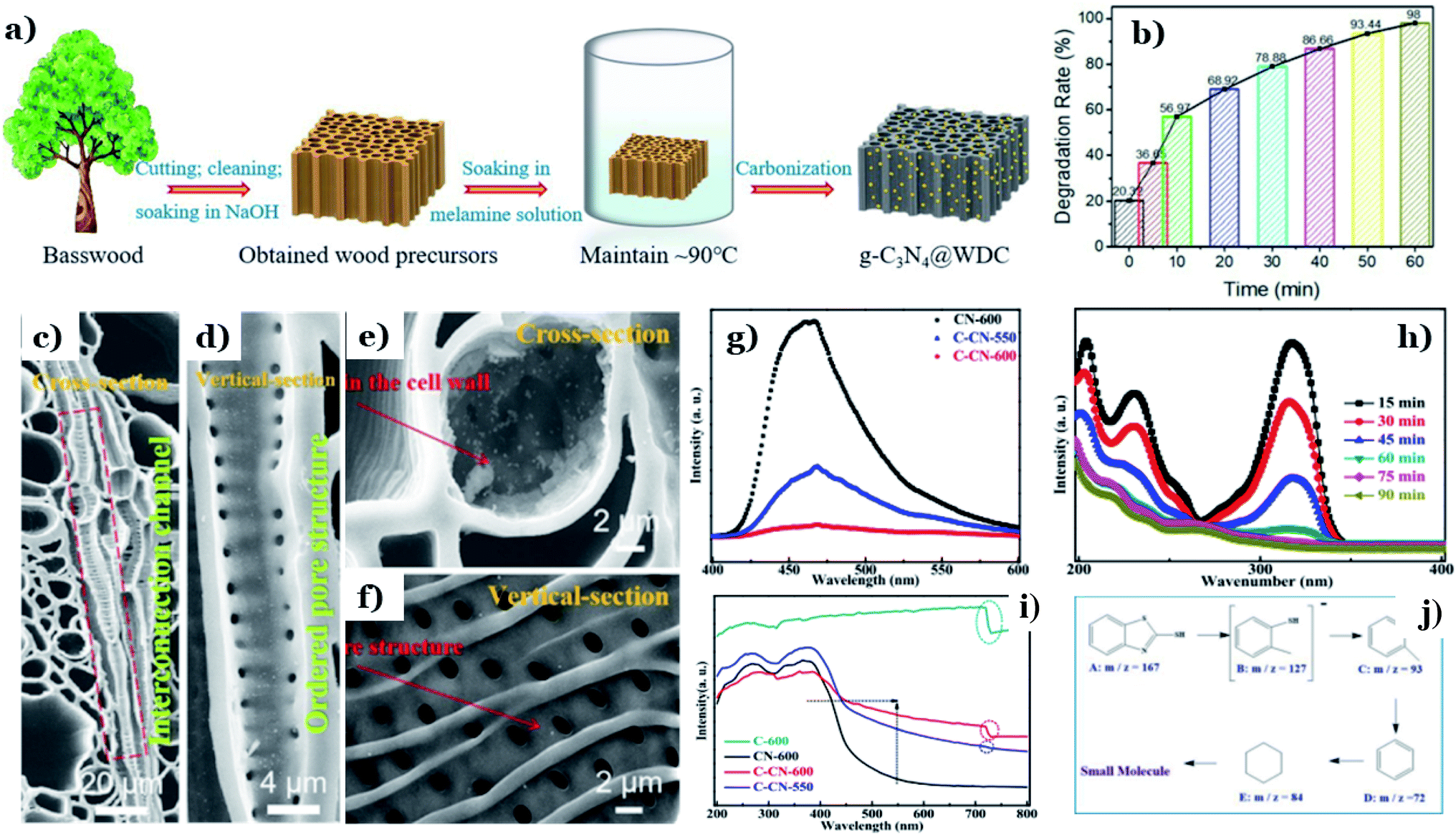 | ||
| Fig. 12 The synthetic pathway to g-C3N4@WDC (a); the photodegradation rate of MB as a function of time (b); and SEM images of (c and e) cross sections and (d and f) vertical sections of g-C3N4@WDC. These figures have been reproduced from ref. 122 with permission from Elsevier, copyright: 2020. PL analysis of CN-600, C-CN-600, and C-CN-550 (g); UV-vis DRS spectra (i) of C-600, CN-600, C-CN-550, and C-CN-600; absorbance variation curves of MBT solution over C-CN-600 (h) within 90 min; and the possible intermediate products (j). These figures have been reproduced from ref. 123 with permission from the Royal Society of Chemistry, copyright: 2020. | ||
In other research, the carbonization temperature used during corncob-derived carbon preparation is proved to have a considerable impact on the g-C3N4 activity.123 Significant red-shifts towards longer wavelengths of 450–700 nm are observed at high carbonization temperatures (550–600 °C), providing evidence of the improved light-harvesting capacity (Fig. 12k). Tight CN–C bonds were formed, and they worked as charge-transfer bridges due to the strong interfacial interactions between the two components. e−/h+ transfer and separation strongly took place at the highest temperature of 600 °C (Fig. 12g). Additionally, the higher carbonization temperature has a positive influence on the photocatalytic activity of g-C3N4. The degradation ability toward 2-mercaptobenzothiazole (20 ppm) using the hybrid system of carbon/C3N4 formed at 600 °C was roughly 2 times higher than that at 550 °C. The degradation mechanism revealed that 2-mercaptobenzothiazole underwent a series of oxidation reactions and was finally decomposed into CO2 and H2O (see Fig. 12h–m). A report by He et al.124 concluded that a chitin-derived carbon heterojunction is proved to effectively trap free electrons, retarding the recombination of electrons and holes. The formation of a heterojunction between chitin-derived carbon and g-C3N4 is favorable for the transfer of charge carriers. However, the authors found that with an excessive amount of chitin, the interactions between chitin (as the carbon precursor) and urea (as the g-C3N4 precursor) might have an adverse impact on thermal polymerization. The formation of many defects results in a lower degree of crystallinity and a higher charge-carrier recombination rate. In this study, the optimal chitin:urea mass ratio was determined to be 1:10. Yeast-derived carbon material was successfully utilized to form a Z-scheme heterojunction with g-C3N4 and Co3O4 (BC/g-C3N4/Co3O4).125 Zhao and coworkers explained that the superior photocatalytic abilities of BC/g-C3N4/Co3O4 are derived from the synergistic effects of two components, g-C3N4 and Co3O4, in a Z-scheme and the impressive charge carrier transmission abilities of the carbon material. The existence of biomass carbon is ascribed to be the driving force promoting the magnetic properties of Co3O4 in the heterojunction, improving the recyclability and therefore promoting the application capabilities of this material in real life. Similarly, Che et al.126 described the irreplaceable role of yeast-derived carbon as a charge carrier bridge in a 2D–2D Cu2WS4/g-C3N4 heterojunction. The authors pointed out three significant benefits of the as-synthesized spherical-shaped carbon in the heterojunction, which were retarding e−/h+ recombination, enhancing the light-harvesting capacity, and increasing the BET surface area. In the visible range (420 nm), the rate constant of Cr(VI) elimination using the g-C3N4/YC/Cu2WS4 heterojunction (corresponding efficiency of up to 98%) was roughly 5.5 times higher than when using pure g-C3N4 (corresponding efficiency of ∼30%).
4.3. Carbon-based CdS
Cadmium sulfide (CdS), a member of the metal sulfide group, has recently been used to fabricate nanomaterials for environmental remediation via photocatalysis. CdS possess an ideal band-gap energy of 2.4 eV, illustrating its excellent activation capacity in the visible range.146,147 However, CdS is known to be unstable and easily converted to toxic Cd2+ ions through photocorrosion, hindering its practical usage. Numerous attempts have been made to overcome these drawbacks, such as elemental doping, morphological/structural modification, and surface sensitization.129 Recently, the deposition of CdS on support systems, such as natural-biomass-based carbon materials, has proved to be an effective strategy for preparing highly stable CdS composite nanomaterials.Xing et al.128 utilized rice straw as a carbon source and Cd-containing plating wastewater as a Cd source to fabricate a CdS@C photocatalyst via facile pyrolysis carbonization and a hydrothermal process (see Fig. 13a). The morphology (Fig. 13b) of CdS@C revealed numerous uniformly distributed particles that were well dispersed on the surfaces of porous and tubular structures. Lattice spacings of 0.298 nm, representing the (111) plane of the mixed cubic phase, and 0.318 nm, representing the (101) plane of the hexagonal phase, were determined, as illustrated in Fig. 13c. The addition of biomass carbon brings about more active performance in the visible region. The band-gap energy of CdS@C was 2.1 eV, which is much lower than that of CdS (2.6 eV) (Fig. 13d). This means that CdS@C possesses remarkably improved visible-light-harvesting capabilities. Additionally, a remarkable enhancement in the separation and transport of charge carriers was demonstrated through the significantly higher photocurrent upon adding the carbon material (see Fig. 13e). Owing to the good conductivity, rice-straw-derived biochar acted as a support system, effectively separating photogenerated electron–hole pairs. Then, reduction–oxidation reactions took place right on the surface of biochar to produce active species, which can decompose pollutant such as dye into CO2 and H2O, as shown in Fig. 13f.
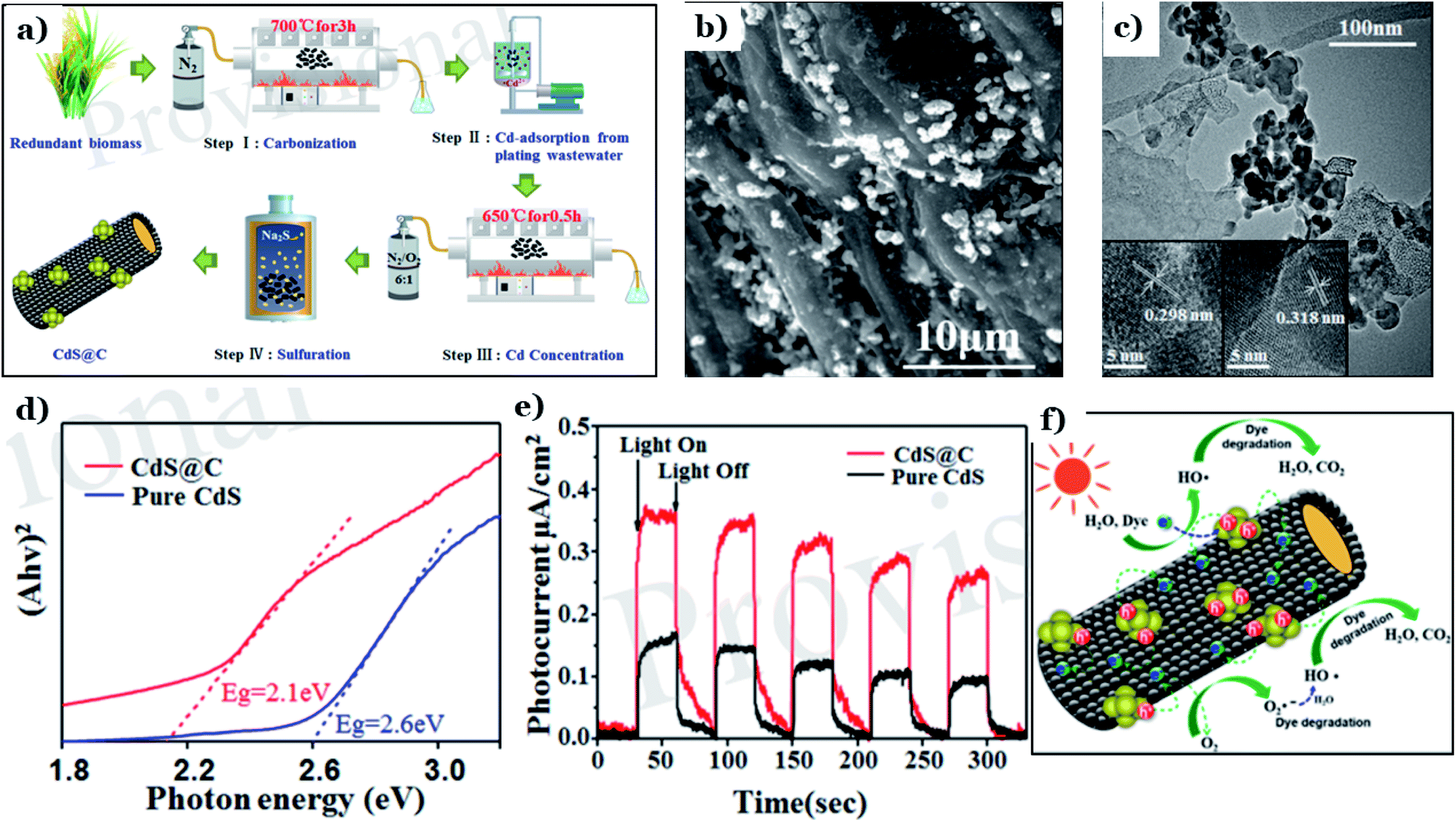 | ||
| Fig. 13 The synthetic pathway to CdS@C (a); SEM images of CdS@C (b); a TEM image of CdS@C (c) (the insets show HRTEM images); DRS spectra of pure CdS and CdS@C (d); transient photocurrent spectra of pure CdS and CdS@C under visible light (e); and a schematic illustration of the proposed mechanism for the photodegradation of dye using CdS@C under visible light (f). These figures have been reproduced from ref. 128 with permission from Frontiers, copyright: 2020. | ||
In other research, the selection of the activation temperature during activated carbon fabrication was proved to have a considerable impact on the final characteristics and photocatalytic performance of CdS supported by lotus-seedpod-derived activated carbon (CdS@SAC).129 With an increase in the activation temperature from 600 to 800 °C (see Fig. 14a), the BET surface area of CdS@SAC increased from 459.7 to 943.2 m2 g−1. The corresponding photodegradation of RhB also increased from 40% to 94% (Fig. 14b). The driving force behind this phenomenon is the formation of smoother SAC surface morphology at higher temperatures (800 °C) (as shown in Fig. 14c–f), which strongly boosted the interfacial contact between CdS and the carbon material. A higher specific surface area is beneficial for enriching RhB molecules on the surfaces of carbon particles. Similarly, CdS was deposited successfully on activated carbon material derived from natural lotus leaves (CdS@LAC-T).130 The formation of highly ordered surface structures took place strongly at a higher activation temperature (800 °C). Under these conditions, an obvious enhancement in the BET surface area was recorded (1245 m2 g−1) compared to at a lower temperature of 600 °C (634.6 m2 g−1). With a high specific surface area, the accumulation of dye molecules occurred strongly at the interfaces of CdS@LAC-T. Additionally, the authors pointed out that the existence of the microporous LAC support promoted the photocatalytic activity of CdS via narrowing its band-gap energy and inhibiting charge-carrier recombination.
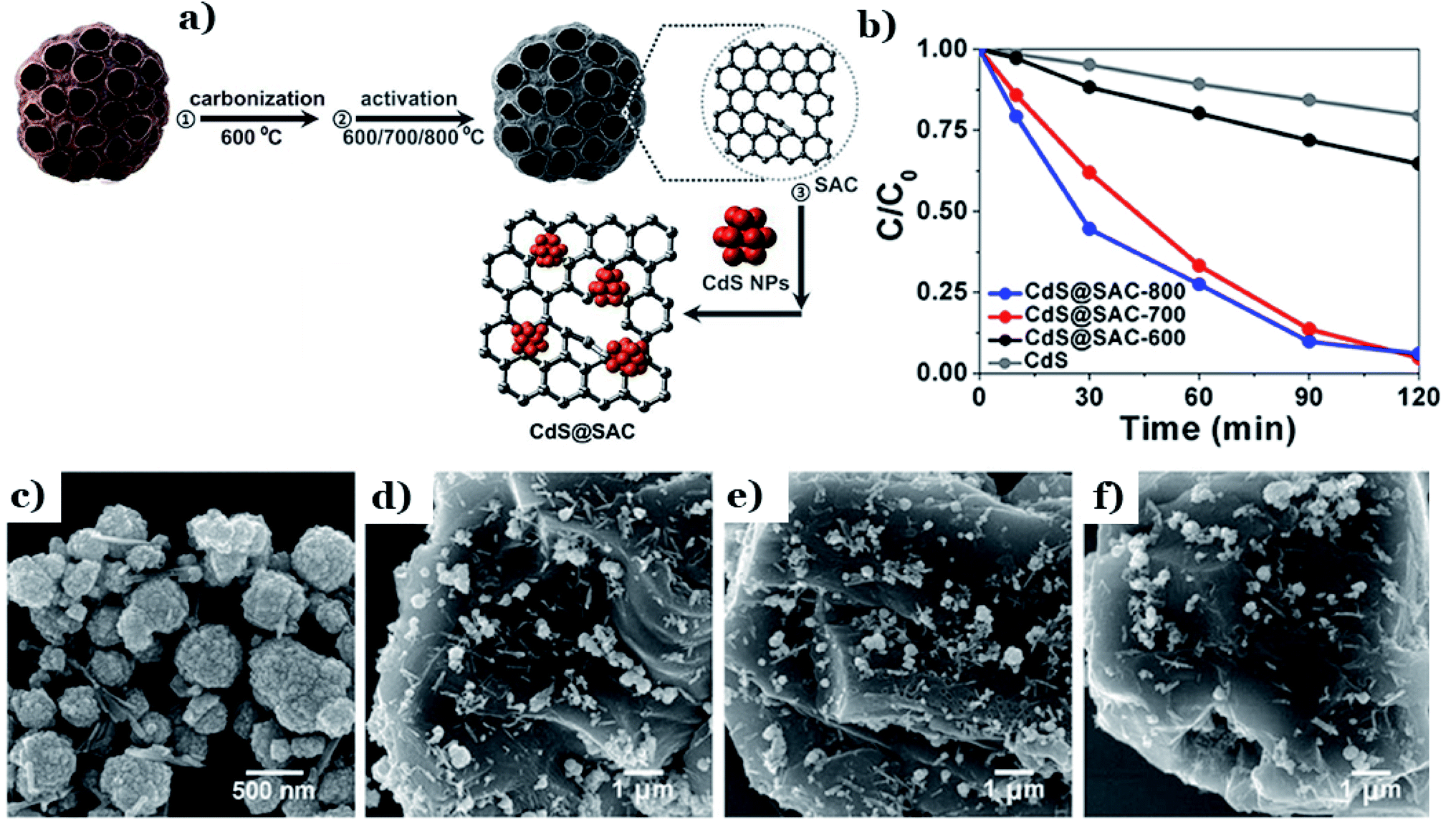 | ||
| Fig. 14 The fabrication method for the CdS@SAC composite material (a); the photocatalytic degradation of RhB using different catalysts (b); SEM images of (c) the CdS precursor, (d) CdS@SAC-600, (e) CdS@SAC-700, and (f) CdS@SAC-800. These figures have been reproduced from ref. 129 with permission from Frontiers, copyright: 2017. | ||
Relevant studies have demonstrated that biomass-derived porous carbon supports effectively boost the release of catalytically active species and retard the recombination of photogenerated electron/hole pairs. The number of studies on synthesizing hybrid CdS/biomass-carbon materials for environmental remediation is still low. However, these results partly open up a new avenue for optimizing the performance of photocatalysts by means of a low-cost and facile method.
4.4. Atom-doping modifications
To improve the sorption capacity and photocatalytic performance of biomass-derived carbon materials, one of the most common strategies is atom-doping modification. This means that chemical functional groups containing oxygen, nitrogen, or sulfur atoms are introduced onto the surfaces of activated carbon particles. Surface functionalization with hybrid atoms, comprising oxygen, sulfur, and nitrogen, has been proved to have a considerable impact on the surface chemistry of the carbon material, improving the surface area and consequently enhancing the adsorption capabilities toward pollutants.148Li et al.132 developed a method coupling adsorption and in situ photodegradation processes to convert Cr(VI) into Cr(III) using N-doped carbon derived from natural biomass cellulose. Herein, N-doped carbon was prepared through hydrothermal treatment via anaerobic pyrolysis at 350 °C and 700 °C, in which urea and natural cellulose served as the nitrogen and carbon precursors, respectively. Upon introducing nitrogen atoms into the carbon structure, a new intermediate energy level was formed in the energy band structure. The band-gap energy of the doped sample was determined to be 1.32 eV, which is lower than the undoped form (1.58 eV), as shown in Fig. 15a. Notably, at a higher temperature, the chemical structure of N-doped carbon collapsed and generated gases (NOx, CO, and CO2), producing a unique porous structure with a high specific surface area. However, the number of functional groups decreased remarkably. In contrast, at a lower pyrolysis temperature (350 °C), the surface of the material was enriched with numerous functional groups containing O and N, such as C–O, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–O, CO, OCO, CN–C, and N–(C)3 (see Fig. 15b), which is beneficial for the photocatalytic activity of the carbon-based material. The obtained results (Fig. 15c) relating to the adsorption capacity and photocatalytic performance also revealed that N-doped biomass carbon possesses a better adsorption capacity at a higher pyrolysis temperature (700 °C) and superior photocatalytic abilities at a lower temperature of 350 °C. In other research, Zhu et al.133 fabricated S and Cl co-doped carbon dots (Bio-CDs) via one-step hydrothermal treatment. Palm veneer was used as the carbon source, and thionyl chloride served as the dopant precursor (Fig. 15e). The well-dispersed Bio-CDs nanoparticles exhibited a quasi-spherical shape and possessed well-resolved lattice fringes, indicating a high degree of crystallinity (see Fig. 15f). In carbon-dot-based catalysts, the formation of energy levels is attributed to distinct defect states derived from functional groups containing oxygen on the surface of the carbon material and to heteroatom doping effects, as illustrated in Fig. 15g. Codoping with S and Cl generated distinct defect states in the carbon-dot structure, hindering the transition of excited electron back to the ground state and thereby inhibiting the recombination of electron/hole pairs. Owing to synergistic effects from S and Cl doping on the carbon dot electronic structure, Bio-CDs exhibited superior photocatalytic performance towards the removal of rhodamine B (roughly 72%) and methylene blue (94.2%) under visible light irradiation. Similarly, the green algae Dunaliella salina were utilized for the first time as a carbon source to synthesize carbon dots co-doped with nitrogen and sulfur (N/S-CDs) via a one-step hydrothermal reaction.134 The authors underlined that the formation of surface carbonyl groups on the N/S-CDs surface is the main driving force behind the enhancement of the photocatalytic activity. The surface carbonyl groups might act as active sites, facilitating the separation and transfer of charge carriers in N/S-CDs. Strongly oxidizing holes are released on the reaction surface of N/S-CDs under visible light. The photodegradation rates of methylene blue and methyl violet using N/S-CDs were more than two times higher than when using the undoped form. In another approach, Wang et al.135 developed a facile one-step carbonization method to fabricate vanadium-doped carbon/Bi2O3 composites via using sodium lignosulfonate, an extract from the bamboo pulping process, as the carbon source and catalyst. Bi2O3 served as the host semiconductor and the lignin-based carbon material was utilized as the support. V doping played an irreplaceable role in improving the properties of this hybrid system. V4+ can act as a charge carrier trapping center, while V5+ might work as an electron acceptor. After absorbing photons, excited electrons moved to the lignin-derived carbon material and left positive holes in the valence band of Bi2O3. Due to the V doping, the electrons were trapped at the V activation centers. Hence, the separation of photogenerated electron and hole pairs took place effectively. Additionally, the formation of CC and CO bonds promoted the visible light absorption capacity of the lignin-based carbon material.
C–O, CO, OCO, CN–C, and N–(C)3 (see Fig. 15b), which is beneficial for the photocatalytic activity of the carbon-based material. The obtained results (Fig. 15c) relating to the adsorption capacity and photocatalytic performance also revealed that N-doped biomass carbon possesses a better adsorption capacity at a higher pyrolysis temperature (700 °C) and superior photocatalytic abilities at a lower temperature of 350 °C. In other research, Zhu et al.133 fabricated S and Cl co-doped carbon dots (Bio-CDs) via one-step hydrothermal treatment. Palm veneer was used as the carbon source, and thionyl chloride served as the dopant precursor (Fig. 15e). The well-dispersed Bio-CDs nanoparticles exhibited a quasi-spherical shape and possessed well-resolved lattice fringes, indicating a high degree of crystallinity (see Fig. 15f). In carbon-dot-based catalysts, the formation of energy levels is attributed to distinct defect states derived from functional groups containing oxygen on the surface of the carbon material and to heteroatom doping effects, as illustrated in Fig. 15g. Codoping with S and Cl generated distinct defect states in the carbon-dot structure, hindering the transition of excited electron back to the ground state and thereby inhibiting the recombination of electron/hole pairs. Owing to synergistic effects from S and Cl doping on the carbon dot electronic structure, Bio-CDs exhibited superior photocatalytic performance towards the removal of rhodamine B (roughly 72%) and methylene blue (94.2%) under visible light irradiation. Similarly, the green algae Dunaliella salina were utilized for the first time as a carbon source to synthesize carbon dots co-doped with nitrogen and sulfur (N/S-CDs) via a one-step hydrothermal reaction.134 The authors underlined that the formation of surface carbonyl groups on the N/S-CDs surface is the main driving force behind the enhancement of the photocatalytic activity. The surface carbonyl groups might act as active sites, facilitating the separation and transfer of charge carriers in N/S-CDs. Strongly oxidizing holes are released on the reaction surface of N/S-CDs under visible light. The photodegradation rates of methylene blue and methyl violet using N/S-CDs were more than two times higher than when using the undoped form. In another approach, Wang et al.135 developed a facile one-step carbonization method to fabricate vanadium-doped carbon/Bi2O3 composites via using sodium lignosulfonate, an extract from the bamboo pulping process, as the carbon source and catalyst. Bi2O3 served as the host semiconductor and the lignin-based carbon material was utilized as the support. V doping played an irreplaceable role in improving the properties of this hybrid system. V4+ can act as a charge carrier trapping center, while V5+ might work as an electron acceptor. After absorbing photons, excited electrons moved to the lignin-derived carbon material and left positive holes in the valence band of Bi2O3. Due to the V doping, the electrons were trapped at the V activation centers. Hence, the separation of photogenerated electron and hole pairs took place effectively. Additionally, the formation of CC and CO bonds promoted the visible light absorption capacity of the lignin-based carbon material.
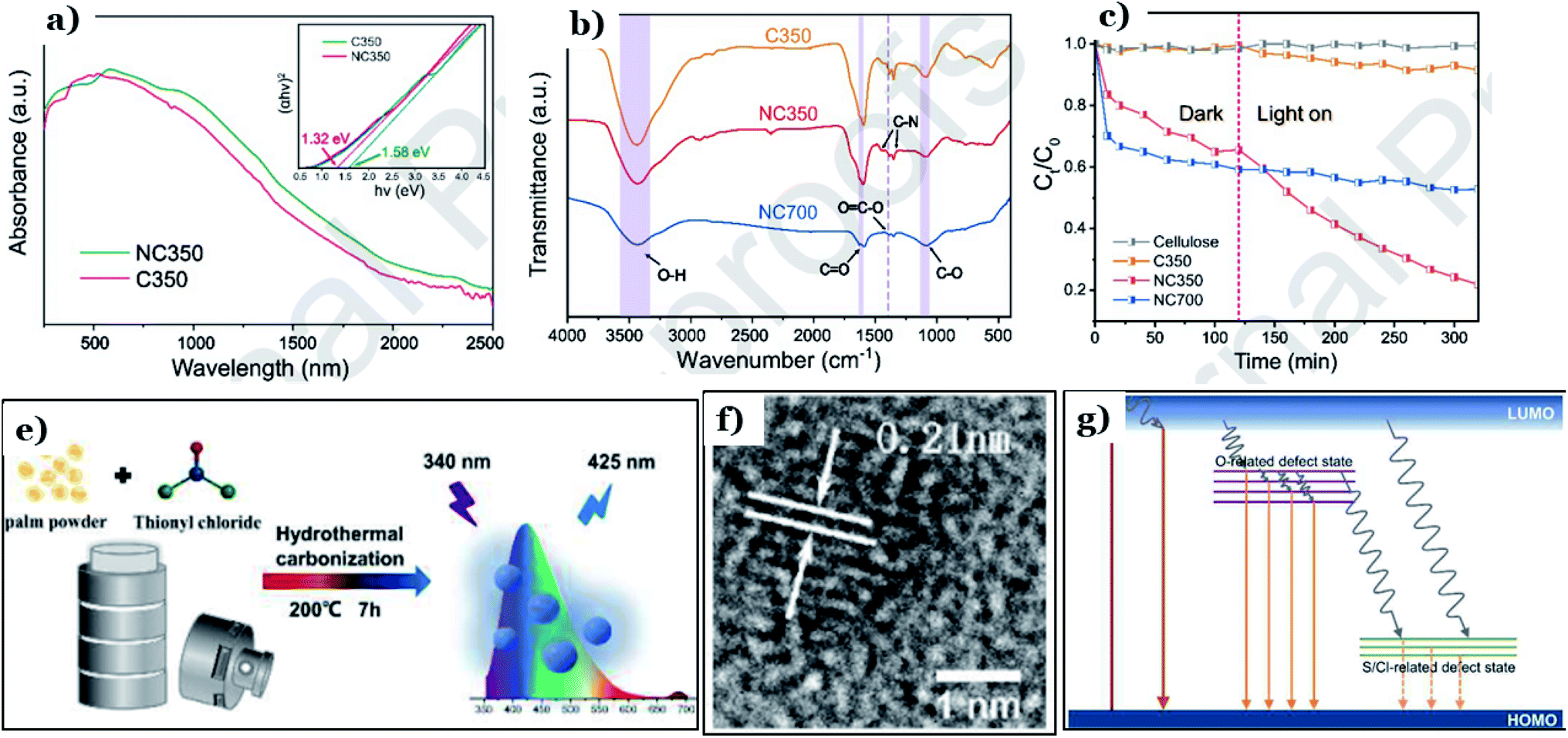 | ||
| Fig. 15 The UV-vis DRS spectra of NC350 and C350 (the inset shows the calculated band-gap energies) (a); the FT-IR spectra of C350, NC350, and NC700 (b); and the synergistic effects during adsorption and photo-reduction shown by different samples (c). This figure has been reproduced from ref. 132 with permission from Elsevier, copyright: 2019. A schematic illustration of the synthesis of Bio-CDs (e); a HR-TEM image of Bio-CDs (f); and a schematic illustration of the electronic transitions in the Bio-CDs structure (g). This figure has been reproduced from ref. 133 with permission from Elsevier, copyright: 2019. | ||
5. Conclusions and perspectives
This work highlights recent advances relating to the application of biomass-derived carbon-based photocatalytic materials for the photodegradation of pollutants based on the considerable number of relevant published articles so far. The authors have systematically presented the following key points: (i) the characteristics and compositions of biomass sources; (ii) the various synthetic routes and physicochemical properties of carbon materials; and (iii) material design strategies, mechanisms of action, and photocatalytic performance data for biomass-carbon-based photocatalysts.Biomass-carbon-based photocatalysts offer various attractive features, such as being non-toxic, abundant, low-cost, and highly porous. The unique structures of activated carbon materials can result in outstanding adsorption capacities and improved oxidation capacities toward various organic pollutants. The interesting combination of biomass-derived carbon and catalysts in the form of hybrid photocatalysts has proven to be a perfect combination, providing synergistic effects in both adsorption and photodegradation processes. In addition, biomass-carbon materials can also act as independent electronic bridges, extending the lifetimes of charge carriers.
However, there are several research gaps that need to be paid further attention. Firstly, high temperature carbonization is an irreplaceable step and, therefore, biomass-carbon preparation greatly depends on high levels of energy consumption. Consequently, the high cost of producing carbon remains a challenge preventing scaling-up. Secondly, there are no studies focusing on the economic aspects of the application of biomass-derived-carbon-modified semiconductors to date. Economic feasibility and environmental impact are two key factors that need to be comprehensively assessed. Additionally, most current studies are only limited to the lab-scale. It should be noted that apart from abundant biomass obtained from nature, large amounts of biomass in the form of industrial and agricultural waste are discarded annually, and this biomass can be utilized as low-cost carbon-rich precursors for environmental applications. Hence, the large-scale production of biomass carbon must be further developed in the near future. Another point is that the use of biomass feedstocks with various characteristics and different synthetic conditions can remarkably influence the properties of the obtained carbon materials. Therefore, further studies are needed to determine the ideal composition of the initial biomass and the optimal corresponding synthetic parameters. The specific relationships between the morphologies and physicochemical properties of carbon materials and their photocatalytic performances require research in future works. Finally, it is imperative to investigate environmental risks when using biomass precursors, particularly industrial biomass waste, which may contain harmful substances such as heavy metals. As-synthesized carbon materials may be influenced if the biomass precursor has a harmful composition, inhibiting the photocatalytic degradation of pollutants. Despite the aforementioned challenges that presently exist, biomass-derived-carbon-based photocatalysts offer an alternative approach with considerable advantages over traditional semiconductors for effectively degrading pollutants from wastewater under visible light irradiation. Undoubtedly, the transformation of naturally abundant biomass to carbon materials can also contribute to global waste minimization strategies. Therefore, significant progress in the field of environmental remediation can be expected in the future. This work is expected to inform readers of appropriate, facile, cheap, and environmentally friendly approaches involving biomass utilization, enabling the further development of superior photocatalysts for environmental remediation.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge Thu Dau Mot University for the financial support and required journal access during the construction of this work.References
- A. Malathi, J. Madhavan, M. Ashokkumar and P. Arunachalam, Appl. Catal., A, 2018, 555, 47–74 CrossRef CAS.
- Y. L. Pang, C. W. Lim, K. P. Y. Shak, S. Lim, W. C. Cheam, C. H. Koo and A. Z. Abdullah, in Nanophotocatalysis and Environmental Applications, Springer, 2020, pp. 55–86 Search PubMed.
- W. Sun, H. Li, H. Li, S. Li and X. Cao, Chem. Eng. J., 2019, 360, 645–653 CrossRef CAS.
- J. G. Brody, A. Aschengrau, W. McKelvey, C. H. Swartz, T. Kennedy and R. A. Rudel, Environ. Health, 2006, 5, 1–11 CrossRef PubMed.
- M. Phillips, J. Eastwood, J. Curtis, P. Gower and H. De Wardener, Br. Med. J., 1974, 2, 149–151 CrossRef CAS PubMed.
- T. S. Bui, P. Bansal, B.-K. Lee, T. Mahvelati-Shamsabadi and T. Soltani, Appl. Surf. Sci., 2020, 506, 144184 CrossRef CAS.
- S. Wang, Y. Hu and J. Wang, J. Environ. Manage., 2018, 217, 240–246 CrossRef CAS PubMed.
- J. B. Sluiter, R. O. Ruiz, C. J. Scarlata, A. D. Sluiter and D. W. Templeton, J. Agric. Food Chem., 2010, 58, 9043–9053 CrossRef CAS PubMed.
- T. Hou, H. Du, Z. Yang, Z. Tian, S. Shen, Y. Shi, W. Yang and L. Zhang, Sep. Purif. Technol., 2019, 223, 123–132 CrossRef CAS.
- M. Kamrani and A. Akbari, J. Environ. Chem. Eng., 2018, 6, 583–587 CrossRef CAS.
- N. Suriyanon, J. Permrungruang, J. Kaosaiphun, A. Wongrueng, C. Ngamcharussrivichai and P. Punyapalakul, Chemosphere, 2015, 136, 222–231 CrossRef CAS PubMed.
- J. Ouyang, L. Zhou, Z. Liu, J. Y. Heng and W. Chen, Sep. Purif. Technol., 2020, 253, 117536 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- J. Zhang, B. Tian, L. Wang, M. Xing and J. Lei, in Photocatalysis, Springer, 2018, pp. 1–15 Search PubMed.
- A. Fujishima, X. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- N. C. S. Selvam, A. Manikandan, L. J. Kennedy and J. J. Vijaya, J. Colloid Interface Sci., 2013, 389, 91–98 CrossRef CAS PubMed.
- R. Saravanan, V. Gupta, E. Mosquera and F. Gracia, J. Mol. Liq., 2014, 198, 409–412 CrossRef CAS.
- X. Wang, M. Sun, M. Murugananthan, Y. Zhang and L. Zhang, Appl. Catal., B, 2020, 260, 118205 CrossRef CAS.
- X. Chen, Z. Wu, D. Liu and Z. Gao, Nanoscale Res. Lett., 2017, 12, 1–10 CrossRef CAS PubMed.
- P. Lisowski, J. C. Colmenares, O. Mašek, D. Łomot, O. Chernyayeva and D. Lisovytskiy, J. Anal. Appl. Pyrolysis, 2018, 131, 35–41 CrossRef CAS.
- A. Zawawi, R. M. Ramli and N. Yub Harun, Technologies, 2017, 5, 82 CrossRef.
- Q. Yao, H. Wang, C. Wang, C. Jin and Q. Sun, ACS Sustainable Chem. Eng., 2018, 6, 4695–4704 CrossRef CAS.
- P. Thomas, C. W. Lai and M. R. B. Johan, J. Anal. Appl. Pyrolysis, 2019, 140, 54–85 CrossRef CAS.
- Y. Liu, J. Chen, B. Cui, P. Yin and C. Zhang, Journal of Carbon Research, 2018, 4, 53 CrossRef CAS.
- E. N. Yargicoglu, B. Y. Sadasivam, K. R. Reddy and K. Spokas, Waste Management, 2015, 36, 256–268 CrossRef CAS PubMed.
- W.-J. Liu, L. Ling, Y.-Y. Wang, H. He, Y.-R. He, H.-Q. Yu and H. Jiang, Environ. Sci.: Nano, 2016, 3, 745–753 RSC.
- K. Bilba, H. Savastano Junior and K. Ghavami, Mater. Res., 2013, 16, 903–923 CrossRef.
- A. R. Reed and P. T. Williams, Int. J. Energy Res., 2004, 28, 131–145 CrossRef CAS.
- T. Räisänen and D. Athanassiadis, 2013, (http://biofuelregion.se/wp-content/uploads/2017/01/1_2_IS_2013-01-31_Basic_chemical_composition.pdf).
- J. Nurmi, Heating values of mature trees, The Society of Forestry in Finland, 1997 Search PubMed.
- M. Ioelovich, Plant biomass as a renewable source of biofuels and biochemicals, LAP LAMBERT Academic Publishing, 2013 Search PubMed.
- R. Biswas, H. Uellendahl and B. K. Ahring, BioEnergy Res., 2015, 8, 1101–1116 CrossRef CAS.
- N. Curvetto, D. Figlas and R. Gonzalez Matute, Mushroom's Grower's Handbook, 2005, vol. 2 Search PubMed.
- E. Chauvet, Hydrobiologia, 1987, 148, 35–44 CrossRef CAS.
- B. Cagnon, X. Py, A. Guillot, F. Stoeckli and G. Chambat, Bioresour. Technol., 2009, 100, 292–298 CrossRef CAS PubMed.
- J. Zhang, Y. S. Choi, C. G. Yoo, T. H. Kim, R. C. Brown and B. H. Shanks, ACS Sustainable Chem. Eng., 2015, 3, 293–301 CrossRef CAS.
- J. U. Hernández-Beltrán, H.-D. Lira, I. Omar, M. M. Cruz-Santos, A. Saucedo-Luevanos, F. Hernández-Terán and N. Balagurusamy, Appl. Sci., 2019, 9, 3721 CrossRef.
- T. Wei, X. Wei, L. Yang, H. Xiao, Y. Gao and H. Li, J. Power Sources, 2016, 331, 373–381 CrossRef CAS.
- J. Chang, Z. Gao, X. Wang, D. Wu, F. Xu, X. Wang, Y. Guo and K. Jiang, Electrochim. Acta, 2015, 157, 290–298 CrossRef CAS.
- X. Zhu, S. Yu, K. Xu, Y. Zhang, L. Zhang, G. Lou, Y. Wu, E. Zhu, H. Chen and Z. Shen, Chem. Eng. Sci., 2018, 181, 36–45 CrossRef CAS.
- Q. Wang, Y. Zhang, J. Xiao, H. Jiang, T. Hu and C. Meng, J. Alloys Compd., 2019, 782, 1103–1113 CrossRef CAS.
- X.-L. Su, M.-Y. Cheng, L. Fu, J.-H. Yang, X.-C. Zheng and X.-X. Guan, J. Power Sources, 2017, 362, 27–38 CrossRef CAS.
- G. Yang and S.-J. Park, J. Alloys Compd., 2018, 741, 360–367 CrossRef CAS.
- Z. Xiao, W. Chen, K. Liu, P. Cui and D. Zhan, Int. J. Electrochem. Sci., 2018, 13, 5370–5381 CrossRef CAS.
- X. Zhu, X. Wang and Z. Xiong, Forest by-product and speciality in China, 2007, 2, pp. 9–11 Search PubMed.
- M. Abou Raya, M. Shalaby, S. Hafez and A. M. Hamouda, Journal of Food and Dairy Sciences, 2014, 5, 421–434 CrossRef.
- X.-M. Wang, J. Zhang, L.-H. Wu, Y.-L. Zhao, T. Li, J.-Q. Li, Y.-Z. Wang and H.-G. Liu, Food Chem., 2014, 151, 279–285 CrossRef CAS PubMed.
- V. Dıez and A. Alvarez, Food Chem., 2001, 75, 417–422 CrossRef.
- J. Yin and L. Zhou, Food Res. Dev., 2008, 29, 133–136 Search PubMed.
- J. A. Pineda-Insuasti, C. P. Soto-Arroyave and L. B. Ramos-Sánchez, Biotecnol. Apl., 2014, 31, 43–47 Search PubMed.
- W. Arbia, L. Arbia, L. Adour and A. Amrane, Food Technol. Biotechnol., 2013, 51, 12–25 Search PubMed.
- C. Chen, W. Ge, L. Jia, C. M. Pedersen, Y. Qiao, S. Jia, X. Guo, Y. Wang and X. Hou, ChemistrySelect, 2016, 1, 6540–6545 CrossRef.
- M. J. Hülsey, H. Yang and N. Yan, ACS Sustainable Chem. Eng., 2018, 6, 5694–5707 CrossRef.
- T. T. L. Chau, T. P. Vu, H. T. Le, C. X. Nguyen, T. D. Luong, P. T. A. Le, N. T. T. Dang, L. V. Nguyen and N. D. Cuong, Opt. Mater., 2021, 116, 111100 CrossRef CAS.
- J. P. Latgé, Mol. Microbiol., 2007, 66, 279–290 CrossRef PubMed.
- C. R. Robbins, in Chemical and physical behavior of human hair, Springer, 2012, pp. 105–176 Search PubMed.
- J. Wang, X. Zhang, Z. Li, Y. Ma and L. Ma, J. Power Sources, 2020, 451, 227794 CrossRef CAS.
- W. Qian, F. Sun, Y. Xu, L. Qiu, C. Liu, S. Wang and F. Yan, Energy Environ. Sci., 2014, 7, 379–386 RSC.
- J. R. Rangel-Mendez, R. Monroy-Zepeda, E. Leyva-Ramos, P. E. Diaz-Flores and K. Shirai, J. Hazard. Mater., 2009, 162, 503–511 CrossRef CAS PubMed.
- R. Yang, Y. Su, K. B. Aubrecht, X. Wang, H. Ma, R. B. Grubbs, B. S. Hsiao and B. Chu, Polymer, 2015, 60, 9–17 CrossRef CAS.
- A. Shavandi, T. H. Silva, A. A. Bekhit and A. E.-D. A. Bekhit, Biomater. Sci., 2017, 5, 1699–1735 RSC.
- J. McKittrick, P.-Y. Chen, S. Bodde, W. Yang, E. Novitskaya and M. Meyers, Jom, 2012, 64, 449–468 CrossRef.
- K. Gell, J. van Groenigen and M. L. Cayuela, J. Hazard. Mater., 2011, 186, 2017–2025 CrossRef CAS PubMed.
- Z. Zhu and Z. Xu, Renewable Sustainable Energy Rev., 2020, 134, 110308 CrossRef CAS.
- M. Van de Velden, J. Baeyens, A. Brems, B. Janssens and R. Dewil, Renewable energy, 2010, 35, 232–242 CrossRef CAS.
- V. Dhyani and T. Bhaskar, Renewable energy, 2018, 129, 695–716 CrossRef CAS.
- H. Yang, R. Yan, H. Chen, D. H. Lee and C. Zheng, Fuel, 2007, 86, 1781–1788 CrossRef CAS.
- A. Bridgwater, H. Hofbauer and S. Van Loo, Thermal biomass conversion, CPL Press, 2009 Search PubMed.
- A. Hornung, Transformation of biomass: theory to practice, John Wiley & Sons, 2014 Search PubMed.
- S. Vitolo, B. Bresci, M. Seggiani and M. Gallo, Fuel, 2001, 80, 17–26 CrossRef CAS.
- A. Bridgwater and G. Peacocke, Renewable Sustainable Energy Rev., 2000, 4, 1–73 CrossRef CAS.
- S. D. Stefanidis, K. G. Kalogiannis, E. F. Iliopoulou, C. M. Michailof, P. A. Pilavachi and A. A. Lappas, J. Anal. Appl. Pyrolysis, 2014, 105, 143–150 CrossRef CAS.
- A. V. Bridgwater, Biomass Bioenergy, 2012, 38, 68–94 CrossRef CAS.
- Z. Zhang, Z. Zhu, B. Shen and L. Liu, Energy, 2019, 171, 581–598 CrossRef CAS.
- R. Zanzi, K. Sjöström and E. Björnbom, Biomass Bioenergy, 2002, 23, 357–366 CrossRef CAS.
- F. Ronsse, S. Van Hecke, D. Dickinson and W. Prins, GCB Bioenergy, 2013, 5, 104–115 CrossRef CAS.
- T. Chen, Y. Zhang, H. Wang, W. Lu, Z. Zhou, Y. Zhang and L. Ren, Bioresour. Technol., 2014, 164, 47–54 CrossRef CAS PubMed.
- Y. Yue, Q. Lin, Y. Xu, G. Li and X. Zhao, J. Anal. Appl. Pyrolysis, 2017, 124, 355–361 CrossRef CAS.
- D. Mohan, H. Kumar, A. Sarswat, M. Alexandre-Franco and C. U. Pittman Jr, Chem. Eng. J., 2014, 236, 513–528 CrossRef CAS.
- C. A. Mullen, A. A. Boateng, N. M. Goldberg, I. M. Lima, D. A. Laird and K. B. Hicks, Biomass Bioenergy, 2010, 34, 67–74 CrossRef CAS.
- Y. Lee, J. Park, C. Ryu, K. S. Gang, W. Yang, Y.-K. Park, J. Jung and S. Hyun, Bioresour. Technol., 2013, 148, 196–201 CrossRef CAS PubMed.
- J. Lee, K. Lee, D. Sohn, Y. M. Kim and K. Y. Park, Energy, 2018, 153, 913–920 CrossRef CAS.
- J. A. Libra, K. S. Ro, C. Kammann, A. Funke, N. D. Berge, Y. Neubauer, M.-M. Titirici, C. Fühner, O. Bens and J. Kern, Biofuels, 2011, 2, 71–106 CrossRef CAS.
- W. Yan, J. T. Hastings, T. C. Acharjee, C. J. Coronella and V. R. Vásquez, Energy Fuels, 2010, 24, 4738–4742 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Energy Environ. Sci., 2011, 4, 1765–1771 RSC.
- E. Sabio, A. Álvarez-Murillo, S. Román and B. Ledesma, Waste Management, 2016, 47, 122–132 CrossRef CAS PubMed.
- S. K. Hoekman, A. Broch and C. Robbins, Energy Fuels, 2011, 25, 1802–1810 CrossRef CAS.
- M. Sevilla and A. B. Fuertes, Chem.–Eur. J., 2009, 15, 4195–4203 CrossRef CAS PubMed.
- A. J. Romero-Anaya, M. Ouzzine, M. A. Lillo-Rodenas and A. Linares-Solano, Carbon, 2014, 68, 296–307 CrossRef CAS.
- F. Salvador, M. J. Sánchez-Montero and C. Izquierdo, J. Phys. Chem. C, 2007, 111, 14011–14020 CrossRef CAS.
- J. M. Calderon-Moreno, S. S. Swamy and M. Yoshimura, Solid State Ionics, 2002, 151, 205–211 CrossRef CAS.
- A. Orozco, M. Ahmad, D. Rooney and G. Walker, Process Saf. Environ. Prot., 2007, 85, 446–449 CrossRef CAS.
- M. Nüchter, B. Ondruschka, W. Bonrath and A. Gum, Green Chem., 2004, 6, 128–141 RSC.
- M. Guiotoku, C. Rambo, F. Hansel, W. Magalhaes and D. Hotza, Mater. Lett., 2009, 63, 2707–2709 CrossRef CAS.
- M.-M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204–1212 RSC.
- R. Azargohar and A. Dalai, Microporous Mesoporous Mater., 2008, 110, 413–421 CrossRef CAS.
- I. Ghouma, M. Jeguirim, S. Dorge, L. Limousy, C. M. Ghimbeu and A. Ouederni, C. R. Chim., 2015, 18, 63–74 CrossRef CAS.
- A. Silvestre-Albero, M. Gonçalves, T. Itoh, K. Kaneko, M. Endo, M. Thommes, F. Rodríguez-Reinoso and J. Silvestre-Albero, Carbon, 2012, 50, 66–72 CrossRef CAS.
- B. Cabal, T. Budinova, C. O. Ania, B. Tsyntsarski, J. B. Parra and B. Petrova, J. Hazard. Mater., 2009, 161, 1150–1156 CrossRef CAS PubMed.
- Z.-q. Zheng, H.-y. Xia, C. Srinivasakannan, J.-h. Peng and L.-b. Zhang, Chem. Eng. Process., 2014, 82, 1–8 CrossRef CAS.
- Q. Chen, X. Tan, Y. Liu, S. Liu, M. Li, Y. Gu, P. Zhang, S. Ye, Z. Yang and Y. Yang, J. Mater. Chem. A, 2020, 8, 5773–5811 RSC.
- M. F. González-Navarro, L. Giraldo and J. C. Moreno-Piraján, J. Anal. Appl. Pyrolysis, 2014, 107, 82–86 CrossRef.
- R. Chauveau, G. Grévillot, S. Marsteau and C. Vallières, Chem. Eng. Res. Des., 2013, 91, 955–962 CrossRef CAS.
- J. Feng, K. Qiao, L. Pei, J. Lv and S. Xie, Ecological Engineering, 2015, 84, 209–217 CrossRef.
- A.-N. A. El-Hendawy, A. J. Alexander, R. J. Andrews and G. Forrest, J. Anal. Appl. Pyrolysis, 2008, 82, 272–278 CrossRef CAS.
- Y. Gao, W. Zhang, Q. Yue, B. Gao, Y. Sun, J. Kong and P. Zhao, J. Power Sources, 2014, 270, 403–410 CrossRef CAS.
- V. Njoku, M. A. Islam, M. Asif and B. Hameed, Chem. Eng. J., 2014, 251, 183–191 CrossRef CAS.
- Y. Lv, F. Zhang, Y. Dou, Y. Zhai, J. Wang, H. Liu, Y. Xia, B. Tu and D. Zhao, J. Mater. Chem., 2012, 22, 93–99 RSC.
- W. Xing, C. Huang, S. Zhuo, X. Yuan, G. Wang, D. Hulicova-Jurcakova, Z. Yan and G. Lu, Carbon, 2009, 47, 1715–1722 CrossRef CAS.
- M. Zabihi, A. H. Asl and A. Ahmadpour, J. Hazard. Mater., 2010, 174, 251–256 CrossRef CAS PubMed.
- M. Smits, D. Huygh, B. Craeye and S. Lenaerts, Catal. Today, 2014, 230, 250–255 CrossRef CAS.
- M. Shi, W. Wei, Z. Jiang, H. Han, J. Gao and J. Xie, RSC Adv., 2016, 6, 25255–25266 RSC.
- X. Fu, H. Yang, G. Lu, Y. Tu and J. Wu, Mater. Sci. Semicond. Process., 2015, 39, 362–370 CrossRef CAS.
- Z. Hu, T. Xu and B. Fang, Appl. Surf. Sci., 2017, 420, 34–42 CrossRef CAS.
- E. O. Oseghe and A. E. Ofomaja, J. Environ. Manage., 2018, 223, 860–867 CrossRef CAS PubMed.
- M. A. Faiz, C. C. Azurahanim, Y. Yazid, A. Suriani and M. S. N. Ain, Mater. Res. Express, 2020, 7, 015613 CrossRef CAS.
- M. Faisal, M. Alsaiari, M. A. Rashed and F. A. Harraz, J. Taiwan Inst. Chem. Eng., 2021, 120, 313–324 CrossRef CAS.
- H. Wang, X. Qiu, W. Liu and D. Yang, Appl. Surf. Sci., 2017, 426, 206–216 CrossRef CAS.
- M. Chen, C. Bao, D. Hu, X. Jin and Q. Huang, J. Anal. Appl. Pyrolysis, 2019, 139, 319–332 CrossRef CAS.
- N. Gao, Z. Lu, X. Zhao, Z. Zhu, Y. Wang, D. Wang, Z. Hua, C. Li, P. Huo and M. Song, Chem. Eng. J., 2016, 304, 351–361 CrossRef CAS.
- L. Wu, Y. Chen, Y. Li, Q. Meng and T. Duan, Appl. Surf. Sci., 2021, 540, 148440 CrossRef CAS.
- X. Yu, Z. Liu, Y. Wang, H. Luo and X. Tang, New J. Chem., 2020, 44, 15908–15918 RSC.
- B. He, M. Feng, X. Chen, D. Zhao and J. Sun, Appl. Surf. Sci., 2020, 527, 146737 CrossRef CAS.
- X. Zhao, Z. Lu, R. Ji, M. Zhang, C. Yi and Y. Yan, Catal. Commun., 2018, 112, 49–52 CrossRef CAS.
- H. Che, G. Che, P. Zhou, C. Liu and H. Dong, J. Colloid Interface Sci., 2019, 546, 262–275 CrossRef CAS PubMed.
- Y. Yu, Z. Zhu, W. Fan, Z. Liu, X. Yao, H. Dong, C. Li and P. Huo, J. Taiwan Inst. Chem. Eng., 2018, 89, 151–161 CrossRef CAS.
- R.-Z. Xing, J.-X. Li, X.-G. Yang, Z.-W. Chen, R. Huang, Z.-X. Chen, S.-G. Zhou and Z. Chen, Front. Chem., 2020, 8, 140 CrossRef CAS PubMed.
- H.-B. Huang, Y. Wang, F.-Y. Cai, W.-B. Jiao, N. Zhang, C. Liu, H.-L. Cao and J. Lü, Front. Chem., 2017, 5, 123 CrossRef PubMed.
- H.-B. Huang, Y. Wang, W.-B. Jiao, F.-Y. Cai, M. Shen, S.-G. Zhou, H.-L. Cao, J. Lü and R. Cao, ACS Sustainable Chem. Eng., 2018, 6, 7871–7879 CrossRef CAS.
- Z. Chen, J. Wang, R. Xing, Y. Wang, S. Wang, D. Wei, J. Li, Z. Chen and J. Lü, Inorg. Chem. Commun., 2019, 109, 107559 CrossRef CAS.
- R. Li, D. Hu, K. Hu, H. Deng, M. Zhang, A. Wang, R. Qiu and K. Yan, Sci. Total Environ., 2020, 704, 135284 CrossRef CAS PubMed.
- Z. Zhu, P. Yang, X. Li, M. Luo, W. Zhang, M. Chen and X. Zhou, Spectrochim. Acta, Part A, 2020, 227, 117659 CrossRef CAS PubMed.
- Y. Li, F. Liu, J. Cai, X. Huang, L. Lin, Y. Lin, H. Yang and S. Li, Microchem. J., 2019, 147, 1038–1047 CrossRef CAS.
- T. Wang, X. Liu, D. Han, C. Ma, M. Wei, P. Huo and Y. Yan, Environ. Res., 2020, 182, 108998 CrossRef CAS PubMed.
- Z. Zhu, C. Ma, K. Yu, Z. Lu, Z. Liu, P. Huo, X. Tang and Y. Yan, Appl. Catal., B, 2020, 268, 118432 CrossRef CAS.
- O. Pliekhov, O. Pliekhova, Y. O. Donar, A. Sınağ, N. N. Tušar and U. L. Štangar, Catal. Today, 2017, 284, 215–220 CrossRef CAS.
- K. Palanivelu, J.-S. Im and Y.-S. Lee, Carbon Lett., 2007, 8, 214–224 CrossRef.
- R. Daghrir, P. Drogui and D. Robert, Ind. Eng. Chem. Res., 2013, 52, 3581–3599 CrossRef CAS.
- J. Wu, B. Liu, Z. Ren, M. Ni, C. Li, Y. Gong, W. Qin, Y. Huang, C. Q. Sun and X. Liu, J. Colloid Interface Sci., 2018, 517, 80–85 CrossRef CAS PubMed.
- D.-d. Ouyang, L.-b. Hu, G. Wang, B. Dai, F. Yu and L.-l. Zhang, New Carbon Mater., 2021, 36, 350–372 CrossRef.
- Z. Bi, Q. Kong, Y. Cao, G. Sun, F. Su, X. Wei, X. Li, A. Ahmad, L. Xie and C.-M. Chen, J. Mater. Chem. A, 2019, 7, 16028–16045 RSC.
- Y. Song, Y. Peng, N. V. Long, Z. Huang and Y. Yang, Appl. Surf. Sci., 2021, 542, 148584 CrossRef CAS.
- H. Wang, J. Zhang, X. Yuan, L. Jiang, Q. Xia and H. Chen, Chem. Eng. J., 2020, 392, 123638 CrossRef CAS.
- H. Dong, X. Zhang, J. Li, P. Zhou, S. Yu, N. Song, C. Liu, G. Che and C. Li, Appl. Catal., B, 2020, 263, 118270 CrossRef CAS.
- R.-M. Ma, L. Dai and G.-G. Qin, Nano Lett., 2007, 7, 868–873 CrossRef CAS PubMed.
- N. Zhang, Y. Zhang, X. Pan, X. Fu, S. Liu and Y.-J. Xu, J. Phys. Chem. C, 2011, 115, 23501–23511 CrossRef CAS.
- G. Ma, Q. Yang, K. Sun, H. Peng, F. Ran, X. Zhao and Z. Lei, Bioresour. Technol., 2015, 197, 137–142 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2021 |