
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical probes targeting the kinase CK2: a journey outside the catalytic box
Jessica
Iegre†
 a,
Eleanor L.
Atkinson†
a,
Paul D.
Brear
b,
Bethany M.
Cooper
a,
Marko
Hyvönen
b and
David R.
Spring
*a
a,
Eleanor L.
Atkinson†
a,
Paul D.
Brear
b,
Bethany M.
Cooper
a,
Marko
Hyvönen
b and
David R.
Spring
*a
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: spring@ch.cam.ac.uk
bDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge, CB2 1GA, UK
First published on 29th March 2021
Abstract
CK2 is a protein kinase that plays important roles in many physio-pathological cellular processes. As such, the development of chemical probes for CK2 has received increasing attention in the past decade with more than 40 lead compounds developed. In this review, we aim to provide the reader with a comprehensive overview of the chemical probes acting outside the highly-conserved ATP-site developed to date. Such probes belong to different classes of molecules spanning from small molecules to peptides, act with a range of mechanisms of action and some of them present themselves as promising tools to investigate the biology of CK2 and therefore develop therapeutics for many disease areas including cancer and COVID-19.
 Jessica Iegre | Jessica Iegre was born in Italy and obtained her MSci in Medicinal Chemistry and Pharmaceutical Technology at the University of Pisa, Italy in 2013. The same year, she joined the AstraZeneca IMED Graduate programme in Göteborg, Sweden where she spent 2 years working across three different departments: medicinal chemistry, DMPK and computational chemistry. In 2015 she joined the Spring group at the University of Cambridge, and she obtained her PhD in chemical biology in 2019. Jessica is currently a Postdoctoral Research Associate in the group, and she is developing novel stapled peptides to inhibit medicinally relevant protein–protein interactions. |
 Eleanor L. Atkinson | Eleanor L. Atkinson received her MChem degree from the University of Oxford in 2018 after completing her Masters’ project under the supervision of Professor Angela Russell, investigating the metabolism of novel drug candidates. In 2018, she started her PhD studies at the University of Cambridge under the supervision of Professor David Spring, where her research focuses on the development of novel stapled peptides for the inhibition of protein–protein interactions. |
 Paul D. Brear | Paul Brear obtained his Mchem degree from the University of Durham in 2008 and obtained his PhD from the University of St Andrews in 2012 under the supervision of Prof. Nicholas Westwood and Dr Rupert J. Russell. He joined the group of Dr Marko Hyvönen at the University of Cambridge in 2012 and from 2018 is also the X-ray crystallography facility manager at the biochemistry department of the University of Cambridge. |
 Bethany M. Cooper | Bethany M. Cooper received her MChem degree from the University of Leeds in 2018, having completed her 3rd year of study at Lubrizol Ltd, Hazelwood. On return to the University of Leeds her final year was spent under the supervision of Professor Steve Marsden. In 2019, she started her PhD studies at the University of Cambridge under the supervision of Professor David Spring and industrial supervisor Dr Maria Ölwegård-Halvarsson, where her research has focused on peptide stapling methodologies for use within therapeutics. |
 Marko Hyvönen | Marko Hyvönen is a research group head at the University of Cambridge. He got his undergraduate degree from University of Helsinki and did his PhD work at EMBL in Heidelberg, Germany, graduating in 1997. He moved to Cambridge in 1998 as a postdoctoral fellow in the group of Tom Blundell. In 2001 he was awarded a BBSRC David Phillips Fellowship, appointed to a University Lectureship in 2008 and became a Reader in 2018. Dr Hyvönen and his group use structural biology techniques to study the structure and function of proteins and for structure-guided and fragment-based drug discovery. |
 David R.Spring | David Spring is currently Professor of Chemistry and Chemical Biology at the University of Cambridge within the Chemistry Department. He received his DPhil (1998) at Oxford University under Sir Jack Baldwin. He then worked as a Wellcome Trust Postdoctoral Fellow at Harvard University with Stuart Schreiber (1999–2001), after which he joined the faculty at the University of Cambridge. His research programme is focused on the use of chemistry to explore biology. |
Introduction
Protein kinases are considered the master regulators of the cell machinery. They exert their functions through the phosphorylation of other proteins with consequent activation or deactivation of crucially important cellular signaling pathways.1 The essential cellular processes that kinases regulate comprise cell growth, proliferation, differentiation, and death. Therefore, it is unsurprising that gene mutations that alter the function or expression levels of protein kinases lead to the insurgence of several diseases including neurological, immunological, hematological, endocrine, and skeletal disorders, as well as cancer.2 The implication of protein kinases in disease generation and progression has been widely recognized by the pharmaceutical community, and, as of today, there are 75 FDA approved drugs targeting this class of proteins.3Among the >500 protein kinases identified in the human proteome, there is CK2s (formerly referred to as Casein Kinase 2).4 CK2α and its very similar paralog CK2α′ are a serine/threonine kinases belonging to the CMCG family. Unlike other eukaryotic kinases, CK2α is constitutively active and as such, does not require upstream phosphorylation to exert its function.5 They form a tetrameric complex with a non-catalytic scaffolding subunit CK2β. CK2 phosphorylates more than 300 substrates and is thus one of the most promiscuous kinase we know.6 CK2 is found in both healthy and cancerous cells – whilst the protein levels in healthy cells increase during proliferation only, cancerous cells are characterized by overexpression of the different subunits which contributes to their rapid growth, proliferation, and their ability to escape apoptosis.7 Breast, lung, prostate, colorectal, renal cancer, leukemia, and glioblastoma are among the cancerous tissues featuring particularly high CK2 content, and hence, CK2 inhibitors could prove particularly useful as therapeutic agents for the treatment of these types of cancer.8 Indeed, an orally available CK2α inhibitor, named CX4945 (silmitasertib), is currently undergoing clinical studies for the treatment of cholangiocarcinoma paving the way for CK2 to be validated as an anticancer target.9
Although the role of CK2 in cancer progression is the most studied, it should be noted that the kinase is also involved in other pathological processes. For example, recently CK2 has been found to be interacting with the nucleocapsid of SARS-CoV-2, possibly contributing to COVID-19.10,11 Studies probing this relationship are in their infancy, however, it is possible that CK2 may represent a valid target for the treatment of coronaviruses in the future.
Many are the mechanisms by which CK2 can be inhibited, and therefore, there is a large number of inhibitors and chemical probes developed to date: from small molecules to peptide and peptidomimetics, to polyoxometalates.12 Similar to other kinases, the most studied mode of inhibition is ATP-competitive, where a ligand competes with ATP and hence prevents substrate phosphorylation. Though effective, one of the drawbacks of this strategy is that the ATP binding site is highly conserved across the kinome, leading to numerous challenges when developing a selective chemical probe for the target kinase.13,14 Most CK2α inhibitors serve as an example to this problem, including the clinical trial candidate CX4945.9 In addition, cellular concentration of ATP is high and therefore high-affinity ATP-competitive inhibitors are needed to effectively reduce the activity of the target. To overcome this issue, more selective strategies of CK2 inhibition have been explored including targeting of sites outside the ATP pocket, inhibition of the formation of the holoenzyme, and displacement of endogenous substrates.
In this review, we aim to provide a comprehensive overview of the molecular and structural biology of CK2, mechanisms of inhibition, and chemical probes for CK2 developed to date with emphasis on those acting outside the catalytic box (Fig. 1). In addition to describing their discovery and applications, this review provides a critical view of the challenges and opportunities of this promising class of chemical probes.
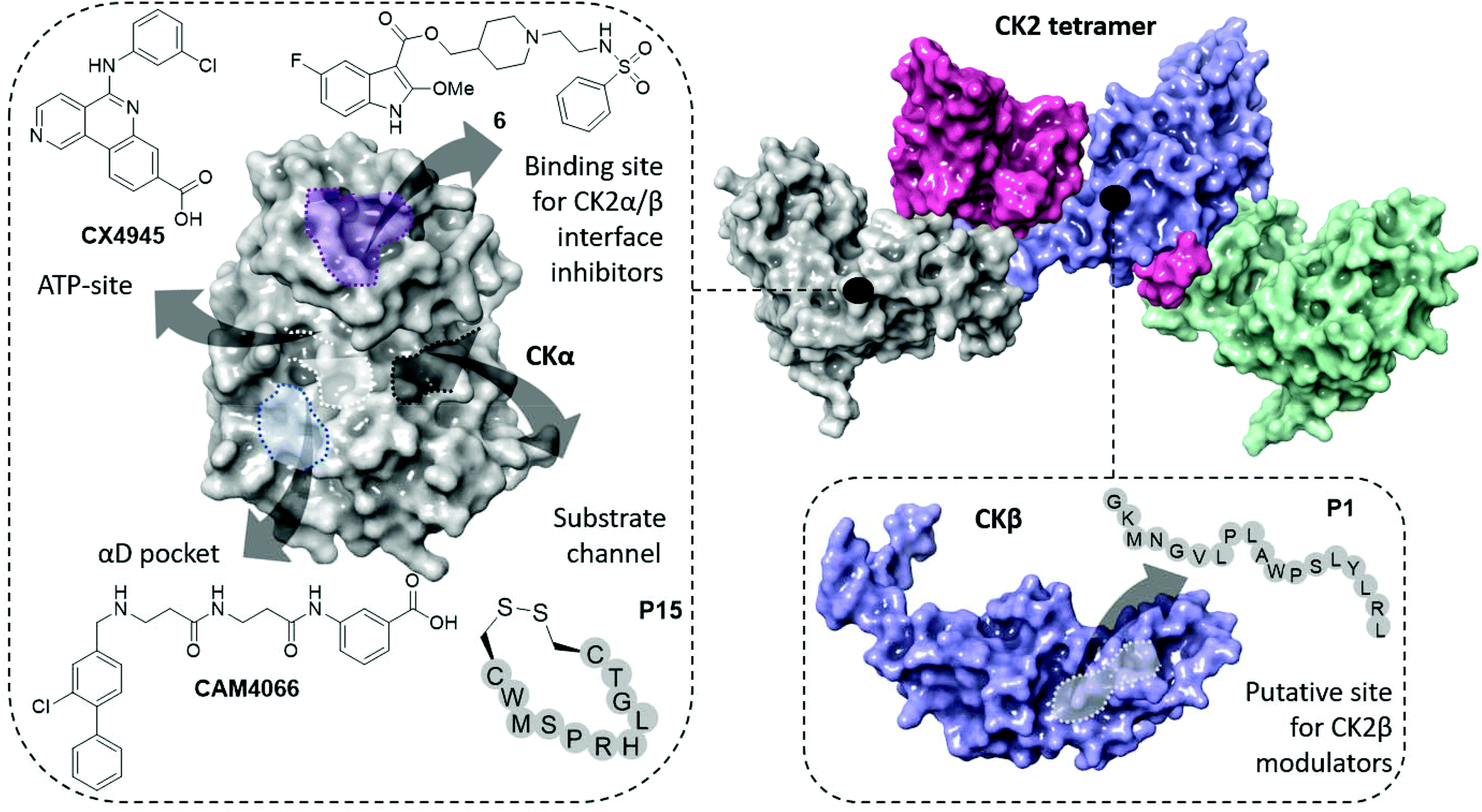 | ||
| Fig. 1 The structure of the CK2 holoenzyme is shown on the top right (PDB: 1JWH). The surfaces of two catalytic subunits are shown in grey and green; the surfaces of the two regulatory subunits are shown in pink and purple. Key binding sites and inhibitors of CK2α are shown on the left (ATP-site in white, α/β interface site in purple, substrate channel in black and αD pocket in light blue). Key binding site and inhibitor of CK2β is shown on the bottom right. | ||
CK2 kinase
Protein structure and characteristics
The CK2 holoenzyme is a tetramer constituted by two catalytic subunits, α and/or α′ connected by a dimer of the two regulatory subunits, β (Fig. 1). The α subunit presents 20 additional amino acids at the C-terminus that are absent in α′. The catalytic domain (∼48 kDa) is responsible for the phosphorylation of protein substrates and comprises the ATP binding site between the N- and C-terminal domains, linked via the so-called αD region. The N-terminal domain comprises of one α-helix, named αC and five β-sheets, β1–β5. The αC helix contacts the kinase substrates and, due to its lysine-rich content, imparts to the kinase a preference for acidic substrates. The C-terminal domain of CK2α comprises seven α-helices and two β-sheets that form the floor of the ATP binding site and the activation loop. The αD pocket, a pocket that has been recently discovered and that can be exploited to achieve selective CK2 inhibition, is located near the αD helix of the C-terminal domain.15,16 It is the conformation of the αC helix and the DWG motif in the activation segment that make the kinase constitutively active. In particular, the protein's N-terminus makes extensive contact with the αC helix which remains locked in an open conformation and allows for substrate binding. Unlike other closely related kinases, CK2 features a DWG motif (Asp175–Trp176–Gly177) in the activation loop rather than a DFG motif. The Phe to Trp mutation allows the creation of an additional H-bond between the indole of Trp176 and the Leu173 which maintains the kinase in an active state. This constitutive activity means that, notably, the majority of CK2 substrates do not require the presence of CK2β to be phosphorylated.16,17Although not required for CK2α activation, the regulatory β subunit (∼28 kDa) enhances the catalytic activity of CK2α by making the kinase more thermostable.12 Moreover, CK2β regulates the activity of the kinase by allowing the holoenzyme to shuttle between intracellular compartments, to dock to and penetrate the nucleus where the majority of the substrates are located.18–20 Furthermore, CK2β acts as a docking station for substrates including p53, eIF2β, Nopp140, FGF-II amongst others.19 CK2β forms an obligate dimer, each protomer of which comprises of a Zn2+ coordinating globular domain with the elongated C-terminus wrapping around the opposite protomer's globular part. A short, linear epitope in the C-terminus forms a β-turn and is essential for the formation of the holoenzyme assembly, with residues Arg186, Tyr188, Phe190, and His193 being crucial for this interaction.15,19,21
In cells, the formation of the holoenzyme involves the assembly of the two regulatory subunits into a dimer, followed by their interaction with a surface of approximately ∼830 Å2 in the N-lobe of the catalytic subunits.22 Consequent to the formation of the protein–protein interaction (PPI) between the two subunits, conformational changes occur in CK2α. Specifically, the β4–β5 loop is found to be in a closed form in isolated CK2α whereas it switches to an open form upon complexation with CK2β. The latter, on the other hand, does not undergo drastic conformational changes upon holoenzyme formation.23
Physiopathology of CK2
CK2 is involved in a multitude of cellular functions, the most important of which are cell survival, apoptosis, and cell cycle regulation. This can be easily understood if we consider the wide-spread localisation of CK2 within the cell compartment including in the nucleus, cytoplasm, Golgi, ER, ribosomes, and plasma membrane.19 The mechanisms by which CK2 regulates the cell decisions of life and death are extremely complex and, in part, not fully understood.Briefly, CK2 is thought to regulate various stages of the cell cycle by phosphorylating cell cycle regulators such as cdc34, topoisomerase II and p34.19 In addition, CK2 is believed to interact with multiple signalling pathways within the apoptotic cascade including the cysteine-rich glycoproteins (Wnts), inhibitor of apoptosis proteins (IAPs), production of reactive oxygen species (ROS), activation or regulations of caspases (through interactions with ARC, Bcl-2 protein and Max), TNF pathway and PI3K/AKT.24 An exemplified overview of CK2 involvement in cell signalling controlling survival and apoptosis can be found in Fig. 2.
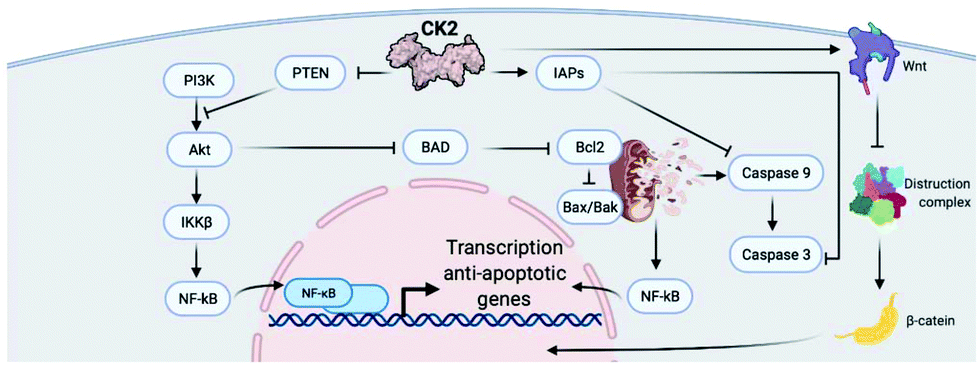 | ||
| Fig. 2 Simplified schematic of the involvement of CK2 in intracellular signalling pathways that regulate cell survival and apoptosis. CK2 inhibits PTEN activity, promoting the PI3K-Akt pathway of which PTEN is a negative repressor, and phosphorylates AKT, preventing BAD-induced cell death. CK2 also promotes the NFκB pathway by promoting IκB degradation. This facilitates the transcription of anti-apoptotic genes, preventing cell death. Additionally, CK2 promotes IAPs, preventing caspase 9 activation and subsequent cell death. CK2 is a positive regulator of the Wnt signalling pathway, preventing the formation of the destruction complex and thus leading to increased levels of β-catenin within the cell, resulting in cell proliferation, survival and migration. Created with BioRender.com. | ||
Expression levels and activity of CK2 are highly regulated in healthy cells, keeping cell survival and apoptosis in balance. Indeed, increased levels of the protein are found during cell proliferation only. It is not surprising that dysregulation of the expression levels of such a crucial protein leads to the generation and progression of several diseases. The role of CK2 in cancer is undoubtedly the most studied; however, an increasing number of studies suggest that CK2 plays a part in other serious diseases such as cystic fibrosis, multiple sclerosis, chronic intestinal inflammation, neurodegenerative diseases such as Alzheimer's and Parkinson's, atherosclerosis, thrombosis, and diabetes mellitus.25–29 Furthermore, CK2 has been reported to be important in the regulation of viral RNA binding activity, transcription, replication, and virus assembly of certain RNA viruses.30 Recently, CK2 inhibitors have been found to show anti-SARS-Cov2 activity, making CK2 a potential target in the fight against COVID-19.30,31
The next two sections will focus on the role of CK2 in cancer and COVID-19.
Importance of CK2 in cancer
Cancer is one of the main causes of mortality worldwide, and it is defined as the uncontrolled proliferation of cells. Hanahan and Weinberg outlined six hallmarks in cancer: proliferative signalling, resisting cell death, evading growth suppressors, inducing angiogenesis, enabling replicative immortality and activating invasion and mortality (Fig. 3).32,33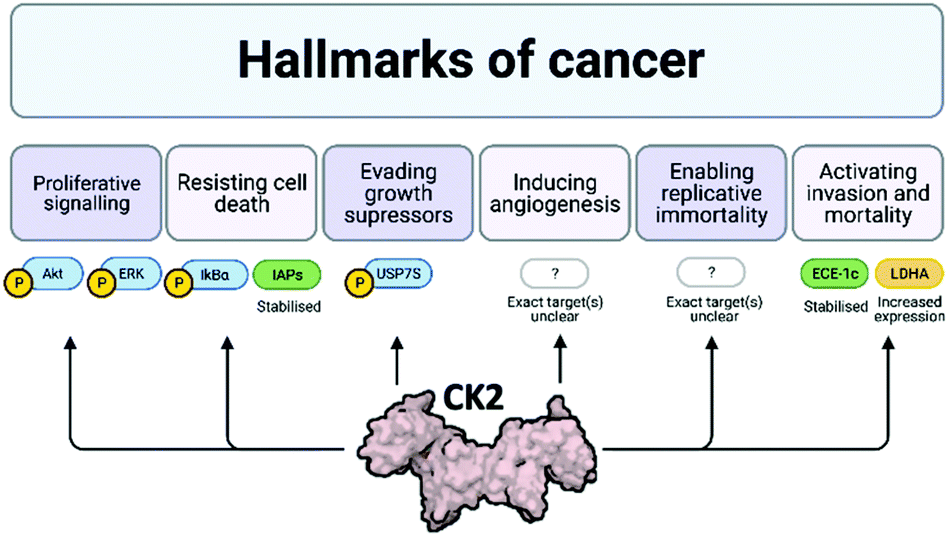 | ||
| Fig. 3 Schematic of the six hallmarks in cancer and the involvement of CK2 in each of them. Where specific proteins are known to be affected by CK2 with respect to each of these hallmarks, examples are given: proteins attached to a P are phosphorylated by CK2, proteins in green are stabilised by CK2 and proteins in yellow have increased expression due to CK2. Despite evidence that CK2 induces angiogenesis and enables replicative immortality, the specific pathways affected by CK2 are not currently known.24,32–39 Created with BioRender.com. | ||
The discovery of CK2's oncogenic potential dates back to the 1980s, evidenced by reports indicating that, within tumours, the level of CK2 expression was significantly greater than that found in normal cells.7 For many cancers, CK2 expression levels are sustained at a newer higher level, regardless of the stage of the cell cycle and such a change in basal level occurs when the cells morph into cancer cells.33 Furthermore, it has been reported that the increased levels of CK2 found within the nucleus of cancer cells including head and neck carcinoma, prostate and colon cancers, are an important indication of the complexity of the dysplasia and can be used as a prognostic marker.34–39 Further evidence of the involvement of CK2 in cancer generation and progression is summarised by Trembley et al.24 In particular, it was found that CK2 is responsive to modulations of mitogenic signals; downregulation of CK2 positively impacts inflammation, angiogenesis and drug efflux; downregulation of CK2 results in inhibition of cell growth, proliferation and an increase in apoptotic activity with no redundant pathways able to compensate for such downregulation.
Here, we summarise key findings that support the relevance of CK2 in different types of cancer. The reader is directed to several reviews for more information on the importance of CK2 in other cancer types.7,40
Importance of CK2 in COVID-19
CK2 is emerging within the infectious disease field as an attractive target. There are validated studies regarding CK2 phosphorylation and modulation of viral proteins’ function in human immunodeficiency virus (HIV), vesicular stomatitis virus (VCV), hepatitis C virus (HCV), herpes simplex-1 (HSV-1) and human papilloma virus (HPV).30 CK2 also plays a key role in a number of signalling cascades that viruses hijack including JAK/STAT, PTEN/PI3K/Akt-PKB and NF-κB.30 It is thought that if CK2 was inhibited, viral replication would be impacted. A recent study by Bouhaddou et al. used a combination of 87 kinase inhibitors to identify those with antiviral efficacy. Kinases p38, CK2, CDK, PIKFYVE and AXL were identified as having antiviral potential. The conducted experiments demonstrated the instrumental role of CK2 inhibition for the SARS-CoV2 infection in vitro.30,48 SARS-CoV2 nucleocapsid protein has been shown to directly interact with CK2. By enabling co-localization along the filopodia protrusions, this interaction promotes several functions including virus egress and cell-to-cell spread through infected cells’ epithelial monolayers.30,48 Particularly for CK2, the virus-host PPI was associated with increased activity of CK2 as documented by the enhanced phosphorylation of CK2 substrates such as STAT1, HMGN 1, HMGA 1, HDAC 2 and CTNNA 1.48The potential of CK2 as an anti-COVID-19 target is documented by two small clinical trials using previously developed CK2 inhibitors: CX4945 and CIGB-325. In particular, it was found that CX4945 was able to improve the oxygen level of patients with COVID-19-induced pneumonia within 24 hours of starting treatment, and a further five days later the first patient was discharged.31 Similarly, a small study was conducted on twenty SARS-CoV-2 positive patients using the anti-CK2 peptide CIGB-325 (formerly CIGB-325). It was found that 50% of COVID-19 positive patients that were treated with CIBG-325 had a reduction in the number of pulmonary lesions by day 7 in comparison to only 20% in the standard-of-care only treatment.30
Inhibition mechanism and CK2 inhibitors
Approaches to the discovery of CK2 inhibitors used so far
The first CK2 inhibitors were identified by the investigation into biologically active compounds and natural products, such as heparin and emodin.49,50 However, with the rapid growth in the field of computational chemistry and structural biology, more recently inhibitors have been designed specifically for CK2 using structural-based approaches such as fragment-based drug discovery and phage display. The rapid growth in technologies facilitating the guided and rational design of protein modulators has had a drastic impact upon the development of CK2 inhibitors, as is self-evident from the rapid growth in the field over the past 20 years.From the many inhibitor development studies targeting CK2, multiple different binding sites have been identified on CK2α. These sites all have different advantages and disadvantages which have led to them being utilised with varying success for the inhibition of CK2. These sites can be initially grouped into four broad categories: the ATP site, CK2β protein–protein interaction site, the substrate binding channel and potential allosteric sites outside of the classic ATP site (Fig. 4a).
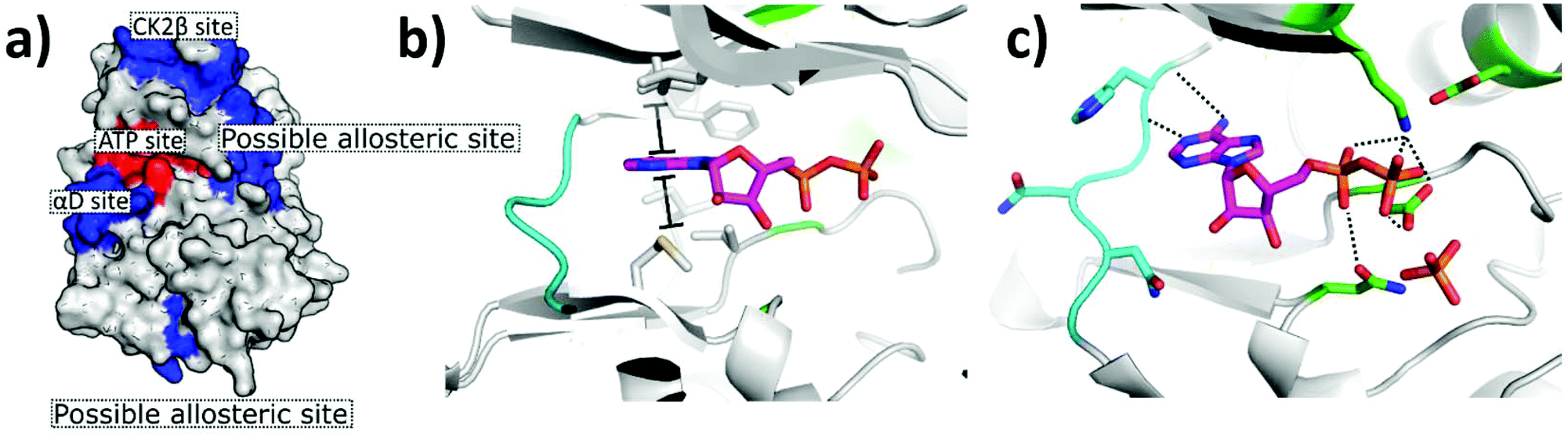 | ||
| Fig. 4 Crystal structure of CK2α in complex with ADP (PDB: 6YPN). (a) The small-molecule binding sites on CK2α. The ATP site is shown in red and the sites outside the ATP pocket are shown in blue. (b) and (c) The ATP site of CK2α. The hinge residues are shown in cyan, the polar phosphate binding residues are shown in green and the hydrophobic sandwiching residues are shown in grey. | ||
In the next sections, we will focus on the approaches used to discover CK2 inhibitors targeting sites outside the catalytic box and we aim to provide a comprehensive overview of the molecules reported to date. A brief overview of the ATP competitive inhibitors is also provided.
Overview of ATP competitive inhibitors
The site where the most development has happened is the ATP site. This site is the where the protein naturally binds the small molecule cofactor, and as such, is seen as the easiest site to target due to its characteristics. Firstly, the site is pre-formed in the apo structure meaning that there is no barrier to ligand binding. Secondly, the site is composed of a deep hydrophobic cleft sandwiched by Lys68 and the hinge region at opposite ends. This layout is ideally set up for sandwiching an aromatic ring system with hydrogen bonding substituents protruding off either side (Fig. 4b).In a recent crystallographic fragment screen conducted by Brear et al., the ATP site was the only site identified as binding fragments.51 This is significant as fragment screens are often used to probe the surface of proteins for small-molecule binding sites. The ATP site has been used with great success in identifying ligands that bind with high affinity and displace the endogenous ATP hence preventing the phosphorylation of the substrates. Indeed, a number of small molecules with picomolar affinities have been identified for this site.52
CK2 ATP-competitive inhibitors can broadly be divided into four main classes: halogenated compounds such as DRB and its derivatives,53,54 condensed polyphenolic compounds such as emodin and its derivatives,50,55 pyrazolo-triazines and pyrazolo-pyrimidines56,57 and indoloquinazolines such as CX4945.9 The chronology of the most influential inhibitors developed thus far is detailed in Fig. 5.
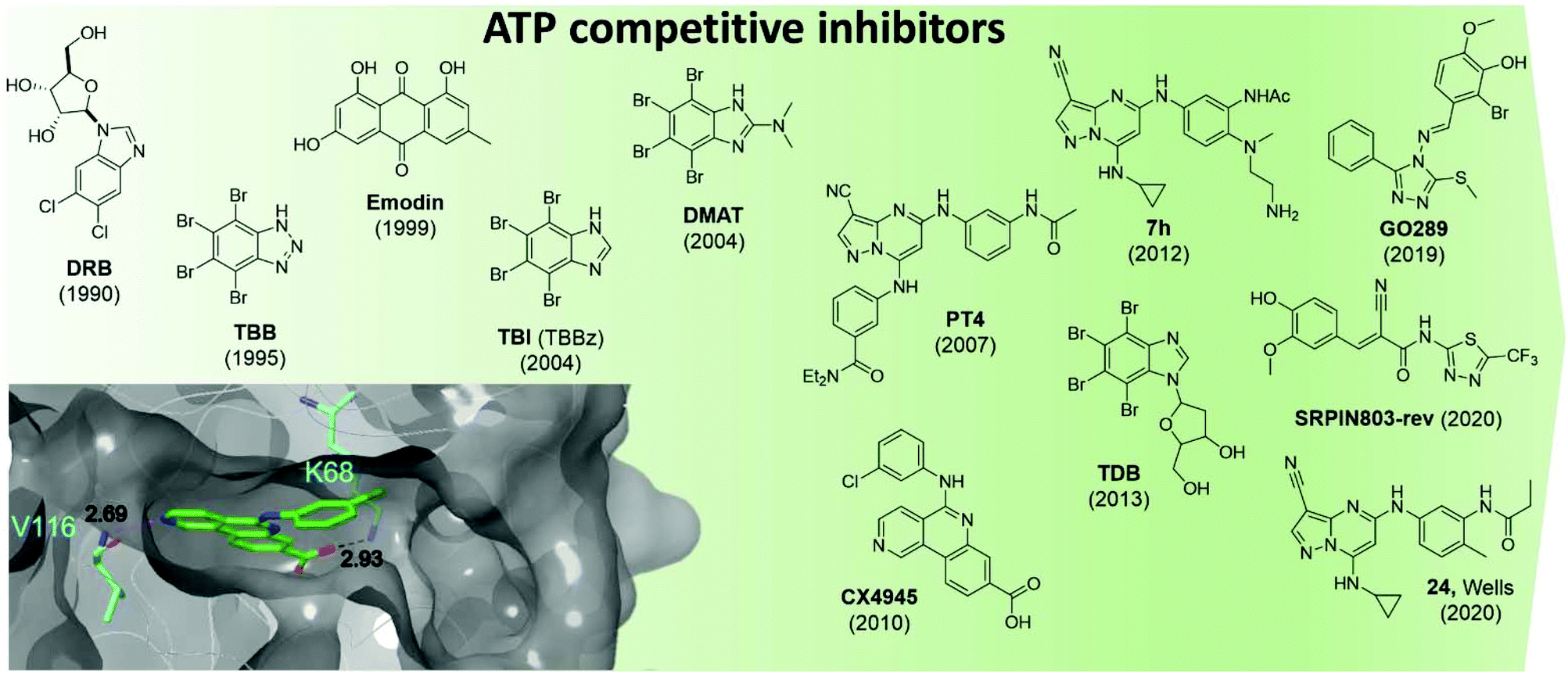 | ||
| Fig. 5 Structures of the most influential ATP-competitive inhibitors of CK2 reported in chronological order. The X-ray co-crystal structure of CX4945 and CK2 is shown on the bottom left (PDB: 3NGA). Residues which hydrogen bond with CX4945 are shown in cyan alongside their hydrogen bond lengths. | ||
The reader is redirected to a recent review for an up to date comprehensive review on ATP-competitive inhibitors for CK2.58
Unfortunately, there are a number of drawbacks associated with the use of this site, with the primary issue being selectivity. The ATP site has evolved to bind ATP which is a very common cofactor amongst the genome. Indeed, Uniprot annotates 1391 human genes as binding ATP. Therefore, any small molecule that targets just the ATP site is likely to bind and inhibit any number of these other ATP-binding proteins. An example highlighting the selectivity issue of this class of compounds is given by the small molecule clinical candidate CX4945 which is frequently described in the literature as highly selective for CK2 with an IC50 of 1 nM. However, when screened in a selectivity panel of 238 kinases, CX4945 also inhibits another seven kinases more than 90% (tested at 500 nM). The most significant is the effect seen with Dyrk1A and 1B where CX4945 showed IC50 values of 6.8 and 6.4 nM, respectively.9 A number of efforts are currently ongoing to identify new selective series of CK2 ATP-competitive inhibitors.
In 2016, Dowling et al. identified compound 7h as the most promising CK2α inhibitors out of a pyrazolepyrimidine series. The compound showed picomoloar activity in an SPR assay and was found to inhibit the Wnt pathways with an IC50 of 50 nM. Despite its limited oral bioavailability, 7h presented promising pharmacokinetic properties after IV administration, and it was effective as monotherapy in both HCT116 cells and SW-620 xenografts. The compound also showed limited off-target activity when screened in a panel of 402 kinases with 12 members of the CMGC family being inhibited more than 50%.52
Oshima et al. identified GO289 in 2019 as having excellent activity in vitro (IC50 = 7 nM) as well as high selectivity for CK2; the second most inhibited protein, PIM2, is much more weakly inhibited by GO289 than CK2 (PIM2 IC50 = 13 μM).59 In 2020, SRPIN803-rev-derivatives were found to have moderate inhibitory activity against CK2 (IC50 = 0.28 μM for the lead compound) as well as good selectivity (only CK2α inhibited over 50% at 1 μM in a 320 kinase panel) and cellular activity (DC50 = 12.8 μM, Jurkat cells).60
Following on from Dowling's work on pyrazolopyrimidines, in 2020 Wells et al. developed compound 24.61 The lead compound proved to be selective against a panel of 402 kinases and showed an IC50 of 36 nM and 16 nM against CK2α and CK2α′ respectively in a nanoBRET assay carried out in Hek-293 cells. While compound 24 showed target-specific inhibition of Akt Ser129 phosphorylation, it did not activate Caspase 3/7 as been seen with other CK2α inhibitors and had variable effects on proliferation in a panel of cancer cell lines.61
Most of the newly identified series of inhibitors require further optimisation, characterisation, and evaluation before highly potent, selective and fully understood cell-permeable CK2 inhibitors are obtained, but all classes represent promising starting points for this to take place. Importantly, whilst minor off target effects could be beneficial in cancer treatment, they are not desirable while investigating signalling pathways related to a targeted protein.
In order to overcome selectivity issues associated with the use of ATP competitive inhibitors, developers of kinase inhibitors are looking for alternative mechanisms of inhibition. This is also true for CK2 inhibition. Within CK2α alternative binding sites can be split into four categories: the substrate-binding site, the CK2β interface site, the αD site, and possible allosteric sites. As of yet, only the αD site, the CK2β interface site and the substrate-binding channel have been successfully utilised and validated for the development of inhibitors (Fig. 4a). As of today, no allosteric, non-competitive inhibitors are known for CK2.
Substrate channel inhibitors
An alternative strategy for CK2 inhibition is through blocking of the substrate-binding site only. One of CK2's distinctive features is its substrate preferences – it phosphorylates only acidophilic substrates.62 Therefore, greater selectivity of CK2 modulation may be achieved through the development of substrate-competitive inhibitors.Polyglutamyl peptides were identified as CK2 substrate-competitive inhibitors in 1983 by Meggio et al. It was found that (Glu)70 was able to inhibit CK2 in vitro (Ki = 0.11 μM) but was inactive towards the closely related CK1 and cAMP-dependent protein kinase.63 They subsequently identified the hexapeptide RSEEEVE as a very weak substrate-competitive inhibitor of CK2 (Ki = 3.98 M)64 as well as the random heteropolymers Glu/Tyr (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) and Glu/Tyr (1:1) which had good activities against CK2 (Ki = 25 nM, 50 nM respectively).65 Successive work by Tellez et al. in the early 1990s further reinforced the notion that polymers incorporating Glu and Tyr were able to inhibit CK2.66,67
:1) and Glu/Tyr (1:1) which had good activities against CK2 (Ki = 25 nM, 50 nM respectively).65 Successive work by Tellez et al. in the early 1990s further reinforced the notion that polymers incorporating Glu and Tyr were able to inhibit CK2.66,67
Through screening of a random cyclic peptide phage display library, Perea et al. identified peptide P15 (WMSPRHLGT) which prevented CK2 phosphorylation in vitro in a substrate-competitive manner.68 Addition of two cysteine residues to either end of the peptide, as well as attachment of the known cell-penetrating peptide TAT to the N-terminus and cyclisation with a disulfide bridge afforded the cell-permeable CK2 substrate-competitive inhibitor P15-TAT (subsequently renamed and hereafter referred to as CIGB-325). CIGB-325 showed moderate inhibition of cell viability against a range of tumour cell lines with an EC50 of between 20 and 136 μM depending on the cell line.69 Furthermore, it was found that daily intra-tumoural administration of CIGB-325 into TC-1 lung epithelial tumours (C57BL6 mice) led to a significant reduction in tumour growth.69
Proteomics studies also revealed the up- and down-regulation of proteins involved in the apoptotic intrinsic pathways in the presence of this peptide. In 2008, the first-in-man clinical trial with CIGB-325 was conducted: 75% of the patients (31 women with cervical cancer) had significant lesion reduction and, most promisingly, 19% of the patients exhibited full histological regression. Additionally, the effect of CIGB-325 on spontaneous lung metastasis was investigated; systemic treatment with CIGB-325 prevented breast cancer colonization in the lung.70 In 2019, Winiewska-Szajewska et al. identified a 6-mer peptide (KESEEE-NH2) which weakly binds to the CK2α substrate binding-site (Kd = 0.39 mM).71 They showed that both the peptide and the ATP-competitive inhibitor TBI could bind simultaneously to CK2 without a drastic loss in binding affinity (Kd = 0.45 mM for KESEEE-NH2 with TBI present).71 Therefore, the novel peptide KESEEE-NH2 represents a potential substrate-competitive inhibitor.
CIGB-325 currently represents the only highly potent substrate-competitive inhibitor of CK2 that is effective in vivo on multiple cancer cell lines. In particular, the promising results shown against cancer cell metastasis make CIGB-325 a promising candidate for limiting metastatic cancer spread. It should be noted that the lack of structural data on the substrate-binding makes the development of inhibitors at this site harder to achieve.
Bi-specific ATP/substrate competitive inhibitors
The substrate-binding channel of CK2α has been defined in various publications as the groove highlighted in Fig. 6a based on other kinases structures.72 To date, no publications have shown any substrates of CK2α binding in this groove. An alternative strategy sees linking of inhibitors bound in the ATP site with substrate peptide mimics that bind in the substrate-binding channel (bi-specific inhibitors). This technique has generated selective inhibitors as described below. However, the structural data presented for these compounds has shown good density only for the ATP site ligand, but no significant density for the substrate-binding site portion of the ligand (despite the fact that the peptide portion has been modelled to the structure). It is therefore not clear how much the binding in the substrate channel contributes to the binding of the inhibitor.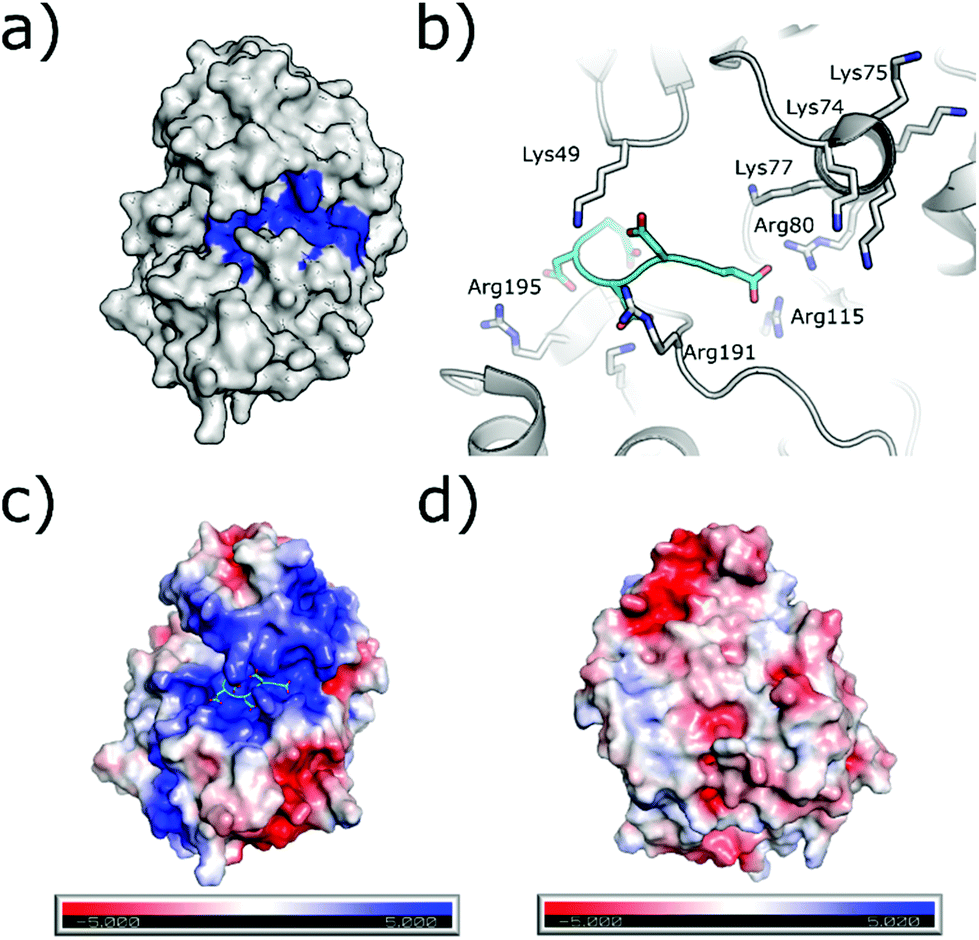 | ||
| Fig. 6 (a) Surface of CK2α with substrate binding channel highlighted in blue (PDB: 5CU6). (b) Modelled peptide mimic (cyan) bound in the substrate-binding channel (PDB: 6SPW). The basic substrate binding residues are shown. (c) The surface electrostatics (generated by APBS Electrostatics) of CK2α are shown. The basic substrate-binding grove can be clearly seen in blue. (d) The electrostatics of the surface rotated by 180°. | ||
The first bi-substrate inhibitor developed was ARC-1502.73 It consisted of the ATP-competitive inhibitor TBI conjugated to an acidophilic peptide (Fig. 7). The most potent inhibitor in the series, ARC-1502, had a strong binding affinity for CK2α (Ki = 0.5 nM) alongside good selectivity; when tested at 1 μM against a panel of 140 kinases, 9 other enzymes were inhibited over 50%.73
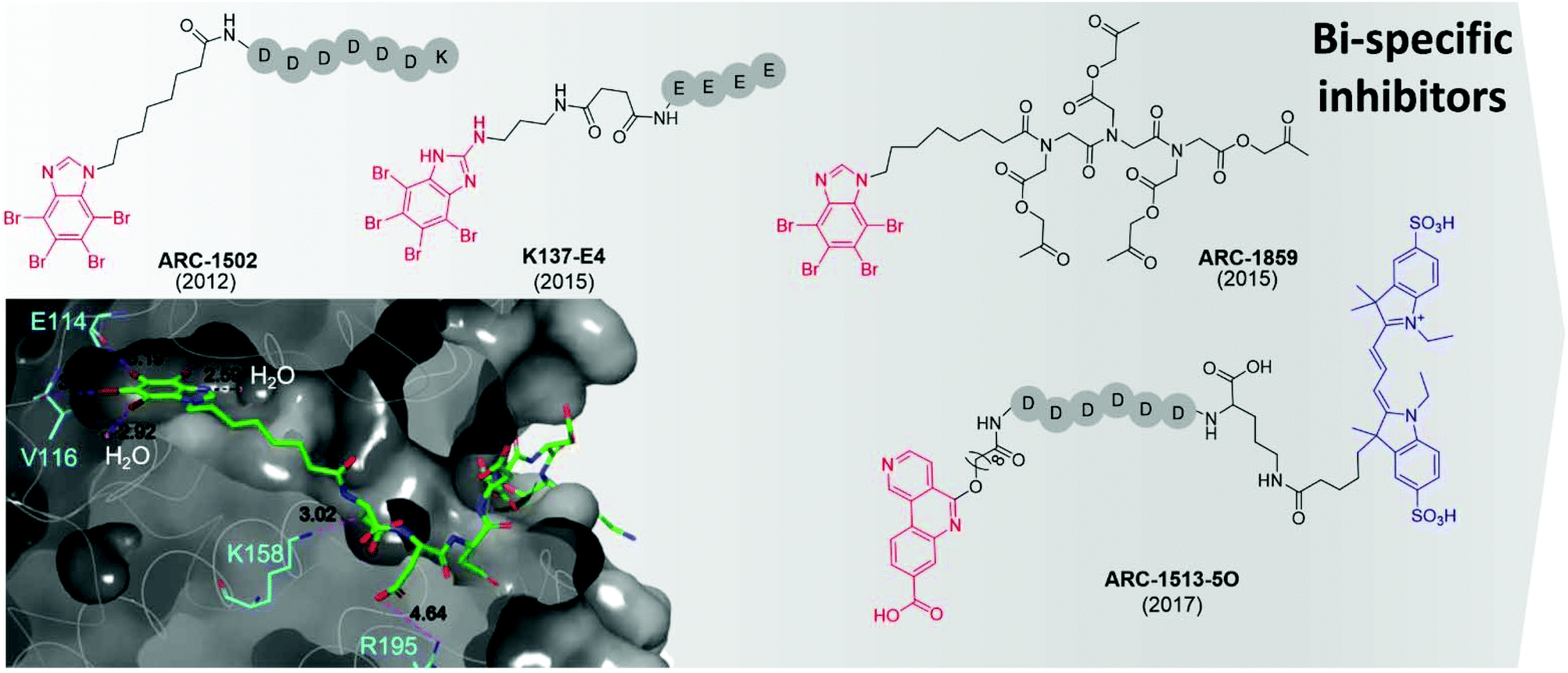 | ||
| Fig. 7 Bi-specific ATP/substrate competitive inhibitors discovered to date. The co-crystal structure of ARC1502 and CK2α is shown in the bottom left (PDB: 6SPX). Residues which hydrogen bond to the ligand are shown in cyan alongside the hydrogen bond lengths. Portion of the molecule binding in the ATP site is shown in red, the motif binding to the substrate channel is shown in black. For ARC-1513-5O the fluorophore is shown in blue. | ||
However, ARC-1052 is not proteolytically stable or cell-permeable, limiting its utility as chemical probes. In 2015, Viht et al. improved upon ARC-1502 by creating a cell-permeable and stable bi-substrate inhibiting “pro-drug” named ARC-1859 (Fig. 7).74 The stability of the peptide portion of the inhibitor was improved by the replacement of amino acids with a peptoid chain (poly-N-substituted glycines) which is much less susceptible to proteolytic degradation. Additionally, the inhibitor was rendered cell-permeable by the masking of the aspartic acid side chains as acetoxymethyl esters.74 Once inside the cells, the acetoxymethyl esters are cleaved to reveal the free carboxylic acids and the active compound is trapped in the cell and able to reduce the phosphorylation of two CK2 substrates (Cdc37 S13 and NFκB S529.63) in a concentration-dependent manner.74,75
At the same time as the ARC-series of compounds were being developed, Cozza et al. identified the bi-substrate inhibitor K137-E4 (Fig. 7).72 The ATP-competitive inhibitor N1-(4,5,6,7-tetrabromo-1H-benzimidazole-2-yl)-propane-1,3-diamine (K137) was functionalised with a chain of four glutamic acid residues, attached to the primary amine of the ATP site inhibitor. K137-E4 shows a significantly improved activity compared to the parent inhibitor K137 (IC50 = 25 nM and 130 nM respectively) as well as exhibiting greater selectivity for CK2; K137 inhibits 35 kinases more potently than CK2 whereas K137-E4 inhibited CK2 the most strongly when tested against 140 other enzymes. However, K137-E4 is not cell permeable and therefore exhibits negligible biological effects in cellular assays.73
In 2017, Vahter et al. developed a series of bi-substrate inhibitors using a fragment derived from CX4945 as the ATP-competitive inhibiting portion of the compound.76ARC-1513-5O showed exceptional binding ability to CK2 (Kd = 16–37 pM, Fig. 7). Similarly to CX4945, ARC-1424-5O showed moderate selectivity when tested in a panel of 140 protein kinases; when tested at 1 μM, 23 kinases were inhibited more than 50%, including 6 which were strongly inhibited (>90%).76 Despite its promiscuity, ARC-1424-5O remains one of the best candidates for the measurement of binding affinities of other CK2 inhibitors in competition experiments due to its picomolar activity.
The field of bi-substrate inhibitors of CK2 is relatively new compared to ATP-competitive inhibitors. However, the compounds identified so far highlight that it is a promising strategy of inhibition for the development of both potent and selective inhibitors of CK2, but pharmacological properties need to be improved before these can be considered as viable developmental candidates.
Inhibitors of the holoenzyme assembly
An alternative method to modulate the activity of CK2 is via disruption of the PPI at the CK2α/β interface. CK2α is rare for a kinase in that it is constitutively active.6 However, it forms a hetero-tetrameric complex with CK2β which is believed to regulate the activity and substrate selectivity of CK2α. CK2 substrates can be divided into three classes: Class I, II and III.77 Class I can be phosphorylated by the CK2α/CK2β complex or by CK2α alone; Class II can be phosphorylated by CK2α alone; Class III can be phosphorylated only by the CK2α/CK2β complex. It has been shown that preventing the formation of the complex prevents the phosphorylation of the Class III substrates, affects the localisation of the complex and destabilises CK2α.77 There are some reports of weak inhibition of the phosphorylation of Class I and II substrates by ligands binding at the CK2β interface site.78 However, higher affinity compounds that bind more selectively in the interface site and have been more reliably validated do not show this inhibition.79 Therefore, it seems unlikely that this site can be used for the direct inhibition of all CK2 substrates. The dominant feature of the interface site is a pocket in which Phe190 binds, a shallow grove in which Tyr125 binds and a flexible loop (Val101-Pro109 loop) that opens and closes the interface site (Fig. 8a).80 The size of this site varies depending on which ligands are bound in the site. For example, the Val101-Pro109 loop can be seen in a closed conformation in the structure (Fig. 8b grey cartoon, c grey surface). Whereas, in the CK2β-bound structure the loop folds back to give a significantly more open pocket (Fig. 8b cyan loop, d grey surface). This range of movement allows the pocket to accommodate a large range of ligands and provides more potential for optimisation. The main advantage of the interface site over the ATP site is that this site is less conserved amongst kinases. This means there is a greater possibility of generating selective inhibitors using this site. However, there are also a number of drawbacks to this site. Firstly, generating high-affinity small molecule inhibitors using this site is less facile than the ATP site for which it is possible to access small molecules with a pico-molar affinity.52,81 This has a lot to do with the depth of the ATP site where inhibitors can make extensive contacts with the target, shielded from the solvent. In contrast, the interface site is a shallow pocket, or rather a dent, on the surface of the protein, which offers little three-dimensionality and contact. The highest affinity reported so far for a small molecule binding at the interface site is 30 μM (ref. 82) and higher affinity inhibitors for this site are peptides or stapled peptides. This is not surprising if we consider that bigger molecules like peptides better interact with the big and shallow pocket found at the interface.83 The indirect nature of the inhibition from this pocket may be an advantage or a disadvantage to this site; however, more advanced chemical tools and testing are required to confirm this.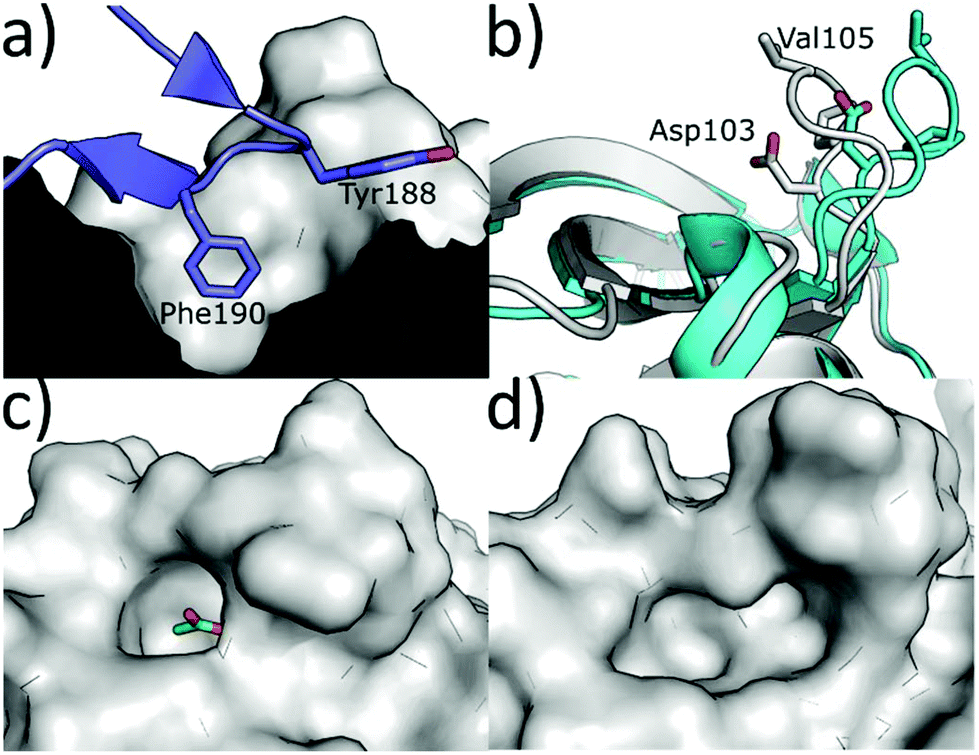 | ||
| Fig. 8 (a) CK2β interface loop (purple) binding in the CK2α interface site (grey) (PDB: 4MD7). (b) Flexibility of the interface site. Open/peptide bound structure in cyan (PDB: 6Q4Q), closed/small molecule bound structure in grey (PDB: 5CU6). (c) Surface of the closed/small molecule bound structure (PDB: 5CU6) and (d) surface of the open/peptide bound structure (PDB: 6Q4Q). Ligand is shown in green and the surface in grey. | ||
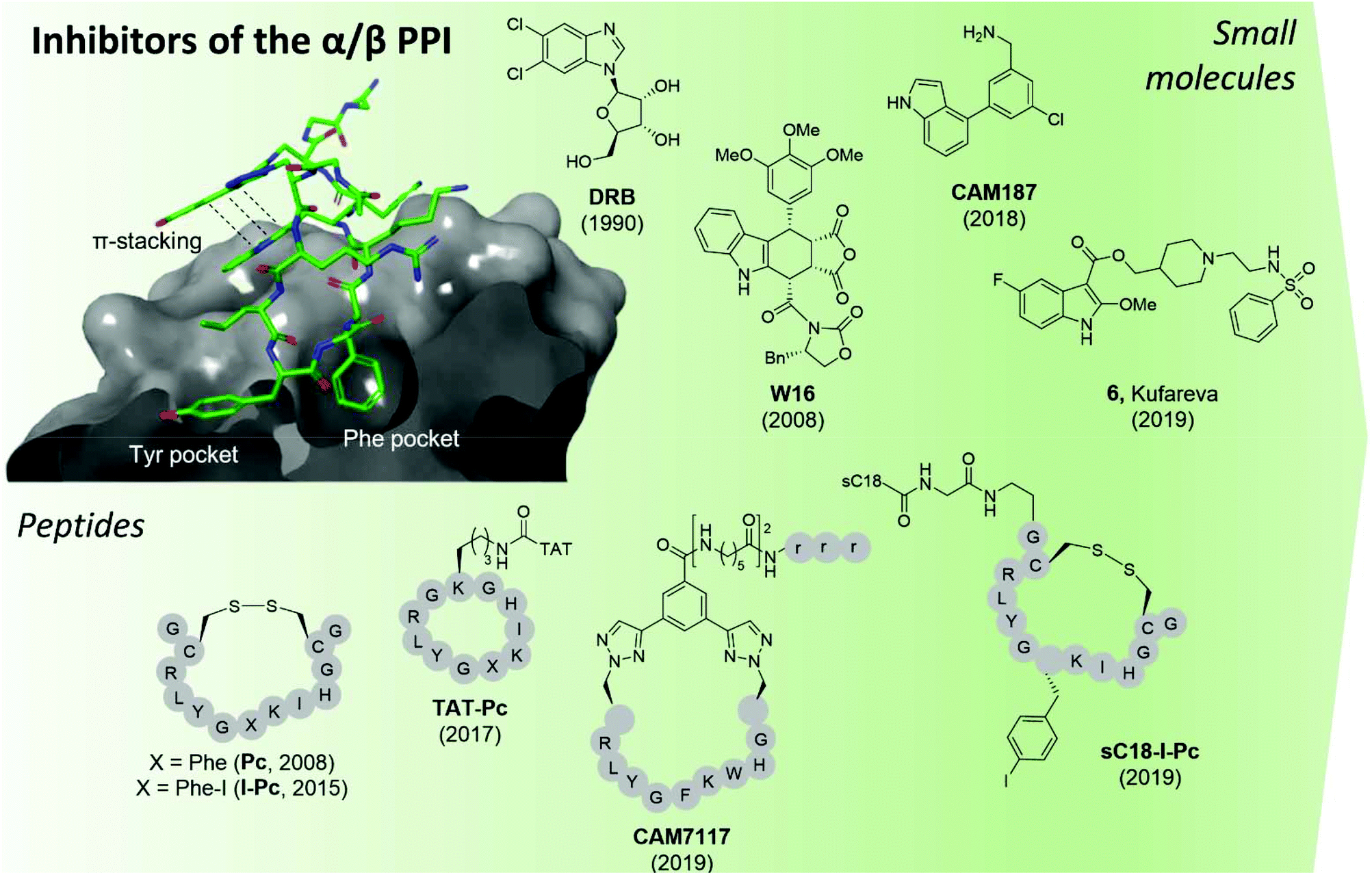 | ||
| Fig. 9 CK2 inhibitors reported in chronological order and acting at the protein interface between the catalytic and regulatory subunit with the co-crystal structure of CK2α and CAM7117 shown on the left (PDB: 6Q4Q). The hydrophobic Tyr and Phe pockets key to binding are highlighted, alongside the pi–pi stacking interactions of the ligand. sC18 = H2N-GLRKRLRKFRNKIKEK-NH2;93 TAT = GRKKRRQRRRPPQ.96 Compounds have been labelled as in the related papers. r = D-Arg. | ||
The second small molecule modulator of the PPI was the podophyllotoxin indole-analogue W16, identified as having a modest IC50 of 30–40 μM in enzymatic assays (Fig. 9).86 Kinetic analysis showed that W16 is a non-competitive inhibitor of CK2α and that this inhibition was reversed upon the addition of CK2β or the CK2β-mimicking peptide Pc, suggesting that W16 binds in or close to the CK2β-binding pocket on CK2α. The inhibition of CK2α may result from small conformational changes of the protein upon binding of W16.86
Recently, Kröger et al. found that the enantiomer of W16 was six times more potent than its analogue (Ki = 4.9 μM vs. 31 μM respectively).87 They also replaced the labile anhydride group of W16 with a more stable imide and N-methylimide, increasing stability without the loss of binding affinity (Ki = 3.6 μM vs. 2.8 μM respectively). However, the increased inhibition of the PPI did not result in a greater biological effect as the new analogues of W16 were not seen to inhibit CK2. This clearly highlights that perturbation to the equilibrium of the CK2 holoenzyme complex formation does not necessarily lead to enzyme inhibition.87 Due to the large aromatic structures of the podophyllotoxin indolo-analogues, the compounds suffer from poor solubility making the likelihood of developing therapeutics derived from W16 low.86,87
A high-concentration X-ray crystallography fragment screen identified 1 (in Brear et al.) binding at both the CK2α/β interface and in the ATP-pocket of CK2 with a weak IC50 (900 μM).88 Iterative design-test cycles resulted in the development of the small molecule CAM187 which had greatly improved bioactivity (IC50 = 44 μM) and did not show promiscuity (Fig. 9). Due to its significantly smaller size and, as such, more favourable physical properties than W16, the fragment CAM187 is a much-improved starting point for the development of selective small molecule drug-like compounds to target the CK2 PPI. However, further analyses of its activity and selectivity against other kinases, including evaluation of its biological effect in cells are required.
With a virtual screening of a compound library followed by surface-plasmon resonance (SPR), NMR and crystallography, Kufareva et al. identified compound 6 (Kd = 30 μM) a novel CK2 PPI inhibiting molecule (Fig. 9).82 Compound 6 (in Kufareva et al.) successfully inhibited the phosphorylation of CK2β-dependent substrates, suppressed MDA-MB231 triple-negative breast cancer cell growth and induced apoptosis in the μM range.82 As such, compound 6 is the first example of a small molecule inhibitor of the CK2α/β interaction which has been shown to be effective in cells. Alongside CAM187, compound 6 represents an excellent starting point for the development of selective and potent small molecule therapeutics for the inhibition of CK2 via the perturbation of the CK2 PPI.
In 2015, Hochscherf et al. altered the Phe residue in the structure of Pc to include para-halogens. The iodinated-Pc peptide was the most effective, showing a slight improvement in Kd compared to Pc (0.24 vs. 0.56 μM)95 In an attempt to make I-Pc cell-permeable, in 2019, the same research group introduced a cell-penetrating tag, namely, sC1896 to translocate the peptide into cells. Although the introduction of sC18 resulted in a 4-time increase of the Ki, sC18-I-Pc successfully inhibited the phosphorylation of CK2β-dependent substrates as well as evoking cell death in the mid-μM range (Fig. 9).93
Tang et al. used structure-based computational design in combination with experimental evaluation to develop point mutated Pc derivatives.108 The most promising mutant, I192F exhibited a 10-fold improvement in the predicted binding affinity to CK2α compared to Pc (0.5 vs. 8.9 μM). When the TAT-conjugated cell-permeable derivative of the peptide was synthesised, TAT-I192F showed comparable anti-proliferative effects against HepG2 cells to TAT-Pc (20.1 μM vs. 30.4 μM).96,108
The development of CAM7117 took a different approach to the aforementioned peptides in that it was the cyclic constraint that was the main site of optimisation due to the lability of the disulfide bond in the Pc derivatives. Using a structure-based approach, a variety of covalent constraints were designed and tested to determine which held the peptide in the optimal conformation. In addition, molecular modelling and X-ray crystallography of the peptide sequence directed a point mutation in the central sequence of Pc, namely Ile 192 to Trp. Movement of one of the cyclising residues by one position and the use of CuAAC chemistry gave CAM7117 (Fig. 9).79 The peptide had an improved Kd value compared to Pc (0.2 vs. 1 μM respectively) in an isothermal titration calorimetry (ITC) assay, was stable in human serum and cell-permeable. CAM7117 was able to inhibit cell growth with a GI50 of 33 μM in U2OS cells. It was postulated that the difference between the enzymatic and the cellular activity was due to partial trapping in the Golgi apparatus.79
In just over a decade, there has been a movement from the identification of CK2α/CK2β interaction site as a potential target for small molecule-directed inhibition of CK2 to the development of a handful of potent CK2 PPI inhibitors. Both the peptide and small molecule inhibitors have their limitations and require further optimisation before a sufficiently stable, extremely potent and highly selective cellular chemical probe or therapeutic is obtained for this site. However, the most promising inhibitors currently known are an excellent place for optimisation to continue from.
Inhibitors of CK2α acting outside the ATP pocket
Other strategies to inhibit CK2 target binding sites outside of the ATP pocket. Upon ligand binding, conformational modifications take place in the catalytic subunits resulting in the inhibition of its phosphorylating activity. Today, the only pockets outside the ATP site identified within CK2 are the so-called αD pocket and a pocket located at the interface between the αC helix and the glycine-rich loop, although recent findings question the existence of the latter.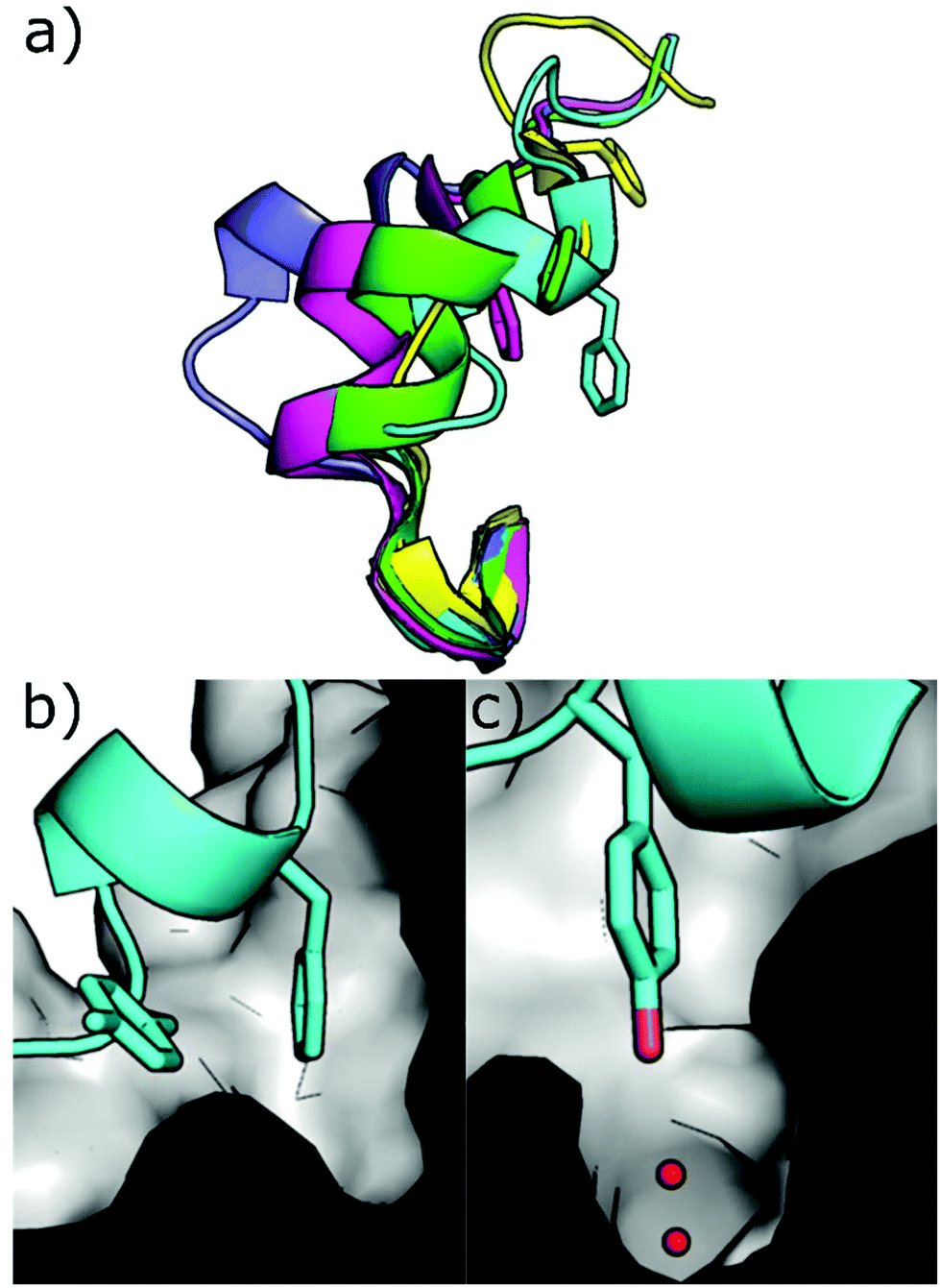 | ||
| Fig. 10 (a) Different conformations of the αD loop (cyan = 6YPK, green = 5CU6, purple = 5CLP, yellow = 5CVG, magenta = 5CS6); (b) cross-section of the αD pocket in the apo conformation showing the surface of the pocket and loop in cartoon format with Phe121 and Tyr125 shown (PDB: 6YPK); (c) cross-section of the αD pocket in the apo conformation showing the surface of the pocket and loop in cartoon format with Tyr125 shown in the water channel. (PDB: 6YPK). | ||
Using a fragment-based drug discovery approach, a series of benzylamine biaryl molecules binding in the αD pocket have been developed in a collaboration between our groups.51,97,99 Starting from high-concentration X-ray crystallography of commercially available fragments, compound 1 (in Brear et al.) was found to bind to the cryptic pocket in addition to other binding sites, namely the ATP binding site and in a hydrophobic pocket located at the interface with CK2β.97 Interestingly, compound 1 was the first ligand to reveal the full size of the αD pocket. Despite the lack of inhibitory effect on the catalytic activity of CK2, 1 set the basis for the development of molecules with improved binding affinity and potentially inhibitory activity (Fig. 11).
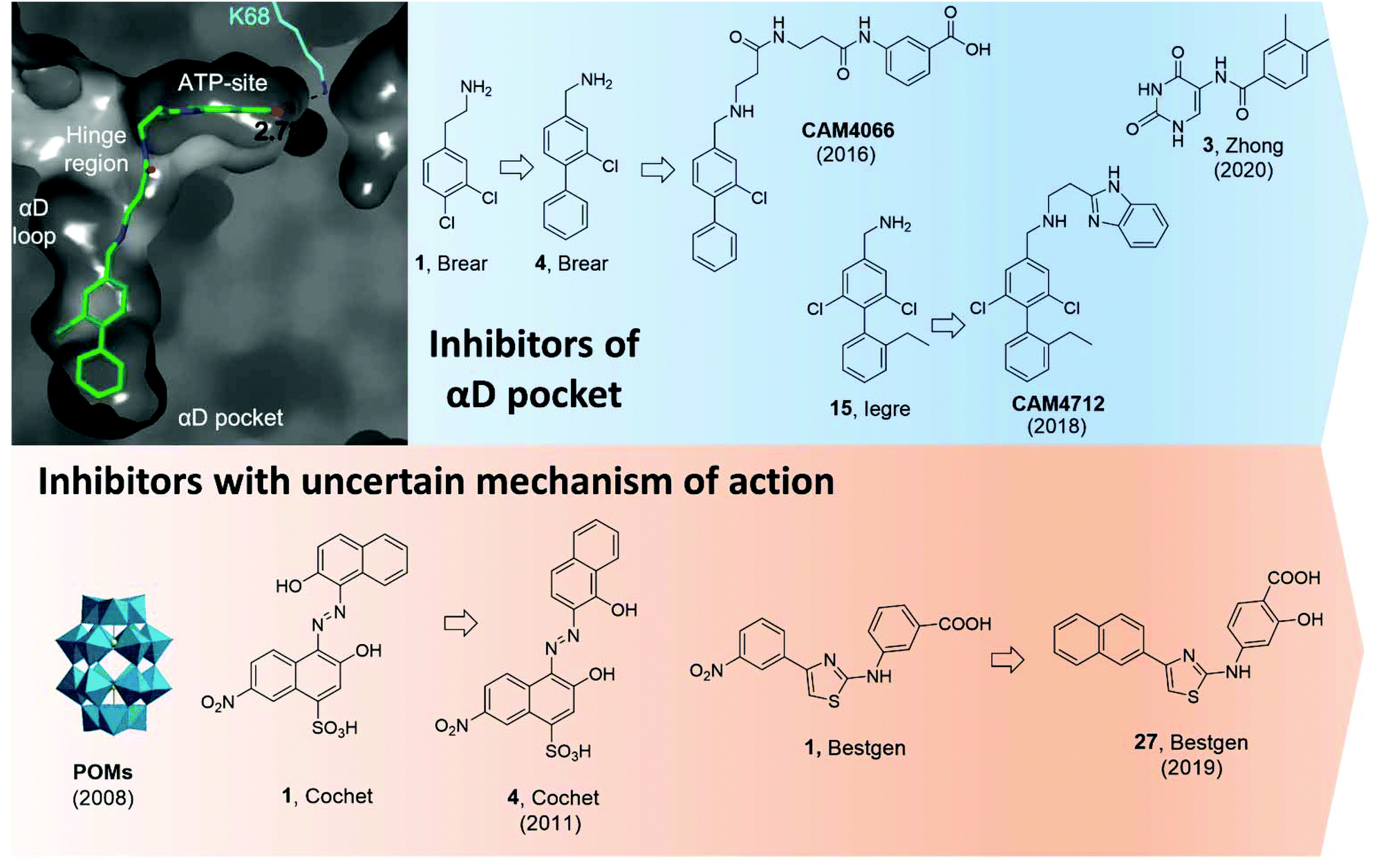 | ||
| Fig. 11 CK2 inhibitors reported in chronological order and acting on the catalytic subunit either on allosteric pockets or at dubious binding sites. In blue, CK2 inhibitors acting at the αD pocket with the co-crystal structure of CAM4066 and CK2α on the left (PDB: 5CU3). Lys68, which forms a hydrogen bond to CAM4066, is shown in cyan alongside the hydrogen bond length. The key binding pockets are labelled. In orange, CK2 inhibitors with reported binding in a pocket located between the αC helix and the activation loop but later reported to bind in the ATP-site. Compounds have been labelled as in the related papers. | ||
Initially, it was decided to optimise the fragment by growing towards vacant parts of the pocket. This led to compound 4 with a Kd of 270 μM but yet, no inhibitory effects. Considering the proximity of the αD pocket to the ATP binding site, it was decided to link fragment 4 with a weakly binding ATP-site fragment. It was envisioned that selectivity for CK2 among other kinases could be obtained since the binding affinity of the linked molecules was driven by the portion binding to the αD pocket which is poorly conserved among the kinases. Indeed, this was the case for the lead inhibitor CAM4066 (Fig. 11) which showed a Kd of 320 nM, an IC50 of 370 nM, and a GI50 of 8.8 μM (for the methyl-ester prodrug), and it proved to be selective when screened in a panel of 52 closely related kinases with a Gini coefficient of 0.82.51 Following up from CAM4066, efforts went into developing a pure allosteric inhibitor that did not interact with the ATP binding site and that featured improved physicochemical properties. A fragment growing strategy starting from 15 (Kd = 6.5 μM) lead to the development of CAM4172 (Fig. 11) which retained the binding mode of CAM4066 in the αD pocket but inhibited CK2 without reaching the ATP binding site.99CAM4172 showed a Kd of 4 μM, an IC50 of 7 μM, and a GI50 of 10 μM.
More recently, Zhong et al. used a structure-based virtual screening approach to identify modulators of CK2. Among the compounds screened, compound 3 (Fig. 11) was identified as the most promising non-ATP competitive inhibitor of CK2α with an IC50 of 13.0 μM and it was shown to stop the proliferation of A549 cancer cells (GI50 of 23.1 μM).98 The authors found that the IC50 value did not depend on the concentration of ATP used and concluded that such a compound was acting via a non-ATP-competitive mechanism. Computational studies suggested the compound was binding in the αD pocket,98 but this is still to be demonstrated experimentally.
In 2008, Prudent et al. utilised high-throughput screening of the National Cancer Institute Diversity Set and the Mechanistic Diversity Set chemical libraries to identify non-ATP competitive CK2 inhibitors. The screening identified polyoxometalates (POMs) as the most promising inhibitors with IC50 < 10 nM and the highest inhibition obtained for K6[P2Mo18O62].103 Doubts remained around the mechanism of action of this probe which is known to hydrolyse in the assay media into the individual MoO42− and PO43− components which proved inactive when tested individually. Interestingly, degradation was not observed when POM was in the presence of CK2. The lead POM compound showed exquisite selectivity for CK2 when tested in a panel of 29 Ser/Thr kinases. When the mechanism of action of POMs was investigated more closely with steady-state kinetic analysis, it was found that POMs were not ATP site or peptide site-directed inhibitors. Kinase assays with CK2 holoenzyme, affinity chromatography, trypsin proteolysis, and site-directed mutagenesis excluded binding of POMs to the α/β interface and confirmed lack of binding at the ATP/peptide-binding pocket. In addition, it was found that mutations around the Gly-rich loop, helix αC, and the activation segment weakened the sensitivity of CK2 to POM inhibition. Despite all these studies, a clear binding site for POMs has not yet been identified. Similarly, the efficacy of POMs on CK2 inhibition in a cellular context remains to be seen.103
In 2011, a screening of some 3000 small molecules from the National Cancer Institute Developmental Therapeutics Program led Cochet et al. to the identification of azanapthalene compounds as hits against CK2α.86 Through SAR studies, hit compound 1 (in Cochet et al.) was modified into compound 4 which showed an IC50 of 0.4 μM and it was found not to act via an aggregation mechanism despite its chemical structure (Fig. 11). Furthermore, steady-state kinetic analysis showed that the compound was non-competitive towards ATP and peptide substrate. Mutagenesis studies indicated that residues located on helix αC and the activation segment might be part of compound 4's binding site. As seen for the POM inhibitors, the lack of further conclusive evidence (NMR or X-crystallography) of the inhibitor in complex with the protein leaves uncertainty around the actual binding site. The effect of compound 4 in cells was also investigated and it was found to promote cell cycle arrest. The compound proved to be selective for CK2 with a Gini coefficient of 0.803 when screened at 5 μM in a panel of 42 related kinases.86
Recently, Bestgen et al. reported aryl aminothiazole derivatives as allosteric modulators of CK2 by targeting the interface between the αC helix and the glycine-rich loop (G-loop).100,101 Starting from a virtual ligand screening campaign of around 2 million compounds, compound 1 (in Bestgen et al.) was identified as a hit and it was able to inhibit CK2 with an IC50 of 28 μM. SAR studies initially led to the discovery of compound 2 (IC50 of 7 μM) and later to the lead compound 27 (in Bestgen et al.) with an IC50 of 0.6 μM, Kd of 0.3 μM, and EC50 of 5 μM (Fig. 11). Enzymatic kinetic studies, native mass spectroscopy, circular dichroism experiments together with STD NMR studies hinted that compound i was acting as an allosteric inhibitor with a non-ATP-competitive mechanism of action able to stabilise the inactive conformation of CK2.100,101 However, Brear et al. used a combination of crystal structures, competitive ITC and NMR, hydrogen–deuterium exchange (HDX) mass spectrometry, and computational analyses of the amino thiazole compounds to confirm that these molecules were binding to the ATP binding site of the kinase, and hence acted as type II inhibitors.104 Independently, Lindenblatt et al. also obtained co-crystal structures of these compounds with CK2 showing binding to the ATP site and used kinase enzyme assay to demonstrate that these inhibitors do indeed act via an ATP-competitive mechanism.105
Modulators of the CK2β subunit
Lastly, another approach to inhibit CK2 outside its catalytic pocket is to interact with the CK2β subunit.Back in 2006, Cochet et al. made use of a yeast two-hybrid approach to identify molecules that bind at the N-terminus of CK2β.103 The peptide aptamer identified was an 18-mer unconstrained peptide (GKMNGVLPLAWPSLYLRL) showing a Kd of 0.4 μM in an SPR assay. Interestingly, P1 had high sequence homology with the cytomegalovirus IE2 protein, known to interact with CK2β and consequently arrest the cell cycle and trigger apoptosis (Fig. 1). The peptide identified did neither disrupt nor prevent the formation of the CK2 holoenzyme. ELISA assays with truncated versions of CK2β suggested that it was interacting at a site in between residues 1 and 55 of the N-terminus. Treatment of NIH3T3, HCT116, and MCF-10A cells transfected with GFP-P1 showed typical signs of apoptosis and cell cycle arrest. It was postulated that P1 was able to induce apoptosis by interacting with the p53-dependent apoptosis pathways.106 Although P1 represented the first peptide of this kind of inhibitor, it should be noted that more mechanistic studies would be needed to fully assess its potential. In addition, cell-permeable variants will also be needed for P1 to be regarded as a chemical probe.
Conclusions
In this review we described the key features of CK2, its involvement in a variety of diseases with particular focus on cancer and COVID-19, and we aimed to provide the reader with a comprehensive review on all the chemical probes reported to date.Despite the numerous efforts gone into developing chemical tools to inhibit such a crucial kinase, clinical candidates are yet to reach the market. Only one molecule, CX4945, is currently undergoing clinical studies; however, due to its limited selectivity, even if approved, there will still be a long way to go before CK2 can be considered a truly validated target in oncology. For this reason, scientists have recently shifted their attention to the development of CK2 inhibitors that interact selectively with CK2. Strategies have seen the development of chemical probes acting at sites located outside the ATP-pocket on CK2α, with the site at the interface with CK2β and the αD site being the most validated. With the increasing amount of information around these sites and advances in biochemical techniques, it is reasonable to believe that potent and selective CK2 inhibitors with suitable pharmacokinetic and pharmacodynamic properties will soon be discovered. In addition, recent advances in chemistry make it possible to foresee that different types of chemical probes will soon be developed including PROTACs acting outside the catalytic box, covalent inhibitors and small molecule/peptide hybrids to target multiple sites simultaneously.
Based upon the explosion of interest in the field of CK2 inhibition over the past two decades, it is not then unrealistic to imagine that CK2 inhibition will remain a key and active area of research over the coming years, as the search for novel therapeutics for cancer, COVID-19 and a multitude of other diseases in which CK2 is implicated continues.
Conflicts of interest
There are no conflicts to declare.Notes and references
- F. Ardito, M. Giuliani, D. Perrone, G. Troiano and L. Lo Muzio, Int. J. Mol. Med., 2017, 40, 271–280 CrossRef CAS PubMed.
- P. Lahiry, A. Torkamani, N. J. Schork and R. A. Hegele, Nat. Rev. Genet., 2010, 1, 60–74 CrossRef PubMed.
- http://www.kinase-screen.mrc.ac.uk/phosphorylation-ubiquitylation-drug-discovery Accessed January 2021.
- G. Manning, D. B. Whyte, R. Martinez, T. Hunter and S. Sudarsanam, Science, 2002, 298, 1912–1934 CrossRef CAS PubMed.
- N. N. Singh and D. P. Ramji, J. Mol. Med., 2008, 86, 227–897 CrossRef PubMed.
- L. A. Pinna, J. Cell Sci., 2002, 115, 3873–3878 CrossRef CAS PubMed.
- C. E. Ortega, Y. Seidner and I. Dominguez, PLoS One, 2014, 9, e115609 CrossRef PubMed.
- J. Zhang, P. L. Yang and N. S. Gray, Nat. Rev. Cancer, 2009, 9, 28–39 CrossRef CAS PubMed.
- A. Siddiqui-Jain, D. Drygin, N. Streiner, P. Chua, F. Pierre, S. E. O′ Brien, J. Bliesath, M. Omori, N. Huser and C. Ho, et al. , Cancer Res., 2010, 70, 10288–10299 CrossRef CAS PubMed.
- M. Bouhaddou, D. Memon, B. Meyer, K. M. White, V. V. Rezelj, M. Correa Marrero, B. J. Polacco, J. E. Melnyk, S. Ulferts and R. M. Kaake, et al. , Cell, 2020, 182, 685–712 CrossRef CAS PubMed.
- D. E. Gordon, G. M. Jang, M. Bouhaddou, J. Xu, K. Obernier, K. M. White, M. J. O'Meara, V. V. Rezelj, J. Z. Guo and D. L. Swaney, et al. , Nature, 2020, 583, 459–468 CrossRef CAS PubMed.
- R. Prudent and C. Cochet, Chem. Biol., 2009, 16, 112–120 CrossRef CAS PubMed.
- Z. A. Knight and K. M. Shokat, Chem. Biol., 2005, 12, 621–637 CrossRef CAS PubMed.
- M. I. Davis, J. P. Hunt, S. Herrgard, P. Ciceri, L. M. Wodicka, G. Pallares, M. Hocker, D. K. Treiber and P. P. Zarrinkar, Nat. Biotechnol., 2011, 29, 1046–1051 CrossRef CAS PubMed.
- K. Niefind, B. Guerra, I. Ermakowa and O. G. Issinger, EMBO J., 2001, 20, 5320–5331 CrossRef CAS PubMed.
- K. Niefind, J. Raaf and O. G. Issinger, Cell. Mol. Life Sci., 2009, 66, 1800–1816 CrossRef CAS PubMed.
- E. Papinutto, A. Ranchio, G. Lolli, L. A. Pinna and R. Battistutta, J. Struct. Biol., 2012, 177, 382–391 CrossRef CAS PubMed.
- A. C. Bibby and D. W. Litchfield, Int. J. Biol. Sci., 2005, 1, 67–79 CrossRef CAS PubMed.
- D. W. Litchfield, Biochem. J., 2003, 369, 1–15 CrossRef CAS PubMed.
- O. Filhol, J. L. Martiel and C. Cochet, EMBO Rep., 2004, 5, 351–355 CrossRef CAS PubMed.
- G. Poletto, J. Vilardell, O. Marin, M. A. Pagano, G. Cozza, S. Sarno, A. Falqués, E. Itarte, L. A. Pinna and F. Meggio, Biochemistry, 2008, 32, 8317–8325 CrossRef PubMed.
- J. Raaf, N. Bischoff, K. Klopffleisch, E. Brunstein, B. B. Olsen, G. Vilk, D. W. Litchfield, O.-G. Issinger and K. Niefind, Biochemistry, 2011, 50, 512–522 CrossRef CAS PubMed.
- J. Raaf, E. Brunstein, O.-G. Issinger and K. Niefind, Protein Sci., 2008, 12, 2180–2186 CrossRef PubMed.
- J. H. Trembley, Z. Chen, G. Unger, J. Slaton, B. T. Kren, C. Van Waes and K. Ahmed, BioFactors, 2010, 36, 187–195 CrossRef CAS PubMed.
- S. A. Gibson and E. N. Benveniste, Trends Immunol., 2018, 38, 82–85 CrossRef PubMed.
- J. Castello, A. Ragnauth, E. Friedman and H. Rebholz, Pharmaceuticals, 2017, 10(1), 7 CrossRef PubMed.
- S. Koch, C. T. Capaldo, R. S. Hilgarth, B. Fournier, C. A. Parkos and A. Nusrat, Mucosal Immunol., 2013, 6, 136–145 CrossRef CAS PubMed.
- E. Ampofo, L. Nalbach, M. D. Menger, M. Montenarh and C. Götz, Int. J. Mol. Sci., 2019, 18, 4398 CrossRef PubMed.
- K. J. Treharne, R. M. Crawford, Z. Xu, J. H. Chen, O. G. Best, E. A. Schulte, D. C. Gruenert, S. M. Wilson, D. N. Sheppard, K. Kunzelmann and A. Mehta, J. Biol. Chem., 2007, 282, 10804–10813 CrossRef CAS PubMed.
- L. R. Cruz, I. Baladrón, A. Rittoles, P. A. Díaz, R. Santana, M. M. Vázquez, A. García, D. Chacón, G. Perera and A. González, et al. , ACS Oharmacol. Transl. Sci., 2021, 4(1), 206–212 CrossRef CAS PubMed.
- R. Turner, BeingWell Online, https://medium.com/beingwell/is-silmitasertib-the-covid-19-treatment-breakthrough-weve-been-waiting-for-263cf9c696d5 (accessed January 2021).
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2000, 100, 57–70 CrossRef CAS PubMed.
- R. A. Faust, G. Niehans, M. Gapany, D. Hoistad, D. Knapp, D. Cherwitz, A. Davis, G. L. Adams and K. Ahmed, Int. J. Biochem. Cell Biol., 1999, 31, 941–949 CrossRef CAS PubMed.
- J. H. Trembley, G. Wang, G. Unger, J. Slaton and K. Ahmed, Cell. Mol. Life Sci., 2009, 66, 1858–1867 CrossRef CAS PubMed.
- R. A. Faust, M. Gapany, P. Tristani, A. Davis, G. L. Adams and K. Ahmed, Cancer Lett., 1996, 101, 31–35 CrossRef CAS PubMed.
- S. Yenice, A. T. Davis, S. A. Goueli, A. Akdas, C. Limas and K. Ahmed, Prostate, 1994, 24, 11–16 CrossRef CAS PubMed.
- G. Seitz, U. Münstermann, H. R. Schneider and O. G. Issinger, Biochem. Biophys. Res. Commun., 1989, 163, 635–641 CrossRef CAS PubMed.
- M. Gapany, R. A. Faust, S. Tawfic, A. Davis, G. L. Adams and K. Ahmed, Mol. Med., 1995, 1, 659–666 CAS.
- M. Chua, C. Ortega, A. Sheikh, M. Lee, H. Abdul-Rassoul, K. Hartshorn and I. Dominguez, Pharmaceuticals, 2017, 10, 18 CrossRef PubMed.
- P. O-Charoenrat, V. Rusch, S. G. Talbot, I. Sarkaria, A. Viale, N. Socci, I. Ngai, P. Rao and B. Singh, Clin. Cancer Res., 2004, 10, 5792–5803 CrossRef CAS PubMed.
- M. J. Ku, J. W. Park, B. J. Ryu, Y. J. Son, S. H. Kim and S. Y. Lee, Bioorg. Med. Chem. Lett., 2013, 23, 5609–5613 CrossRef CAS PubMed.
- F. Benavent Acero, C. S. Capobianco, J. Garona, S. M. Cirigliano, Y. Perera, A. J. Urtreger, S. E. Perea, D. F. Alonso and H. G. Farina, Lung Cancer, 2017, 107, 14–21 CrossRef PubMed.
- K. Y. Lin, C. L. Fang, Y. Chen, C. F. Li, S. H. Chen, C. Y. Kuo, C. Tai and Y. H. Uen, Ann. Surg. Oncol., 2010, 17, 1695–1702 CrossRef PubMed.
- J. Zou, H. Luo, Q. Zeng, Z. Dong, D. Wu and L. Liu, J. Transl. Med., 2011, 9, 97 CrossRef CAS PubMed.
- F. Zhou, J. Xu, G. Ding and L. Cao, Pathol. Oncol. Res., 2014, 20, 73–79 CrossRef CAS PubMed.
- G. Di Maira, A. Gentilini, M. Pastore, A. Caligiuri, B. Piombanti, C. Raggi, E. Rovida, M. Lewinska, J. B. Andersen and C. Borgo, et al. , Oncogenesis, 2019, 8, 61 CrossRef PubMed.
- M. Bouhaddou, D. Memon, B. Meyer, K. M. White, V. V. Rezelj, M. C. Marrero, B. J. Polacco, J. E. Melnyk, S. Ulferts and R. M. Kaake, et al. , Cell, 2020, 182, 685–712 CrossRef CAS PubMed.
- G. M. Hathaway, T. H. Lubben and J. A. Traugh, J. Biol. Chem., 1980, 255, 8038–8041 CrossRef CAS.
- H. Yim, Y. H. Lee, C. H. Lee and S. K. Lee, Planta Med., 1999, 65, 009–013 CrossRef CAS PubMed.
- C. De Fusco, P. Brear, J. Iegre, K. H. Georgiou, H. F. Sore, M. Hyvönen and D. R. Spring, Bioorg. Med. Chem., 2017, 13, 3471–3482 CrossRef PubMed.
- J. E. Dowling, M. Alimzhanov, L. Bao, C. Chuaqui, C. R. Denz, E. Jenkins, N. A. Larsen, P. D. Lyne, T. Pontz, Q. Ye, G. A. Holdgate, L. Snow, N. O'Connell and A. D. Ferguson, ACS Med. Chem. Lett., 2016, 7, 300–305 CrossRef CAS PubMed.
- R. Zandomeni, M. C. Zandomeni, D. Shugar and R. Weinmann, J. Biol. Chem., 1986, 261, 3414–3419 CrossRef CAS.
- S. Sarno, E. de Moliner, M. Ruzzene, M. A. Pagano, R. Battistutta, J. Bain, D. Fabbro, J. Schoepfer, M. Elliott, P. Furet, F. Meggio, G. Zanotti and L. A. Pinna, Biochem. J., 2003, 374, 639–646 CrossRef CAS PubMed.
- F. Meggio, M. A. Pagano, S. Moro, G. Zagotto, M. Ruzzene, S. Sarno, G. Cozza, J. Bain, M. Elliott, A. D. Deana, A. M. Brunati and L. A. Pinna, Biochemistry, 2004, 43, 12931–12936 CrossRef CAS PubMed.
- Z. Nie, C. Perretta, P. Erickson, S. Margosiak, R. Almassy, J. Lu, A. Averill, K. M. Yager and S. Chu, Bioorg. Med. Chem. Lett., 2007, 17, 4191–4195 CrossRef CAS PubMed.
- J. E. Dowling, C. Chuaqui, T. W. Pontz, P. D. Lyne, N. A. Larsen, M. H. Block, H. Chen, N. Su, A. Wu and D. Russell, et al. , ACS Med. Chem. Lett., 2012, 3, 278–283 CrossRef CAS PubMed.
- E. L. Atkinson, J. Iegre, P. D. Brear, E. A. Zhabina, M. Hyvönen and D. R. Spring, Molecules, 2021, 26, 1977 CrossRef CAS PubMed.
- T. Oshima, Y. Niwa, K. Kuwata, A. Srivastava, T. Hyoda, Y. Tsuchiya, M. Kumagai, M. Tsuyuguchi, T. Tamaru, A. Sugiyama, N. Ono, N. Zolboot, Y. Aikawa, S. Oishi, A. Nonami, F. Arai, S. Hagihara, J. Yamaguchi, F. Tama, Y. Kunisaki, K. Yagita, M. Ikeda, T. Kinoshita, S. A. Kay, K. Itami and T. Hirota, Sci. Adv., 2019, 5, 9060–9083 CrossRef PubMed.
- A. Dalle Vedove, F. Zonta, E. Zanforlin, N. Demitri, G. Ribaudo, G. Cazzanelli, A. Ongaro, S. Sarno, G. Zagotto, R. Battistutta, M. Ruzzene and G. Lolli, Eur. J. Med. Chem., 2020, 195, 112267 CrossRef CAS PubMed.
- C. Wells, D. H. Drewry, J. E. Pickett, D. W. Litchfield, S. Knapp and A. D. Axtman, Cell Chem. Biol., 2021 DOI:10.1016/j.chembiol.2020.12.013.
- B. Kobe, T. Kampmann, J. K. Forwood, P. Listwan and R. I. Brinkworth, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1754, 200–209 CrossRef CAS PubMed.
- F. Meggio, L. A. Pinna, F. Marchiori and G. Borin, FEBS Lett., 1983, 162, 235–238 CrossRef CAS PubMed.
- F. Meggio, F. Marchiori, G. Borin, G. Chessa and L. A. Pinna, J. Biol. Chem., 1984, 259, 14576–14579 CrossRef CAS.
- F. Meggio and L. A. Pinna, Biochim. Biophys. Acta, Mol. Cell Res., 1989, 1010, 128–130 CrossRef CAS.
- R. Tellez, M. Gatica, C. C. Allende and J. E. Allende, FEBS Lett., 1990, 265, 113–116 CrossRef CAS PubMed.
- R. Tellez, C. C. Allende and J. E. Allende, FEBS Lett., 1992, 308, 113–115 CrossRef CAS PubMed.
- Y. Perera, H. G. Farina, I. Hernandez, O. Mendoza, J. M. Serrano, O. Reyer, D. E. Gomez, R. E. Gomez, B. E. Acevedo, D. F. Alfonso and S. E. Perea, Int. J. Cancer, 2008, 122, 57–62 CrossRef CAS PubMed.
- S. E. Perea, O. Reyes, Y. Puchades, O. Mendoza, N. S. Vispo, I. Torrens, A. Santos, R. Silva, B. Acevedo, E. López, V. Falcón and D. F. Alonso, Cancer Res., 2004, 64, 7127–7129 CrossRef CAS PubMed.
- M. F. Gottardo, C. S. Capobianco, J. E. Sidabra, J. Garona, Y. Perera, S. E. Perea, D. F. Alonso and H. G. Farina, Sci. Rep., 2020, 10, 14689 CrossRef CAS PubMed.
- M. Winiewska-Szajewska, D. Płonka, I. Zhukov and J. Poznański, Sci. Rep., 2019, 9, 11018 CrossRef PubMed.
- G. Cozza, S. Zanin, S. Sarno, E. Costa, C. Girardi, G. Ribaudo, M. Salvi, G. Zagotto, M. Ruzzene and L. A. Pinna, Biochem. J., 2015, 471, 415–430 CrossRef CAS PubMed.
- E. Enkvist, K. Viht, N. Bischoff, J. Vahter, S. Saaver, G. Raidaru, O. G. Issinger, K. Niefind and A. Uri, Org. Biomol. Chem., 2012, 10, 8645–8653 RSC.
- K. Viht, S. Saaver, J. Vahter, E. Enkvist, D. Lavogina, H. Sinijärv, G. Raidaru, B. Guerra, O. G. Issinger and A. Uri, Bioconjugate Chem., 2015, 26, 2324–2335 CrossRef CAS PubMed.
- R. Y. Tsien, Nature, 1981, 290, 527–528 CrossRef CAS PubMed.
- J. Vahter, K. Viht, A. Uri and E. Enkvist, Bioorg. Med. Chem., 2017, 25, 2277–2284 CrossRef CAS PubMed.
- T. N. de Villavicencio-Diaz, Y. Mazola, Y. P. Negrín, Y. C. García, O. G. Cruz and S. E. Perea Rodríguez, Biochem. Biophys. Rep., 2015, 4, 20–27 Search PubMed.
- F. Meggio, D. Shugar and L. A. Pinna, Eur. J. Biochem., 1990, 187, 89–94 CrossRef CAS PubMed.
- J. Iegre, P. Brear, D. J. Baker, Y. S. Tan, E. L. Atkinson, H. F. Sore, D. H. O'Donovan, C. S. Verma, M. Hyvönen and D. R. Spring, Chem. Sci., 2019, 10, 5056–5063 RSC.
- G. Lolli, A. Ranchio and R. Battistutta, ACS Chem. Biol., 2014, 9, 366–371 CrossRef CAS PubMed.
- S. Zanin, C. Borgo, C. Girardi, S. E. O'Brien, Y. Miyata, L. A. Pinna, A. Donella-Deana and M. Ruzzene, PLoS One, 2012, 11, e49193 CrossRef PubMed.
- I. Kufareva, B. Bestgen, P. Brear, R. Prudent, B. Laudet, V. Moucadel, M. Ettaoussi, C. F. Sautel, I. Krimm, M. Engel, O. Filhol, M. Le Borgne, T. Lomberget, C. Cochet and R. Abagyan, Sci. Rep., 2019, 9, 15893 CrossRef PubMed.
- J. C. Fuller, N. J. Burgoyene and R. M. Jackson, Drug Discovery Today, 2009, 14, 155–161 CrossRef CAS PubMed.
- J. Raaf, E. Brunstein, O. G. Issinger and K. Niefind, Chem. Biol., 2008, 2, 111–117 CrossRef PubMed.
- B. Laudet, V. Moucadel, R. Prudent, O. Filhol, Y. S. Wong, D. Royer and C. Cochet, Mol. Cell. Biochem., 2008, 316, 63–69 CrossRef CAS PubMed.
- V. Moucadel, R. Prudent, C. F. Sautel, F. Teillet, C. Barette, L. Lafanechere, V. Receveur-Brechot and C. Cochet, Oncotarget, 2011, 2, 997–1010 CrossRef PubMed.
- L. Kröger, C. G. Daniliuc, D. Ensan, S. Borgert, C. Nienberg, M. Lauwers, M. Steinkrüger, J. Jose, M. Pietsch and B. Wünsch, ChemMedChem, 2020, 15, 871–881 CrossRef PubMed.
- P. Brear, A. North, J. Iegre, K. Hadje Georgiou, A. Lubin, L. Carro, W. Green, H. F. Sore, M. Hyvonen and D. R. Spring, Bioorg. Med. Chem., 2018, 26, 3016–3020 CrossRef CAS.
- L. Nevola and E. Giralt, Chem. Commun., 2015, 51, 3302–3315 RSC.
- J. A. Wells and C. L. McClendon, Nature, 2007, 450, 1001–1009 CrossRef CAS.
- B. Laudet, C. Barette, V. Dulery, O. Renaudet, P. Dumy, A. Metz, R. Prudent, A. Deshiere, O. Dideberg, O. Filhol and C. Cochet, Biochem. J., 2007, 408, 363–373 CrossRef CAS PubMed.
- B. Bestgen, Z. Belaid-Choucair, T. Lomberget, M. Le Borgne, O. Filhol and C. Cochet, Pharmaceuticals, 2017, 1, 16 CrossRef PubMed.
- D. Lindenblatt, M. Horn, C. Götz, K. Niefind, I. Neundorf and M. Pietsch, ChemMedChem, 2019, 14, 833–841 CrossRef CAS PubMed.
- S. Tang, N. Zhang, Y. Zhou, W. A. Cortopassi, M. P. Jacobson, L. Zhao and R. Zhong, Mol. Inf., 2019, 38, 1800089 CrossRef PubMed.
- J. Hochscherf, D. Lindenblatt, M. Steinkrüger, E. Yoo, Ö. Ulucan, S. Herzig, O.-G. G. Issinger, V. Helms, C. Götz, I. Neundorf, K. Niefind and M. Pietsch, Anal. Biochem., 2015, 468, 4–14 CrossRef CAS PubMed.
- I. Neundorf, R. Rennert, J. Hoyer, F. Schramm, K. Löbner, I. Kitanovic and S. Wölfl, Pharmaceuticals, 2009, 2, 49–65 CrossRef CAS PubMed.
- P. Brear, C. De Fusco, K. Hadje-Georgiou, N. J. Francis-Newton, C. J. Stubbs, H. F. Sore, A. R. Venkitaraman, C. Abell, D. R. Spring and M. Hyvönen, Chem. Sci., 2016, 7, 6839–6845 RSC.
- C. Li, X. Zhang, N. Zhang, Y. Zhou, G. Sun, L. Zhao and R. Zhong, Molecules, 2020, 25, 237 CrossRef CAS PubMed.
- J. Iegre, P. Brear, C. De Fusco, M. Yoshida, S. L. Mitchell, M. Rossmann, L. Carro, H. F. Sore, M. Hyvönen and D. R. Spring, Chem. Sci., 2018, 9, 3041–3049 RSC.
- B. Bestgen, I. Krimm, I. Kufareva, A. A. M. Kamal, W. G. Seetoh, C. Abell, R. W. Hartmann, R. Abagyan, C. Cochet, M. Le Borgne, M. Engel and T. Lomberget, J. Med. Chem., 2019, 62, 1803–1816 CrossRef CAS.
- B. Bestgen, I. Kufareva, W. Seetoh, C. Abell, R. W. Hartmann, R. Abagyan, M. Le Borgne, O. Filhol, C. Cochet, T. Lomberget and M. Engel, J. Med. Chem., 2019, 62, 1817–1836 CrossRef CAS PubMed.
- X. Chen, C. Li, D. Wang, Y. Chen and N. Zhang, Molecules, 2020, 25, 870 CrossRef CAS.
- R. Prudent, V. Moucadel, B. Laudet, C. Barette, L. Lafanechère, B. Hasenknopf, J. Li, S. Bareyt, E. Lacôte, S. Thorimbert, M. Malacria, P. Gouzerh and C. Cochet, Chem. Biol., 2008, 15, 683–692 CrossRef CAS PubMed.
- P. Brear, D. Ball, K. Stott, S. D'Arcy and M. Hyvönen, J. Med. Chem., 2020, 21, 12786–12798 CrossRef PubMed.
- D. Lindenblatt, A. Nickelsen, V. M. Applegate, J. Jose and K. Niefind, J. Med. Chem., 2020, 14, 7766–7772 CrossRef.
- V. Martel, O. Filhol, P. Colas and C. Cochet, Oncogene, 2006, 25, 7343–7353 CrossRef CAS PubMed.
- B. Laudet, C. Barette, V. Dulery, O. Renaudet, P. Dumy, A. Metz, R. Prudent, A. Deshiere, O. Dideberg, O. Filhol and C. Cochet, Biochem. J., 2007, 408, 363–373 CrossRef CAS PubMed , 17714077.
- S. Tang, N. Zhang, Y. Zhou, W. A. Cortopassi and M. P. Jacobson, Mol. Inf., 2018, 38, 1800089 CrossRef PubMed.
Footnote |
| † These authors contributed equally to the work. |
| This journal is © The Royal Society of Chemistry 2021 |