
Carbon nanotubes coupled with layered graphite to support SnTe nanodots as high-rate and ultra-stable lithium-ion battery anodes†
Huanhui
Chen‡
ab,
Guanxia
Ke‡
a,
Xiaochao
Wu‡
a,
Wanqing
Li
a,
Hongwei
Mi
 a,
Yongliang
Li
a,
Lingna
Sun
a,
Qianling
Zhang
a,
Chuanxin
He
a and
Xiangzhong
Ren
*a
a,
Yongliang
Li
a,
Lingna
Sun
a,
Qianling
Zhang
a,
Chuanxin
He
a and
Xiangzhong
Ren
*a
aCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen, Guangdong 518060, P.R. China. E-mail: renxz@szu.edu.cn; Tel: +86-755-26558134; Fax: +86-755-26558134
bShenzhen Engineering Laboratory of Flexible Transparent Conductive Films, School of Materials Science and Engineering, Harbin Institute of Technology, Shenzhen 518055, China
First published on 10th February 2021
Abstract
SnTe exhibits a layered crystal structure, which enables fast Li-ion diffusion and easy storage, and is considered to be a promising candidate for an advanced anode material. However, its applications are hindered by the large volume variation caused by intercalation/deintercalation during the electrochemical reaction processes. Herein, topological insulator SnTe and carbon nanotubes (CNTs) supported on a graphite (G) carbon framework (SnTe-CNT-G) were prepared as a new, active and robust anode material for high-rate lithium-ion batteries by a scalable ball-milling method. Remarkably, the SnTe-CNT-G composite used as a lithium-ion battery anode offered an excellent reversible capacity of 840 mA h g−1 at 200 mA g−1 after 100 cycles and high initial coulombic efficiencies of 76.0%, and achieved a long-term cycling stability of 669 mA h g−1 at 2 A g−1 after 1400 cycles. The superior electrochemical performance of SnTe-CNT-G is attributed to the stable design of its electrode structure and interesting topological transition of SnTe, combined with multistep conversion and alloying processes. Furthermore, in situ X-ray diffraction and ex situ X-ray photoelectron spectroscopy were employed to study the reaction mechanism. The results presented here provide new insights to design and reveal the reaction mechanisms of transition metal telluride materials in various energy-storage materials.
1. Introduction
Developing high-capacity and high-density rechargeable batteries is the key to meeting increasing demand for energy systems.1,2 Despite their commercial success in the portable electronics market, the application of lithium-ion batteries (LIBs) is still limited in many fields due to the low theoretical capacity of commercial graphite anodes.3–5 Thus, it is imperative to develop new anode materials with high energy density and long cycle life.6 A multitude of anode materials, such as transition metal oxides, transition metal sulfides and transition metal selenides,7–9 have been investigated for LIBs. Unfortunately, their intrinsic thermodynamic restrictions and kinetic limitations plague the practical applications of many of these anode materials in LIBs.10,11 In addition, their intrinsic low conductivity, agglomeration and pulverization in circulation lead to poor cycling performances of many anode materials.12,13 Recently, transition metal telluride has shown a sufficient potential for high-performance lithium-ion battery anodes due to its high conductivity and large theoretical specific capacities.14,15Topological insulators are a new quantum state. Due to their unique physical characteristics, they have huge and important application value in spintronics and quantum computing.16,17 In particular, the topological insulator SnTe with high energy density can be used in related fields of national defense and the military industry, even if this material is not so inexpensive.18 In addition, elemental Te possesses high metallic characteristics (2 × 10−4 mS m−1), making SnTe have extremely high conductivity.19 Moreover, SnTe has a typical layered structure, providing excellent structural flexibility for rapid Li+ diffusion. Furthermore, the density of SnTe (6.445 g cm−3) is much higher than that of other Sn-based materials, which allows SnTe to exhibit a large volumetric capacity when used in LIB electrodes.20 Nonetheless, similar to other alloying-type anode materials, the practical application of SnTe is still restrained by its inherent large volume variation, resulting in the pulverization of active particles, which produces an unstable solid electrolyte interface (SEI) layer and thus results in an undesirable electrochemical performance.21 Designing a stable electrode structure with short and efficient Li+-ion and electron pathways, a large contact area with the electrolyte and determining the barrier to alleviate the significant volume strain are key to solving these problems.22
In an effort to tackle the foregoing drawbacks, various strategies have been proposed, and much research progress has been achieved. For example, Son et al.23 designed a SnTe-TiC-C composite by heat-treated and high energy ball-milling methods, and this composite was evaluated as high-performance anodes for lithium-ion batteries and showed improved electrochemical performances. Further, Son et al. demonstrated that increasing the TiC contents resulted in better performance. This study identified that modifying the large volume expansion material with an inert material is a feasible pathway to enhance the electrochemical performance and achieve a long cycle stability. However, the synthetic approach usually involves complex multi-step processes, and the literature lacks a mechanism. Another effective strategy to improve the performance is via the effect of mixed cations combining to give inert component protection. For instance, Wu et al.24 designed SnSb2Te4 nanodots with highly doped graphene as the anode for LIBs, and owing to the rapid ion transport accelerated by the p–n heterojunction, they exhibited an impressive long cycle stability. However, the random distribution of particles causes poor ion and electron transport, thus restricting the high-rate property of these anodes. Up to now, there are few reports on transition metal telluride electrodes that pay special attention to its electrochemical performance and reaction mechanism. In particular, the topological insulation performance is still not well resolved, which limits the application of such materials in LIBs.25,26
In this work, we demonstrate a robust and stable SnTe-carbon nanotube-graphite (SnTe-CNT-G) electrode material via a simple and scalable ball-milling process. Owing to its structural advantages, the smart SnTe-CNT-G material exhibited the following synergistic effects. On one hand, the multistep conversion reactions and alloying process used to prepare the SnTe composites resulted in sufficient reversible capacity. On the other hand, graphite can provide a barrier to buffer volume expansion, thus achieving a long cycle life. More importantly, the insertion of CNTs into the carbon framework not only increases the structural elasticity, but also facilitates the penetration of electrolyte and enhances the transport of ions and electrons thus increasing the Coulomb efficiency.27,28 Stimulated by these merits, when employed as anodes for lithium-ion batteries, the SnTe-CNT-G exhibited a high discharge capacity of 840 mA h g−1 at 200 mA g−1 after 100 cycles and delivered a superior lithium storage capacity of 669 mA h g−1 after 1400 cycles at 2 A g−1. The galvanostatic intermittent titration (GITT) technique confirms that SnTe-CNT-G possesses the best reaction kinetics. In addition, the electrochemical reaction mechanism of SnTe is revealed by in situ X-ray diffraction (XRD) and ex situ X-ray photoelectron spectroscopy (XPS) analysis. Furthermore, ex situ scanning electron microscopy (SEM) and transmission electron microscopy (TEM) confirmed that this material has good structural stability. It is envisioned that the materials prepared herein could open up a possible new route for commercial lithium-ion battery anode materials.
2. Experimental section
2.1. Preparation of SnTe-CNT-G materials
The SnTe-CNT-G powder was prepared by ball milling. In detail, 1.0 g SnTe powder (Macklin, 99.999%), 0.8 g graphite (Aldrich, 99.95%) and 0.2 g multi-walled carbon nanotubes (Macklin, >95%) were added into a hardened-steel container. Then, stainless steel balls with diameters of 10 mm were used, and the powder-to-ball mass ratio was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30. Subsequently, these powder mixtures were assembled in a glove box under an Ar atmosphere, and the ball-milling process was conducted at 500 rpm for 15 h. For comparison, SnTe-G was prepared under the same conditions with the weight ratio SnTe:graphite = 1:1, and SnTe was ball-milled under the same conditions without any additives. In addition, SnTe-CNT-G was prepared under the same conditions with different contents (adding 0.6 and 1.0, respectively).
:30. Subsequently, these powder mixtures were assembled in a glove box under an Ar atmosphere, and the ball-milling process was conducted at 500 rpm for 15 h. For comparison, SnTe-G was prepared under the same conditions with the weight ratio SnTe:graphite = 1:1, and SnTe was ball-milled under the same conditions without any additives. In addition, SnTe-CNT-G was prepared under the same conditions with different contents (adding 0.6 and 1.0, respectively).
2.2. Physical characterization
The crystalline structure was characterized by X-ray powder diffraction (XRD, PANalytical Empyrean, Netherlands) with CuKα radiation (λ = 0.154 nm ) using an operating voltage of 45 kV and a current of 40 mA. The nanostructure was observed by field emission scanning electron microscopy (FESEM, JSM-7800F and TEAM Octane Plus, 10 kV, Japan). Transmission electron microscopy (TEM, JEM-2100 and X-Max80, Japan) was used to determine the high-resolution morphology, and energy dispersive X-ray spectroscopy (EDX) was performed. The elemental composition and chemical valence state were determined by X-ray photoelectron spectroscopy (XPS, Thermo Scientific K-Alpha+, UK). Nitrogen adsorption/desorption curves were analysed by the Brunauer–Emmett–Teller (BET) method (NOVA1200e, Japan). The structural evolution during the electrochemical process was investigated by XRD. The structural stability was verified by ex situ SEM and TEM.2.3. Electrochemical measurements
The working electrode was prepared with a slurry composed of 70% active material, 15% super P and 15% carboxymethyl cellulose (CMC) in deionized water. The resultant slurry was coated on a copper foil and vacuum-dried at 80 °C for 24 h. The mass loading of the active material was 1.5 mg cm−2. CR2032 coin-type cells were assembled in an argon-filled glove box, where the as-prepared samples were used as the anode, 1 mol L−1 LiPF6 EC/DMC/EMC (1:1:1 vol ratio) was applied as the electrolyte, lithium film served as the counter and reference electrodes, and a Celgard 2400 polymer membrane was used as the separator. Cyclic voltammetry (CV) was conducted between 0.01 to 3.0 V with an electrochemical workstation (CHI760E, China). Electrochemical impedance spectroscopy (EIS) was conducted by an electrochemical workstation in the frequency range 0.01 Hz to 100 kHz. The galvanostatic discharge–charge curve was recorded on a LAND CT2001A instrument (China) testing system. The in situ battery was assembled with a beryllium sheet as the current collector and used during the in situ X-ray powder diffraction experiment.
3. Results and discussion
The hierarchical structure of SnTe-CNT-G was prepared by milling the pure SnTe powder, carbon nanotubes and graphite. As schematically shown in Fig. 1, SnTe and few-layer graphite are physically exfoliated by the strong interaction among the layered bulk SnTe, graphite powders and steel balls during the ball-milling process, where CNTs are embedded between layers by interleaving.29,30 Permeability of the electrolyte is a prerequisite condition for surface storage charge and ion diffusion in multiscale pores. High specific surface area and reasonable pore-size distribution are beneficial to increase the energy density and power density of carbon-based lithium-ion batteries.31,32 On the one hand, the CNT and SnTe particles are confined to the cross-linked layered carbon layer. The entire carbon surface may be exposed to the electrolyte and may participate in charge storage.33 On the other hand, the engineering design of CNTs in carbon materials can optimize the porosity to promote ion transport. | ||
| Fig. 1 Schematic illustration of the SnTe-CNT-G composite synthesis process. | ||
Typical SEM images of SnTe-CNT-G are shown in Fig. 2a and b. It can be clearly observed that the carbon nanotubes are embedded into few-layer graphite, which can generate channels between the graphite layers and SnTe and improve the vertical conduction of electrons. In addition, the layered cross-linked structure can contribute to the electrochemical performance of topological materials. Notably, the SEM images of pure SnTe are shown in Fig. S1a and b,† and it can be seen that a massive structure is formed by agglomeration of the layers. In addition, the SEM images of the SnTe-G composites obtained at different magnifications are shown in Fig. S1c and d,† which reveal that this composite is a bulk structure formed by small layers. For comparison, the morphology of SnTe-CNT-G materials with different graphite content are also different. Excessive graphite will cause uneven dispersion of CNTs, and a small amount of graphite will cause CNTs to agglomerate (Fig. S2†). These massive structures are not conducive to effectively storing lithium, and the easy generation of by-products hinders further lithium storage. The detailed microstructure of the SnTe-CNT-G hybrids was further examined by TEM, and the images are shown in Fig. 2c–e. The tiny SnTe nanosheets were distributed on the surface of the few-layer graphite sheets, and the carbon nanotubes are evenly cross-linked with the interlayer sheets. This cross-linking feature can not only improve the conductivity, but also alleviate the volume effect of SnTe in the electrochemical process. The high-resolution TEM (HRTEM) image of SnTe-CNT-G shows a crystal lattice with a spacing of 0.316 nm, which corresponds to the (200) plane of SnTe.23 Furthermore, the other interlayer spacings of approximately 0.335 and 0.337 nm correspond to the (002) plane of the CNTs and few-layer graphite, respectively.34,35 In addition, the energy dispersive spectroscopy (EDS) mapping results of SnTe-CNT-G (Fig. 2f) clearly reveal that Sn, Te and C were uniformly dispersed on the composites.
 | ||
| Fig. 2 (a and b) SEM images of the SnTe-CNT-G samples. (c–e) TEM and HRTEM images of the SnTe-CNT-G composites. (f) TEM mapping of the SnTe-CNT-G. | ||
The XRD patterns of SnTe, SnTe-G and the SnTe-CNT-G hybrids are presented in Fig. 3a. Both materials illustrate similar diffraction peaks that can be well assigned to the SnTe phase (JCPDS no. 46-1210),18 indicating that the ball-milling process does not change the crystal structure of SnTe. In addition, an extra peak at 26.4° in the XRD patterns of the SnTe-G and SnTe-CNT-G composites can be ascribed to the (002) plane of graphite (JCPDS no. 41-1487).36 The Raman spectra of all samples are shown in Fig. 3b; the increase in the D/G intensity ratio of SnTe-CNT-G suggests the increasing number of vacancy-type defects in the structure, and these characteristics could improve the conductivity and boost the reaction kinetics.37
 | ||
| Fig. 3 (a) XRD patterns of SnTe-CNT-G, SnTe-G and SnTe. (b) Raman spectra of SnTe-CNT-G, SnTe-G and SnTe. Core-level XPS spectra of Sn 3d (c), Te 3d (d) and C 1s (e). (f) N2 adsorption and desorption isotherm curves of all samples, where the inset shows the pore-size distribution. | ||
XPS was applied to analyse the elemental composition and the chemical valence states of the SnTe-CNT-G composite. Fig. S3† shows that the Te, Sn and C elements coexist in the samples, which is consistent with the EDS mapping results. The peaks located at 486.2 and 494.6 eV in Fig. 3c are characteristic of Sn 3d5/2 and Sn 3d3/2.38 The Te 3d spectrum (Fig. 3d) can be deconvoluted into Te–Sn (575.7/586.1 eV), and the pairs of small peaks are assigned to Te–C bonds (572.8/583.2 eV).15,20 In addition, the peak of the high-resolution C 1s spectrum of the SnTe-CNT-G composite in Fig. 3e can be split into four distinct peaks centred at 284.3 284.5, 284.8 and 286.0 eV, which correspond to C–Te, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–C and C–O bonds, respectively.15,20 These results confirm the existence of C–Te bonding between SnTe and carbonaceous materials. To study the electronic interaction between each component, XPS was also performed on SnTe and SnTe-G. Fig. S4 and S5† show the survey scan spectrum and core-level spectrum obtained for the SnTe and SnTe-G composites, respectively. The high-resolution Sn 3d spectra of pure SnTe show a peak located at 495.3/486.8 eV, and the Te 3d spectrum can be deconvoluted into two peaks at 586.0/576.8 eV. Besides, the Sn 3d spectrum of SnTe-G shows a peak located at 494.3/485.9 eV, and the Te 3d spectrum can be deconvoluted into two pairs of peaks at 585.8/575.4 eV and 583.2/572.8 eV. Interestingly, the binding energies (BEs) of the Sn 3d peak of the SnTe-CNT-G spectrum are blue-shifted relative to the Sn 3d peak of the SnTe spectrum. In addition, the Te–Sn peak shifts slightly towards a higher BE. The observed electron transfer may be ascribed to the energy band offset of the heterojunction electric field between SnTe and the carbon material, which could play a critical role in facilitating the surface reaction kinetics and enhancing the electrochemical performance.10 Nitrogen adsorption and desorption isothermal methods were used to study the specific surface areas and pore-size distribution of SnTe, SnTe-G and SnTe-CNT-G (Fig. 3f). The SnTe-CNT-G, SnTe-G and SnTe materials exhibit a BET surface area of 61.33, 14.86 and 0.53 m2 g−1, respectively. The pore-size distributions of all the samples show that the SnTe-CNT-G composite possesses more mesoporous channels than SnTe or SnTe-G, which are mainly in the size range between 1 and 50 nm (Fig. 3f, inset). The large specific surface area and pore-size distribution of SnTe-CNT-G can be attributed to the introduction of carbon nanotubes in the structure, which not only increases the channel size on the composites, but also enhances the stripping efficiency during the ball-milling process. Moreover, such structural features can facilitate the electrochemical performance in batteries.
C, C–C and C–O bonds, respectively.15,20 These results confirm the existence of C–Te bonding between SnTe and carbonaceous materials. To study the electronic interaction between each component, XPS was also performed on SnTe and SnTe-G. Fig. S4 and S5† show the survey scan spectrum and core-level spectrum obtained for the SnTe and SnTe-G composites, respectively. The high-resolution Sn 3d spectra of pure SnTe show a peak located at 495.3/486.8 eV, and the Te 3d spectrum can be deconvoluted into two peaks at 586.0/576.8 eV. Besides, the Sn 3d spectrum of SnTe-G shows a peak located at 494.3/485.9 eV, and the Te 3d spectrum can be deconvoluted into two pairs of peaks at 585.8/575.4 eV and 583.2/572.8 eV. Interestingly, the binding energies (BEs) of the Sn 3d peak of the SnTe-CNT-G spectrum are blue-shifted relative to the Sn 3d peak of the SnTe spectrum. In addition, the Te–Sn peak shifts slightly towards a higher BE. The observed electron transfer may be ascribed to the energy band offset of the heterojunction electric field between SnTe and the carbon material, which could play a critical role in facilitating the surface reaction kinetics and enhancing the electrochemical performance.10 Nitrogen adsorption and desorption isothermal methods were used to study the specific surface areas and pore-size distribution of SnTe, SnTe-G and SnTe-CNT-G (Fig. 3f). The SnTe-CNT-G, SnTe-G and SnTe materials exhibit a BET surface area of 61.33, 14.86 and 0.53 m2 g−1, respectively. The pore-size distributions of all the samples show that the SnTe-CNT-G composite possesses more mesoporous channels than SnTe or SnTe-G, which are mainly in the size range between 1 and 50 nm (Fig. 3f, inset). The large specific surface area and pore-size distribution of SnTe-CNT-G can be attributed to the introduction of carbon nanotubes in the structure, which not only increases the channel size on the composites, but also enhances the stripping efficiency during the ball-milling process. Moreover, such structural features can facilitate the electrochemical performance in batteries.
The lithium storage performances were investigated in a voltage range from 0.01 to 3.0 V. Fig. 4a shows the cycling stability performance of the as-prepared samples at 200 mA g−1. The initial charge/discharge capacities are 1169/1539 mA h g−1 for SnTe-CNT-G, 1151.5/1545 mA h g−1 for SnTe-G and 1084/1450 mA h g−1 for SnTe, with an initial Coulomb efficiency of 76.0%, 74.5% and 74.7%, respectively. In addition, as shown in Fig. S6,† the SnTe-CNT-G material with 0.8 g graphite content has higher specific capacity and better cycle stability. The SnTe-CNT-G composite demonstrated a satisfactory cycling stability and a capacity of 840 mA h g−1 after 100 cycles. In stark contrast, the pure SnTe-G electrode presented an initial capacity similar to that of SnTe-CNT-G and a lower capacity retention than SnTe-CNT-G, which was 235 mA h g−1 after 100 cycles. SnTe had a specific capacity of approximately 446 mA h g−1 after 100 cycles, far below the capacity of SnTe-CNT-G. The excellent cycling stability and unmatched coulombic efficiency of SnTe-CNT-G materials can be ascribed to carbon nanotubes and graphite having high conductivity, good structural stability, adjustable surface functionality and excellent mechanical properties, which can provide SnTe-CNT-G with better lithium battery performance. The rate performances of all the samples, with stepwise increases from 100 to 200, 500, 1000 and eventually 2000 mA g−1, are shown in Fig. 4b. The SnTe-CNT-G electrodes delivered excellent rate stabilities of 907, 812, 720, 607 and 490 mA h g−1 after 10 cycles. Notably, a discharge capacity of 801 mA h g−1 was obtained when the current density was returned to 100 mA g−1. When evaluated under similar conditions, the capacity of the SnTe electrode cannot be restored when the current density returned to the initial value due to the decay of the electrodes. In addition, the SnTe-G electrode showed a stable performance and low capacity.
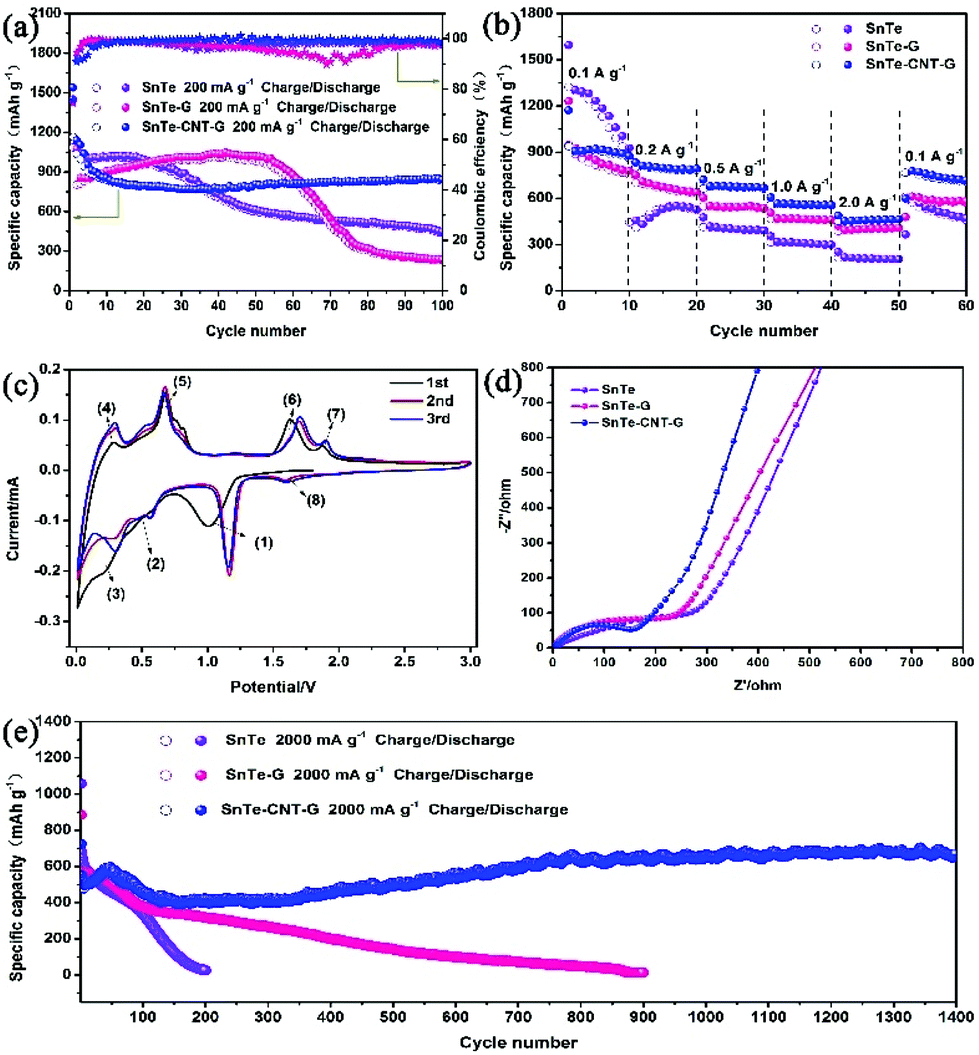 | ||
| Fig. 4 (a) Cycling performance and coulombic efficiency of the three samples at 200 mA g−1. (b) Rate performance of all samples. (c) CV curves of the SnTe-CNT-G anodes at 0.2 mV s−1. (d) Nyquist plots of all electrodes before cycling. (e) Cycling performance at 2000 mA g−1. | ||
CV was used to detect the electrochemical reaction process. Fig. 4c shows the CV curves of the SnTe-CNT-G electrode for the first three cycles at a scan rate of 0.2 mV s−1. During the initial lithiation process, there are three peaks at 1.06 V, 0.51 V and 0.23 V, which correspond to the phase transformation of SnTe to Li2Te and Sn (1), the alloying reaction of Sn with Li+-ions to form LixSn and the formation of an SEI film (2) and the multistep alloying process, forming Li4.4Sn (3).39,40 In the following anodic sweep process, two oxidation peaks appearing at 0.30 and 0.69 V can be associated with the delithiation of LixSn (4) and (5).41 In addition, other peaks at 1.65 V and 1.89 V correspond to the phase transformation of Li2Te and Sn to SnTe and the reversible conversion of Li2Te into Te, respectively (6) and (7).42 In the second cycle, the reductive peak at 1.9 V is related to the lithiation process of Te to form Li2Te (8).43 According to the CV analyses, the cycling mechanism of SnTe-CNT-G is proposed and is briefly summarized as follows:
| SnTe + 2Li+ + 2e− → Li2Te + Sn | (1) |
| Sn + xLi+ + xe− → LixSn (0 ⩽ x ⩽ 4.4) | (2) |
| LixSn → Li4.4Sn | (3) |
| Li4.4Sn → LixSn | (4) |
| LixSn → Sn + xLi+ + xe− | (5) |
| Li2Te + Sn → SnTe + 2Li+ + 2e− | (6) |
| Li2Te → Te + 2Li+ + 2e− | (7) |
| Te + 2Li+ + 2e− → Li2Te | (8) |
This result will be further discussed later with in situ XRD. In addition, the CV curves of the SnTe and SnTe-G electrodes are presented in Fig. S7a and S7b.† The characteristic peaks are similar, confirming that the two electrodes undergo the same redox processes. As shown in Fig. S8,† the discharge platform of the samples is consistent with the CV diagram. The current density increases with the decrease of specific capacitance of the materials. However, SnTe-CNT-G shows a higher specific capacity, which is consistent with the rate performance graph.
To further demonstrate the excellent properties of the SnTe-CNT-G material, the reaction kinetics were analysed by EIS (Fig. 4d and Fig. S9†). Obviously, the small diameter of the semicircle suggests that SnTe-CNT-G possesses a reduced Li-ion diffusion resistance.44 Long-term cycling at 2 A g−1 also indicates the stable and robust performance of SnTe-CNT-G (Fig. 4e). Specifically, SnTe-CNT-G exhibits a discharge capacity of 669 mA h g−1 after 1400 cycles, and the initial capacity decay can be attributed to degeneration of the active material structure and the reversible growth of an SEI film on the surface.2 It is worth noting that the slight increase in capacity during cycling is mainly due to the activation process of SnTe nanoparticles. This process is caused by repeated lithiation/delithiation processes and capacitive lithium storage behaviour.45,46The robust electrochemical performance of SnTe-CNT-G indicate that CNTs plays a significant role in SnTe-CNT-G composites. The electrochemical performance of the SnTe-CNT-G electrode is superior to that of previously reported Te-based or Sn-based electrodes (Table S1†).
To study the Li-ion storage mechanism, CV was performed at different scan speeds of 0.2, 0.4, 0.6, 0.8 and 1.0 mV s−1 (Fig. 5a). The relationship can be determined from the following equations:47,48
| i = avb |
| i(V) = k1v + k2v1/2 |
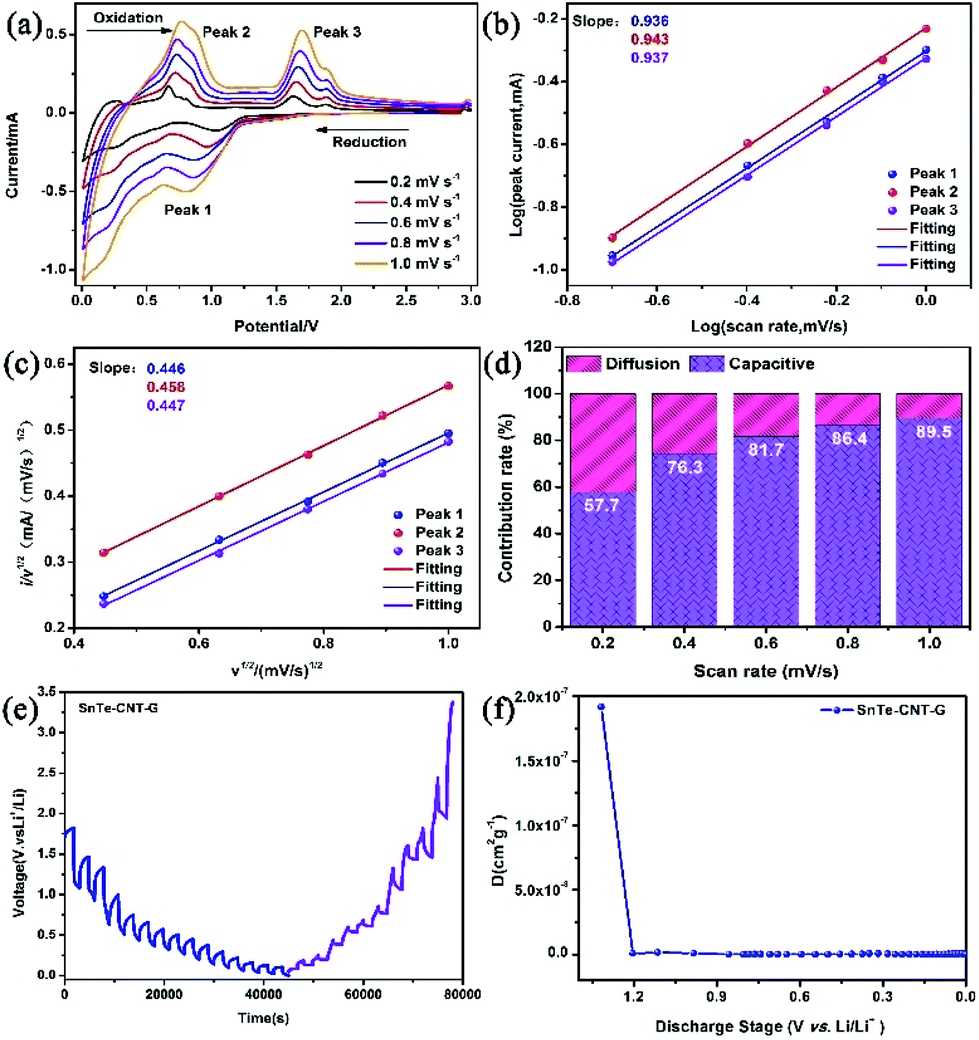 | ||
| Fig. 5 (a) CV curves obtained at different scan rates. (b) The corresponding log(i) versus log(v) plots and (c) the corresponding i/v1/2versus v1/2 plots of the SnTe-CNT-G electrodes. (d) The normalized ratio of the capacitive- and diffusion-controlled contributions at different scan rates. GITT curves of the SnTe-CNT-G electrode (e) and their Li+ diffusion coefficient for the initial discharge states (f). | ||
More importantly, the lithium reaction kinetics of SnTe-CNT-G, SnTe-G and SnTe were further investigated by a GITT technique. The Li+ diffusion coefficients of all the electrodes were calculated from the following equation:52

The reaction mechanism of the SnTe-CNT-G anode was further investigated by performing in situ XRD at 100 mA g−1 between 0.01 and 3 V. The first cycle of the SnTe-CNT-G electrode at the selected charged/discharged states is shown in Fig. 6a. The corresponding phase transformations are shown in Fig. 6b; the signal at nearly 39°, which appeared first, is ascribed to the conversion of SnTe and the formation of Li2Te. As the discharge process progresses, there are two diffraction peaks at 34.2° and 36.0° that belong to the emerged Sn. Specifically, as lithiation proceeds, the presence of Li4.4Sn suggests the multistep alloying of Sn. Additionally, the disappearance of the Li4.4Sn phases and the new emergence of the Sn phase also confirms the further dealloying reaction of Li4.4Sn. Besides, the 3D contour plots of the in situ XRD patterns obtained for the SnTe-CNT-G anode clearly confirmed these reaction processes (Fig. S13†). The reversible reaction of the SnTe-CNT-G anode is in excellent agreement with the previous CV analysis. In addition, the element valence state of the electrode material after cycling was investigated by ex situ XPS after the fully charged and fully discharged states were obtained. As shown in Fig. 6c, the Sn 3d spectrum of the SnTe-CNT-G anode was divided into two fitted peaks, which is fairly consistent with the spectrum of the fresh composites. This result confirmed the high reversibility of the SnTe-CNT-G electrode. However, the Sn 3d spectrum of the SnTe electrode substantially changed and presented two new peaks at 487.0 and 496.2 eV, confirming the formation of Sn4+ species.56,57 This phenomenon may be attributed to partial oxidation during the electrochemical process. Additionally, the Te 3d spectra of SnTe-CNT-G and SnTe are also shown in Fig. 6d, and a newly formed pair of small peaks at 572.4/582.9 eV were attributed to the formation of Te.58 These results are in excellent agreement with the results obtained from the CV and in situ XRD analysis.
 | ||
| Fig. 6 (a) The first cycle performed at 0.2 A g−1 between 0.01 and 3 V. (b) The contour plots of the in situ XRD patterns obtained for the SnTe-CNT-G anodes during the initial cycle. XPS core-level spectra of (c) Sn 3d and (d) Te 3d for the SnTe-CNT-G and SnTe electrodes at a current density of 200 mA g−1 after 100 cycles. (e) SEM images of the SnTe-CNT-G electrode at 200 mA g−1 after 100 cycles. (f) Schematic diagram of the storage mechanism of the SnTe-CNT-G anode. | ||
To investigate the structural stability of all the samples, FESEM was used to detect the structure of the SnTe, SnTe-G and SnTe-CNT-G electrodes at 200 mA g−1 in the fully charged and fully discharged states after 100 cycles. Fig. 6e shows the SEM images of the SnTe-CNT-G electrode; note that the electrode still maintains a hierarchical structure. Compared with the fresh electrode (Fig. S14†), the SnTe-CNT-G electrode lacks structural damage and has fewer by-products. However, the structure of the SnTe electrode changed dramatically, due to reunion and crushing (Fig. S15†), which ultimately resulted in a low electron transport efficiency, poor rate capacity and large capacity attenuation. Similarly, the SEM images of the SnTe-G electrode are compared in Fig. S16;† the relatively complete structure of the SnTe-G electrode after discharge indicated that the carbon layer can provide a sufficient barrier to relieve volume expansion. Unfortunately, there are still many discharge products, which may be caused by the lack of ion transmission channels.
Based on the above discussion, the notable lithium-ion battery performance of the SnTe-CNT-G anode could be attributed to contributions from its unique hierarchical structure. As illustrated in Fig. 6f, SnTe-CNT-G composite material has the following advantages. (1) SnTe is an intermetallic compound of Sn and Te, which increases the conductivity of active nanoparticles and has a large volume as an electrode of LIBs.59 (2) Carbon nanotubes and graphite have high conductivity, good structural stability, adjustable surface functionality and excellent mechanical properties, which can provide better lithium battery performance.60 (3) The three-dimensional cross-linked structure the CNT-G hybrid matrix promotes electron transport and Li+-ion diffusion, thereby enhancing the electrochemical performance of the SnTe-CNT-G electrode.61
4. Conclusions
In summary, a robust and novel hierarchical nanostructure of a SnTe-CNT-G electrode has been successfully fabricated via a facile ball-milling means. Interestingly, carbon nanotubes coupled with graphite to support the SnTe architecture are able to buffer volume expansion, facilitate the penetration of electrolyte, enhance the transfer of ions/electrons and protect the layered SnTe from being cracked and pulverized. In addition, SnTe with a layered structure and its topological transformation provides the electrode with more active sites and increases the reversible capacity. Due to these advantages, the SnTe-CNT-G electrode shows an unparalleled initial Coulomb efficiency (75.8%, 200 mA g−1), remarkable rate property and long cycling stability (669 mA h g−1 after 1400 cycles). In addition, we systematically confirmed the electrochemical reaction mechanism and electrode structure stability. This work not only opens up opportunities to design electrodes for different alloy-type materials, but also provides a promising strategy to investigate the reaction mechanisms of various energy-storage materials.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21671136 and 21878189), Guangdong Basic and Applied Basic Research Foundation (2020A1515010379), Shenzhen Science and Technology Project Program (JCYJ20190808144413257, JCYJ20180305125729925, JCYJ20170818092720054 and JCYJ20190808145203535), China Postdoctoral Science Foundation (2019M663074) and the Instrumental Analysis Center of Shenzhen University.Notes and references
- H. Li, Z. Wang, L. Chen and X. Huang, Adv. Mater., 2009, 21, 4593–4607 CrossRef CAS.
- J. Chen, H. Zhou, H. Chen, B. An, L. Deng, Y. Li, L. Sun, X. Ren and P. Zhang, J. Electrochem. Soc., 2019, 166, A3820–A3829 CrossRef CAS.
- H. Chen, L. Deng, S. Luo, X. Ren, Y. Li, L. Sun, P. Zhang, G. Chen and Y. Gao, J. Electrochem. Soc., 2018, 165, A3932–A3942 CrossRef CAS.
- H. Jiang, H. Zhang, L. Chen, Y. Hu and C. Li, Small, 2020, 16, 2002351 CrossRef CAS.
- Y. Dong, Z. Zhu, Y. Hu, G. He, Y. Sun, Q. Cheng, I. P. Parkin and H. Jiang, Nano Res., 2020, 14, 74–80 CrossRef.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- L. Fan, X. Li, B. Yan, X. Li, D. Xiong, D. Li, H. Xu, X. Zhang and X. Sun, Appl. Energy, 2016, 175, 529–535 CrossRef CAS.
- M. Huang, H. Chen, J. He, B. An, L. Sun, Y. Li, X. Ren, L. Deng and P. Zhang, Electrochim. Acta, 2019, 317, 638–647 CrossRef CAS.
- Z. Huang, B. Liu, D. Kong, Y. Wang and H. Yang, Energy Storage Mater., 2018, 10, 92–101 CrossRef.
- H. Chen, J. He, G. Ke, L. Sun, J. Chen, Y. Li, X. Ren, L. Deng and P. Zhang, Nanoscale, 2019, 11, 16253–16261 RSC.
- L. Chen, H. Jiang, H. Jiang, H. Zhang, S. Guo, Y. Hu and C. Li, Adv. Energy Mater., 2017, 7, 1602782 CrossRef.
- H. Chen, J. He, Y. Li, S. Luo, L. Sun, X. Ren, L. Deng, P. Zhang, Y. Gao and J. Liu, J. Mater. Chem. A, 2019, 7, 7691–7700 RSC.
- Z. Deng, H. Jiang and C. Li, Small, 2018, 14, 1800148 CrossRef.
- H. Kim, M. Kim, Y. H. Yoon, Q. H. Nguyen, I. T. Kim, J. Hur and S. G. Lee, Electrochim. Acta, 2019, 293, 8–18 CrossRef CAS.
- Y. Wei, L. Huang, J. Chen, Y. Guo, S. Wang, H. Li and T. Zhai, ACS Appl. Mater. Interfaces, 2019, 11, 41374–41382 CrossRef CAS.
- R. Gusmao, Z. Sofer, J. Luxa and M. Pumera, ACS Sustainable Chem. Eng., 2019, 7, 15790–15798 CrossRef CAS.
- B. R. Dahal, R. P. Dulal, I. L. Pegg and J. Philip, Solid State Commun., 2017, 253, 42–45 CrossRef CAS.
- A. R. Park and C. M. Park, ACS Nano, 2017, 11, 6074–6084 CrossRef CAS.
- K.-H. Nam, G.-K. Sung, J.-H. Choi, J.-S. Youn, K.-J. Jeon and C.-M. Park, J. Mater. Chem. A, 2019, 7, 3278–3288 RSC.
- Y. Wei, J. Chen, S. Wang, X. Zhong, R. Xiong, L. Gan, Y. Ma, T. Zhai and H. Li, ACS Appl. Mater. Interfaces, 2020, 12, 16264–16275 CrossRef CAS.
- W. Li, X. Sun and Y. Yu, Small Methods, 2017, 1, 1600037 CrossRef.
- M. Liu, P. Zhang, Z. Qu, Y. Yan, C. Lai, T. Liu and S. Zhang, Nat. Commun., 2019, 10, 3917 CrossRef.
- S. Y. Son, J. Hur, K. H. Kim, H. B. Son, S. G. Lee and I. T. Kim, J. Power Sources, 2017, 365, 372–379 CrossRef CAS.
- Z. Wu, G. Liang, W. K. Pang, T. Zhou, Z. Cheng, W. Zhang, Y. Liu, B. Johannessen and Z. Guo, Adv. Mater., 2020, 32, e1905632 CrossRef.
- N. Ma, X. Y. Jiang, L. Zhang, X. S. Wang, Y. L. Cao and X. Z. Zhang, Small, 2018, 14, e1703680 CrossRef.
- J. E. Moore, Nature, 2010, 464, 194–198 CrossRef CAS.
- Y. Wang, Y. Zhang, H. Li, Y. Peng, J. Li, J. Wang, B.-J. Hwang and J. Zhao, Chem. Eng. J., 2018, 332, 49–56 CrossRef CAS.
- S.-K. Park, G. D. Park and Y. C. Kang, Nanoscale, 2018, 10, 11150–11157 RSC.
- M. Buzaglo, I. P. Bar, M. Varenik, L. Shunak, S. Pevzner and O. Regev, Adv. Mater., 2017, 29, 1603528 CrossRef.
- D. Sun, D. Ye, P. Liu, Y. Tang, J. Guo, L. Wang and H. Wang, Adv. Energy Mater., 2018, 8, 1702383 CrossRef.
- H. Chen, G. Ke, X. Wu, W. Li and X. Ren, Chem. Eng. J., 2020, 406, 126775 CrossRef.
- G. Ke, H. Chen, J. He, X. Wu, Y. Gao, Y. Li, H. Mi, Q. Zhang, C. He and X. Ren, Chem. Eng. J., 2021, 403, 126251 CrossRef CAS.
- X. Liu, C. Lai, Z. Xiao, S. Zou, K. Liu, Y. Yin, T. Liang and Z. Wu, ACS Appl. Energy Mater., 2019, 2, 3185–3193 CrossRef CAS.
- Y. Liu, C. Yang, Q. Pan, Y. Li, G. Wang, X. Ou, F. Zheng, X. Xiong, M. Liu and Q. Zhang, J. Mater. Chem. A, 2018, 6, 15162–15169 RSC.
- Z. Wu, X. Cheng, D. Tian, T. Gao, W. He and C. Yang, Chem. Eng. J., 2019, 375, 121997 CrossRef CAS.
- J. Hu, Z. Xu, X. Li, S. Liang, Y. Chen, L. Lyu, H. Yao, Z. Lu and L. Zhou, J. Power Sources, 2020, 462, 228098 CrossRef CAS.
- X. Hu, B. Shang, T. Zeng, Q. Peng, G. Li, Y. Zou and Y. Zhang, Nanoscale, 2019, 11, 23268–23274 RSC.
- Y. Cheng, Z. Yi, C. Wang, Y. Wu and L. Wang, Chem. Eng. J., 2017, 330, 1035–1043 CrossRef CAS.
- H. X. Dang, K. C. Klavetter, M. L. Meyerson, A. Heller and C. B. Mullins, J. Mater. Chem. A, 2015, 3, 13500–13506 RSC.
- J. Zhang, Y.-X. Yin, Y. You, Y. Yan and Y.-G. Guo, Energy Technol., 2014, 2, 757–762 CrossRef CAS.
- L. Xu, C. Kim, A. K. Shukla, A. Dong, T. M. Mattox, D. J. Milliron and J. Cabana, Nano Lett., 2013, 13, 1800–1805 CrossRef CAS.
- J. U. Seo, G. K. Seong and C. M. Park, Sci. Rep., 2015, 5, 7969 CrossRef CAS.
- Y. Liu, J. Wang, Y. Xu, Y. Zhu, D. Bigio and C. Wang, J. Mater. Chem. A, 2014, 2, 12201–12207 RSC.
- W. Li, J. Huang, L. Feng, L. Cao, Y. Liu and L. Pan, J. Power Sources, 2018, 398, 91–98 CrossRef CAS.
- B. Zhang, G. Xia, W. Chen, Q. Gu, D. Sun and X. Yu, ACS Nano, 2018, 12, 12741–12750 CrossRef CAS.
- D. A. Grishanov, A. A. Mikhaylov, A. G. Medvedev, J. Gun and O. Lev, Energy Technol., 2017, 6, 127–133 CrossRef.
- Z. Lei, L. Xu, Y. Jiao, A. Du, Y. Zhang and H. Zhang, Small, 2018, 14, e1704410 CrossRef.
- M. Li, Q. Deng, J. Wang, K. Jiang, Z. Hu and J. Chu, Nanoscale, 2018, 10, 741–751 RSC.
- X. Li, X. Sun, Z. Gao, X. Hu, R. Ling, S. Cai, C. Zheng and W. Hu, Nanoscale, 2018, 10, 2301–2309 RSC.
- L. Shi, D. Li, P. Yao, J. Yu, C. Li, B. Yang, C. Zhu and J. Xu, Small, 2018, 14, 1802716 CrossRef.
- B. Sheng, L. Wang, H. Huang, H. Yang, R. Xu, X. Wu and Y. Yu, Small, 2020, 16, 2005272 CrossRef CAS.
- D. Gao, Z. Zeng, H. Mi, L. Sun, X. Ren, P. Zhang and Y. Li, J. Mater. Chem. A, 2019, 7, 23964–23972 RSC.
- K. Wang, Y. Wang, Y. Zhang, F. Liu, J. Shi, S. Liu, X. Xie, G. Cao and A. Pan, Nanoscale, 2020, 12, 12623–12631 RSC.
- S. W. Fan, G. D. Li, F. P. Cai and G. Yang, Chem. – Eur. J., 2020, 26, 8579–8587 CrossRef CAS.
- H. Bi, X. Li, J. Chen, L. Zhang and L. Bie, J. Mater. Sci.: Mater. Electron., 2020, 31, 22224–22238 CrossRef CAS.
- G. D. Park, J.-K. Lee and Y. Kang, J. Mater. Chem. A, 2017, 5, 25319–25327 RSC.
- H. Liu, X. Zhang, Y. Zhu, B. Cao, Q. Zhu, P. Zhang, B. Xu, F. Wu and R. Chen, Nano-Micro Lett., 2019, 11, 65 CrossRef CAS.
- H. Yin, W. Shen, H.-Q. Qu, C. Li and M.-Q. Zhu, Nano Energy, 2020, 70, 104468 CrossRef CAS.
- M. V. Kovalenko, W. Heiss, E. V. Shevchenko, J.-S. Lee, H. Schwinghammer, A. P. Alivisatos and D. V. Talapin, J. Am. Chem. Soc., 2007, 129, 11354–11355 CrossRef CAS.
- S. Zeng, X. Zhou, B. Wang, Y. Feng, R. Xu, H. Zhang, S. Peng and Y. Yu, J. Mater. Chem. A, 2019, 7, 15774–15781 RSC.
- J. Zhang, Z. Xie, W. Li, S. Dong and M. Qu, Carbon, 2014, 74, 153–162 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: SEM images, XPS spectra, CV curves and discharge/charge profiles, etc. See DOI: 10.1039/d0nr07355e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |