
Coordination environment engineering on nickel single-atom catalysts for CO2 electroreduction†
Mengbo
Ma
,
Fuhua
Li
and
Qing
Tang
 *
*
School of Chemistry and Chemical Engineering, Chongqing Key Laboratory of Theoretical and Computational Chemistry, Chongqing University, Chongqing 401331, China. E-mail: qingtang@cqu.edu.cn
First published on 20th October 2021
Abstract
Coordination engineering has recently emerged as a promising strategy to boost the activity of single atom catalysts (SACs) in electrocatalytic CO2 reduction reactions (CO2RR). Understanding the correlation between activity/selectivity and the coordination environment would enable the rational design of more advanced SACs for CO2 reduction. Herein, via density functional theory (DFT) computations, we systematically studied the effects of coordination environment regulation on the CO2RR activity of Ni SACs on C, N, or B co-doped graphene. The results reveal that the coordination environments can strongly affect the adsorption and reaction characteristics. In the C and/or N coordinated Ni–BXCYNZ (B-free, X = 0), only Ni acts as the active site. While in the B, C and/or N coordinated Ni–BXCYNZ (X ≠ 0), the B has transition-metal-like properties, where B and Ni function as dual-site active centers and concertedly tune the adsorption of CO2RR intermediates. The tunability in the adsorption modes and strengths also results in a weakened linear scaling relationship between *COOH and *CO and causes a significant activity difference. The CO2RR activity and the adsorption energy of *COOH/*CO are correlated to construct a volcano-type activity plot. Most of the B, C, and/or N-coordinated Ni–BXCYNZ (X ≠ 0) are located in the left region where *CO desorption is the most difficult step, while the C and/or N coordinated Ni–BXCYNZ (X = 0) are located in the right region where *COOH formation is the potential-determining step. Among all the possible Ni–BXCYNZ candidates, Ni–B0C3N1 and Ni–B1C1N2-N-oppo are predicted to be the most active and selective catalysts for the CO2RR. Our findings provide insightful guidance for developing highly effective CO2RR catalysts based on a codoped coordination environment.
 Qing Tang | Qing Tang is a Professor of Chemistry at Chongqing University. She received her B.S. in Chemistry from Shenzhen University in 2009, and her Ph.D. in Chemistry from Nankai University in 2014. After three years of postdoctoral research at the University of California Riverside (UCR), she joined the faculty at the school of chemistry and chemical engineering at Chongqing University in 2018. Her current research interests focus on the application of multi-scale computational methods to understand, design and discover new materials for electrocatalytic applications, such as hydrogen evolution, oxygen reduction and CO2 conversion. |
Introduction
With the excessive consumption of non-renewable fossil fuels (coal, oil and natural gas), the increasing emission of carbon dioxide (CO2) into nature has caused severe ecological and environmental problems.1–3 As an alternative way, CO2 can be reduced into useful chemical products, such as carbon monoxide, methane, ethylene and methanol. However, the carbon element in CO2 is in the highest +4 oxidation state and the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is highly stable, which makes it very difficult to be reduced.4–7 Among the variety of developed solutions, the electrochemical reduction of CO2 is one of the most promising strategies and has attracted widespread attention due to its low energy consumption, mild reaction conditions, and simple operation, which relies on efficient catalysts to accelerate the reaction.8–10 The promising electrocatalysts for the electrochemical CO2 reduction reaction (CO2RR) should be able to operate at a low overpotential and can effectively control the product selectivity, inhibit the competitive hydrogen evolution reaction (HER) and maintain high electrochemical stability.11–14
O bond is highly stable, which makes it very difficult to be reduced.4–7 Among the variety of developed solutions, the electrochemical reduction of CO2 is one of the most promising strategies and has attracted widespread attention due to its low energy consumption, mild reaction conditions, and simple operation, which relies on efficient catalysts to accelerate the reaction.8–10 The promising electrocatalysts for the electrochemical CO2 reduction reaction (CO2RR) should be able to operate at a low overpotential and can effectively control the product selectivity, inhibit the competitive hydrogen evolution reaction (HER) and maintain high electrochemical stability.11–14
Among the various types of investigated electrocatalysts,15,16 single-metal-atom catalysts have developed rapidly and emerged as a promising class of catalysts for the CO2RR.17–20 Compared to the metal bulk and nanoparticle counterparts, the atomically dispersed single metal catalysts have largely exposed active sites and exhibit effective atom utilization and high selectivity for the electrochemical CO2RR.21,22 Moreover, their well-defined structures serve as an ideal model to establish the relationship between the structure and catalytic properties at an atomic level. To stabilize the single metal center from agglomeration, the single-metal-atom catalysts are usually coordinated and immobilized on nitrogenated carbon-based conductive substrates, such as carbon nanotubes, graphene and amorphous or porous carbon.23–25 In particular, the graphene matrix has been widely used due to its high electronic conductivity and large surface area for metal loading.
Many transition metals, such as Mn, Fe, Co, Ni, and Cu,26–30 have been embedded into nitrogen-doped carbon systems and investigated as CO2RR electrocatalysts. The results demonstrate that the intrinsic CO2RR activity of Fe–N–C and especially Ni–N–C catalysts to yield CO is higher than that of the Co-, Mn-, and Cu-based moieties, which even rival the state-or-the-art Au- and Ag-based catalysts.26,31–34 However, the relatively strong binding of CO* over Fe–Nx and Mn–Nx single sites could lead to lower selectivity for CO formation.
Particularly, apart from being cost effective, Ni single atom catalysts have displayed exceptional activity, selectivity and high stability.6,35–37 Additionally, several recent experimental advances have demonstrated that the CO2RR activity of Ni–N–C entities can be effectively manipulated by controlling the local coordination environment of the Ni active center.19,38 For example, Li et al.22 realized a specific Ni–N4 structure through a topo-chemical transformation strategy by carbon layer coating, which shows excellent activity and remarkable stability for the CO2RR to CO. Bao et al.39 reported that coordinatively unsaturated Ni–N sites within porous carbon had higher selectivity and activity for the CO2RR than the Ni–N4 sites. Joo et al.40 and Lu et al.41 both showed that the Ni–N3V (V: vacancy) sites with a shrunk Ni–N–C local structure incorporated into the graphene lattice exhibit enhanced CO2RR performance compared to the Ni–N4 sites. Moreover, Jiang et al.42 prepared a series of Ni single atom catalysts with controlled Ni–N coordination numbers (Ni–Nx–C, x = 2, 3, 4) by varying the pyrolysis temperature of the polypyrrole@MgNi-MOF-74 precursor. They revealed that the Ni–N2–C catalyst with Ni coordinated by two N and two C shows far superior CO faradaic efficiency and turnover frequency compared to the Ni–N3–C and Ni–N4–C counterparts. Cheng et al.43 fabricated Ni-based catalysts with various N/C coordination numbers (Ni@NxCy) through pyrolysis of carbon substrates at different temperatures and achieved an optimal catalytic performance for the Ni@N2C2 catalysts. Theoretically, Zhang et al.11 suggested that the graphene embedded Ni–N5 site with additional ligated axial N atoms exhibits lower CO2RR onset overpotential than the Ni–N4 site.
This recent progress has evidenced the strong potential of coordination environment regulation in tuning the CO2RR activity of a single metal center. In principle, due to the difference in electronegativity and atomic size, changing the surrounding coordination elements will modify the electronic structure of the metal center, which would essentially affect the adsorption strength of the reaction intermediates and in turn, modify the activity and selectivity. It is noteworthy that, in addition to the widely studied Ni–NxCy catalysts with Ni–N and Ni–C coordination environments, many recent studies have shown that boron can be doped into the carbon matrix and the incorporation of boron can greatly accelerate the electrocatalytic reactions.44–46 The boron (∼0.82 Å) has a comparable covalent radius with C (∼0.77 Å) and N (∼0.75 Å), which can form rich compounds with C (e.g., BC3),47 N (BN)48,49 and Ni (e.g., NiB, Ni2B, Ni3B, Ni4B3).50 Moreover, B, in many cases, functions like a transition metal since the hybridization of its non-fully occupied valence electrons supplies a great chance to accept and donate lone-pair electrons. Hence, B can also be a potential element to dope and adjust the coordination environment.
In this research, we investigate the CO2RR performance of Ni-centered graphene with various B/C/N coordination environments from density functional theory (DFT) calculations. Recently, Goddard and Luo et al.51 reported a comprehensive study of the CO2RR mechanism on C/N coordinated Ni SAC by taking kinetics, the solvation effect, and experiment comparison into consideration. The CO2RR performance varies remarkably by differences in the number of C or N bonded to Ni. Herein, we used the computational hydrogen electrode model with a number of simplifications and approximations for fast screening the potential catalysts by calculating the thermodynamic energetics. This simplified model, in most cases, allows the reliable alignment of theoretical electrochemical potentials to those measured in experiment. Compared to the prior theoretical studies that mainly focus on the N and C coordination,51,52 the hybrid coordination by non-metal C, N and transition-metal-like B leads to more versatile adsorption characteristics and diverse activities. Our results showed that most of the Ni–BXCYNZ (X + Y + Z = 4) catalysts (25 out of 27 possible candidates) have high thermodynamic and electrochemical stability. The adsorption modes of CO2, *COOH and *CO are versatile. In the case of C and/or N coordinated Ni–BXCYNZ (B-free, X = 0), only the Ni center acts as the active site, and the CO2 hydrogenation to *COOH is the potential-determining step. While in the case of B-coordinated Ni–BXCYNZ (X ≠ 0), the B can actively participate in the reaction. As a result, the Ni and B function as dual sites to flexibly tune the adsorption of CO2RR intermediates, which facilitate the CO2 hydrogenation but increase the adsorption strength of *CO, making *CO desorption the most difficult step. Because of the versatile single- and dual-site adsorption modes, the scaling relationship between the adsorption energy of *COOH and *CO is significantly weakened. Moreover, we build the correlation between the adsorption energy and the limiting potential to describe the activity and selectivity. Compared to the pure N-coordinated Ni, the hybrid coordination by C–N, B–N or B–C–N can lead to much higher electrocatalytic activity, and several highly promising candidates stand out with high CO2RR activity and selectivity, including Ni–B0C3N1, Ni–B1C0N3, Ni–B2C0N2-B-hex, Ni–B1C1N2-N-oppo, and Ni–B1C1N2-N-hex. These results provide useful insights into the understanding of the coordination effect on the CO2RR and offer a reference for further research on advanced electrocatalysts.
Computational details
All the spin-unrestricted DFT computations are carried out in the DMol3 code.53 The generalized gradient approximation (GGA)54 of the Perdew–Burke–Ernzerhof (PBE)55 functional is used to describe the exchange–correlation interactions and the core electrons are treated with semi-core pseudopotentials (DSPPs).56 The double numerical plus polarization (DND) basis is adopted and the global orbital cut off value is set as 4.0 Å. The convergence tolerance of geometry optimization is set as 2.0 × 10−5 Ha in energy, 0.004 Ha Å−1 in force, and the force exerted on each atom is set to be 0.005 Å. To simulate the aqueous solvent environment, a conductor-like screening model (COSMO) with a dielectric constant of 78.54 is used.57 The k-points mesh of 5 × 5 × 1 and 9 × 9 × 1 using the Monkhorst–Pack method58 is utilized for the structural optimization and electronic property calculations, respectively.The absorption free energy of CO2 on Ni–BxCyNz is calculated by: ΔGads = ΔEads + ΔZPE − TΔS, where ΔEads is the adsorption energy obtained by subtracting the total energy of the isolated Ni–BxCyNz catalyst and free CO2 molecule from the adsorbed system. Due to the inaccurate description of CO2 molecule by the PBE functional, we added a correction of +0.10 eV for CO2.59
The change of Gibbs free energy (ΔG) for each elementary reaction step is calculated using the equation: ΔG = ΔE + ΔZPE − TΔS + ΔGU, where ΔE is the electronic reaction energy of a certain reaction step directly obtained from DFT energies, ΔGU = −eU, with U being the electrode potential of the electrochemical step. ΔZPE and TΔS are the contributions of the zero-point energy and entropy, respectively, which originate from the calculation of vibrational frequencies of the adsorbed intermediates at T = 298.15 K. The entropy of gas phase molecules is derived from the NIST database. The detailed data are provided in the ESI.†
Results and discussion
Catalyst models and stability
As shown in Fig. 1, we use a 6 × 6 × 1 graphene supercell with two C vacancies as the original substrate to construct the model catalysts, and then a single Ni atom is placed into the vacancy center and binds to four pyridine-like coordination atoms (B, C or N). The corresponding catalyst models are named Ni–BXCYNZ (X + Y + Z = 4). Note that there are three possible coordination configurations when X, Y or Z = 2, denoted as Ni–BXCYNZ-oppo, Ni–BXCYNZ-pen, and Ni–BXCYNZ-hex, in which the same two atoms occupy the opposite coordination sites or neighboring coordination sites in penta-atomic and hex-atomic rings, respectively. Ultimately, 27 catalyst structures are constructed.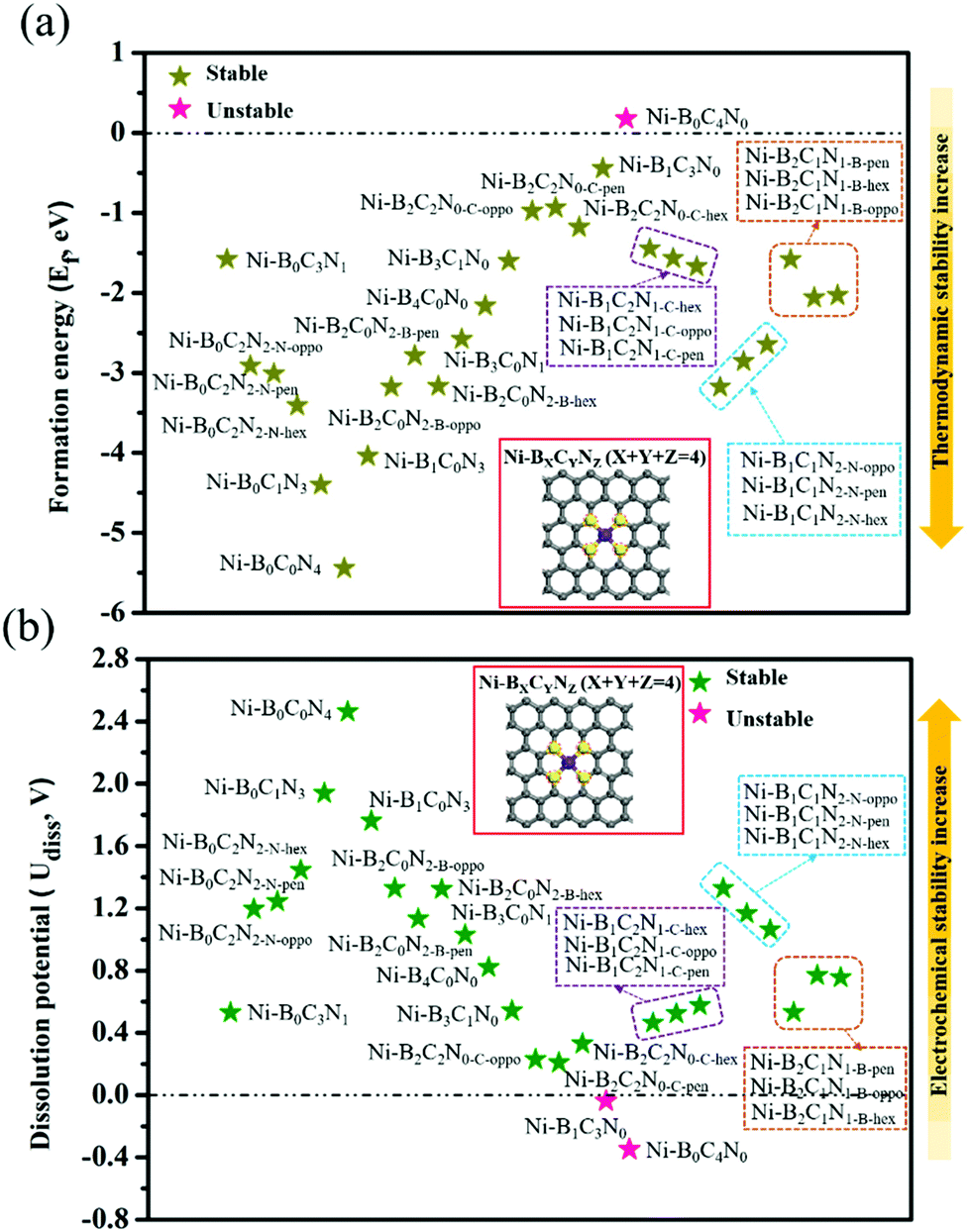 | ||
| Fig. 1 The DFT-computed formation energy (a) and dissolution potential (b) of Ni–BXCYNZ (X + Y + Z = 4). The inset shows the top view of the catalyst model, where the highlighted yellow atoms represent the coordination atoms of the Ni center (purple atom). | ||
In order to characterize the thermodynamic stability, we calculated the formation energy (Ef) of Ni–BXCYNZ systems, defined as Ef = Etotal − nBμB − nCμC − nNμN − ENi. Here, Etotal is the total energy of Ni–BXCYNZ. μB, μC and μN represent the chemical potential of B, C, and N, which corresponds to the energy of a single carbon atom in graphene, a single boron atom in planar hexagonal B36,60 and half of the energy of N2 molecules, respectively. While nB, nC, and nN stand for the number of corresponding coordination atoms in Ni–BXCYNZ, ENi is the energy of the isolated Ni atom. From Fig. 1a, one can see that except for the pure carbon coordinated Ni–B0C4N0, the calculated Ef values of the other 26 Ni–BXCYNZ catalysts are all negative (−0.94 to −5.44 eV), indicating their high thermodynamic stabilities and high synthetic potential in experiments. In addition, the dissolution potential Udiss is another important parameter to evaluate the electrochemical stabilities in a realistic electrochemical environment.61,62 The Udiss is calculated as 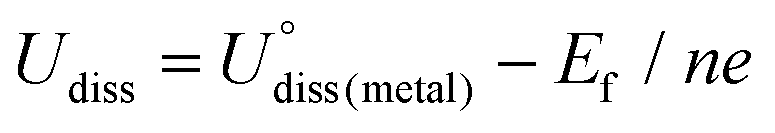 , where
, where 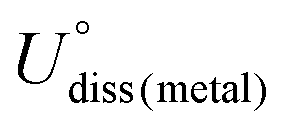 is the standard dissolution potential of the Ni metal, Ef is the formation energy, and n is the number of electrons involved in the dissolution (herein n = 2). According to this definition, only a catalyst with Udiss > 0 V vs. SHE can stably exist under acidic electrochemical conditions (the exact values of Ef and Udiss are listed in Table S1†).61,62 Among the 27 Ni–BXCYNZ systems, only Ni–B1C3N0 and Ni–B0C4N0 are excluded due to their negative Udiss and electrochemical instability (Fig. 1b). Finally, after the stability screening, there are about 25 Ni–BXCYNZ structures that both meet the criteria of thermodynamic and electrochemical stability. The corresponding structures of the 25 Ni–BXCYNZ are displayed in Fig. 2, which are further investigated as the candidate catalysts of the CO2RR.
is the standard dissolution potential of the Ni metal, Ef is the formation energy, and n is the number of electrons involved in the dissolution (herein n = 2). According to this definition, only a catalyst with Udiss > 0 V vs. SHE can stably exist under acidic electrochemical conditions (the exact values of Ef and Udiss are listed in Table S1†).61,62 Among the 27 Ni–BXCYNZ systems, only Ni–B1C3N0 and Ni–B0C4N0 are excluded due to their negative Udiss and electrochemical instability (Fig. 1b). Finally, after the stability screening, there are about 25 Ni–BXCYNZ structures that both meet the criteria of thermodynamic and electrochemical stability. The corresponding structures of the 25 Ni–BXCYNZ are displayed in Fig. 2, which are further investigated as the candidate catalysts of the CO2RR.
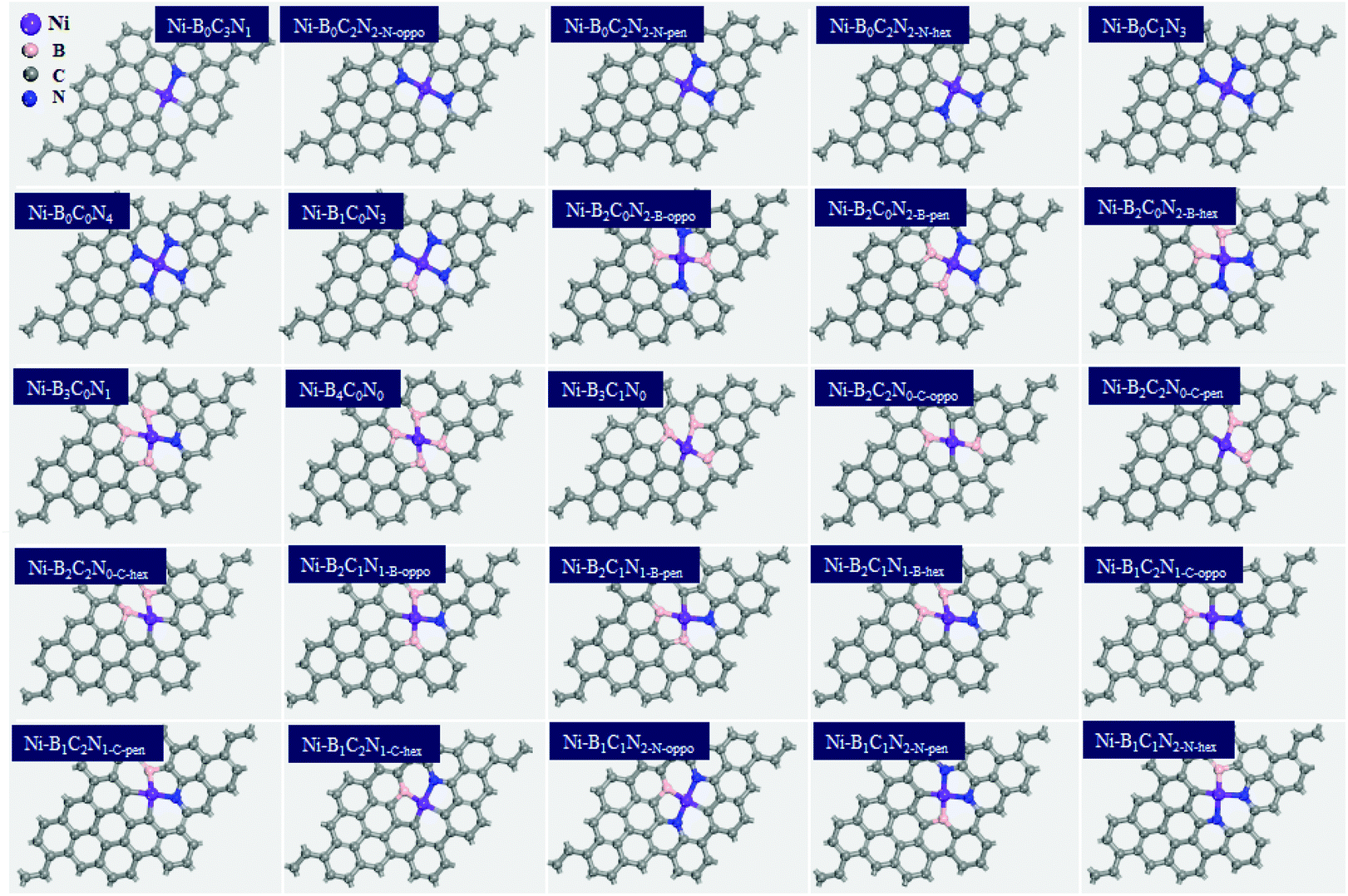 | ||
| Fig. 2 The optimized structures (top view) of the screened 25 Ni–BXCYNZ (X + Y + Z = 4), the color modes: purple for Ni, pink for B, blue for N, and grey for C. | ||
Adsorption of reaction species
The activation of CO2 as well as the adsorption of reaction intermediates (e.g., *COOH, *CO) onto the surface of catalysts plays a critical role during the electrocatalytic CO2RR. Hence, we firstly investigated the adsorption behaviors, and the most stable adsorption configurations for *CO2, *COOH and *CO are shown in Fig. 3 (the detailed adsorption free energy and charge transfer values are listed in Table S2†). From Fig. 3a, the *CO2 adsorption has two types of adsorption modes: physical adsorption and chemical adsorption. Among them, about 22 Ni–BXCYNZ compositions form physisorption. On Ni–BXCYNZ (X = 0), Ni–BXCYNZ (X = 1, 2(pen); Y = 0), Ni–BXCYNZ (X = 1; Y = 2; Z = 1) and Ni–BXCYNZ (X = 1; Y = 1; Z = 2(hex)), the CO2 is physically adsorbed with the C atom of CO2 pointed above the Ni center. While for Ni–BXCYNZ (X = 2(oppo, hex), 3, 4; Y = 0), Ni–BXCYNZ (Y = 1, 2; Z = 0) and Ni–BXCYNZ (X = 2(oppo, hex); Y = 1; Z = 1), the CO2 is physically adsorbed with the O atom of CO2 pointed above the Ni center. In the case of physical adsorption, the calculated CO2 adsorption free energy is around −0.09–0.20 eV, and the charge transfer between CO2 and Ni–BXCYNZ is around 0.004–0.019|e| from Bader charge analysis. In the case of chemical adsorption, there also exist two types of binding modes. On Ni–B2C1N1-B-pen, CO2 is chemically adhered with both the C and O atoms bonded to the Ni atom, leading to a favorable adsorption free energy of −0.18 eV, substantial charge transfer of 0.36|e|, and curved O–C–O bond angle of 145.8°. Differently, on Ni–B1C1N2-N-oppo and Ni–B1C1N2-N-pen, the C atom of chemisorbed CO2 is bonded to the Ni atom, while the O atom is bonded to the coordinated B atom. The dual-site activation of CO2 results in stronger adsorption energies (−0.25 and −0.39 eV), larger charge transfer (0.76 and 0.96|e|), and stronger deviation of the O–C–O bond angle from linearity (130.8° and 129.3°). The charge density difference of Ni–B2C1N1-B-pen, Ni–B1C1N2-N-oppo and Ni–B1C1N2-N-pen with chemically captured CO2 is shown in Fig. S1.† One can see that the Ni center and the directly coordinated N, C or B atom are the main player in the activation and orbital interaction with the CO2 reactant.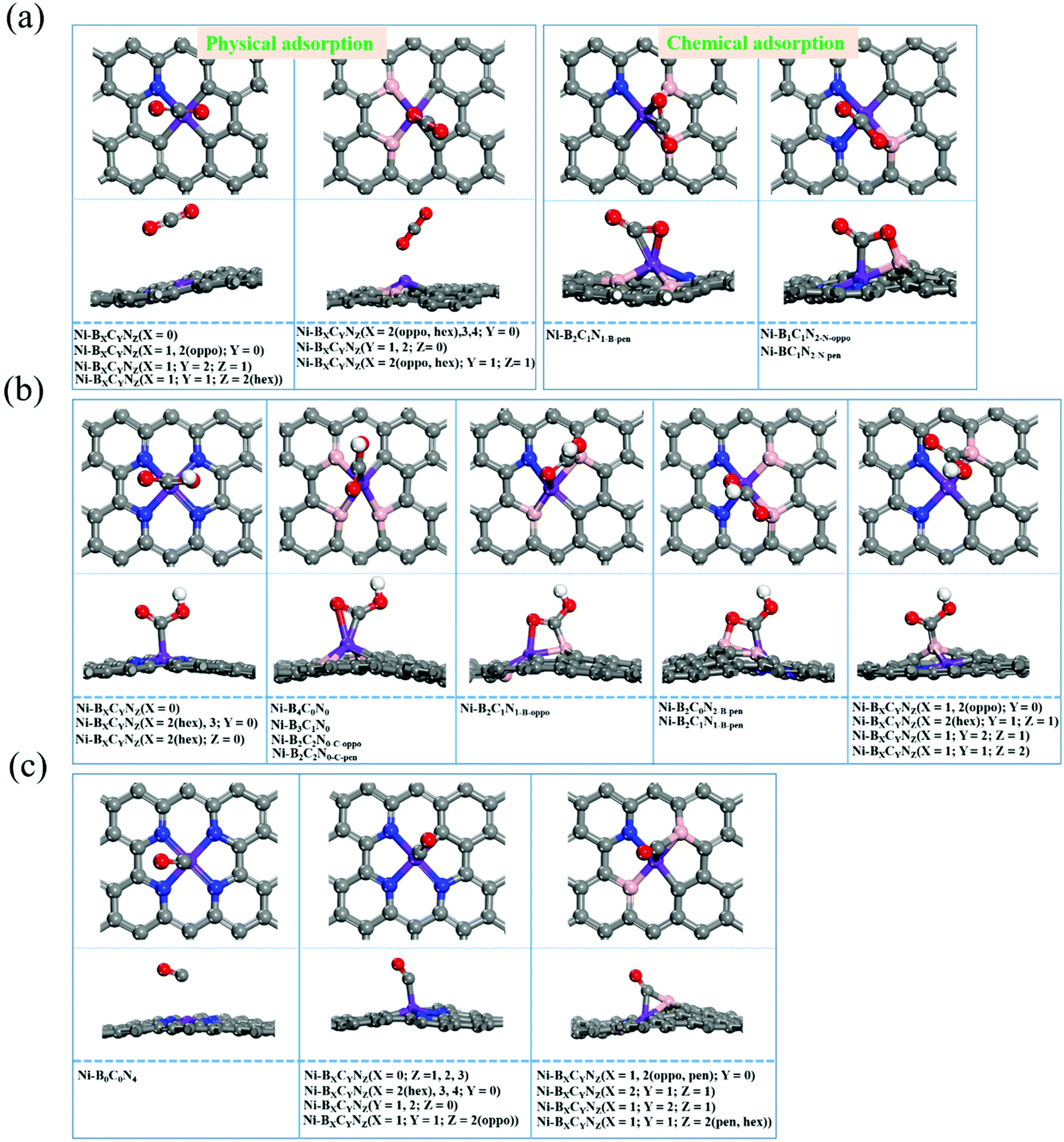 | ||
| Fig. 3 The different adsorption modes of *CO2 (a), *COOH (b), and *CO (c) intermediates on Ni–BXCYNZ. | ||
From Fig. 3b, the *COOH has versatile adsorption modes, which can be divided into five types. Specifically, (i) on Ni–BXCYNZ (X = 0), Ni–BXCYNZ (X = 2(hex), 3; Y = 0) and Ni–BXCYNZ (X = 2(hex); Z = 0), the C atom of *COOH is singly bonded to the Ni active center; (ii) on Ni–B4C0N0, Ni–B3C1N0, Ni–B2C2N0-C-oppo and Ni–B2C2N0-C-pen, both the C and O atoms of *COOH are co-adsorbed onto the Ni atom; (iii) on Ni–B2C1N1-B-oppo, the C atom of *COOH is bonded to a B atom while the O atom is bonded to the Ni atom; (iv) conversely, on Ni–B2C0N2-B-pen and Ni–B2C1N1-B-pen, the C of *COOH is bonded with Ni while the O atom binds with the B atom; (v) on Ni–BXCYNZ (X = 1, 2(oppo); Y = 0), Ni–BXCYNZ (X = 2(hex); Y = 1; Z = 1), Ni–BXCYNZ (X = 1; Y = 2; Z = 1) and Ni–BXCYNZ (X = 1; Y = 1; Z = 2), the C atom of *COOH is singly bonded to the B atom, which indicates that the B acts as the active center. Moreover, from Fig. 3c, the *CO can have three types of adsorption modes. (i) On Ni–B0C0N4, the *CO is physically adsorbed on the catalyst; (ii) on Ni–BXCYNZ (X = 0; Z = 1, 2, 3), Ni–BXCYNZ (X = 2(hex), 3, 4; Y = 0), Ni–BXCYNZ (Y = 1, 2; Z = 0) and Ni–BXCYNZ (X = 1; Y = 1; Z = 2(oppo)), the *CO is chemically adsorbed at the Ni center forming Ni–C single bonds; (iii) on Ni–BXCYNZ (X = 1, 2(oppo, pen); Y = 0), Ni–BXCYNZ (X = 2; Y = 1; Z = 1), Ni–BXCYNZ (X = 1; Y = 2; Z = 1) and Ni–BXCYNZ (X = 1; Y = 1; Z = 2(pen, hex)), the *CO forms bridging coordination with the Ni and B atoms. These flexible and versatile adsorption modes of CO2, *COOH and *CO indicate that not only the Ni center but also the non-metal coordination atoms, B, in particular, play a vital role in tuning and stabilizing the reaction intermediates.
Scaling relations
The reduction of CO2 to CO is a two-electron reaction, which includes two step hydrogenations of carbonaceous intermediates, i.e., *CO2 + H+ + e− → *COOH and *COOH + H+ + e− → *CO + H2O. On the traditional metal surfaces, the adsorption energies of *COOH and *CO are usually linearly correlated,63 and the weak *COOH or *CO adsorption (*COOH formation becomes unfavorable) can lead to a low CO2RR activity, while a strong *COOH or *CO adsorption makes the release of the CO product become difficult. Therefore, how to break or weaken the linear scaling relationship between *COOH and *CO has long been pursued in the CO2RR study. In our case, we also examined the correlation between *COOH and *CO. The adsorption energy (Eads) of *COOH and *CO on Ni–BXCYNZ is calculated using the equation:| Eads(*COOH) = E(Ni–BXCYNZ+COOH) − E(Ni–BXCYNZ) − ECO2(g) − 1/2EH2 | (1) |
| Eads(*CO) = E(Ni–BXCYNZ+CO) − E(Ni–BXCYNZ) − ECO(g) | (2) |
The calculated adsorption energies are provided in Table S3,† and the correlation between Eads (*COOH) and Eads (*CO) is shown in Fig. 4. From Fig. 4, the adsorption energy of *COOH and *CO follows a fitted linear relationship Eads (*COOH) = 0.95Eads (*CO) − 1.86, and the scaling relations (R ≈ 0.87) between them are slightly weakened with scattered points compared to those of pure metal surfaces (R ≈ 0.96).63 A special case is Ni–B1C1N2-N-oppo, which deviates greatly from the overall linear correlation. Note that on the pure metal surfaces, the *COOH and *CO are uniformly adsorbed to the surface metal atoms via single coordination mode (C–M bond). However, in our Ni–BXCYNZ systems, the change of coordination environment, especially with the introduction of B, leads to versatile coordination modes (e.g., single or dual-site coordination) that vary greatly with the type and number of coordination elements. The versatility in the adsorption structures could be the main reason for the weakened linear scaling between *COOH and *CO in Ni–BXCYNZ.
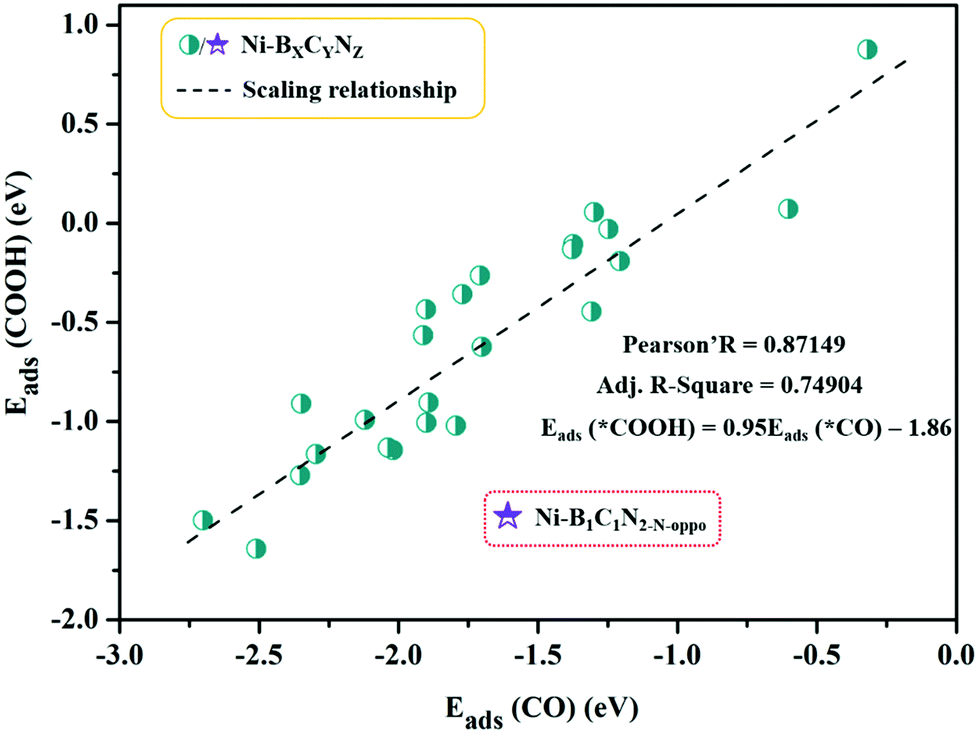 | ||
| Fig. 4 Scaling relationship between the adsorption energies of *COOH (Eads(*COOH)) and *CO (Eads(*CO)). | ||
CO2RR activity and selectivity
As discussed above, the adsorption characteristics of CO2, *COOH and CO varied with the change of coordination environments, which indicates that the activity and product selectivity would be significantly affected. Thus, we further explored the activity of the CO2RR to CO. The CO2RR pathways can be divided into four elementary steps (Fig. 5a): (i) CO2 adsorption: CO2 (g) + * → *CO2; (ii) *COOH formation: *CO2 + H+ + e− → *COOH; (iii) *CO formation: *COOH + H+ + e− → *CO + H2O; and (iv) *CO desorption: *CO → CO (g) + *. The Gibbs free energy changes (ΔG) for each reaction step are provided in Tables S4–S6,† and the ΔG (ΔG = max (ΔGi, ΔGii, ΔGiii, ΔGiv)) of the most difficult step, or the most sluggish step, dictates the reaction activity. The activity as a function of the binding energy of COOH and CO is shown in Fig. 5b. It is worth noting that the activity diagram is divided into two regions. In the right region, the *CO adsorption is relatively weak, and the activity is determined by the potential-dependent protonation of *CO2 to *COOH (step ii); while in the left region, the *CO adsorption is strong, and the activity is determined by the thermodynamic-dependent *CO desorption (*CO → * + CO(g), step iv). Clearly, most of the B-coordinated BXCYNZ (X ≠ 0) catalysts are located in the left region, where ΔG (*CO) varies from −0.35 to −1.42 eV, and the *CO desorption becomes the most difficult step. While Ni–B2C2N0-C-oppo, Ni–B2C2N0-C-hex, and all the C and/or N coordinated Ni–BXCYNZ (X = 0) systems are located in the right region, where the *COOH formation becomes the potential-determining step, and the corresponding free energy change of *CO2 hydrogenation to *COOH varies from 0.36 to 1.49 eV. Note that for the B-free Ni–BXCYNZ (X = 0), the CO2RR activity is gradually decreased as the number of coordinated nitrogen atoms increases (Ni–B0C3N1 > Ni–B0C2N2-N-oppo ≈ Ni–B0C2N2-N-pen > Ni–B0C2N2-N-hex ≈ Ni–B0C1N3 > Ni–B0C0N4). Moreover, from the volcano plot, Ni–B0C3N1 (the bottom of the right region) and Ni–B1C1N2-N-oppo (the bottom of the left region) stand out as the most active catalysts for the CO2RR. Along the right region, the Ni–B0C3N1 has the lowest free energy (0.36 eV) for the reaction-limiting step (*CO2 + H+ + e− → *COOH); and along the left region, the Ni–B1C1N2-N-oppo has the lowest desorption energy for *CO release (−0.35 eV). The detailed free energy diagram of CO2 reduction on Ni–B0C3N1 and Ni–B1C1N2-N-oppo is shown in Fig. 5c.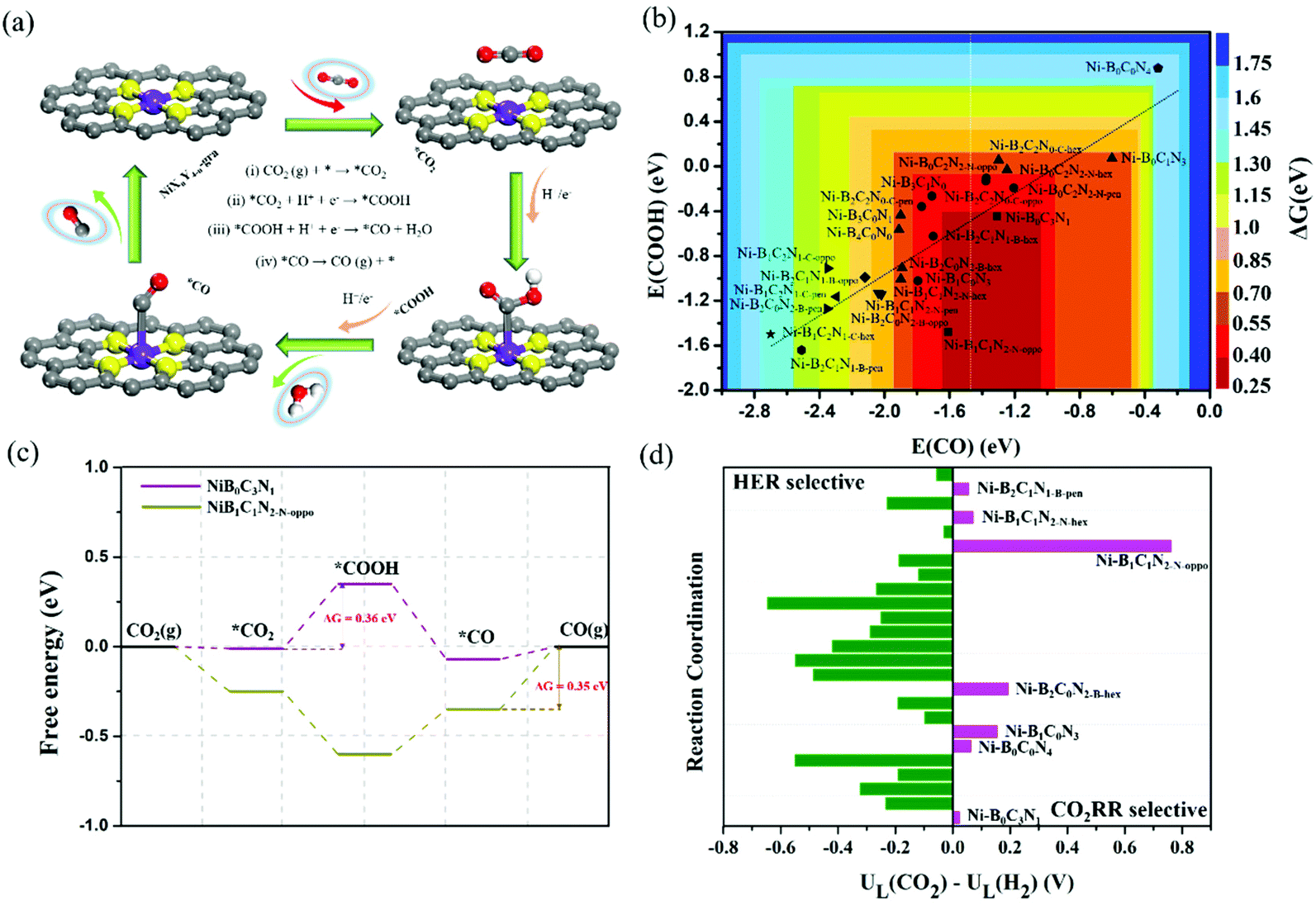 | ||
| Fig. 5 Schematic of the four-step reaction processes of CO2 reduction to CO on Ni–BXCYNZ (a). 2D volcano plot for CO evolution on Ni–BXCYNZ catalysts (b). Free energy diagrams for the CO2RR on Ni–B0C3N1 and Ni–B1C1N2-N-oppo (c). The limiting potential difference between CO2 reduction (UL(CO2)) and HER (UL(H2)) on various Ni–BXCYNZ catalysts (d). | ||
Furthermore, to compare the computational accuracy of DMol3 with the plane-wave basis method in the VASP code, we choose the six N/C coordinated Ni–BXCYNZ (X = 0) systems to calculate the free energy diagram for CO evolution (Fig. S2†), and the free energy change for the potential-determining step (*COOH formation) is summarized in Table S7.† The free energy change predicted by the two methods differs by about 0.06–0.2 eV, and the trend of the predicted activity is similar except for Ni–B0C2N2 and Ni–B0C1N3 (DMol3 predicts a higher activity of Ni–B0C2N2-N-oppo/Ni–B0C2N2-N-pen over Ni–B0C1N3, while VASP predicts the opposite). Recent studies by Luo et al.51 applied a grand canonical potential kinetics method to predict the reaction mechanism and rates for the CO2RR over Ni–N2C2, Ni–N3C1, and Ni–N4 sites in graphene. They revealed that Ni–N2C2 leads to the lowest onset potential (−0.84 V to achieve 10 mA cm−2 current density), followed by Ni–N3C1 (−0.92 V) and Ni–N4 (−1.03 V). Moreover, Liu et al.52 applied a “slow-growth” sampling approach to evaluate the reaction barriers and showed that the Ni-atom coordinated with one N and three C atoms (Ni–N1C3) is most active and selective for the CO2RR. These indicate that the CO2RR activity tends to decrease with the increase of coordinated nitrogen, which is qualitatively in good agreement with our DMol3 results based on the simplified computational hydrogen electrode model.
In addition, since the HER usually competes with the CO2RR, it is thus also important to evaluate the selectivity between CO2 reduction and HER by comparing their limiting potential (UL), which is defined as −ΔG/e (ΔG refers to the most sluggish step). According to this definition, a more positive ΔUL (UL(CO2) − UL(H2)) means a higher selectivity toward the CO2RR (the limiting free energy change of the competitive HER, ΔGHER, is provided in Table S6†). From Fig. 5d, most of the Ni–BXCYNZ catalysts are selective to the HER except for Ni–B0C3N1, Ni–B0C0N4, Ni–B1C0N3, Ni–B2C0N2-B-hex, Ni–B1C1N2-N-oppo, Ni–B1C1N2-N-hex and Ni–B2C1N1-B-pen. Among the 7 CO2RR selective catalysts, Ni–B0C3N1 and Ni–B0C0N4 are potentially determined by *CO2 hydrogenation. However, the ΔG for *COOH formation on Ni–B0C0N4 is very high (1.49 eV), making Ni–B0C0N4 selective but less active for the CO2RR. A recent report by Zhang et al.64 showed that single Ni2+ atoms fourfold coordinated by N and dispersed over a carbon black support displayed excellent CO Faraday efficiency (above 90%) and a low faradaic efficiency to H2. The higher CO selectivity of the Ni–N4 catalyst over the HER is in agreement with our predictions of the CO2RR selective Ni–B0C0N4. Moreover, for the screened Ni–B1C0N3, Ni–B2C0N2-B-hex, Ni–B1C1N2-N-oppo, Ni–B1C1N2-N-hex and Ni–B2C1N1-B-pen, the *CO desorption is the most sluggish step, and the corresponding ΔG (*CO) is −0.48 eV, −0.63 eV, −0.35 eV, −0.64 eV, and −1.25 eV, respectively. Since the CO2RR process requires rapid desorption of *CO to improve the CO selectivity, Ni–B2C1N1-B-pen is thus excluded as a viable CO2RR catalyst due to its over strong *CO adsorption. Note that CO2 electroreduction can produce many possible products. Previous experimental studies have verified that the N/C coordinated Ni produces CO as the main product, hence we only focus on the CO pathway for the B-free Ni–BXCYNZ (X = 0). With regard to the B-coordinated systems, we select the 5 CO2RR selective catalysts (Ni–B1C0N3, Ni–B2C0N2-B-hex, Ni–B1C1N2-N-oppo, Ni–B1C1N2-N-hex and Ni–B2C1N1-B-pen) to further examine the possible HCOOH pathway. From Fig. S3,† the limiting potential required for HCOOH formation is much higher, which indicates that CO is the more preferred product. In addition, since the *CO adsorption on Ni–B2C1N1-B-pen is very strong (−1.25 eV), we further studied its potential to produce more deep-reduced products. From Fig. S3e,† the *CO can be further reduced to CH4 and CH3OH, and the potential-determining step corresponds to hydrogenation of *CHO to *OCH2 (0.92 eV).
Activity origin
The activity of an electrocatalyst is known to be governed by its intrinsic electronic structure. To decode the underlying activity origin, we first analyzed the electronic structures, and the projected density of states (PDOS) of Ni and the coordination atoms (B, C and N) on pure Ni–BXCYNZ catalysts is shown in Fig. S4.† Since all the investigated 25 catalysts are non-magnetic, there is no spin-polarization in the calculated electronic states; we thus only plotted one of the spin channels. In the C- and N-coordinated Ni–BXCYNZ (B-free, X = 0), the electronic states around the Fermi level are mainly contributed by the Ni atom for Ni–B0C3N1, Ni–B0C2N2-N-oppo, Ni–B0C2N2-N-pen, and Ni–B0C2N2-N-hex (Fig. S4a, 2b, 2c and 2d†). While in the case of Ni–B0C1N3 (Fig. S4e†) and Ni–B0C0N4 (Fig. S4f†), the Ni electronic states shifted downward, and the Fermi levels are mainly contributed by the N atoms. This can qualitatively explain why Ni–B0C1N3 and Ni–B0C0N4 with higher N-content show lower CO2RR activity. We further analyzed the Bader charge of the Ni active center in Ni–BXCYNZ (B-free, X = 0) (Table S8†), where the Ni atom carries positive charge between +0.625|e| and +0.830|e|. Particularly, we note that the charge of the Ni atom in Ni–B0C3N1 (+0.625|e|) is the lowest, indicating that the Ni atom has more electrons in the outer shell. The previous research studies reveal that the larger magnitude of electrons in the outer shell would facilitate the adsorption of CO2 and *COOH intermediates thereby accelerating the CO2RR.65,66On the other hand, the electronic structures of B-coordinated Ni–BXCYNZ (X ≠ 0) are much more complicated (Fig. S4g–2y†). Depending on the various coordination environments, the electronic states at the Fermi level are mainly contributed by the Ni atoms (Ni–B1C0N3, Ni–B2C0N2-B-pen, Ni–B2C0N2-B-hex, Ni–B1C2N1-C-oppo, Ni–B1C1N2-N-pen and Ni–B1C1N2-N-hex), the B atoms (Ni–B2C0N2-B-oppo, Ni–B2C2N0-C-hex and Ni–B2C1N1-B-pen), the Ni and B atoms (Ni–B3C0N1, Ni–B4C0N0, Ni–B3C1N0, Ni–B2C1N1-B-oppo, Ni–B2C1N1-B-hex and Ni–B1C1N2-N-oppo), or the Ni and C atoms (Ni–B2C2N0-C-oppo, Ni–B2C2N0-C-pen, Ni–B1C2N1-C-pen and Ni–B1C2N1-C-hex), respectively. In the B-coordinated systems, both the Ni and B atoms are positively charged (except for Ni–B4C0N0 where the Ni atom has a negative charge of −0.0411|e|), and Ni is found to carry much less positive charge (+0.07–+0.49|e|) than B (+1.22–+1.51|e|). Compared to the B-free Ni–BXCYNZ (X = 0) (Ni atomic charge: +0.625–+0.830|e|), the charge of the Ni atom is significantly reduced with the introduction of B (Table S8†). It is known that B behaves like a transition metal and can be the active site in electrocatalysis. The valence electronic configuration of B atoms is 2s22p1 and the sp3 hybridization of these orbitals can accept and donate the lone-pair electron, which is similar to the d orbital of transition metals.45,67,68 In addition, the electronegativity of B (2.04) is close to that of Ni (1.91) and lower than C (2.55) and N (3.04), which also means that the B has transition-metal-like properties. This indicates that Ni and B can work synergistically to function as dual-active sites, which can explain the dual-site adsorption of CO2, *COOH and *CO in some of the Ni–BXCYNZ (X ≠ 0) catalysts, leading to more favorable *COOH formation and stronger *CO adsorption.
Fig. 6 shows the correlation between the calculated Bader charge of the active center and the adsorption free energy of *COOH and *CO species. In the B-free Ni–BXCYNZ (X = 0), only the Ni acts as the active site, and both the ΔG (*COOH) and ΔG (*CO) display linear correlation (R2(*COOH) ≈ 0.83 and R2(*CO) ≈ 0.78) with the Ni Bader charge (Fig. 6a and b). However, in the B-coordinated BXCYNZ (X ≠ 0), the active site varies with the coordination environment, and either the single Ni site, single B site or dual Ni–B site can be the catalytic active center. Due to the complexity in the bonding behaviors between CO2RR intermediates and the active sites, the correlation between the atomic charge of Ni or B (Table S9†) with the adsorption free energy of *COOH and *CO is very poor (Fig. 6c–f). Compared to the single metal atom catalyst, the dual-site or multifunctional site catalysts need a more complex descriptor to explain the intrinsic activity, which would stimulate the future research on the dual- or multi-site electrocatalysts.
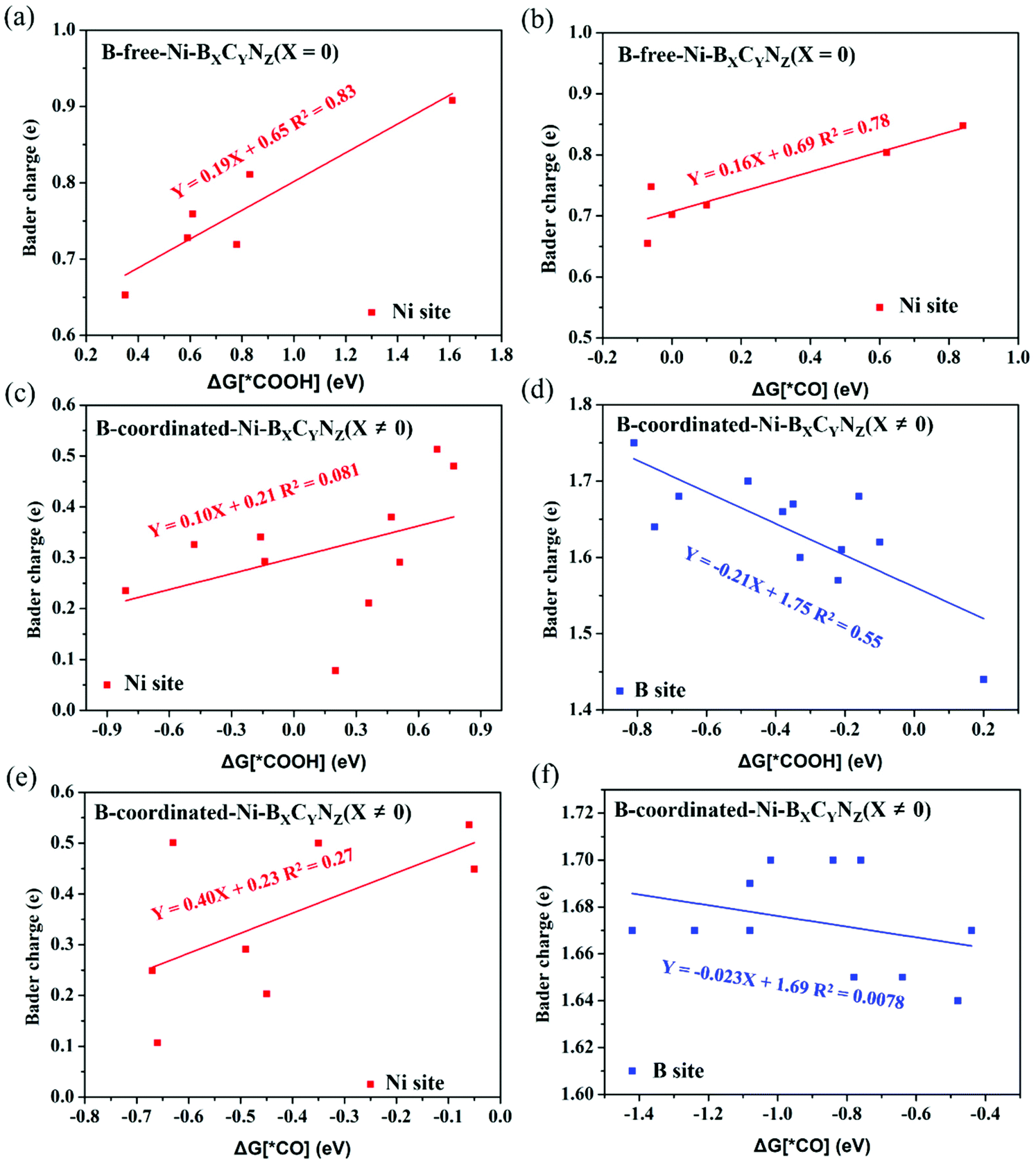 | ||
| Fig. 6 Bader charge analysis of active sites (Ni or B atom) on *COOH and *CO adsorbed Ni–BXCYNZ. Correlations of the adsorption free energy of *COOH (a) and *CO (b) intermediates with Ni Bader charge on B-free Ni–BXCYNZ (X = 0). Correlations of the adsorption free energy of *COOH and *CO intermediates with Ni (c, e) and B Bader charge (d, f) on B-coordinated Ni–BXCYNZ (X ≠ 0). | ||
Conclusion
In summary, based on DFT computations, we explored the coordination effect on the electrocatalytic activity of Ni SACs towards CO2 reduction, where the coordination atoms are C, N or B. Our results showed that the coordination environments of the metal atom have a remarkable influence on the adsorption and reaction characteristics. In the C and/or N coordinated Ni–BXCYNZ (B-free, X = 0), only the Ni atom acts as the active site, which features as a single-site adsorption mode for CO2RR intermediates. Differently, with the introduction of B (Ni–BXCYNZ, X ≠ 0), the coordinated B atom and the Ni metal function as a dual-site and lead to versatile coordination modes of reaction species (single or dual-site adsorption) that varies with the type and number of coordination elements. The versatility in the adsorption modes also results in a weakened linear scaling relationship between *COOH and *CO. Moreover, we established the volcano-type activity plot. It is found that most of the B-coordinated Ni–BXCYNZ (X ≠ 0) catalysts are located in the left region where *CO desorption becomes the most difficult step, while all the C and/or N coordinated Ni–BXCYNZ (X = 0) are located in the right region where the *COOH formation becomes the potential-determining step. Our results predict that Ni–B0C3N1 and Ni–B1C1N2-N-oppo stand out as the most active catalysts for the CO2RR and have high selectivity over the HER. Hence, by precisely controlling the coordination environment, the experimentally available Ni SACs can be utilized as highly active and selective CO2RR electrocatalysts. This work provides significant inspiration on boosting the electrocatalytic CO2RR activity of Ni or other metal catalysts by regulating the coordination environment.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21903008), the Chongqing Science and Technology Commission (cstc2020jcyj-msxmX0382), and the Fundamental Research Funds for the Central Universities (2020CDJQY-A031, 2020CDJ-LHZZ-063). This research used resources of the National Supercomputer Center in Guangzhou.References
- H. H. Chen, X. Guo, X. D. Kong, Y. L. Xing, Y. Liu, B. L. Yu, Q. X. Li, Z. G. Geng, R. Si and J. Zeng, Green Chem., 2020, 22, 7529–7536 RSC.
- S. E. Schwartz, Energy Environ. Sci., 2008, 1, 430–453 RSC.
- W. Choi, M. Kim, B. J. Kim, Y. Park, D. S. Han, M. R. Hoffmann and H. Park, Appl. Catal., B, 2020, 265, 118607 CrossRef CAS.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef CAS PubMed.
- X. Zhang, Z. S. Wu, X. Zhang, L. W. Li, Y. Y. Li, H. M. Xu, X. X. Li, X. L. Yu, Z. S. Zhang, Y. Y. Liang and H. L. Wang, Nat. Commun., 2017, 8, 14675 CrossRef PubMed.
- Q. Fan, P. F. Hou, C. H. Choi, T. S. Wu, S. Hong, F. Li, Y. L. Soo, P. Kang, Y. S. Jung and Z. Y. Sun, Adv. Energy Mater., 2020, 10, 1903068 CrossRef CAS.
- Y. W. Yue, Y. Y. Sun, C. Tang, B. Liu, Z. Ji, A. Q. Hu, B. Shen, Z. Z. Zhang and Z. Z. Sun, Carbon, 2019, 154, 108–114 CrossRef CAS.
- S. Dou, J. J. Song, S. B. Xi, Y. H. Du, J. Wang, Z. F. Huang, Z. C. J. Xu and X. Wang, Angew. Chem., Int. Ed., 2019, 58, 4041–4045 CrossRef CAS PubMed.
- T. T. Zheng, K. Jiang and H. T. Wang, Adv. Mater., 2018, 30, 1802066 CrossRef PubMed.
- H. Q. Zhou, X. L. Zou, X. Wu, X. Yang and J. Li, J. Phys. Chem. Lett., 2019, 10, 6551–6557 CrossRef CAS PubMed.
- X. L. Zhang, W. C. Wang and Z. X. Yang, ACS Sustainable Chem. Eng., 2020, 8, 6134–6141 CrossRef CAS.
- M. M. Zhao, H. Tang, Q. M. Yang, Y. L. Gu, H. Zhu, S. C. Yan and Z. G. Zou, ACS Appl. Mater. Interfaces, 2020, 12, 4565–4571 CrossRef CAS PubMed.
- K. P. Kuhl, T. Hatsukade, E. R. Cave, D. N. Abram, J. Kibsgaard and T. F. Jaramillo, J. Am. Chem. Soc., 2014, 136, 14107–14113 CrossRef CAS PubMed.
- C. Kim, F. Dionigi, V. Beermann, X. L. Wang, T. Moller and P. Strasser, Adv. Mater., 2019, 31, 1805617 CrossRef PubMed.
- R. Shi, J. H. Guo, X. R. Zhang, G. I. N. Waterhouse, Z. J. Han, Y. X. Zhao, L. Shang, C. Zhou, L. Jiang and T. R. Zhang, Nat. Commun., 2020, 11, 3028 CrossRef CAS PubMed.
- Y. R. Wang, R. X. Yang, Y. F. Chen, G. K. Gao, Y. J. Wang, S. L. Li and Y. Q. Lan, Sci. Bull., 2020, 65, 1635–1642 CrossRef CAS.
- F. P. Pan, B. Y. Li, E. Sarnello, S. Hwang, Y. Gang, X. H. Feng, X. M. Xiang, N. M. Adli, T. Li, D. Su, G. Wu, G. F. Wang and Y. Li, Nano Energy, 2020, 68, 104384 CrossRef CAS.
- R. Sui, J. J. Pei, J. J. Fang, X. J. Zhang, Y. F. Zhang, F. J. Wei, W. X. Chen, Z. Hu, S. Hu, W. Zhu and Z. B. Zhuang, ACS Appl. Mater. Interfaces, 2021, 13, 17736–17744 CrossRef CAS PubMed.
- T. N. Nguyen, M. Salehi, Q. V. Le, A. Seifitokaldani and C. T. Dinh, ACS Catal., 2020, 10, 10068–10095 CrossRef.
- F. P. Pan, B. Y. Li, E. Sarnello, Y. H. Fei, X. H. Feng, Y. Gang, X. M. Xiang, L. Z. Fang, T. Li, Y. H. Hu, G. F. Wang and Y. Li, ACS Catal., 2020, 10, 10803–10811 CrossRef CAS.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef CAS PubMed.
- X. G. Li, W. T. Bi, M. L. Chen, Y. X. Sun, H. X. Ju, W. S. Yan, J. F. Zhu, X. J. Wu, W. S. Chu, C. Z. Wu and Y. Xie, J. Am. Chem. Soc., 2017, 139, 14889–14892 CrossRef CAS PubMed.
- Y. Hou, Y.-L. Liang, P.-C. Shi, Y.-B. Huang and R. Cao, Appl. Catal., B, 2020, 271, 118929 CrossRef CAS.
- H. N. Zhang, J. Li, S. B. Xi, Y. H. Du, X. Hai, J. Y. Wang, H. M. Xu, G. Wu, J. Zhang, J. Lu and J. Z. Wang, Angew. Chem., Int. Ed., 2019, 58, 14871–14876 CrossRef CAS PubMed.
- C. Zhang, Z. Fu, Q. Zhao, Z. Du, R. Zhang and S. Li, Electrochem. Commun., 2020, 116, 106758 CrossRef CAS.
- W. Ju, A. Bagger, G. P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 944 CrossRef PubMed.
- J. K. Li, P. Prslja, T. Shinagawa, A. J. M. Fernandez, F. Krumeich, K. Artyushkova, P. Atanassov, A. Zitolo, Y. C. Zhou, R. Garcia-Muelas, N. Lopez, J. Perez-Ramirez and F. Jaouen, ACS Catal., 2019, 9, 10426–10439 CrossRef CAS.
- T. Asset, S. T. Garcia, S. Herrera, N. Andersen, Y. C. Chen, E. J. Peterson, I. Matanovic, K. Artyushkova, J. Lee, S. D. Minteer, S. Dai, X. Q. Pan, K. Chavan, S. C. Barton and P. Atanassov, ACS Catal., 2019, 9, 7668–7678 CrossRef CAS.
- W. Z. Zheng, J. Yang, H. Q. Chen, Y. Hou, Q. Wang, M. Gu, F. He, Y. Xia, Z. Xia, Z. J. Li, B. Yang, L. C. Lei, C. Yuan, Q. G. He, M. Qiu and X. L. Feng, Adv. Funct. Mater., 2020, 30, 1907658 CrossRef CAS.
- C. Xu, X. Zhi, V. Anthony, D. Wang, B. Jin, Y. Jiao, Y. Zheng and S.-Z. Qiao, Small Struct., 2020, 2, 2000058 CrossRef.
- F. P. Pan, W. Deng, C. Justiniano and Y. Li, Appl. Catal., B, 2018, 226, 463–472 CrossRef CAS.
- X. M. Hu, H. H. Hval, E. T. Bjerglund, K. J. Dalgaard, M. R. Madsen, M. M. Pohl, E. Welter, P. Lamagni, K. B. Buhl, M. Bremholm, M. Beller, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2018, 8, 6255–6264 CrossRef CAS.
- T. Moller, W. Ju, A. Bagger, X. L. Wang, F. Luo, T. N. Thanh, A. S. Varela, J. Rossmeisl and P. Strasser, Energy Environ. Sci., 2019, 12, 640–647 RSC.
- W. Zheng, F. Chen, Q. Zeng, Z. Li, B. Yang, L. Lei, Q. Zhang, F. He, X.-L. Wu and Y. Hou, Nanomicro Lett., 2020, 12, 108 CrossRef CAS PubMed.
- C. M. Zhao, X. Y. Dai, T. Yao, W. X. Chen, X. Q. Wang, J. Wang, J. Yang, S. Q. Wei, Y. E. Wu and Y. D. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS PubMed.
- K. Jiang, S. Siahrostami, A. J. Akey, Y. B. Li, Z. Y. Lu, J. Lattimer, Y. F. Hu, C. Stokes, M. Gangishetty, G. X. Chen, Y. W. Zhou, W. Hill, W. B. Cai, D. Bell, K. R. Chan, J. K. Norskov, Y. Cui and H. T. Wang, Chem, 2017, 3, 950–960 CAS.
- H. B. Yang, S. F. Hung, S. Liu, K. D. Yuan, S. Miao, L. P. Zhang, X. Huang, H. Y. Wang, W. Z. Cai, R. Chen, J. J. Gao, X. F. Yang, W. Chen, Y. Q. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef CAS.
- P. F. Yao, J. W. Zhang, Y. L. Qiu, Q. Zheng, H. M. Zhang, J. W. Yan and X. F. Li, ACS Sustainable Chem. Eng., 2021, 9, 5437–5444 CrossRef CAS.
- C. C. Yan, H. B. Li, Y. F. Ye, H. H. Wu, F. Cai, R. Si, J. P. Xiao, S. Miao, S. H. Xie, F. Yang, Y. S. Li, G. X. Wang and X. H. Bao, Energy Environ. Sci., 2018, 11, 1204–1210 RSC.
- Y. J. Sa, H. Jung, D. Shin, H. Y. Jeong, S. Ringe, H. Kim, Y. J. Hwang and S. H. Joo, ACS Catal., 2020, 10, 10920–10931 CrossRef CAS.
- X. Rong, H. J. Wang, X. L. Lu, R. Si and T. B. Lu, Angew. Chem., Int. Ed., 2020, 59, 1961–1965 CrossRef CAS PubMed.
- Y. N. Gong, L. Jiao, Y. Y. Qian, C. Y. Pan, L. R. Zheng, X. C. Cai, B. Liu, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS PubMed.
- X. Yang, J. Cheng, X. Yang, Y. Xu, W. F. Sun, N. Liu and J. Z. Liu, ACS Sustainable Chem. Eng., 2021, 9, 6438–6445 CrossRef CAS.
- Y. Xia, X. Zhao, C. Xia, Z.-Y. Wu, P. Zhu, J. Y. T. Kim, X. Bai, G. Gao, Y. Hu, J. Zhong, Y. Liu and H. Wang, Nat. Commun., 2021, 12, 4225 CrossRef CAS PubMed.
- C. Y. Ling, X. H. Niu, Q. Li, A. J. Du and J. L. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- M. M. He, W. An, Y. Q. Wang, Y. Men and S. Liu, Small, 2021, 8, 2104445 CrossRef PubMed.
- X. Luo, J. Yang, H. Liu, X. Wu, Y. Wang, Y. Ma, S.-H. Wei, X. Gong and H. Xiang, J. Am. Chem. Soc., 2011, 133, 16285–16290 CrossRef CAS PubMed.
- X. Li, X. Yong, M. Wu, S. Lu, H. Liu, S. Meng, J. S. Tse and Y. Li, J. Phys. Chem. Lett., 2019, 10, 2554–2560 CrossRef CAS PubMed.
- A. Abengózar, D. Sucunza, P. García-García, D. Sampedro, A. Pérez-Redondo and J. J. Vaquero, J. Org. Chem., 2019, 84, 7113–7122 CrossRef PubMed.
- S. Carenco, D. Portehault, C. Boissiere, N. Mezailles and C. Sanchez, Chem. Rev., 2013, 113, 7981–8065 CrossRef CAS PubMed.
- M. D. Hossain, Y. F. Huang, T. H. Yu, W. A. Goddard and Z. T. Luo, Nat. Commun., 2020, 11, 2256 CrossRef CAS PubMed.
- X. H. Zhao and Y. Y. Liu, J. Am. Chem. Soc., 2020, 142, 5773–5777 CrossRef CAS PubMed.
- B. Delley, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- M. Ernzerhof and G. E. Scuseria, J. Chem. Phys., 1999, 110, 5029–5036 CrossRef CAS.
- B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- Q. Tang, Y. J. Lee, D. Y. Li, W. Choi, C. W. Liu, D. Lee and D. E. Jiang, J. Am. Chem. Soc., 2017, 139, 9728–9736 CrossRef CAS PubMed.
- Z. A. Piazza, H. S. Hu, W. L. Li, Y. F. Zhao, J. Li and L. S. Wang, Nat. Commun., 2014, 5, 3113 CrossRef PubMed.
- S. Wang, L. Li, J. Li, C. Z. Yuan, Y. Kang, K. S. Hui, J. T. Zhang, F. Bin, X. Fan, F. M. Chen and K. N. Hui, J. Phys. Chem. C, 2021, 125, 7155–7165 CrossRef CAS.
- F. H. Li and Q. Tang, J. Mater. Chem. A, 2021, 9, 8761–8771 RSC.
- H. A. Hansen, J. B. Varley, A. A. Peterson and J. K. Norskov, J. Phys. Chem. Lett., 2013, 4, 388–392 CrossRef CAS PubMed.
- H. Z. Yang, L. Shang, Q. H. Zhang, R. Shi, G. I. N. Waterhouse, L. Gu and T. R. Zhang, Nat. Commun., 2019, 10, 4585 CrossRef PubMed.
- S. Fang, X. R. Zhu, X. K. Liu, J. Gu, W. Liu, D. H. Wang, W. Zhang, Y. Lin, J. L. Lu, S. Q. Wei, Y. F. Li and T. Yao, Nat. Commun., 2020, 11, 14848 Search PubMed.
- M. H. Sun, J. P. Ji, M. Y. Hu, M. Y. Weng, Y. P. Zhang, H. S. Yu, J. J. Tang, J. C. Zheng, Z. Jiang, F. Pan, C. D. Liang and Z. Lin, ACS Catal., 2019, 9, 8213–8223 CrossRef CAS.
- F. H. Li and Q. Tang, Nanoscale, 2019, 11, 18769–18778 RSC.
- M. A. Legare, G. Belanger-Chabot, R. D. Dewhurst, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–899 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Computed formation energy, dissolution potential, adsorption free energy, charge transfer, charge density difference, reaction free energy change of the elementary reaction step of CO2RR, projected density of states, and Bader charge analyses of Ni–BXCYNZ systems. See DOI: 10.1039/d1nr05742a |
| This journal is © The Royal Society of Chemistry 2021 |