
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAzo synthesis meets molecular iodine catalysis†
Rozhin Rowshanpour and
Travis Dudding *
*
Brock University, 1812 Sir Isaac Brock Way, St. Catharines, Ontario L2S 3A1, Canada. E-mail: tdudding@brocku.ca
First published on 11th February 2021
Abstract
A metal-free synthetic protocol for azo compound formation by the direct oxidation of hydrazine HN–NH bonds to azo group functionality catalyzed by molecular iodine is disclosed. The strengths of this reactivity include rapid reaction times, low catalyst loadings, use of ambient dioxygen as a stoichiometric oxidant, and ease of experimental set-up and azo product isolation. Mechanistic studies and density functional theory computations offering insight into this reactivity, as well as the events leading to azo group formation are presented. Collectively, this study expands the potential of main-group element iodine as an inexpensive catalyst, while delivering a useful transformation for forming azo compounds.
Introduction
Azo compounds have captivated chemists ever since their debut in the 1860's and use as an industrial manufactured color, shortly after Peter Griess's synthesis of Manchester Bismarck brown.1 Nowadays, the commercial, academic and industrial uses of azo compounds are widespread, including applications as organic dyes and pigments,2 free-radical initiators,3 acid–base indicators4 and chemosensors.5 Beyond this mélange of usages, the switchable and spatiotemporal (light/thermal)-driven control of E- vs. Z-azo group double geometry of many azo compounds continues to prompt their use in photoresponsive soft materials functioning as liquid crystals,6 smart polymers,7 photochromic ligands for optochemical genetics8 and photoswitches.9As to be expected, this revered status has ushered in the advancement of various synthetic strategies for accessing (hetero)aryl functionalized azo compounds, yet practical synthetic methods for preparing these valuable compounds is needed. Among the most widely used methods for forming azo compounds are oxidative dimerization of aromatic amines,10 reductive dimerization of nitrobenzenes,11 Mills reaction,12 diazonium coupling13 and oxidative dehydrogenation14 (Fig. 1, top); however, each of these synthetic methods have inherent drawbacks, such as employing toxic and expensive transition metals and/or hazardous reagents. For instance, the oxidative dimerization of aromatic amines frequently relies on the use of stoichiometric AgO,15 ferrate salts,16 tert-butyl hypoiodite17 or environmentally unfriendly heavy-metal oxidants BaMnO4,18 Pb(OAc)4,19 and HgO.20 Moreover, the Mills reaction12 and diazonium coupling13 require the use of toxic nitrosoarenes or explosive diazonium salt substrates. Similarly, reductive dimerization of nitroarenes11 is plagued by the use of toxic and often carcinogenic nitro-containing compounds.
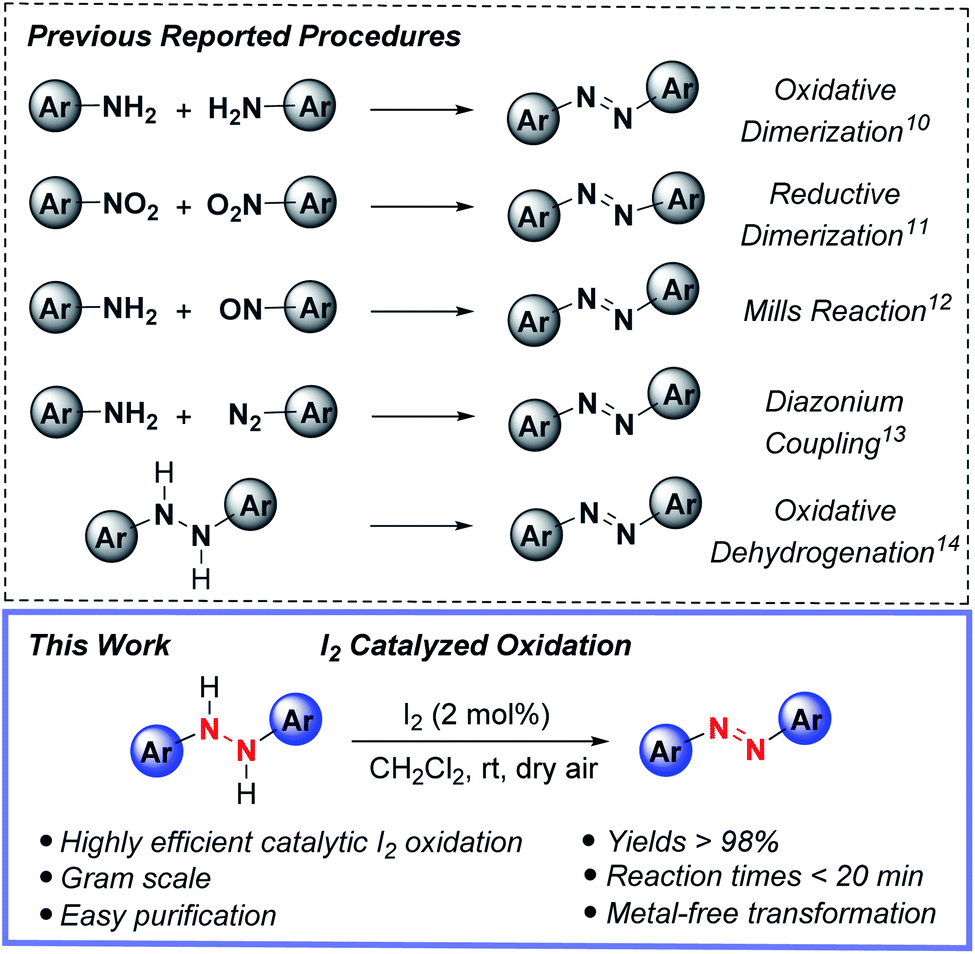 | ||
| Fig. 1 Previous reported procedures for azo synthesis (top) and current iodine catalyzed synthesis of azo compounds (bottom).10–14 | ||
From this precedent along with other works,21,22 and with it, a heavy reliance upon transition metals and/or hazardous reagents, the prospect of employing the oxidative capacity of main-group elements for azo group construction captured our attention. This possibility was especially appealing in light of a growing interest in replacing, or at the very least, complementing metal-based procedures with sustainable main group element-catalyzed protocols.23 Driving this interest, the key parameters of cost, dwindling supplies of precious metals and associated political factors, and importantly global sustainability enabling productive harmony, stability and resilience to support present and future generations. Towards this end, we report herein the first metal-free, synthetic protocol for azo compound formation by the direct oxidation of hydrazine HN–NH bonds to azo group functionality catalyzed by substoichiometric amounts of molecular iodine (Fig. 1, bottom). The strengths of this reactivity include rapid reaction times, low catalyst loadings, use of ambient dioxygen as a stoichiometric oxidant, and ease of experimental set-up and azo product isolation.
Results and discussion
At the outset of this study with an eye towards using easy-to-handle, environmentally friendly and relatively inexpensive solid molecular iodine (e.g., over the last 5 years the bulk price of iodine has been $19–27 per kg) as a catalyst for azo double bond formation, we sought to delineate to what degree, if any, halogen bonding might have in triggering hydrazine HN–NH bond oxidation. Prompting this interest, among other factors, was the prominence of iodine-catalyzed reactions, many of which catalytically pivot around the formation of a “halogen bond” (X–B)—defined by IUPAC as the association between the electrophilic region of a polarized halogen atom and a Lewis basic unit.24 The electrophilic region, in this case, commonly being referred to as a sigma hole (σ).25 For molecular iodine this feature is clearly visible from the calculated electrostatic potential surface, displaying significant anisotropic electron distribution with positive polarization on the iodine atoms (Fig. 2, left-hand side, top).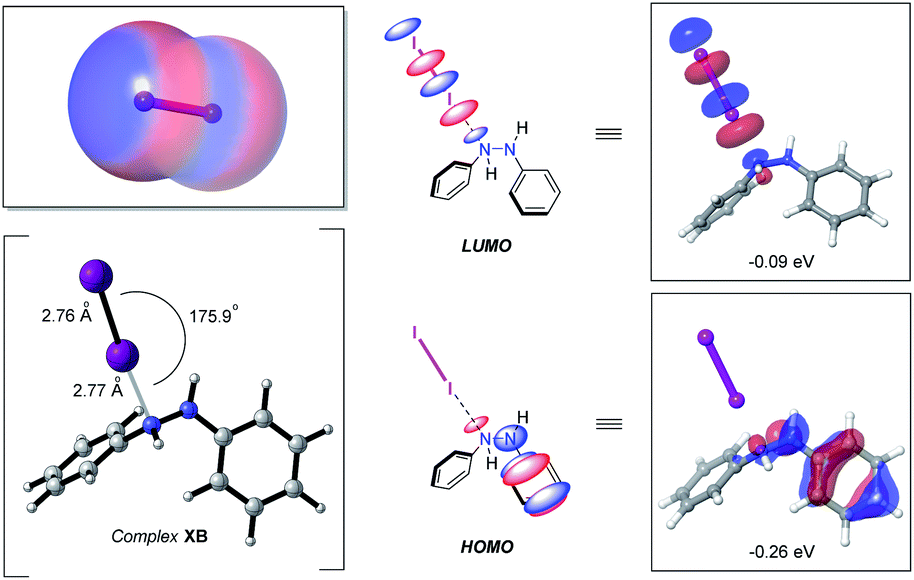 | ||
| Fig. 2 Electrostatic potential surface of I2 (electronegative (red) and electropositive (blue) regions (left-hand side, top)) computed at the B3LYP-D3/lacv3p**++ level of theory (isovalue = −0.003 (max = 0.3, min = −0.3)). Halogen bond complex XB (left-hand side, bottom) computed at the (IEFPCMDCM)UAPFD/def2-TZVP//UAPFD/DGDZVP level of theory. Chemdraw and computed HOMO and LUMO of complex XB with orbital energies in eV (middle and right-hand side). | ||
With this bearing in mind, and in reflecting upon the history of the halogen bond dating back more than two-centuries to the first reported synthesis of a halogen bond complex NH3⋯I2 by Colin in 1814,26 it occurred to us that the formation of a hydrazine–I2 complex should be a facile process. Indeed, formation of computed n-type halogen bond complex XB from diphenyl hydrazine and I2 was computed to be exergonic by −0.1 kcal mol−1 at the (IEFPCMDCM)UAPFD/def2-TZVP//UAPFD/DGDZVP level of theory. This complex displayed an almost linear N⋯I–I angle of 175.9° with nitrogen-iodine and iodine–iodine bond distances of 2.77 Å and 2.76 Å (Fig. 2, bottom left-hand side). Moreover, in foreshadowing reactivity, the highest occupied molecular orbital (HOMO, −0.26 eV) of this halogen bond complex was localized upon the hydrazine fragment, while the lowest unoccupied molecular orbital (LUMO, −0.09 eV), dominated by anti-bonding σ*-character, resided on I2 (Fig. 2, middle and right-hand side).
Optimization studies
With this insight, yet cautiously aware of the lack of a reported metal-free molecular iodine-catalyzed oxidation of hydrazine HN–NH bonds to azo N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bonds,27 we commenced our study by investigating the reaction of diphenyl hydrazine (1a) in various solvents using catalytic iodine loadings (2–10 mol%) as summarized in Table 1. To this end, performing the reaction with a tenth of an equivalent of molecular iodine at room temperature in the presence of light and dry air as a stoichiometric co-oxidant resulted in very little azo product (2a) formation in polar protic solvent MeOH (Table 1, entry 1). Further, nonpolar solvents hexane and toluene proved even worse (Table 1, entries 2 and 3), whereas polar aprotic solvents, including acetone, acetonitrile, ethyl acetate, and tetrahydrofuran (THF) resulted in negligible conversions (Table 1, entries 4–7). Suspecting this poor reactivity was not a systemic problem, but rather an intrinsic feature of (1) hydroxyl, nitrile or carbonyl binding to I2, that is, a solvent–solute sequestering effect or (2) insolubility of I2 in nonpolar solvents, we performed the reaction in dichloromethane (DCM) and chloroform (CHCl3). The logic for choosing these solvents stemming from their weak ability to engage in halogen–halogen or hydrogen-halogen bonding with iodine, while offering good solubility. Indeed, this hypothesis proved productive as full conversion to product azobenzene 2a occurred in an hour with DCM as the solvent and high conversion was observed in CHCl3 (Table 1, entries 8 and 9). Moreover, decreasing the loading of catalyst I2 afforded high conversion (Table 1, entries 10 and 11). Meanwhile, the use of dry air in the absence of light dramatically reduced the reaction time (Table 1, entry 12). Lastly, demonstrating the importance of iodine as a catalyst, no conversion was observed in the absence of I2 (Table 1, entry 13).
N double bonds,27 we commenced our study by investigating the reaction of diphenyl hydrazine (1a) in various solvents using catalytic iodine loadings (2–10 mol%) as summarized in Table 1. To this end, performing the reaction with a tenth of an equivalent of molecular iodine at room temperature in the presence of light and dry air as a stoichiometric co-oxidant resulted in very little azo product (2a) formation in polar protic solvent MeOH (Table 1, entry 1). Further, nonpolar solvents hexane and toluene proved even worse (Table 1, entries 2 and 3), whereas polar aprotic solvents, including acetone, acetonitrile, ethyl acetate, and tetrahydrofuran (THF) resulted in negligible conversions (Table 1, entries 4–7). Suspecting this poor reactivity was not a systemic problem, but rather an intrinsic feature of (1) hydroxyl, nitrile or carbonyl binding to I2, that is, a solvent–solute sequestering effect or (2) insolubility of I2 in nonpolar solvents, we performed the reaction in dichloromethane (DCM) and chloroform (CHCl3). The logic for choosing these solvents stemming from their weak ability to engage in halogen–halogen or hydrogen-halogen bonding with iodine, while offering good solubility. Indeed, this hypothesis proved productive as full conversion to product azobenzene 2a occurred in an hour with DCM as the solvent and high conversion was observed in CHCl3 (Table 1, entries 8 and 9). Moreover, decreasing the loading of catalyst I2 afforded high conversion (Table 1, entries 10 and 11). Meanwhile, the use of dry air in the absence of light dramatically reduced the reaction time (Table 1, entry 12). Lastly, demonstrating the importance of iodine as a catalyst, no conversion was observed in the absence of I2 (Table 1, entry 13).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Catalyst loading | Reaction conditions | Reaction time | Yieldb (%) |
| a Reaction conditions: 1a (1 mmol), solvent (5.0 mL) and I2 (2–10 mol%) at room temperature under a dry air atmosphere in the absence/presence of ambient light.b Yields are determined by quantitative 1H NMR using CDCl3 as the internal standard. | |||||
| 1 | MeOH | 10 mol% | Room light | 2 hours | >5 |
| 2 | Toluene | 10 mol% | Room light | 2 hours | 0 |
| 3 | Hexane | 10 mol% | Room light | 2 hours | 0 |
| 4 | Acetone | 10 mol% | Room light | 2 hours | 0 |
| 5 | Acetonitrile | 10 mol% | Room light | 2 hours | 0 |
| 6 | Ethyl acetate | 10 mol% | Room light | 2 hours | 0 |
| 7 | Dry THF | 10 mol% | Room light | 2 hours | >5 |
| 8 | Dry DCM | 10 mol% | Room light | 1 hour | 100 |
| 9 | Dry chloroform | 10 mol% | Room light | 1 hour | 92 |
| 10 | Dry DCM | 5 mol% | Room light | 1 hour | 100 |
| 11 | Dry DCM | 2 mol% | Room light | 1 hour | 100 |
| 12 | Dry DCM | 2 mol% | Dark | 20 min | 100 |
| 13 | Dry DCM | — | Dark | 48 hours | 0 |
Substrate scope
With the optimal reaction conditions established, we briefly examined the scope of this azo-forming protocol. To this end, various 1,2-disubstituted hydrazines with electron-rich and electron-poor aromatic groups smoothly reacted to furnish the target azo compounds in good-to-excellent yields (2a–u, Table 2). For instance, the reaction of o- and/or p-alkyl substituted aryl hydrazines (1a–i) successfully afforded aromatic azo compounds 2a–i in good-to-high yields, though sterically hindered hydrazines 1i provided a slightly reduced yield of azo compound 2i. Meanwhile, the presence of inductively-withdrawing and weak electron-donating p-substituted halogens on the hydrazine aryl rings had little effect on the reaction efficiency. For instance, p-iodo-, p-fluoro-, and p-chloro-substituted aryl hydrazines (1j–l) smoothly reacted to afford azo compounds 2j–l. Similarly, o-chloro-substituted aryl hydrazine 1m was compatible, furnishing azo compound 2m in high yield. Moreover, p-Alkyl and m-chloro- or m- or p-bromo-substituted aryl hydrazines (1n–p) underwent efficient oxidation to provide azo compounds 2n–p. Further, the presence of electron-withdrawing trifluoromethyl- and trifluoromethyl ether groups had no effect on the reaction with azo compounds 2q–s formed in high yields. Likewise, the reaction of electron-donating p-methoxy-substituted aryl hydrazine 1t afforded azo product 2t in high yield. Nitro functionality was also tolerated with azo compound 2u formed in high yield. Lastly, to demonstrate the synthetic utility of this methodology, we performed a gram-scale reaction using 2.15 grams of 1a to afford 2a in 94% isolated yield (Table 2, bottom).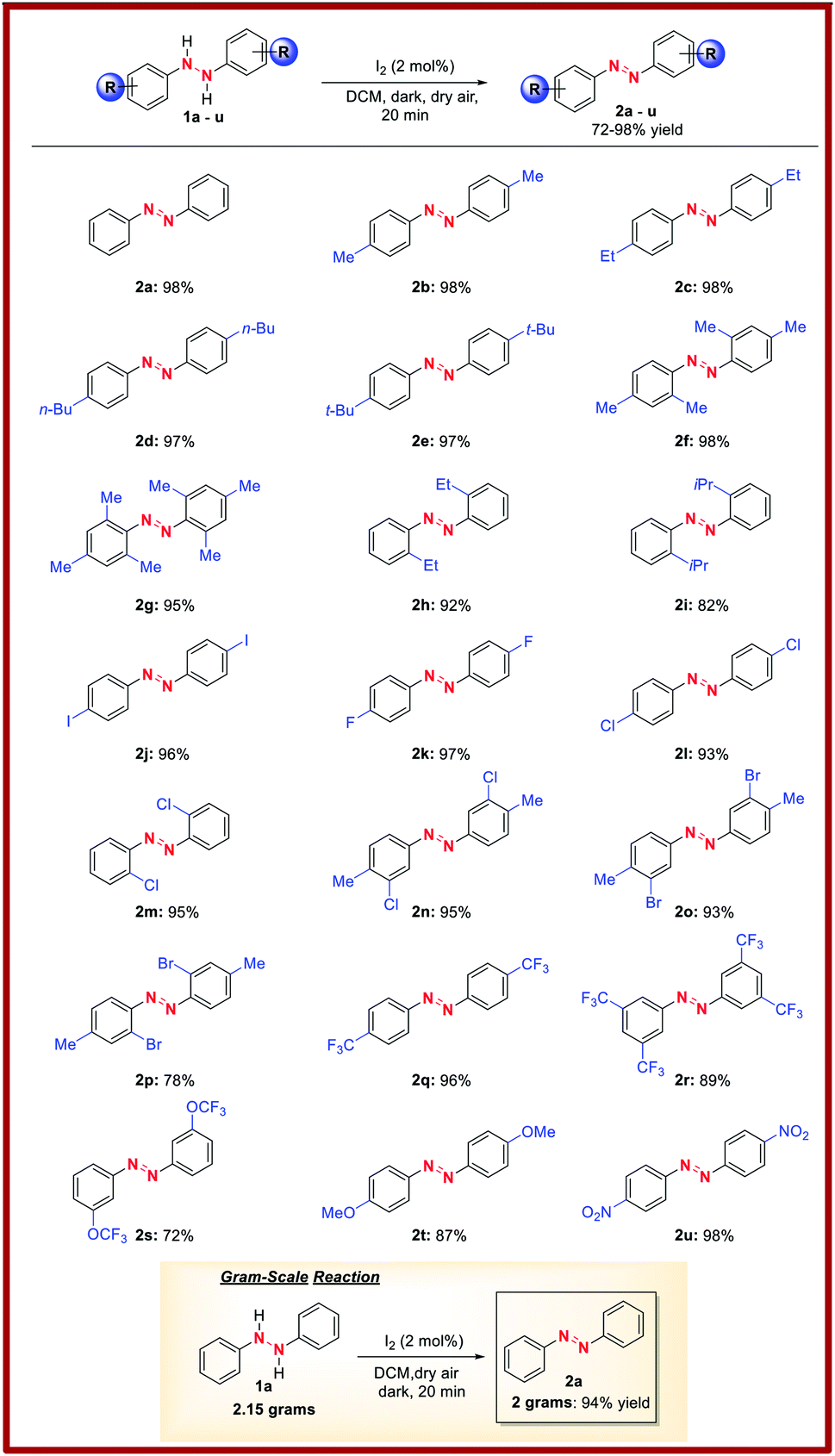
Control studies
Our interest next shifted to understanding the mechanistic details of this simple iodine-catalyzed azo NN double bond forming reaction. An initial reaction using catalytic iodine (2 mol%) in the absence of air negatively affected azo product formation, suggesting the oxidation of molecular iodine or iodide ion to higher oxidation states might have a role in the reaction (Table 3, entry 1). To explore this aspect further, the role of higher iodine oxidation states were investigated using hydrogen peroxide (entry 2) resulting in quantitative conversion of 1a to azobenzene (2a). Moreover, when we added DMSO (5 mol%, entry 3), a very good iodonium binding solvent,23 to the original iodine (2 mol%) catalyzed reaction conditions, a good conversion was obtained, thus suggesting iodonium is not involved in the reaction. On the other hand, this finding is not at all surprising and can be explained by the fact that DMSO is known to oxidize iodine, thus implicating oxidative pathway(s). On the other hand, in the presence of 10 mol% KOH (entry 4), which could form potassium hypoiodite (KOI) in situ, no azo product was formed suggesting that a hypoiodous/hypoiodite pathway is unlikely.
| Entry | Catalyst (2 mol%) | Additive | Conditions | Reaction time | Conversionb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 1a (1 mmol), dry DCM (5.0 mL) and catalyst I2 (2 mol%) in presence/absence of additives at room temperature, in the absence of light and under dry air atmosphere or N2(g).b Yields were determined by 1H NMR. | |||||
| 1 | I2 | — | N2, dark | 2 hours | 0 |
| 2 | I2 | H2O2 | Dry air, dark | 20 min | 100 |
| 3 | I2 | DMSO (5 mol%) | Dry air, dark | 20 min | 100 |
| 4 | I2 | KOH (10 mol%) | Dry air, dark | 2 hours | 0 |
| 5 | I2 | TEMPO (5 mol%) | Dry air, dark | 2 hours | 74 |
| 6 | I2 | TEMPO (100 mol%) | Dry air, dark | 1 hour | 100 |
| 7 | — | HI (10 mol%) | Dry air, dark | 20 min | 100 |
| 8 | — | HCl (10 mol%) | Dry air, dark | 2 hours | 0 |
| 9 | — | KI (10 mol%) | Dry air, dark | 2 hours | 8 |
| 10 | Br2 | — | Dry air, dark | 2 hours | 0 |
| 11 | I2 | Water (5 mol%) | Dry air, dark | 1 hour | 40 |
| 12 | I2 | Water (10 mol%) | Dry air, dark | 1 hour | 68 |
Further, addition of radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) to the reaction had little effect, hinting that a radical pathway was not involved (entries 5 and 6).28 Next, exchanging elemental iodine for HI still provided product suggesting that hydrogen iodide might be an intermediate in the reaction (entry 7). Conversely, the use of HCl resulted in no conversion establishing the essential role of iodine ion in this reaction (entry 8). Meanwhile, the addition of potassium iodide (10 mol%), negatively affected the reaction outcome (entry 9), which might be due to the formation of potassium triiodide. The use of molecular bromine instead iodine as a catalyst was also attempted resulting in no observed azo product formation and clearly demonstrating the importance of I2 as a catalyst (entry 10). Lastly, moisture had a negative effect on reactivity, suggesting in situ derived hypoiodous acid (HOI) formed by disproportionation of molecular iodine and water had a negative effect on azo product formation (entries 11 and 12).
Mechanistic studies
As a last tool for understanding this reactivity we turned to DFT computations at the (IEFPCMDCM)UAPFD/def2-TZVP//UAPFD/DGDZVP level of theory using diphenyl hydrazine as a representative substrate. Ultimately, this led to the tentative computed pathway depicted in Fig. 3 initiating from hydrazine 1a that upon binding to I2 forms halogen bond complex XB.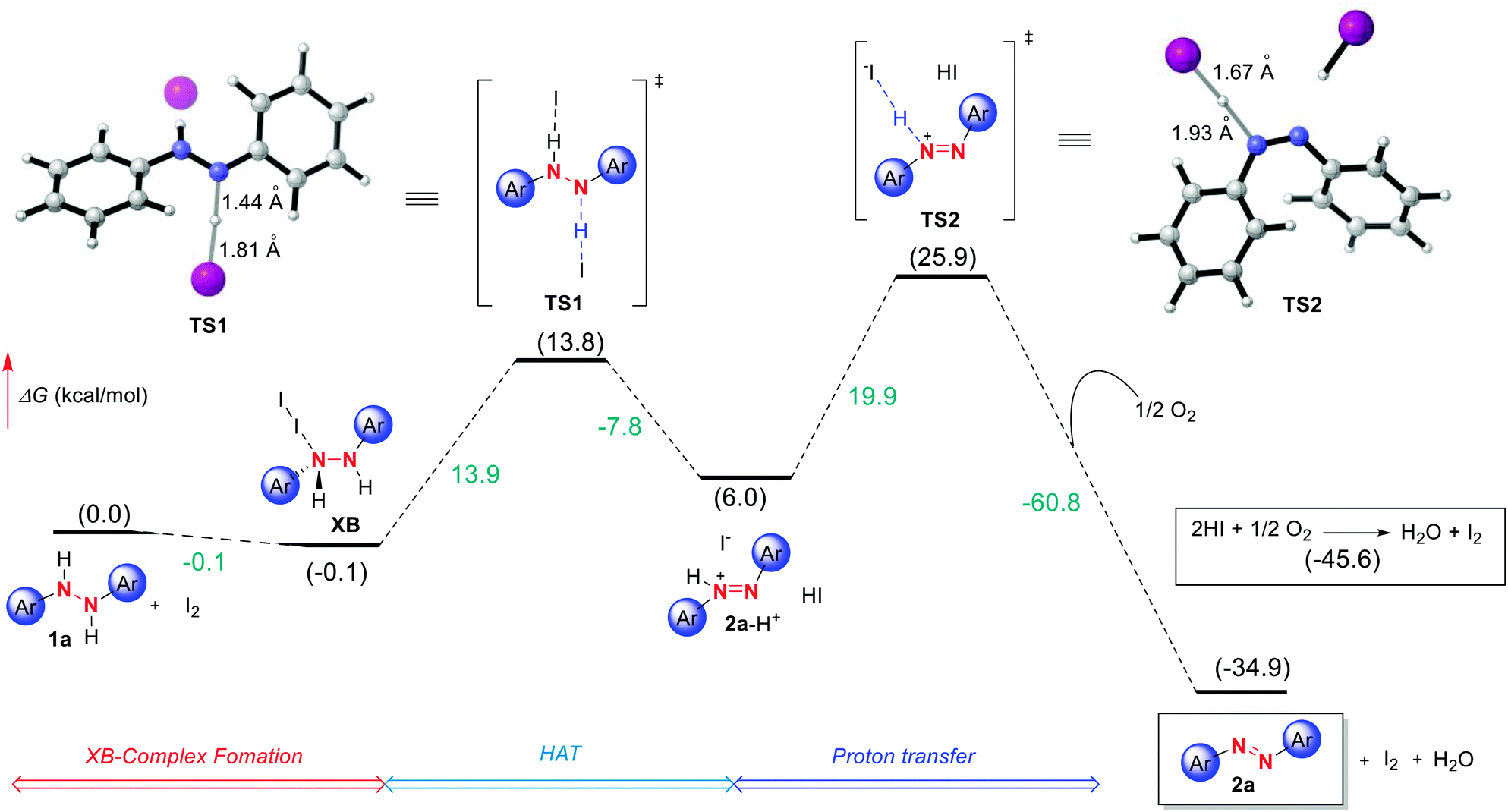 | ||
| Fig. 3 Free energy profile for I2 catalyzed azo formation computed at the (IEFPCMDCM)UAPFD/def2-TZVP//UAPFD/DGDZVP level of theory. Free energies are given in kcal mol−1. | ||
Next, following slight structural reorganization subsequent first-order saddle point TS1 with a Gibbs free activation barrier (ΔG‡) of 13.9 kcal mol−1 is surmounted. Defining this transition state involving Hydrogen Atom Transfer (HAT) and electron transfer type events were bond-breaking and bond-making distances of 1.44 Å and 1.81 Å. The resulting cationic azo intermediate 2a-H+ of TS1 then reacts by proton transfer transition state TS2 with an activation barrier of 19.9 kcal mol−1 and bond-breaking and bond-making distances of 1.67 Å and 1.93 Å. Desired azo product (2a) formation follows concomitant with the reaction of dioxygen and in situ derived HI to afford water and catalytic I2. Collectively, these transformations result in an overall exergonic process of −34.9 kcal mol−1. Meanwhile, in terms of the energetic span model and turnover frequency (TOF), halogen bond complex XB corresponds to the TOF determining intermediate (TDI), while the TOF determining transition state (TDTS) is TS2, thus resulting in an overall energy span of 26.0 kcal mol−1 (TON = 5.5 × 10−8, TOF = 2.9 × 10−9 s−1).29
Greenness measurements of the procedure
Lastly, to highlight the overall greenness and efficiency of our molecular iodine catalyzed azo forming protocol we employ green metrics.30 In this regard, as no one green metric is capable of accounting for every possible parameter influencing the environmental impact of a process, we use several green metrics and the standard reaction metric yield for this analysis. Further, to showcase the favorability of our protocol comparisons are made to previous literature reports for oxidizing hydrazine HN–NH bonds to azo groups. For this analysis two metrics relating to mass efficiency were selected; (1) Sheldon's E-factor was selected as an easy measure of relative waste and (2) GlaxoSmith-Klein's (GSK) metric was used to gauge reaction mass efficiency. Carbon efficiency (CE) was also calculated to determine the efficiency of the transfer of organic material. E-Factors and RME were calculated by eqn (1) and (2) respectively, such that msm is the mass of starting materials and mp is the mass products.31
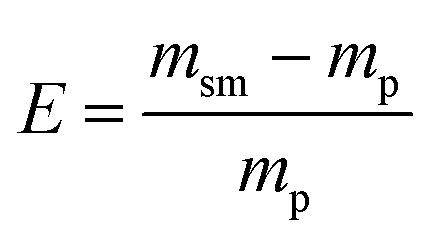 | (1) |
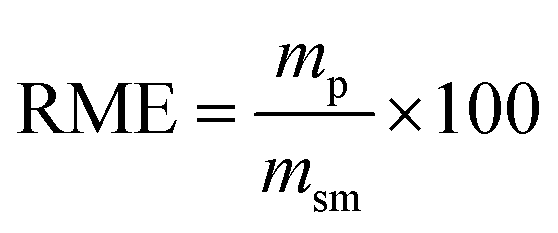 | (2) |
The mass of all consumable starting reagents and catalysts were incorporated, while solvents and silica gel were considered recoverable and aqueous solutions were considered benign, and therefore excluded from the calculation. These assumptions were made for the sake of comparing bench scale chemistries, where solvent choices, quantities and lifecycles can be expected to change significantly during scale-up to an industrial process. CE was determined according to eqn (3), such that np is the moles of product, nsm is the moles of each reagent, Csm is the number of carbons of that reagent and Cp is the number of carbons in the product.
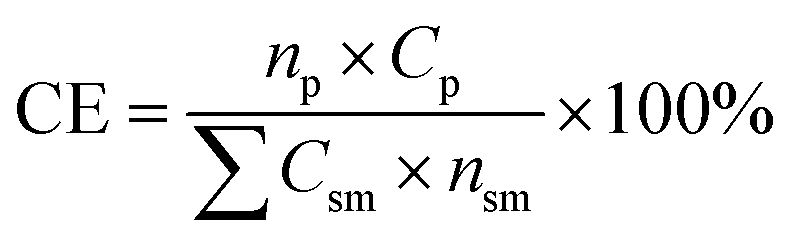 | (3) |
Overall, the green metrics and yield of our reaction are high, if not, excellent as seen from Table 4. This is clearly seen by comparison with reported t-BuOCl and t-BuOK procedures for oxidizing hydrazine HN–NH bonds to azo groups.17,32 In terms of yield and Reaction Mass Efficiency (RME) all three procedures are high yielding with respectable RME values. Though the reported quantitative isolated product yield for the dehydrohalogenation procedure of Okumura et al.17 is remarkable given the need for several purification steps, including silica gel flash chromatography. Meanwhile, our iodine-catalyzed oxidation procedure benefited from significantly higher carbon efficiency (CE) compare to the two other reported procedures, in addition to having a much lower E-factor making it a greener chemistry.
Finally, in speaking to sustainability and in light of the new twelve green chemistry principles promulgated recently by Zimmerman et al.,33 it is notably our reported molecule iodine catalyzed synthesis of azo compounds fulfils the majority of these twelve principles.
Conclusion
To recap, in harnessing the oxidative power of molecular iodine, we have reported an innovative catalytic method for converting hydrazine HN–NH bonds to azo group NN double bond functionally. The significance of this work is manifold, including the use of commercially available, cheap, and easy-to-handle molecular iodine as a non-metal catalyst. In addition, the synthetic protocol described in this letter allows for the preparation of privileged azo group functionality from plentiful hydrazine precursors, while offering underlying mechanistic understanding into iodine catalysis. Ongoing efforts in our lab are expanding this unique reactivity to a repertoire of uses and related findings will be reported in due course.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
T. D. acknowledges financial support from the Natural Science and Engineering Research Council (NSERC) Discovery grant (2019-04205). Computations were carried out using facilities at SHARCNET (Shared Hierarchical Academic Research Computing Network: http://www.sharcnet.ca) and Compute/Calcul Canada. T. D. and R. R. thank the “Brock Library Open Access Publishing Fund”.References
- S. V. Heins, J. Chem. Educ., 1958, 35, 187 CrossRef.
- R. G. Anderson and G. Nickless, Analyst, 1967, 92, 207 RSC.
- (a) L. Androvic, J. Bartacek and M. Sedlak, Res. Chem. Intermed., 2016, 42, 5133 CrossRef CAS; (b) D. Braun, Int. J. Polym. Sci., 2009, 2009, 1 CrossRef.
- N. A. Naser, K. H. Kahdim and D. N. Taha, J. Oleo Sci., 2012, 61, 387 CrossRef CAS.
- N. DiCesare and J. R. Lakowicz, Org. Lett., 2001, 3, 3891 CrossRef CAS.
- H. K. Bisoyi and Q. Li, Chem. Rev., 2016, 116, 15089 CrossRef CAS.
- S. Xie, A. Natansohn and P. Rochon, Chem. Mater., 1993, 5, 403 CrossRef CAS.
- M. A. Kienzler and E. Y. Isacoff, Curr. Opin. Neurobiol., 2017, 45, 202 CrossRef CAS.
- (a) S. Kadota, K. Aoki, S. Nagano and T. Seki, J. Am. Chem. Soc., 2005, 127, 8266 CrossRef CAS; (b) Y. Zhao and J. He, Soft Matter, 2009, 5, 2686 RSC.
- S. Okumura, Y. Takeda and S. Minakata, Angew. Chem., 2012, 124, 7924 CrossRef.
- (a) L. Hu, X. Cao, L. Shi, F. Qi, Z. Guo, J. Lu and H. Gu, Org. Lett., 2011, 13, 5640 CrossRef CAS; (b) N. Sakai, K. Fujii, S. Nabeshima, R. Ikeda and T. Konakahara, Chem. Commun., 2010, 46, 3173 RSC.
- J. H. Boyer, in The Chemistry of the Nitro and Nitroso Groups, Part 1, ed. H. Feuer, Interscience, New York, 1969, pp. 278–283 Search PubMed.
- (a) H. A. Dabbagh, A. Teimouri and A. N. Chermahini, Dyes Pigm., 2007, 73, 239 CrossRef CAS; (b) M. Barbero, S. Cadamuro, S. Dughera and C. Giaveno, Eur. J. Org. Chem., 2006, 21, 4884 CrossRef; (c) K. Haghbeena and E. W. Tan, J. Org. Chem., 1998, 63, 4503 CrossRef.
- (a) Y. Zhu and Y. Shi, Org. Lett., 2013, 15, 1942 CrossRef CAS; (b) Z. Hu and F. M. Kerton, Org. Biomol. Chem., 2012, 10, 1618 RSC; (c) W. Lu and C. Xi, Tetrahedron Lett., 2008, 49, 4011 CrossRef CAS.
- B. Ortiz, P. Villanueva and F. Walls, J. Org. Chem., 1972, 37, 2748 CrossRef CAS.
- (a) H. Firouzabadi, D. Mohajer and M. Entezari-Moghadam, Bull. Chem. Soc. Jpn., 1988, 61, 2185 CrossRef CAS; (b) H. Huang, D. Sommerfeld, B. C. Dunn, C. R. Lloyd and E. M. Eyring, J. Chem. Soc., Dalton Trans., 2001, 8, 1301 RSC.
- S. Okumura, C. H. Lin, Y. Takeda and S. Minakata, J. Org. Chem., 2013, 78, 12090 CrossRef CAS.
- H. Firouzabadi and Z. Mostafavipoor, Bull. Chem. Soc. Jpn., 1983, 56, 914 CrossRef CAS.
- E. Baer and A. L. Tosoni, J. Am. Chem. Soc., 1956, 78, 2857 CrossRef CAS.
- (a) S. Farhadi, P. Zaringhadam and R. Z. Sahamieh, Acta Chim. Slov., 2007, 54, 647 CAS; (b) K. Orito, T. Hatakeyama, M. Takeo, S. Uchiito, M. Tokuda and H. Suginome, Tetrahedron, 1998, 54, 8403 CrossRef CAS.
- (a) G. Jurmann, O. Tsubrik, K. Tammeveski and U. Maeorg, J. Chem. Res., 2005, 2005, 661 CrossRef; (b) M. J. S. Dewar, J. Chem. Soc., 1946, 160, 777 RSC; (c) W. Wang, G. Tan, R. Feng, Y. Fang, C. Vhen, H. Ruan, Y. Zhao and X. Wang, Chem. Commun., 2020, 56, 3285 RSC.
- (a) H. Lv, R. D. Laishram, Y. Yang, J. Li, D. Xu, Y. Zhan, Y. Luo, Z. Su, S. More and B. Fan, Org. Biomol. Chem., 2020, 18, 3471 RSC; (b) G. Jo, M. H. Kim and J. Kim, J. Org. Chem., 2020, 7, 834 CAS; (c) Y. Xu, X. Shi and L. Wu, RSC Adv., 2019, 9, 24025 RSC.
- (a) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS; (b) T. A. Engesser, M. R. Lichtenthaler, M. Schleep and I. Krossing, Chem. Soc. Rev., 2016, 45, 789 RSC; (c) R. L. Melen, Science, 2019, 363, 479 CrossRef CAS; (d) J. R. Lawson and R. L. Melen, Inorg. Chem., 2017, 56, 8627 CrossRef CAS.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711 CAS.
- (a) T. Clark, M. Hennemann, J. S. Murray and P. Politzer, J. Mol. Model., 2007, 13, 291 CrossRef CAS; (b) P. Politzer, J. S. Murray and T. Clark, Phys. Chem. Chem. Phys., 2013, 15, 11178 RSC.
- (a) M. Colin and H. Gaultier de Claubry, Ann. Chim., 1814, 90, 87 Search PubMed; (b) M. Colin, Ann. Chim., 1814, 91, 252 Search PubMed.
- (a) D. S. Barak, S. U. Dighe, I. Avasthi and S. Batra, J. Org. Chem., 2018, 83, 3537 CrossRef CAS; (b) O. Prakash, H. K. Gujral, N. Rani and S. P. Syn, Com, 2000, 30, 417 CAS; (c) H. Liu, Y. Wei and C. Cai, New J. Chem., 2016, 40, 674 RSC.
- Recently, Fan et al. reported the use of TEMPO as an organocatalyst for the oxidative dehydrogenation of hydrazobenzenes to azobenzenes, see ref. 22a (vide supra). Notably, compared to this reported procedure our I2 catalyzed protocol has the advantages of using lower catalyst loadings and much shorter reaction times, in addition to not requiring heating.
- (a) S. A. Kozuch, Comput. Mol. Biosci., 2012, 2, 795 CrossRef CAS; (b) S. Kozuch and S. Shaik, Acc. Chem. Res., 2011, 44, 101 CrossRef CAS; (c) A. Uhe, S. Kozuch and S. Shaik, J. Comput. Chem., 2011, 32, 978 CrossRef CAS.
- (a) R. A. Sheldon, Chem. Commun., 2008, 3352 RSC; (b) R. A. Sheldon, Green Chem., 2007, 9, 1273 RSC; (c) For a recent comparison of ‘Green’ metrics see: D. J. C. Constable, A. D. Curzons and V. L. Cunningham, Green Chem., 2002, 4, 521 RSC; (d) For other ‘Green’ metrics see: T. Hudlicky, D. A. Frey, L. Koroniak, C. D. Claeboe and L. E. Brammer, Green Chem., 1999, 57 RSC; (e) B. M. Trost, Science, 1991, 254, 1471 CrossRef CAS; (f) A. D. Curzons, D. J. C. Constable, D. N. Mortimer and V. L. Cunningham, Green Chem., 2001, 3, 1 RSC.
- P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998, p. 30 Search PubMed.
- L. Wang, A. Ishida, Y. Hashidoka and M. Hashimoto, Angew. Chem., Int. Ed., 2017, 56, 870 CrossRef CAS.
- J. B. Zimmerman, P. T. Anastas, H. C. Erythropel and W. Leitner, Science, 2020, 367, 397 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00369k |
| This journal is © The Royal Society of Chemistry 2021 |