
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in the use of diaziridine-based sweetener derivatives to elucidate the chemoreception mechanism of the sweet taste receptor
Makoto Hashimoto
 *a,
Tomoya Nakagita
b and
Takumi Misaka
c
*a,
Tomoya Nakagita
b and
Takumi Misaka
c
aDivision of Applied Science, Graduate School of Agriculture, Hokkaido University, Kita 9, Nishi 9, Kita-ku, Sapporo 060-8589, Japan. E-mail: hasimoto@abs.agr.hokudai.ac.jp
bDepartment of Agricultural Chemistry, School of Agriculture, Meiji University, Kawasaki, Kanagawa 214-8571, Japan
cDepartment of Applied Biological Chemistry, Graduate School of Agricultural and Life Sciences, The University of Tokyo, 1-1-1 Yayoi, Bunkyo-ku, Tokyo 113-8657, Japan
First published on 1st October 2021
Abstract
All sweeteners are recognized by the sweet taste receptor (T1R2–T1R3). The elucidation of the chemoreception mechanism of receptor–ligand interactions is an attractive topic for researchers. Molecular biology and computational biology techniques can reveal the proposed mechanisms for this topic. Other approaches, including chemical biology (bioorganic chemistry), have helped to identify mechanisms on the basis of molecular structure. In this mini-review, we have summarized the recent progress in the synthesis of sweetener derivatives, which includes the use of photoaffinity labeling of diazirine-based derivatives to elucidate the chemoreception of sweeteners.
 Makoto Hashimoto | Makoto Hashimoto was born in Hokkaido, Japan in 1966. He received both his B.S. (1989) and PhD (1995) from Hokkaido University and conducted his postdoctoral research at Toyama Medical and Pharmaceutical University (1995–1997) as a Research Fellow of the Japan Society for the Promotion of Science, and at the University of Bath, UK (1999–2000) as a Research Officer. In 1997, he joined the bioorganic laboratory at Obihiro University of Agriculture and Veterinary Medicine until 2010. He was appointed associate professor from 2010 and professor from 2021 at Graduate School of Agriculture, Hokkaido University. His research is concentrated on defining and understanding the mechanisms of bioactive compounds with bioorganic methods. |
 Tomoya Nakagita | Tomoya Nakagita was born in Mie, Japan in 1988. He received his B.S. (2010) from Tokyo University of Science and PhD (2015) from the University of Tokyo. He conducted his postdoctoral research at Kyoto University (2015–2019), and at Ehime University (2019–2021). He learned structural biology and antibodies production at these labs. In 2021, he joined a food science laboratory at Meiji University as an assistant professor. His research is focused on food science, especially on the molecular mechanism of taste reception. |
 Takumi Misaka | Takumi Misaka was born in Hyogo, Japan in 1971. He received both his B.S. (1993) and PhD (1998) from the University of Tokyo. He worked in a food company (Nissin Food Products. Co. Ltd. in Tokyo, 1998–2000), and then became a postdoctoral fellow at the University of Tokyo (2000–2001). He became a research fellow of the Japan Society for the Promotion of Science, and then became a research associate of National Institute for Physiological Sciences in Okazaki. In 2005, he moved to the University of Tokyo, and was appointed an associate professor from 2008 at Graduate School of Agricultural and Life Sciences, the University of Tokyo. His research is focused on food science, especially on the molecular mechanism of taste reception. |
1. Introduction
Five taste modalities have been identified in humans: sweet, sour, bitter, salty, and umami.1 Sweetness provides a means to identify an energy source in the form of carbohydrates. A greater understanding of the chemoreception mechanism of sweet taste and the way in which sweet molecules interact with the sweet receptor will provide food scientists with the knowledge required to develop new sweeteners.There are three major approaches to this research, as summarized in Fig. 1: (I) molecular biology; (II) computational biology; and (III) chemical biology (bioorganic chemistry).
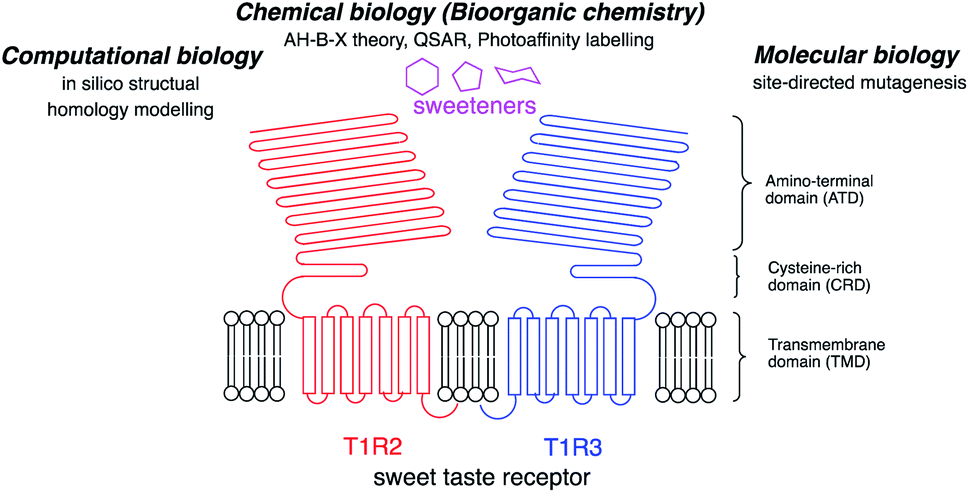 | ||
| Fig. 1 Schematic structure of the sweet taste receptor and approaches to the structural analysis of the sweetener–sweet taste receptor interactions. | ||
(I) Molecular biology: the sweet taste receptor is a member of the family of class C G protein-coupled receptors (GPCRs) and forms a heterodimeric structure with T1R2 and T1R3 subunits. Each subunit has a large amino-terminal domain (ATD) linked by a cysteine-rich domain (CRD) at the extracellular site to a seven transmembrane helical domain (TMD).9 The human heterodimeric sweet taste receptor (hT1R2–hT1R3) responds to a wide variety of chemical substances, including naturally occurring sugars, glycosides, D-amino acids, and artificial chemical compounds, such as sucralose, aspartame, and saccharin.10 Although these sweeteners have various chemical structures, all the compounds bind to the same sweet taste receptor.8,11
The sweet taste assay in combination with site-directed mutagenesis of the sweet taste receptors revealed the important amino acid residues for the chemoreception of each sweetener.
The structural features of the homodimeric metabotropic glutamate type 1 receptor (mGluR1) have been identified from X-ray crystal structure analysis, and this was the first example of the structure of a class C GPCR.12
(II) Computational biology: computational biology, based on homology modeling of the metabolic glutamate receptor (mGluR), can predict the 3D structure for the sweetener-sweet taste receptor complex.13 The structural homology between the sweet taste receptor and mGluR1 can predict important amino acid residues for the sweetener binding site from the mutagenesis studies and in silico structure modeling. Recent progress in the in silico analysis of the ligand–receptor binding modes afforded the useful information for the development of new sweeteners.
(III) Chemical biology (bioorganic chemistry): structure-based elucidation of sweeteners was performed to reveal the interactions of sweeteners with their binding sites. In 1967, Shallenberger and Acree proposed the AH–B theory for the structures of sweeteners. The sweetener must contain a glycophore, which consists of a hydrogen bond donor (AH) and a hydrogen bond acceptor (B) at a distance of 2.5 to 4 Å, and the glycophore can react with a complementary AH–B site on the receptor with a pair of hydrogen bonds.2 The theory was expanded by a third binding site (x) by Kier3 in 1972 and, subsequently, eight recognition sites were proposed by Nofre and Tinti in 1996.4
Based on these findings, the quantitative structure–activity relationship (QSAR) of sweeteners in silico is one of the methods for prediction.5–7 The models are based on the structure of binding ligands, but do not take into consideration the structure of the sweet receptor.8
The development of analytical methods for the study of the mature ligand–receptor binding complex may be one of the complementary methods to understand the mode of sweet receptor chemoreception. Chemical biology/bioorganic chemistry methods are used to elucidate the ligand–receptor binding complex.
The elucidation of protein function based on the structure–activity relationship is a major goal for scientists trying to understand the mechanisms of biological homeostasis. The understanding of the mechanism of the molecular interactions between small bioactive ligands and proteins is an important for rational drug design and discovery. Chemical methods provide an alternative route for the direct identification of target proteins in mixtures of biomolecules, as well as their ligand binding site structure, because these analyses are based on the affinity between the ligand and target protein.
Photoaffinity labeling14–17 significantly increased the specific capabilities of tagging. Photochemically generated highly reactive species introduce covalent bonds between the ligand and protein in a nonselective manner and any amino acid in the binding site can be tagged.
From these results, the researchers can select the most suitable approach to determine the sweet taste chemoreception mechanism, as summarized in Fig. 1.
The choice of photophore for effective photoaffinity labelling is important. Typically, aryl azide, benzophenone, and aryl diazirine are used (Fig. 2). Aryl azide is photoactivated by wavelengths below 300 nm, which sometimes causes damage to biomolecules, and generates nitrene18 as an active species, which is sometimes rearranged to the undesirable side product of ketenimine.19 Benzophenone20 is photoreactivated at wavelengths over 350 nm and generates the reactive triplet states of carbonyl, which regenerates the ground-state carbonyl compounds and can be re-used for photolabeling; however, the photophore sometimes requires a long photoirradiation period for labeling.
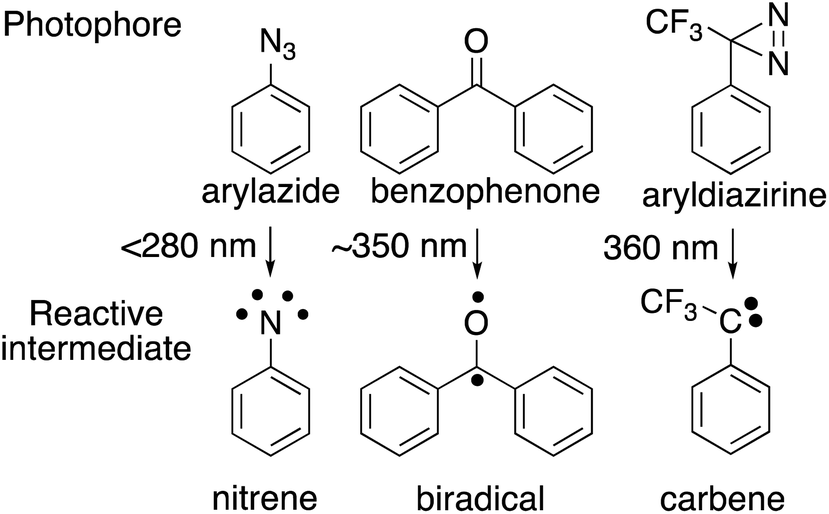 | ||
| Fig. 2 Photochemical reactions of major photophores used for photoaffinity labeling. | ||
Trifluoromethylphenyldiazirine21 is also photoreactivated with by wavelengths over 350 nm and generates carbene, which is more reactive than other photophores species, rapidly forming crosslinks to biomolecules with short photoirradiation. Comparative irradiation studies of these three photophores in living cells suggested that the irradiation to generate the active species from azide and benzophenone caused cell death during photolysis because of the long irradiation time needed to incorporate the photophore to the cell membrane surface. In contrast, the carbene precursor (3-trifluoromethyl)phenyldiazirine (TPD) did not promote cell death during the generation of active species. These results indicated that photoaffinity labeling with TPD was the most promising strategy for the investigation of labeled components.
In this mini-review, we have summarized the recent progress in the synthesis of sweetener derivatives, which includes the use of the photoaffinity labeling of diazirine-based derivatives to elucidate the chemoreception of sweeteners.
2. Photoaffinity probes for sweetener derivatives
2.1. Sucrose
Sucrose, which is found in an abundance of natural sources, is recognized as sweet; however, not all sucrose and fructose derivatives are sweet, especially if a substitution is made on one hydroxyl group. The detailed relationship between the structure and biological activity has not been completely clarified. Chemical designs are important when photoaffinity labeling is used to perform a functional analysis of sweetness. It was found that several sucrose-based oligosaccharides have a sweet taste and 1-kestose (GF2), which has an additional fructose, was linked at the 1′-position of the fructose unit of sucrose and acted as a sweetener.22 The results indicated that the 1′-position of the sucrose could accept substitutions. Although selective esterification of 1′-position was achieved by biotransformation,23 the ester linkage was easily hydrolyzed under physiological conditions and metabolized during analysis. In contrast, ether (phenoxy) linkages appear more stable than esters for functional analysis.24 In general, the reactivity of sucrose primary alcohols toward halogenation was greater for 6- and/or 6′-modification, and lower for the neopentyl-like 1′-position.25The Mitsunobu reaction is one of the most popular reactions for the formation of ether linkages between different alcohols.26 The o- or p-nitro substituted phenols, which contain strong electron-withdrawing functional groups, were better substrates for phenoxy modifications at the 1′-position of heptaacetyl sucrose (1) over 85% yield. Although the trifluoromethyldiazirinyl phenol derivatives (2 and 3) had moderate electron-withdrawing properties, the Mitsunobu reaction conditions promoted the decomposition of the diazirinyl three-membered ring and the desired products (4 and 5) were afforded at a yield of less than 10%. No improvement was observed for phenoxylation at the 1′-position of heptaacetyl sucrose with various reaction conditions (temperature, amount of reagents, and reaction time) (Fig. 3A).27
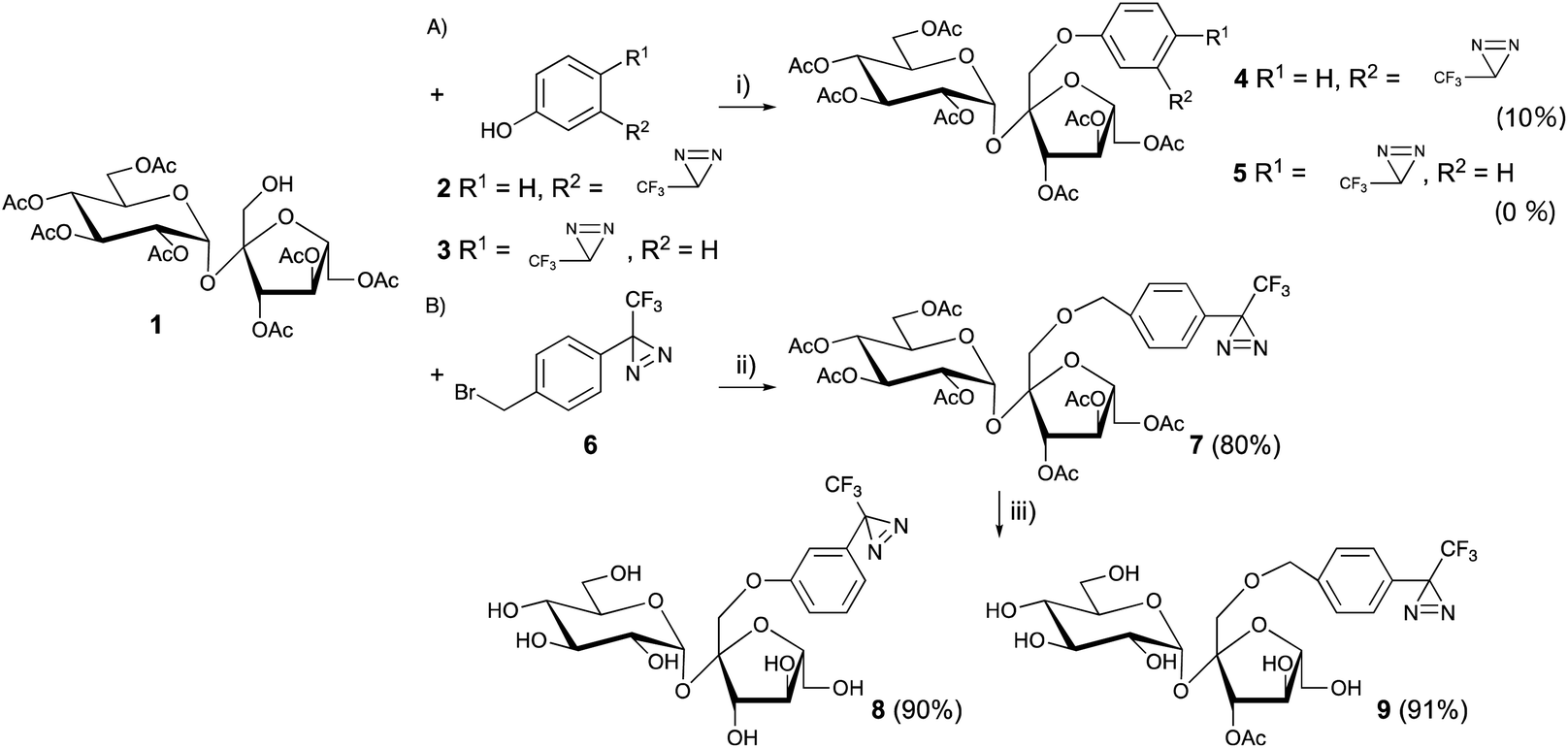 | ||
Fig. 3 Synthesis of trifluoromethylphenyldiazirine-modified sucrose derivatives at the 1′-position. (i) 2 or 3, DIAD, Ph3P (each 7.5 eq.), toluene, 60 °C, 24 h, (ii) 4 (2 eq.), Ag2O (3 eq.), MS-4A, CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n-hexane = 1:4, 60 °C, N2, 16 h, (iii) NH3–CH3OH, rt, 12 h. :n-hexane = 1:4, 60 °C, N2, 16 h, (iii) NH3–CH3OH, rt, 12 h. | ||
However, it has been reported that the more sterically hindered neopentyl-like 1′-hydroxyl modification of sucrose exhibited the lowest reactivity among the primary hydroxyls in many reactions,25,28,29 which confers difficulties to the modification of the 1′-position of sucrose.
Benzyl ether modifications at the 1′-position with benzyl halide and silver oxide were performed to overcome the lower reactivity of the 1′-hydroxyl group in sucrose.
1′-Hydroxy heptaacetyl sucrose 1 and the diazirinyl benzyl halide derivative 6 were reacted in the presence of Ag2O in CH2Cl2, which is a common solvent in carbohydrate chemistry. A large excess of diazirinyl benzyl halide derivative (10 eq.) and Ag2O (15 eq.) at 60 °C for 24 h afforded a moderate yield (∼60%) for benzylation.30 However, the conditions with large excess were not suitable for rare benzyl halide derivatives. A detailed analysis of the reaction mixture revealed that the benzyl alcohol derivatives were generated under the reaction conditions. The hydrolysis was inhibited in the hydrocarbon solvents of (cyclo)hexane and n-pentane. However, the solubility of the carbohydrate derivatives was quite low in these solvents. The use of a co-solvent in (cyclo)hexane and CH2Cl2 (4:1) improved the benzylation over 80% in the presence of diazirinyl benzyl halide derivatives (2 eq.) and Ag2O (3 eq.) at 60 °C under an N2 atmosphere (Fig. 3B).31 Subsequently, deprotection of the acetyl groups in both 5 and 7 afforded the 1′-diazrinyl sucrose derivatives 8 and 9.
2.2. D-Amino acids
As the receptor distinguishes D- and L-amino acids, with a preference for D-amino acids,32–34 the structural relationships between D-amino acids and other sweeteners and the structural features of D-amino acid derivatives that favor the activation of the sweet taste receptor have been studied using conformational analysis by crystallography, NMR analysis, and molecular modeling. However, it is still difficult to understand the receptor-bound conformations of the sweeteners, as the structural information on the ligands complexed with the receptor is limited.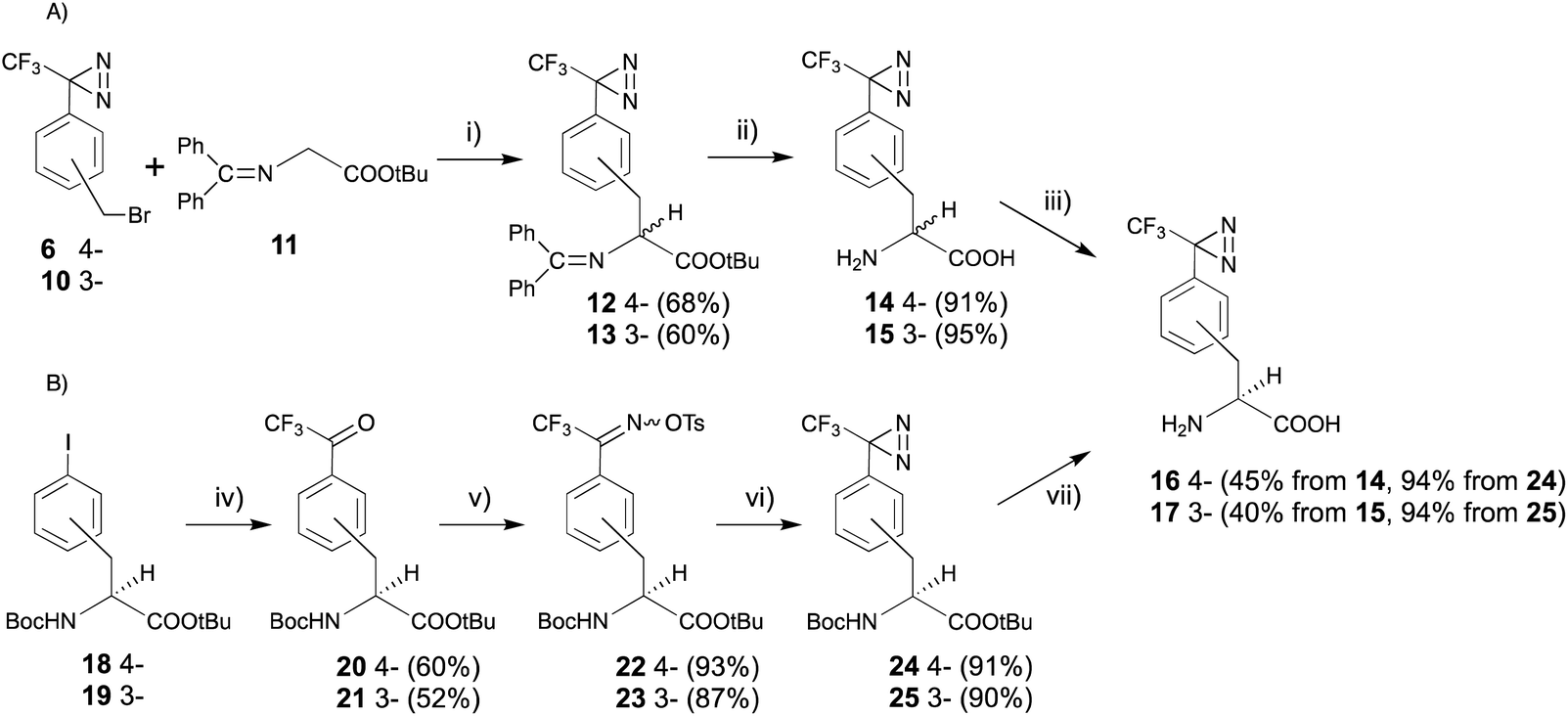 | ||
| Fig. 4 Synthesis of (3- and 4-trifluoromethyldiazirinyl)-D-phenylalanine derivatives. (i) CsOH, Bu4NBr, CH2Cl2, rt, 12 h, (ii) TFA, rt, 2 h, (iii) L-amino acid oxidase, pH 7.0, 37 °C, 12 h, (iv) (1) MeLi, (2) tBuLi, (3) CF3CO2Et, toluene, −78 °C, 12 h, (v) (1) NH2OH·HCl, pyridine, 70 °C, 1.5 h, (2) TsCl, TEA, acetone, rt, 1 h, (vi) NH3 (l), ether, 80 °C, 9 h, (vii) TFA, rt, 8 h. | ||
Recently, our group reported a trifluoromethyldiazirinyl moiety constructed on aromatic chiral phenylalanine derivatives in a stereocontrolled manner. The 3- or 4-iodo derivatives with an alkaline-stable protecting group at both the N- and C-termini (18 and 19) were converted to the trifluoroacetyl derivatives (20 and 21), which were subjected to oximation and o-tosylation (22 and 23), successively. One-pot conversion, which consisted of the construction and oxidation of the diaziridine in liquid ammonia can be applied to the phenylalanine derivatives without racemization (24 and 25). The one-pot reaction with improved reagents, LiNH2, promoted racemization of the α-amino acid moiety. Acidic deprotection afforded desired products (16 and 17) (Fig. 4B).42 The synthetic route was applied for the synthesis of deuterated derivatives from the deuterated phenylalanine derivatives.43 The trifluoromethyldiazirinyl D-phenylalanine derivatives (16 and 17) had stronger sweetness than unmodified D-phenylalanine and tryptophan, and the same as that of 1.25 mM aspartame. The responses were drastic decrease in the presence of lactisole, which is an inhibitor of sweetness (Table 1).44
| Normalized response | |
|---|---|
| Aspartame | 0.83 ± 0.20 |
| D-Tryptophane | 0.57 ± 0.06 |
| D-Phenylalanine | 0.14 ± 0.12 |
| D-Phenylalanine (4-dia) 16 | 0.72 ± 0.12 |
| D-Phenylalanine (3-dia) 17 | 0.97 ± 0.02 |
| 16 + lactisole | 0 |
| 17 + lactisole | 0 |
A complex model of D-phenylalanine in the ATD of the hT1R2 has been constructed using the crystal structure of the ATD of the metabotropic glutamate receptor type-1 (mGluR1).12 D-Phenylalanine was docked into the binding site with a guide of the salt bond between the amino group of the ligand and Glu302 of the receptor, which is conserved and crucial in binding in mGluR1 and was suggested to be crucial for the binding of some sweeteners, such as aspartame, by mutation experiments.11 The phenyl group bound a hydrophobic pocket, which also binds a larger indole moiety of D-tryptophan. The docking of the designed diazirinyl D-phenylalanines at the same site suggested that the receptor accepts the diazirinyl moiety in the binding pocket.44
Tryptophan has been synthesized from an indole with serine in acetic acid and acetic anhydride under reflux conditions.49 In the original report, the active species were generated at high temperatures in the presence of indole derivatives. The diazirinyl moieties of 36 and 37 were also decomposed in acetic acid under reflux conditions. The tryptophan skeletons were constructed following a detailed analysis of the reaction mixture. The active species were generated from L-serine, acetic anhydride, and acetic acid in reflux conditions without indole derivatives. The generated active species could react with diazirinyl indoles at low temperature to prevent the decomposition of the diazirinyl ring. The racemate of diazirinyl N-acetyl tryptophan derivatives 38 and 39 was subjected to enzymatic resolution with D-aminoacylase to afford optically pure diazirinyl D-tryptophan (40 and 41) without the decomposition of the diazirinyl moiety (Fig. 5).50
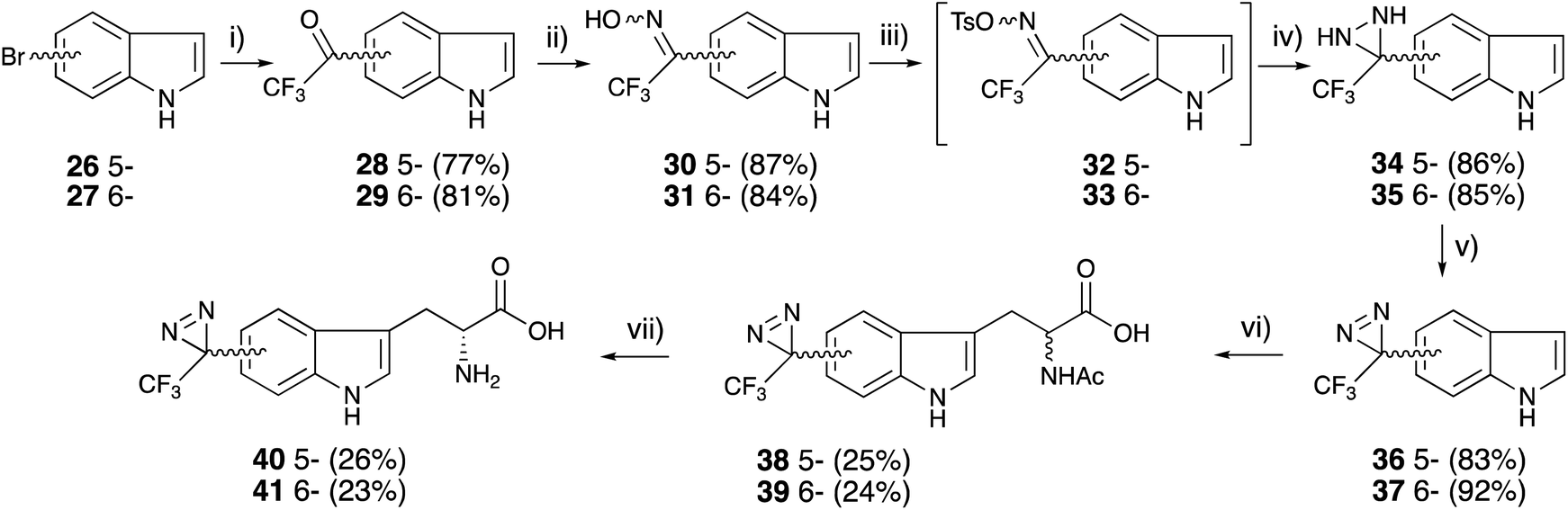 | ||
| Fig. 5 Synthesis of (5- and 6-trifluoromethyldiazirinyl)-D-tryptophane derivatives. (i) (1) KH, tert-BuLi, (2) trifluoroacetyl piperidine, THF, −78 °C, 2 h, (ii) NH2OH·HCl, pyridine, 80 °C, 3 h, (iii) TsCl, TEA, acetone, 0 °C, 1 h, (iv) (1) NH3 (l), ether, −78 °C, (2) rt, 6 h, (v) MnO2, ether, rt, 1 h, (vi) (1) L-serine, acetic anhydride, acetic acid, 75 °C, 1 h, (2) rt, 1.5 h, (vii) D-aminoacylase, sodium phosphate pH 7.6, CoCl2, 37 °C, 24 h. | ||
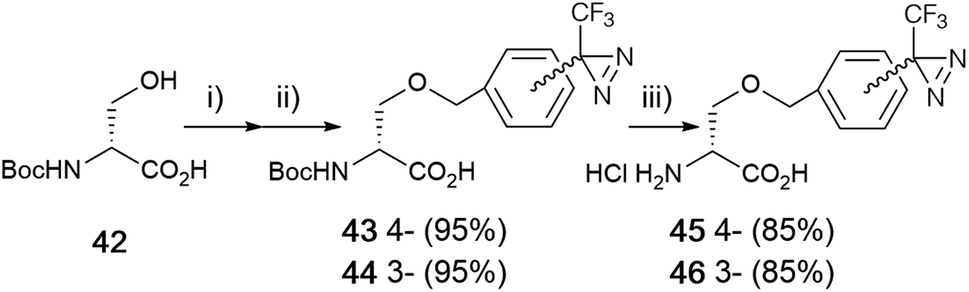 | ||
| Fig. 6 Synthesis of o-(3- and 4-trifluoromethyldiazirinyl)benzyl-D-serine derivatives. (i) NaH (3.3 eq.), DMF, 0 °C, 1 h, (ii) diazirinyl benzyl bromide 4 or 10 (1.5 eq.), DMF, rt, 15 min, (iii) 4 M HCl–dioxane, rt, 1.5 h. | ||
Boc-D-serine (42) was chosen for the starting material for the preparation of the o-benzyl serine derivative. First, the alkoxide ion generated from the hydroxyl group of a side chain, followed by the reaction with benzyl halide derivatives. A detailed analysis revealed that the chemical yield was influenced by the proportion of reagents and reaction time for each step. Sodium hydride treatment was preferred over an hour with 3.3 equivalents and the benzylation step was optimized within 15 min with 1.5 equivalents of benzyl bromide derivatives at room temperature. There was no difference in the benzylation between stereoisomers. Boc-D-serine (42) was treated with NaH (3.3 eq.) in DMF for 60 min, followed by the reaction with diazirinyl benzyl bromides 4 or 10 (1.5 eq.) within 15 min to afford o-benzyl-D-serine derivatives 43 and 44 with good yields (>85%) (Fig. 5).57
2.2. Aspartame
Aspartame (L-Asp-L-Phe-OMe, 47) is one of the most well-known artificial sweeteners. Based on structure–activity relationships, the substitution of a methoxy group at the 2-position of phenylalanine (48) did not reduce the sweetness and 3-substitution also had less influence on sweet potency compared with aspartame.58 In contrast, substitution at the 4-position (49) dramatically decreased the sweetness, and the 4-diazirinyl aspartame derivative (50) had no sweet taste, as shown in a pioneering work on diazirine-based photoaffinity labeling.36 These results indicated that the substitution of 2-methoxy-5-trifluoromethyldiazirinyl on phenylalanine (51) maintained sweetness (Fig. 7).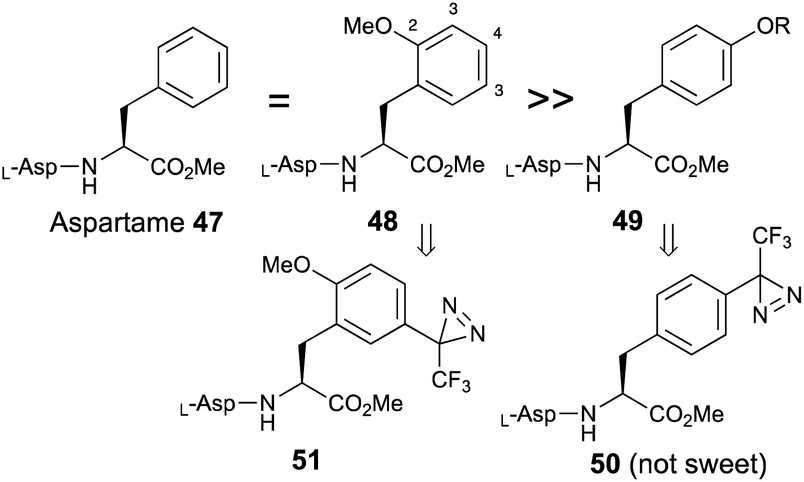 | ||
| Fig. 7 Effects of substitution of an aromatic moiety on phenylalanine on the sweetness of aspartame. | ||
4-Methoxy-trifluoromethylphanyldiazirine (52) was prepared from 4′-methoxy-2,2,2-trifluoroacetophenone with general methods for diazirine construction.59,60 The formyl group was introduced by a Friedel–Crafts reaction with dichloromethyl methyl ether in the presence of titanium chloride at 0 °C. Benzaldehyde aldehyde derivative (53) was reduced with sodium borohydride to afford benzyl alcohol (54), which was converted to benzyl bromide derivatives (55) with carbon tetrabromide and triphenylphosphine. The diazirinyl benzyl bromide (55) was subjected to asymmetric synthesis with glycine derivative (9 → 11) in the presence of the phosphazene base BTPP and catalytic amounts of o-allyl-N-(9-anthracenylmethyl)chinchonidium bromide at −78 °C.61 The condensed product (12) was slightly decomposed during silica gel-column chromatography, so the reaction mixture was directly deprotected with TFA to afford a new photoreactive L-phenylalanine derivative (57). The chirality of the synthesized 57 was analyzed using chiral HPLC, to reveal an ee of over 96%. The diazirinyl phenylalanine (57) was converted to the methyl ester (58) with thionyl chloride in methanol, then reacted with N-Boc-L-Asp(OtBu)-OSu in the presence of diisopropyl ethyl amine at room temperature, followed by the deprotection of protecting groups with TFA to afford the new photoreactive aspartame derivative (51) in good yield (Fig. 8). The synthesized photoreactive aspartame derivative 51 was subjected to preliminary sensory evaluations using a filter-paper disk test.62 The synthetic compound had lower sweet potency than the parent compound aspartame, but a slightly higher sweet potency than sucrose.
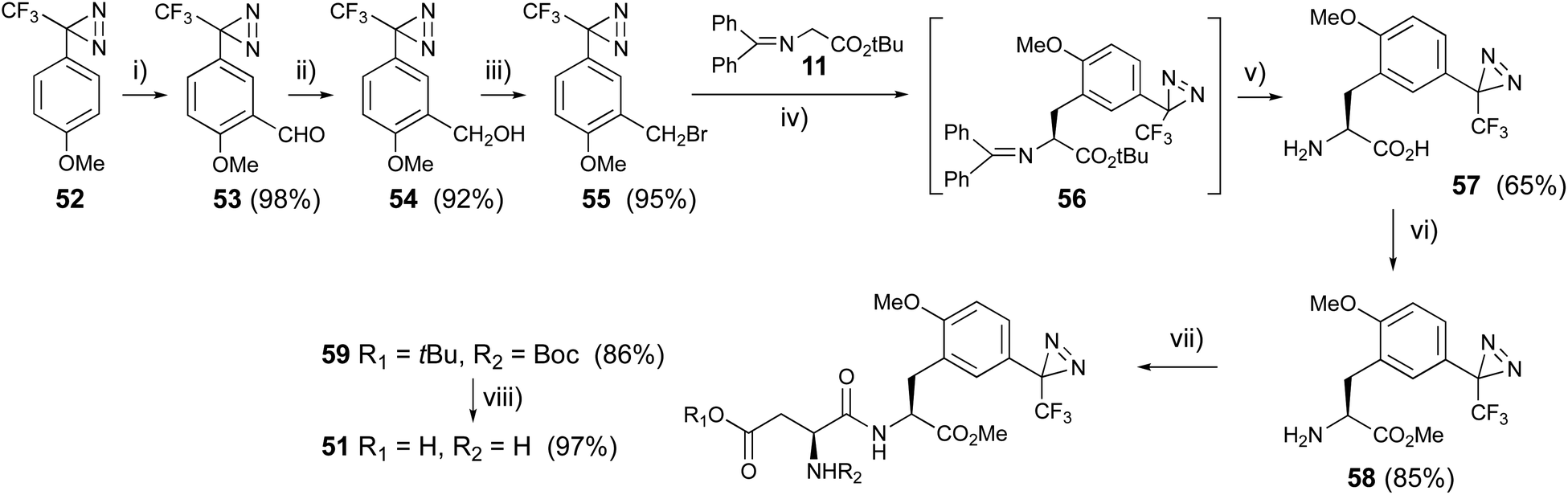 | ||
| Fig. 8 Synthesis of trifluoromethyldiazirinyl aspartame derivative. (i) Cl2CHOMe, TiCl4, 0 °C, 1 h, (ii) NaBH4, EtOH, rt, 4 h, (iii) CBr4, Ph3P, CH2Cl2, rt, 4 h, (iv) phosphazene base BTPP, o-allyl-N-(9-anthracenylmethyl)chinchonidium bromide, CH2Cl2, −78 °C, 8 h, (v) TFA, 0 °C, 2 h, (vi) SOCl2, MeOH, rt, 8 h, (vii) N-Boc-L-Asp(OtBu)OSu, (i-Pr)2NEt, THF, rt, 8 h, (viii) TFA, 0 °C, 3 h. | ||
2.3. Saccharin
Saccharin is one of the most common artificial sweeteners in the world and is several hundred times sweeter than sucrose. Suami et al. reported on several structure–activity relationships for saccharin, and found that substitutions at the 5- and 6-positions in saccharin were tolerated with regards to its biological activities.63 In addition, some sweet compounds also interact with other taste modalities. 3-(p- or m-Tolyl)-3-(trifluoromethyl)diazirines (60 and 61) were subjected to aromatic chlorosulfonation at adjacent positions of the toluene methyl group with chlorosulfonic acid at −20 °C. The addition of the strong acid at a higher temperature promoted decomposition of the diazirinyl ring.43,64 The sulfonyl chloride was converted to N-alkylated sulfonamide using tBuNH2 at room temperature. The reaction in the triethylamine and tBuNH2 system both 2 equivalents; experimental conditions reported65 afforded a more complex mixture than that in tBuNH2 only (4 equivalents). Oxidative cyclization with H5IO6 and CrO3 between the methyl group and o-oriented N-tert-butyl sulfonamide synthesized the saccharin skeleton (62 and 63).The purification for each step generated lower isolated yields for the cyclized compounds 62 and 63 (less than 10%). We performed these three steps as one-pot reactions (∼30% yields). The deprotection of the tBu group with TFA under reflux conditions afforded diazirinyl saccharin derivatives (64 and 65, Fig. 9). The synthesized photoreactive saccharin derivatives were insoluble in water. We performed further purification of the synthesized saccharin derivatives by converting them to the corresponding sodium salts with aqueous sodium hydroxide.
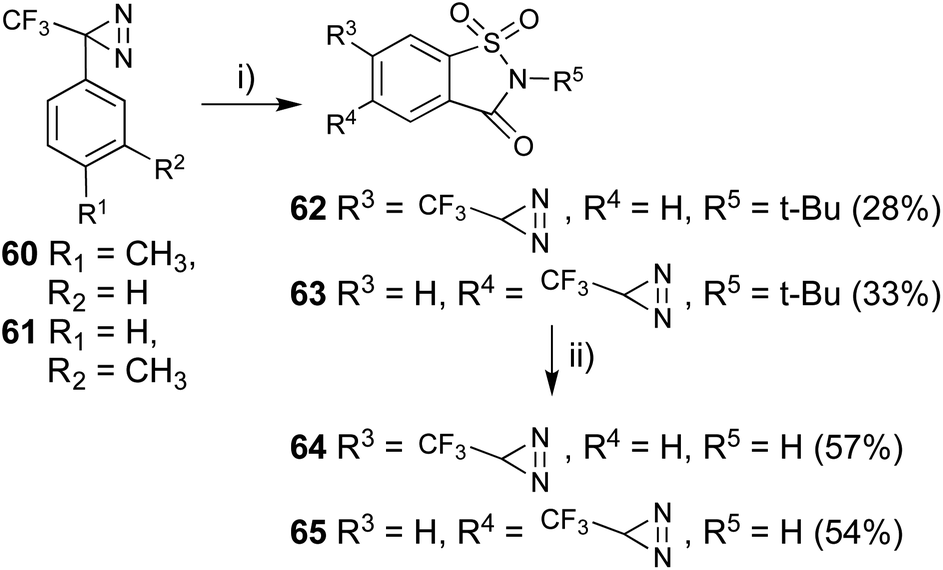 | ||
| Fig. 9 Synthesis of 5- or 6-trifluoromethyldiazirinyl saccharin derivatives. (i) (1) ClSO3H, TiCl4, −20 °C, 1 h, then rt, 4 h, (2) t-BuNH2, CH2Cl2, 0 °C, 2 h, then rt, 22 h, (3) H5IO6, CrO3, Ac2O, CH3CN, 0 °C, 0.5 h, then rt, 48 h, (ii) CF3COOH, reflux, 24 h. | ||
The synthesized photoreactive saccharin derivatives were subjected to preliminary gustatory receptor assays at 10 mM. Saccharin, compounds 64 and 65 have 90%, 80%, and 65% relative sweetness activity, respectively, against the same concentration of aspartame.52,66 The response to the synthetic compounds was completely inhibited by addition of lactisole, which is one of the specific inhibitors in the sweet taste assay (Table 2).67
| Normalized response | |
|---|---|
| Saccharin | 0.86 ± 0.17 |
| Saccharin (6-dia) 64 | 0.64 ± 0.30 |
| Saccharin (5-dia) 65 | 0.81 ± 0.05 |
| 64 + lactisole | 0 |
| 65 + lactisole | 0 |
2.4. 2-Propoxyaniline
5-Nitro-2-propoxyaniline (66, Fig. 9) has a sweetness intensity that is approximately 4000-times greater than that of sucrose and is commonly considered as one of the strongest artificial sweeteners.68 Despite previous use as an artificial sweetener, it has been banned because of its possible toxicity.69 The mechanism of sweetness and toxicity of 5-nitro-2-propoxyaniline has still not been studied. There have been only a few studies using homology modeling to determine the mechanism of action of 5-nitro-2-propoxyaniline at the gustatory receptor.70,71 According to a previous report,72 3-amino-4-propoxybenzonitrile (67), in which the nitro group of 66 was substituted by a cyano group, resulted in almost no decrease in sweetness.To the best of our knowledge, the synthesis of photoreactive-2-propoxyaniline derivatives for photoaffinity labeling has not yet been reported. The introduction of the diazirinyl photophore at the 5-position of 2-propoxyaniline may be one possible reaction to the introduce photophores into the ligand skeleton (Fig. 10).
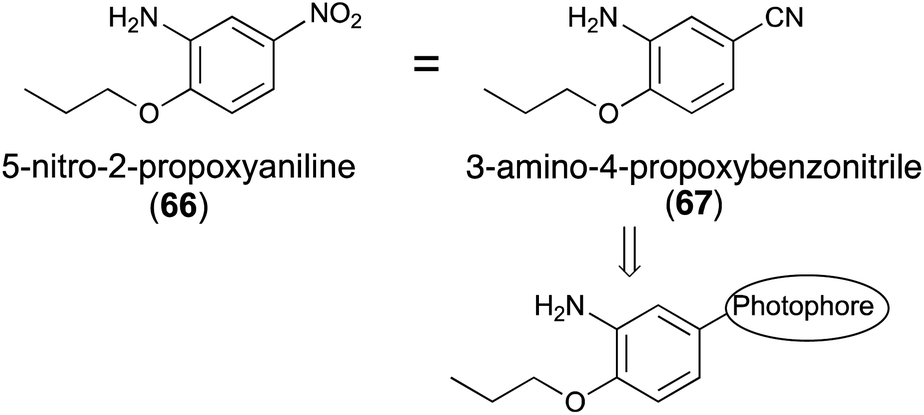 | ||
| Fig. 10 Substitution effects of the aromatic moiety of the sweetness of 5-nitro-2-propoxyaniline (66) and its derivatives. | ||
4-Propoxybenzaldehyde (68) was treated with CF3-TMS73 and worked up with 1 M HCl to construct the 2,2,2-trifluoro-1-phenylethan-1-ol skeleton (69), then oxidized with Dess–Martin periodinane74 to afford the trifluoroacetophenone derivative (70) in excellent yield. Nitration of compound (70) with fuming HNO3 in acetic anhydride at 0 °C proceeded selectively (71). The trifluoroacetyl moiety was converted to the corresponding oxime (72) and o-tosyloxime (73) to diaziridine (74) using a general method.75 The reduction of the nitro group with Na2S2O4 (ref. 76) to amine (74) occurred more quickly than oxidation of diaziridine (75) to diazirine (76) (Fig. 11).77
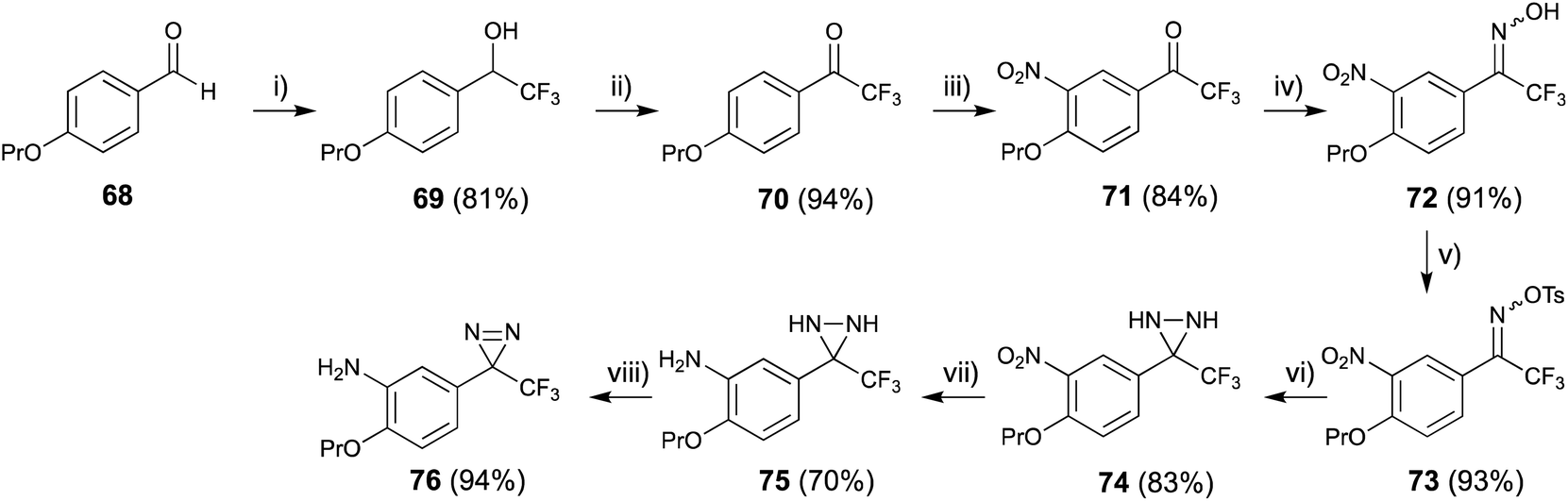 | ||
| Fig. 11 Synthesis of 2-propoxy-5-trifluoromethyldiazirinyl aniline derivative. (i) (1) TMS-CF3, CsF, THF, rt, 4 h, (2) 1 M HCl, rt, 6 h, (ii) Dess–Martin periodinane, TFA, CH2Cl2, rt, 12 h, (iii) HNO3, Ac2O, 0 °C, 40 min, (iv) NH2OH·HCl, pyridine, 80 °C, 3 h, (v) TsCl, TEA, 0 °C, 1 h, (vi) NH3 (l), ether, rt, 6 h, (vii) Na2S2O4, THF, EtOH, H2O, rt, 1 h, (viii) MnO2, ether, rt, 1 h. | ||
3. Photoaffinity probes for inhibition (negative allosteric modulation) of the sweet taste receptor
3.1. Lactisole ((±)-2-(4-methoxyphenoxy)-propionic acid)
Lactisole, which is known as an inhibitor (negative allosteric modulator; NAMs) of the human sweet taste receptor, has a 2-phenoxypropionic acid skeleton and has been shown to interact with the transmembrane domain of the T1R3 subunit (T1R3-TMD) of the receptor. Lactisole was first isolated from roasted coffee beans,78 and has naturally occurring optical isomers owing to the lactic acid structure present in the molecule. In general, L-lactic acid [(S)-lactic acid] is more abundant in plants than D-lactic acid, so the (S)-isomer of lactisole is predominant in roasted coffee beans.79The parent skeleton, 2-phenoxypropanoic acid, is found in several phenoxy herbicides, such as 2-(2,4-dichlorophenoxy)propanoic acid (dichlorprop, 2,4-DP) 2-(4-chlorophenoxy)propanoic acid (4-CPP),80 and the (R)-isomers that have predominantly herbicide activities. Racemic lactisole and 2,4-DP (2-(2,4-dichlorophenoxy)propionic acid) are also used as NAMs for the sweet taste receptors owing to their specific interaction with T1R3-TMD;80 thus, the detailed functional analysis of the stereocenter may be important to elucidate the mechanism of sweet taste chemoreception.
The key steps in the construction of a lactisole and 2,4-DP skeleton in an asymmetric manner are achieved by the Mitsunobu reaction of 4-methoxyphenol (77) or 2,4-dichlorophenol (78) and optically pure methyl lactate (79), followed by hydrolysis to afford optically pure lactisole (82) and 2,4-DP (83), respectively (Fig. 12).
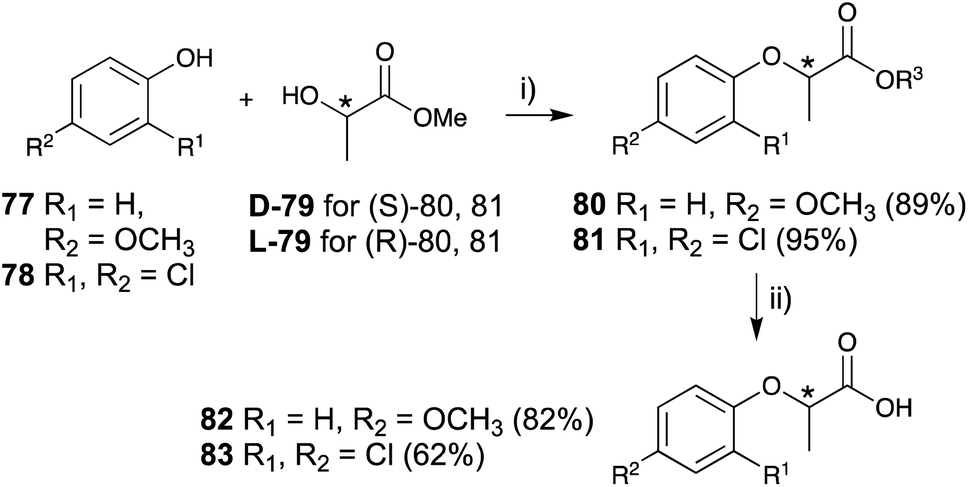 | ||
| Fig. 12 Asymmetric synthesis of lactisole and (2-(2,4-dichlorophenoxy)propionic acid) derivative. (i) (1) PPh3, CH2Cl2, 0 °C, 10 min, (2) DEAD, rt, 12 h, (ii) K2CO3, CH3OH, H2O, reflux, 2 h. | ||
The (S)-isomers of both lactisole and 2,4-DP showed potent inhibitory activities with the following IC50 values: 20 μM for (S)-lactisole, 2.6 μM for (S)-2,4-DP, and 45 μM for (R)-2,4-DP. (R)-2,4-DP was over 10-fold less effective than (S)-2,4-DP, and (R)-lactisole exerted almost no inhibition at millimolar concentrations. These results were consistent with both point mutations of T1R3 affinity measurements and simulations for molecular dynamics-based energy minimization in silico. The detailed in silico analysis indicated that the o-substitution of the phenol moiety may play an important role in the strong inhibitory activity.81
Trifluoromethyldiazirine-based lactisole derivatives have not yet been reported. The three membered azi (N N) partial structure of trifluoromethyldiazirine is not stable in the Mitsunobu conditions because the reactant, DEAD, also has azo group in the structure. The reaction with diazirinyl phenol derivatives and 1′-hydroxy peracetylsucrose with DEAD or Tsuda reagents did not proceed in our previous study.27 The synthetic plan was based on the preparation of trifluoroacetyl modified on the aromatic ring of lactisole, and then the construction of a diazirinyl-three membered ring on the trifluoroacetyl group. The liquid ammonia reagent is essential for the construction of diazirine moiety, but the reagent also reacts with a methyl ester to form amide; tert-butyl ester was utilized for this purpose.
p-Trifluoroacetyl anisole 84 was treated with lithium chloride under reflux condition to obtain trifluoroacetyl phenol 86.77 m-Trifluoroacetyl phenol 85 was synthesized with boron tribromide at room temperature. The Mitsunobu reactions for these phenols with chiral methyl lactate were performed in an identical manner to construct lactisole methyl ester skeletons 88 and 89. The hydrolysis of methyl esters with K2CO3 under reflux conditions yielded the corresponding trifluoroacetyl-substituted carboxylic acids 90 and 91, which were difficult to convert diazirine directly. The carboxylic acids were converted to tert-butyl esters with tert-butyl bromide in the presence of K2CO3 and TBAB under reflux conditions. Although the reactions were not completed within 5 h, the decomposition of the diazirinyl moiety of the starting material was observed over 5 h. tert-Butyl derivatives 92 and 93 were obtained in moderate yields and the starting materials were recovered from the reaction mixture (Fig. 13). Mitsunobu reactions for trifluoroacetyl phenol 86 and 87 with the chiral tert-butyl lactate were also conducted, but no reactions were observed.
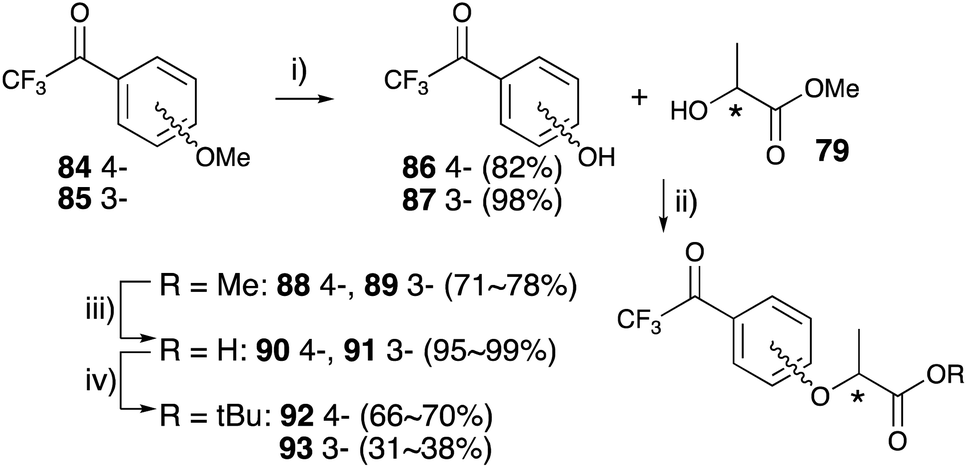 | ||
| Fig. 13 Asymmetric synthesis of 3- and 4-trifluoroacetyl lactisole derivatives. (i) LiCl, DMF, reflux, 5 h for 86, BBr3, CH2Cl2, rt, 2 h for 87, (ii) (1) PPh3, CH2Cl2, 0 °C, 10 min, (2) DEAD, CH2Cl2, rt, 8 h, (iii) K2CO3, MeOH, H2O, reflux, 2 h, (iv) t-BuBr, K2CO3, tetrabutylammonium bromide (TBAB), DMA, 55 °C, 5 h. | ||
2-Trifluoroactyl phenol, which was prepared by the Fries rearrangement of phenyl 2,2,2-trifluoroacetate with AlCl3,82 was subjected to the Mitsunobu reaction with chiral methyl lactate 79 and did not afford 2-trifluoroacetyl lactisole skeleton. The Mitsunobu reaction of salicylaldehyde and methyl lactate 79 followed by conversion of the aldehyde to the trifluoroacetyl group also failed owing to the instability of the methyl ester under the conditions for trifluoroacetyl construction.
Thioacetal-protected salicylaldehyde 94 (ref. 83) was selected for trifluoroacetyl substitution at the 2-position of lactisole. Compound 94 was subjected to the Mitsunobu reaction with chiral methyl lactate 79 to construct the lactisole methyl ester 95 in an identical manner to that described above. After hydrolysis of methyl esters with methanolic NaOH under reflux condition, the carboxylic acids 96 were converted to lactisole tert-butyl ester 97, and then deprotected with methyl iodide. The aldehyde 98 was treated TMS-CF3 and then oxidized with Dess–Martin periodinane to afford 2-trifluoroacetyl-substituted lactisole tert-butyl ester 99 (Fig. 14).
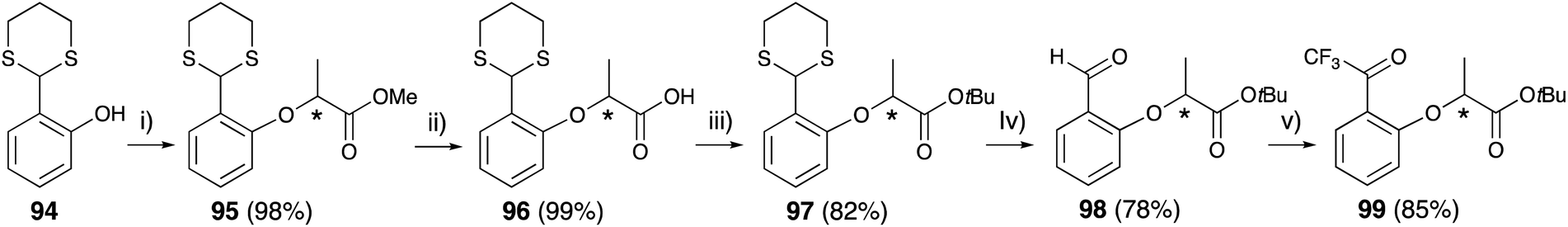 | ||
| Fig. 14 Asymmetric synthesis of 2-trifluoroacetyl lactisole derivative. (i) (1) 79, PPh3, CH2Cl2, 0 °C, 10 min, (2) DEAD, rt, 8 h, (ii) K2CO3, MeOH, H2O, reflux, 2 h, (iii) t-BuBr, K2CO3, TBAB, DMA, 55 °C, 5 h, (iv) CH3I, NaHCO3, CH3CN, H2O, rt, 24 h, (v) (1) TMS-CF3, K2CO3, DMF, rt, 4 h, (2) 1 M HCl, rt, 4 h, (3) Dess–Martin periodinane, CH2Cl2, rt, 3 h. | ||
Each trifluoroacetyl-substituted lactisole tert-butyl ester (92, 93, 99) was subjected to diazirine construction, oximation with hydroxylamine hydrochloride 100–102, tosylation of the oxime 103–105, diaziridine formation in liquid ammonia 106–108, and oxidation with activated MnO2 to obtain 109–111, which were examined for their chiral center configuration. The tert-butyl esters were hydrolyzed with TFA to afford trifluoromethyldiazirine-based lactisole derivatives 112–114 in good yields (Fig. 15).
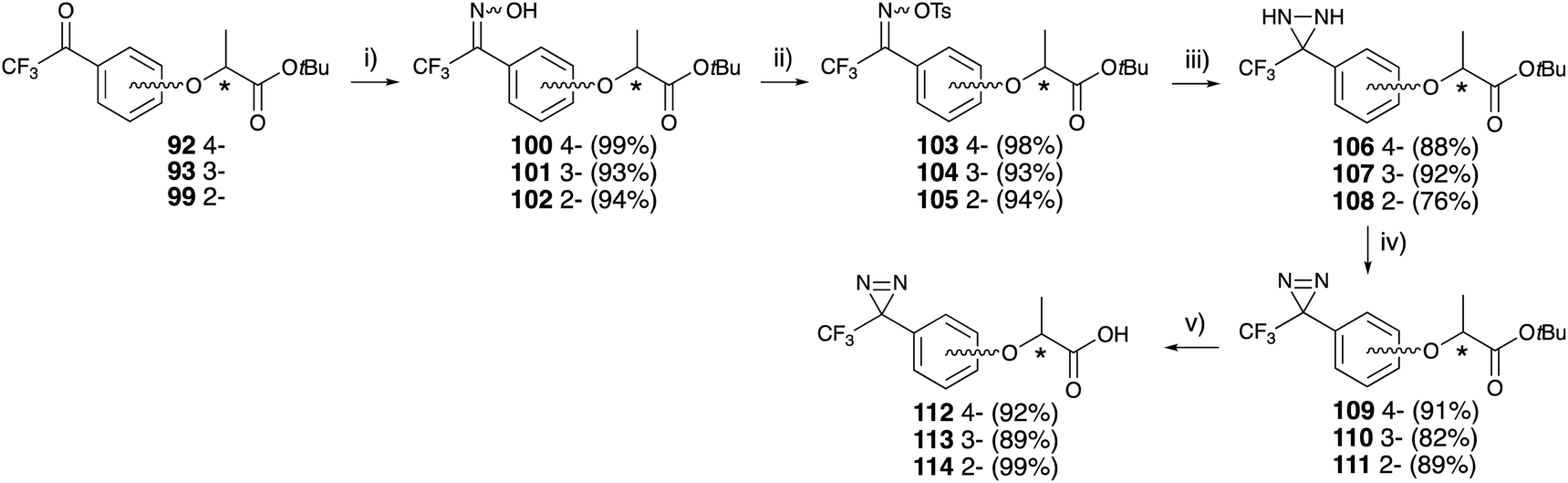 | ||
| Fig. 15 Asymmetric synthesis of trifluoromethyldiazirinyl lactisole derivatives. (i) H2NOH·HCl, pyridine, EtOH, 60 °C, 12 h, (ii) tosyl chloride, DMAP, CH2Cl2, rt, 45 min, (iii) NH3 (l), Et2O, rt, 12 h, (iv) MnO2, CH2Cl2, rt, 2 h, (v) CF3COOH, CH2Cl2, rt, 4 h. | ||
Human sweet receptor assays for diazirine-containing lactisole derivatives revealed that (S)-isomers at the 4- and 3-position of lactisole derivatives 112 and 113 had identical affinity to optically pure (S)-lactisole 82. These results indicated that photoaffinity labeling with synthetic lactisole derivatives would be useful for the functional analysis of the sweet taste receptor (Table 3).84
| (RS)-lactisole 82 | 82 ± 0.44 | (S)-112 | 12 ± 0.067 |
| (S)-82 | 20 ± 4.3 | (R)-112 | 250 ± 1.8 |
| (R)-82 | nd | (S)-113 | 19 ± 0.14 |
| (RS)-2,4-DP 83 | 7.9 ± 0.067 | (R)-113 | 250 ± 1.7 |
| (S)-83 | 2.6 ± 0.51 | (S)-114 | 37 ± 0.28 |
| (R)-83 | 45 ± 6.1 | (R)-114 | 300 ± 1.8 |
4. Conclusions
There are many challenges to elucidating the sweet taste chemoreception mechanism using photoaffinity labeling, which is one of the chemical biology/bioorganic chemistry methods. Other photophores (phenylazide and benzophenone) have also been utilized for phenylalanine 115 (ref. 85) o-benzyl-D-serine 116–119 (ref. 57) 2-propoxyaniline 120, 121 (ref. 77) and lactisole 122–127 (ref. 84) (Fig. 16).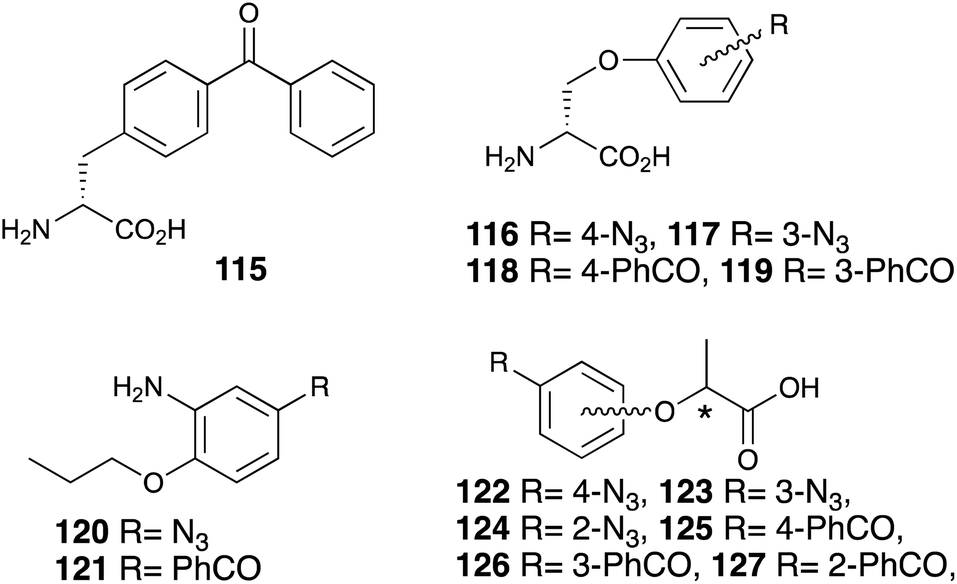 | ||
| Fig. 16 Photoreactive sweetener derivatives containing azide or benzophenone. | ||
It is often observed that photophore substitution reduces the sweet taste properties of the photoreactive benzophenone and phenylazide derivatives. The introduction of the trifluoromethyl diazirinyl group on the aromatic moiety of sweeteners has become the first choice for this functionality.
Photoaffinity labeling strategies have also been developed to elucidate the bitter taste receptors, which are mediated by the T2R family of GPCRs. Photoreactive salicin 128–129 (ref. 86, 87) and phenylthiourea 131–136 (ref. 88) and highly concentrated saccharine derivatives will be used in bitter taste assays to elucidate the chemoreception mechanism (Fig. 17).
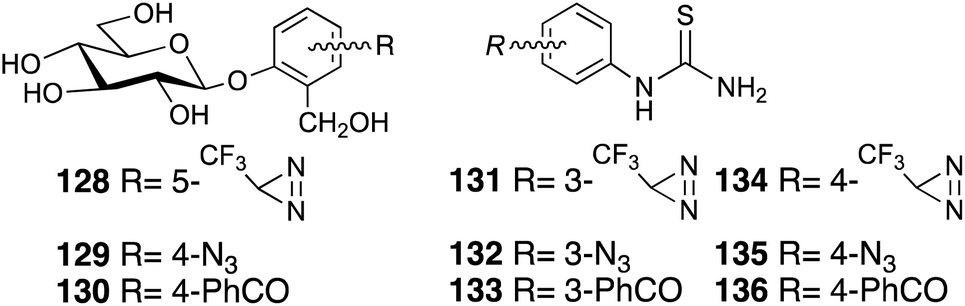 | ||
| Fig. 17 Photoreactive bitter substance derivatives. | ||
The combination of analysis techniques from molecular biology, computational biology, and chemical biology will play a key part in our understanding of gustatory chemoreception.
Author contributions
M. H. conceived the concept and idea of the present review and worked on the study design strategy. T. N. performed the literature searches and analyzed the data. T. M. selected the topics to be discussed. All authors have read and approved the final draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Part of this work was performed under the Cooperative Research Program of “Network Joint Research Center for Materials and Devices”.Notes and references
- P. Temussi, J. Mol. Recognit., 2006, 19, 188 CrossRef CAS PubMed.
- R. S. Shallenberger and T. E. Acree, Nature, 1967, 216, 480 CrossRef CAS PubMed.
- L. B. Kier, J. Pharm. Sci., 1972, 61, 1394 CrossRef CAS PubMed.
- C. Nofre and J. M. Tinti, Food Chem., 1996, 56, 263 CrossRef CAS.
- X. Yang, Y. Chong, A. Yan and J. Chen, Food Chem., 2011, 128, 653 CrossRef CAS.
- M. Zhong, Y. Chong, X. Nie, A. Yan and Q. Yuan, J. Food Sci., 2013, 78, S1445 CrossRef CAS PubMed.
- C. Rojas, D. Ballabio, V. Consonni, P. Tripaldi, A. Mauri and R. Todeschini, Theor. Chem. Acc., 2016, 135, ARTN 66 Search PubMed.
- M. Cui, P. H. Jiang, E. Maillet, M. Max, R. F. Margolskee and R. Osman, Curr. Pharm. Des., 2006, 12, 4591 CrossRef CAS PubMed.
- J. Chandrashekar, M. A. Hoon, N. J. Ryba and C. S. Zuker, Nature, 2006, 444, 288–294 CrossRef CAS PubMed.
- X. Li, L. Staszewski, H. Xu, K. Durick, M. Zoller and E. Adler, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4692–4696 CrossRef CAS PubMed.
- H. Xu, L. Staszewski, H. Tang, E. Adler, M. Zoller and X. Li, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14258–14263 CrossRef CAS PubMed.
- N. Kunishima, Y. Shimada, Y. Tsuji, T. Sato, M. Yamamoto, T. Kumasaka, S. Nakanishi, H. Jingami and K. Morikawa, Nature, 2000, 407, 971–977 CrossRef CAS PubMed.
- K. Masuda, A. Koizumi, K. Nakajima, T. Tanaka, K. Abe, T. Misaka and M. Ishiguro, Plos One, 2012, 7, e35380 CrossRef CAS PubMed.
- J. Brunner, Annu. Rev. Biochem., 1993, 62, 483–514 CrossRef CAS PubMed.
- T. Tomohiro, M. Hashimoto and Y. Hatanaka, Chem. Rec., 2005, 5, 385–395 CrossRef CAS PubMed.
- M. Hashimoto and Y. Hatanaka, Eur. J. Org. Chem., 2008, 2513–2523 CrossRef CAS.
- Y. Hatanaka, Chem. Pharm. Bull., 2015, 63, 1–12 CrossRef CAS PubMed.
- M. S. Platz, Acc. Chem. Res., 2002, 28, 487–492 CrossRef.
- W. L. Karney and W. T. Borden, J. Am. Chem. Soc., 1997, 119, 3347–3350 CrossRef CAS.
- R. E. Galardy, L. C. Craig and M. P. Printz, Nat. New Biol., 1973, 242, 127–128 CrossRef CAS PubMed.
- R. A. Smith and J. R. Knowles, J. Am. Chem. Soc., 1973, 95, 5072–5073 CrossRef CAS PubMed.
- T. Oku, T. Tokunaga and N. Hosoya, J. Nutr., 1984, 114, 1574–1581 CrossRef CAS PubMed.
- G. Carrea, S. Riva, F. Secundo and B. Danieli, J. Chem. Soc., Perkin Trans., 1989, 1, 1057–1061 RSC.
- R. Matsumi, H. Atomi, A. J. Driessen and J. van der Oost, Res. Microbiol., 2011, 162, 39–52 CrossRef CAS PubMed.
- S. Jarosz and M. Mach, Eur. J. Org. Chem., 2002, 2002, 769–780 CrossRef.
- O. Mitsunobu, Synthesis, 1981, 1981, 1–28 CrossRef.
- Y. Tsunekawa, K. Masuda, M. Muto, Y. Muto, Y. Murai, Y. Hashidoko, Y. Orikasa, Y. Oda, Y. Hatanaka and M. Hashimoto, Heterocycles, 2012, 84, 283–290 CrossRef CAS.
- C. Chauvin and D. Plusquellec, Tetrahedron Lett., 1991, 32, 3495–3498 CrossRef CAS.
- D. M. Clode, D. McHale, J. B. Sheridan, G. G. Birch and E. B. Rathbone, Carbohydr. Res., 1985, 139, 141–146 CrossRef.
- L. Wang, Z. P. Tachrim, N. Kurokawa, F. Ohashi, H. Wakasa, Y. Sakihama, Y. Hashidoko, T. Suzuki and M. Hashimoto, Arkivoc, 2018, 58–65 Search PubMed.
- L. Wang, Y. Hashidoko and M. Hashimoto, J. Org. Chem., 2016, 81, 4464–4474 CrossRef CAS PubMed.
- T. Kaneko, Nippon Kagaku Kaishi, 1938, 59, 433–439 CrossRef CAS.
- T. Kaneko, Nippon Kagaku Kaishi, 1939, 60, 531–538 CrossRef CAS.
- J. W. Finley and M. Friedman, J. Agric. Food Chem., 1973, 21, 33–34 CrossRef CAS PubMed.
- M. Nassal, J. Am. Chem. Soc., 1984, 106, 7540–7545 CrossRef CAS.
- L. B. Shih and H. Bayley, Anal. Biochem., 1985, 144, 132–141 CrossRef CAS PubMed.
- G. Baldini, B. Martoglio, A. Schachenmann, C. Zugliani and J. Brunner, Biochemistry, 1988, 27, 7951–7959 CrossRef CAS PubMed.
- C. W. G. Fishwick, J. M. Sanderson and J. B. C. Findlay, Tetrahedron Lett., 1994, 35, 4611–4614 CrossRef CAS.
- M. Hashimoto, Y. Hatanaka, Y. Sadakane and K. Nabeta, Bioorg. Med. Chem. Lett., 2002, 12, 2507–2510 CrossRef CAS PubMed.
- H. Nakashima, M. Hashimoto, Y. Sadakane, T. Tomohiro and Y. Hatanaka, J. Am. Chem. Soc., 2006, 128, 15092–15093 CrossRef CAS PubMed.
- M. T. Stankovich and B. G. Fox, Biochim. Biophys. Acta, 1984, 786, 49–56 CrossRef CAS.
- L. Wang, Y. Murai, T. Yoshida, A. Ishida, K. Masuda, Y. Sakihama, Y. Hashidoko, Y. Hatanaka and M. Hashimoto, Org. Lett., 2015, 17, 616–619 CrossRef PubMed.
- L. Wang, Y. Murai, T. Yoshida, M. Okamoto, K. Masuda, Y. Sakihama, Y. Hashidoko, Y. Hatanaka and M. Hashimoto, Biosci., Biotechnol., Biochem., 2014, 78, 1129–1134 CrossRef CAS PubMed.
- K. Masuda, A. Koizumi, T. Misaka, Y. Hatanaka, K. Abe, T. Tanaka, M. Ishiguro and M. Hashimoto, Bioorg. Med. Chem. Lett., 2010, 20, 1081–1083 CrossRef CAS PubMed.
- S. Bittner, R. Scherzer and E. Harlev, Amino Acids, 2007, 33, 19–42 CrossRef CAS PubMed.
- J. Ma, W. Yin, H. Zhou, X. Liao and J. M. Cook, J. Org. Chem., 2009, 74, 264–273 CrossRef CAS PubMed.
- P. S. Baran, B. D. Hafensteiner, N. B. Ambhaikar, C. A. Guerrero and J. D. Gallagher, J. Am. Chem. Soc., 2006, 128, 8678–8693 CrossRef CAS PubMed.
- H. Eto, C. Eguchi and T. Kagawa, Bull. Chem. Soc. Jpn., 1989, 62, 961–963 CrossRef CAS.
- H. R. Snyder and J. A. MacDonald, J. Am. Chem. Soc., 2002, 77, 1257–1259 CrossRef.
- Y. Murai, K. Masuda, Y. Sakihama, Y. Hashidoko, Y. Hatanaka and M. Hashimoto, J. Org. Chem., 2012, 77, 8581–8587 CrossRef CAS PubMed.
- S. S. Schiffman, K. Sennewald and J. Gagnon, Physiol. Behav., 1981, 27, 51–59 CrossRef CAS PubMed.
- K. Nakajima, T. Asakura, J. Maruyama, Y. Morita, H. Oike, A. Shimizu-Ibuka, T. Misaka, H. Sorimachi, S. Arai, K. Kitamoto and K. Abe, Appl. Environ. Microbiol., 2006, 72, 3716–3723 CrossRef CAS PubMed.
- G. Barcelo, J.-P. Senet, G. Sennyey, J. Bensoam and A. Loffet, Synthesis, 1986, 1986, 627–632 CrossRef.
- J. T. Spletstoser, P. T. Flaherty, R. H. Himes and G. I. Georg, J. Med. Chem., 2004, 47, 6459–6465 CrossRef CAS PubMed.
- N. Umezawa, N. Matsumoto, S. Iwama, N. Kato and T. Higuchi, Bioorg. Med. Chem., 2010, 18, 6340–6350 CrossRef CAS PubMed.
- M. Sechi, F. Carta, L. Sannia, R. Dallocchio, A. Dessi, R. I. Al-Safi and N. Neamati, Antiviral Res., 2009, 81, 267–276 CrossRef CAS PubMed.
- M. Hashimoto, T. Yoshida, Z. P. Tachrim, Y. Sakihama, Y. Hashidoko, Y. Hatanaka and Y. Kanaoka, Heterocycles, 2017, 95, 462–473 CrossRef CAS.
- M. Kawai, M. Chorev, J. Marin-Rose and M. Goodman, J. Med. Chem., 1980, 23, 420–424 CrossRef CAS PubMed.
- Y. Hatanaka, M. Hashimoto, H. Kurihara, H. Nakayama and Y. Kanaoka, J. Org. Chem., 1994, 59, 383–387 CrossRef CAS.
- M. Hashimoto, Y. Kato and Y. Hatanaka, Tetrahedron Lett., 2006, 47, 3391–3394 CrossRef CAS.
- E. J. Corey, F. Xu and M. C. Noe, J. Am. Chem. Soc., 1997, 119, 12414–12415 CrossRef CAS.
- K. Berling, J. Knutsson, A. Rosenblad and M. von Unge, Acta Otolaryngol, 2011, 131, 488–493 CrossRef PubMed.
- T. Suami, L. Hough, T. Machinami, T. Saito and K. Nakamura, Food Chem., 1998, 63, 391–396 CrossRef CAS.
- R. A. Moss, J. M. Fede and S. Yan, Org. Lett., 2001, 3, 2305–2308 CrossRef CAS PubMed.
- L. Xu, H. Shu, Y. Liu, S. Zhang and M. L. Trudell, Tetrahedron, 2006, 62, 7902–7910 CrossRef CAS.
- T. Ueda, S. Ugawa, H. Yamamura, Y. Imaizumi and S. Shimada, J. Neurosci., 2003, 23, 7376–7380 CrossRef CAS PubMed.
- L. Wang, T. Yoshida, Y. Muto, Y. Murai, Z. P. Tachrim, A. Ishida, S. Nakagawa, Y. Sakihama, Y. Hashidoko, K. Masuda, Y. Hatanaka and M. Hashimoto, Eur. J. Org. Chem., 2015, 2015, 3129–3134 CrossRef CAS.
- W. S. Cox, J. AOAC Int., 1953, 36, 749–750 CrossRef CAS.
- O. G. Fitzhugh, A. A. Nelson and J. P. Frawley, J. Am. Pharm. Assoc., 1951, 40, 583–586 CrossRef CAS PubMed.
- A. Bassoli, M. G. Drew, L. Merlini and G. Morini, J. Med. Chem., 2002, 45, 4402–4409 CrossRef CAS PubMed.
- G. Morini, A. Bassoli and P. A. Temussi, J. Med. Chem., 2005, 48, 5520–5529 CrossRef CAS PubMed.
- H. Iwamura, J. Med. Chem., 1980, 23, 308–312 CrossRef CAS PubMed.
- A. Chatterjee, D. J. Oh, K. M. Kim, K. S. Youk and K. H. Ahn, Chem.–Asian J., 2008, 3, 1962–1967 CrossRef CAS PubMed.
- R. J. Linderman and D. M. Graves, J. Org. Chem., 2002, 54, 661–668 CrossRef.
- Y. Hatanaka, M. Hashimoto, H. Nakayama and Y. Kanaoka, Chem. Pharm. Bull., 1994, 42, 826–831 CrossRef CAS.
- S. Kosemura, H. Emori, S. Yamamura, T. Anai, K. Tomita and K. Hasegawa, Tetrahedron Lett., 1997, 38, 2125–2128 CrossRef CAS.
- Y. Murai, T. Yoshida, L. Wang, K. Masuda, Y. Hashidoko, K. Monde, Y. Hatanaka and M. Hashimoto, Synlett, 2016, 27, 946–950 CrossRef CAS.
- E. B. Rathbone, G. D. Patel, R. W. Butters, D. Cookson and J. L. Robinson, J. Agric. Food Chem., 1989, 37, 54–58 CrossRef CAS.
- E. B. Rathbone, R. W. Butters, D. Cookson and J. L. Robinson, J. Agric. Food Chem., 1989, 37, 58–60 CrossRef CAS.
- E. L. Maillet, R. F. Margolskee and B. Mosinger, J. Med. Chem., 2009, 52, 6931–6935 CrossRef CAS PubMed.
- T. Nakagita, A. Ishida, T. Matsuya, T. Kobayashi, M. Narukawa, T. Hirokawa, M. Hashimoto and T. Misaka, PLoS One, 2019, 14, e0213552 CrossRef CAS PubMed.
- D. V. Sevenard, M. Vorobyev, V. Y. Sosnovskikh, H. Wessel, O. Kazakova, V. Vogel, N. E. Shevchenko, V. G. Nenajdenko, E. Lork and G.-V. Röschenthaler, Tetrahedron, 2009, 65, 7538–7552 CrossRef CAS.
- A. Neogi, T. P. Majhi, B. Achari and P. Chattopadhyay, Eur. J. Org. Chem., 2008, 2008, 330–336 CrossRef.
- T. Nakagita, A. Ishida, Z. P. Tachrim, L. Wang, T. Misaka and M. Hashimoto, Molecules, 2020, 25, 2790 CrossRef CAS PubMed.
- Y. Murai, L. Wang, Y. Muto, Y. Sakihama, Y. Hashidoko, Y. Hatanaka and M. Hashimoto, Heterocycles, 2013, 87, 2119–2126 CrossRef CAS.
- M. Sakurai, T. Yoshida, L. Wang, Y. Murai, K. Masuda, Y. Sakihama, Y. Hashidoko, Y. Hatanaka and M. Hashimoto, Heterocycles, 2015, 90, 698–705 CrossRef CAS.
- T. Yoshida, Y. Hashidoko and M. Hashimoto, Heterocycles, 2016, 93, 355–361 CrossRef CAS.
- A. Ishida, L. Wang, Z. P. Tachrim, T. Suzuki, Y. Sakihama, Y. Hashidoko and M. Hashimoto, Chemistryselect, 2017, 2, 160–164 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |