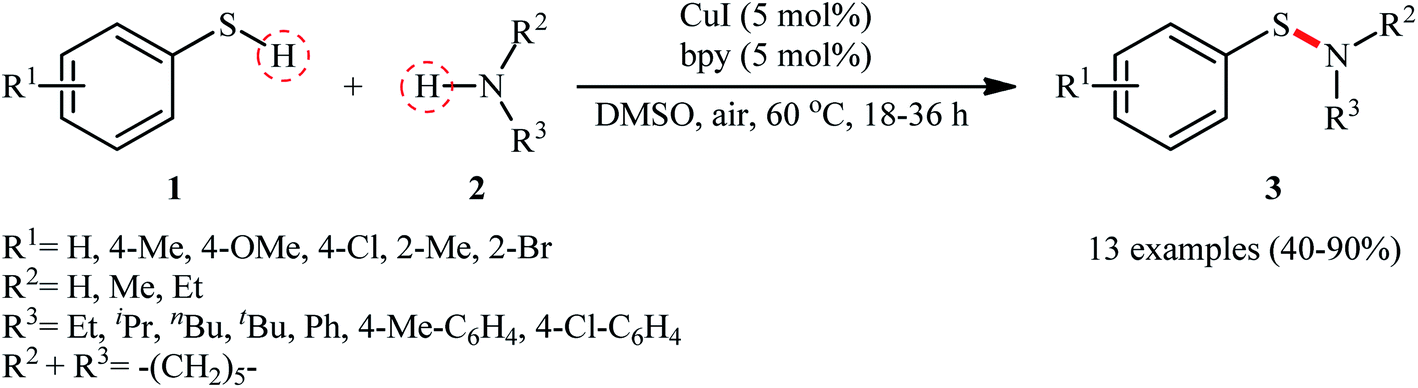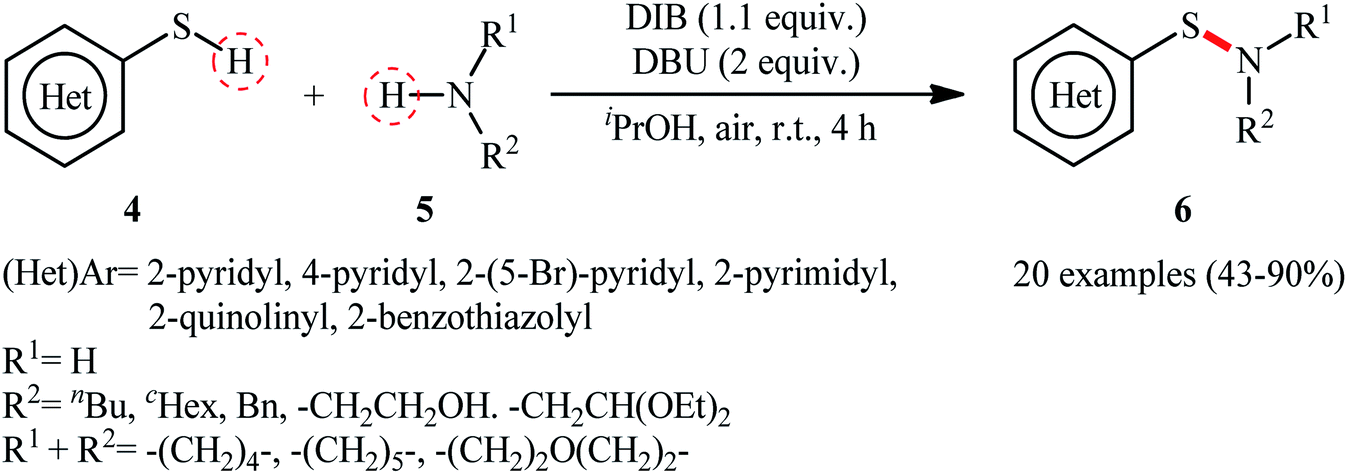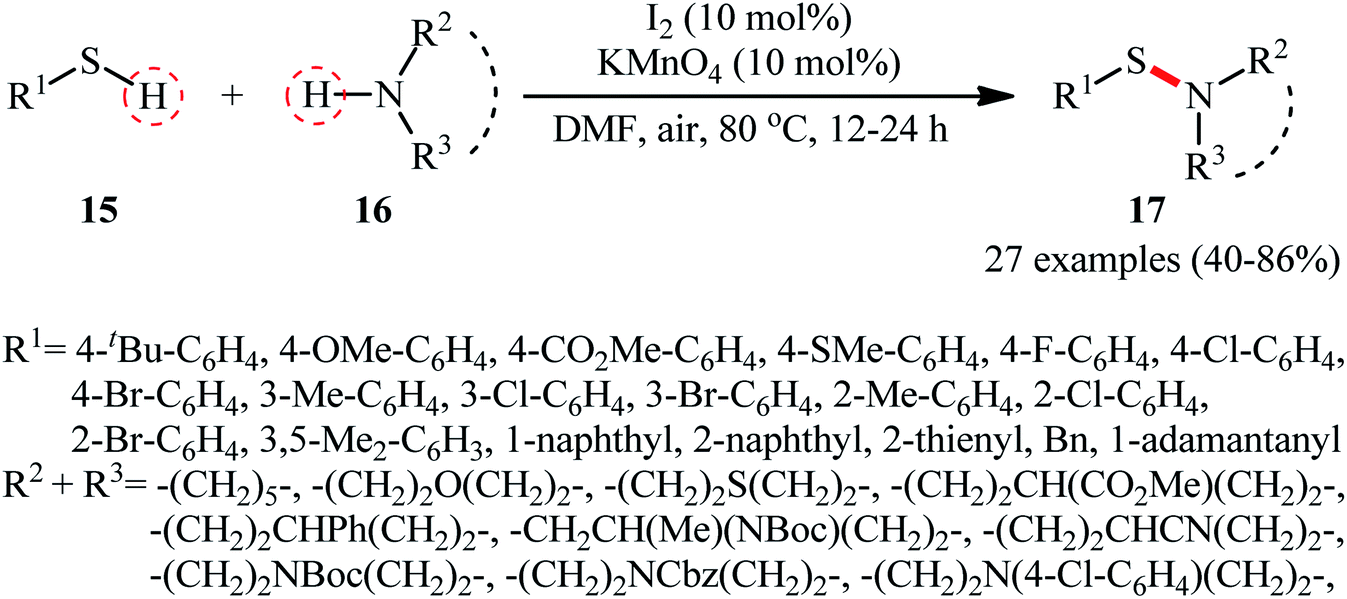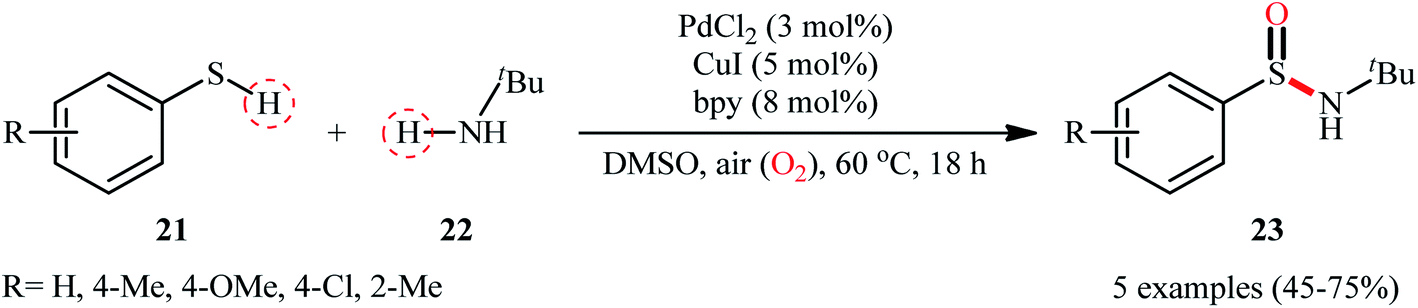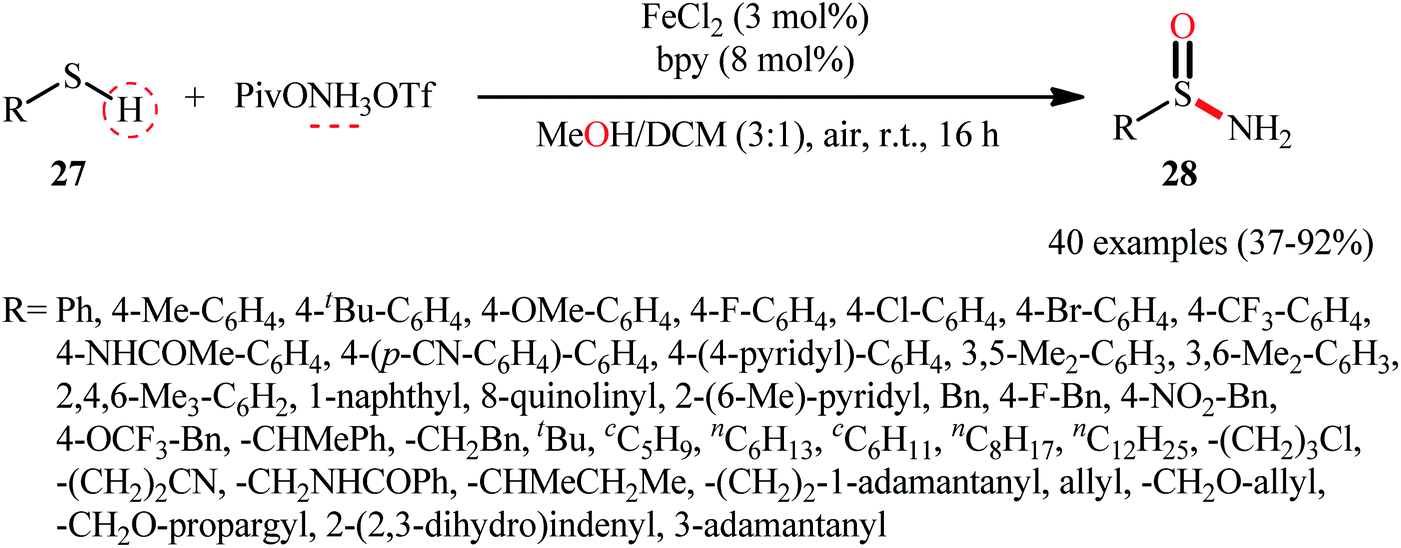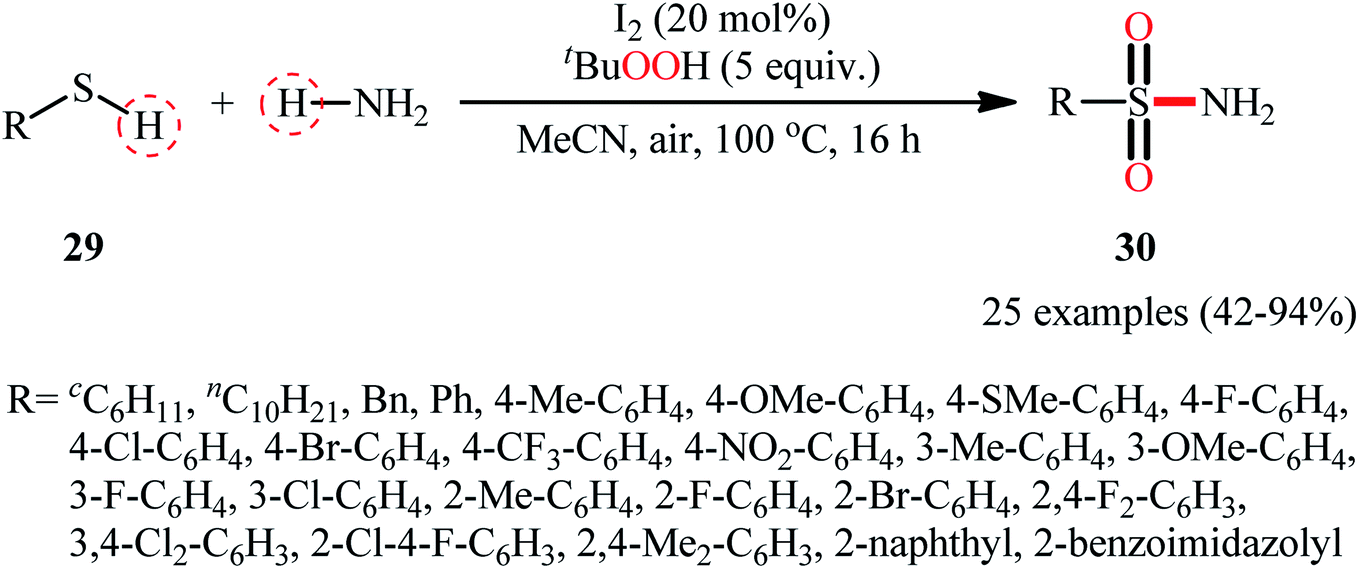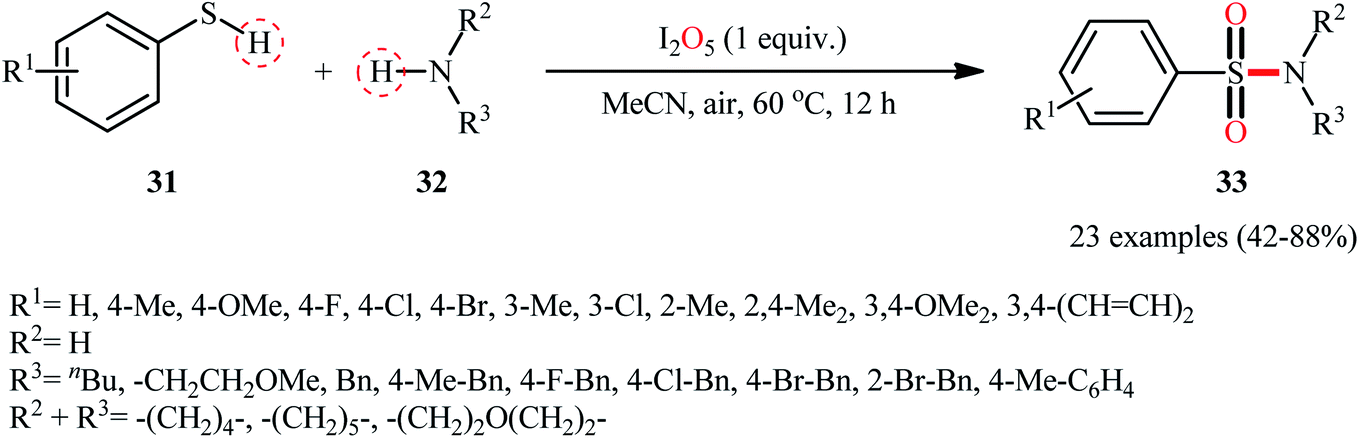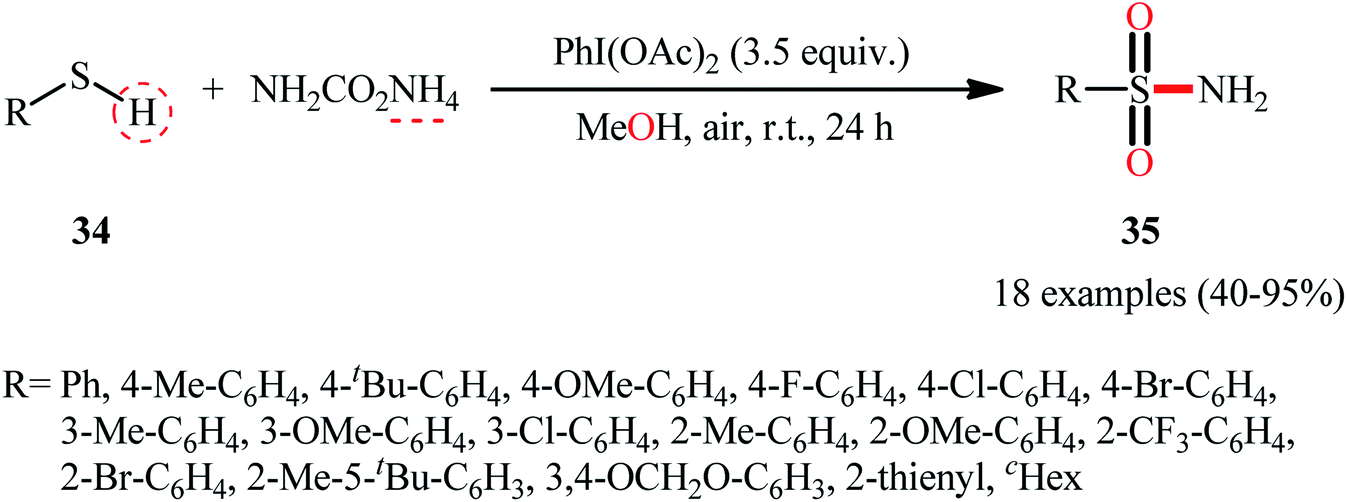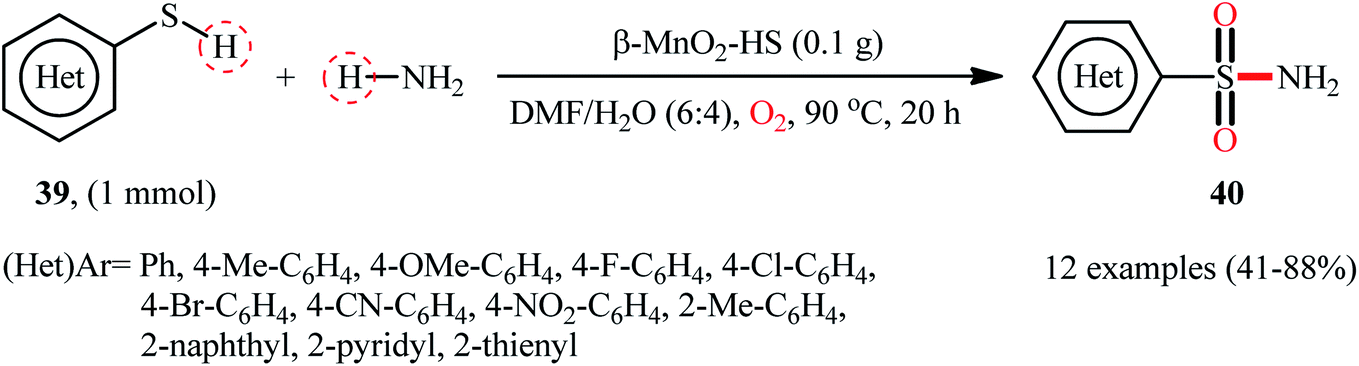DOI:
10.1039/D1RA04368D
(Review Article)
RSC Adv., 2021,
11, 32394-32407
Direct synthesis of sulfenamides, sulfinamides, and sulfonamides from thiols and amines
Received
6th June 2021
, Accepted 13th September 2021
First published on 1st October 2021
Abstract
Needless to say that organosulfur compounds with sulfur–nitrogen bonds have found various applications in diverse fields such as pharmaceuticals, agrochemicals, polymers, and so forth. Three major groups of such compounds are sulfenamides, sulfinamides, and sulfonamides which have been widely applied as building blocks in medical chemistry. Owing to their significant role in drug design and discovery programs, the search for and development of efficient, environmentally friendly, and economic processes for the preparation of the title compounds is of great importance in the pharmaceutical industry. Recently, oxidative coupling of thiols and amines, two readily available low-cost commodity chemicals, has emerged as a highly useful method for synthesizing structurally diverse sulfenamides, sulfinamides, and sulfonamides in a single step. Since this strategy does not require additional pre-functionalization and de-functionalization steps, it considerably streamlines synthetic routes and substantially reduces waste generation. This review will focus on recent advances and achievements in this attractive research arena.
 Shahrzad Abdolmohammadi | Shahrzad Abdolmohammadi was born in Iran, in 1976. She received her BSc. degree in chemistry from Alzahra University (AU), Tehran, Iran, in 1999, her MSc. degree in organic chemistry from the Tarbiat Modarres University (TMU), Tehran, Iran under the supervision of Professor Issa Yavari in 2002 and her PhD degree in organic chemistry from the Tehran University (TU), Tehran, Iran, under the supervision of Professor Hooshang Pirelahi and Professor Saeed Balalaie, in 2008. She is associate professor in East Tehran Branch, Islamic Azad University, Tehran, Iran. Her research interests include organic synthesis, heterocyclic synthesis, multicomponent reactions, nanocatalysis, organocatalysis, and synthetic methodology. |
 Roya Ahmadi | Dr Roya Ahmadi began her bachelor's degree in pure chemistry in 1992 at Shahid Beheshti University in Tehran, where she completed her master's degree in inorganic chemistry at Kharazmi University of Tehran (Teacher Training) in 2000 and received her PhD in mineral chemistry from Islamic Azad University, Tehran Science and Research branch in 2007. In the same year, he worked as an assistant professor at Islamic Azad University, Imam Khomeini's Yadegar branch, and in 2014 he was promoted to associate professorship in the field of experimental and computational chemistry over the years, more than 115 articles in chemistry journals and 163 papers in national and international conferences as posters and lectures. Guidance and counseling of more than 75 graduate students, master's and doctoral degrees. |
 Abdolghaffar Ebadi | Dr Abdol Ghaffar Ebadi finished his doctoral degree in Environmental Biotechnology (Algology) from Tajik Academy of Sciences. Now he is a researcher in TAS in Tajikistan and faculty member at the Islamic Azad University of Jouybar in Mazandaran. Dr Ebadi published more than 400 scientific papers in qualified international journals and attended more than 50 international conferences. He has cooperation with many research project teams around world such as China, Malaysia, and Thailand. His interests are Environmental Biotechnology, Biochemistry, Gene pathways in the phytoremediation processes. |
 Esmail Vessally | Esmail Vessally was born in Sharabiyan, Sarab, Iran, in 1973. He received his B.S. degree in pure chemistry from university of Tabriz, Tabriz, Iran, and his M.S. degree in organic chemistry from Tehran university, Tehran, Iran, in 1999 under the supervision of Prof. H. Pirelahi. He completed his PhD degree in 2005 under the supervision of Prof. M. Z. Kassaee. Now he is working at Payame Noor University as Professor in organic chemistry. His research interests include theoretical organic chemistry, new methodologies in organic synthesis and spectral studies of organic compounds. |
1. Introduction
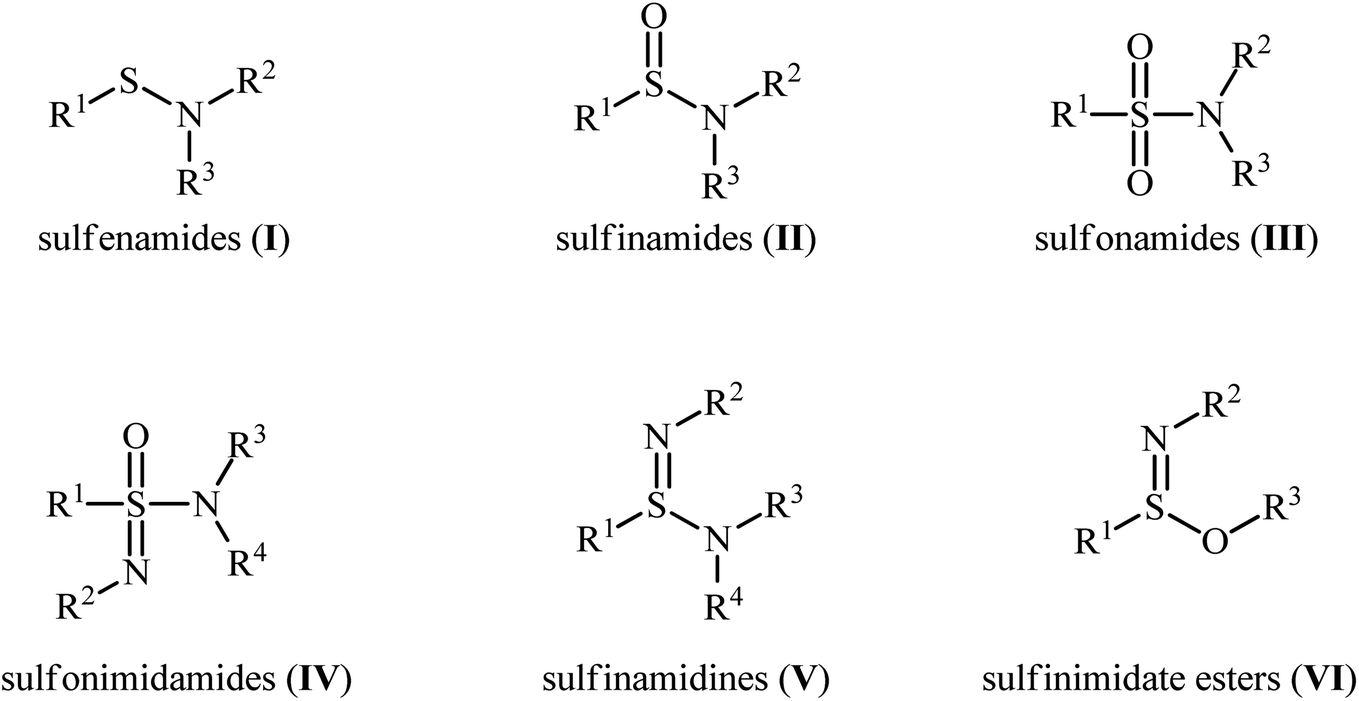
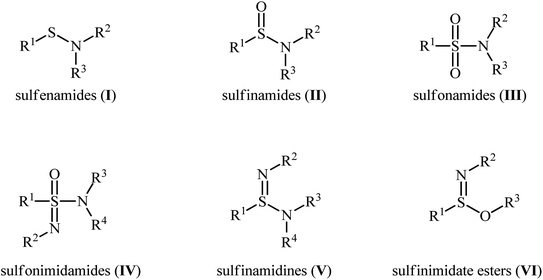
Sulfenamides (Fig. 1, structure I) are versatile reagents that are used in the synthesis of various types of valuable organosulfur compounds such as sulfides,1 sulfonimidamides,2 sulfinamidines and sulfinimidate esters.3 Moreover, due to the high lability of their S–N bond, they are widely used as vulcanization accelerators in the rubber industry.4 In addition to their synthetic and industrial applications, sulfenamides are considered as a potential scaffold for promising antihypertensive, anticancer, and diuretic agents.5 In a similar way, sulfinamides (Fig. 1, structure II) are not only prevalent in a wide variety of natural products6 and commercially available chiral auxiliaries7 but also used as a valuable synthetic block in organic synthesis.8 Sulfonamides (Fig. 1, structure III) are another family of sulfur–nitrogen bond-containing organosulfur compounds that widely used as medicines,9 dyes,10 and plasticizers for fiber-reinforced composites materials.11
 |
| | Fig. 1 General structure of sulfenamides (I), sulfinamides (II), sulfonamides (III), and some related compounds. | |
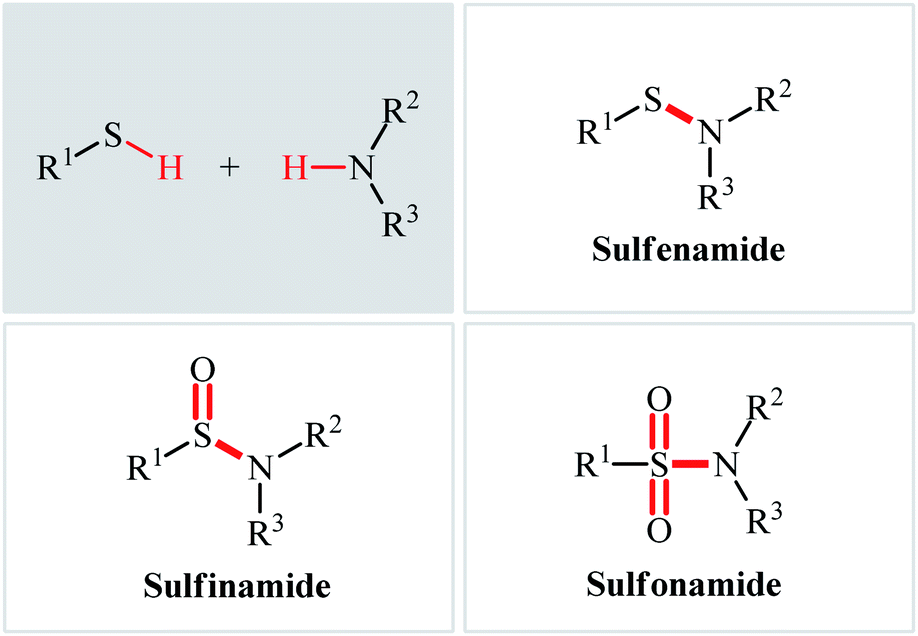
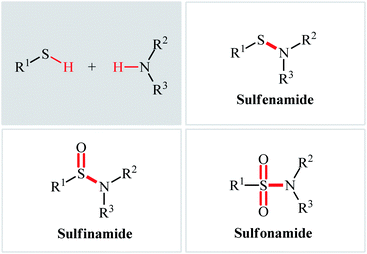
The classical methods to synthesize N-sulfenyl-, sulfinyl-, and sulfonylamine derivatives involve the condensation of sulfenyl, sulfinyl, and sulfonyl chlorides, respectively, and amines.5a,12–14 However, corrosion and instability of most sulfenyl and sulfinyl chlorides, and some sulfonyl chlorides makes their handling troublesome.15 In order to bypass these limitations, over the years, several convenient strategies have developed.16–18 Among them, oxidative coupling of thiols and amines has been the focus of considerable attention due to its straightforward manner with high atom- and step-economy, providing concise and efficient methods for the fabrication of the titled compounds from easily available starting materials. In light of the increasing interest on this chemistry, we concluded that it was timely to summarize the available literature on this domain in a comprehensive review paper. In continuation of our preceding works on organosulfur chemistry19 and modern organic synthesis,20–26 we summarize here the most important contributions toward the synthesis of N-sulfenyl-, sulfinyl-, and sulfonylamines through the controllable oxidative coupling of thiols and amines (Fig. 2), by hoping that it will inspire researchers to make further progress in the field.
 |
| | Fig. 2 Direct synthesis of sulfenamides, sulfinamides, and sulfonamides from thiols and amines. | |
2. Synthesis of sulfenamides
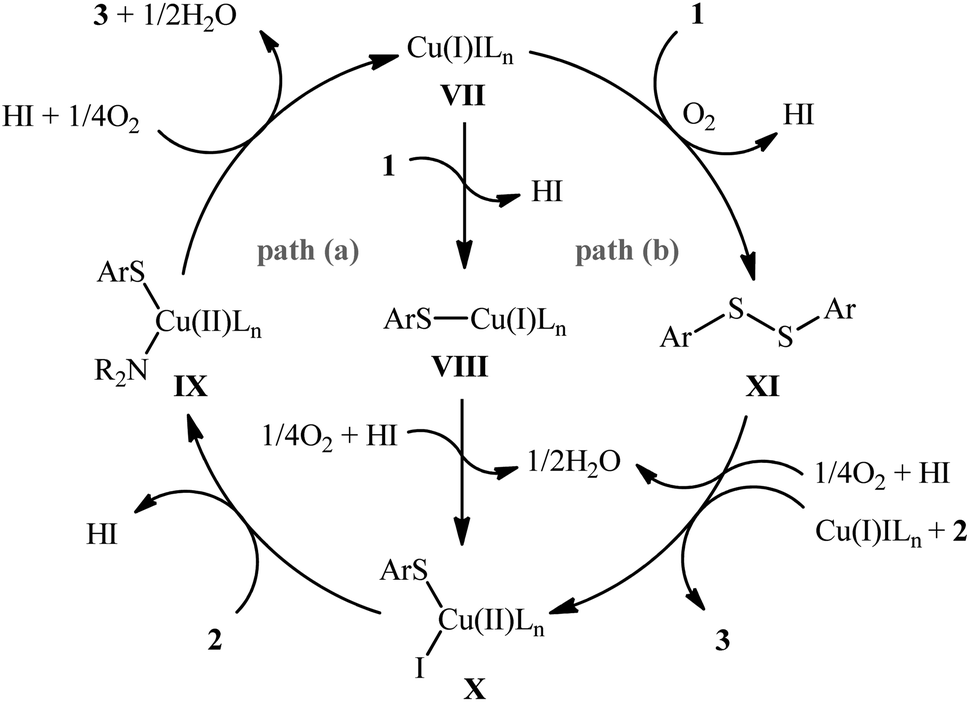
The possibility of synthesizing sulfenamides through the oxidative coupling of thiols and amines was first realized by Taniguchi in 2010.27 By considering the coupling of p-thiocresol with tert-butylamine as the model reaction, the author carefully evaluated the reaction variables such as catalyst, ligand, and solvent. The optimal system was identified using inexpensive CuI catalyst in combination with 2,2′-bipyridine (bpy) ligand in DMSO at 60 °C, which gave the expected sulfenamide in a yield of 90%. Under the optimized conditions, various thiophenol derivatives 1 reacted efficiently with both aromatic and aliphatic amines 2 to give the corresponding sulfenamides 3 in 40–90% yields (Scheme 1). However, aliphatic and NO2-substituted aromatic thiols failed to participate in this transformation. Regarding the scope of amines, aliphatic amines gave considerably higher yields compared to the aromatic ones. Intriguingly, when the reaction was performed under an oxygen atmosphere, instead of sulfenamides, the respective sulfonamides were obtained in modest to excellent yields. Furthermore, when the merge of PdCl2 and CuI with bpy was applied as the catalytic system, it was found that the corresponding sulfinamides were selectively produced. The mechanism proposed to explain the formation of sulfenamides 3 starts with the generation of ArS–CuILn complex VIII via oxidative addition of thiophenol 1 to the in situ generated active copper catalyst VII. Subsequently, oxidation of this intermediate with air oxygen gives ArS–CuII(I)Ln complex IX, which after reaction with amine 2 leads to the intermediate X. Finally, reductive elimination of this intermediate X in the presence of oxygen provides the expected product 3 and regenerates catalyst VII (Scheme 2, path A). In another possibility, oxidative coupling of thiophenol 1 forms diaryl disulfide XI that, after reaction with amine 2 produces the final product 3 and releases the intermediate IX (Scheme 2, path B).
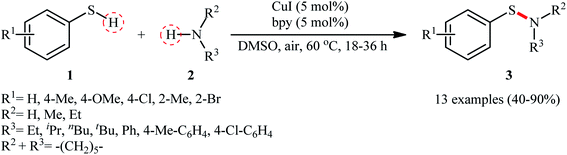 |
| | Scheme 1 Taniguchi's synthesis of sulfenamides 3. | |
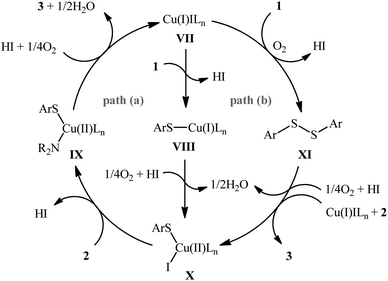 |
| | Scheme 2 Plausible mechanism of Cu-catalyzed cross-coupling of thiols 1 and amines 2. | |
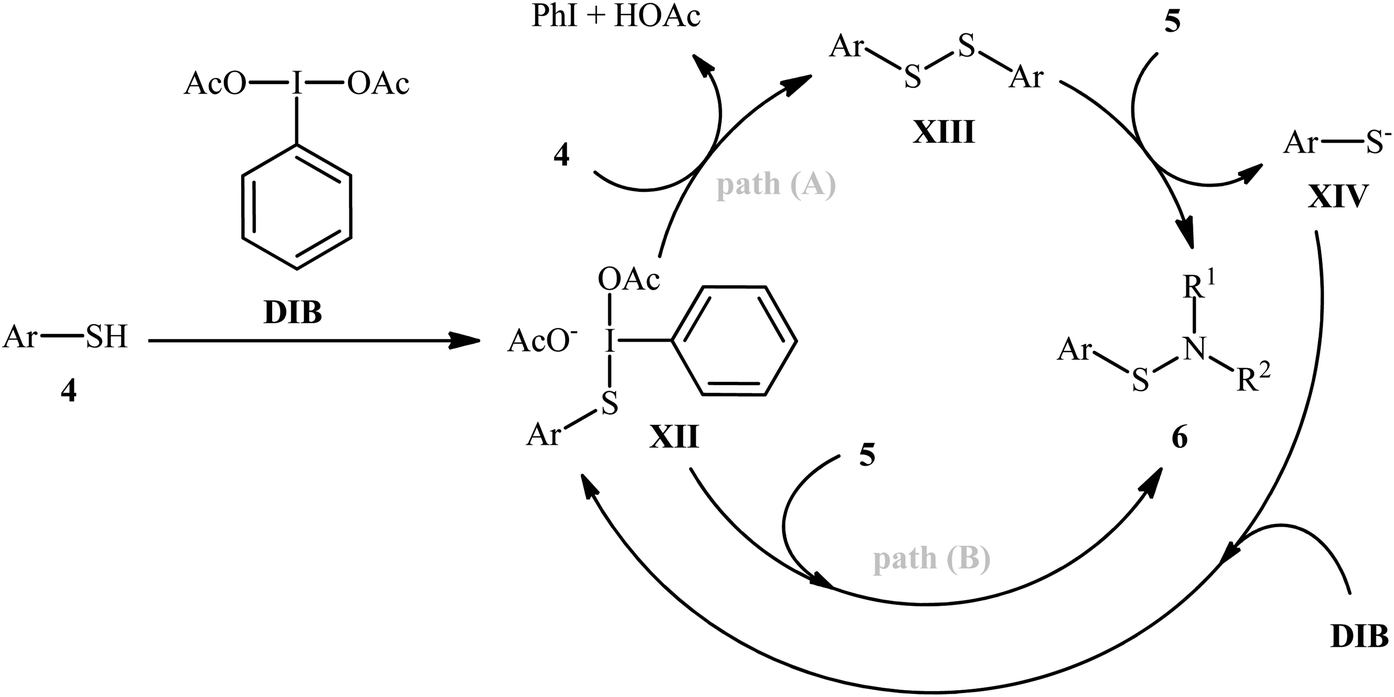
Shortly afterwards, Wacharasindhu and co-workers further expanded the scope of this chemistry to heteroaromatic thiols using hypervalent iodine(III) reagent (diacetoxyiodo)benzene (DIB) as promoter and 1,8-diazabicyclo[5.4.0]-undec-7-ene (DBU) as base.28 A relatively broad range of N-heterocyclic thiols 4 could be coupled with various primary and secondary aliphatic amines 5 by this method, and effectively furnished the corresponding S-heteroaryl sulfenamides 6 in fair to excellent yields (Scheme 3). However, thiols with the insufficient electrophilicity of the sulfur atom (e.g., unsubstituted pyridine thiols) underwent competitive homocoupling side reactions, thereby provided a mixture of the desired sulfenamides and corresponding disulfides. Furthermore, sterically hindered (e.g., tert-butylamine) and less nucleophilic amines (e.g., anilines) were inert in this reaction and only disulfides were isolated using these nucleophiles. Based on the previous literature report, the authors proposed two possible pathways for this transformation as depicted in Scheme 4. The thiol 4 reacts with DIB to form the reactive key intermediate XII, followed by the nucleophilic attack on the sulfur atom by the SH-group of another molecule of thiol 4 with the removal of acetic acid and iodobenzene resulting in the disulfide intermediate XIII, which undergoes reaction with amine 5 to form the target product 6 and sulfide anion XIV. Finally, simple ligand exchange of DIB reagent with the anionic intermediate XIV regenerates the key intermediate XII (Scheme 4, path A). In another possibility, the direct nucleophilic substitution of intermediate XII with amine 5 affords the final product 6 (Scheme 4, path B).
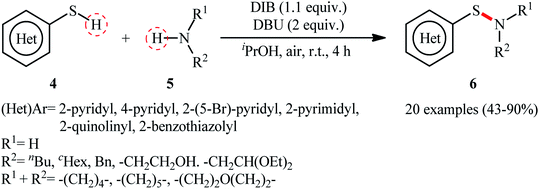 |
| | Scheme 3 DIB-promoted S–N coupling of N-heterocyclic thiols 4 with amines 5. | |
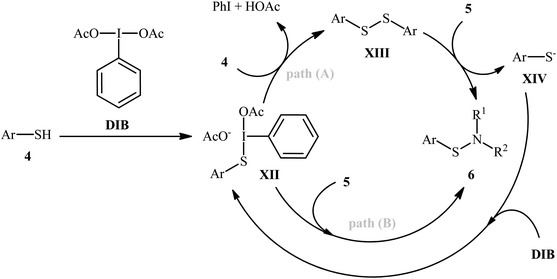 |
| | Scheme 4 Proposed reaction mechanism for the formation of S-heteroaryl sulfenamides 6. | |
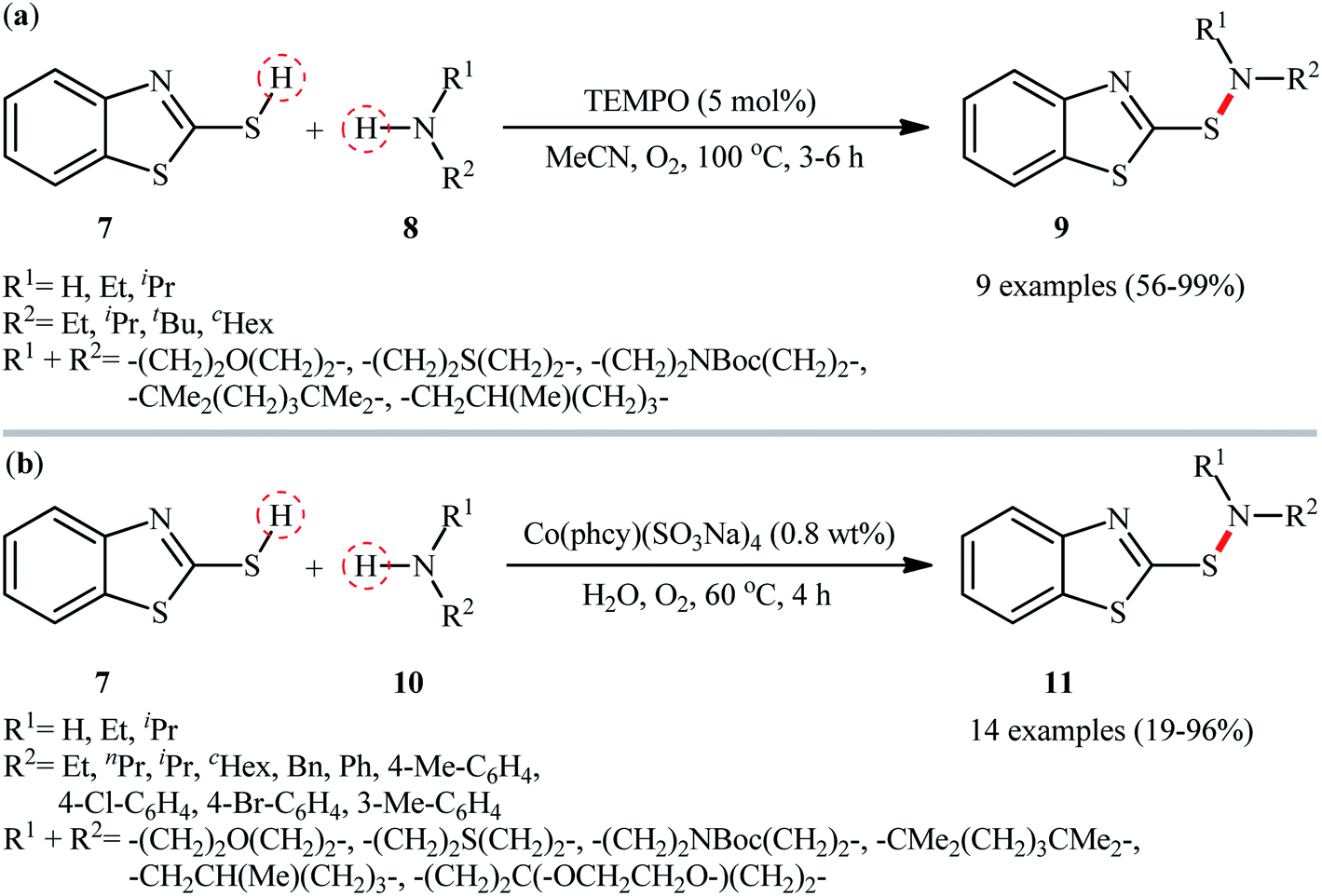
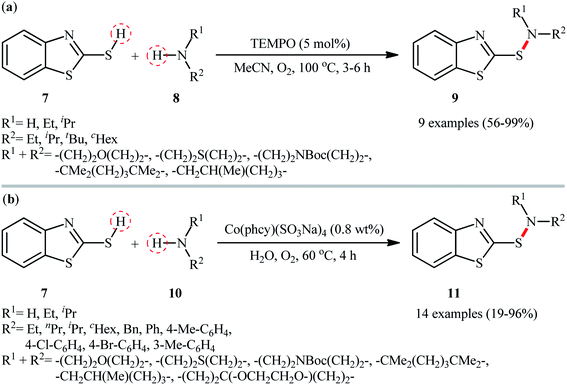
At the outset of 2017, a straightforward and environmentally approach for the synthesis of 2-benzothiazolesulfenamides 9 by the reaction between 2-mercaptobenzothiazole 7 and alkylamines 8 was well reported by Yuan and colleagues (Scheme 5a).29 This S–N bond formation reaction required 2,2,6,6-(tetramethylpiperidin-1-yl)oxyl (TEMPO) as catalyst and molecular oxygen as the terminal oxidant in acetonitrile. Various primary and secondary alkylamines including sterically hindered ones suitably participated in this reaction, delivering good to quantitative yield of the coupled products within 3–6 h. However, the applicability of arylamines was not investigated in this study. The reaction, moreover, appears to be limited to unsubstituted 2-mercaptobenzothiazole. In the same year, utilized a conceptually similar strategy to fabrication of 2-benzothiazolesulfenamide derivatives 11 in acceptable yields employing phthalocyanine-tetra-sodium cobalt(II) sulfonate [Co(phcy)(SO3Na)4] as a reusable catalyst and water as the solvent (Scheme 5b).30 Particularly, the reaction tolerated both alkyl and aryl amines 10. However, like Yuan's work, except for 2-mercaptobenzothiazole 7, other thiols, such as phenyl thiols failed in the system to provide the corresponding sulfenamides. Mechanistically, the authors speculated that the reaction proceeded through a free radical pathway via formation of a thiyl radical as the key step. Very recently, the group of Ko also synthesized a library of 2-benzothiazolesulfenamides (11 examples; 42–96% yield) in a similar manner employing dibenzyl azodicarboxylate (DBAD) as a promoter under ambient conditions.31 Similarly, 2-mercaptobenzoxazole was also compatible and reacted well with the same set of amines to give the corresponding 2-benzoxazolesulfenamides with slightly diminished yields as compared with the 2-mercaptobenzothiazole.
 |
| | Scheme 5 (a) TEMPO-catalyzed oxidative coupling of 2-mercaptobenzothiazole 7 with alkylamines 8; (b) synthesis of 2-benzothiazolesulfenamide derivatives 11 via co-catalyzed reaction of 2-mercaptobenzothiazole 7 with amines 10. | |
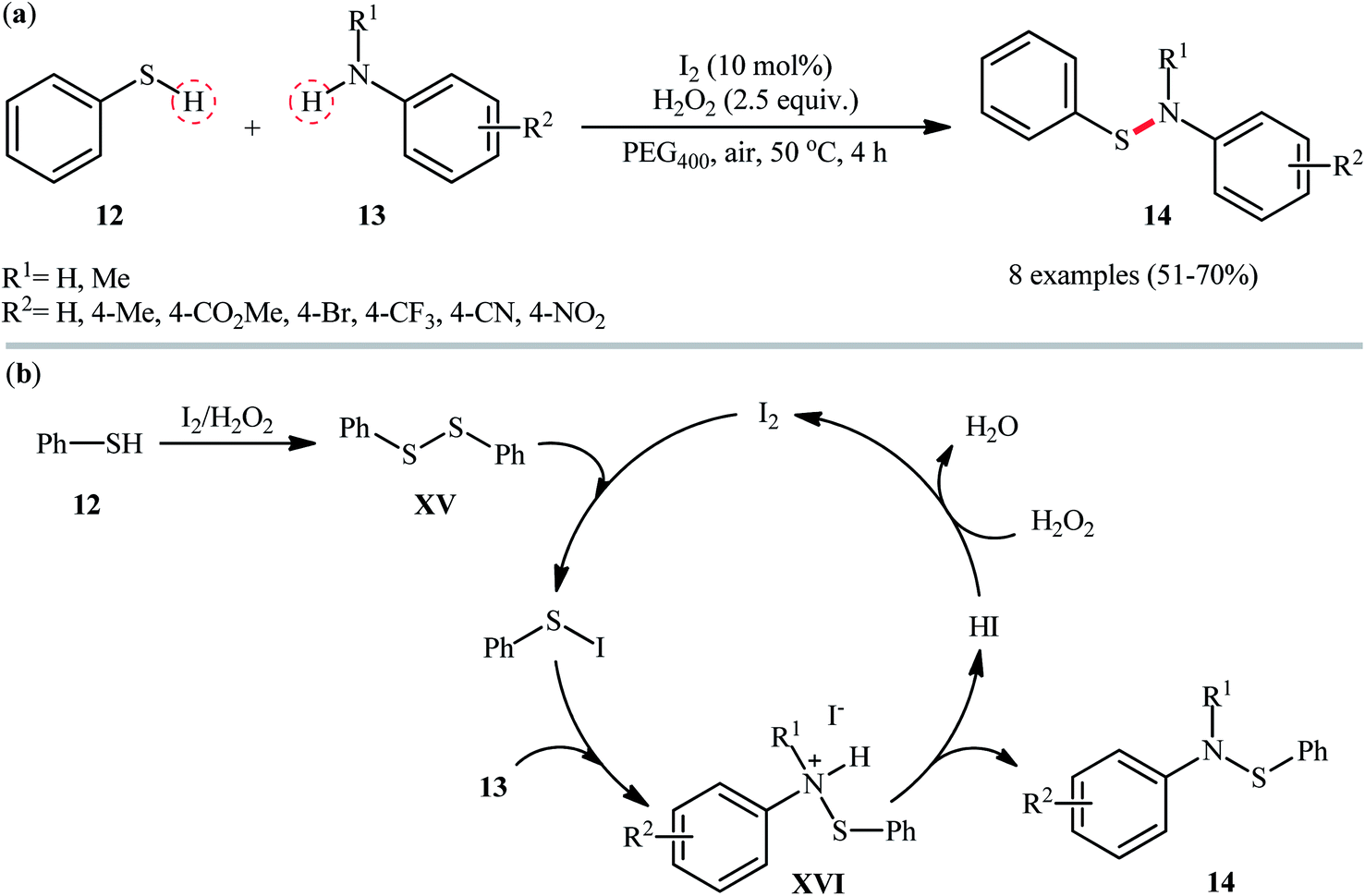
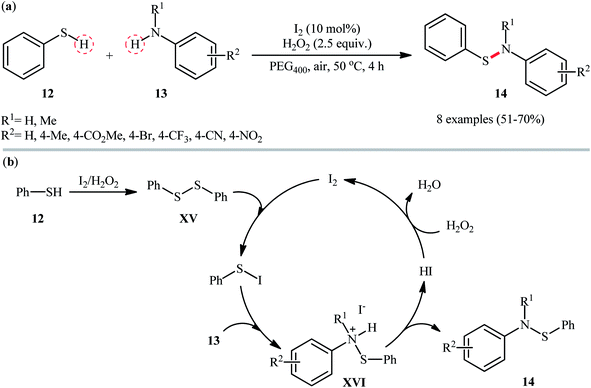
In 2018, Zeng and co-workers exploited the I2/H2O2 catalytic system for cross-dehydrogenative coupling reaction between thiophenol 12 and anilines 13 with diverse steric and electronic properties, providing expedite access to a range of N,S-diaryl sulfenamides 14 (Scheme 6a).32 This metal free transformation was carried out in presence of 10 mol% I2 and 2.5 equiv. of H2O2 in green and biodegradable polyethylene glycol (PEG-400) under mild conditions, tolerated various synthetically useful functionalities such as Br, CF3, CN, NO2, CO2Me, and generally provided the respective products in moderate to good yields. Notably, this protocol also proved to be applicable to the use of sulfoximines as coupling partners. According to the author's proposed mechanism (Scheme 6b), presumably, the reaction initiated with the formation of the disulfide intermediate XV by homocoupling of thiophenol 12. Next, the disulfide reacted with I2 and generated the active electrophilic species, Ph–S–I which was readily attacked by aniline 13 to provide ionic intermediate XVI and deliver the desired product 14 followed by a deprotonation process. Finally, hydriodic acid (HI) is oxidized by H2O2 to restore the I2 catalyst.
 |
| | Scheme 6 (a) I2-catalyzed cross-dehydrogenative coupling of thiophenol 12 with anilines 13; (b) putative mechanism for the formation of N,S-diaryl sulfenamides 14. | |
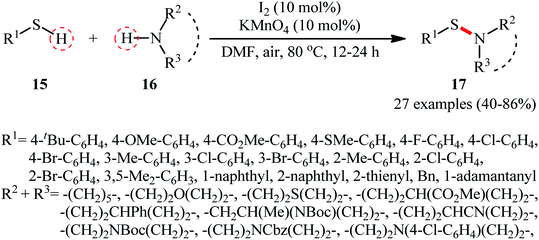
Following these works, Zhang and Qian along with their co-workers used the I2/KMnO4 dual catalyst system for the oxidative dehydrogenation of thiols 15 and amines 16 to afford a wide range of sulfenamides 17 in moderate to high yields (Scheme 7).33 Although various aliphatic and (hetero)aromatic thiols with either electron-donating or electron-withdrawing substituents were well tolerated under the reaction conditions, the protocol was restricted to only secondary cyclic amines, however, the method was successfully explored for the synthesis of a drug-like sulfenamide with antihypertensive activity. It should be mentioned that under the identical conditions, due to their higher reactivity, the primary amines were converted to sulfinamides rather than sulfenamides. Based on the mechanistic studies conducted, the author speculated that the activated MnO2 (generated in situ through the heating of KMnO4 under O2 atmosphere) was an electron transfer bridge for assisting iodine in completing the catalytic cycle.
 |
| | Scheme 7 Zhang–Qian's synthesis of sulfenamides 17. | |
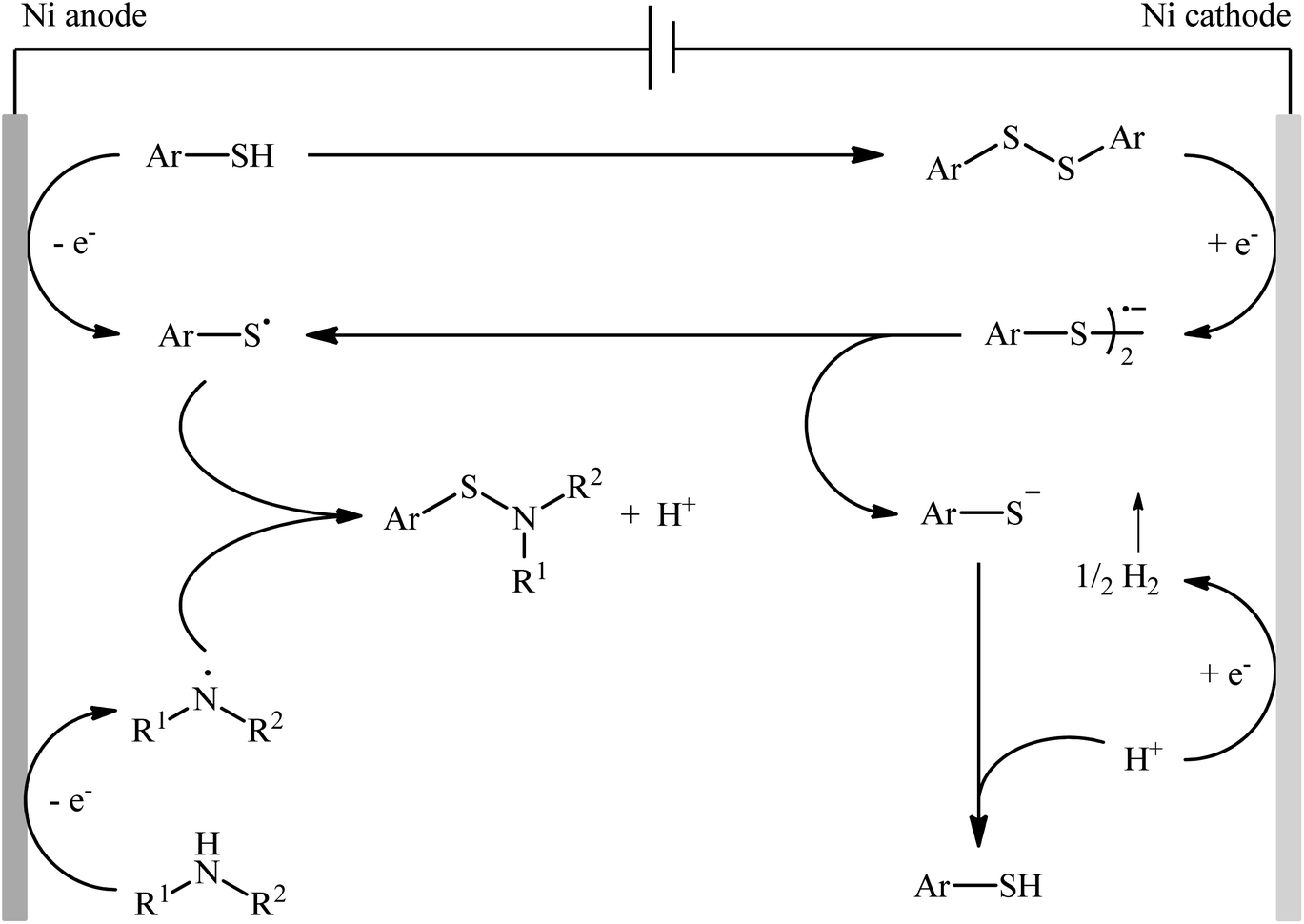
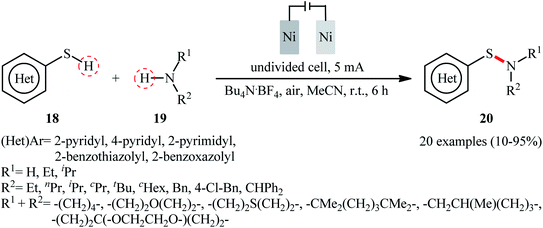
Recently, the Li–Yuan group developed an efficient electrochemical synthesis of S-heteroaryl sulfenamides 20 via dehydrogenative S–H/N–H cross-coupling of the corresponding heteroaryl thiols 18 with alkyl amines 19 under oxidant- and catalyst-free conditions.34 Screening of various electrolytes such as Bu4N·Br, Bu4N·ClO4, Bu4N·BF4, and LiClO4, conclusively led to the use of Bu4N·BF4 as the most promising electrolyte, whereas MeCN was found to be the most suitable solvent. The reaction was conducted in an undivided cell with inexpensive nickel foam electrodes under constant current of 5 mA at room temperature for 6 h to deliver the coupled products in poor to excellent yields (Scheme 8). Concerning the substrate scope, the reaction is strongly dependent on the steric- and electronic-factors of amines. Sterically demanding amines such as diisopropylamine and 2,2,6,6-tetramethylpiperidine afforded less satisfactory results and less nucleophilic aryl amines completely failed in the system to provide the corresponding products. Notably, the reaction was scalable as exemplified by the formation of N-tert-butyl-2-benzothiazolesulfenamide on a 50 g scale (94%) while the Ni sheets could be reused. A plausible catalytic radical pathway for the formation of sulfenamides 20 was proposed as shown in Scheme 9. Initially, the thiyl radical and the amino radical were generated from thiol and amine, respectively, through the single-electron-transfer oxidation on the anode surface. Then the coupling of these radicals produced the desired product. Meanwhile, the thiyl radical underwent dimerization to form a disulfide that after the single-electron reduction at the cathode afforded the corresponding radical anion. Finally, the cleavage of this radical anion reproduced the thiyl radical and a thiyl anion.
 |
| | Scheme 8 Electrochemical oxidant- and catalyst-free synthesis of S-heteroaryl sulfenamides 20 through dehydrogenative S–H/N–H coupling of heteroaryl thiols 18 with alkyl amines 19. | |
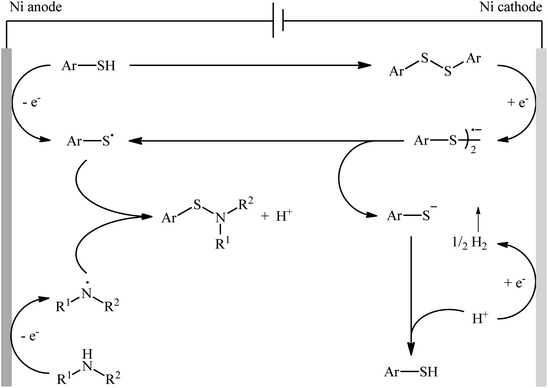 |
| | Scheme 9 A plausible mechanism for the reaction in Scheme 8. | |
3. Synthesis of sulfinamides
In 2010, in the same paper describing Cu-catalyzed cross-dehydrogenative coupling of thiols and amines to sulfenamides, Taniguchi also utilized a dual Cu/Pd catalytic system towards the formation of sulfinamides from the same starting materials.27 Detailed investigations revealed that the suitable conditions for this chemical transformation were: 5 mol% of CuI, 3 mol% of PdCl2, and 8 mol% of bpy under open air in DMSO. Under optimal conditions, a small series of aryl thiols 21 were treated with tert-butylamine 22 for 18 h leading to the corresponding sulfinamides 23 in moderate to good yields (Scheme 10). However, in this preliminary work, only one amine was examined, without any substrate scope exploration. It is worth noting to mention that both CuI and PdCl2 had a profound impact on this catalytic regime. As mentioned by the author, when the reaction was carried out with only the CuI-bpy catalyst, no sulfinamide is produced and sulfenamides are obtained selectivity instead. Similarly, the palladium catalyst only also did not afford the desired sulfinamides and the corresponding disulfides are obtained as the major products under this regime. Unfortunately, no comment was made by the author regarding the plausible mechanistic course of this reaction.
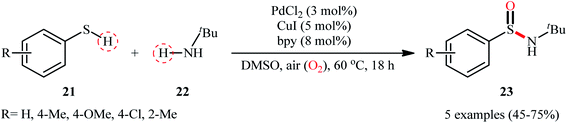 |
| | Scheme 10 Synthesis of sulfinamides 23 from aryl thiols 21 and tert-butylamine 22 catalyzed by PdCl2–CuI binary catalyst. | |
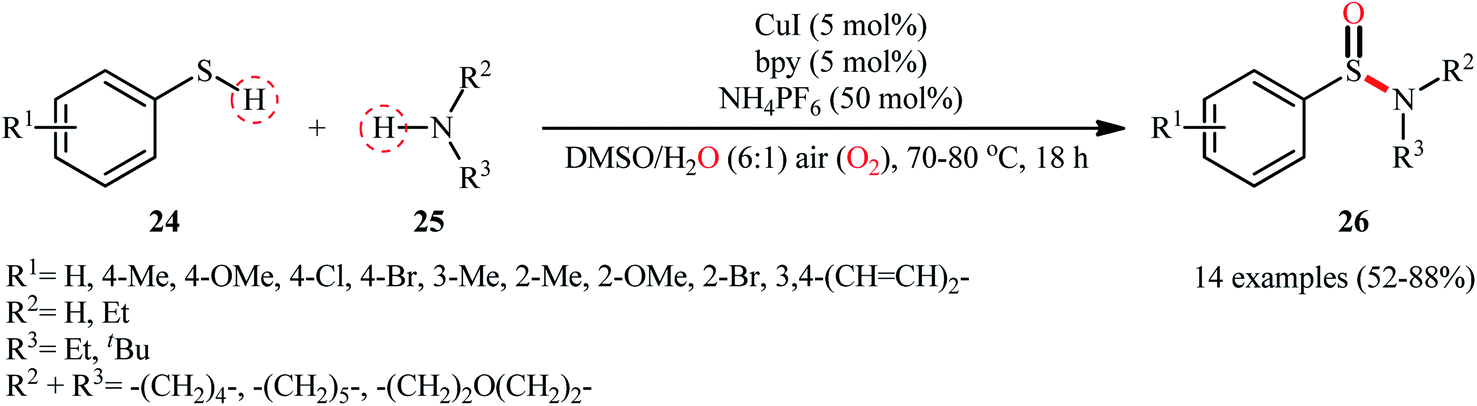
With the aim of designing a more practical and substrate compatible protocol to sulfinamides through oxidative coupling of the corresponding thiols with amines, the innovative group of Taniguchi also unveiled a highly efficient Cu-catalyzed dual S–H/N–H activation methodology to synthesize a relatively broad array of aryl sulfinamides 26 by combination of aromatic thiols 24 with various primary and secondary aliphatic amines 25 in a 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of DMSO and H2O in presence of 5 mol% CuI as catalyst, 5 mol% bpy as a ligand, and 0.5 equiv. of NH4PF6 as additive under air at 70–80 °C (Scheme 11).35 The results demonstrated that the reaction was equally efficient for both electron-rich and electron-poor aryl thiols. Howbeit, amino-group bearing aryl thiols turned out to be ineffective coupling partners under the standard reaction condition. Additionally, neither aliphatic thiols nor aromatic amines could react in this system. Based on a series of mechanistic studies conducted, the author proposed that through a similar catalytic cycle depicted in Scheme 2, a sulfenamide intermediate is initially formed which underwent further oxidation into the corresponding sulfinamide under the reaction condition. To shed light on the source of oxygen atom of the sulfinamide products, the author performed isotope labelling experiments with H218O which indicated that the oxygen atom originated from both water and molecular oxygen as a mixture of both 18O- and 16O-containing products in the ratio of 3:2, respectively, was formed.
:1 mixture of DMSO and H2O in presence of 5 mol% CuI as catalyst, 5 mol% bpy as a ligand, and 0.5 equiv. of NH4PF6 as additive under air at 70–80 °C (Scheme 11).35 The results demonstrated that the reaction was equally efficient for both electron-rich and electron-poor aryl thiols. Howbeit, amino-group bearing aryl thiols turned out to be ineffective coupling partners under the standard reaction condition. Additionally, neither aliphatic thiols nor aromatic amines could react in this system. Based on a series of mechanistic studies conducted, the author proposed that through a similar catalytic cycle depicted in Scheme 2, a sulfenamide intermediate is initially formed which underwent further oxidation into the corresponding sulfinamide under the reaction condition. To shed light on the source of oxygen atom of the sulfinamide products, the author performed isotope labelling experiments with H218O which indicated that the oxygen atom originated from both water and molecular oxygen as a mixture of both 18O- and 16O-containing products in the ratio of 3:2, respectively, was formed.
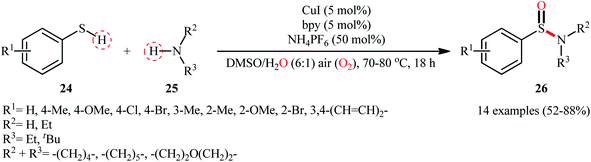 |
| | Scheme 11 Cu-catalyzed synthesis of aryl sulfinamides 26 from aryl thiols 24 and amines 25 in the presence of air. | |
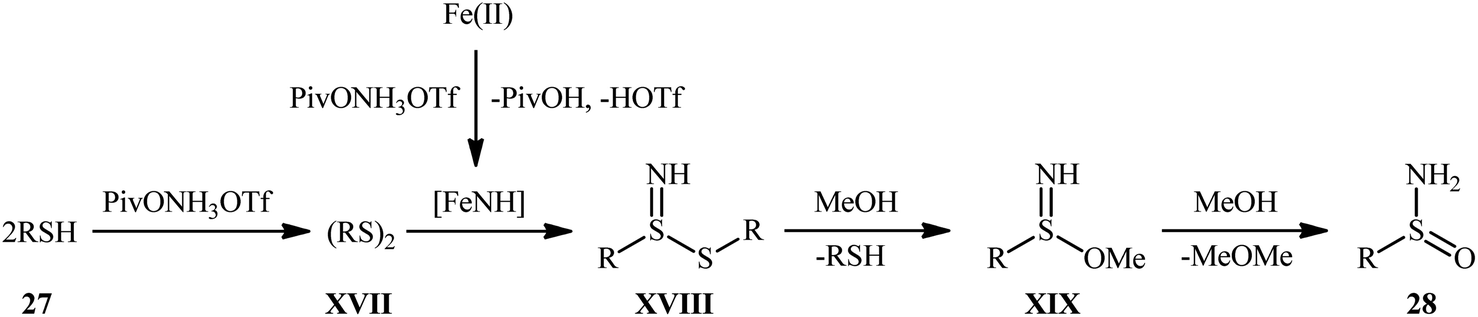
Inspired by these pioneering works, very recently, Morandi and co-workers disclosed an interesting iron catalyzed protocol for the preparation of valuable unprotected sulfinamides 28 from thiols 27 and O-pivaloyl hydroxylamine triflic acid (PivONH3OTf), where the hydroxylamine derivative was consumed as both oxidant and the aminating reagent in the presence of bidentate chelating bpy ligand.36 The reaction was performed in the binary solvent MeOH/DCM with ratio 3:1 at room temperature, tolerated a wide variety of aliphatic, benzylic, aromatic, and heteroaromatic thiols, and accomplished the desired products in moderate to excellent yields within 16 h (Scheme 12). Notably, the author also showcased the synthetic versatility of this method through amino-oxidation of natural product derived 2-pinanthiol and thiocholesterol in modest yields. Intriguingly, mechanistic studies affirmed that the oxygen atom present in the resulting sulfinamides came from the alcoholic solvent and not from air oxygen. The plausible mechanism proposed by the authors for this transformation is illustrated in Scheme 13.
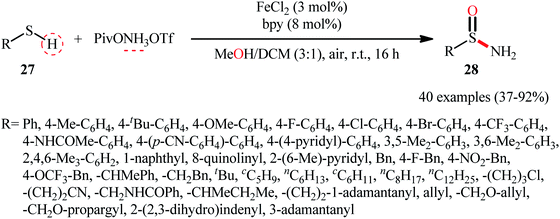 |
| | Scheme 12 Morandi's synthesis of unprotected sulfinamides 28. | |
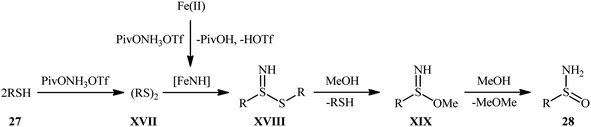 |
| | Scheme 13 Proposed mechanism for Fe-catalyzed amino-oxidation of thiols 27. | |
4. Synthesis of sulfonamides
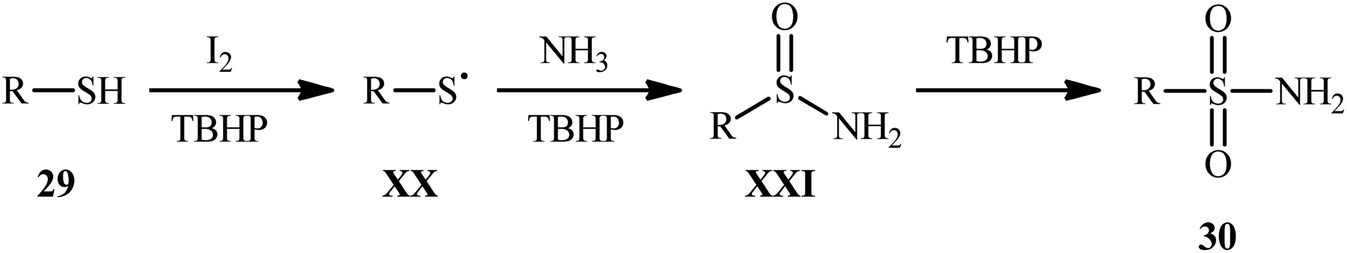
Drawing inspiration from the extensive research efforts on one-pot two step synthesis of sulfonamides from the respective thiols and amines through oxidative chlorination of thiols to sulfonyl chlorides followed by reaction with amines,37,38 the first general report on the formation of three new bonds (two S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and one S–N) in a single step to chemoselectively form sulfonamide products was published by Feng and Wu in 2016.39 In this investigation, a diverse array of N-unsubstituted sulfonamides 30 were obtained in reasonable yields by reaction of various alkyl and aryl thiols 29 with aqueous ammonia in refluxing acetonitrile through the action of I2/tBuOOH (Scheme 14). A wide panel of synthetically useful functional groups such as –OMe, –SMe, –F, –Cl, –Br, –NO2 were perfectly tolerated by this reaction, thus promising further modifications of the final products. In addition to ammonia, tert-butylamine was applied as well and gave the desired product in excellent yield. Based on the preliminary control experiments, the authors proposed a plausible mechanism that consists of the following key steps (Scheme 15): (i) initial formation of thiyl radical XX through the one-electron oxidation of thiol 29 in the presence of I2/tBuOOH; (ii) oxidative combination of radical XX with ammonia to give sulfinamide intermediate XXI; and (iii) oxidation of sulfinamide XXI with tBuOOH to afford the final sulfonamide 30.
O and one S–N) in a single step to chemoselectively form sulfonamide products was published by Feng and Wu in 2016.39 In this investigation, a diverse array of N-unsubstituted sulfonamides 30 were obtained in reasonable yields by reaction of various alkyl and aryl thiols 29 with aqueous ammonia in refluxing acetonitrile through the action of I2/tBuOOH (Scheme 14). A wide panel of synthetically useful functional groups such as –OMe, –SMe, –F, –Cl, –Br, –NO2 were perfectly tolerated by this reaction, thus promising further modifications of the final products. In addition to ammonia, tert-butylamine was applied as well and gave the desired product in excellent yield. Based on the preliminary control experiments, the authors proposed a plausible mechanism that consists of the following key steps (Scheme 15): (i) initial formation of thiyl radical XX through the one-electron oxidation of thiol 29 in the presence of I2/tBuOOH; (ii) oxidative combination of radical XX with ammonia to give sulfinamide intermediate XXI; and (iii) oxidation of sulfinamide XXI with tBuOOH to afford the final sulfonamide 30.
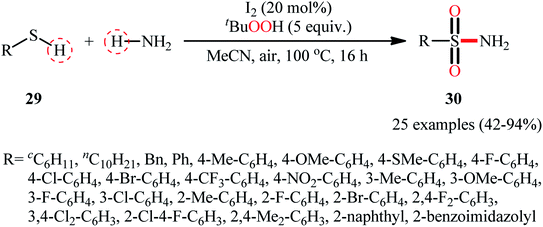 |
| | Scheme 14 I2-mediated synthesis of N-unsubstituted sulfonamides 30 from thiols 29 and ammonia. | |
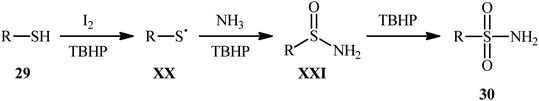 |
| | Scheme 15 Mechanism of reaction between thiols 29 and ammonia in the presence of I2 and tBuOOH. | |
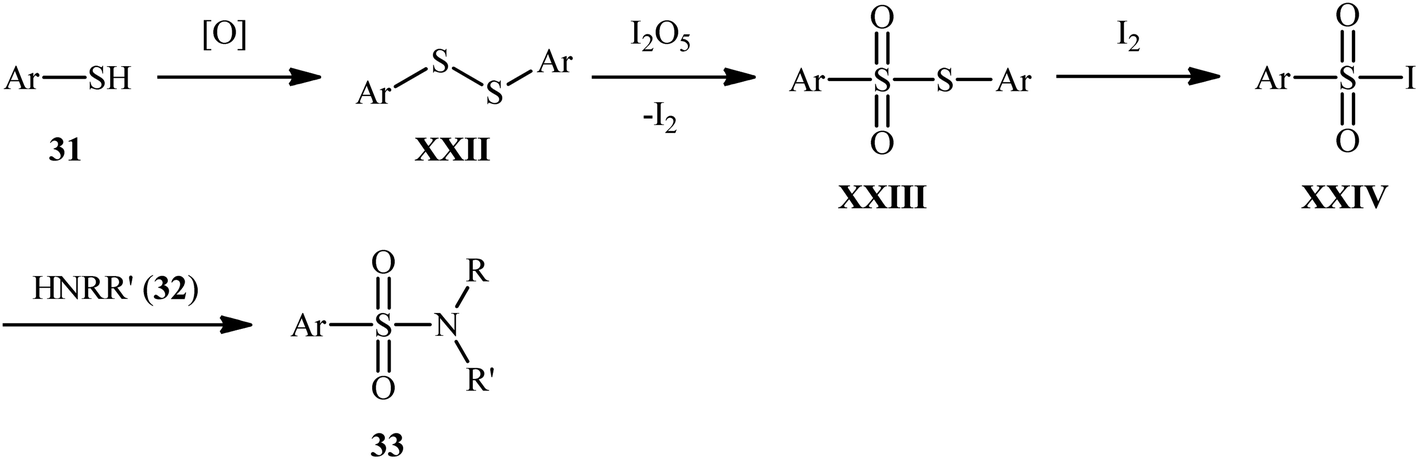
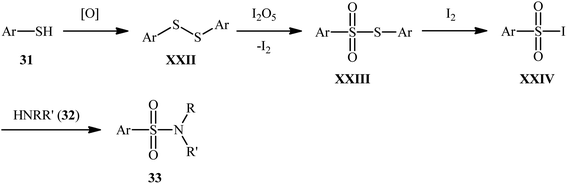
Subsequently, Wei and Wang along with their colleagues unfolded the I2O5-mediated direct sulfonylation of amines 32 with arylthiols 31 to synthesize N-mono- and N,N-di-substituted sulfonamide derivatives 33 under metal-free condition (Scheme 16).40 The reaction showed broad substrate scope, irrespective of whether electron-donating or withdrawing groups were at different positions of phenyl rings of aryl thiols, and both the aliphatic and aromatic amines exhibited good applications under standard conditions and afforded the target products in moderate to high yields. Moreover, a tolerance for 4-pyridinethiol (a heteroaromatic thiol) was also demonstrated. It should be mentioned that in the cases of primary amines, the use of stoichiometric amounts of DBU as the base was necessary because the yields of the corresponding products were obviously reduced when only I2O5 was used in the reaction. The authors nicely highlighted the synthetic utility of their methodology by high yielding gram-scale synthesis of 4-tosylmorpholine (81% yield on 10 mmol scale). With regard to the mechanism (Scheme 17), the reaction initiates by the formation of disulfide XXII through the oxidative dimerization of the thiol 31, followed by its oxidation with I2O5 to give benzenesulfonothioate XXIII and molecular iodine. Next, the interaction of benzenesulfonothioate XXIII with I2 generates the sulfonyl iodide intermediate XXIV, which after nucleophilic substitution by amine 32 leads to the formation of the observed sulfonamide 33.
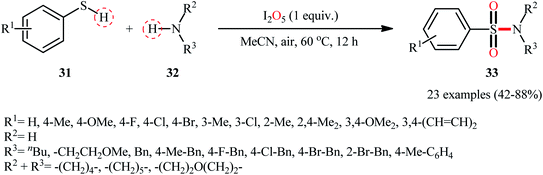 |
| | Scheme 16 Direct synthesis of sulfonamides 33 from thiols 31 and amines 32 through the action of I2O5. | |
 |
| | Scheme 17 Proposed mechanism of I2O5-mediated sulfonylation of amines 32 with arylthiols 31. | |
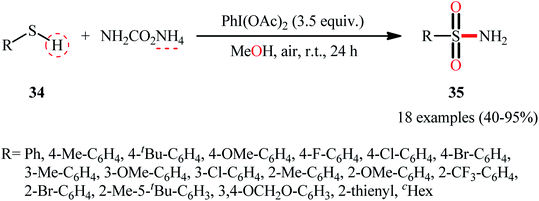
In 2018, the Luisi–Bull group disclosed a novel route to synthesize primary sulfonamides 35 from thiols 34 through a one-pot NH and O transfer process using ammonium carbamate (NH2CO2NH4) as the N-source and MeOH as the source of the O-atom as well as the reaction medium.41 The reaction was performed in the presence of a hypervalent iodine reagent at room temperature, and a series of (hetero)aromatic and aliphatic thiols showed good compatibilities under standard conditions, affording corresponding N-unsubstituted sulfonamide products 35 in good to almost quantitative yields (Scheme 18). Notably, when the reaction was carried out in the presence of higher ammonia concentrations (4 equiv.) and reduced reaction times (3 h), related sulfonimidates were observed as the sole products, which was an interesting example. Mechanistically, the reaction was speculated to be preceded through a similar type of mechanism as depicted in Scheme 13.
 |
| | Scheme 18 Luisi–Bull's synthesis of primary sulfonamides 35. | |
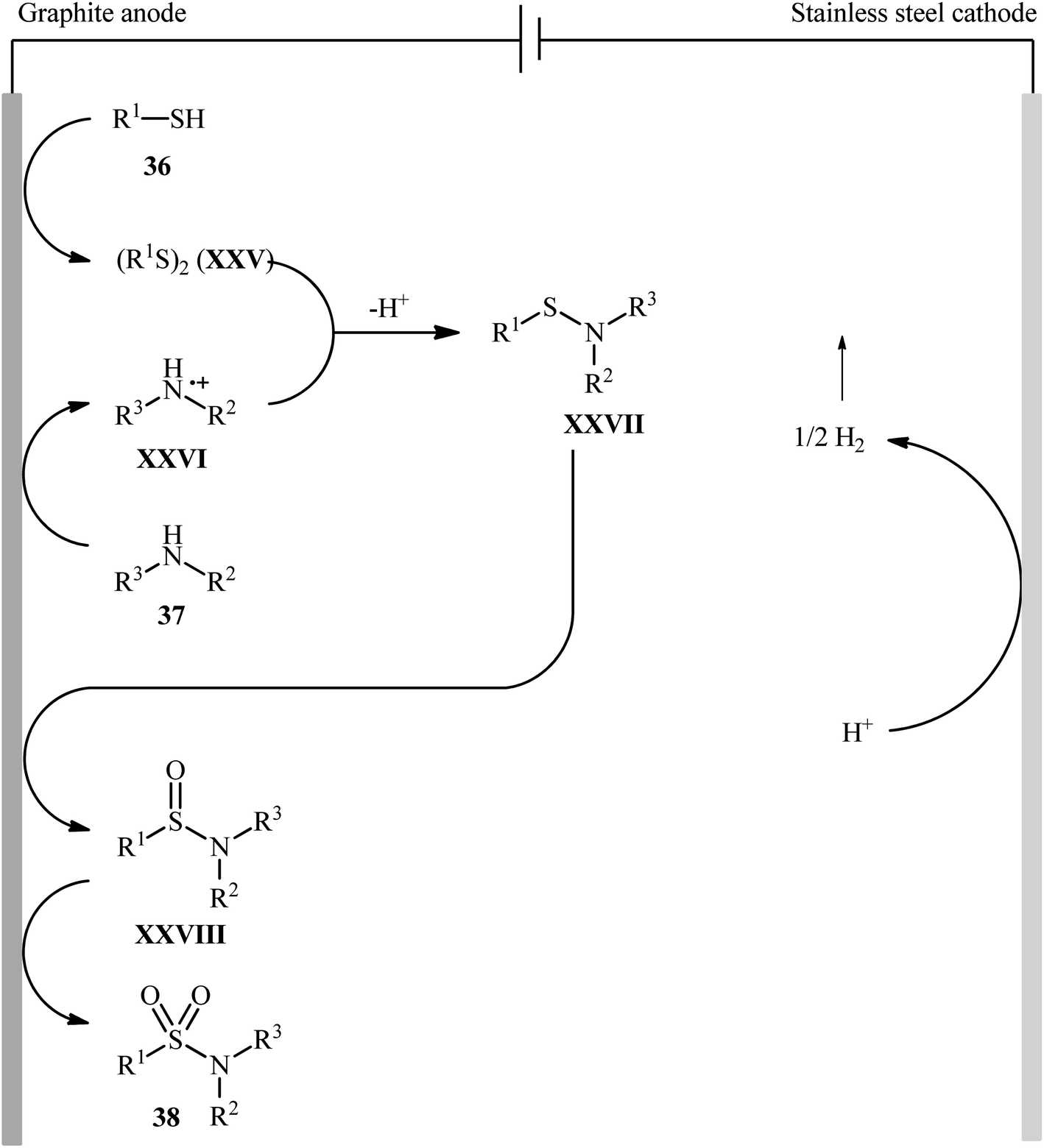
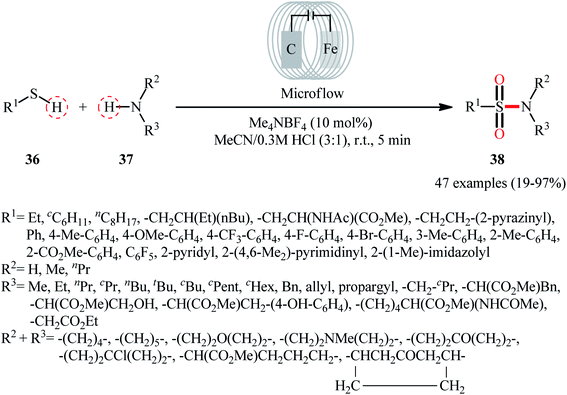
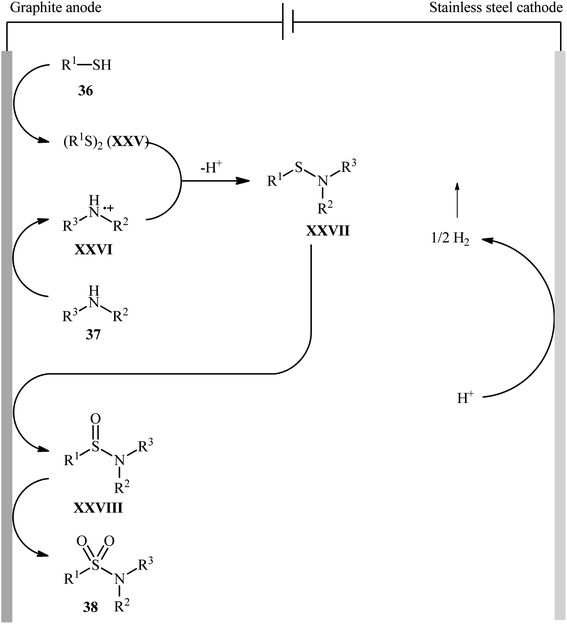
Recently, Noël and co-workers have made significant contributions to the field by developing a highly efficient electrochemical oxidative coupling of thiols and amines in a flow cell without the use of a catalyst or external oxidizing agent.42 They showed that the treatment of thiols 36 with either primary or secondary amines 37 in an electrochemical flow microreactor with graphite anode and stainless steel cathode in a 3:1 mixture of MeCN/0.3 M HCl containing 0.01 M Me4NBF4 at room temperature produced the corresponding sulfonamides 38 in poor to high yields (Scheme 19). Although various alkyl, aryl, and heteroaryl thiols were well tolerated under the reaction conditions, the substrate scope of amines was merely limited to aliphatic derivatives. However, by adding an equivalent of pyridine as an electron-mediator, heteroaryl amines became competent coupling partners but attempts to expand the reaction scope to aniline derivatives were unsuccessful. Notably, carrying out this interesting electrosynthesis in a batch electrochemical cell was also possible but required higher loadings (100 mol%) of the supporting electrolyte and longer reaction times (24 h). The authors attributed the acceleration effect in flow to the shorter diffusion distances, the large electrode surface to volume ratio and the intensified mass transport due to the formation of hydrogen gas, which induces turbulence. A plausible mechanism based on several control experiments was outlined in Scheme 20. The first step involves formation of a disulfide intermediate XXV from the thiol substrate 36 via anodic oxidation. Then electro-oxidation of amine 37 generates the aminium radical intermediate XXVI, which reacts with the disulfide XXV to form the sulfenamide XXVII. Next, two consecutive oxidation steps of the sulfenamide occurs and the targeted sulfonamide 38 produces via a sulfinamide intermediate XXVIII.
 |
| | Scheme 19 Electrochemical oxidative coupling of thiols 36 and amines 37 in flow cells. | |
 |
| | Scheme 20 Proposed mechanism for the electrosynthesis of sulfonamides 38. | |
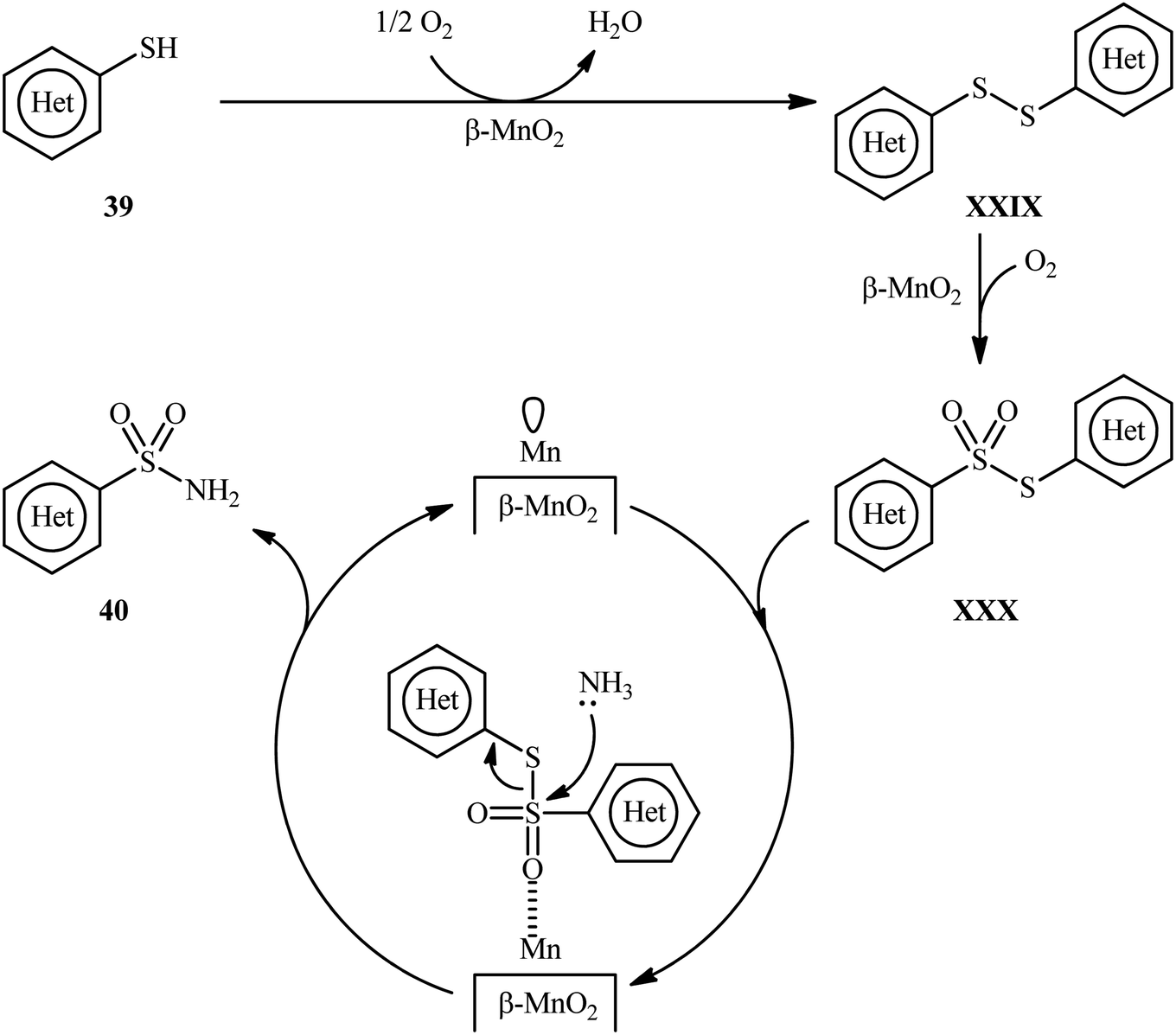
Very recently, the research group of Kamata and Hara explored the catalytic application of dual-functional high-surface-area β-MnO2 (β-MnO2-HS) nanoparticles for the direct sulfonylation of ammonia with thiols to synthesize primary sulfonamides.43 Various aromatic and heteroaromatic thiols 39 have been reacted with aqueous NH3 in the presence of a catalytic amount of β-MnO2-HS under O2 atmosphere to give (hetero)aryl sulfonamides 40 in moderate to high yields (Scheme 21). The reaction works better with electron rich than with electron poor substituents on the aryl thiols (88% yield for 4-OMe-substituted substrate compared to 58% for the 4-nitro derivative). It is worthwhile mentioning that the authors displayed the applicability of this methodology by performing a scale-up reaction, affording 89% of toluenesulfonamide. Unfortunately, no desired product was obtained when aliphatic thiols were employed under these reaction conditions. The recycling test established that the catalyst could be recovered by filtration and after washing with DMF and calcination reused for several consecutive runs without detrimental loss of catalytic activity. In this report, several control experiments and density functional theory (DFT) calculations were performed for the insight of the reaction mechanism, which indicated the formation of key thiosulfonate intermediate XXX in the catalytic cycle (Scheme 22).
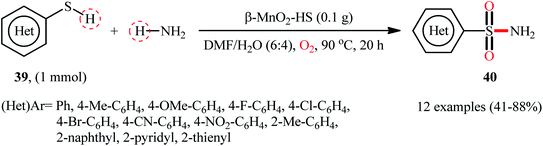 |
| | Scheme 21 β-MnO2-catalyzed oxidative sulfonamidation of (hetero)aromatic thiols 39 with ammonia. | |
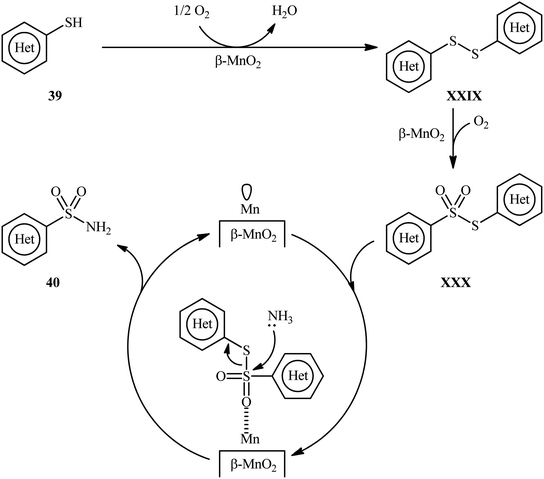 |
| | Scheme 22 Plausible mechanistic pathway for the reaction in Scheme 21. | |
5. Conclusion
Sulfenamide, sulfinamides, and sulfonamides are among the most important subclasses of sulfur–nitrogen bond-containing organosulfur compounds that are widely used in diverse fields such as pharmaceutical, pesticide, and polymer industries. Owing to their versatile applications, the development of new, cost-efficient, and nearly waste-free methods for their preparation, from simple commercially available starting materials is highly desirable. As illustrated, the direct synthesis of titled compounds via condition-controlled oxidative coupling of thiols and amines, two inexpensive and readily available commodity chemicals, constitutes an atom-efficient and step-economic strategy, since no pre-functionalization of substrates is required. Despite remarkable achievements over the past few years in this appealing research arena, several great challenges still remain to be overcome. For example: (i) the majority of reported examples in the field have been performed in the presence of large amounts of molecular or hypervalent iodines. Therefore, development of related processes employing sub-stoichiometric amounts of other inexpensive reagents and catalysts will be attractive; (ii) the scope of amine partners was largely limited to aliphatic ones, thus expanding of the substrate scope of these transformations to aromatic amines are necessary; and (iii) the number of reported examples in this chemistry (specially for the synthesis of sulfinamides) are narrow and there is a further need to study the scope and limitations of these reactions. We conclude this focus-review by hoping that it may serve as an inspiration for future studies and research on the field.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) M. Shimizu, S. Y. Suzuki, S. Tanaka, W. Ando and N. Sakai, Phosphorus, Sulfur Silicon Relat. Elem., 2019, 194, 764–767 CrossRef CAS;
(b) S. Majedi and S. Majedi, J. Chem. Lett., 2020, 1, 2–8 Search PubMed.
- E. L. Briggs, A. Tota, M. Colella, L. Degennaro, R. Luisi and J. A. Bull, Angew. Chem., Int. Ed., 2019, 58, 14303–14310 CrossRef CAS PubMed.
- M. Andresini, M. Spennacchio, G. Romanazzi, F. Ciriaco, G. Clarkson, L. Degennaro and R. Luisi, Org. Lett., 2020, 22, 7129–7134 CrossRef CAS PubMed.
-
(a) G. Rong, Y. Chen, L. Wang, J. Li, J. Wang, M. J. Panzer and Y. Pang, J. Appl. Polym. Sci., 2014, 131, 39699 CrossRef;
(b) P. Charoeythornkhajhornchai, C. Samthong and A. Somwangthanaroj, J. Appl. Polym. Sci., 2017, 134, 44822 CrossRef.
-
(a) L. Craine and M. Raban, Chem. Rev., 1989, 89, 689–712 CrossRef CAS;
(b) S. S. Klioze, R. C. Allen, J. C. Wilker and D. L. Woodward, J. Med. Chem., 1980, 23, 677–679 CrossRef CAS PubMed;
(c) Z. Li, Y. Shi, A. Zhu, Y. Zhao, H. Wang, B. P. Binks and J. Wang, Angew. Chem., Int. Ed., 2021, 60, 3928–3933 CrossRef CAS PubMed;
(d) L. Zhang, M. Zhang, S. You, D. Ma, J. Zhao and Z. Chen, Sci. Total Environ., 2021, 780, 146505 CrossRef CAS PubMed;
(e) H. Wang, J. Cui, Y. Zhao, Z. Li and J. Wang, Green Chem., 2021, 23, 405–411 RSC;
(f) L. Zhang, J. Zheng, S. Tian, H. Zhang, X. Guan, S. Zhu and Z. Li, J. Environ. Sci., 2020, 91, 212–221 CrossRef PubMed;
(g) X. Wang, P. Gao, Y. Liu, H. Li and F. Lu, Curr. Bioinf., 2020, 15(10), 493–502 CrossRef CAS;
(h) M. Niu, Y. Lin and Q. Zou, Plant Mol. Biol., 2021, 105(4–5), 483–495 CrossRef CAS PubMed;
(i) S. Sun, L. Xu, Q. Zou, G. Wang and J. Gorodkin, Bioinformatics, 2021, 37, 1319–1321 CrossRef CAS PubMed.
-
(a) J. J. Petkowski, W. Bains and S. Seager, J. Nat. Prod., 2018, 81, 423–446 CrossRef CAS PubMed;
(b) Q. Wang, S. Sun, X. Zhang, H. Liu, B. Sun and S. Guo, Bioresour. Technol., 2021 DOI:10.17582/journal.pjz/20190715000752;
(c) Z. Congfen, L. Yingxiao, H. Linfeng, S. Fengjun, Y. Weitang and L. Xuegang, Pak. J. Zool., 2021, 1–8 Search PubMed.
- C. Achuenu, S. Carret, J. F. Poisson and F. Berthiol, Eur. J. Org. Chem., 2020, 5901–5916 CrossRef CAS.
-
(a) N. Gigant, E. Drège, P. Retailleau and D. Joseph, Chem.–Eur. J., 2015, 21, 15544–15547 CrossRef CAS PubMed;
(b) C. S. Richards-Taylor, C. Martínez-Lamenca, J. E. Leenaerts, A. A. Trabanco and D. Oehlrich, J. Org. Chem., 2017, 82, 9898–9904 CrossRef CAS PubMed;
(c) L. J. Ma, G. X. Li, J. Huang, J. Zhu and Z. Tang, Tetrahedron Lett., 2018, 59, 4255–4258 CrossRef CAS.
-
(a) K. A. Scott and J. T. Njardarson, Top. Curr. Chem., 2018, 376, 1–34 CrossRef CAS PubMed;
(b) S. Shafiei and S. Davaran, Chem. Rev. Lett., 2020, 3, 19–22 Search PubMed;
(c) F. Nareetsile, J. T. P. Matshwele, S. Ndlovu and M. Ngaski, Chem. Rev. Lett., 2020, 3, 140–160 CAS.
-
(a) Y. Zhang, Y. Liu, X. Ma, X. Ma, B. Wang, H. Li, Y. Huang and C. Liu, Dyes Pigm., 2018, 158, 438–444 CrossRef CAS;
(b) D. K. Sharma, S. T. Adams Jr, K. L. Liebmann, A. Choi and S. C. Miller, Org. Lett., 2019, 21, 1641–1644 CrossRef CAS PubMed.
- D. F. Cadogan and C. J. Howick, Plasticizers, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012, vol. 27, pp. 599–618 Search PubMed.
- I. V. E. Koval', Russ. Chem. Rev., 1995, 64, 731–751 CrossRef.
- D. A. Cogan, G. Liu, K. Kim, B. J. Backes and J. A. Ellman, J. Am. Chem. Soc., 1998, 120, 8011–8019 CrossRef CAS.
- K. K. Anderson, in Sulfonic acids and their derivatives in comprehensive organic chemistry, ed. D. H. R. Barton, W. D. Ollis and D. N. Jones, Pergamon Press, Oxford, 1979, vol. 3, p. 331 Search PubMed.
- H. Konishi, H. Tanaka and K. Manabe, Org. Lett., 2017, 19, 1578–1581 CrossRef CAS PubMed.
-
(a) M. Bao and M. Shimizu, Tetrahedron, 2003, 59, 9655–9659 CrossRef CAS;
(b) N. Taniguchi, Synlett, 2007, 1917–1920 CrossRef CAS.
- E. Wojaczyńska and J. Wojaczyński, Chem. Rev., 2020, 120, 4578–4611 CrossRef PubMed.
-
(a) M. Ashfaq, S. S. Shah, T. Najjam, S. Shaheen and G. Rivera, Mini-Rev. Org. Chem., 2013, 10, 160–170 CrossRef CAS;
(b) D. Joseph, M. A. Idris, J. Chen and S. Lee, ACS Catal., 2021, 11, 4169–4204 CrossRef CAS.
-
(a) S. Arshadi, E. Vessally, L. Edjlali, R. Hosseinzadeh-Khanmiri and E. Ghorbani-Kalhor, Beilstein J. Org. Chem., 2017, 13, 625–638 CrossRef CAS PubMed;
(b) A. Hosseinian, S. Ahmadi, F. A. H. Nasab, R. Mohammadi and E. Vessally, Top. Curr. Chem., 2018, 376, 1–32 CrossRef PubMed;
(c) F. A. H. Nasab, L. Z. Fekri, A. Monfared, A. Hosseinian and E. Vessally, RSC Adv., 2018, 8, 18456–18469 RSC;
(d) A. Hosseinian, P. D. K. Nezhad, S. Ahmadi, Z. Rahmani and A. Monfared, J. Sulfur Chem., 2019, 40, 88–112 CrossRef CAS;
(e) M. Hamzehloo, A. Hosseinian, S. Ebrahimiasl, A. Monfared and E. Vessally, J. Fluorine Chem., 2019, 224, 52–60 CrossRef CAS;
(f) M. R. J. Sarvestani, N. Mert, P. Charehjou and E. Vessally, J. Chem. Lett., 2020, 1, 93–102 Search PubMed;
(g) L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 9–18 Search PubMed;
(h) S. Majedi, L. Sreerama, E. Vessally and F. Behmagham, J. Chem. Lett., 2020, 1, 25–31 Search PubMed.
- Z. Liu, A. Ebadi, M. Toughani, N. Mert and E. Vessally, RSC Adv., 2020, 10, 37299–37313 RSC.
- B. Azizi, M. R. P. Heravi, Z. Hossaini, A. Ebadi and E. Vessally, RSC Adv., 2021, 11, 13138–13151 RSC.
- S. Ahmadi, A. Hosseinian, P. D. Kheirollahi Nezhad, A. Monfared and E. Vessally, Iran. J. Chem. Chem. Eng., 2019, 38, 1–19 CAS.
- E. Vessally, S. Mohammadi, M. Abdoli, A. Hosseinian and P. Ojaghloo, Iran. J. Chem. Chem. Eng., 2020, 39, 11–19 CAS.
- X. Ma, Z. Kexin, W. Yonggang, A. G. Ebadi and M. Toughani, Iran. J. Chem. Chem. Eng., 2021 DOI:10.30492/ijcce.2021.529010.4694.
- R. T. Kareem, B. Azizi, M. Asnaashariisfahani, A. Ebadi and E. Vessally, RSC Adv., 2021, 11, 14941–14955 RSC.
-
(a) A. Hosseinian, S. Farshbaf, L. Z. Fekri, M. Nikpassand and E. Vessally, Top. Curr. Chem., 2018, 376, 1–19 CrossRef PubMed;
(b) W. Peng, E. Vessally, S. Arshadi, A. Monfared, A. Hosseinian and L. Edjlali, Top. Curr. Chem., 2019, 377, 1–22 CrossRef CAS PubMed;
(c) Y. Yang, D. Zhang and E. Vessally, Top. Curr. Chem., 2020, 378, 1–32 CrossRef PubMed;
(d) Z. He, D. Wu and E. Vessally, Top. Curr. Chem., 2020, 378, 1–30 CrossRef PubMed;
(e) L. Feng, X. Li, B. Liu and E. Vessally, J. CO2 Util., 2020, 40, 101220 CrossRef CAS;
(f) W. Xu, A. G. Ebadi, M. Toughani and E. Vessally, J. CO2 Util., 2020, 43, 101358 CrossRef;
(g) A. Bakhtiary, M. R. P. Heravi, A. Hassanpour, I. Amini and E. Vessally, RSC Adv., 2020, 11, 470–483 RSC.
- N. Taniguchi, Eur. J. Org. Chem., 2010, 2670–2673 CrossRef CAS.
- E. Rattanangkool, W. Krailat, T. Vilaivan, P. Phuwapraisirisan, M. Sukwattanasinitt and S. Wacharasindhu, Eur. J. Org. Chem., 2014, 4795–4804 CrossRef CAS.
- L. Yang, S. Li, Y. Dou, S. Zhen, H. Li, P. Zhang, B. Yuan and G. Yang, Asian J. Org. Chem., 2017, 6, 265–268 CrossRef CAS.
- Y. Dou, X. Huang, H. Wang, L. Yang, H. Li, B. Yuan and G. Yang, Green Chem., 2017, 19, 2491–2495 RSC.
- S. H. Ryu, J. Ra and H. M. Ko, Asian J. Org. Chem., 2020, 9, 933–938 CrossRef CAS.
- L. Yang, J. Feng, M. Qiao and Q. Zeng, Org. Chem. Front., 2018, 5, 24–28 RSC.
- S. Liu, Z. Qi, Z. Zhang and B. Qian, Org. Lett., 2019, 21, 7722–7725 CrossRef CAS PubMed.
- S. Tang, Y. Liu, L. Li, X. Ren, J. Li, G. Yang, H. Li and B. Yuan, Org. Biomol. Chem., 2019, 17, 1370–1374 RSC.
- N. Taniguchi, Eur. J. Org. Chem., 2016, 2157–2162 CrossRef CAS.
- S. Chatterjee, S. Makai and B. Morandi, Angew. Chem., Int. Ed., 2021, 60, 758–765 CrossRef CAS PubMed.
-
(a) S. W. Wright and K. N. Hallstrom, J. Org. Chem., 2006, 71, 1080–1084 CrossRef CAS PubMed;
(b) J. D. Bonk, D. T. Amos and S. J. Olson, Synth. Commun., 2007, 37, 2039–2050 CrossRef CAS;
(c) K. Bahrami, M. M. Khodaei and M. Soheilizad, J. Org. Chem., 2009, 74, 9287–9291 CrossRef CAS PubMed;
(d) K. Bahrami, M. M. Khodaei and M. Soheilizad, Tetrahedron Lett., 2010, 51, 4843–4846 CrossRef CAS;
(e) H. Veisi, R. Ghorbani-Vaghei, S. Hemmati and J. Mahmoodi, Synlett, 2011, 2315–2320 CrossRef CAS;
(f) K. Bahrami, M. M. Khodaei and J. Abbasi, Tetrahedron, 2012, 68, 5095–5101 CrossRef CAS;
(g) H. Veisi, Bull. Korean Chem. Soc., 2012, 33, 383–386 CrossRef CAS;
(h) A. R. Massah, S. Sayadi and S. Ebrahimi, RSC Adv., 2012, 2, 6606–6616 RSC;
(i) B. Maleki, S. Hemmati, R. Tayebee, S. Salemi, Y. Farokhzad, M. Baghayeri, F. M. Zonoz, E. Akbarzadeh, R. Moradi, A. Entezari and M. R. Abdi, Helv. Chim. Acta, 2013, 96, 2147–2151 CrossRef CAS.
-
(a) A. Gucchait, K. Jana and A. K. Misra, RSC Adv., 2017, 7, 32478–32487 RSC;
(b) R. Parnian, E. Soleimani and K. Bahrami, ChemistrySelect, 2019, 4, 8554–8557 CrossRef CAS;
(c) S. Sohrabnezhad, K. Bahrami and F. Hakimpoor, J. Sulfur Chem., 2019, 40, 256–264 CrossRef CAS.
- J. B. Feng and X. F. Wu, Org. Biomol. Chem., 2016, 14, 6951–6954 RSC.
- M. Zhu, W. Wei, D. Yang, H. Cui, L. Wang, G. Meng and H. Wang, Org. Biomol. Chem., 2017, 15, 4789–4793 RSC.
- A. Tota, S. St John-Campbell, E. L. Briggs, G. O. Estévez, M. Afonso, L. Degennaro, R. Luisi and J. A. Bull, Org. Lett., 2018, 20, 2599–2602 CrossRef CAS PubMed.
- G. Laudadio, E. Barmpoutsis, C. Schotten, L. Struik, S. Govaerts, D. L. Browne and T. Noël, J. Am. Chem. Soc., 2019, 141, 5664–5668 CrossRef CAS PubMed.
- E. Hayashi, Y. Yamaguchi, Y. Kita, K. Kamata and M. Hara, Chem. Commun., 2020, 56, 2095–2098 RSC.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *b,
Roya Ahmadi
c,
Alibek Issakhovde,
Abdol Ghaffar Ebadi
f and
Esmail Vessally
*b,
Roya Ahmadi
c,
Alibek Issakhovde,
Abdol Ghaffar Ebadi
f and
Esmail Vessally