
A current overview of the oxidative desulfurization of fuels utilizing heat and solar light: from materials design to catalysis for clean energy
Xian Bin
Lim
 ab and
Wee-Jun
Ong
*abc
ab and
Wee-Jun
Ong
*abc
aSchool of Energy and Chemical Engineering, Xiamen University Malaysia, Selangor Darul Ehsan 43900, Malaysia. E-mail: weejun.ong@xmu.edu.my
bCenter of Excellence for NaNo Energy & Catalysis Technology (CONNECT), Xiamen University Malaysia, Selangor Darul Ehsan 43900, Malaysia
cCollege of Chemistry and Chemical Engineering, Xiamen University, Xiamen 361005, China
First published on 6th May 2021
Abstract
The ceaseless increase of pollution cases due to the tremendous consumption of fossil fuels has steered the world towards an environmental crisis and necessitated urgency to curtail noxious sulfur oxide emissions. Since the world is moving toward green chemistry, a fuel desulfurization process driven by clean technology is of paramount significance in the field of environmental remediation. Among the novel desulfurization techniques, the oxidative desulfurization (ODS) process has been intensively studied and is highlighted as the rising star to effectuate sulfur-free fuels due to its mild reaction conditions and remarkable desulfurization performances in the past decade. This critical review emphasizes the latest advances in thermal catalytic ODS and photocatalytic ODS related to the design and synthesis routes of myriad materials. This encompasses the engineering of metal oxides, ionic liquids, deep eutectic solvents, polyoxometalates, metal–organic frameworks, metal-free materials and their hybrids in the customization of advantageous properties in terms of morphology, topography, composition and electronic states. The essential connection between catalyst characteristics and performances in ODS will be critically discussed along with corresponding reaction mechanisms to provide thorough insight for shaping future research directions. The impacts of oxidant type, solvent type, temperature and other pivotal factors on the effectiveness of ODS are outlined. Finally, a summary of confronted challenges and future outlooks in the journey to ODS application is presented.
 Xian Bin Lim | Xian Bin Lim is currently a research student at the School of Energy and Chemical Engineering at Xiamen University, Malaysia under the supervision of Assoc. Prof. Dr Wee-Jun Ong. His current research interests include the design of catalysts for oxidative desulfurization, photocatalysis and energy conversion. |
 Wee-Jun Ong | Wee-Jun Ong received his BEng and PhD in Chemical Engineering from Monash University. He is an Associate Professor at the School of Energy and Chemical Engineering at Xiamen University, Malaysia. In 2021, he became a Director of “CONNECT” research center. Previously, he was a scientist at the Agency for Science, Technology and Research (A*STAR), Singapore. In 2019, he was a visiting scientist at Technische Universität, Dresden and a visiting professor at Lawrence Berkeley National Laboratory. At present, he serves as the Chief Editor of Frontiers in Nanotechnology, Associate Editor of Frontiers in Chemistry, Beilstein Journal of Nanotechnology and Engineering Reports, and Board Member of Materials Horizons, Nano Research, Langmuir, Chinese Journal of Catalysis, Scientific Reports and Nanotechnology. His research interests include nanomaterials for advanced oxidative reactions, photocatalytic, photoelectrocatalytic and electrochemical processes, and energy storage devices. For more details, refer to https://sites.google.com/site/wjongresearch/. |
1. Introduction
Moving into the 21st century, meteoric population growth and industrialization have kindled the incessant proliferation of global energy demand. As the paramount energy source under current technologies, fossil fuels have been substantially consumed for energy production and transportation, leading to the colossal release of hazardous gases, including sulfur oxides (SOx), nitrogen oxides (NOx) and carbon oxides, into the atmosphere. Such increased emissions have signaled the advent of an environmental crisis, eliciting the attention of the global community. Currently, SOx emission is a terrifying environmental issue because of its contribution to acid rain and particle pollution, which engender unceasing and deleterious impacts on the entire ecosystem and human health.1–3 In catalysis, the existence of sulfur contaminants in fossil fuels also triggers the deactivation of valuable metallic catalysts such as nickel and palladium that are typically utilized in automobile exhaust purification and the catalytic reforming process, thereby causing poor catalytic performance as well as enormous financial damage.4 More significantly, the combustion of heavy fuel (e.g. diesel with a higher degree of sulfur content) greatly aggravates these impacts on the world.5 Embracing the preservation of biodiversity as well as the health and welfare of human communities, developed countries like the United States and European Union members have gradually tightened environmental regulations to control the sulfur content in diesel fuels and gasolines and enforce the utilization of ultra-low-sulfur fuels (≤10 ppm) in transportation and industry.6 To safeguard the vitality of our planet and future generations and fulfill stringent regulations, an efficacious desulfurization technology is mandatory in the oil refinery industry to curb the sulfur level in fuels.Over the past few decades, hydrodesulfurization (HDS) has been extensively applied to attenuate environmental stress and the effect of sulfur poisoning on oil processing.7,8 In the HDS of gasoline, the organosulfur constituents such as thiols, sulfides and disulfides are fully converted into hydrocarbons and hydrogen sulfide (H2S) gas by interacting with H2 gases over transition metal catalysts at 300–450 °C and 10–250 atm.9,10 However, in the case of diesel fuel, a lower HDS efficiency is obtained due to the emergence of refractory sulfur compounds, namely thiophene (Th), benzothiophene (BT), dibenzothiophene (DBT) and other thiophene derivatives, which possess outstanding stability in their aromatic chemical structure as well as strong steric hindrance effects on the active sites of the catalyst.11 Accordingly, the removal of these aromatic sulfur compounds from heavy fuels through HDS necessitates harsher reaction conditions and a more active catalyst to overcome the strong C–S bond energy. The reactivity order of various sulfur compounds in fuels is presented in Fig. 1. Obviously, the harsh operational conditions and the release of hazardous H2S gas in HDS have limited its prospects in sustainable chemistry. Also, the mediocre efficiency of sulfur removal, the immense energy expenses and investment capital required and the dangerous process have set a firm barrier against the industrial production of ultra-low-sulfur diesels via HDS. Therefore, a productive, economical, streamlined and low-risk desulfurization method is continually being pursued to replace HDS at an industrial scale.
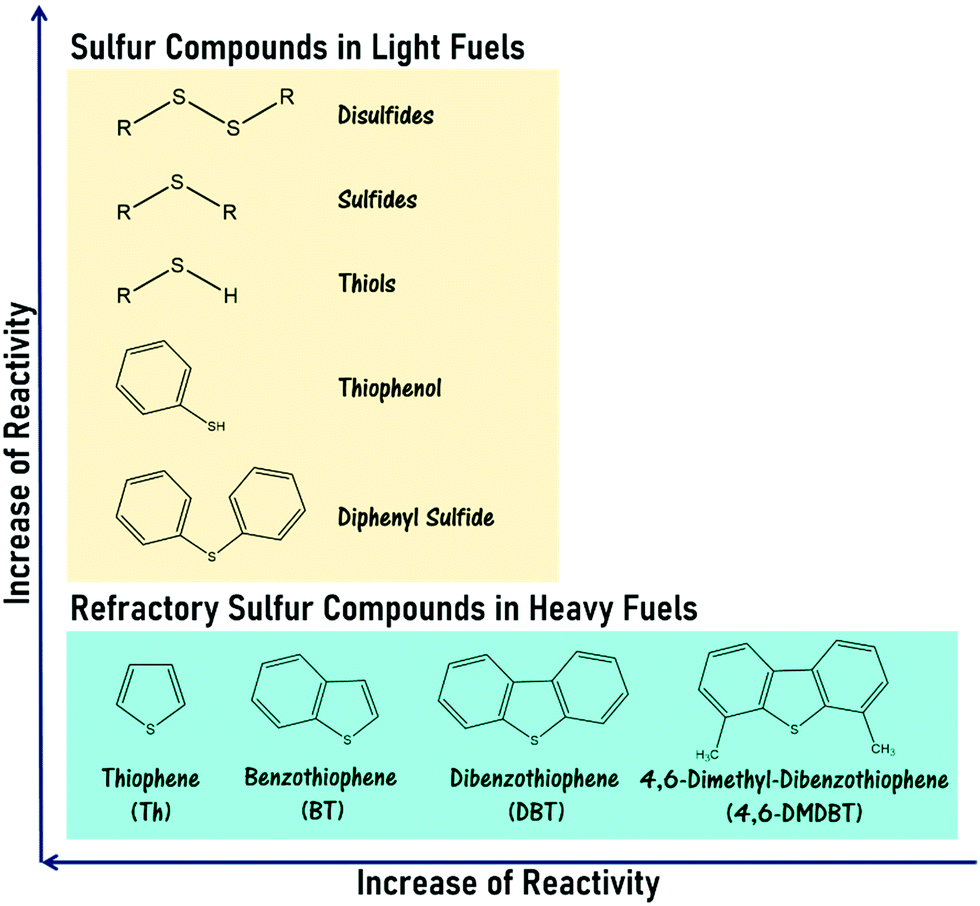 | ||
| Fig. 1 Sulfur compounds and refractory sulfur compounds, which are found in light fuels and heavy fuels, respectively, in the order of reactivity in HDS.12–14 | ||
Benefitting from the arduous efforts of researchers, several potential fuel desulfurization technologies have been discovered, paving the way toward a pollution-free Earth. The alternative desulfurization methods are adsorptive desulfurization (ADS),15–17 biodesulfurization (BDS),18 extractive desulfurization (EDS)19,20 and oxidative desulfurization (ODS),21 which have been thoroughly reviewed in the literature to date. Among the frontline desulfurization routes, ODS has recently become a research hotspot, attributed to its attractive advantages such as moderate operating conditions, magnificent selectivity, utilization of safe and low-priced oxidant and prominent efficiency in removing aromatic sulfur constituents from diesel fuels.22–24 For instance, scientists showed that the utilization of ODS to purify diesel fuel achieved up to 92% sulfur removal, which was far superior to the EDS method which attained only 45%.25 In addition to the appealing merits, the burgeoning market demand for ultra-low-sulfur fuels due to strict environmental regulations and the limitations of the ultra-deep HDS process have ignited an abrupt surge of research interest in the ODS of fuels, especially in the past 10 years. This can be seen in the Web of Science database with a search keyword of “oxidative desulfurization”. As depicted in Fig. 2, the number of publications on ODS has progressively grown from 2011 to 2020. The citations of ODS works have also increased drastically since 2012. It is anticipated that the soaring trend of ODS research will persist in the future. This has also unambiguously highlighted the significance of ODS and the rising urgency for more environmentally-friendly and sustainable desulfurization technologies in the field of environmental remediation. However, there is still progress to be made to commercialize ODS given the limited understanding of the modification strategies to revamp the intrinsic features of ODS catalysts to achieve maximum catalytic activity. Additionally, several drawbacks, such as the transient stability and life cycles of catalysts, lengthy reaction time and high consumption of oxidant, have markedly hindered the progress of ODS commercialization. In this context, additional studies on ODS are urgently needed to alleviate these issues and create a more sustainable ODS process.
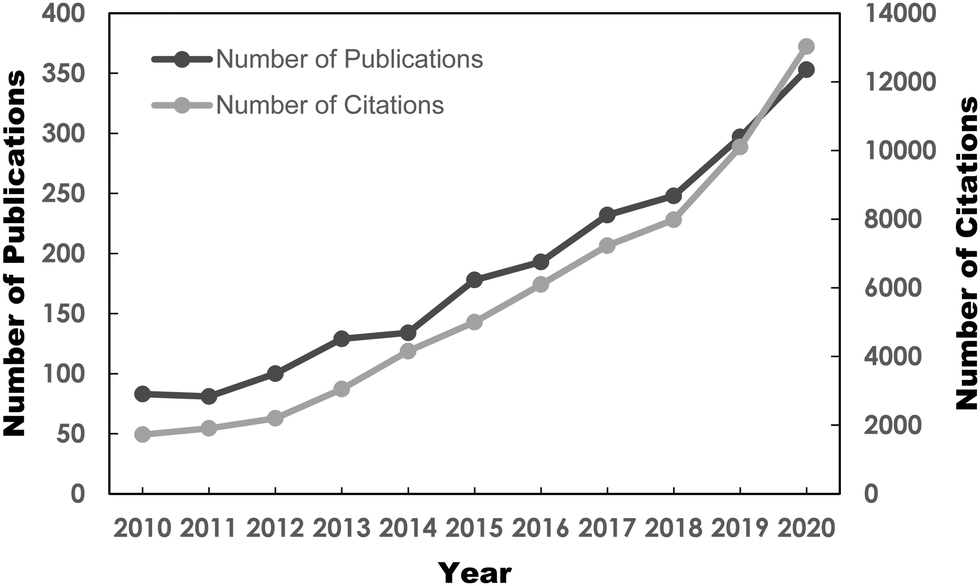 | ||
| Fig. 2 Number of published reports and citations on ODS from 2010 to 2020, sourced from Web of Science database with the topic keyword “oxidative desulfurization” on 3rd May 2021. | ||
The fact that catalysts have been denoted as the pivotal factor in accomplishing an efficient ODS process has caused scholars in catalytic science to dive deep into the intrinsic properties of myriad materials to seek prospective materials with outstanding catalytic activity in ODS. To date, copious review papers have been published to provide mechanistic insights into the application of different materials in ODS driven by thermal or light energy. Rajendran et al. systematically described the significant relationships between the catalysts, operating parameters and reaction mechanisms in ODS under different classifications.26 In the work of Zhou and co-workers, the salient challenges in photocatalytic ODS, including rapid charge carrier recombination rate and deficient light absorption capability, along with the countermeasures were reported to guide the future development of photocatalytic ODS.27 The catalytic performances of various homogeneous and heterogeneous ODS catalysts have also been critically discussed and compared in several reports.28–30 The applications of ionic liquids (ILs),31–33 polyoxometalates (POMs),34 metal–organic frameworks (MOFs)35,36 and graphene11 in ODS have also been individually reviewed. Although these scientific reviews have encapsulated a wide panorama of the innumerable ODS applications, the structure–activity connection of multiple materials, which is of vital importance to direct material engineering for shaping catalysts with extraordinary catalytic activity and stability in ODS, has not yet been completely unraveled. To the best of our knowledge, an encyclopedic summary of the synergistic effect on ODS performance from the hybridization of many materials with unique characteristics has not been published in the literature of ODS.
Additionally, a limited review article targets the configuration of the catalyst for ODS. To develop a pragmatic and effective ODS process for industrial application, rational design of the catalyst is a crucial piece of the puzzle. The reaction parameters in a synthesis approach have a huge impact on the physico-chemical properties of catalysts and further influence their catalytic performance in ODS. None of the previous scholars have covered thermal-driven and light-driven ODS in a single review paper. This is very useful to lay a substantial knowledge foundation on light-driven ODS, contributing a groundbreaking reference for designing versatile catalysts which are applicable for both thermal-driven and light-driven ODS processes. More importantly, the incorporation of inexhaustible photon energy in various applications, especially in the realms of environmental protection and energy production, was reported as one of the future trending research directions in the World Economic Forum 2020.37 These knowledge gaps have greatly invigorated us to compose another useful ODS review.
In this review, the core theories and fundamental mechanisms of thermal-driven and light-driven ODS are first spotlighted to create an overall picture of ODS. In the subsequent sections, the focal point is the influences of synthesis pathways and modification strategies toward ameliorated physico-chemical properties, including the morphologies, compositions, topographies and optical properties of myriad catalysts together with their corresponding catalytic performances in ODS. The ODS catalysts are categorized according to their species as metal oxides, ILs, deep eutectic solvents (DESs), POMs, MOFs, metal-free nanomaterials and their hybrid heterostructured composites, which will be thoroughly discussed in their respective sections. Moreover, theoretical insights into the reaction mechanisms of different materials in thermal-driven and light-driven ODS will be elucidated to explore the structure–activity connection. This current review concludes with a concise summary, an overview of the encountered challenges in ODS based on the research works, accompanied by a number of constructive recommendations and viewpoints to proffer fruitful guidance which will be conducive to the commercialization of ODS in years to come. In short, the predominant objectives of this critical review are to recount the ongoing innovations in ODS in the topics of synthesis approach, catalyst design, properties, reaction pathways and catalytic performance, and to serve as a useful scaffold for future research directions on ODS (Fig. 3).
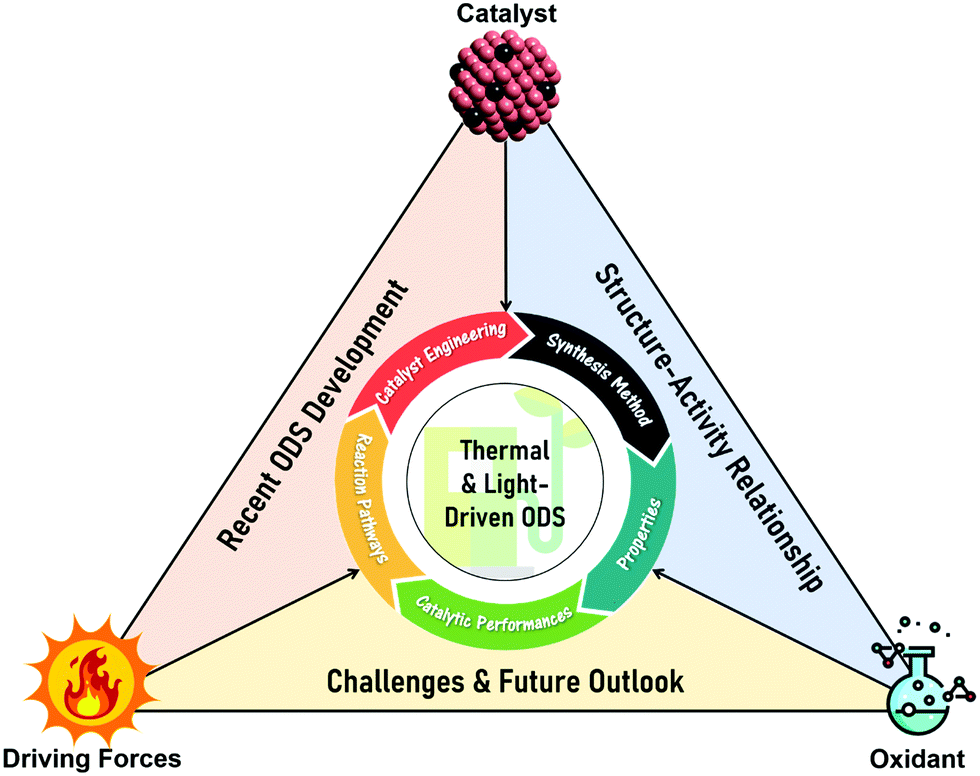 | ||
| Fig. 3 Graphical synopsis of the five predominant focal points, including engineering, synthesis, properties, catalytic performance and reaction mechanisms of catalysts, in contributing insights into recent ODS advancements, the structure–activity relationship of catalysts and the challenges and future outlook in thermal and light-driven ODS systems. | ||
2. Oxidative desulfurization (ODS) process
ODS is a two-step process that begins with the oxidation of aromatic sulfur compounds (Fig. 4) in the presence of an appropriate oxidizing agent such as H2O2 and O2, followed by the separation of the oxidized sulfur species from the fuels through solvent extraction or distillation.29 Owing to the sluggish oxidation rate, catalysis is typically integrated into ODS to boost the reaction kinetics by lowering the requisite activation energy for the conversion of the oxidant into the corresponding reactive oxygen species. The theory of ODS involves the chemical transformation of sulfur compounds into sulfones, which have an easily removable structure, by modifying their chemical properties. ODS endows increased polarity and reduced boiling point to sulfur compounds by attaching O2 at the S atom.38 This can greatly enhance the miscibility of sulfur compounds in polar solvents and increase the boiling point difference between hydrocarbons and sulfur compounds, thereby facilitating the downstream separation process to yield ultra-low-sulfur fuels. After the incorporation of oxidizing agents and catalysts, the driving force both initiates and accelerates the ODS process. Ascribed to their superiority in accessibility, economy and environmental friendliness, thermal and light energies are the front-runners and are capable of offering abundant dynamism to drive the ODS process under current technology. Additionally, different operational parameters, such as reaction temperature, dosage of catalyst and amount of oxidant, impact the efficiency of ODS and the quality of the purified fuels. The fundamental concepts of thermal-driven ODS and photo-driven ODS are discussed in the next sections.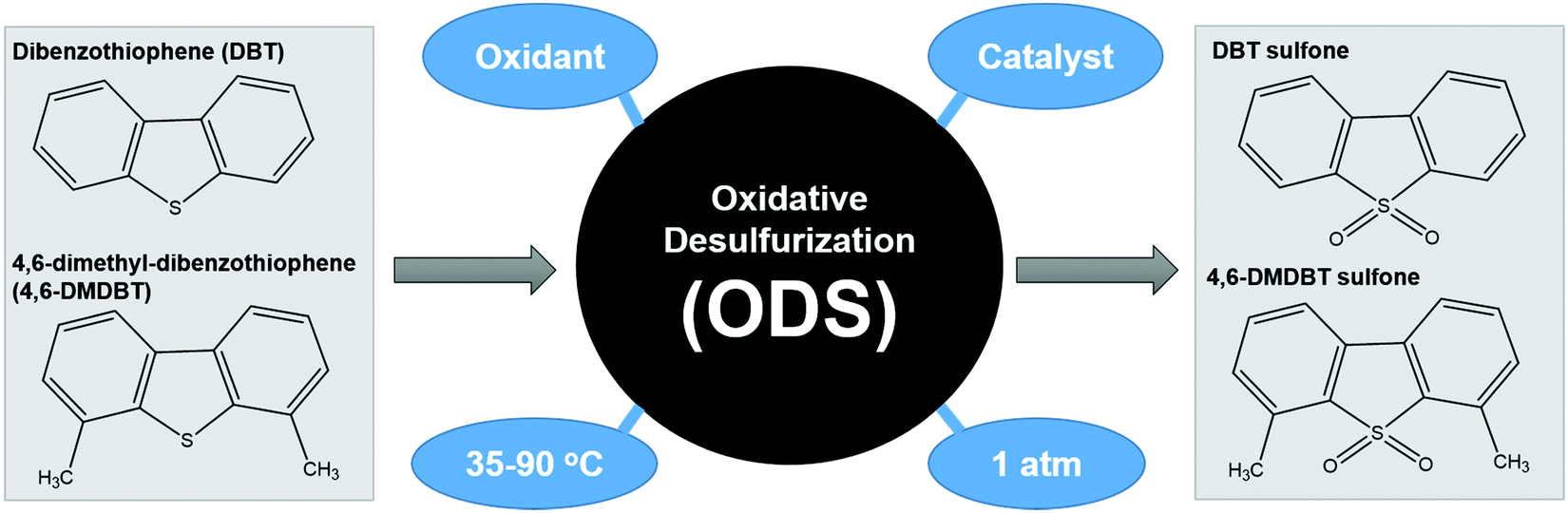 | ||
| Fig. 4 Typical reaction route for ODS of DBT and 4,6-DMDBT. | ||
2.1 Thermal-driven ODS
During thermal-driven ODS, heat energy is incorporated into the ODS process as the source of activation energy to propel the reaction. To summarize the overall idea of thermal-driven ODS, the mechanism for the oxidation of DBT using O2 as an example oxidant is illustrated in Fig. 5a. Following kinetic molecular theory, the kinetic energies of oxidants, sulfur compounds and catalyst are intensified after absorbing thermal energy from the surroundings, which further stimulates effective collisions between particles. The rapid particle collisions and ample supply of activation energy (1) prompt the adsorption of O2 on the surface of the catalyst (2), leading to the conversion of O2 into highly reactive oxygen species. Due to the vigorous affinity of the sulfur atom towards oxygen, DBT is then activated by the active oxygen groups on the surface of catalyst to form S–O bonding, creating DBT sulfoxide (3). As DBT sulfoxide is thermodynamically unstable, it reacts with another active oxygen species to yield DBT sulfone as the final product (4). Lastly, DBT sulfone is desorbed from the catalyst's surface. As mentioned previously, the resulting fuels undergo a specific downstream purification process to eliminate the oxidized sulfur compounds. In comparison with other desulfurization techniques, the reaction temperature of thermal-driven ODS prevalently stays in the range of 35 to 90 °C,38 which is much lower than those in the conventional HDS (300–360 °C)39–41 and ADS approaches (∼350 °C).42 The energy efficiency of thermal-driven ODS tends to preserve operational cost and minimize the safety considerations for the desulfurization process, opening a prospective door to future industrial application.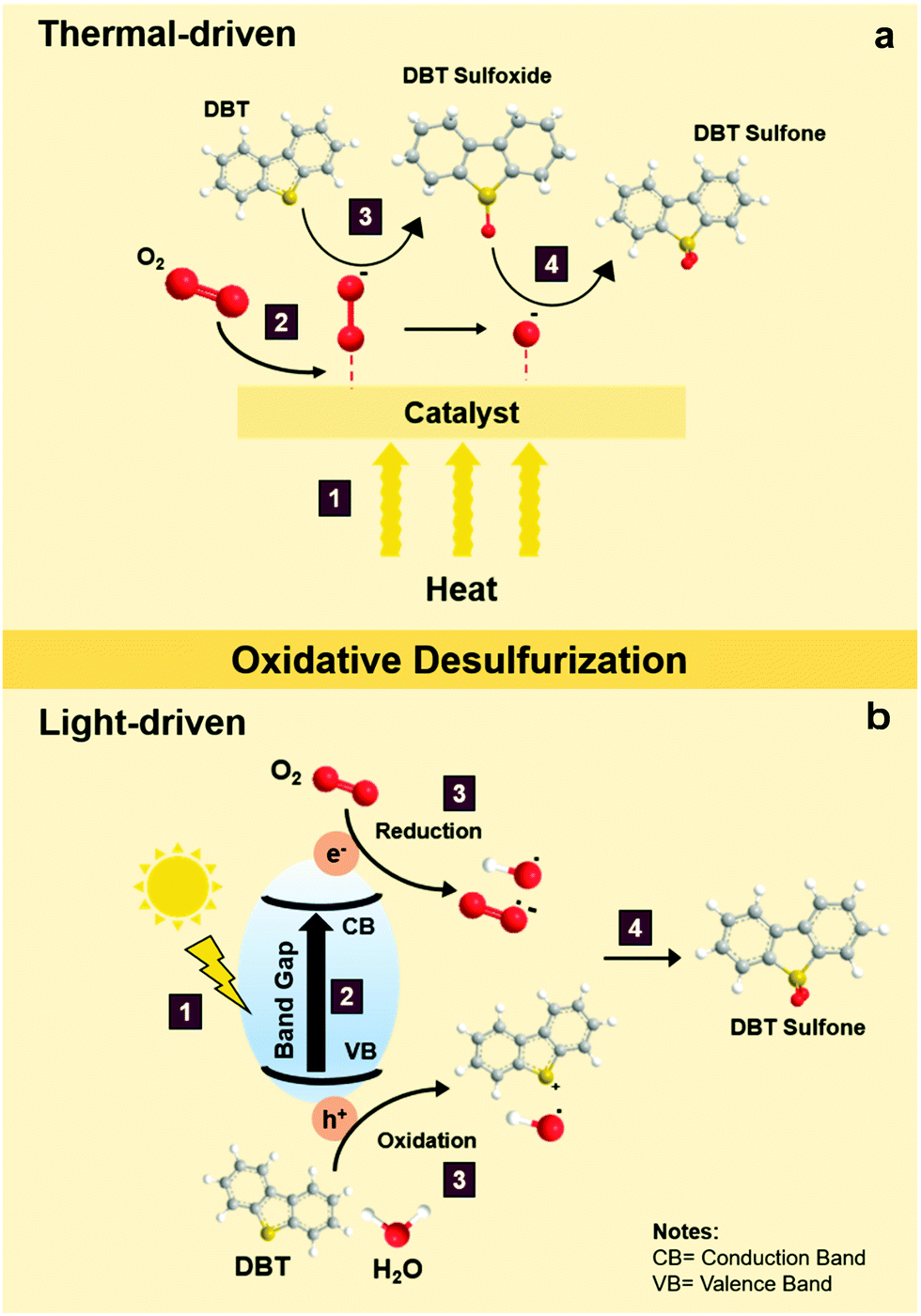 | ||
| Fig. 5 Reaction mechanisms of sulfur removal by DBT via (a) thermal-driven ODS and (b) light-driven ODS. | ||
2.2 Light-driven ODS
In general, light-driven ODS can be described as a combination of photocatalysis and conventional ODS. Certainly, knowledge of photocatalysis is indispensable to unravel the mechanistic insights on light-driven ODS. To build an explicit understanding, the operation of light-driven ODS of DBT with O2 as the oxidant is depicted in Fig. 5b. To commence photocatalysis, a material with light-harvesting property is demanded. Moreover, the photon energy in the incoming light must be equivalent to or higher than the band gap of the photocatalyst to initiate photocatalysis.43 Once the above criteria are fulfilled, light absorption by the photocatalyst takes place (1) upon the illumination of solar light. After the stable electrons in the valence band (VB) receive enough energy from the photons, they are excited and lifted to the conduction band (CB) at a higher energy level (2).44,45 Under this circumstance, electron–hole pairs are momentarily induced and move from the bulk of photocatalyst to the surface to generate electron-rich and hole-rich sites. At the same time, the reactants in the bulk liquid phase diffuse and absorb onto the surface of the photocatalyst.46,47 The adsorbed DBT and H2O molecules readily react with a hole in the VB and form reactive intermediates composed of a radical cation and hydroxyl radicals (˙OH), respectively, through the oxidation process (3).27 Meanwhile, O2 undergoes a reduction process in the CB (3) and transforms into energetic oxygen species including superoxide radicals (˙O2−) and ˙OH radicals (3).48–52 Consequently, the redox reaction is accomplished, followed by the desorption of the products from the surface of the photocatalyst. Ultimately, a reaction between the DBT reactive intermediate and oxygen radicals occurs to produce DBT sulfoxide which is subsequently converted to DBT sulfone (4).In contrast to thermal-driven ODS, light-driven ODS employs photon energy to energize the catalyst in order to carry out redox reactions. Over the last decade, attention has turned to light-driven ODS which is presumed to be the leading edge of desulfurization technology and a viable replacement for conventional desulfurization techniques because of its benefits in economy, sustainability and safety.53,54 Nevertheless, the emergence of several difficult challenges, such as rapid electron–hole recombination, a narrow light absorption range and sluggish light response, has hampered the industrialization of photo-driven ODS. In a later section, the strenuous efforts by previous researchers to tackle these handicaps will be critically discussed.
3. Catalyst design and mechanisms for thermal-driven ODS
To date, impressive progress and outcomes have been gained by adopting the thermal-driven ODS process with a variety of catalysts to desulfurize fuels. Generally, the application of a heterogeneous catalyst in thermal-driven ODS is more favorable than a homogenous catalyst such as acetic acid55 and phosphotungstic acid56,57 due to the remarkable trait of easing the downstream separation process, which reduces the overall capital expenditure for the ODS process. Even so, homogeneous catalysts cannot be neglected completely as they can constitute an extensive mass transfer phase for the ODS reaction, thereby accelerating the rate of reaction. Interestingly, every catalyst exhibits different competence in ODS, which is attributed to differences in natural characteristics and reaction mechanisms. The synthesis approaches, structure–activity relationships and reaction pathways of various catalysts in thermal-driven ODS will be evaluated extensively in the following sections.3.1 Application of metal oxides in ODS
In the field of catalysis, metal oxides with a Lewis acid characteristic are prominent and inveterate catalysts that speed up most important reactions, such as oxidation and acid–base reactions, compensating for the drawback of poor selectivity.58,59 Significantly, metal oxides have been advocated to replace precious metals such as Pt60 and Au61 in performing ODS to counter the high price and restricted availability that are stumbling blocks to their industrial application. In line with previous research, various relatively economical metal oxides, such as MoO3, WO3, Co3O4, MnO2 and so on, have been highlighted as promising candidates for fuel purification via ODS.62–71For the synthesis of metal oxide catalysts, the calcination method has been broadly adopted because of its facile and controllable steps.72 Basically, the formulation of metal oxide catalysts via calcination can be referred to as the activation of metal precursors in the presence of oxygen or air under elevated temperature, resulting in the oxidation of the metal. Due to the variations in metal precursors, other synthesis techniques, such as solvothermal and hydrothermal methods, have been utilized to shape certain metal oxide catalysts, like WO3-based catalysts.66,70 In contrast to the calcination method, the solvothermal and hydrothermal routes exhibit comparably intricate processes but are carried out under lower temperatures. In these synthesis routes, the heating temperature and duration play decisive roles in controlling the nucleation and growth of metallic nanoparticles that crucially affect the morphologies of the resulting metal oxide catalysts. As revealed by Thalgaspitiya et al., WO3 fabricated at a higher calcination temperature and shorter reaction time exhibited a smaller specific surface area because of sintering.73 Eventually, the decrement of active sites brings about lower catalytic activity in ODS. Recently, Zhang et al. impregnated W precursors in the structure of poly(methyl methacrylate) and performed calcination to eliminate the template and oxidize the W to engineer 3D nanoporous WO3 with oxygen defects (3DOM WOx).74 The 3DOM WOx fabricated at different calcination temperatures demonstrated different rates of 4,6-DMDBT oxidation owing to morphology variations. As seen in the SEM images (Fig. 6a and b), 3DOM WOx calcined at 500 °C (3DOM WOx-500) possessed a more disordered porous structure compared with that calcined at 400 °C (3DOM WOx-400) due to the breakdown of the structure from overheating. This led to a lower pore volume and reduced the number of available active sites for ODS. The mass diffusion of sulfur compounds toward the active sites was also restricted. As a result, 3DOM WOx-400, with greater morphological benefits, achieved 99% oxidation of 4,6-DMDBT in 4 h with the continuous injection of air at 120 °C (Fig. 6c). Accordingly, optimization of the calcination temperature is imperative to preserve the advantageous textual properties of metal oxide catalysts and attain outstanding desulfurization activity in ODS.
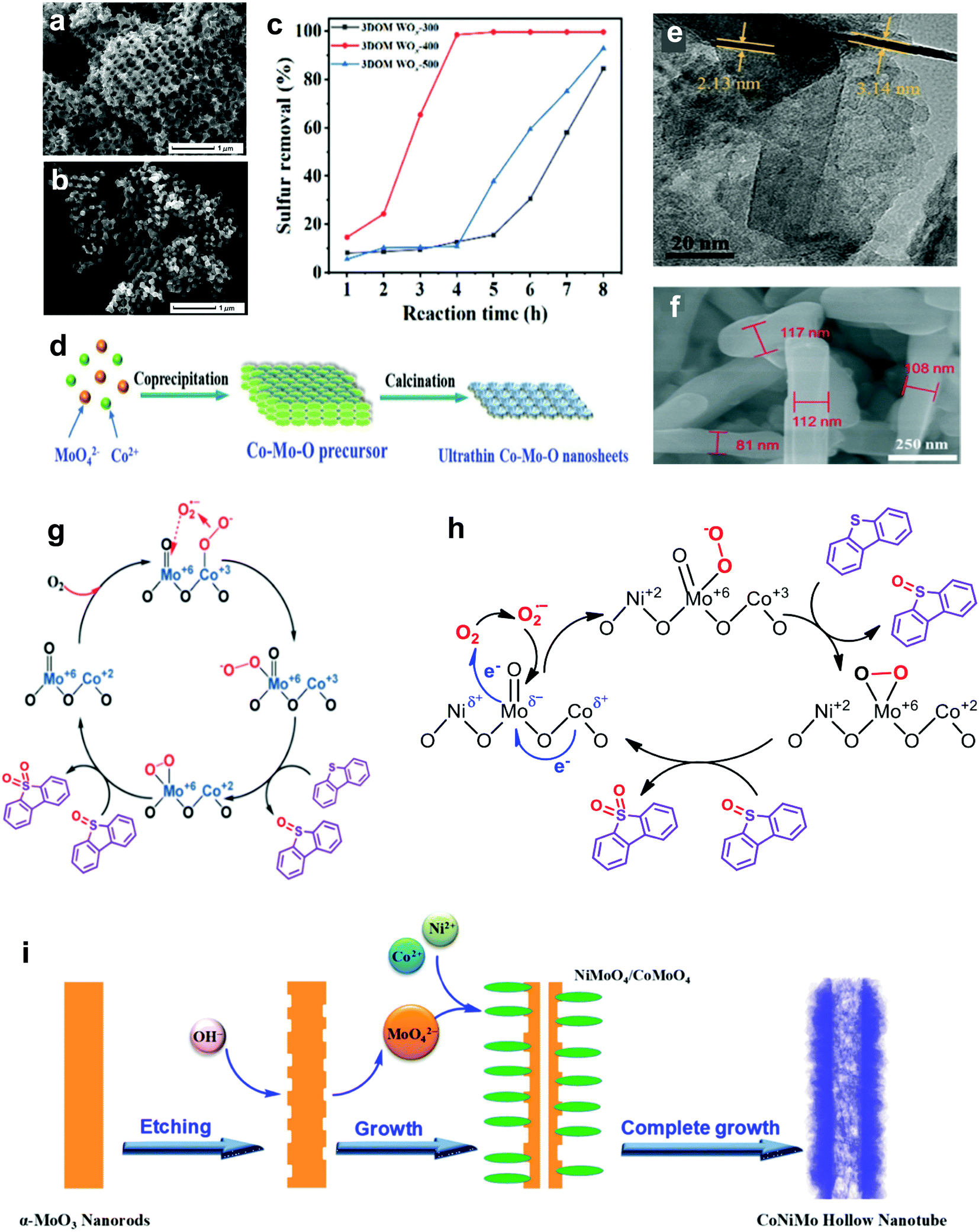 | ||
| Fig. 6 SEM images of (a) 3DOM WOx-400 and (b) 3DOM WOx-500. (c) Removal efficiencies of 4,6-DMDBT using O2 at 120 °C over 3DOM WOx-300, 3DOM WOx-400 and 3DOM WOx-500, synthesized at calcination temperatures of 300, 400 and 500 °C, respectively. Reprinted with permission.74 Copyright 2021, American Chemical Society. (d) Synthesis mechanism and (e) TEM image of Co–Mo–O nanosheet. Reprinted with permission.77 Copyright 2019, Royal Society of Chemistry. (f) SEM image of Co–Mo–O nanorod and (g) its proposed reaction pathway in the ODS of DBT. Reprinted with permission.75 Copyright 2019, Royal Society of Chemistry. (h) Plausible reaction mechanism for ODS of DBT by Co–Ni–Mo–O nanotube and (i) its fabrication route. Reprinted with permission.76 Copyright 2020, American Chemical Society. | ||
3.1.2.1 Aluminum oxide supported metal oxides. In 2011, Jia's group reported the complete oxidation of DBT by MoO3/γ-Al2O3 in merely 15 min at 60 °C.78 In their findings, the utilization of solvents like acetonitrile and methanol crucially reduced the oxidation of DBT (Fig. 7a) due to the existence of adsorption competition between the solvent molecules and DBT on the active sites of MoO3/γ-Al2O3. From the outlook of sustainability, the omission of solvent in ODS is also helpful in cutting down the production of chemical waste and alleviating pollution risk. Thus, Akbari et al. studied the viability of using ultrasound for a similar function as solvent in ODS to enhance the accessibility of the catalyst to the sulfur reactants.63 Impressively, above 98% DBT oxidation was attained using MoO3/Al2O3 and H2O2 in the system. Also, the stability of MoO3/Al2O3 in the ultrasound system (>6 runs) was higher than that in a conventional stirring system (<3 runs). This could be imputed to the elimination of impurities from the surface of MoO3/Al2O3 by the ultrasound, thus inhibiting deactivation of the catalyst due to pore blockage.
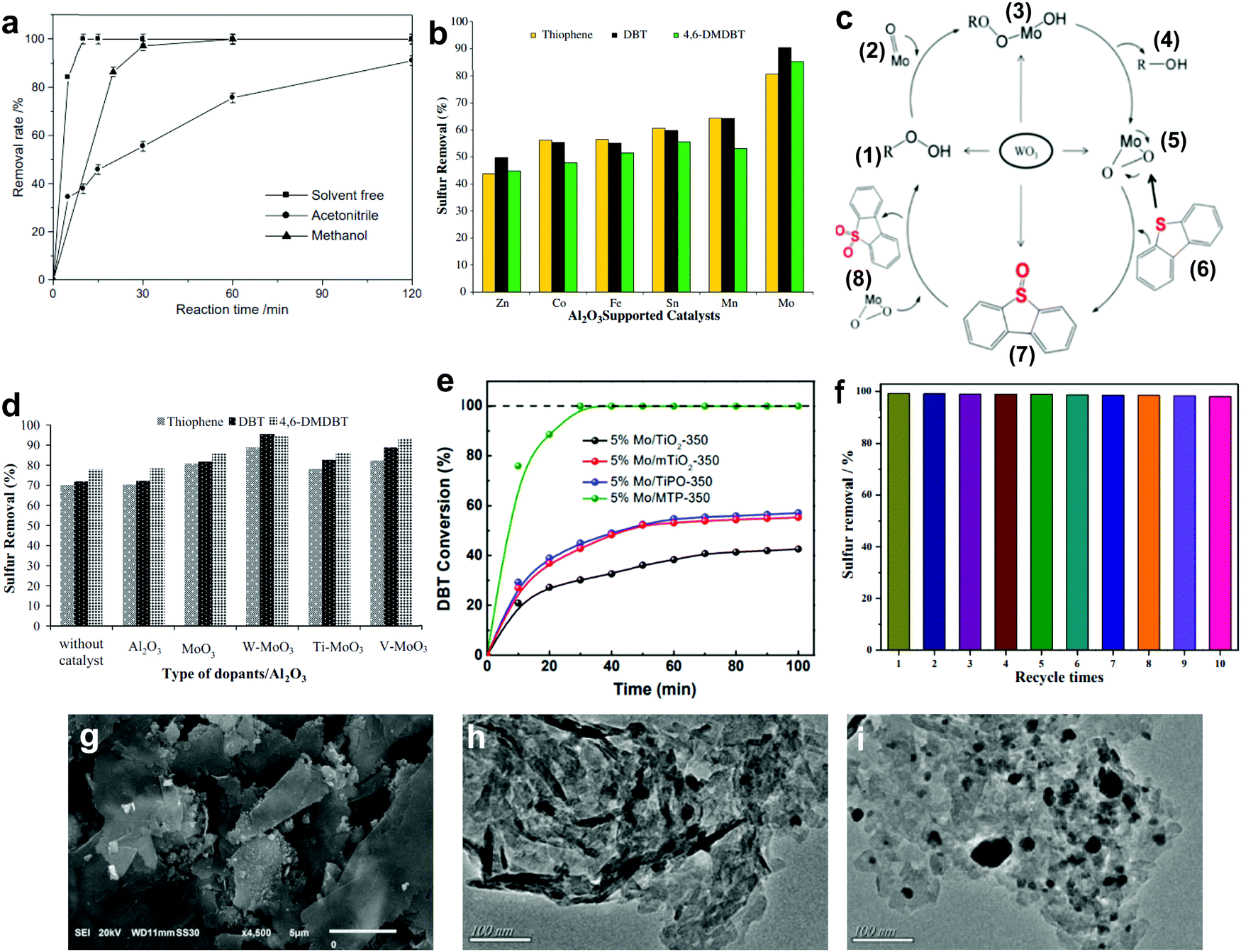 | ||
| Fig. 7 (a) Effect of different solvents on the removal rate of DBT over MoO3/γ-Al2O3 utilizing H2O2 at 60 °C. Reprinted with permission.78 Copyright 2011, Elsevier B.V. (b) Sulfur removal rate in ODS using various metal oxides supported on Al2O3 catalysts at 60 °C in the presence of TBHP. (c) ODS mechanism of DBT for WO3/MoO3/Al2O3 catalyst. Reprinted with permission.62 Copyright 2012, Elsevier B.V. (d) Sulfur removal efficiency of different dopants on MoO3/Al2O3 catalyst at 60 °C in the presence of TBHP. Reprinted with permission.79 Copyright 2018, Elsevier B.V. (e) Conversion of DBT using MoO3 with different supports as catalyst in the presence of TBHP at 40 °C. Reprinted with permission.86 Copyright 2021, Elsevier Inc. (f) DBT removal for ten runs, at 1 h per run, using TiO2/GC with H2O2 at 50 °C and (g) the corresponding SEM image. Reprinted with permission.87 Copyright 2020, American Chemical Society. TEM images of WO3 on (h) bulk g-C3N4 and (i) layered g-C3N4. Reprinted with permission.89 Copyright 2019, Elsevier B.V. | ||
Returning to the work of Abu Bakar et al., the ODS activities of various metal oxides, including Mo, Mn, Sn, Fe, Co and Zn, supported on Al2O3 were examined.62 Among all the prepared catalysts, MoO3/Al2O3 was the best catalyst in ODS with the highest ODS activity in the presence of tert-butyl hydroperoxide (TBHP) at 60 °C (Fig. 7b). Likewise, MoO3/Al2O3 also attained the highest DBT conversion with TBHP compared with other metal oxides at 110 °C in the report of Wang et al.81 The marvelous performance of MoO3/Al2O3 in ODS stems from its superior Lewis acidity and lower oxidation potential in the highest oxidation state of Mo compared to other metal oxides, facilitating the conversion of H2O2 and resulting in abundant active oxygen species for ODS.82 Taking O2 as an example, the stronger Lewis acid feature allows rapid interaction between the Mo and O atoms, leading to partial dissociation of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and accelerating the rate of ODS. Therefore, MoO3 can be labelled the most competent metal oxide catalyst for desulfurizing fuels, attributed to its impressive strength in catalyzing the activation of oxidants which is conducive to the oxidation of aromatic sulfur compounds.
O bond and accelerating the rate of ODS. Therefore, MoO3 can be labelled the most competent metal oxide catalyst for desulfurizing fuels, attributed to its impressive strength in catalyzing the activation of oxidants which is conducive to the oxidation of aromatic sulfur compounds.
In order to develop a breakthrough in ODS activity, several attempts have been made to dope TiO2, V2O5 and WO3 onto MoO3/Al2O3.62,79,83 Among these dopants, only WO3 enhanced the sulfur removal in MoO3/Al2O3, which was about 10% higher than that of the pure MoO3/Al2O3 under the same operating conditions (Fig. 7d). This can be explained by the transfer of electrons from WO3 to MoO3 strengthening the nucleophilicity of MoO3.62,79 From the perspective of catalyst structure, WO3/MoO3/Al2O3, with a smaller particle size than MoO3/Al2O3, possessed a greater proportion of MoO3 particles on the Al2O3 surface which developed robust active sites for ODS. With respect to the reaction mechanism (Fig. 7c), the enhancement of active sites and nucleophilic property promoted the transformation of molybdate (2) to the peroxomolybdate species (5) after the nucleophilic attack of TBHP (1). With this rich active oxygen species, the DBT (6) in diesel fuel was readily oxidized to DBT sulfoxide (7) and DBT sulfone (8), resulting in a higher rate of oxidation. In other words, selecting an appropriate dopant, which is beneficial to improving the nucleophilicity and morphology of pure metal oxides, is pivotal for efficient ODS.
3.1.2.2 Zirconium oxide supported metal oxides. Instead of applying conventional Al2O3 as the support, Muñoz et al. immobilized WO3 on ZrO2 by applying the equilibrium adsorption method and examined the performance of the prepared catalyst in ODS.84 During the catalyst synthesis process, the pH of the solution was well controlled to create a charge difference on the surfaces of WO3 and ZrO2 to facilitate the formation of chemical bonds and the entire adsorption process. The introduction of WO3 on ZrO2 tremendously improved the conversion of diphenyl sulfide into diphenyl sulfone, from 45% to 100%, with H2O2. This impressive enhancement manifested the paramount role of surface acidity in governing the catalytic activity in ODS, as WO3 possesses a higher acid strength. In the report of Xun et al., a hydrothermal and calcination approach was adopted to support WO3 on ZrO2 to catalyze the ODS of model diesel. This work revealed that the increase of calcination temperature constructed a more mesoporous structure in WO3/ZrO2, resulting in the increment of active sites for ODS and the subsequent enhancement of sulfur removal.70 On the downside, the structure of WO3/ZrO2 suffered collapse at extreme calcination temperatures (800 °C) because the excessive thermal energy broke the molecular bonding between the particles. As a consequence, the porosity advantage of WO3/ZrO2 was forfeited, leading to a lower ODS performance.
3.1.2.3 Metal phosphonate supported metal oxides. As an emerging porous nanomaterial, metal phosphonate has attracted enormous research interest in the area of catalysis owing to its tailorable structure, vast surface area and beneficial surface properties.85 The propitious properties of metal phosphonate inspired Chen et al. to support MoO3 with mesoporous titanium phosphonates (MTP) via a simple hydrothermal process to enhance the dispersion of MoO3 and guarantee its catalytic activity in ODS.86 Interestingly, the organophosphonic ligands in MTP served as essential coordinators for anchoring MoO3 nanoparticles to form a uniform dispersion. In comparison with other Ti-based supports, MoO3/MTP achieved the highest DBT conversion rate in the presence of TBHP at 40 °C (Fig. 7e). This was attributed to the valuable substrate–support interaction, which was created by organophosphonic ligands, prevented the agglomeration of MoO3 and simultaneously generated smaller MoO3 nanoparticles with a higher exposure of catalytic sites for ODS. This work not only highlights the importance of substrate–support interactions on the ODS activity of metal oxide-based catalysts but also proposes a novel support for metal oxides.
3.1.2.4 Metal-free nanomaterial supported metal oxides. Xun et al. decorated TiO2 nanoparticles on graphitic carbon (GC) via annealing and calcination.87 From the view of morphology, TiO2 was uniformly anchored on the surface of 2D layered GC (Fig. 7g). Impressively, the activity of TiO2/GC in oxidizing DBT with H2O2 remained at above 98% (Fig. 7f) for ten consecutive runs. This suggests the capability of GC to stabilize or grasp TiO2 nanoparticles firmly through strong chemical interactions, thereby hindering the aggregation of particles and ensuring durability in ODS. Zhao et al. deposited WO3 nanoparticles on g-C3N4 through the calcination method to address the active site shortage in WO3 and applied the prepared composite in ODS.88 Unfortunately, the occurrence of severe WO3 aggregation on the surface of g-C3N4 led to a low specific surface area and limited number of surface active sites. In ODS, WO3/g-C3N4 took 3 h to achieve 91.2% DBT removal with H2O2 at 60 °C. To further enlarge the specific surface area, Ma and co-workers developed a layered g-C3N4 by thermal decomposition to support WO3 in ODS.89 Compared with the morphology of bulk g-C3N4 (Fig. 7h), a relatively minor agglomeration of WO3 nanoparticles occurred on layered g-C3N4 (Fig. 7i) as a result of the beneficial 2D morphology that offers innumerable anchor sites for WO3. Evidently, WO3/layered g-C3N4 showed a higher specific surface area (186.32 m2 g−1) than WO3/bulk g-C3N4 (129.71 m2 g−1). Consequently, WO3/layered g-C3N4, with a greater number of active sites, achieved a higher DBT conversion (100%) with H2O2 at 50 °C in 40 min than did WO3/bulk g-C3N4 (91%). Recently, Wang et al. constructed 2D/2D V2O5/BN nanosheets through the solvothermal method.90 Owing to the high surface to volume ratio in 2D morphology, the V2O5 nanosheets were well-dispersed on the BN nanosheets, which contributed to the increment of surface active sites. The V2O5/BN nanosheet attained higher DBT oxidation (99.6%) than did V2O5/bulk BN (62.3%). The large contact area between V2O5 and BN also established strong 2D/2D chemical interactions, inhibiting the deactivation of the catalyst due to catalyst loss during ODS. Indeed, the fabrication of ultrathin 2D nanomaterials, especially the assembly of 2D/2D nanocomposites, is a rational and effective strategy for improving catalytic activity and stability in ODS.
Support introduction and metal oxide doping are promising strategies for intensifying the catalytic activity of metal oxides in ODS by elevating the exposure of active sites as well as the electron density. Moreover, the operating conditions of the synthesis method play a vital role in controlling the morphology of metal oxides. Hence, metal oxide characteristics should be well tuned to engineer a metal oxide with excellent ODS catalytic performance. Table 1 summarizes the performances of various metal oxide-based catalysts in ODS and the operating parameters.
| Catalysts | Reaction temperature (°C) | Oxidant | Extractant | Performances | Ref. | ||
|---|---|---|---|---|---|---|---|
| Sulfur removal (reactant) | Reaction time (h) | Stability (h) | |||||
| TBHP: tert-butyl hydroperoxide; DMF: dimethylformamide; DBT: dibenzothiophene; WOx–OAm: amphiphilic tungsten oxide with oxygen vacancies (synthesized with oleylamine solvent); WOx–OEt: amphiphilic tungsten oxide with oxygen vacancies (synthesized with ethanol solvent); NMP: N-methyl-2-pyrrolidone; C16–WO3/ZrO2: [C16H33N(CH3)3]4SiW12O40 (ionic liquid)–tungsten trioxide/zirconium dioxide; MTP: mesoporous titanium phosphonates; CNTs: carbon nanotubes; CHYPO: cyclohexanone peroxide. | |||||||
| WO3/MoO3/Al2O3 | 60 | TBHP (70 wt%) | DMF | 100% (DBT) | 1 | >150 | 62 |
| MoO3/Al2O3 | 110 | TBHP (70 wt%) | N/A | 86.13% (DBT) | 3 | N/A | 81 |
| WO3/Al2O3 | 78.76% (DBT) | ||||||
| CrO3/Al2O3 | 3.47% (DBT) | ||||||
| V2O5/Al2O3 | 53.34% (DBT) | ||||||
| Nb2O5/Al2O3 | 43.42% (DBT) | ||||||
| ZrO2/Al2O3 | 33.87% (DBT) | ||||||
| MoO3/Al2O3 | 45 | H2O2 (30 wt%) | N/A | 98% (DBT) | 0.5 | >3 | 63 |
| MoO3/Al2O3 | 70 | H2O2 (30 wt%) | Acetonitrile | ∼80% (DBT) | 2.5 | N/A | 91 |
| WO3/TiO2 | 50 | H2O2 (30 wt%) | N/A | 100% (DBT) | 1 | >7 | 92 |
| V2O5/TiO2 | 60 | H2O2 (30 wt%) | Acetonitrile | 99.5% (DBT) | 0.5 | N/A | 64 |
| V2O5/Al2O3–TiO2 | 97.9% (DBT) | ||||||
| V2O5/SiO2 | 59.8% (DBT) | ||||||
| V2O5/Nb2O5 | 97.7% (DBT) | ||||||
| Ni–MoO3 | 70 | H2O2 (30 wt%) | Acetonitrile | 99.8% (DBT) | 2 | <8 | 93 |
| Mo–Ni–Cu–Zn–Co–OX | 120 | O2 | N/A | 100% (DBT) | 3 | >18 | 65 |
| WOx–OAm | 60 | H2O2 (30 wt%) | N/A | 58.8% (DBT) | 0.67 | >10 | 66 |
| WOx–OAm/HCl | 100% (DBT) | N/A | |||||
| WOx–Et | 9.9% (DBT) | N/A | |||||
| WOx–Et/HCl | 35.1% (DBT) | N/A | |||||
| Ca/MoO3/Al2O3 | 55 | H2O2 (30 wt%) | N/A | ∼100% (DBT) | 0.13 | <1.07 | 67 |
| SnO2 | 60 | H2O2 (30 wt%) | Methanol | 100% (DBT) | 2 | >28 | 68 |
| C16–WO3/ZrO2 | 50 | H2O2 (30 wt%) | Acetonitrile | 100% (DBT) | 1 | >10 | 70 |
| MnO2/Al2O3 | 150 | O2 | DMF | ∼80% (DBT) | 8 | N/A | 71 |
| Acetonitrile | |||||||
| NMP | |||||||
| Methanol | |||||||
| Na2WO4/Al2O3 | 70 | H2O2 (30 wt%) | Acetonitrile | ∼82% (DBT) | 3 | N/A | 80 |
| Na2WO4P1.5/Al2O3 | ∼98% (DBT) | <12 | |||||
| V/W/MoO3/Al2O3 | 60 | TBHP (70 wt%) | DMF | 100% (DBT) | 0.5 | >2.5 | 79 |
| Ti/W/MoO3/Al2O3 | 94.2% (DBT) | N/A | |||||
| W/MoO3/Al2O3 | 95.6% (DBT) | N/A | |||||
| MoO3/Al2O3 | 84.7% (DBT) | N/A | |||||
| MoO3/γ-Al2O3 | 60 | H2O2 (30 wt%) | N/A | 100% (DBT) | 0.25 | <4.5 | 78 |
| V2O5–MoO3/Al2O3 | 60 | TBHP (70 wt%) | Acetonitrile | ∼25% (DBT) | 1 | N/A | 83 |
| 3DOM WOx | 120 | O2 | N/A | 99% (4,6-DMDBT) | 4 | >20 | 74 |
| MoO3/MTP | 40 | TBHP (70 wt%) | N/A | 100% (DBT) | 0.5 | >5 | 86 |
| ZrO2 | 75 | H2O2 (30 wt%) | N/A | 70% (diphenyl sulfide) | 1 | N/A | 84 |
| WO3/ZrO2 | 100% (diphenyl sulfide) | >2 | |||||
| Co2–Mo–O | 120 | O2 | N/A | 100% (DBT) | 3 | >15 | 75 |
| Co2–Mo–O | 100 | 83% (DBT) | 6 | N/A | |||
| Co–Mo–O | 100 | 46% (DBT) | 6 | N/A | |||
| Co–Mo2–O | 100 | 29% (DBT) | 6 | N/A | |||
| Co–Ni–Mo–O | 110 | O2 | N/A | 100% (DBT) | 2 | >14 | 76 |
| Co–Ni–Mo–O | 100 | 100% (DBT) | 4 | N/A | |||
| Ni–Mo–O | 100 | ∼16% (DBT) | 4 | N/A | |||
| Co–Mo–O | 100 | ∼65% (DBT) | 4 | N/A | |||
| Co–Mo–O (nanosheet) | 100 | O2 | N/A | 100% (DBT) | 4 | >18 | 77 |
| Co–Mo–O (bulk) | 21.5% (DBT) | N/A | |||||
| Co–O | 59.3% (DBT) | N/A | |||||
| TiO2/graphitic carbon | 50 | H2O2 (30 wt%) | N/A | 100% (DBT) | 1 | >10 | 87 |
| Pt/bulk V2O5 | 110 | O2 | N/A | 19.7% (DBT) | 5 | N/A | 94 |
| Pt/V2O5 nanosheet | 99.2% (DBT) | >45 | |||||
| 2D V2O5/bulk BN | 120 | O2 | N/A | 62.3% (DBT) | 4 | N/A | 90 |
| 2D/2D V2O5/BN nanosheet | 99.6% (DBT) | >32 | |||||
| WO3/CNTs | 50 | H2O2 (30 wt%) | Acetonitrile | 90.73% (DBT) | 1 | <5 | 95 |
| WO3/g-C3N4 | 60 | H2O2 (30 wt%) | Ionic liquids | 91.2% (DBT) | 3 | >15 | 88 |
| WO3/bulk g-C3N4 | 50 | H2O2 (30 wt%) | N/A | 91% (DBT) | 0.67 | N/A | 89 |
| WO3/layered g-C3N4 | 100% (DBT) | >4 | |||||
| MoO3/MOF-199 | 50 | O2 | N/A | 94.8% (DBT) | 2 | N/A | 96 |
| Fe3O4–MoO3/MOF-199 (spherical MoO3) | 95.1% (DBT) | 1 | N/A | ||||
| Fe3O4–MoO3/MOF-199 (clubbed MoO3) | 98.9% (DBT) | 0.75 | N/A | ||||
| Fe3O4–MoO3/MOF-199 (fibroid MoO3) | 100% (DBT) | 0.75 | <9 | ||||
| MoO3–TiO2/MCM-22 | 100 | CHYPO (50 wt%) | N/A | 99.96% (DBT) | 0.25 | >2 | 97 |
| MoO2/C | 55 | H2O2 (30 wt%) | Acetonitrile | 56% (DBT) | 1 | <1 | 98 |
| N–MoO3/SiC | 99.9% (DBT) | <4 | |||||
| MoO3/MOF-199 | 40 | O2 | [Bmim]BF4 | 76.7% (DBT) | 1 | N/A | 99 |
| W–MoO3/MOF-199 | 100% (DBT) | N/A | |||||
| Fe3O4–W–MoO3/MOF-199 | 100% (DBT) | <20 | |||||
3.2 Application of ionic liquids (ILs) in ODS
Ionic liquids (ILs) are defined as liquids built up from a cation and an anion through ionic bonding with a melting point not exceeding 100 °C.100 Since Bösmann and co-workers discovered the superior ability of ILs to remove aromatic sulfur compounds from model diesel in 2011,101 there has been growing attention on the application of ILs in ODS. Crucially, their valuable intrinsic properties, including non-volatility, robust thermal stability, notable catalytic activity and high polarity, have contributed to the low-hazard and effortlessly recyclable features of ILs.102 With increasing interest in multifunctional materials, ILs have been proposed to play the roles of both catalyst and extractive solvent in ODS systems.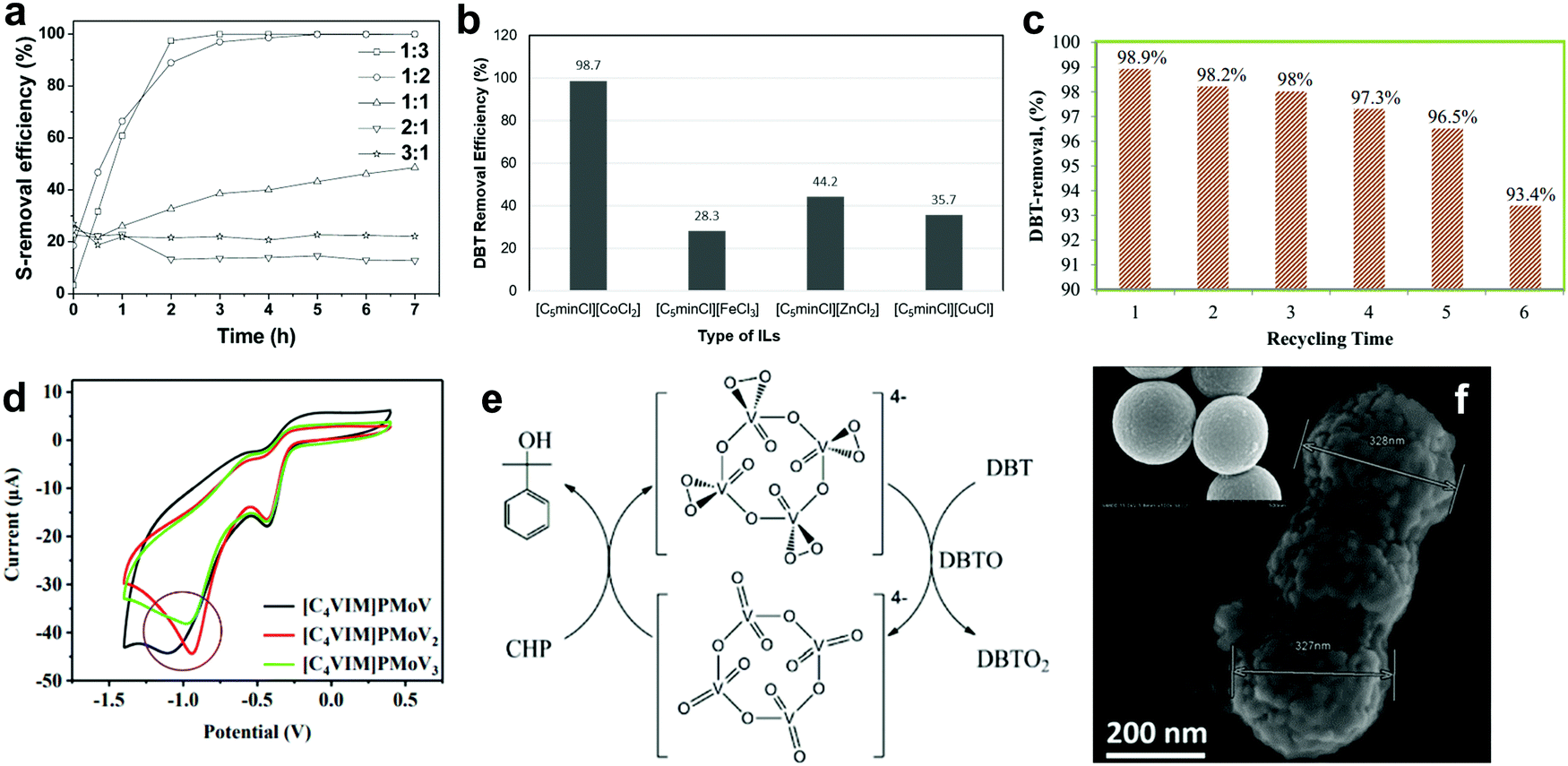 | ||
| Fig. 8 (a) Effect of [Hnmp]Cl to ZnCl2 molar ratio on the DBT removal efficiency in the presence of H2O2 at 30 °C. Reprinted with permission.103 Copyright 2015, American Chemical Society. (b) ODS activity of different metal chloride ILs at 30 °C in the presence of [C5min]2[S2O8] after 65 min. Redrawn with permission.104 Copyright 2019, American Chemical Society. (c) DBT removal efficiency of [BMPy][(C6H5)COO] for six successive cycles at 90 min per cycle and 60 °C using H2O2 as oxidant. Reprinted with permission.106 Copyright 2020, Elsevier B.V. (d) Cyclic voltammetry curves of oxygen reduction reaction on different types of POM-based IL in 0.1 M oxygen-saturated KOH solution. Reproduced with permission.107 Copyright 2020, Elsevier B.V. (e) Reaction mechanism for ODS of DBT using V-PIL and CHP. Reprinted with permission.109 Copyright 2016, American Chemical Society. (f) SEM image of [(C8H17)3NCH3]3PMo12O40 on the rough-surfaced MMS and the smooth-surfaced MMS (inset). Reproduced with permission.111 Copyright 2019, Elsevier B.V. | ||
Guo et al. developed a task-specific IL, namely [C5min]2[S2O8] as the oxidant for ODS via metathesis reaction.104 The application of [C5min]2[S2O8] in the ODS of model diesel with [C5minCl][CoCl2] as the catalyst and extractant achieved 98.7% DBT removal, better than that achieved utilizing pure K2S2O8 (36%). This revealed that the substitution of 1-pentyl-3-methylimidazolium for potassium successfully lowered the activation barrier of [C5min]2[S2O8] and promoted the supply of reactive oxygen species for ODS. In addition to [C5minCl][CoCl2], the researchers also investigated the catalytic activity of other metal chloride ILs, including [C5minCl][FeCl3], [C5minCl][ZnCl2] and [C5minCl][CuCl], in identical ODS systems. The results showed that [C5minCl][CoCl2] exhibited the highest DBT removal due to its stronger ability to activate [C5min]2[S2O8] (Fig. 8b). The reaction pathway for the ODS process using [C5min]2[S2O8] remains obscure, but is of vital significance for mechanistic understanding of the activation of [C5min]2[S2O8]. This could substantially help in widening the application of [C5min]2[S2O8] in ODS, as it possesses higher oxidative ability than other conventional oxidants like H2O2 and O2. Similar to [C5min]2[S2O8], [OMIM]2[S2O8] was also adopted as a bi-functional IL in extractive ODS and achieved remarkable performance in oxidizing and extracting aromatic sulfur compounds from fuel.105
Although the employment of task-specific ILs in ODS can eliminate the need for external oxidants and extractants, task-specific ILs are non-reusable after the ODS reaction, which is an uneconomical and unsustainable approach to industrial application. To address this, Elwan et al. recently produced a pyridinium-based IL, [BMPy][(C6H5)COO], for the ODS of model diesel in the presence of H2O2.106 Because of the nature of the pyridinium cation, aromatic sulfur compounds were extracted into the IL phase for oxidation by strong π–π interactions between the pyridinium ring of [BMPy][(C6H5)COO] and sulfur compounds. The prominent extraction capability of [BMPy][(C6H5)COO] brought about 98.9% DBT removal at 60 °C within 90 min. However, the DBT removal efficiency began to drop significantly after the fifth run (Fig. 8c) due to an irreversible accumulation of DBT sulfone in [BMPy][(C6H5)COO] that hindered the extraction of DBT. Therefore, a novel modification strategy is demanded to ameliorate the chemical properties of ILs and increase their stability in ODS applications in the future. Furthermore, Zhang et al. synthesized a POM-based IL, [C4VIM]PMoV2, by ion-exchange method and unveiled the influence of the V number to its catalytic activity in ODS.107 As seen in the cyclic voltammetry curves (Fig. 8d), [C4VIM]PMoV2 exhibited a greater positive potential than [C4VIM]PMoV and [C4VIM]PMoV3, which revealed the presence of higher electrophilicity in [C4VIM]PMoV2. As a result, [C4VIM]PMoV2 activated O2 more efficaciously, leading to increased generation of oxygen radicals for the ODS of sulfur compounds. [C4VIM]PMoV2 achieved the highest DBT conversion (98.9%) after 5 h at 120 °C.
Despite the fact that ILs are capable of remarkable ODS activity, the oxidation of sulfur compounds entails considerable reaction time. In order to boost the reaction rate, ultrasound irradiation was integrated into an ODS system with a Brønsted acidic IL, [Omin][H2SO4], as the catalyst and extractant.108 With ultrasound, the reaction time for [Omin][H2SO4] to attain 100% DBT oxidation was shortened from 70 min to 3 min under the same operating conditions. This was ascribed to the intense mixing that enhanced the mass transfer of sulfur compounds into the IL phase and propelled the interactions between catalyst and reactants. However, additional settling time after the ODS reaction was unavoidable to ensure complete phase separation for removing the IL and the oxidized sulfur compounds from the fuel. This report suggested a constructive method for developing a rapid and efficient ODS process with the aid of ultrasonication.
Anchoring ILs on porous materials is also a common and efficient strategy to heterogenize ILs, enhance their catalytic activity and reduce their usage amounts in catalysis. In the report of Xun et al., a POM-based IL, [(C8H17)3NCH3]3PMo12O40, was immobilized on a magnetic mesoporous silica nanosphere (MMS), Fe3O4–SiO2–mesoporous SiO2, to enlarge the specific surface area of IL and facilitate the separation process in ODS.110 In the ODS experiment, 100% DBT removal was achieved by [(C8H17)3NCH3]3PMo12O40/MMS in 5 h with O2 as the oxidant at 120 °C. However, only 79.5% DBT removal could be attained when non-porous SiO2 replaced the mesoporous SiO2. This indicates the significance of support porosity to the ODS activity, as the emergence of enormous pores creates more adsorption sites for the reactants, thereby promoting the oxidation of sulfur compounds. In order to further enhance the desulfurization efficiency, the same research group performed morphology modification on MMS by introducing hexane into the precursor solution to form a rough surface (Fig. 8f).111 As a result, the wettability of the catalyst to the organic phase, or hydrophobicity, was improved and contributed to a higher accessibility of the catalyst for sulfur compounds compared to the smooth-surfaced MMS. The [(C8H17)3NCH3]3PMo12O40 on the rough surface MMS demonstrated better DBT conversion (98.3%) than that on the smooth surface MMS (53.1%) after 50 min in an ODS system with H2O2 at 60 °C. However, excessive increment of hydrophobicity could lead to the reduction of ODS activity, which was proved in the work by utilizing a strongly hydrophobic rough-surfaced MMS. Additionally, it increases the difficulty of the subsequent catalyst regeneration process. Optimal hydrophobicity should be pursued to maximize the desulfurization performance in ODS. On the whole, the porosity and morphology of the support are the predominant factors governing the catalytic activity of supported ILs in ODS.
The application of ILs in ODS has contributed numerous benefits, including high efficiency in producing ultra-low-sulfur fuels, an easy recycling process and a low temperature reaction. Nonetheless, the shortcomings of ILs, such as relatively expensive precursors, considerable toxicity, poor regeneration efficiency and high viscosity, should be resolved by screening economical and harmless anion and cation sources and enhancing their chemical properties, especially the solubility in organic media, in order to improve their industrial compatibility. Table 2 summarizes the performances of various ILs in different ODS systems.
| Catalysts | Reaction temperature (°C) | Oxidant | Extractant | Performances | Ref. | ||
|---|---|---|---|---|---|---|---|
| Sulfur removal (reactant) | Reaction time (h) | Stability (h) | |||||
| PIL: poly-ionic liquid; MMS: magnetic mesoporous silica sphere; SS-MMS: smooth surface mesoporous silica sphere; RS-MMS: rough surface mesoporous silica sphere; DBN: 1,5-diazabicyclo[4.3.0]-non-3-ene. | |||||||
| [Hnmp]Cl/ZnCl2 | 75 | H2O2 (30 wt%) | [Hnmp]Cl/ZnCl2 | 99.9% (DBT) | 0.33 | >2.33 | 103 |
| [C5minCl][FeCl3] | 30 | [C5min]2[S2O8] | [C5minCl][FeCl3] | 28.3% (DBT) | 1.08 | N/A | 104 |
| [C5minCl][ZnCl2] | [C5minCl][ZnCl2] | 44.2% (DBT) | N/A | ||||
| [C5minCl][CuCl] | [C5minCl][CuCl] | 35.7% (DBT) | N/A | ||||
| [C5minCl][CoCl2] | [C5minCl][CoCl2] | 98.7% (DBT) | >7.58 | ||||
| [OMIM]2[S2O8] | 60 | [OMIM]2[S2O8] | [OMIM]2[S2O8] | 97.8% (DBT) | 3 | N/A | 105 |
| [BMPy][(C6H5)COO] | 60 | H2O2 (30 wt%) | [BMPy][(C6H5)COO] | 98.9% (DBT) | 1.5 | >7.5 | 106 |
| [Omin][H2SO4] | 25 | H2O2 (30 wt%) | [Omin][H2SO4] | 100% (DBT) | 1.17 | >7 | 108 |
| [Omin][H2SO4] (with ultrasonic) | 100% (DBT) | 0.05 | >0.3 | ||||
| [C4VIM]PMoV | 120 | O2 | [C4VIM]PMoV | 21.2% (DBT) | 5 | N/A | 107 |
| [C4VIM]PMoV2 | [C4VIM]PMoV2 | 98.9% (DBT) | >30 | ||||
| [C4VIM]PMoV3 | [C4VIM]PMoV3 | 44.8% (DBT) | N/A | ||||
| V-PIL | 50 | CHP | N/A | 98.0% (DBT) | 1 | >12 | 109 |
| [(C8H17)3NCH3]3PMo12O40/MMS | 120 | O2 | N/A | 100% (DBT) | 5 | >20 | 110 |
| [(C8H17)3NCH3]3PMo12O40/SS-MMS | 60 | O2 | N/A | 53.1% (DBT) | 0.83 | N/A | 111 |
| [(C8H17)3NCH3]3PMo12O40/RS-MMS | N/A | 98.2% (DBT) | >4.17 | ||||
| [HDBN]Cl/ZnCl2 | 40 | H2O2 (30 wt%) | [HDBN]Cl/ZnCl2 | 99.9% (DBT) | 2 | >10 | 112 |
3.3 Application of deep eutectic solvents (DESs) in ODS
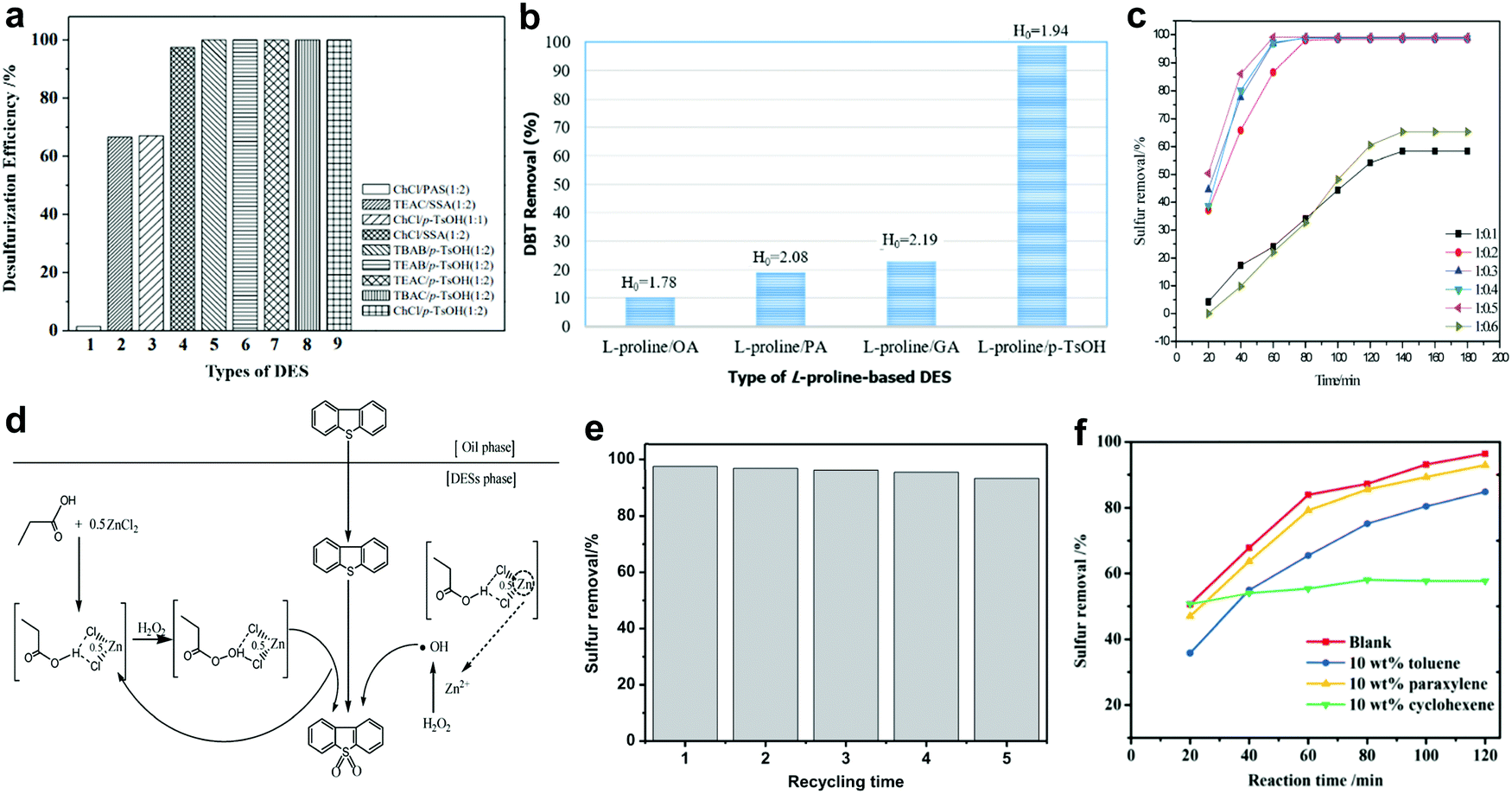
In 2003, the discovery of deep eutectic solvents (DESs) by Abbott et al. created a new research horizon in chemistry.113 Different from ILs, DESs are generally synthesized from a hydrogen bond donor (HBD) with a hydrogen bond acceptor (HBA) or a quaternary ammonium salt through hydrogen bonding.114 Therefore, the physico-chemical properties of DESs can be facilely tuned to the application purpose by selecting the appropriate precursors or modifying the composition of DESs. Apart from having unique characteristics analogous to ILs, DESs are also biodegradable, non-toxic and comparatively cheap.115 This has attracted widespread attention from researchers to dive deep into their applications owing to the rising demand for green solvents in industry. In view of their relative advantages, DESs have been appointed as a prospective alternative to ILs in the arena of fuel desulfurization technology.116,117Currently, the argument on whether the acidity of DESs is the most decisive factor for achieving remarkable catalytic activity in ODS has not come to a substantive conclusion. The connection between the acidity of DESs and the ODS efficiency was revealed by Yin et al.118 In their work, various acidic DESs were prepared by mixing different species of HBA and HBD for the ODS of model fuel. The experimental results disclosed that the employment of HBDs with greater acidity, like p-toluenesulfonic acid (p-TsOH), could significantly improve the conversion of BT using H2O2 as oxidant (Fig. 9a). Notably, the conversion of BT remained at 99% when p-TsOH was coupled with different HBAs. Obviously, HBD exhibited a greater influence on the ODS activity of DESs compared with HBA. However, Hao et al. expressed their disagreement on the positive correlation between the acidity of DESs and desulfurization efficiency after examining the activity of several L-proline-based DESs in an ODS system with H2O2.119 It was discovered that L-proline/oxalic acid (OA) with the strongest acidity demonstrated the lowest DBT removal efficiency after 30 min at 60 °C, whereas L-proline/p-TsOH with a Hammett acidity function (H0) of 1.94 achieved 99% DBT removal in an identical ODS system (Fig. 9b). Therefore, the effect of the acidity of DESs on ODS efficiency greatly depends on the types of HBD and HBA but is not absolutely important for all cases.
 | ||
| Fig. 9 (a) Catalytic activities of different DESs in oxidizing BT with H2O2 as oxidant at 25 °C in 1 h. Reprinted with permission.118 Copyright 2015, Royal Society of Chemistry. (b) DBT removal efficiency and Hammett acidity function (H0) of different L-proline-based DESs in ODS system with H2O2 at 60 °C in 30 min. Reproduced with permission.119 Copyright 2017, Elsevier B.V. (c) DBT removal efficiency using C3H6O2/XZnCl2 (X = 0.1 to 0.6) and H2O2 at 40 °C in 1 h. (d) Reaction mechanism for oxidation of DBT using C3H6O2/0.5ZnCl2 as catalyst and extractant with H2O2 as oxidant. Reprinted with permission.120 Copyright 2017, Royal Society of Chemistry. (e) DBT oxidation for five runs, at 3 h per run, using [PSTEtA]Cl/OA as catalyst and extractant at 50 °C in the presence of H2O2. Reprinted with permission.123 Copyright 2020, Elsevier B.V. (f) Effects of toluene, paraxylene and cyclohexene on the removal of DBT in the ODS system with ChCl/PEG/BA and H2O2 at 60 °C. Reprinted with permission.124 Copyright 2019, Royal Society of Chemistry. | ||
The molar ratio of HBA to HBD also has great influence on the ODS activity of DESs. Mao and co-workers investigated the ability of propionic acid-based DESs with different compositions of ZnCl2 as HBD to convert DBT into DBT sulfone in the presence of H2O2.120 Interestingly, the conversion of DBT increased with the change of the C3H6O2-to-ZnCl2 molar ratio from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1 to 1:0.5, but decreased drastically when the molar ratio reached 1:0.6 (Fig. 9c). The improvement of ODS activity was attributed to the escalation of Lewis acidity, which was conducive to the acceleration of H2O2 activation to form more reactive oxygen species for ODS. Nevertheless, the excessive Lewis acidity could also lead to the decomposition of H2O2 into unreactive H2O and O2, which substantially diminished the reaction kinetics of ODS. With respect to the reaction pathway (Fig. 9d), DBT was oxidized by the hydroxyl radicals generated from the activation of H2O2 by DES and by the peroxy-C3H6O2/0.5ZnCl2 generated from the oxidation of C3H6O2/0.5ZnCl2 by H2O2 after the extraction of DBT from the oil phase to DES phase. In the report of Hao et al., the increase of the HBA-to-HBD molar ratio from 1:1 to 3:1 in caprolactam (CPL)/OA also markedly enhanced the DBT removal efficiency from 62.6% to 98.4% with H2O2 as oxidant at 60 °C.121 This was due to the strengthening of the hydrogen bonding in CPL/OA that was essential for weakening the aromaticity of DBT by developing vigorous interactions between CPL/OA and DBT, thus boosting the activity of CPL/OA in ODS. This was supported by Lü et al. who studied the effect of the HBA-to-HBD molar ratio on the ODS performances of oxalate-based DESs.122 By and large, optimization of the HBA-to-HBD molar ratio is necessary to regulate the physico-chemical properties of DESs to maximize the ODS activity of DESs.
:0.1 to 1:0.5, but decreased drastically when the molar ratio reached 1:0.6 (Fig. 9c). The improvement of ODS activity was attributed to the escalation of Lewis acidity, which was conducive to the acceleration of H2O2 activation to form more reactive oxygen species for ODS. Nevertheless, the excessive Lewis acidity could also lead to the decomposition of H2O2 into unreactive H2O and O2, which substantially diminished the reaction kinetics of ODS. With respect to the reaction pathway (Fig. 9d), DBT was oxidized by the hydroxyl radicals generated from the activation of H2O2 by DES and by the peroxy-C3H6O2/0.5ZnCl2 generated from the oxidation of C3H6O2/0.5ZnCl2 by H2O2 after the extraction of DBT from the oil phase to DES phase. In the report of Hao et al., the increase of the HBA-to-HBD molar ratio from 1:1 to 3:1 in caprolactam (CPL)/OA also markedly enhanced the DBT removal efficiency from 62.6% to 98.4% with H2O2 as oxidant at 60 °C.121 This was due to the strengthening of the hydrogen bonding in CPL/OA that was essential for weakening the aromaticity of DBT by developing vigorous interactions between CPL/OA and DBT, thus boosting the activity of CPL/OA in ODS. This was supported by Lü et al. who studied the effect of the HBA-to-HBD molar ratio on the ODS performances of oxalate-based DESs.122 By and large, optimization of the HBA-to-HBD molar ratio is necessary to regulate the physico-chemical properties of DESs to maximize the ODS activity of DESs.
In 2020, Jiang et al. prepared an innovative Brønsted acidic DES with dual active sites for ODS by assembling (3-sulfopropyl)triethylammonium chloride ([PSTEtA]Cl) and OA.123 Through DFT computations, the sulfonic group and the carboxylic group in [PSTEtA]Cl and OA, respectively, were found to be the active sites that interacted with H2O2 to form peroxy-intermediates for ODS. This contributed to a higher efficiency of DBT removal in the ODS system with [PSTEtA]Cl/OA (97.7%) compared to that with tetrabutylammonium chloride (TBAC)/OA (72.9%), which possessed only a carboxylic group as the catalytic site. Moreover, [PSTEtA]Cl/OA demonstrated outstanding stability in ODS (Fig. 9e) and could be regenerated via simple water washing owing to its hydrophilic property. In another pioneering work of Jiang et al., polyethylene glycol (PEG) and boric acid (BA) were adopted as HBDs along with choline chloride (ChCl) as an HBA to create a ternary DES for ODS.124 It was noteworthy that the increment of BA composition positively influenced the ODS activity of ChCl/PEG/BA but the increase of PEG composition led to the decline of ODS activity. This indicated the paramount role of BA in catalyzing the oxidation of sulfur compounds. Here, the replacement of ChCl with TBAC, which has a long carbon chain structure, resulted in an improved extractive ability while decreasing the catalytic activity in ODS. Apparently, the tuning of DES composition greatly affects the performances as an extractant and a catalyst in ODS due to discrepancies in the structure and properties of different HBAs and HBDs. In addition, the effect of various hydrocarbons in real diesel on the ODS performances of DES was addressed in the same report. As shown in Fig. 9f, the addition of cyclohexene into model fuel drastically reduced the DBT removal efficiency, whereas the introductions of toluene and paraxylene brought about little influence on the oxidation of DBT. This phenomenon is produced by the competitive oxidation reaction between hydrocarbons and DBT. The detrimental impact of cyclohexene on the ODS reaction was also ascertained by Liu et al., who studied the ODS system with L-pyroglutamic acid/trifluoroacetic acid.125 Therefore, further research should be performed to strengthen the selectivity towards aromatic sulfur compounds by modulating the intrinsic characteristics of DESs to secure the outstanding ODS activity of DESs in treating commercial diesel.
DESs are a promising catalyst and extractant in ODS due to their ability to achieve ultra-deep desulfurization of fuels at a relatively mild reaction temperature, which is highly desirable for future industrial applications. Proper screening of the HBA and HBD as well as alteration of their molar ratio must be performed to engineer DESs with excellent desulfurization performances in ODS. As a suggestion, additional study on the utilization of cheaper and environmentally benign O2 as the oxidant in the ODS system with DESs should be conducted to proffer valuable insights into the development of a sustainable ODS process. Table 3 presents the performances of various DESs in different ODS systems.
| Catalysts | Reaction temperature (°C) | Oxidant | Extractant | Performances | Ref. | ||
|---|---|---|---|---|---|---|---|
| Sulfur removal (reactant) | Reaction time (h) | Stability (h) | |||||
| TEAC: tetraethylammonium chloride; SSA: 5-sulfosalicylic acid; ChCl: choline chloride; TBAB: tetrabutylammonium bromide; p-TsOH: p-toluenesulfonic acid; TEAB: tetraethylammonium bromide; TBAC: tetrabutylammonium chloride; OA: oxalic acid; PA: propanedioic acid; GA: glutaric acid; TMAC: tetramethylammonium chloride; L-Pyro: L-pyroglutamic acid; TFA: trifluoroacetic acid; [PSTEtA]Cl: (3-sulfopropyl)triethylammonium chloride; AA: acetic acid; BA: boric acid; PEG: polyethylene glycol; PMS: peroxymonosulfate; Pr: propionic acid; EG: ethylene glycol. | |||||||
| TEAC/SSA | 25 | H2O2 (30 wt%) | TEAC/SSA | ∼67% (BT) | 1 | N/A | 118 |
| ChCl/SSA | ChCl/SSA | ∼97% (BT) | N/A | ||||
| TBAB/p-TsOH | TBAB/p-TsOH | ∼99% (BT) | N/A | ||||
| TEAB/p-TsOH | TEAB/p-TsOH | ∼99% (BT) | N/A | ||||
| TEAC/p-TsOH | TEAC/p-TsOH | ∼99% (BT) | N/A | ||||
| TBAC/p-TsOH | TBAC/p-TsOH | ∼99% (BT) | N/A | ||||
| ChCl/p-TsOH | ChCl/p-TsOH | ∼99% (BT) | >4 | ||||
| L-Proline/OA | 60 | H2O2 (30 wt%) | L-Proline/OA | 10% (DBT) | 0.5 | N/A | 119 |
| L-Proline/PA | L-Proline/PA | 19% (DBT) | N/A | ||||
| L-Proline/GA | L-Proline/GA | 23% (DBT) | N/A | ||||
| L-Proline/p-TsOH | L-Proline/p-TsOH | 99% (DBT) | >3 | ||||
| C3H6O2/0.5ZnCl2 | 30 | H2O2 (30 wt%) | C3H6O2/0.5ZnCl2 | 99.42% (DBT) | 1 | >5 | 120 |
| Caprolactam/OA | 60 | H2O2 (30 wt%) | Caprolactam/OA | 98.4% (DBT) | 0.5 | N/A | 121 |
| TBAC/OA | 50 | H2O2 (30 wt%) | TBAC/OA | 91% (DBT) | 3 | N/A | 122 |
| ChCl/OA | ChCl/OA | 71% (DBT) | N/A | ||||
| TMAC/OA | TMAC/OA | 41% (DBT) | N/A | ||||
| L-Pyro/TFA | 60 | H2O2 (30 wt%) | L-Pyro/TFA | 99.7% (DBT) | 3 | >4.5 | 125 |
| L-Pyro/formic acid | L-Pyro/formic acid | 97.0% (DBT) | N/A | ||||
| L-Proline/TFA | L-Proline/TFA | 99.6% (DBT) | N/A | ||||
| L-Proline/formic acid | L-Proline/formic acid | 4.7% (DBT) | N/A | ||||
| [PSTEtA]Cl/OA | 50 | H2O2 (30 wt%) | [PSTEtA]Cl/OA | 97.7% (DBT) | 3 | >15 | 123 |
| TBAC/OA | TBAC/OA | 72.9% (DBT) | N/A | ||||
| [PSTEtA]Cl/AA | [PSTEtA]Cl/AA | 95.1% (DBT) | N/A | ||||
| ChCl/BA | 60 | H2O2 (30 wt%) | ChCl/BA | 34.0% (DBT) | 2 | N/A | 124 |
| ChCl/PEG | ChCl/PEG | 27.4% (DBT) | N/A | ||||
| ChCl/PEG/BA | ChCl/PEG/BA | 96.4% (DBT) | >4 | ||||
| TBAC/PEG/BA | TBAC/PEG/BA | 56.4% (DBT) | N/A | ||||
| 2Acetamide/GA | 60 | H2O2 (30 wt%) | 2Acetamide/GA | 57% (DBT) | 0.5 | N/A | 126 |
| Acetamide/GA | Acetamide/GA | 95% (DBT) | N/A | ||||
| Acetamide/2GA | Acetamide/2GA | 99% (DBT) | N/A | ||||
| Acetamide/3GA | Acetamide/3GA | 95% (DBT) | N/A | ||||
| CoCl2–ChCl/2PEG | 20 | PMS | CoCl2–ChCl/2PEG | 100% (DBT) | 1 | >6 | 127 |
| CoCl2–ChCl/2Pr | CoCl2–ChCl/2Pr | 83% (DBT) | N/A | ||||
| CoCl2–ChCl/2EG | CoCl2–ChCl/2EG | 73% (DBT) | N/A | ||||
| CoCl2–ChCl/2glycerol | CoCl2–ChCl/2glycerol | 55% (DBT) | N/A | ||||
3.4 Application of polyoxometalates (POMs) in ODS
To date, an appreciable amount of literature has been published on the ODS of diesel fuel by utilizing POMs, chosen for their tunable properties, ample exposure of active sites and exceptional performances in redox reactions.128–135 Typically, POMs are cluster form materials belonging to the metal oxide family, which is comprised of numerous transition metal oxoanions and oxygen atoms, frequently with heteroatoms.136 In regard to their structure, POMs can be classified into several types, including Anderson–Evans,134 Keggin,133,137–139 Dawson140 and so forth, which respectively possess the coordination of a single heteroatom surrounded by 6 transition metal atoms ([XM6O24]n−),141 one heteroatom surrounded by 12 transition metal atoms ([XM12O40]n−)140 and two heteroatoms surrounded by 18 transition metal atoms ([X2M18O62]n−).140 Among the POM types, Keggin-based POMs have been popularly investigated in ODS by previous researchers on account of their superiority in industrial applicability, thermal resistance and simplicity of synthesis procedures.26,130 Owing to their poor miscibility in the organic phase, homogeneous POMs are invariably coupled with an extractant such as ILs, DESs or organic polar solvents to accelerate the desulfurization process by facilitating the mass diffusion of sulfur compounds in fuels with POMs.129,133,134,137,138,142In the past, Wang and co-workers highlighted phosphotungstic acid (HPW) as a promising POM for desulfurizing model diesel because it proficiently transformed 99.23% of DBT to DBT sulfone with the aid of H2O2 within 60 min at 60 °C.132 This was not in agreement with the research of Trakarnpruk and Rujiraworawut, which manifested that HPW achieved merely 93% conversion of DBT after 150 min under the identical reaction temperature.139 The performance discrepancy originated from a difference in the applied oxygen to sulfur (O/S) molar ratio, as the first report utilized 15:1 compared to 10:1 in the second report. In order to construct a comprehensive picture of the correlation between the O/S ratio and ODS activity, the proposed ODS mechanism of HPW is shown in Fig. 10a. In fact, the increment of the O/S ratio appreciably increased the ODS efficiency. The excess H2O2 was able to promote the generation of an unstable peroxo-HPW intermediate by accelerating the rate of nucleophilic attack towards the W atom of HPW in the aqueous phase (1). With the help of a polar extractant, DBT was transferred from the organic phase to the aqueous phase and readily reacted with the peroxo-HPW to yield DBT sulfoxide (2). Eventually, DBT sulfoxide was oxidized to DBT sulfone through the same pathway (3 and 4). Meanwhile, DBT sulfone was sealed in the aqueous phase due to the increase of polarity and was removed through a further separation process. However, Wang et al. also revealed that the O/S ratio no longer significantly intensified the ODS activity when it was increased to 1:18.132 Moreover, the excessive oxidants triggered the rapid oxidation of alkenes and aromatic hydrocarbons in the diesel fuel. Consequently, the yield of clean diesel fuel greatly declined, which could exacerbate the operational cost for ODS.57 Therefore, knowledge of the optimum O/S ratio for different ODS systems is essential to economically maximize the ODS activity and bring it a step closer to industrial application.
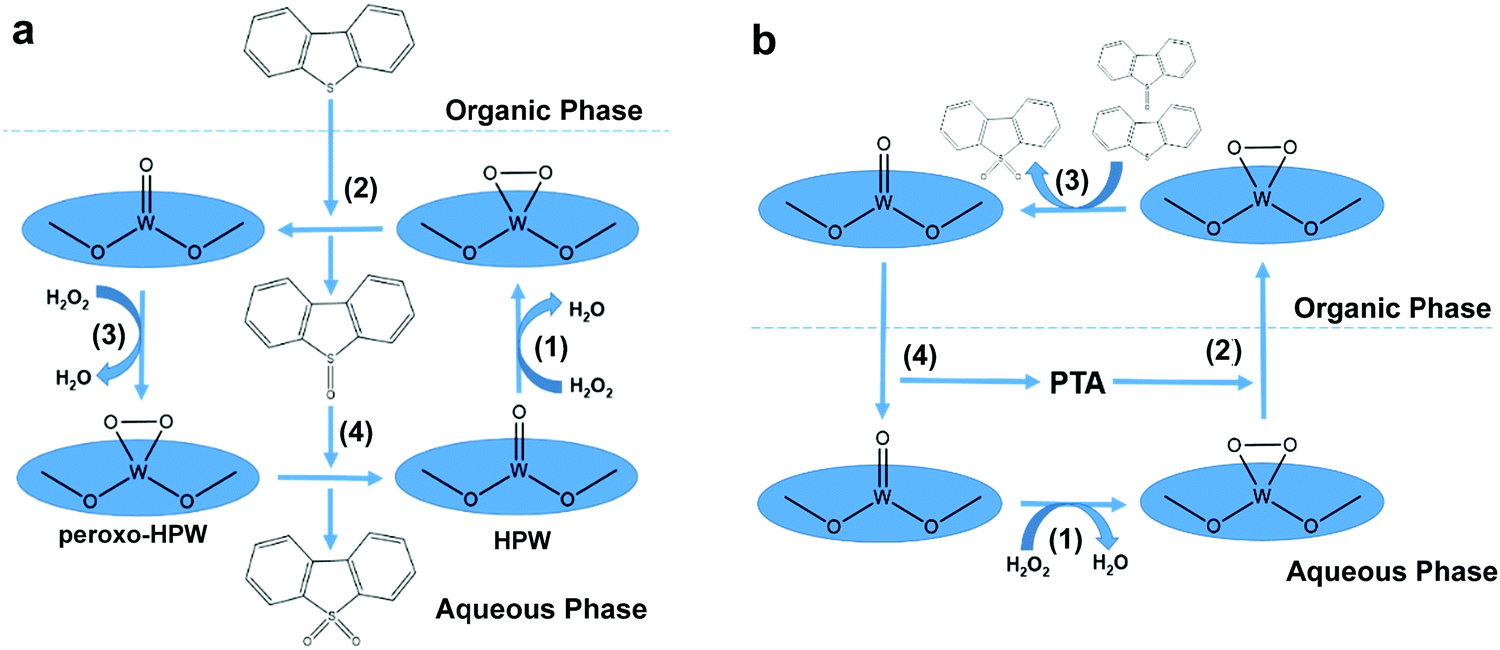 | ||
| Fig. 10 ODS mechanisms of DBT over HPW POM using (a) extractant and (b) phase transfer agent (PTA). | ||
Instead of utilizing extractant, Choi et al. applied a phase transfer agent (PTA) to perform ODS with POMs with the assistance of vigorous mixing and ultrasound.137,138 While an extractant and PTA share the same objective in promoting mass transfer between catalyst and reactant, their mechanisms in ODS were contrary. In the ODS system with PTA and HPW (Fig. 10b), the oxidation of DBT (3) arose in the organic phase rather than in aqueous phase due to the PTA conveying active peroxo-HPW. After the oxidation process, the peroxo-HPW was degraded to HPW, detached from PTA and transported back to the aqueous phase (4). At the same time, the oxidized DBT was endowed with a higher miscibility in aqueous phase, which facilitated its dissociation from the fuels.
Apart from traditional Keggin-type POMs, POMs modified by heteroatom substitution have been investigated for their catalytic activity in ODS. First, the [PV2Mo10O40]4− complex prepared by Komintarachat and Trakarnpruk achieved 86% DBT conversion in gas oil after 5 h, which was mainly attributed to the higher tendency of Mo–O–V species to generate a more unstable peroxo-POMs lattice after the replacement of Mo with V.142 Nevertheless, a few years later, Wang et al. reported that the replacement of Mo with W in phosphomolybdic acid (HPMo) demonstrated better ODS efficiency (>98%) with H2O2 in 1 h.132 The increase of W in HPMo improved the ODS activity in the order of W6 (99.79%) > W3 (99.22%) > W1 (98.80%) owing to the function of the W atom in promoting the Brønsted acidity of HPMo. Recently, Bertleff et al. produced H8PV5Mo7O40 by substituting Mo with V in HPMo, which attained up to 99% sulfur removal with the consumption of O2 at 140 °C and 20 bar oxygen pressure within 6 h.131 Despite the harsh operating conditions and long reaction time, the hazardous and uneconomical H2O2 was supplanted by O2 in the study, contributing a valuable insight into environmentally-friendly ODS. The application of a green ODS process on an industrial scale without compromising the rate and operating conditions of ODS is a momentous challenge for the future. In the above studies, the development of POMs in ODS is clearly illustrated, from improving the ODS activity and rate of reaction by attempting various heteroatom substitution to establishing an eco-friendly ODS process by utilizing safe and free ingredients.
3.4.2.1 Heterogeneous POMs synthesized by precipitation. For the precipitation of POMs with insoluble cations, the protons or hydrogen atoms of POMs are replaced with large insoluble metal ions to improve the heterogeneity. Recently, Akbari et al. synthesized Cs salts of HPW (CsPW) by co-precipitation method (CP), reversed emulsion (RE) method and reversed microemulsion (RME) method and applied them in the ODS of model diesel.145 The utilization of surfactant in RME led to the attachment of the surfactant on the surface of CsPW and fostered an amphiphilic property in CsPW. The surfactant also ameliorated the total surface energy of CsPW by building up more counter ions on the surface. This weakened the van der Waals forces between CsPW particles, hampering the agglomeration of particles and generating separated particles (Fig. 11a). In contrast, the CsPW synthesized by CP agglomerated and formed a large granule (Fig. 11b) due to the absence of surfactant. As a result, the CsPW obtained via RME exhibited the highest activity (50%) in eliminating DBT (Fig. 11c), which was attributed to the increase of active sites exposed to the sulfur reactants for ODS. In spite of that, the achieved ODS activity is still quite far from ultra-deep desulfurization and is crucially restricted by the scarcity of active sites.
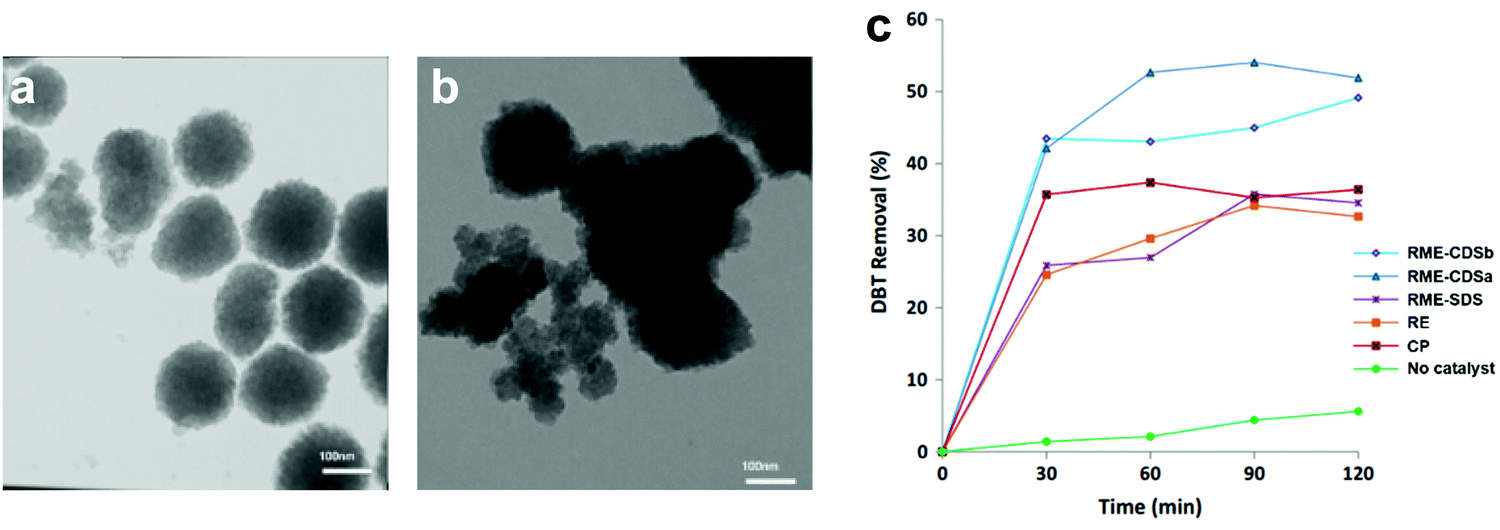 | ||
| Fig. 11 TEM images of CsPW synthesized via (a) reverse microemulsion (RME) method and (b) co-precipitation (CP) method. (c) DBT removal with TBHP at 60 °C over various CsPW created by CP, reversed emulsion (RE) method and RME with sodium dodecyl sulfate (SDS) or caesium dodecyl sulfate (CDS) as surfactant. Reprinted with permission.145 Copyright 2020, Springer Nature. | ||
In addition to Keggin-type POMs, Li's research team recently used the Anderson–Evans type POM Na6TeW6O24·22H2O (TeW6) in ODS.146 They also incorporated Cu ions into the structure of TeW6 to endow a heterogeneous feature in organic fuels. The results showed that the conversion efficiency of thioanisole over Te2W8Cu2 attained 99% with H2O2 after 6 h at room temperature, which was slightly higher than those of TeW6Cu (89%) and conventional TeW6 (89%). It was demonstrated that the introduction of Cu ions successfully developed new active sites for TeW6, leading to the improvement of ODS activity. Despite the sluggish ODS kinetics, this research proved that it is viable to achieve notable ODS performance without inputting external heat energy to the system.
3.4.2.2 Heterogeneous POMs synthesized by anchoring on support. Alternatively, POMs can also be impregnated into an appropriate support with a large surface area and high porosity characteristics. In this regard, Julião and coworkers fabricated a peroxophosphotungstate-based catalyst (PW4) on trimethylammonium group-functionalized mesoporous silica (TMA-SBA-15) through an impregnation method and studied its activity in a biphasic ODS system with model and real diesel fuels.57 Both PW4 and PW4/TMA-SBA-15 attained complete desulfurization in model diesel within 2 h under the same operating conditions, but PW4/TMA-SBA-15 demonstrated higher stability (>30 h) than PW4 (<20 h). The robustness of the catalyst can be observed in Fig. 12a and b, which display the unchanged morphology and structure of PW4/TMA-SBA-15 after catalyst recovery. This phenomenon was ascribed to the functionalization of the SBA-15 surface with TMA organic groups which resulted in stronger intermolecular bonds between PW4 and SBA-15, inhibiting the occurrence of leaching. Mirante et al. also implemented a similar strategy in functionalizing large-pore mesoporous silica nanospheres (LPMS) with TMA, which later served as the support for a sandwich-type samarium-phosphomolybdic catalyst (SmPMo) through the impregnation method (Fig. 12c).147 In the FTIR analysis (Fig. 12d and e), the similar spectral profiles of initial and recovered SmPMo/TMA-LPMS disclosed the robust structure and outstanding stability of the catalyst which further contributed to the 100% desulfurization of model diesel for three consecutive cycles. Both studies confirmed the key role of surface functional groups, like TMA, in developing POM composites with a stable structure and prolonged lifetime in ODS.
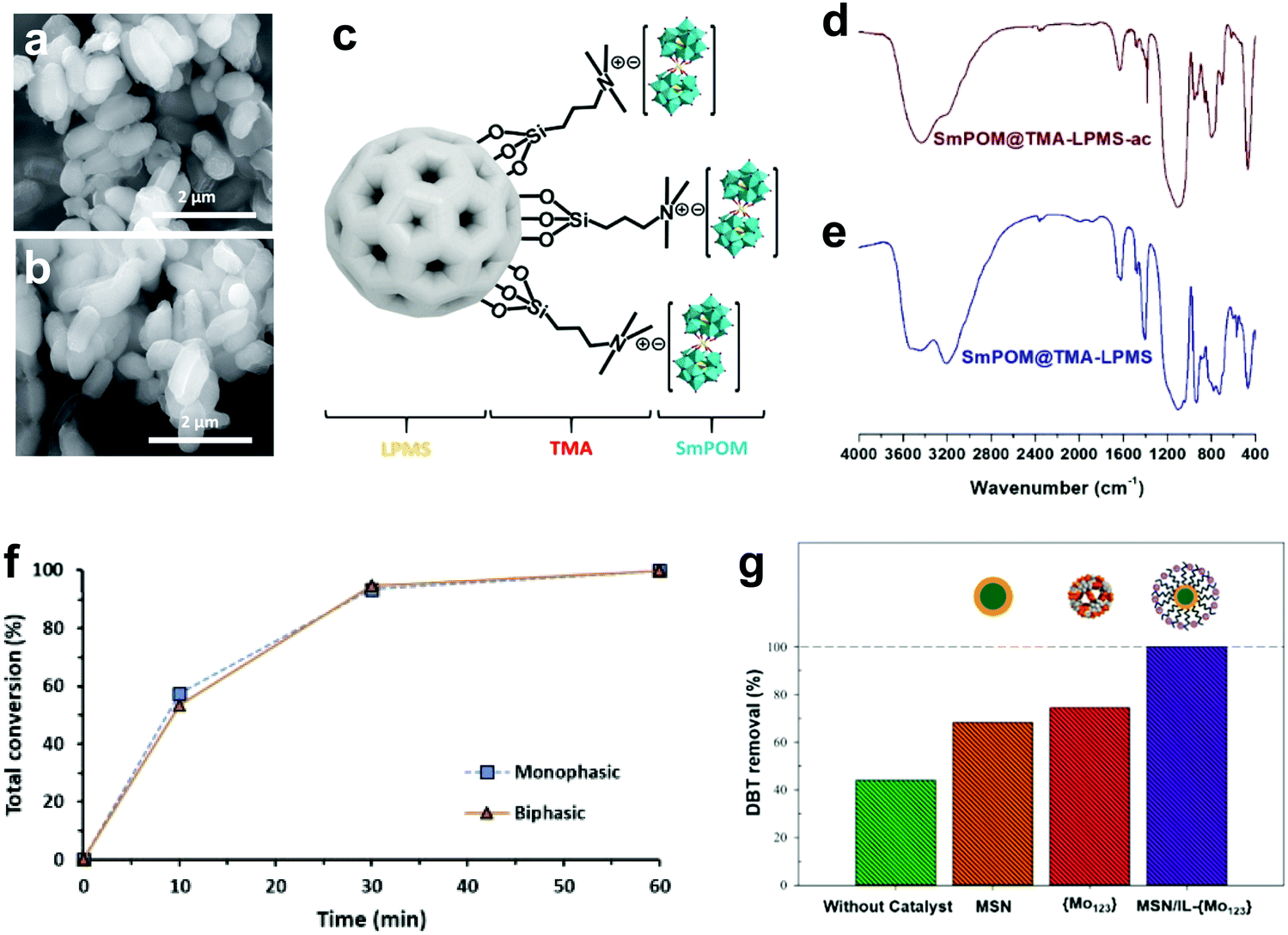 | ||
| Fig. 12 SEM images of (a) initial and (b) recovered PW4/TMA-SBA-15. Reprinted with permission.57 Copyright 2019, Elsevier Ltd. (c) Schematic of SmPMo/TMA-LPMS. FTIR spectra of (d) recovered SmPMo/TMA-LPMS and (e) initial SmPMo/TMA-LPMS. Reprinted with permission.147 Copyright 2020, Elsevier Ltd. (f) ODS performances of PW11/aptes-SBA-15 in monophasic and biphasic systems with model diesel and H2O2 at 70 °C. Reprinted with permission.133 Copyright 2019, Elsevier B.V. (g) DBT removal over MSN and Mo123/IL-MSN for 0.5 h with H2O2 at 62.5 °C. Reprinted with permission.148 Copyright 2021, Elsevier Ltd. | ||
Ribeiro and coworkers employed the amine functional group (3-aminopropyl)triethoxysilane (aptes) and the tributylammonium (TBA) functional group to functionalize the surface of SBA-15 in order to enhance the dative bonding between SBA-15 and lacunary phosphotungstic catalyst (PW11).133 Interestingly, the aptes-functionalized composite attained a higher desulfurization efficiency (100%) than the composite with TBA (80%) in 70 min. This phenomenon stemmed from the blockage of pores in SBA-15 by TBA with its larger particle size, which immensely inhibited the interactions of reactants and active sites, thereby diminishing the catalytic performance in ODS. PW11/aptes-SBA-15 exhibited identical ODS activity in biphasic and monophasic systems with model diesel (Fig. 12f) due to the improvement of heterogeneity, which contributed to the good dispersion of the catalyst in the organic phase. The remarkable performance of POMs in the absence of extractive solvent can be noted as a breakthrough in ODS and a valuable insight for future industrial application.
Mojaverian Kermani's research group reported an ingenious strategy to deal with the burdensome catalyst recovery challenge in ODS systems with POMs.148 They magnetized a Keplerate-type POM, isopolyoxomolybdate (Mo132), by embedding it into IL-functionalized magnetic silica nanoparticles (IL-MSN) to ease the recycling of the catalyst after ODS with a magnet. Moreover, IL-MSN served as a valuable dispersion platform for Mo132 to create more surface active sites for catalyzing ODS. This contributed to a tremendous enhancement of DBT conversion at 62.5 °C utilizing H2O2 as oxidant in 30 min. However, light leaching of Mo132 emerged after 1 h of ODS owing to the poor chemical bonding between Mo123 and IL-MSN, resulting in significant ODS activity loss (Fig. 12g). In other words, the IL functional group on the surface of the MSN was insufficient to strongly bond Mo123 and prevent its detachment in long-time ODS. Thus, the substitution of the IL with a relatively robust functional group would prolong the lifetime and strengthen the recyclability of POMs nanocomposites in ODS.
Besides mesoporous silica-based supports, carbonaceous materials with extensive surfaces have also been nominated as supports for POMs in ODS. According to the pioneering work by Xiao et al., HPW was immobilized on activated carbon (AC) as the supporting material by wet impregnation method.149 The enlargement of the specific surface area from 10 m2 g−1 to 478.4 m2 g−1 in HPW after impregnation contributed 90% Th conversion in model fuel with H2O2 at 90 °C. However, the existence of readily oxidized hydrocarbons like xylene, octadiene and cyclohexene as competitors to Th significantly decreased the effectiveness of HPW/AC in desulfurizing real fuel. A possible explanation for this event was that hydrocarbons with particle sizes smaller than the pore size of catalyst were able to access the pore channels and occupy the active sites, further curtailing the availability of adsorption and oxidation platforms for Th. Since the long hydrocarbons in commercial diesel can vastly influence the activity of catalysts, this should be considered during ODS and countermeasures addressing this issue would be a fruitful area for further study.
Beyond the impregnation method, Liao and co-workers proposed an innovative electrochemical method to embed HPW into rGO (HPW/rGO-Ele).150 With a stronger reduction capability, the electrochemical method eliminated more oxygen-containing functional groups during the reduction process of GO into rGO. Advantageously, the aggregation problem from the strong interactions between HPW and oxygen-containing functional groups was efficaciously minimized. Hence, HPW/rGO-Ele displayed uniform dispersion (Fig. 13a), but not the agglomerated structure which could be observed in the morphology of impregnation method-prepared HPW/rGO (HPW/rGO-Imp) (Fig. 13b). As a result, HPW/rGO-Ele possessed larger specific surface area and richer surface catalytic sites. The stronger interaction forces between HPW and rGO, which stemmed from the substantial interfacial charge transfer, protected the structure of HPW/rGO-Ele from dissociation, giving rise to the consistent and impressive DBT removal rate for five successive runs (Fig. 13c). Although the electrochemical method exhibits more advantages in fabricating hybrid materials than the conventional chemical approach, its application in large scale production still remains uncertain because the demand for an incessant supply of electrons to drive the synthesis process is a major challenge.
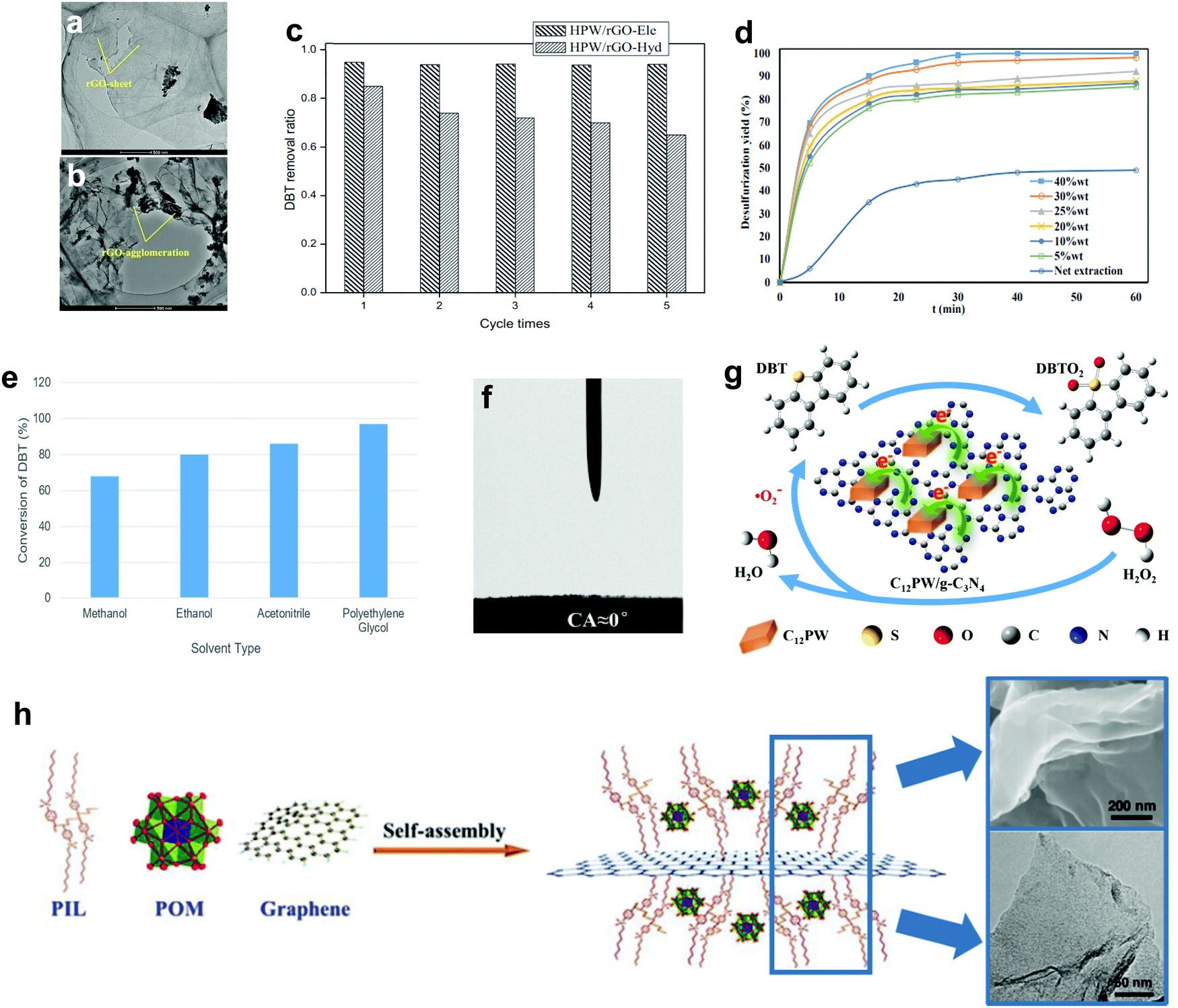 | ||
| Fig. 13 TEM images of (a) HPW/rGO-Ele and (b) HPW/rGO-Imp. (c) DBT removal with H2O2 by HPW/rGO-Ele and HPW/rGO-Imp at 60 °C for five cycles, with 8 h per cycle. Reprinted with permission.150 Copyright 2019, Elsevier B.V. (d) Oxidation of DBT over HPW/GO with different HPW contents at 60 °C in the presence of H2O2. Reprinted with permission.151 Copyright 2019, Elsevier Ltd. (e) Conversion of DBT by applying TBAPMo11Cu/CuO in ODS system with different solvents and H2O2 at 35 °C. Redrawn with permission.153 Copyright 2020, American Chemical Society. (f) Contact angle for model oil droplet on the surface of C12PW/g-C3N4. (g) Proposed ODS mechanism for C12PW/g-C3N4 in the presence of H2O2. Reprinted with permission.154 Copyright 2020, Elsevier B.V. (h) Synthesis route for POM/PIL/Gr along with the morphology of POM/PIL/Gr in SEM (top) and TEM images (bottom). Reproduced with permission.155 Copyright 2020, Elsevier B.V. | ||
In another work, different quantities of HPW were grafted onto GO to examine the effect of HPW loading on the oxidation activity of HPW/GO.151 The outcome of the study revealed that HPW/GO with higher loadings of HPW possessed better oxidation ability on DBT in the presence of H2O2 at 60 °C (Fig. 13d). This was ascribed to the increment of active sites for ODS. Likewise, Zhu et al. reported the positive effect of increasing the HPW loadings on g-C3N4 in the conversion of DBT.152 Nevertheless, the DBT conversion of 30 wt% HPW/g-C3N4 (99.5%) was slightly lower than that of 20 wt% HPW (100%) after 2.5 h. This was attributed to saturation of the anchor sites on the surface of g-C3N4 which caused the blockage and overlapping of active sites by the additional HPW. Therefore, an optimum loading of catalyst is highly essential for elevating the ODS activity.
As in the previous works, the employment of methanol as a solvent in ODS can create extra expenses for separation, the recovery process and waste management due to the poisonous and flammable properties of solvents. Hence, Shokri Aghbolagh et al. adopted the relatively hazardless polyethylene glycol as the solvent in the ODS system with Keggin-type TBAPMo11Cu on CuO, which was synthesized through the sol–gel method.153 The utilization of polyethylene glycol was conducive to the efficacy of TBAPMo11Cu/CuO in oxidizing DBT and achieved the highest performance compared to ODS systems with conventional solvents including methanol, ethanol and acetonitrile (Fig. 13e). The effect of solvent on the rate of ODS is not clearly understood and would be an interesting research topic to guide the selection of ODS solvents in the future. The coupling of TBAPMo11Cu with CuO not only improved the heterogeneity but also enlarged the specific surface area and later developed more active sites for ODS. As a result, TBAPMo11Cu/CuO attained a greater DBT oxidation (98%) in the presence of H2O2 at 35 °C than did pristine TBAPMo11Cu (61%) after 1 h.
Instead of applying a safer solvent, Yu et al. enhanced the amphiphilic feature of HPW by doping IL to eliminate the extractant and supported the modified HPW on g-C3N4via solvothermal method.154 By substituting long carbon chains for the polar hydrogen atoms, the resulting C12PW/g-C3N4 gained hydrophobic character. This allowed dispersion of the catalyst in the organic phase and accelerated the mass transfer of the catalyst in contact with reactant. This modification was affirmed by the wettability test (Fig. 13f), where C12PW/g-C3N4 possessed a zero contact angle with model diesel. In addition, the fast electron migration from g-C3N4 to C12PW that promoted the activation of H2O2 brought about the complete oxidation of DBT in model fuel at 60 °C after 1 h through the ODS mechanism shown in Fig. 13g. However, safety concerns arising from the utilization of hazardous H2O2 in ODS prompted Ma and colleagues to investigate the possibility of O2 as the oxidizing agent.155 Through the bottom-up self-assembly approach, poly-IL (PIL) was a vital bridging agent, holding the POM anionic clusters and graphene (Gr) together (Fig. 13h) by creating a series of vigorous interactions composed of electrostatic force, van der Waals force, π–π interaction and hydrophobic interaction. As a result, a mesoporous POM/PIL/Gr with 2D structure and large surface area was synthesized and achieved 100% DBT conversion for 3 h at 100 °C. Overall, these studies suggest that the hybridization of POMs with carbon materials can realize 100% desulfurization of fuels.
3.4.2.3 Heterogeneous POMs synthesized by encapsulating in MOFs. In addition to carbonaceous materials, highly porous MOFs with vast surface area and tunable morphology are expected to be a robust support for POMs to address the recycling barrier and leaching phenomenon in POMs. Peng et al. encapsulated HPW in a Zr-based MOF (UiO-67) via solvothermal method and tested its ODS activity with the addition of acetonitrile.156 In spite of HPW/UiO-67 possessing the same degree of DBT conversion as HPW (99.5%), the narrow aperture size in UiO-67 barricaded the escape of HPW from the host, further reinforcing the robustness and recyclability of HPW/UiO-67. The absence of leaching is illustrated in Fig. 14a, in which the degree of DBT conversion remained constant at 85% after the removal of the catalyst at 30 min. Similarly, the encapsulation of HPMo in UiO-67 also showed promising catalytic activity and stability in ODS.157 To further strengthen the ODS activity, Wang and co-workers treated HPMo/UiO-66 with HCl to induce ligand defects in the structure.158 The removal of ligands increased the aperture size in UiO-66 and directly led to the increment of pore volume and specific surface area. In consequence, defective HPMo/UiO-66 achieved 100% DBT conversion in the presence of H2O2 after 1 h, which was around 25% more than pristine HPMo/UiO-66 achieved. The remarkable ODS activity enhancement was attributed to the creation of more active sites as well as the higher accessibility of the pores of UiO-66 for sulfur compounds. In fact, the large aperture size could give rise to activity loss over time owing to the dissociation of POM from the inner surface of the MOF. Impressively, in this case, elimination of ligands did not significantly impact the stability of the catalyst in ODS as the defective HPMo/UiO-66 retained its ODS activity for ten cycles. This could be ascribed to the robust chemical interactions between HPMo and UiO-66 on the internal surface. In other work, Wei and coworkers confirmed the chemical stability of the Keggin-based POM (PMoV)/rht-MOF-1 through PXRD analysis (Fig. 14b).159 However, the ODS efficiency of PMoV/rht-MOF-1 decreased gradually over seven tests, mainly stemming from pore blockage by sulfone compounds. Lu's group reported the confinement of the Weakley-type POM Na7H2LaW10O36 (LaW10) inside mesoporous MIL-101 using the impregnation method and vacuum annealing (Fig. 14c).160 The strong interactions between LaW10 and MIL-101 through the W
O–Cr bond coordinated LaW10 in the cavity of MIL-101, as indicated by the peak shift of Cr 2p3/2 to a lower bonding energy due to the electron transfer (Fig. 14d). This endowed LaW10/MIL-101 with excellent stability and it further achieved above 96% DBT conversion with H2O2 at 60 °C for 7 runs. Generally speaking, the window size of MOFs and the bonding between POMs and MOFs are the crucial factors affecting the stability and activity of POM/MOF catalysts in ODS.
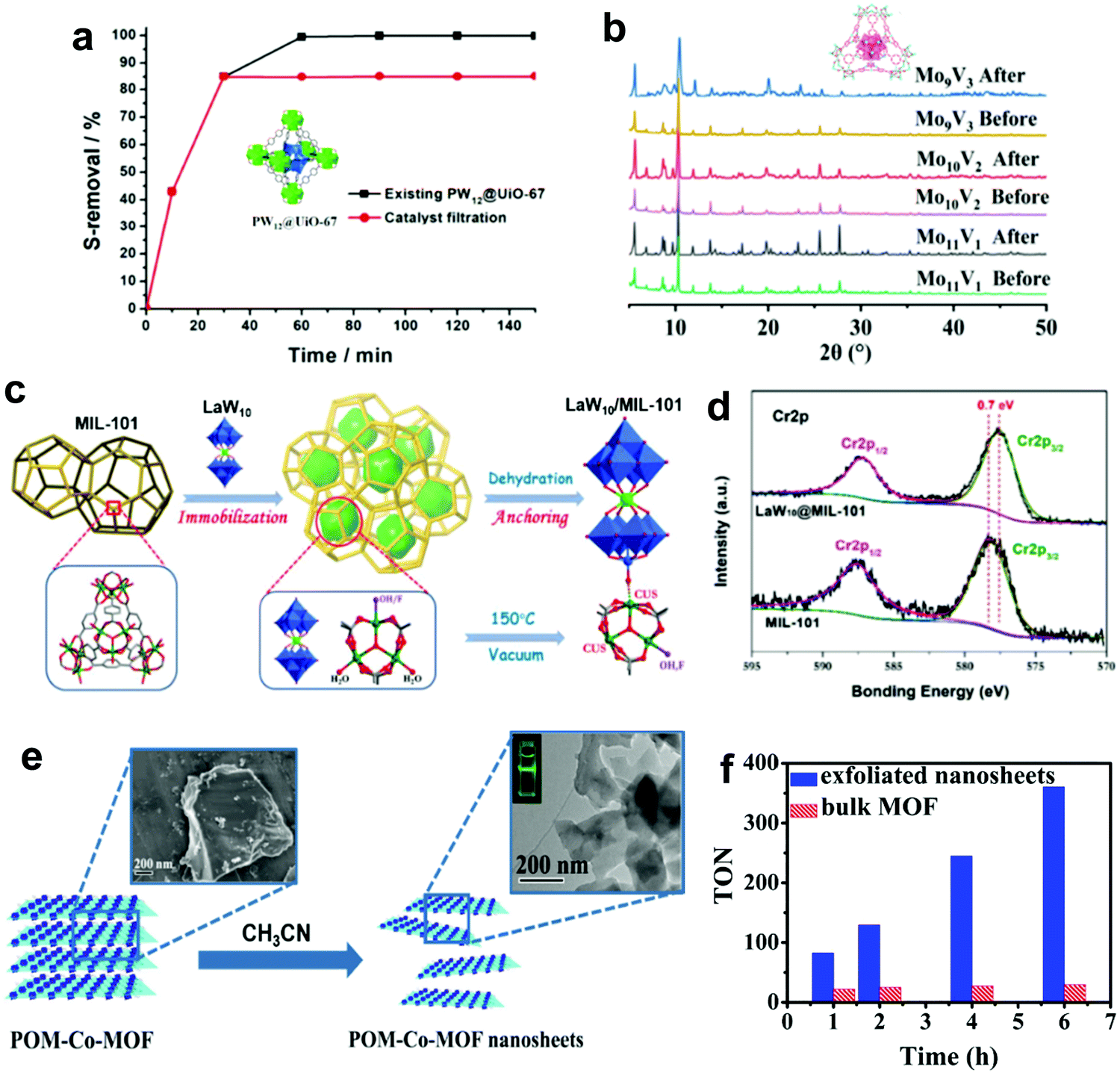 | ||
| Fig. 14 (a) DBT removal with and without the elimination of HPW/UiO-67 at 70 °C in the presence of H2O2. Reproduced with permission.156 Copyright 2018, Royal Society of Chemistry. (b) PXRD spectra of Mo11V1, Mo10V2 and Mo9V3 on rht-MOF-1 before and after ODS. Reproduced with permission.159 Copyright 2020, Elsevier Ltd. (c) Fabrication route for encapsulating LaW10 into MIL-101. (d) High-resolution Cr 2p XPS spectra of LaW10/MIL-101 and MIL-101. Reprinted with permission.160 Copyright 2020, Elsevier Inc. (e) Exfoliation of POM-Co-MOF with the SEM (left) and TEM (right) images. (f) TON of DBT conversion for bulk POM/Co-MOF and POM/Co-MOF nanosheets. Reproduced with permission.161 Copyright 2018, Wiley-VCH. | ||
As is well known, the catalytic activity of a heterogeneous bulk catalyst is substantially restricted by its weak mass transfer in the liquid phase, which is detrimental to interactions with reactants. To address the mass transfer barrier, Xu et al. used a wet ball-milling and sonication-assisted top-down exfoliation approach with acetonitrile to delaminate the layer-structured POM/Co-MOF ([Co2(H2O)4(BTX)3][PMo12O40]) into ultrathin nanosheets (Fig. 14e).161 The turnover number (TON) of the nanosheet structure oxidizing DBT using H2O2 as the oxidant was far beyond that of the bulk structure (Fig. 14f). The fabulous ODS performance of POM/Co-MOF nanosheets in model oil was attributed to the comparably large surface area promoting the development of surface active sites and accelerating the interactions between catalyst and reactant. However, the application of sonication during the exfoliation process would be unfeasible in industrial scale production due to its enormous energy consumption. The preparation of POM/Co-MOF nanosheets via bottom-up approach can overcome this stumbling block but requires profound investigations from researchers in this fast-moving field.
Interestingly, there are also studies relating to the confinement of POM/MOF composites inside another MOF with a larger cage size to increase the heterogeneity and mobility of the catalyst. Li et al. immobilized PMo6W6O40 (PMW)/copper-based MOF (MOF-199) inside the cavities of mesoporous MCM-41 through a one-pot self-assembly hydrothermal process to further enhance the durability of the catalyst in ODS.162 The extensive surface of MCM-41 supplied numerous anchor sites for immobilization of PMW/MOF-199. Hence, PMW/MOF-199/MCM-41 with rich catalytic sites retained 98.5% DBT oxidation in the presence of O2 for five cycles without any activity degradation. Subsequently, researchers employed MCM-41 as the host for surfactant-modified PMW/MOF-199163 and cesium-doped PMW/MOF-199,164 obtaining drastic recyclability enhancements in ODS research. These findings provide an instructive insight into the encapsulation of POM/MOF in MCM-41, which is a useful tactic for enhancing the stability and reusability of catalysts in ODS.
On the whole, incorporation of a support, such as mesoporous silica and metal-free nanomaterials with robust surface areas, as well as encapsulation in MOFs are relatively effective to heterogenize POMs and preserve the excellent catalytic activity in ODS compared with the precipitation approach. Nonetheless, more studies should focus on strengthening the substrate–support interactions of POM-based catalysts to avoid activity loss due to leaching to improve the recyclability in ODS and the industrial practicality. Table 4 summarizes the performances of various POMs in different ODS systems.
| Catalysts | Reaction temperature (°C) | Oxidant | Extractant | Performances | Ref. | ||
|---|---|---|---|---|---|---|---|
| Sulfur removal (reactant) | Reaction time (h) | Stability (h) | |||||
| [C16mim]2Mo2O11: 1-hexadecyl-3-methyl imidazolium peroxomolybdate; [PSTEtA]3PW12O40: trimethylammonium propanesulfonate phosphotungstate; ChCl/2Ac: choline chloride/acetic acid (acidic DES); ODA: trimethyloctadecylammonium; [BMIM]PF6: 1-butyl-3-methylimidazolium hexafluorophosphate; aptes: (3-aminopropyl)triethoxysilane; C32H68BrN: tetraoctylammonium bromide; DMF: dimethylformamide; SmPMo/TMA-LPMS: samarium-coordinated undecamolybdophosphate/trimethylammonium – large pore mesoporous silica; PIL: poly-ionic liquid; HPW: phosphotungstic acid; AC: activated carbon; rGO: reduced graphene oxide; GO: graphene oxide; mpg-C3N4: mesoporous graphitic carbon nitride; C12PW: [C12mim]3PW12O40 (IL modified phosphotungstic acid); POM: H3PMo6W6O40; HPMo: phosphomolybdic acid; MOF-199: microporous copper-based metal–organic framework; MCM-41: mesoporous silica-based metal–organic framework; SRL-POM: [(CH3)2(C14H29)N(CH2)]2[H3PMo6W6O40]; Cs-POM: Cs-PMo6W6O40; POM/Co-MOF: [Co2(H2O)4(BTX)3][PMo12O40] (BTX = 1,4-bis(1,2,-triazol-1-ylmethyl)benzene). | |||||||
| [C16mim]2Mo2O11 | 50 | H2O2 (30 wt%) | N/A | 98.4% (DBT) | 1 | >6 | 128 |
| [PSTEtA]3PW12O40 | 40 | H2O2 (30 wt%) | ChCl/2Ac | 100% (DBT) | 2.5 | N/A | 129 |
| H3PMo12O40 | 50 | H2O2 (30 wt%) | [BMIM]PF6 | 95% (DBT) | 2 | N/A | 130 |
| ODA–PMo12O40 | |||||||
| H8PV5Mo7O40 | 120 | O2 | H2O | 99% (BT) | 6 | >24 | 131 |
| (20 bar oxygen pressure) | |||||||
| H3PWxMo12−xO40 (x = 1, 3, 6) | 60 | H2O2 (30 wt%) | Acetonitrile | >98% (DBT) | 1 | N/A | 132 |
| PW11/aptes-SBA-15 | 70 | H2O2 (30 wt%) | Acetonitrile | 100% (DBT) | 1 | >8 | 133 |
| Na3PW12O40 | 70 | H2O2 (30 wt%) | N/A | 100% (DBT) | 0.5 | N/A | 137 |
| H3PW12O40 | 100% (DBT) | ||||||
| H3PMo12O40 | ∼80% (DBT) | ||||||
| H4SiW12O40 | ∼20% (DBT) | ||||||
| H3PW12O40 | 70 | H2O2 (30 wt%) | C32H68BrN | 100% (DBT) | 0.5 | N/A | 138 |
| H3PMo12O40 | ∼90% (DBT) | ||||||
| H4SiW12O40 | ∼28% (DBT) | ||||||
| Na2HPW12O40 | 60 | H2O2 (30 wt%) | Acetic acid | 98% (DBT) | 2.5 | N/A | 139 |
| [V(VW11)O40]4− | 60 | H2O2 (30 wt%) | DMF | 99% (DBT) | 5 | N/A | 142 |
| [PVW11O40]4− | 88% (DBT) | ||||||
| [W6O19]2− | 87% (DBT) | ||||||
| [PV2Mo10O40]4− | 86% (DBT) | ||||||
| PIL-H2W12O4210− | 30 | H2O2 (30 wt%) | N/A | 95.5% (DBT) | 1.5 | >8 | 165 |
| PW4 | 70 | H2O2 (30 wt%) | Acetonitrile | 100% | 2 | <20 | 57 |
| PW4/TMA-SBA-15 | 100% | >30 | |||||
| TeW6 | Ambient | H2O2 (30 wt%) | Acetonitrile | 89% (thioanisole) | 6 | >30 | 146 |
| TeW6Cu | Ethanol | 89% (thioanisole) | <30 | ||||
| Te2W8Cu2 | 99% (thioanisole) | <30 | |||||
| CsPW12O40 | 60 | TBHP (70 wt%) | N/A | 50% | 2 | <12 | 145 |
| SmPMo/TMA-LPMS | 70 | H2O2 (30 wt%) | [BMIM]PF6 | 100% | 1 | >4.5 | 147 |
| H3PMo12O40/SiO2–C nanospheres | 40 | H2O2 (30 wt%) | Acetonitrile | 99% (DBT) | 3 | >15 | 166 |
| HPW/AC | 90 | H2O2 (30 wt%) | N/A | ∼90% (Th) | 2 | N/A | 149 |
| HPW/rGO (electro-chemical method) | 60 | H2O2 (30 wt%) | Methanol | 95% (DBT) | 8 | >40 | 150 |
| HPW/rGO (chemical reduced method) | 85% (DBT) | <16 | |||||
| HPW/GO | 60 | H2O2 (30 wt%) | Acetonitrile | 100% (DBT) | 0.5 | >8 | 151 |
| HPW/mpg-C3N4 | 60 | H2O2 (30 wt%) | Methanol | 100% (DBT) | 1 | >37.5 | 152 |
| TBAPMo11Cu/CuO | 35 | H2O2 (30 wt%) | Polyethylene glycol | 98% (DBT) | 1 | >5 | 153 |
| C12PW/g-C3N4 | 60 | H2O2 (30 wt%) | N/A | 100% (DBT) | 1 | >5 | 154 |
| Ag/HPW single-walled nanotubes | 20 | H2O2 (30 wt%) | N/A | 98% (diphenyl sulfide) | 1 | >5 | 167 |
| POM/PIL | 100 | O2 | N/A | 66.88% (DBT) | 3 | N/A | 155 |
| POM/PIL/graphene | 100% (DBT) | >18 | |||||
| HPW/UiO-67 | 70 | H2O2 (30 wt%) | Acetonitrile | 99.5% (DBT) | 1 | <8 | 156 |
| HPMo/UiO-67 | 50 | H2O2 (30 wt%) | Acetonitrile | 99.5% (DBT) | 0.5 | >2.5 | 157 |
| HPW/UiO-66 | Ambient | H2O2 (30 wt%) | Acetonitrile | 99.7% (DBT) | 0.42 | >1.67 | 168 |
| Defective HPMo/UiO-66 | 60 | H2O2 (30 wt%) | Acetonitrile | 100% (DBT) | 1 | >15 | 158 |
| PMo11V1/rht-MOF-1 | 70 | H2O2 (30 wt%) | Acetonitrile | 90% (DBT) | 0.83 | >5.83 | 159 |
| PMo10V2/rht-MOF-1 | 92% (DBT) | >5.83 | |||||
| PMo9V3/rht-MOF-1 | 96% (DBT) | >5.83 | |||||
| POM/MOF-199 | 85 | O2 | N/A | 61.4% (DBT) | 3 | <9 | 162 |
| POM/MCM-41 | 42.2% (DBT) | <9 | |||||
| POM/MOF-199/MCM-41 | 98.5% (DBT) | <24 | |||||
| MOF-199/MCM-41 | 60 | O2 | [Bmim]BF4 | 20.2% (DBT) | 2.5 | N/A | 163 |
| SRL-POM/MCM-41 | 45.1% (DBT) | N/A | |||||
| SRL-POM/MOF-199 | 64.9% (DBT) | N/A | |||||
| SRL-POM/MOF-199/MCM-41 | 100% (DBT) | <37.5 | |||||
| Cs-POM/MOF-199/MCM-41 | 80 | O2 | N/A | 99.6% (DBT) | 3 | <45 | 164 |
| LaW10/MIL-101 | 60 | H2O2 (30 wt%) | Acetonitrile | 99.1% (DBT) | 3 | >14 | 160 |
| Bulk POM/Co-MOF | 80 | H2O2 (30 wt%) | Acetonitrile | 100% (DBT) | 4 | >20 | 161 |
| Exfoliated POM/Co-MOF nanosheet | 50% (DBT) | >20 | |||||
| Mo123 | 62.5 | H2O2 (30 wt%) | Acetonitrile | 75% (DBT) | 0.5 | N/A | 148 |
| Mo123/IL-functionalized magnetic silica nanoparticles | 99.97% (DBT) | >2 | |||||
3.5 Application of metal–organic frameworks (MOFs) in ODS
As an arising class of porous and flexible materials, MOFs has been widely explored for numerous applications, especially catalysis, gas processing and storage.169 MOFs are crystalline network structure complexes comprised of inorganic metal-containing units, or secondary building units (SBUs), and rigid organic ligands which act as coordination centers and the significant backbone that links all the SBUs together, respectively.170 By assembling different SBUs and organic ligands, a target MOF with unique topographies and properties in accordance with its functionality for specific chemical reactions can be facilely fabricated. The resulting cage-like structure material is endowed with several auspicious properties, including an abundant surface area, exceptional porosity, unique structural diversity and tailorable morphology, which offer remarkable potential for its integration in the ODS of fuels.170–172Solvothermal method is commonly applied to construct MOFs by heating a mixture of organic solvent, metal precursors and organic ligands to above the boiling point of the solvent in an autoclave under autogenous pressure to stimulate the nucleation and growth of nanoparticles.173 Before application, the generated MOFs undergo thermal activation to remove the guest solvent molecules in the internal pores of the MOFs in order to generate Lewis acid sites for the catalytic reaction.174 By adjusting the synthesis parameters, including temperature, reaction time and so forth, the textural properties and sizes of MOFs can be shaped precisely according to the interest of the creator. Despite the outstanding simplicity of the solvothermal method, its lengthy reaction time, huge demand for relatively expensive solvent and generation of tremendous solvent waste have become a bottleneck for industrial scale production. These arduous challenges have inspired the search for alternative synthesis routes like microwave heating, electrochemical, mechanochemical and sonochemical methods to fabricate MOFs with the desired properties.175 Currently, the alternative synthesis techniques are still not ready for industrial application due to limitations such as high energy consumption, complicated procedures and restricted application. Therefore, extensive research is essential to lay a foundation for manufacturing MOFs through a facile, economical, time-saving and commercially feasible synthesis route.
Drawn by the valuable catalytic properties, Kim and co-workers studied the activity of a titanium-based MOF, MIL-125, and amine-functionalized MIL-125 (NH2-MIL-125) in desulfurizing model fuels in the presence of cumene hydroperoxide (CHP) as the oxidant.176 Both catalysts were prepared by solvothermal method and microwave heating method before activation under vacuum for 12 h at 150 °C. Owing to their microporous features, the performances of MIL-125 and NH2-MIL-125 in DBT conversion were merely 36% and 9%, respectively, as the molecular size of DBT is much larger than the pores of the catalysts. The narrow pores immensely hindered the diffusion of DBT into the internal surfaces of this catalyst with plentiful defect Ti-sites and markedly decreased the interaction between DBT and the active sites. In contrast to NH2-MIL-125, the pore dimensions in MIL-125 were comparably large, which contributed to its higher activity in DBT conversion. Additionally, the functionalization of amine groups on MIL-125 led to the enlargement of particle size, diminishing the amount of vacant active sites on the external surface for ODS and causing the drop in catalytic activity. The inefficient functionalization of the NH2 group was also shown in the work of Valles-Garcia et al.177 It was reported that NH2-MIL-101(Cr) offered similar thiophenol (Thl) conversion (30%) as the parent MIL-101(Cr) in a solvent-free ODS system using O2 as the oxidant. In the same work, the functionalization of the NO2 group on MIL-101(Cr) remarkably enhanced the Thl conversion to about three times greater than pristine MIL-101(Cr). The magnificent enhancement of ODS activity was ascribed to the greater electron-withdrawing property of the NO2 group which drew the electrons of Thl to form the Thl radical. Likewise, Liao et al. discovered that NO2-UiO-66 exhibited the highest DBT oxidation (97%) compared to UiO-66 with Br (∼77%) and NH2 (∼38%) functional groups.178 The functionalization of an electron acceptor group (NO2) in MOFs is preferred over an electron donor group (NH2) to escalate the ODS activity by modifying the Lewis acidity of the parental MOFs.
The poor ODS activity of MIL-125 also motivated Zhang's team to alter its morphology by varying the molar amount of Ti precursors in the solvothermal synthesis.179 The increase of Ti precursors led to excessive Ti cations in solution which accelerated the nucleation of particles, thereby suppressing the coalescence of particles to form larger granules. The MIL-125 with highest molar amount of Ti precursors (MIL-125-S) possessed the smallest particle size. In contrast with other materials, the increase of the surface area in MIL-125 by reduction of particle size failed to significantly intensify the removal of DBT and BT (Fig. 15a). This implies that the micro-sized pores greatly restricted the accessibility of the active sites for the aromatic sulfur constituents. In fact, most of the active sites in MOFs are concentrated on the internal surface, not on the external surface.
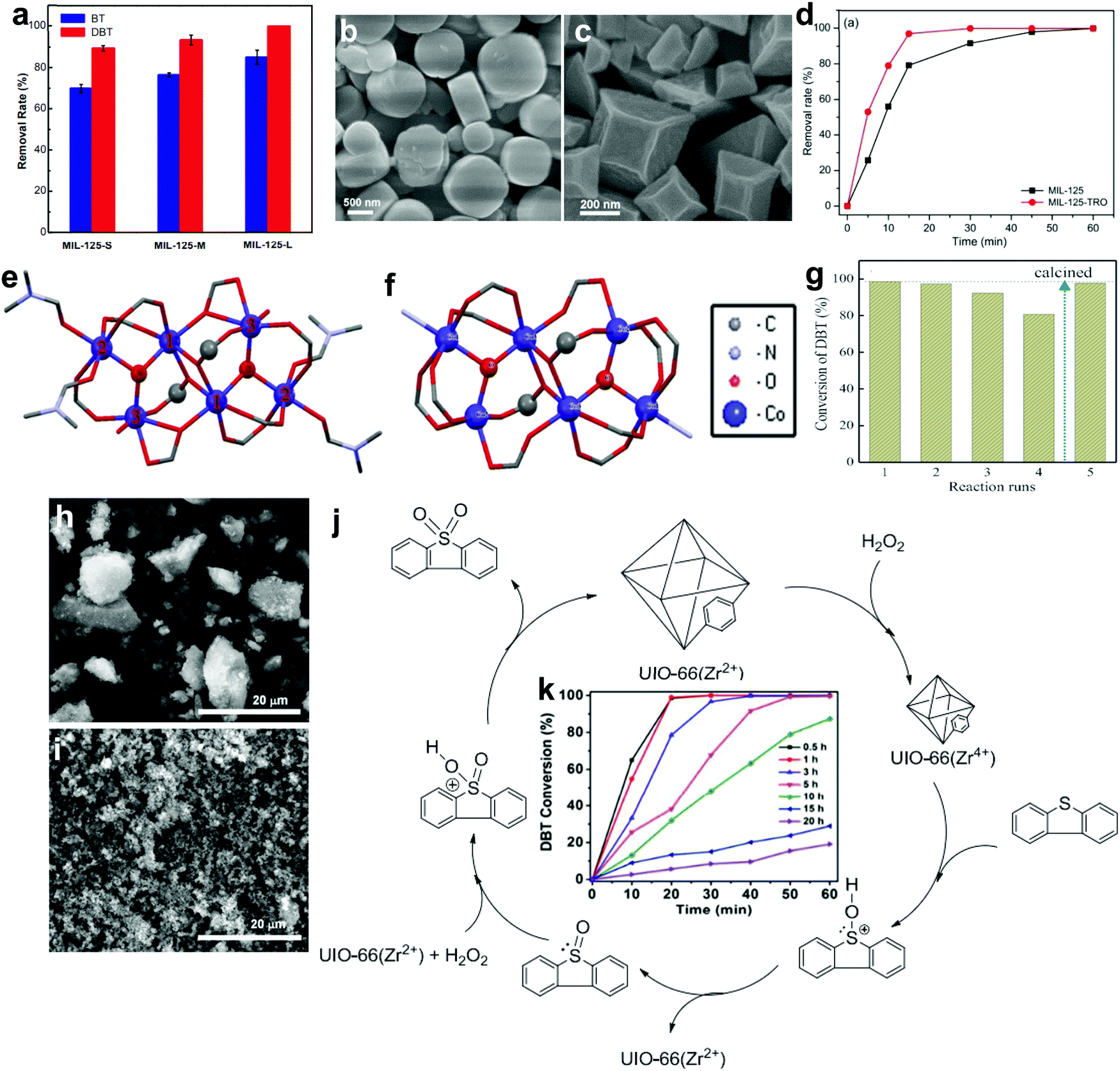 | ||
| Fig. 15 (a) Removal rate of BT and DBT over various MIL-125, namely MIL-125-S, MIL-125-M and MIL-125-L, with 3.6 mmol, 3 mmol and 1.5 mmol Ti(OC4H9)4, respectively, as Ti precursor. Reprinted with permission.179 Copyright 2018, Elsevier Ltd. SEM images of (b) MIL-125 and (c) MIL-125-TRO (truncated octahedral shape) and (d) their DBT removal in ODS at 60 °C using H2O2. Reprinted with permission.180 Copyright 2020, Royal Society of Chemistry. Ball and stick model for the SBUs of (e) TMU-10 and (f) TMU-12. Reprinted with permission.182 Copyright 2015, American Chemical Society. (g) DBT conversion over hierarchical Ti-beta zeolite for five ODS cycles with a duration of 0.5 h per cycle at 60 °C in the presence of TBHP. Reprinted with permission.183 Copyright 2021, Elsevier Inc. SEM images of (h) UiO-66-S and (i) UiO-66-MW. Reprinted with permission.184 Copyright 2019, Multidisciplinary Digital Publishing Institute. (j) ODS mechanism of DBT when utilizing UiO-66 in the presence of H2O2. Reprinted with permission.185 Copyright 2017, Elsevier Ltd. (k) DBT conversion at 60 °C with H2O2 over several UiO-66 with synthesis times of 0.5 h, 1 h, 3 h, 5 h, 10 h, 15 h and 20 h. Reprinted with permission.186 Copyright 2018, Royal Society of Chemistry. | ||
Recently, Yang et al. modified the morphology of MIL-125 (Fig. 15b) through a coordination modulation technique.180 MIL-125 with a truncated octahedral structure (MIL-125-TRO) (Fig. 15c) was created after modulation with butyric acid. The construction of a (101) facet in MIL-125-TRO strengthened its activity in ODS compared to pristine MIL-125 (Fig. 15d). This phenomenon signified a facet-dependence feature of MIL-125 which could significantly affect its ODS activity. Instead of butyric acid, Ye et al. introduced acetic acid in the preparation of titanium terephthalate (Ti-BDC) to modulate the structure.181 Notably, acetic acid created defect sites on Ti-BDC and also played a role in limiting Ti-BDC growth during the solvothermal process. As a result, the improved accessibility of active sites helped Ti-BDC attain complete oxidation of DBT in 10 min at room temperature. While catalyzing ODS, Ti-BDC also acted as the adsorption panel to trap polar sulfone. This is greatly conducive to practical applications because it eliminates the necessity of a further extraction stage to remove sulfone products. The incorporation of an appropriate acid in the synthesis of MOFs can tailor the structure by constructing defect sites, controlling the growth, and removing some organic ligands from the framework of MOFs, thus eventually increasing the exposure of active sites and the catalytic performance in ODS.
In a different work, Masoomi et al. utilized solvothermal method to derive two cobalt-based MOFs, TMU-10 from 4,4′-oxybisbenzoic acid (oba) and TMU-12 from oba and pyrazine organic ligands.182 Impressively, TMU-12 (Fig. 15e) demonstrated a larger void space and window size compared to TMU-10 (Fig. 15f). This further facilitated the passage of aromatic sulfur compounds to the unsaturated active sites near the Co center through the pores. Benefiting from the higher exposure of active sites, TMU-12 achieved better performance in DBT removal (75.2%) after 8 h than TMU-10 (40.5%) in the presence of TBHP. In the research work of Ma et al., hierarchical Ti-beta zeolite was constructed from siliceous FAU zeolite as a catalyst for ODS through interzeolite transformation.183 The application of CTAB as the directing agent to control the crystal growth during the synthesis process contributed to larger crystal sizes and pores in the resulting catalyst. Owing to the presence of innumerable mesopores, the diffusion limitation of sulfur compounds into the inner surface of Ti-beta zeolite was addressed, causing a substantial enhancement of DBT oxidation within 30 min from 34.2% to 98.5%. Despite the unprecedented ODS activity, Ti-beta zeolite underwent severe deactivation after two ODS cycles and required calcination to fully regenerate its catalytic activity (Fig. 15g). Significantly, in order to maximize the ODS activity of MOFs, the expansion of aperture size is necessary to allow the diffusion of sulfur compounds into the interior space, thus facilitating the interactions between sulfur compounds and unoccupied active sites.
Very recently, the application of the microwave-assisted heating method to fabricate MOFs has attracted considerable attention because of its rapid synthesis time and ability to modify the morphologies of MOFs. As reported in the work of Viana et al., Zr-based MOFs (UiO-66) prepared by conventional solvothermal method (UiO-66-S) and microwave-assisted heating method (UiO-66-MW) exhibited dissimilar performances as a result of their structural morphologies.184 Owing to the swift reaction in the microwave-assisted method, the growth of nanoparticles was inhibited, while nucleation was accelerated. Consequently, a smaller particle size was found in UiO-66-MW (Fig. 15i) than in UiO-66-S (Fig. 15h). An identical situation occurred in the research of Kim et al., signifying that the particle dimensions of NH2-MIL-125 synthesized by microwave heating method were smaller than those synthesized by solvothermal method.176 With a larger surface area, UiO-66-MW exhibited significant catalytic activity in ODS (>85%) for three consecutive cycles, whereas the activity of UiO-66-S experienced a sharp decline after the second cycle due to catalyst deactivation. This was because UiO-66-MW provided more active sites for ODS than UiO-66-S did, which contributed to the superior recyclability without serious activity degradation. Furthermore, the shorter synthesis time performed a vital role in generating more defects on the surface of UiO-66 and explicitly boosted the ODS activity. This behavior was revealed in the ODS mechanism (Fig. 15j). The increase of defects in UiO-66, which was the significant factor in liberating unsaturated Zr cation sites with Lewis acidity, contributed to the increment of electron acceptors for H2O2 activation to create Zr-peroxo complexes.185 After the adsorption of DBT on the active site, it was converted to DBT sulfoxide and DBT sulfone, followed by its desorption from UiO-66. A similar synthesis duration effect on ODS activity was also substantiated by Xiao and colleagues, who revealed that the decrease of reaction time in the solvothermal method enhanced the DBT conversion by UiO-66 (Fig. 15k).186 Therefore, it can be concluded that the synthesis time and the morphologies of MOFs, including particle sizes and the number of defect sites, can be tuned and become more favorable toward ODS.
In a recent study, activation of UiO-66 was performed by incorporating anions into the framework via simple mixing with the respective salts, including TiCl4, ZrCl4 and TiBr4.187 Unfortunately, the introduction of Br anions engendered the deactivation of UiO-66, leading to a poorer desulfurization efficiency than pristine UiO-66 in the ODS system with H2O2 at 50 °C (Fig. 16a). On the other hand, Cl anions successfully constructed more defect sites in the framework of UiO-66 and contributed to an essential ODS activity improvement. Despite this work manifesting the capability of Cl anions to elevate the catalytic activity of MOF in ODS, the ODS reaction pathways and the reason for the activity loss after the incorporation of Br anions have not been comprehensively investigated, which is crucial for improving this modification method in the future.
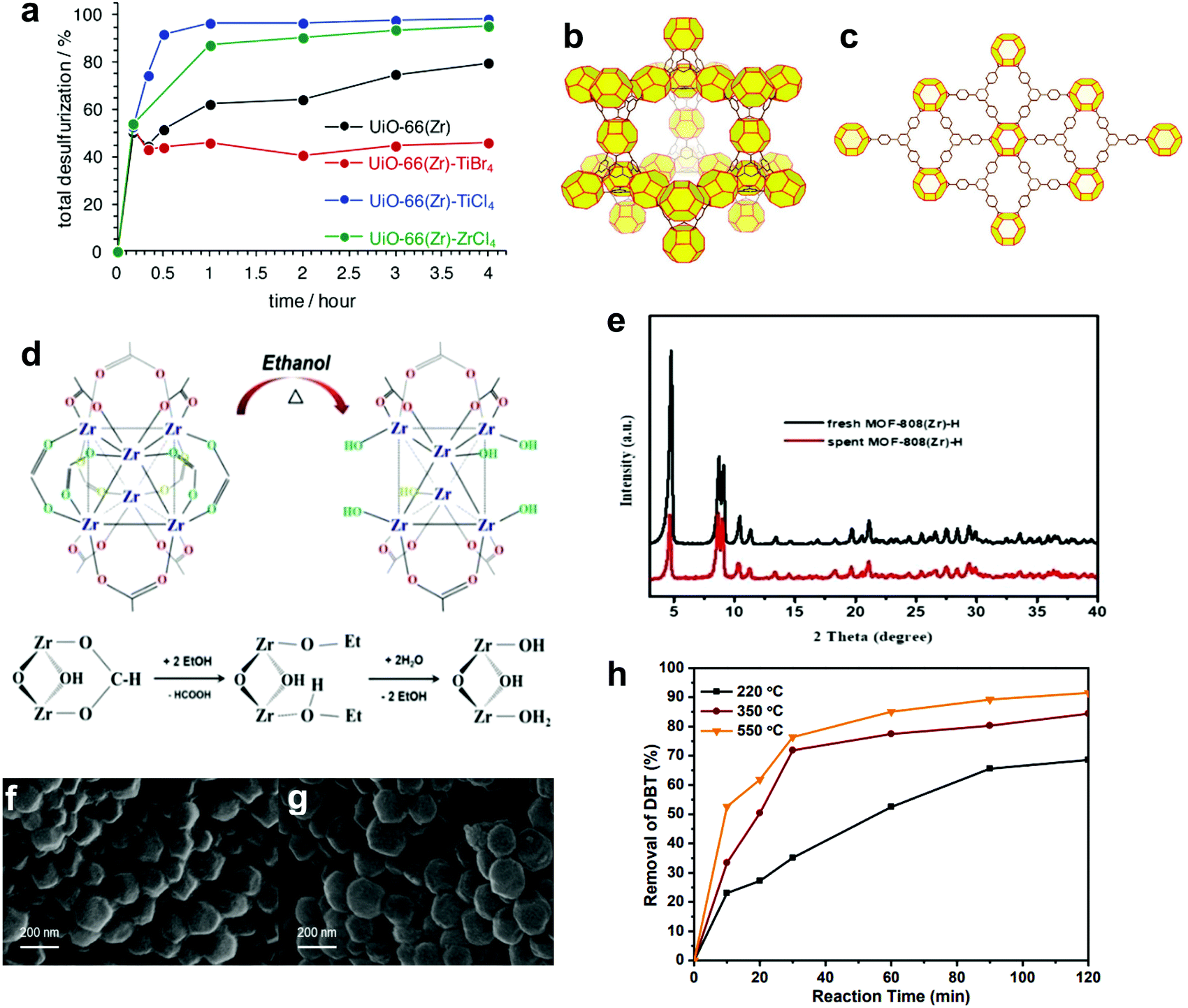 | ||
| Fig. 16 (a) Desulfurization efficiency in ODS with H2O2 at 50 °C using pristine UiO-66(Zr) and post-treated UiO-66(Zr) with various salts, including TiBr4, TiCl4 and ZrCl4. Reprinted with permission.187 Copyright 2021, Elsevier B.V. Structures of (b) MOF-808 and (c) UMCM-309. Reprinted with permission.188 Copyright 2018, Wiley-VCH. (d) Mechanism of the hot ethanol treatment in MOF-808. (e) XRD analysis of the modified MOF-808 before and after the ODS reaction. The SEM images of modified MOF-808 (f) before and (g) after the ODS reaction. Reprinted with permission.190 Copyright 2020, Wiley-VCH. (h) DBT conversion at 60 °C using Ti-modified MIL-101 and CHP synthesized at different calcination temperatures. Reprinted with permission.191 Copyright 2020, Multidisciplinary Digital Publishing Institute. | ||
In another modification route, Fu and fellows carried out postsynthetic ligand exchange (PSLE) to enhance the catalytic activity of Zr-based MOFs, namely MOF-808 and UMCM-309 in ODS.188 In PSLE, the original structural linkers in MOFs are substituted with new linkers to modify the topologies and morphologies.189 In this research, the formate ligands in MOF-808 and UMCM-309 were removed by reacting with methanol to induce more unoccupied Zr open sites for ODS. The increment of missing formate ions in the structure of the MOFs is notably conducive to the conversion of sulfur pollutants. However, UMCM-309 with fewer formate ions achieved a lower conversion rate of BT (18%) than did MOF-808 (60%) after 8 h. The deviation stemmed from the fact that the 3D structured MOF-808 (Fig. 16b) possessed a greater specific surface area, aperture size and number of Lewis acid sites compared to UMCM-309 with its 2D layered structure (Fig. 16c). This accelerated the diffusion of BT into the internal surfaces of MOFs, thereby boosting the reaction between sulfur compounds and Zr active sites. Future work on the application of UMCM-309 in ODS can be strengthened by reconstructing the 2D layered structure into 2D ultrathin sheets through intercalation or exfoliation to unravel the upper limit of ODS activity. Owing to the time-consuming drawback of PSLE, Gu and co-workers amended the strategy by treating MOF-808 with hot ethanol for 4 h instead of methanol for 2 d to detach the formate ligands from the structure (Fig. 16d).190 This action offered crucial enhancement in the ODS activity of MOF-808 by creating more defect Zr sites. The reformed MOF-808 oxidized 99% of DBT after 20 min with CHP, which was significantly beyond the performance of original MOF-808 (20%). In addition, the modified MOF-808 with superior robustness exhibited identical morphologies before and after ODS, proving the absence of leaching (Fig. 16e–g). Therefore, the removal of ligands from the structure of MOFs is essential and efficacious for achieving ultra-deep ODS via increasing the number of open boundaries and defect sites for the adsorption and oxidation of reactants.
Li et al. modified Cr-based MOF MIL-101 with Ti via IWI and calcination method to increase the catalytic sites of MIL-101.191 Fascinatingly, TiO2 and Cr2O3 nanoparticles were created in the pores of MIL-101 during the calcination process. The increase of calcination temperature also led to the enhancement of DBT conversion utilizing CHP at 60 °C (Fig. 16h). This was attributed to the increment of metal oxides with outstanding catalytic activity in the pores of MIL-101. Unfortunately, the catalyst exhibited poor stability and its activity fell significantly after the second run due to loss of active sites. Therefore, the low stability of the nanocomposite requires the development of strong interactions between the host and guest particles to prolong its durability in ODS.
In MOFs, the Lewis acid sites in the internal structure and their pore size are the major decisive factors for ODS activity. In spite of the fact that the direct application of MOFs in the ODS of fuels is viable, only a few species of MOFs are capable of achieving impressive desulfurization and becoming candidates for industrial application. Moreover, most of the MOFs possess microporous characteristics which hinder the accessibility of the unsaturated active sites inside the pores to reactants and requires structural modification before use. On that account, previous researchers have attempted to immobilize or hybridize porous MOFs with massive surface area as supports for various types of materials, including metal oxides and POMs, which possess extraordinary catalytic activity in ODS. Table 5 presents the ODS performances of various MOFs with different operating parameters.
| Catalysts | Reaction temperature (°C) | Oxidant | Extractant | Performances | Ref. | ||
|---|---|---|---|---|---|---|---|
| Sulfur removal (reactant) | Reaction time (h) | Stability (h) | |||||
| CHP: cumene hydroperoxide; TBHP: tert-butyl hydroperoxide. | |||||||
| MIL-125(Ti) | 80 | CHP | N/A | 36% (DBT) | 6 | N/A | 176 |
| NH2-MIL-125(Ti) | 9% (DBT) | ||||||
| NO2-MIL-101(Cr) | 140 | O2 | N/A | 90% (DBT) | 4.5 | >25 | 177 |
| UiO-66(Zr) | 60 | H2O2 (30 wt%) | N/A | ∼69% (DBT) | 2 | N/A | 178 |
| Br-UiO-66(Zr) | ∼77% (DBT) | N/A | |||||
| NH2-UiO-66(Zr) | ∼38% (DBT) | N/A | |||||
| NO2-UiO-66(Zr) | 97% (DBT) | >10 | |||||
| MIL-125(Ti) | 60 | H2O2 (30 wt%) | Methanol | 100% (DBT) | 4 | >20 | 179 |
| NH2-MIL-125(Ti) | 98% (DBT) | >20 | |||||
| MIL-125(Ti) | 60 | H2O2 (30 wt%) | Methanol | ∼90% (DBT) | 0.5 | N/A | 180 |
| MIL-125-TRO(Ti) | 100% (DBT) | >4.5 | |||||
| Defect-rich porous titanium terephthalate | Ambient | CHP | N/A | 100% (DBT) | 0.17 | >0.83 | 181 |
| TMU-10(Co) | 60 | TBHP (70 wt%) | N/A | 40.5% (DBT) | 8 | <24 | 182 |
| TMU-12(Co) | 75.2% (DBT) | >32 | |||||
| Hierarchical Ti-beta zeolite | 60 | TBHP (70 wt%) | N/A | 34.2% (DBT) | 0.5 | >2 | 183 |
| Hierarchical Ti-beta zeolite with CTAB | 98.5% (DBT) | >2 | |||||
| Ta-Beta-Re zeolite | 60 | H2O2 (30 wt%) | N/A | 100% (DBT) | 2 | <8 | 192 |
| UiO-66(Zr) | 50 | H2O2 (30 wt%) | Acetonitrile | 99.5% (DBT) | 3 | <12 | 184 |
| UiO-66(Zr) | 60 | H2O2 (30 wt%) | Acetonitrile | 100% (DBT) | 0.33 | <2 | 186 |
| UiO-66(Zr) | 50 | H2O2 (30 wt%) | Acetonitrile | ∼62% | 1 | N/A | 187 |
| UiO-66(Zr)-TiBr4 | ∼45% | N/A | |||||
| UiO-66(Zr)-TiCl4 | ∼96% | >12 | |||||
| UiO-66(Zr)-ZrCl4 | ∼86% | N/A | |||||
| MOF-808(Zr) | 60 | TBHP (70 wt%) | N/A | 88% (DBT) | 8 | <32 | 188 |
| UMCM-309(Zr) | 59% (DBT) | N/A | |||||
| MOF-808(Zr) | 50 | CHP | N/A | >99% (DBT) | 0.33 | <2 | 190 |
| Ti-Modified MIL-101 | 60 | CHP (70 wt%) | N/A | 90% (DBT) | 0.5 | <6 | 191 |
3.6 Application of metal-free nanomaterials in ODS
Regardless of their spectacular performances in ODS, the inescapable drawbacks of metal-based catalysts, like high toxicity and leaching risk, which are inimical to the environment and fuel oil quality, have induced a significant reduction in the confidence of the petrochemical industry to invest in and upscale ODS.193 The challenges in sustainability and economic and environmental concerns have urged research communities to seek metal-free catalysts which are environmentally friendly and low priced. For their intrinsic properties of large surface area, excellent robustness and high susceptibility to redox reactions, carbonaceous materials stand out as the major species in the metal-free materials field and are deemed to be key for approaching complete desulfurization of diesel fuels in the coming decades.194,195To address environmental friendliness and sustainability, He et al. reported a creative and sustainable technique to derive N,O-doped porous graphene (N,O-PG) from the coke waste of the petroleum refinery industry.199 Basically, a ball-milling process was first conducted in the presence of coke and urea, accompanied by annealing to activate the catalyst and decompose the layered structure graphene into an ultrathin nanosheet structure (Fig. 17a). The auspicious morphology of N,O-PG nanosheets led to 98.5% DBT removal with O2 in 5 h reaction, a value markedly higher than that of commercial graphite (Fig. 17b). Significantly, the presence of nitrogen atoms increased the electron density of porous graphene and enriched the porous graphene surface with electrons, thus directly enhancing the activation of O2 and the ODS performance. Similarly, the significant role of N was also discovered in N-doped porous carbon transformed from waste air-aid paper.200 High specific surface area was another crucial feature that successfully promoted catalytic activity and stability in ODS, as N-doped porous carbon demonstrated complete conversion of H2S with O2 at 190 °C and sustained outstanding ODS activity for more than 40 h. Indeed, the doping of N is a promising strategy to tailor the electronic structure of metal-free nanomaterials with the aim of catalytic activity improvement in ODS.
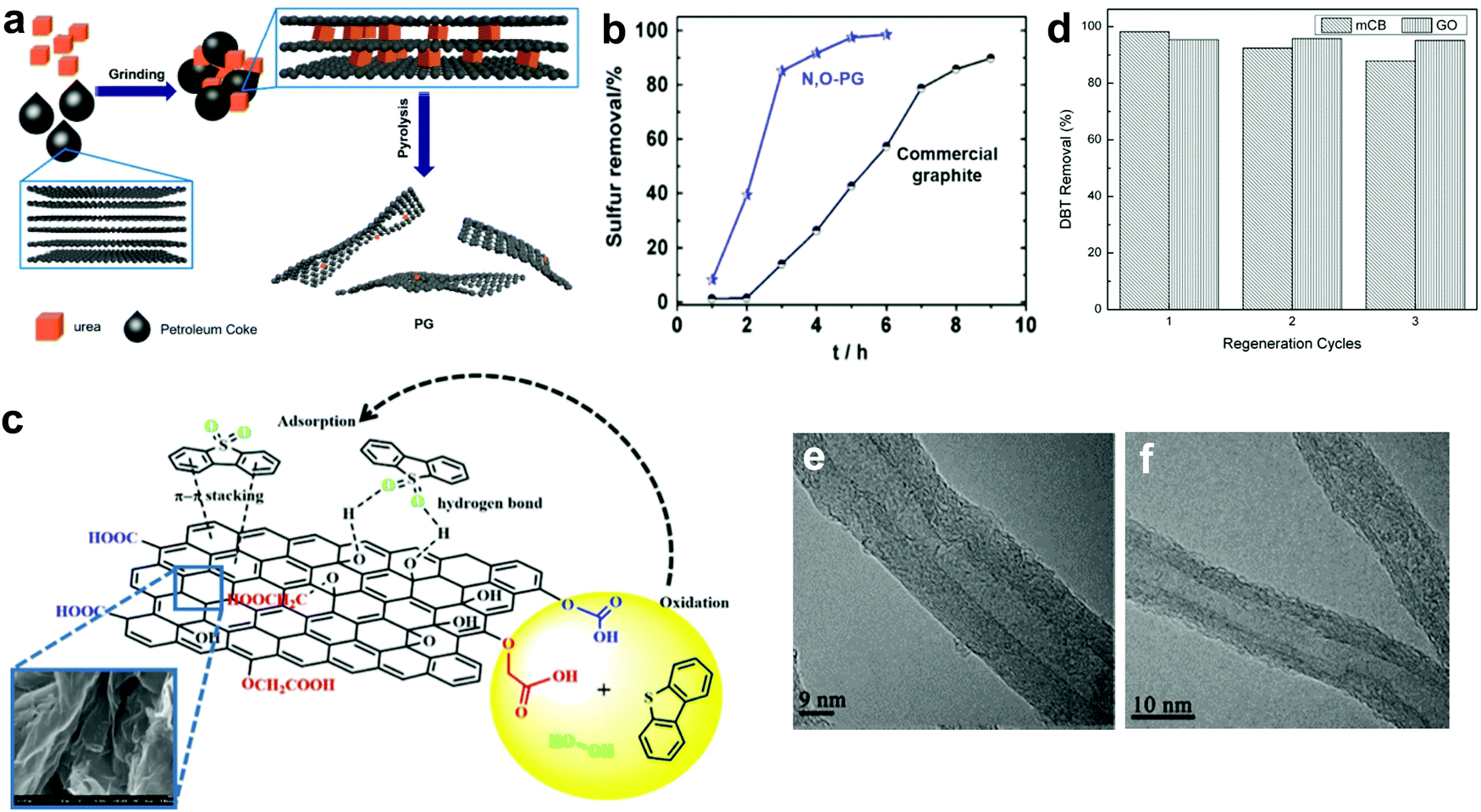 | ||
| Fig. 17 (a) Synthesis pathway of N,O-doped porous graphene. (b) Catalytic activity of N,O-doped porous graphene and commercial graphite in removing DBT using O2 at 120 °C. Reprinted with permission.199 Copyright 2019, American Chemical Society. (c) Proposed route for the oxidation of DBT with H2O2 by GO with a SEM image of GO. Reproduced with permission.203 Copyright 2017, Elsevier Ltd. (d) DBT removal by mCB and GO at 60 °C in the presence of O2 for three regeneration cycles at 5 h per cycle. Reprinted with permission.204 Copyright 2017, Elsevier B.V. HRTEM images of (e) CNT and (f) oxidized CNT. Reprinted with permission.206 Copyright 2019, Elsevier B.V. | ||
In regard to GO, the emergence of rich oxygen-containing functional groups like hydroxyl groups and carboxyl groups on the surface, which are valuable reaction sites for catalysis, has inspired numerous researchers to investigate its activity in ODS.201 For example, Abdi and co-workers prepared GO using the modified Hummers’ method and studied its catalytic activity in oxidizing sulfides to sulfones with H2O2.202 In the absence of extractive solvent, GO achieved 95% conversion of thioanisole in 20 min owing to its amphiphilic property that allowed uniform dispersion in the organic phase. Apart from H2O2 activation, the hydroxyl groups on the surface of GO also acted as the adsorption platform for sulfone to separate sulfur compounds from the fuels. In another work, Abdi et al. comprehensively revealed the function of different oxygen-containing functional groups on the surface of GO grafted with acetic acid moieties in the ODS mechanism (Fig. 17c).203 First, the activation of H2O2 was stimulated by the carboxyl groups at the edge of GO. After DBT oxidation, the produced polar DBT sulfone strongly chemisorbed on the basal plane of GO, which contained hydroxyl groups, via hydrogen bonding and π–π interactions, thus eliminating the need for solvent to separate the DBT sulfone from the organic phase. Taken together, these works underlined the significant dual roles of GO in the desulfurization of fuels.
To compare the ODS activities of various nanocarbon materials, carbon black, modified carbon black (mCB), GO, activated carbon, carbon nanotubes (CNTs) and activated carbon fiber were synthesized by Zhang and Wang and employed in ODS with O2.204 The results disclosed that mCB and GO attained the highest DBT conversion (97%). Despite possessing identical ODS activity, mCB exhibited poorer recyclability than GO (Fig. 17d). This reflects the deficient robustness of mCB against catalyst deactivation stemming from the inefficient removal of sulfone compounds from adsorption sites on the surface. The lack of oxygen-containing functional groups on the surface of mCB was another critical factor, causing notable activity depletion after recovery. Obviously, the presence of oxygen-containing functional groups on the surface of carbon materials is conducive to the maximization of ODS activity and durability.
The role of the GO derivative reduced GO (rGO) in oxidizing various aromatic sulfur compounds with O2 was investigated by Gu and fellows.205 The rGO took 6 h to reach above 90% sulfur removal at 140 °C. The sluggish ODS reaction rate stemmed from the shortage of oxygen-containing functional groups on the rGO surface. Accordingly, the authors proposed a gas-phase oxidative treatment to increase the oxygen content and the number of unstable defect sites (carbonyl groups) in the structure of rGO. As a result, the oxidized rGO accomplished 100% desulfurization in 2 h under the same operating conditions. In another work, Gu et al. functionalized CNTs with oxygen-containing functional groups by means of nitric acid and sulfuric acid treatment.206 The oxidized CNTs achieved complete desulfurization, compared to 34.8% by CNTs, with H2O2 as oxidant and ILs as extractant. The outstanding ODS performance arose from the formation of more carbonyl groups on the surface of CNTs without compromising the original textural properties (Fig. 17e and f). Similarly, Kampouraki et al. tailored the surface chemistry of commercial activated carbon (SX PLUS) via oxidation reaction with sulfuric acid and nitric acid.207 Profiting from the oxidation treatment, the number of acidic functional groups (carboxyl group) on the surface of SX PLUS was increased, which also denoted the enrichment of active sites. On the other hand, the other decisive factors in catalytic ODS, including the porosity and specific surface area of SX PLUS, experienced a significant diminution. Nonetheless, the catalytic activity of modified SX PLUS in oxidizing 4,6-dimethyldibenzothiophene (4,6-DMDBT) still drastically improved by 25% over pristine SX PLUS. These cases shed light on the significance of oxygen-functional groups on the surface of carbon materials for boosting the activity in ODS.
In view of the poor defect sites in graphene, a novel carbon-based material, carbon nitride (C3N4), which is produced by bridging heptazine units together, has gained much attention in the field of catalysis owing to its facile synthesis approach, plentiful surface chemistry and excellent chemical stability.208,209 A few years ago, Shen and colleagues reported the use of thermal polymerization to produce polymeric C3N4 nanomesh from trithiocyanuric acid with a continuous supply of air at different heating temperatures.210 Interestingly, the heating temperature crucially affected the morphology of C3N4, as exemplified in Fig. 18a and b. At elevated temperatures, the dissociation of C3N4 with stacked layer structure was induced by the large supply of thermal energy. The surface energy between every layer was thermodynamically reduced, leading to the generation of ultrathin sheets of 1.1 nm thickness (Fig. 18c and d). Owing to the 2D ultrathin structure of C3N4 with a gigantic specific surface area, the oxidation of H2S was substantially strengthened. A similar phenomenon was also highlighted in the work of Lei et al., who shaped a graphitic C3N4 (g-C3N4) nanosheet from urea through thermal polymerization accompanied by a calcination process at a different temperature to exfoliate the bulk structure into ultrathin sheets (Fig. 18e).211 With greater exposure of surface active sites, the g-C3N4 nanosheet calcined at 600 °C exhibited the highest H2S conversion with air compared to the other g-C3N4 at lower calcination temperatures (Fig. 18f). It also sustained strong ODS activity for more than six successive runs without severe deactivation. Thus, a higher calcination temperature favors the structural decomposition of g-C3N4, resulting in formation of ultrathin sheets with massive surface area. However, the necessity of a high oxidation temperature (>150 °C) during ODS by g-C3N4 to provide ample activation energy to dissociate the powerful covalent bond in O2 exacerbated the energy consumption and the risk of diesel vaporization, resulting in enormous economic losses.
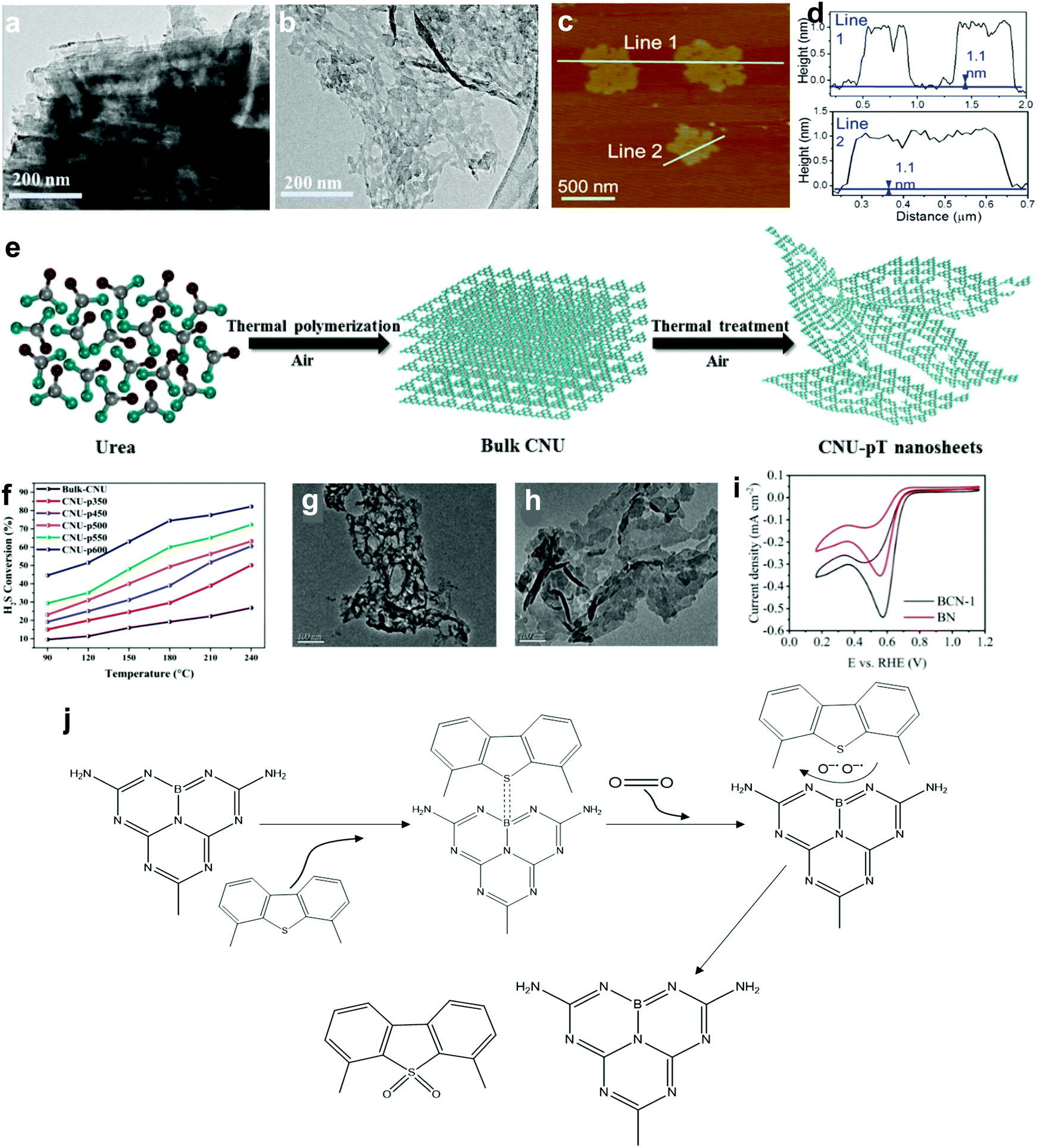 | ||
| Fig. 18 TEM images of C3N4 nanomesh at synthesis temperatures of (a) 500 °C and (b) 600 °C and (c) an AFM image of C3N4 nanomesh at 600 °C with (d) the height profile of lines in the AFM image. Reprinted with permission.210 Copyright 2018, Royal Society of Chemistry. (e) Synthesis route of C3N4 nanosheets from urea. (f) Conversion of hydrogen sulfide with the help of O2 by bulk g-C3N4 and g-C3N4 nanosheets formed at different oxidation temperatures of 350 °C, 450 °C, 500 °C, 550 °C and 600 °C. Reprinted with permission.211 Copyright 2019, American Chemical Society. TEM images of (g) B-C3N4 and (h) g-C3N4. Reprinted with permission.212 Copyright 2019, Elsevier B.V. (i) Cyclic voltammetry curves for B-C3N4 and BN. Reprinted with permission.213 Copyright 2020, Elsevier Inc. (j) Oxidation mechanism of 4,6-DMDBT with O2 molecules by B-C3N4. Redrawn with permission.212 Copyright 2019, Elsevier B.V. | ||
To surmount the unavoidable high energy barrier, Jia et al. attempted to intensify the Lewis acidity of g-C3N4 by doping with boron atoms to increase its catalytic activity.212 Aside from the improvement of the interfacial electronic properties, the boron-doped g-C3N4 (B-C3N4), synthesized from a deep eutectic solvent and urea via annealing (Fig. 18g), was endowed with a higher porosity compared to pristine g-C3N4 (Fig. 18h). This effectively created more active sites on g-C3N4 for ODS. Improved by the morphological changes, the oxidation of 4,6-DMDBT in the presence of air exhibited a sharp increase from 33.6% to 94.1% at only 120 °C. In order to reveal the impact of boron doping, the proposed reaction mechanism for B-C3N4 oxidizing 4,6-DMDBT is shown in Fig. 18j. With its Lewis acid feature, B-C3N4 attracted 4,6-DMDBT and O2 by creating a vigorous Lewis acid–base interaction. After that, the O2 was activated and catalysis proceeded to the oxidation of 4,6-DMDBT to generate 4,6-DMDBT sulfone. Wei et al. also reported the application of B-C3N4 in the ODS of model diesel, but their B-C3N4 was fabricated from a ternary deep eutectic solvent under calcination and spinodal decomposition.213 As expected, B-C3N4 required only 125 °C to achieve 98.4% DBT removal with O2 due to the amelioration of reduction potential in the parent boron nitride that could be observed from the cyclic voltammetry curves (Fig. 18i). In other words, B-C3N4 gained a greater ability to donate electrons to O2 and led to rapid oxygen reduction, forming reactive oxygen species for the oxidation of DBT. Thus, the reaction temperature for activating O2 can be reduced by modifying the electronic property of g-C3N4 through boron doping. During the ODS process, the boron atom was presumed to be the active site for O2 activation, while the carbon atom acted to intensify the delocalization effect of electrons. As evidenced, the vital functions of boron and carbon atoms in ODS were affirmed by Lu's group.214 Although 2D structured B-C3N4 has prospectively become a promising candidate in ODS, its novelty is still limited. Nevertheless, the approach of exfoliating B-C3N4 with a layered structure into ultrathin nanosheets advances the research field. Since an ultrathin structure catalyst has been validated and possesses large specific surface area, this can bring about a crucial breakthrough in catalytic activity while promoting the conservation of thermal energy in ODS.
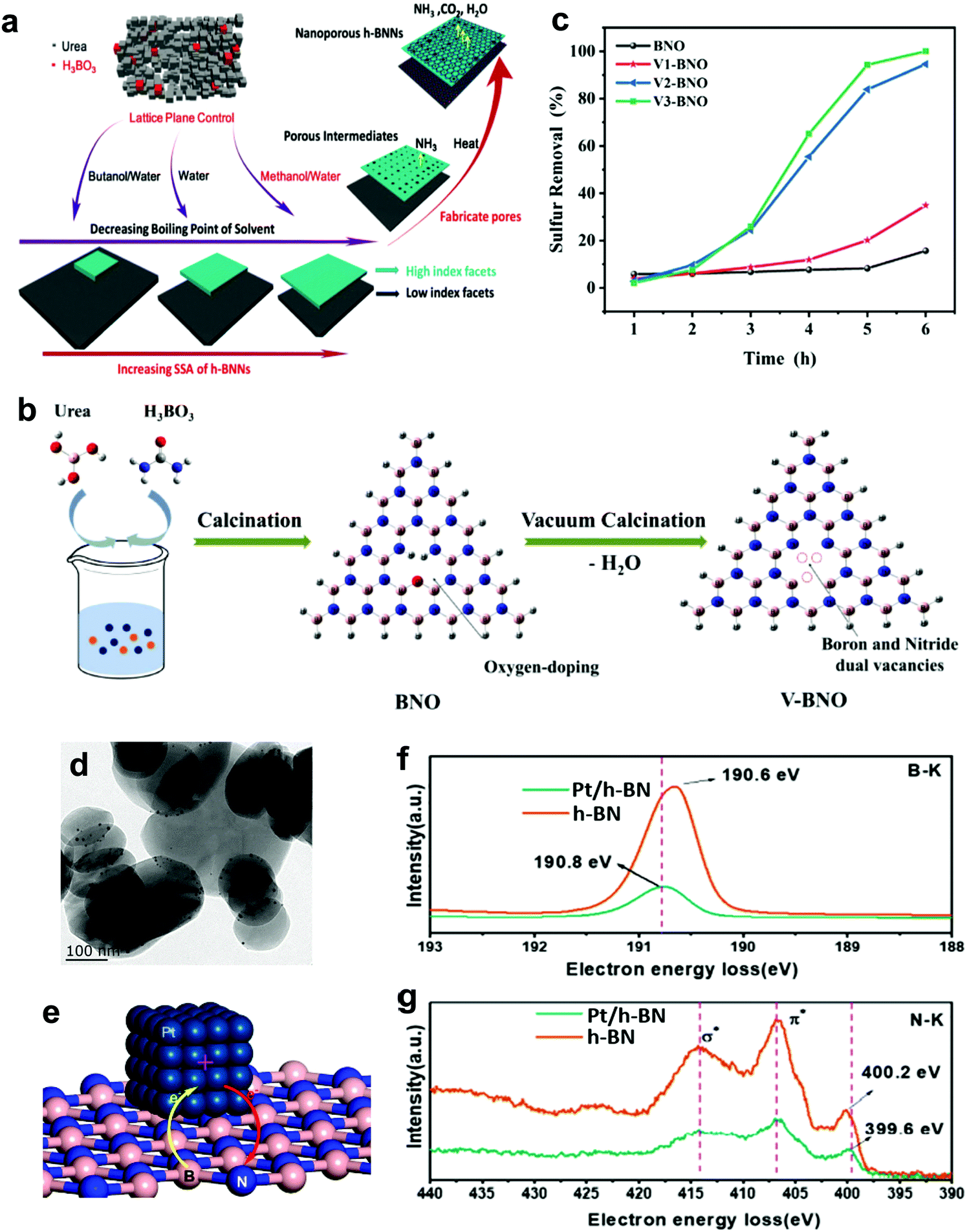 | ||
| Fig. 19 (a) Effects of solvents with different boiling points on the morphology of h-BN. Reprinted with permission.215 Copyright 2016, Royal Society of Chemistry. (b) The route for the fabrication of V-BNO. (c) DBT removal at 125 °C in the presence of O2 over original BNO and V-BNO calcined for 1 h (V1-BNO), 2 h (V2-BNO) and 3 h (V3-BNO). Reprinted with permission.219 Copyright 2020, Wiley-VCH. (d) TEM image and (e) schematic of Pt/h-BN. Electron energy loss spectroscopy (EELS) spectra of Pt/h-BN and bare h-BN for (f) B-K edge and (g) N-K edge. Reprinted with permission.220 Copyright 2020, Elsevier B.V. | ||
Owing to the complexity of the defect sites in BN, Lv et al. revealed the ODS performances and mechanisms of the five major defect sites using O2.218 According to the calculated Gibbs free energies, the activation of O2 decreased in the order of zigzag-B > N-vacancy > zigzag-N > armchair > B-vacancy. With respect to the oxidative activity, zigzag-N was preferable due to its lower energy barrier and exothermic property which promoted the transfer of oxygen radicals to Th. Although previous findings indicated the poor activities of the B-vacancy and N-vacancy, attributed to the blockage of vacant sites by oxygen species, Dai et al. incorporated both B and N vacancies into the structure of oxygen-doped BN (BNO) by detaching some of the hydrogen and oxygen atoms through calcination under vacuum (Fig. 19b).219 The defective BNO (V-BNO) successfully acted as a catalytic promoter to facilitate the activation of O2 in ODS and contributed to the 100% DBT removal. This phenomenon stemmed from the effect of bivacancy sites which enlarge the aperture size of BNO, resolving the blockage of vacant sites by oxygen species. Furthermore, with the increase of the calcination period, more B and N vacancies were constructed in the morphology of BNO and improved the sulfur removal (Fig. 19c). Despite the outstanding ODS performance by V-BNO, profound studies on ameliorating the slow reaction rate are urgently needed to accelerate progress toward industrial application in the future.
In addition to serving as a catalyst, 2D BN, with outstanding stability, very large specific surface area and favorable surface properties, has also become an interesting support to disperse metal nanoparticles in ODS. For instance, Wu et al. supported Pt nanoparticles on 2D h-BN with the objective of improving the catalytic activity in ODS.220 Notably, Pt nanoparticles were immobilized at the edges of h-BN (Fig. 19d) instead of in the center, which is ascribed to the existence of higher electron density at the h-BN edges. The energetic edge interactions stemming from the electron exchange between Pt and h-BN were confirmed by electron energy loss spectroscopy (EELS). The reduction of electron energy loss at the B atom (Fig. 19f) and the increase of electron energy loss at the N atom (Fig. 19g) signified the donation of electrons from the B atom to Pt and the acceptance of electrons at the N atom from Pt (Fig. 19e). In the ODS experiment, Pt/h-BN achieved above 90% sulfur removal with O2 after five cycles. Likewise, He et al. grew PtCu alloy on BN nanosheets via solvothermal method and examined the ability to catalyze the ODS of model fuel.221 Thanks to the robust substrate–support interactions, the aggregation of metal nanoparticles was hindered, resulting in prominent ODS activity. Compared with commercial bulk BN, BN nanosheets provided a larger dispersion platform for PtCu nanoparticles and led to a higher exposure of catalytic sites for ODS. PtCu/BN nanosheets attained 96.1% DBT conversion at 110 °C in 7 h using O2, a result which was substantially better than that of PtCu/commercial BN (51.7%). Overall, BN is a promising support to stabilize metal nanoparticles to achieve outstanding ODS performances.
In summary, metal-free nanomaterials have demonstrated their impressive potential to desulfurize fuels via ODS, but higher reaction temperatures and longer reaction times are required. Accordingly, appropriate modification of the physico-chemical properties must be performed to enhance the catalytic activity of metal-free nanomaterials in ODS to establish a sustainable and rapid ODS process. Table 6 lists representative performances of diverse metal-free nanomaterials in ODS under different operating conditions.
| Catalysts | Reaction temperature (°C) | Oxidant | Extractant | Performances | Ref. | ||
|---|---|---|---|---|---|---|---|
| Sulfur removal (reactant) | Reaction time (h) | Stability (h) | |||||
| BN: boron nitride; WHSV: weight hourly space velocity; GO: graphene oxide; GO/COOH: acetic acid moiety modified graphene oxide; CB: carbon black; AC: activated carbon; rGO: reduced graphene oxide; CNTs: carbon nanotubes [Omim]PF6: 1-methyl-3-octylimidazolium hexafluorophosphate; SX-PLUS: commercial activated carbon; g-C3N4: graphitic carbon nitride; B-C3N4: boron-doped carbon nitride; h-BN: hexagonal boron nitride. | |||||||
| BN | 80 | H2O2 (30 wt%) | N/A | 99.4% (DBT) | 3 | >24 | 217 |
| B4C | 130 | O2 | N/A | 99.5% (DBT) | 8 | >136 | 222 |
| N,O-Doped graphene | 120 | O2 | N/A | 98.5% (DBT) | 5 | <48 | 199 |
| N-Doped porous carbon | 190 | O2 | N/A | 100% (H2S) | 0.6 h−1 (WHSV) | >40 | 200 |
| Acidic GO | 25 | H2O2 (30 wt%) | N/A | 95% (thioanisole) | 0.33 | <4 | 202 |
| GO/COOH | 40 | H2O2 (30 wt%) | N/A | 95% (DBT) | 5 | N/A | 203 |
| GO | 60 | O2 | N/A | 96% (DBT) | 2 | >15 | 204 |
| Modified CB | 97% (DBT) | <15 | |||||
| CB | 90% (DBT) | N/A | |||||
| AC | 78% (DBT) | N/A | |||||
| AC fiber | 71% (DBT) | N/A | |||||
| CNTs | 70% (DBT) | N/A | |||||
| rGO | 140 | O2 | N/A | 100% (DBT) | 6 | >10 | 205 |
| Oxidized CNTs | 70 | H2O2 (30 wt%) | [Omim]PF6 | 100% (DBT) | 1 | >3.33 | 206 |
| CNTs/MOF-199-Mo16V2 | 60 | O2 | N/A | 92.25% (DBT) | 3 | >28 | 223 |
| SX PLUS | 60 | H2O2 (30 wt%) | N/A | 76% (4,6-DMDBT) | 24 | <144 | 207 |
| SX-PLUS-Nox | 100% (4,6-DMDBT) | N/A | |||||
| SX-PLUS-Sox | 100%200 (4,6-DMDBT) | N/A | |||||
| g-C3N4 nanomesh | 180 | O2 | N/A | ∼98% (H2S) | 4 | >24 | 210 |
| N-Doped g-C3N4 | ∼88% (H2S) | <16 | |||||
| Bulk g-C3N4 | ∼72% (H2S) | N/A | |||||
| g-C3N4 nanosheet | 180 | O2 | N/A | ∼75% (H2S) | 4 | >24 | 211 |
| Bulk g-C3N4 | ∼15% (H2S) | N/A | |||||
| g-C3N4 | 120 | O2 | N/A | 33.6% (4,6-DMDBT) | 2 | N/A | 212 |
| B-C3N4 | 94.1% (4,6-DMDBT) | >30 | |||||
| B-C3N4 | 125 | O2 | N/A | 98.2% (DBT) | 4 | >40 | 213 |
| BN | 64.5% (DBT) | N/A | |||||
| BN | 100 | O2 | N/A | 40% (DBT) | 6 | N/A | 214 |
| B-C3N4 nanosheet | 100% (DBT) | >30 | |||||
| h-BN | 150 | O2 | N/A | 28% (DBT) | 1 | N/A | 215 |
| h-BN nanosheet | 98.8% (DBT) | >10 | |||||
| O-Doped BN | 125 | O2 | N/A | 15% (DBT) | 6 | N/A | 219 |
| B,N vacancies O-doped BN | 100% (DBT) | >36 | |||||
| Pt/h-BN | 130 | O2 | N/A | 98.3% (DBT) | 6 | >30 | 220 |
| Pt/BN nanosheet | 110 | O2 | N/A | 22.7% (DBT) | 7 | N/A | 221 |
| PtCu/commercial BN | 51.7% (DBT) | N/A | |||||
| PtCu/BN nanosheet | 96.1% (DBT) | >63 | |||||
4. Photocatalyst design and mechanisms for light-driven ODS
As a state-of-the-art route in ODS, light-driven ODS is seen as a promising approach to actualize green and zero-sulfur fuels in the coming years owing to its direct consumption of free and abundant solar light to degrade hazardous sulfur pollutants. However, the search for appropriate materials with the desired photocatalytic properties, including a broad light absorption range, low electron–hole recombination rate, high surface area and exceptional stability, is a challenge. Researchers have concentrated on the structure–property relation to assist the engineering of photocatalysts with advantageous properties that boost the photocatalytic activity.4.1 Metal oxide-based photocatalysts
Because of its outstanding photoactivity, TiO2 has been utilized as a photocatalyst in the ODS of model oil, but its performance was below expectations.224,225 This circumstance stems from the fast electron–hole recombination rate, limited spectral responsive range and aggregation of the TiO2 nanoparticles which diminished the active surface area for light absorption.226 Therefore, several countermeasures have been adopted to ameliorate the shortcomings. Kalantari et al. adopted an ultrasonic-assisted impregnation method to graft nitrogen atoms on TiO2 in order to enhance its photoactivity in ODS.227 Although there was no crucial difference in the surface structure, the N-doped TiO2 showed an enlarged surface area and reduced band gap (Fig. 20b) compared to pristine TiO2. N-doped TiO2 exhibited an extended light absorption ability within the visible light spectrum (Fig. 20a) and a lower energy requirement to excite electrons from the VB to CB for O2 activation. Correspondingly, the DBT conversion by N-doped TiO2 increased substantially (Fig. 20c). However, the ODS performance of N-doped TiO2 still remains insufficient for fulfilling the basic requirement of ultra-deep ODS owing to the paucity of active sites.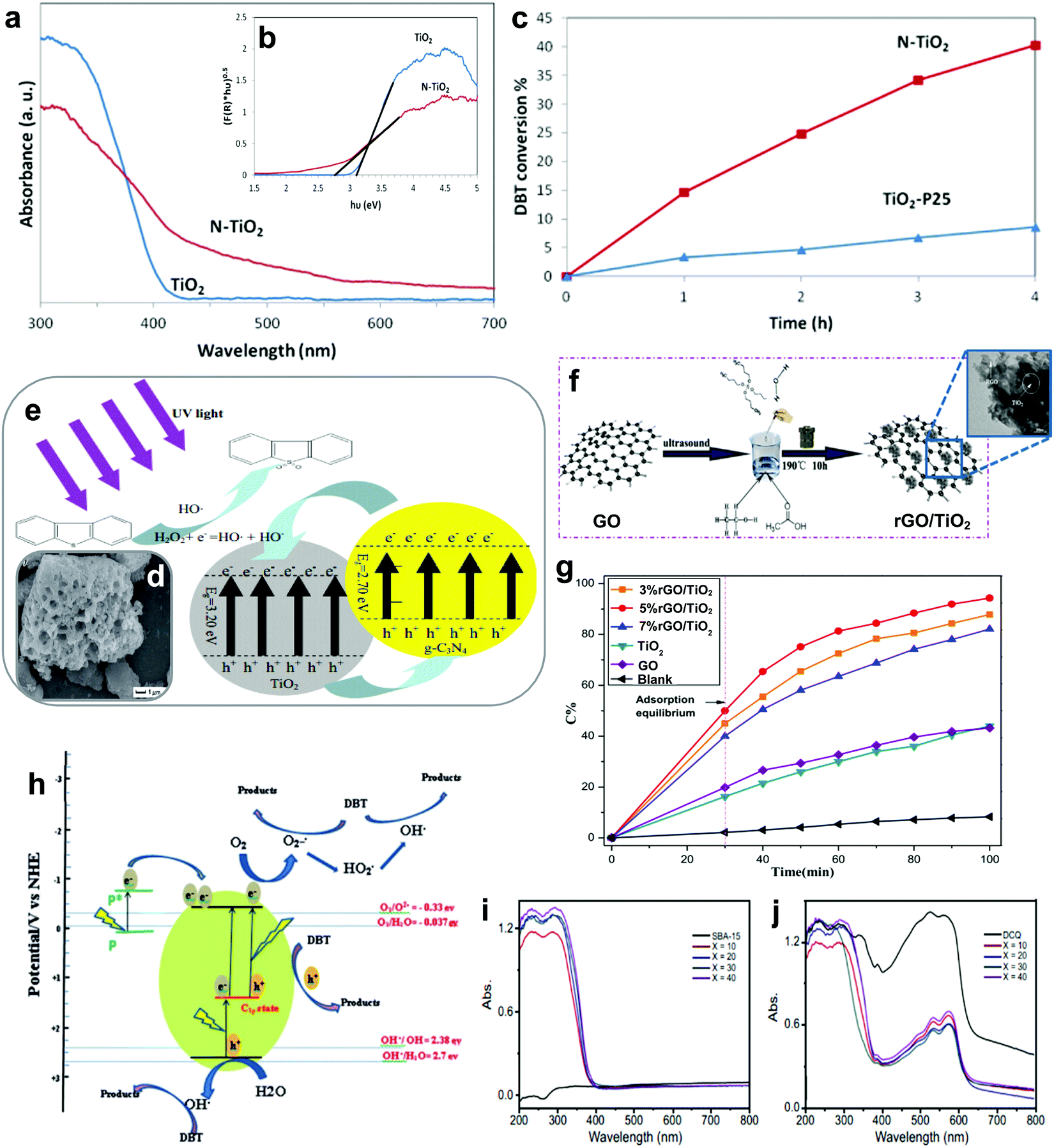 | ||
| Fig. 20 (a) UV-Vis diffusion reflection spectra, (b) Kubelka–Munk function plot and (c) DBT conversion of N-doped TiO2 and TiO2. Reprinted with permission.227 Copyright 2016, Elsevier Ltd. (d) SEM image of TiO2/g-C3N4 and (e) mechanism for DBT oxidation with H2O2. Reproduced with permission.228 Copyright 2014, Elsevier Ltd. (f) Synthesis route of rGO/TiO2 and (g) sulfur removal of rGO/TiO2 with 3%, 5% and 7% rGO, TiO2, GO and a blank with O2 and H2O2. Reproduced with permission.229 Copyright 2019, Elsevier Inc. (h) Proposed mechanism of photo-driven ODS for C-doped TiO2/MCM-41 without oxidants. Reprinted with permission.50 Copyright 2015, Royal Society of Chemistry. UV-Vis diffusion reflection spectra of (i) SBA-15, X%TiO2/SBA-15 and (j) DCQ and DCQ-X%TiO2/SBA-15 (X = 10, 20, 30, 40). Reprinted with permission.230 Copyright 2020, Elsevier B.V. | ||
On account of that, the incorporation of TiO2 with various supports has been intensely studied in ODS by previous scholars. By the impregnation method, TiO2 was embedded onto g-C3N4 with large specific surface area and utilized as the photocatalyst for ODS in the presence of H2O2.228 The resulting TiO2/g-C3N4 possessed a 2D structure with a uniform dispersion of TiO2 (Fig. 20d) which significantly elevated the exposure of TiO2 to the light source. Since g-C3N4 has a narrower band gap, g-C3N4 was more prone to photo-activation compared to TiO2, which triggered the migration of excited electrons and holes across the heterojunction interface following the mechanism in Fig. 20e. In this way, the charge recombination was effectively hampered, preserving more active electrons in the CB of TiO2 to reduce H2O2 into hydroxyl radicals for the oxidation of DBT. Eventually, TiO2/g-C3N4 brought about 98.9% DBT removal in 2 h. Zhang's research group supported TiO2 on rGO via hydrothermal approach to fabricate sheet structured nanocomposites.229 The absence of TiO2 agglomeration (Fig. 20f) was ascribed to the strong interactions between TiO2 and rGO which inhibited the attraction between TiO2 nanoparticles as a result of their high surface energy. Under light irradiation, TiO2/rGO achieved up to 94.3% Th conversion with H2O2 in 100 min, a markedly higher value than that of pristine TiO2 (40%) (Fig. 20g). Similar to g-C3N4, the high electron mobility in rGO accelerated the migration of activated electrons from TiO2 to the rGO surface, suppressing the electron–hole recombination. Hence, the coupling of TiO2 with carbon materials as a photocatalyst not only overcomes the limited surface area issue, but also enhances the electronic property for excellent charge migration.
In another work, Zarrabi et al. employed a non-metal dopant and support to enhance the performance of TiO2 in light-driven ODS.50 Notably, the doping of carbon intensified the visible light absorption and reduced the band gap of TiO2 by creating an interband between CB and VB (Fig. 20h) to reinforce the electron–hole separation. Impregnation on mesoporous MCM-41 also resolved the low specific surface area in TiO2. With these improvements, the obtained catalyst removed 95.6% of DBT without oxidants in 5 h. To discover how the oxidation of DBT can work without oxidants, the authors carried out experiments with several radical scavengers. The results elucidated that hydroxyl radicals and superoxide radicals produced by the breakdown of hydroxyl groups, H2O and O2 on the surface of the catalyst were the major source of oxygen for the ODS process. Likewise, Guo et al. modified TiO2 with organic dye 2,9-dichloroquinacridone (DCQ) and embedded it in SBA-15 molecular sieve through an impregnation-calcination technique to enhance its photoactivity in a biphasic ODS system.230 Owing to the sensitization of TiO2 by DCQ, the optical property in the visible light region was substantially strengthened (Fig. 20i and j), leading to better light absorption for the excitation of electrons. With abundant light absorption sites, DCQ-TiO2/SBA-15 degraded 96.1% of DBT with H2O2 as oxidant within 90 min. Hitam et al. developed ZnO on fibrous nanosilica KCC-1 via in situ microwave-assisted hydrothermal (HM) and impregnation (IM) methods to reveal the influence of the synthesis method on the catalyst properties.231 With respect to textural properties, HM enabled a higher dispersion of ZnO on KCC-1 than IM due to the generation of stronger interactions between ZnO and KCC-1 during the process. This was confirmed by the lower ZnO XRD intensity peaks in ZnO/KCC-1(HM) (Fig. 21a), which indicated a lower concentration of ZnO on the surface of KCC-1. Moreover, the larger specific surface area and pore volume in ZnO/KCC-1(IM), stemming from the poor anchoring of ZnO, also evidenced this fact. With its auspicious structural properties, ZnO/KCC-1(HM) achieved higher DBT conversion (88.9%) in 60 min upon light irradiation due to the existence of more ZnO as light capturing sites. Therefore, the application of HM for immobilizing metal oxide on silica nanoparticles can crucially endow a morphological advantage for nanocomposites, leading to the enhancement of photocatalytic activity in ODS. Overall, the above reports signified that silica nanoparticles play a decisive role by supplying a massive surface area for the immobilization of metal oxides rather than being involved in the light-driven ODS because of their broad band gap and weak light-harvesting property.
 | ||
| Fig. 21 (a) XRD spectra of ZnO/KCC-1(IM), ZnO/KCC-1(HM), KCC-1 and ZnO. Reprinted with permission.231 Copyright 2020, Springer Nature. (b) Graph of (ahv)1/2versus photon energy for g-C3N4 and MoO2/g-C3N4 with different synthesis temperatures. (c) Electronic structure of MoO2/g-C3N4 with the electron movement. Reprinted with permission.232 Copyright 2018, Elsevier B.V. (d) Magnetic hysteresis loops of CuO–Fe3O4 and Fe3O4. Reprinted with permission.233 Copyright 2018, Elsevier Ltd. (e) TEM image of ATP-CeO2/MoS2, (f) DBT oxidation of various catalysts and (g) the recyclability test for ATP-CeO2/MoS2 and CeO2/MoS2 using H2O2. Reprinted with permission.235 Copyright 2016, Elsevier B.V. (h) Reaction pathway of photocatalytic ODS using ATP-CeO2/g-C3N4 and H2O2 under visible light irradiation. Reproduced with permission.237 Copyright 2017, Elsevier B.V. | ||
Recently, Chen et al. investigated the electronic structure of MoO2/g-C3N4 synthesized by impregnation method.232 The decrease of the band gap (Fig. 21b) after the immobilization of MoO2 implied the establishment of a heterojunction structure in MoO2/g-C3N4. The unmatched band gap promoted the movement of excited electrons from g-C3N4 to MoO2 (Fig. 21c) and resulted in rapid charge carrier separation. Although the authors did not examine the photocatalytic activity of MoO2/g-C3N4 in ODS, the discovery of the remarkable light-harvesting property of MoO2/g-C3N4 sheds new light on its potential to be a promising photocatalyst in the coming years. For easy catalyst recovery, light-responsive CuO was coupled with magnetic Fe3O4via impregnation method in the work of Ammar et al.233 As anticipated, the CuO–Fe3O4 nanomaterial inherited the magnetic property of the parent Fe3O4 with negligible loss of magnetization, as detected by magnetometer (Fig. 21d). Thanks to its strong photoactivity and intrinsic DBT adsorption ability, CuO–Fe3O4 converted 95% of DBT into DBT sulfone with H2O2 after 2 h. This study successfully lays the groundwork for future research into the magnetization of nanomaterials to substitute for the cumbersome and environmentally unfriendly chemical extraction methods in ODS for separating the spent catalyst from fuels.
The rare-earth metal oxide CeO2, with semiconductor properties analogous to TiO2, has emerged as an alternative for traditional metal oxides in the photocatalytic decomposition of organic pollutants.234 In this context, Li and co-workers developed a ternary CeO2-based composite (ATP-CeO2/MoS2) by microwave assisted hydrothermal route to unravel its activity in light-driven ODS.235 Through hybridization with 2D structured molybdenum disulfide (MoS2), the undesirable rapid electron–hole recombination in pristine CeO2 was addressed. Furthermore, attributed to the support of the 1D needle-structured attapulgite (ATP) with robust surface area, CeO2 was homogenously distributed in the micropores of ATP (Fig. 21e), prompting the increment of both active sites and thermal mechanical stability. Despite the fact that ATP does not possess any photocatalytic activity, its amazing adsorption tendency facilitated the interaction between DBT and oxygen radicals by gathering them within the catalyst pores.236 Gaining from the above modifications, ATP-CeO2/MoS2 oxidized 98% of DBT with H2O2 in 3 h (Fig. 21f) and demonstrated excellent reusability for ten cycles in contrast to CeO2/MoS2 (Fig. 21g). A similar synergistic effect and remarkable performance in ODS was described in another work of Li et al., wherein the MoS2 support was substituted by metal-free g-C3N4 through an identical synthesis method.237 In regard to the structure–activity interaction, the photocatalytic route in ODS using ATP-CeO2/g-C3N4 is illustrated in Fig. 21h. As g-C3N4 exhibited a narrower band gap compared to CeO2, g-C3N4 served as the engine to initiate the oxidation process upon visible light irradiation, whereas CeO2 played the roles of storage for the excited electrons and the site for H2O2 degradation. Notably, CeO2 is unable to perform photocatalysis alone due to its huge band gap and, hence, a semiconductor with outstanding light-responsive property is imperative for forming a heterojunction structure with CeO2 to accomplish excellent sulfur removal in ODS.
Most recently, BiVO4 was supported on bentonite (BTT) and coupled with CuO to impede the agglomeration of BiVO4 nanoparticles and enhance the photocatalytic activity in ODS.238 Upon 3.5 h of visible light irradiation, CuO–BiVO4/BTT achieved 91.3% DBT oxidation, which was far higher than pristine BiVO4 and BiVO4/BTT. The notable activity improvement was ascribed to the uniform dispersion of BiVO4 on BTT which enormously elevated the exposure of active sites for ODS. Moreover, the formation of a p–n heterojunction by BiVO4 and CuO efficiently promoted the separation of charge carriers and resulted in a higher rate of ODS (Fig. 22a). The photoexcited electrons were transferred from CuO to BiVO4 for the activation of O2, while the generated holes in BiVO4 were transported across the interface to CuO for the oxidation of aromatic sulfur compounds. Aside from introducing a support, the fact that sonication is able to address the agglomeration issue during the synthesis of Cu2O–CeO2 nanocomposites has been discovered recently.239 With the presence of a powerful ultrasonic field, Cu2O was created instead of CuO, due to the reduction of Cu2+ ions to Cu+, and possessed a better light-harvesting property within the visible light range. Moreover, the ultrasound radiation also incurred the shrinkage of Cu2O–CeO2 particles, leading to smaller particle sizes (Fig. 22b) than those of CuO–CeO2 (Fig. 22c) and hence escalating the number of surface active sites. Meanwhile, the heterojunction of Cu2O and CeO2 also significantly hindered the swift electron–hole recombination and promoted the oxidation of sulfur compounds. Benefitting from the synergistic effect, Cu2O–CeO2 achieved a higher desulfurization efficiency (84%) than CuO–CeO2 (39%) under identical reaction conditions. Collectively, this research has revealed the feasibility of the sonochemical approach to fabricate nanocomposites with auspicious morphologies to enhance the photocatalytic activity in ODS.
 | ||
| Fig. 22 (a) Plausible reaction pathway of photocatalytic ODS using CuO–BiVO4/BTT in the presence of O2. Reprinted with the permission.238 Copyright 2021, Elsevier Ltd. SEM images of (b) Cu2O–CeO2 and (c) CuO–CeO2. Reprinted with permission.239 Copyright 2020, Elsevier B.V. (d) TEM image and (e) the DBT removal efficiency for five runs at 1.5 h per run using g-C3N4 and O2via light-driven ODS. Reprinted with permission.245 Copyright 2016, Elsevier B.V. (f) XRD spectra and (g) photoluminescence of g-C3N4 and Na(X)/g-C3N4 (X = 0.3 M, 0.5 M, 1.0 M). (h) Th removal of g-C3N4 and Na(0.3)/g-C3N4 in the presence of O2 under light irradiation. Reprinted with permission.246 Copyright 2019, Elsevier B.V. (i) Reaction route for light-driven ODS using ZnAl-LDH/g-C3N4 (left) and ZnCr-LDH/g-C3N4 (right). Reprinted with permission.249 Copyright 2020, American Chemical Society. (j) Photocatalytic conversion of DBT by ZnAl–(PW12O40)x-LDH in the presence of O2. Reprinted with permission.250 Copyright 2018, Royal Society of Chemistry. | ||
4.2 Metal-free nanomaterial-based photocatalysts
From an economic and environmental point of view, metal-free nanomaterials, like g-C3N4 with its moderate band gap and considerable visible light absorption240–243 and GO with its plentiful oxygen-containing groups and tunable electrical properties,244 have been employed as emerging nanomaterials in photocatalysis. Besides supporting metallic nanomaterials, mesoporous g-C3N4 was applied as the photocatalyst to desulfurize stimulated fuel in the presence of O2 and visible light.245 Marvelously, g-C3N4 oxidized all the DBT within 90 min. This was ascribed to its enormous film-structured surface (Fig. 22d) and appropriate band gap for redox reactions. However, its poor resistance against irreversible deactivation due to the blockage of active sites by aromatic sulfur constituents substantially curtailed the reusability of g-C3N4, leading to a drastic reduction of sulfur removal efficiency starting from the second run (Fig. 22e). To bolster the robustness of g-C3N4, Zhang's team grafted sodium into g-C3N4 through simple mixing followed by calcination to form Na/g-C3N4 composites.246 According to Fig. 22f, the reduction of XRD intensities resulted from the increase of Na loading, which prompted the formation of defect sites in the morphology of g-C3N4. Furthermore, the doping of Na promoted the preservation of electrons and holes, as confirmed by the lower photoluminescence peak intensity in Na/g-C3N4 (Fig. 22g). With these improvements, the Th removal efficiency using Na/g-C3N4 increased spectacularly upon light illumination compared to that using g-C3N4 (Fig. 22h). Relatedly, the photoactivity of GO and its reaction mechanism in light-driven ODS were studied comprehensively by Zeng et al.247 Using DFT computations, the authors proposed the role of defect sites and the zigzag edge as electron suppliers for the generation of reactive oxygen species from O2, hydrogen ions and oxygen-containing functional groups on the surface of GO. With the help of acetonitrile, the transfer of DBT compounds from the organic phase to the polar phase compelled the interaction between oxygen radicals and DBT, resulting in 99.9% DBT conversion under UV irradiation. Despite the exceptional photocatalytic activity demonstrated by metal-free materials, the salient challenges like poor reusability and sluggish reaction kinetics still remain.4.3 Layered double hydroxide (LDH)
Over the past few years, layered double hydroxide (LDH) has gained copious attention in the field of photocatalysis for its unique 2D-layered structure, remarkable light-capturing ability and tailorable band gap.248 Huang et al. hybridized ZnAl- and ZnCr-LDH with g-C3N4 nanosheets through an electrostatic self-assembly approach to establish a Z-scheme heterojunction to improve the efficiency in light-driven ODS.249 The Z-scheme heterojunction recombined excited electrons from LDH and holes from g-C3N4, preserving the free reactive electrons and holes in the CB of g-C3N4 and the VB of LDH, respectively, for redox reactions, which was greatly conducive to the ODS reaction rate (Fig. 22i). From the perspective of textural properties, ZnCr-LDH/g-C3N4 demonstrated comparably more severe aggregation than ZnAl/g-C3N4. This phenomenon caused a slightly lower ODS activity in ZnCr-LDH/g-C3N4 (96.6%) compared to ZnAl-LDH/g-C3N4 (99.8%) due to the emergence of fewer surface active sites. Cai et al. intercalated ZnAl-LDH with PW12O40 by exfoliation-reassembly method to enhance its photocatalytic activity in ODS.250 The robust interactions between ZnAl-LDH and PW12O40 facilitated the electron transfer and electron–hole separation, which extended the lifetime of charge carriers. In the ODS process, PW12O40 acted as the excited electron storage site for the activation of O2 (Fig. 22j). As a result, ZnAl–(PW12O40)0.07-LDH converted 95.3% of DBT using O2 within 3 h of UV irradiation. To achieve an effortless photocatalyst recovery process, Masoumi and Hosseini fabricated magnetic NiCo2-LDH/Fe3O4 and evaluated its photocatalytic performances in ODS.251 Advantageously, the anchoring of NiCo2-LDH on Fe3O4 contributed to a significant increment of specific surface area as well as comparably minor agglomeration. The higher number of surface catalytic sites brought about 74.6% DBT conversion in the presence of O2. These studies manifested LDH photocatalyst as an excellent candidate to catalyze light-driven ODS.In summary, through doping and introducing heterojunctions, the performances of photocatalysts in ODS can be greatly enhanced by the improvement of charge carrier separation, light absorption range and light-capturing sites. Crucially, the previous studies are just the tip of iceberg, as only a few species of nanomaterials have been applied in light-driven ODS. Indeed, it is highly valuable to explore new nanomaterials with light-harvesting property to create a breakthrough in the efficiency of ODS using photon energy. Table 7 illustrates the performances of myriad nanomaterials in light-driven ODS systems with different operating parameters.
| Catalysts | Light source (power) | Oxidant | Extractant | Performances | Ref. | ||
|---|---|---|---|---|---|---|---|
| Sulfur removal (reactant) | Reaction time (h) | Stability (h) | |||||
| rGO: reduced graphene oxide; SBA-15: mesoporous silica-based metal–organic framework; DCQ: 2,9-dichloroquinacridone; MCM-41: mesoporous silica-based metal–organic framework; FCC-1: fibrous silica nanosphere; CeO2: cerium dioxide; ATP: attapulgite; mpg-C3N4: mesoporous graphitic carbon nitride; GO: graphene oxide; DMF: dimethylformamide; LDH: layered double hydroxide. | |||||||
| TiO2 | Hg lamp (250 W) | H2O2 (30 wt%) | N/A | 65.2% (BT) | 2 | N/A | 224 |
| TiO2 | Hg–Xe lamp (200 W) | H2O2 (30 wt%) | Acetonitrile | <40% (DBT) | 10 | N/A | 225 |
| TiO2 | Xe lamp (55 W) | O2 | N/A | 8.6% (DBT) | 4 | N/A | 227 |
| N-Doped TiO2 | 40.3% (DBT) | >12 | |||||
| TiO2 | Hg lamp (250 W) | H2O2 (30 wt%) | N/A | 79.1% (DBT) | 2 | N/A | 228 |
| g-C3N4 | 1.3% (DBT) | N/A | |||||
| TiO2/g-C3N4 | 98.9% (DBT) | >8 | |||||
| TiO2 | Xe lamp | H2O2 (30 wt%) | N/A | 42% (Th) | 1.67 | N/A | 229 |
| GO | 42% (Th) | ||||||
| TiO2/rGO | 94.3% (Th) | ||||||
| TiO2/SBA-15 | Xe lamp (300 W) | H2O2 (30 wt%) | Acetonitrile | 86% (DBT) | 1.5 | N/A | 230 |
| DCQ-TiO2/SBA-15 | 96.1% (DBT) | ||||||
| C/TiO2/MCM-41 | W lamp (300 W) | O2 | N/A | 95.6% (DBT) | 5 | >10 | 50 |
| ZnO | Halide lamp (160 W) | O2 | Acetonitrile | 44.4% (DBT) | 1 | N/A | 231 |
| KCC-1 | 53.9% (DBT) | ||||||
| ZnO/KCC-1 | 88.9% (DBT) | ||||||
| CuO–Fe3O4 | Hg lamp (350 W) | H2O2 (30 wt%) | N/A | 95.2% (DBT) | 2 | >10 | 233 |
| CeO2/MoS2 | Xe lamp (300 W) | H2O2 (30 wt%) | Acetonitrile | 82% (DBT) | 3 | <9 | 235 |
| ATP-CeO2/MoS2 | 98% (DBT) | >30 | |||||
| g-C3N4 | Xe lamp (300 W) | H2O2 (30 wt%) | N/A | 42% (DBT) | 3 | N/A | 237 |
| CeO2/g-C3N4 | 83% (DBT) | <21 | |||||
| ATP-CeO2/g-C3N4 | 98% (DBT) | >24 | |||||
| mpg-C3N4 | Xe lamp (300 W) | O2 | N/A | 100% (DBT) | 1.5 | <7.33 | 245 |
| g-C3N4 nanosheet | Xe lamp (300 W) | O2 | N/A | 52% (Th) | 6 | N/A | 246 |
| Na-Doped g-C3N4 nanosheet | 92.3% (Th) | >24 | |||||
| GO | Hg lamp (100 W) | O2 | Acetonitrile | 99.9% (DBT) | 2.33 | <9.33 | 247 |
| Formic acid | |||||||
| Ti3C2 MXene | Xe lamp (300 W) | O2 | N/A | 29.6% (Th) | 3 | N/A | 252 |
| g-C3N4 | 36.4% (Th) | N/A | |||||
| Ti3C2 MXene/g-C3N4 | 73.6% (Th) | >18 | |||||
| BiVO4 | W lamp (300 W) | O2 | N/A | 49.1% (DBT) | 3.5 | >35 | 238 |
| BiVO4/BTT | 59.8% (DBT) | >35 | |||||
| CuO–BiVO4/bentonite | 91.3% (DBT) | >35 | |||||
| BiOCl | Hg lamp (300 W) | O2 | Acetonitrile | 99.1% (DBT) | 3 | <18 | 253 |
| CuO–CeO2 | Visible lamp (400 W) | O2 | DMF | 39% (Th) | 3 | N/A | 239 |
| Cu2O–CeO2 | 84% (Th) | >12 | |||||
| ZnAl-LDH/g-C3N4 | Hg lamp (500 W) | O2 | Acetonitrile | 99.8% (DBT) | 3 | >15 | 249 |
| ZnCr-LDH/g-C3N4 | 96.6% (DBT) | >15 | |||||
| ZnAl–NO3-LDH | Hg lamp (500 W) | O2 | Acetonitrile | 80.5% (DBT) | 3 | N/A | 250 |
| ZnAl–(PW12O40)0.07-LDH | 95.3% (DBT) | >15 | |||||
| Ni–Co2-LDH/Fe3O4 | W lamp (250 W) | O2 | N/A | 74.6% (DBT) | 3.7 | N/A | 251 |
5. Summary, challenges and future outlook
Over the past few years, the desulfurization of fuels via ODS has gained traction and is anticipated to be the emerging technology in years to come to realize the milestone of environmentally friendly fuels, in contrast to conventional HDS. This has led to explosive interest in ODS in the field of environmental remediation. To this end, the many published works related to the application of catalysts, including metal oxides, ILs, DESs, POMs, MOFs, metal-free materials and their heterostructured composites, in the thermal-driven and light-driven ODS of fuel oils have been reviewed in individual sections and critically discussed in this comprehensive review. With the wide range of discovered ODS catalysts, this review is anticipated to deliver an instructive and interdisciplinary overview of the current progress in thermal-driven and light-driven ODS and methodically covers the intrinsic connections between their synthesis routes, modification approaches, physico-chemical properties, reaction pathways and catalytic performances in ODS. The relationships of the primary factors influencing the ODS processes with respect to different classes of catalysts is pictorially described in Fig. 23.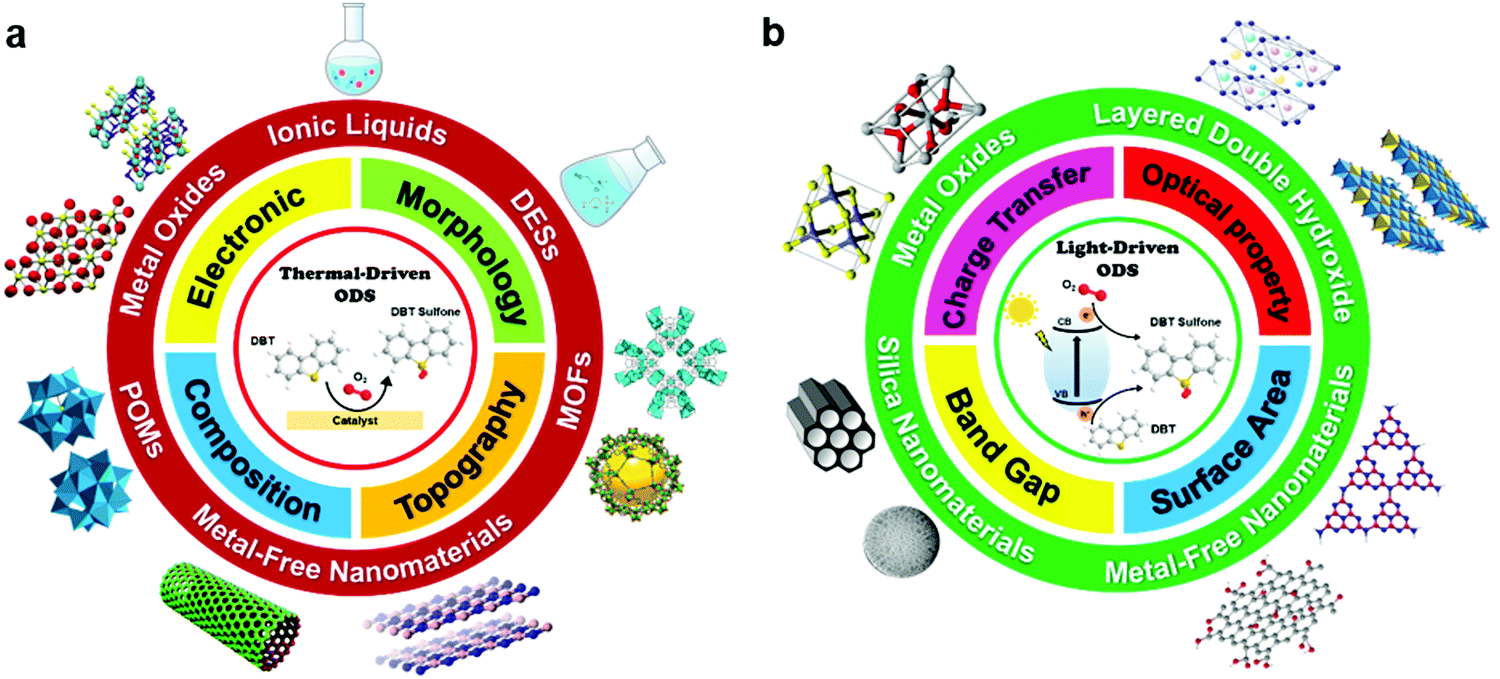 | ||
| Fig. 23 A pictorial overview of (a) thermal-driven ODS and (b) light-driven ODS. | ||
In light of the insufficient surface active sites in metal oxides, modification strategies, such as anchoring on a support, metal oxide doping and morphological tuning, have been carried out to increase the electron density and specific surface area and improve their efficiency in catalyzing ODS. This has invigorated the exploration of POMs, which are capable of extraordinary mass transfer which facilitates the interactions between sulfur pollutants and the catalyst in ODS. However, the costly and intractable downstream separation process has tremendously restricted the further development of POMs until the emergence of various promising schemes, including precipitation with insoluble ions, immobilization on supports and hybridization, with the objective of increasing their heterogeneity in ODS systems. Bifunctional ILs and DESs have also widely served as the catalyst and extractant in ODS because of their facile recyclable properties, excellent catalytic activity and low thermal energy requirements. It is noted that DESs are more favorable than ILs because of their better environmental compatibility, higher biodegradability, cheaper raw materials and lower toxicity. In addition, MOFs have recently garnered considerable attention due to their large specific surface area, which is highly conducive to catalytic activity in ODS. Crucially, the accessibility of the active sites to sulfur compounds is substantially limited by the microporous structure in MOFs, resulting in relatively poor desulfurization rates in ODS. In addition to focusing on the Lewis active sites, the pore dimensions of MOFs are regarded as a pivotal property for attaining remarkable ODS activity. Of the metal-free nanomaterials, carbonaceous nanomaterials g-C3N4 and BN have shown excellent results in ODS application, which are ascribed to their exceptional physico-chemical robustness and tailorable structures. However, their employment in ODS suffers from sluggish reaction kinetics which are far below the basic requirement for industrialization. In order to ameliorate the preceding shortfalls, MOFs and metal-free nanomaterials with robust surface area are often used as the anchoring sites for decoration with metal oxides and POMs. The hybridization of nanomaterials also exhibits an unprecedented synergistic influence on the catalytic performances in ODS. Embracing sustainable chemistry, solar energy as an inexhaustible driving force for ODS has been reported as a novel step toward zero-sulfur clean fuels. Though various explored semiconductors can effectively purify fuels, light-driven ODS is burdened with several daunting challenges, such as a swift charge carrier recombination rate and narrow light absorption range, which have not been entirely resolved.
On top of the foregoing challenges, there are still many issues which must be surmounted to formulate an efficacious and eco-friendly ODS process, thereby accelerating the commercialization progress of ODS. Since a catalyst is an essential booster for ODS, the engineering of nanomaterials is a momentous mission for the materials science community in the search for outstanding ODS activity and recyclability. Until now, knowledge of the optimal operating parameters, including temperature, reaction time, precursor species and others, for the fabrication of ODS catalysts has not been fully exploited, but is of vital importance for customizing favorable catalyst properties to pursue maximum ODS activity. In spite of the numerous studies that have reported successful formation of ODS catalysts, the reproducibility and scalability of the syntheses are still in their infancy. Additionally, some innovative methods like the sonochemical and microwave-assisted routes are not developed enough for the mass production of nanomaterials due to concerns about the safety and controllability of catalyst properties. Therefore, more emphasis should be placed on the optimization and upscaling of synthesis to reveal their exact feasibility and potential in industrial applications. Moreover, the search for relatively streamlined and energy-efficient process routes will work in the long run to replace energy-intensive synthesis techniques.
It is noteworthy that the stability of ODS catalysts remains an urgent hurdle for industrial scale ODS. In this regard, additional investigations must suppress catalytic deactivation to achieve a durable and effectual ODS catalyst. For instance, the reinforcement of substrate–support interactions in nanocomposites and the refinement of pore size are pragmatic practices to respectively hinder the leaching phenomenon and the blockage of pores by oxidized sulfur compounds which are the major root causes of catalytic deactivation in ODS. On the other hand, the critical effect of various hydrocarbons in commercial fuels on the robustness of as-designed ODS catalysts has not been unambiguously explored, as the aforementioned ODS studies were typically performed on a lab scale by employing model fuels. Researchers should grasp the opportunity to thoroughly examine the activity and stability of ODS catalysts in various real fuels with a wide range of olefins and aromatic sulfur compounds, which is essential for the development of state-of-the-art ODS catalysts to adapt to real fuel conditions. Furthermore, it is an arduous task to alleviate the agglomeration of nanoscale particles as the total surface energy or the attraction force between particles increases with the reduction of particle size. To guarantee the highest exposure of catalytic site in ODS, surface functionalization, which ameliorates the vigorous surface charge by introducing appropriate functional groups, will be a fruitful procedure. Further study should be undertaken to shed more light on the effects of multifarious functional groups on the morphology, topography and catalytic performance of ODS catalysts, targeted at easing the functional group screening process to further comprehend the structure–activity relationship.
For green chemistry, a growing number of studies adopt low-priced and hazard-free O2 as the oxidant in ODS. However, the utilization of O2 in ODS requires relatively more thermal or light energy, as well as a longer reaction time to accomplish ultra-deep desulfurization because of its stable molecular structure. As such, improvements are demanded to elevate the activity of ODS catalysts while reducing the energy consumption and boosting the efficiency of ODS systems using O2. Also, the existing separation technologies, such as solvent extraction, for removing the oxidized sulfur compounds from the clean fuels, possess a number of drawbacks, such as the generation of enormous amounts of wastewater and the high risk of fuel contamination, which are great and unceasing concerns for industrial ODS processes. A comparably facile, cost-effective and environmentally safe post-treatment process is pending discovery for its practical advantages and warrants a concerted effort from multidisciplinary researchers. It is also hugely challenging to curtail the competitive impact of the oxidation of hydrocarbons on the oxidation of sulfur compounds in ODS. This necessitates plausible countermeasures to promote the selectivity of ODS catalysts toward sulfur compounds to circumvent undesirable yield loss of fuels.
Given the challenges, there are boundless opportunities for further advancement of this fast-paced research field. On the road to practical ODS applications, a colossal amount of trial and error is mandatory for finding satisfactory materials with prominent catalytic properties, as well as developing viable catalyst modification techniques. To address the time-consuming process, more attention should be paid to computational chemistry methods, namely DFT calculations and molecular dynamics, to assist in the interpretation of experimental data and contribute a mechanistic understanding of the structure–activity relationship, worthwhile information for the rational design of ODS catalysts. In addition, the molecular structure, properties and possible reaction mechanisms in ODS of newly discovered materials can be predicted via computational model or simulation to establish a plentiful material chemical data library. As the world moves into the information age, it is anticipated that the further incorporation of intelligence technology, including artificial intelligence (AI), big data technology, and the Internet of Things (IoTs), to perform in-depth analysis using available databases can prevent the waste of time and research capital in experimental work. Besides expediting the material screening process, encountered bottlenecks in the engineering of ODS catalysts would be addressed by applying these frontline technologies. The growth of 3D and 4D printing has opened a window to the prospective synthesis of ODS catalysts in the future. By means of design software, the properties and geometries of material can be facilely controlled to configure an ODS catalyst with outstanding catalytic activity. More significantly, 3D and 4D printing would be a promising tool to replace existing intricate synthesis techniques and could be a great leap forward for the advancement of ODS. The synergy between computational study, experimental work and smart technologies can constitute useful guidance for the research direction in this trending area and effectively drive the commercialization of ODS.
In the catalysis world, noticeable effort has been directed toward the development of multifunctional catalysts which can carry out multiple catalytic reactions concurrently. It is envisaged that the coupling of thermal and light-harvesting properties into a single catalyst to drive ODS will be very helpful in balancing energy consumption and reaction efficiency, as well as essentially enhancing the industrial practicality of ODS. As an example, a multifunctional ODS catalyst allows an effortless shift from light-driven ODS to thermal-driven ODS to maintain a high ODS reaction rate in poor light intensity conditions. In another stratagem, adsorption capability can be endowed to the ODS catalyst to capture the oxidized sulfur compounds and streamline the post-treatment process. In other words, the oxidized sulfone can be removed together with the ODS catalyst from the fuel through a simple filtration process instead of an environmentally harmful chemical separation process. Moreover, the hybridization of different materials to create ODS catalysts with the ability to simultaneously perform ODS and oxidative denitrogenation (ODN) will establish a new frontier in the arena of fuel purification.254 The one-step degradation of sulfur and nitrogen-containing compounds in fuels kills two birds with one stone, offers enormous economic benefit, and diminishes the deleterious impacts on both the environment and the efficiency of oil processing. The design of multipurpose ODS catalysts is not solely limited to the above-mentioned applications, but many opportunities are still waiting to be uncovered, such as the fusion of different oil refinery processes with the ODS reaction. In summary, a diagrammatic synopsis for the future perspectives of ODS is shown in Fig. 24.
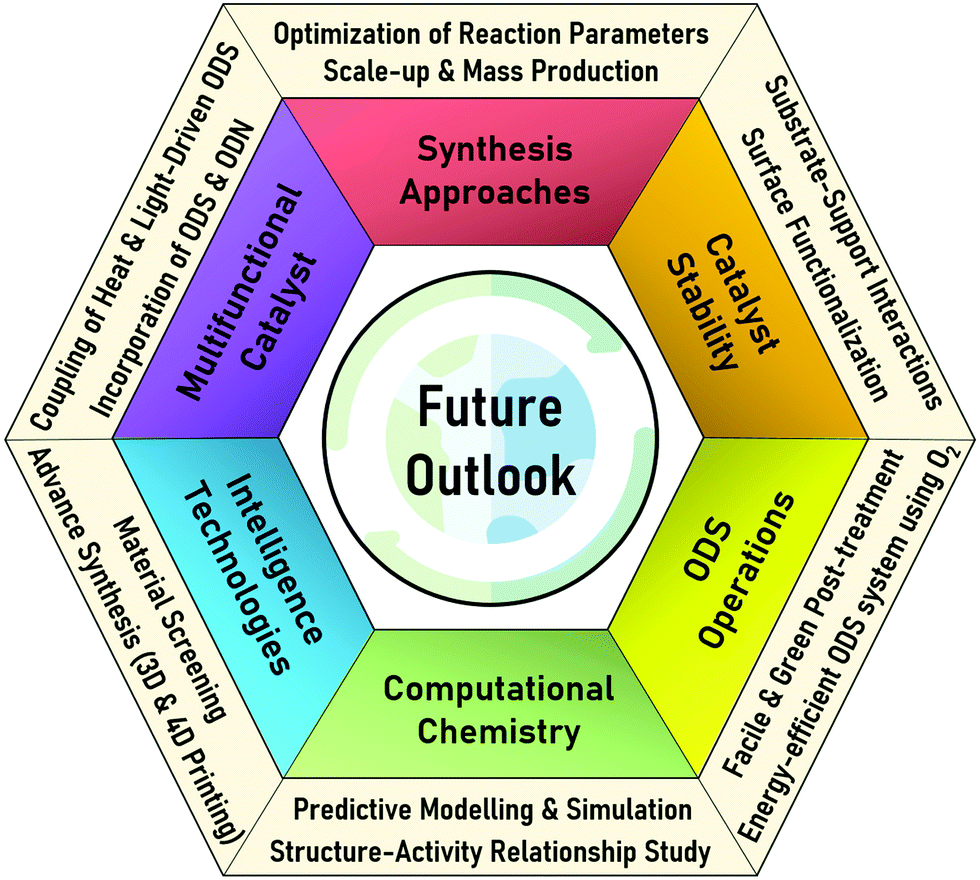 | ||
| Fig. 24 Summary of the future outlook for ODS in the areas of synthesis approaches, improvement of catalyst stability, fine-tuning of operations, integration of computational chemistry, employment of intelligence technologies and engineering of multifunctional catalysts. | ||
The achievement of zero-sulfur fuels via ODS still demands strenuous endeavors from cross-disciplinary researchers all around the world to engineer materials with superlative ODS catalytic performance, controllable and scalable synthesis, and high affordability and availability. Last but not least, collaborative action between local governments, industrial players and scholars is of paramount significance to transform the ODS process from laboratory scale to industrial scale, thereby mitigating the environmental crisis stemming from the combustion of fossil fuel. With such sustainable and efficacious desulfurization technology, we strongly believe that the dawn of a decent and clean environment is no longer out of reach.
Author contributions
X. B. L. was involved in project conceptualization, investigation, writing – original draft and writing – review and editing. W.-J. O. was involved in project administration, resources, supervision, writing – review and editing, as well as funding acquisition.Conflicts of interest
There are no conflicts of interests to declare.Acknowledgements
The authors would like to acknowledge the financial support provided by the Ministry of Higher Education (MOHE) Malaysia under the Fundamental Research Grant Scheme (FRGS) (Ref no. FRGS/1/2020/TK0/XMU/02/1). This work is also funded by Hengyuan International Sdn. Bhd. (Grant no. EENG/0003), Xiamen University Malaysia Investigatorship Grant (Grant no. IENG/0038) and Xiamen University Malaysia Research Fund (XMUMRF/2019-C3/IENG/0013).Notes and references
- C. Nunez, What is acid rain? https://www.nationalgeographic.com/environment/global-warming/acid-rain/, accessed 11 November, 2020.
- U.S. Environmental Protection Agency, Sulfur Dioxide (SO2) Pollution, https://www.epa.gov/so2-pollution/sulfur-dioxide-basics#:~:text=What%20are%20the%20environmental%20effects,which%20can%20harm%20sensitive%20ecosystems, accessed 11 November, 2020.
- U.S. Environmental Protection Agency, Particle Pollution Exposure, https://www.epa.gov/pmcourse/particle-pollution-exposure, accessed 10 November, 2020.
- J. A. Rodriguez and J. Hrbek, Acc. Chem. Res., 1999, 32, 719–728 Search PubMed.
- D. E. Winterbone and A. Turan, in Advanced Thermodynamics for Engineers, ed. D. E. Winterbone and A. Turan, Butterworth-Heinemann, Boston, 2nd edn, 2015, pp. 307–322 DOI:10.1016/B978-0-444-63373-6.00014-9.
- U.S. Environmental Protection Agency, Diesel Fuel Standards and Rulemakings, https://www.epa.gov/diesel-fuel-standards/diesel-fuel-standards-and-rulemakings, accessed 11 November, 2020.
- X. Weng, L. Cao, G. Zhang, F. Chen, L. Zhao, Y. Zhang, J. Gao and C. Xu, Ind. Eng. Chem. Res., 2020, 59, 21261–21274 Search PubMed.
- A. Tanimu and K. Alhooshani, Energy Fuels, 2019, 33, 2810–2838 Search PubMed.
- R. A. Sánchez-Delgado, Organometallic Modeling of the Hydrodesulfurization and Hydrodenitrogenation Reactions, Springer, Netherlands, Dordrecht, 2002, pp. 1–34 DOI:10.1007/0-306-47619-3_1.
- W. Lai, Z. Chen, J. Zhu, L. Yang, J. Zheng, X. Yi and W. Fang, Nanoscale, 2016, 8, 3823–3833 Search PubMed.
- M. A. Betiha, A. M. Rabie, H. S. Ahmed, A. A. Abdelrahman and M. F. El-Shahat, Egypt. J. Pet., 2018, 27, 715–730 Search PubMed.
- X. Ma, K. Sakanishi and I. Mochida, Ind. Eng. Chem. Res., 1994, 33, 218–222 Search PubMed.
- W. Nabgan, M. Rashidzadeh and B. Nabgan, Environ. Chem. Lett., 2018, 16, 507–522 Search PubMed.
- I. Shafiq, S. Shafique, P. Akhter, W. Yang and M. Hussain, Catal. Rev., 2020, 1–86, DOI:10.1080/01614940.2020.1780824.
- P. Tan, Y. Jiang, L.-B. Sun, X.-Q. Liu, K. AlBahily, U. Ravon and A. Vinu, J. Mater. Chem. A, 2018, 6, 23978–24012 Search PubMed.
- K. X. Lee and J. A. Valla, React. Chem. Eng., 2019, 4, 1357–1386 Search PubMed.
- I. Ahmed and S. H. Jhung, J. Hazard. Mater., 2016, 301, 259–276 Search PubMed.
- G. Mohebali and A. S. Ball, Int. Biodeterior. Biodegrad., 2016, 110, 163–180 Search PubMed.
- S. Kumar, V. C. Srivastava and S. M. Nanoti, Sep. Purif. Rev., 2017, 46, 319–347 Search PubMed.
- M. F. Majid, H. F. Mohd Zaid, C. F. Kait, K. Jumbri, L. C. Yuan and S. Rajasuriyan, J. Mol. Liq., 2020, 306, 112870 Search PubMed.
- P. Sikarwar, V. Gosu and V. Subbaramaiah, Rev. Chem. Eng., 2019, 35, 669–705 Search PubMed.
- Z. Ismagilov, S. Yashnik, M. Kerzhentsev, V. Parmon, A. Bourane, F. M. Al-Shahrani, A. A. Hajji and O. R. Koseoglu, Catal. Rev.: Sci. Eng., 2011, 53, 199–255 Search PubMed.
- M. M. H. Mondol, B. N. Bhadra and S. H. Jhung, Appl. Catal., B, 2021, 119988, DOI:10.1016/j.apcatb.2021.119988.
- J. He, P. Wu, L. Chen, H. Li, M. Hua, L. Lu, Y. Wei, Y. Chao, S. Zhou, W. Zhu and H. Li, Chem. Eng. J., 2021, 129022, DOI:10.1016/j.cej.2021.129022.
- M. F. Ali, A. Al-Malki, B. El-Ali, G. Martinie and M. N. Siddiqui, Fuel, 2006, 85, 1354–1363 Search PubMed.
- A. Rajendran, T.-y. Cui, H.-x. Fan, Z.-f. Yang, J. Feng and W.-y. Li, J. Mater. Chem. A, 2020, 8, 2246–2285 Search PubMed.
- X. Zhou, T. Wang, H. Liu, X. Gao, C. Wang and G. Wang, ChemSusChem, 2020, 14, 492–511 Search PubMed.
- M. Haghighi and S. Gooneh-Farahani, Environ. Sci. Pollut. Res., 2020, 27, 39923–39945 Search PubMed.
- M. N. Hossain, H. C. Park and H. S. Choi, Catalysts, 2019, 9, 229 Search PubMed.
- I. Shafiq, S. Shafique, P. Akhter, M. Ishaq, W. Yang and M. Hussain, J. Cleaner Prod., 2020, 125731, DOI:10.1016/j.jclepro.2020.125731.
- M. H. Ibrahim, M. Hayyan, M. A. Hashim and A. Hayyan, Renewable Sustainable Energy Rev., 2017, 76, 1534–1549 Search PubMed.
- F. Liu, J. Yu, A. B. Qazi, L. Zhang and X. Liu, Environ. Sci. Technol., 2021, 55, 1419–1435 Search PubMed.
- H. Zhao and G. A. Baker, Front. Chem. Sci. Eng., 2015, 9, 262–279 Search PubMed.
- J. Li, Z. Yang, S. Li, Q. Jin and J. Zhao, J. Ind. Eng. Chem., 2020, 82, 1–16 Search PubMed.
- C. G. Piscopo, C. M. Granadeiro, S. S. Balula and D. Bošković, ChemCatChem, 2020, 12, 4721–4731 Search PubMed.
- L. Hao, M. J. Hurlock, G. Ding and Q. Zhang, Top. Curr. Chem., 2020, 378, 17 Search PubMed.
- J. G. Martinez, Top 10 Emerging Technologies 2020, World Economic Forum, 2020 Search PubMed.
- S. Houda, C. Lancelot, P. Blanchard, L. Poinel and C. Lamonier, Catalysts, 2018, 8, 344 Search PubMed.
- G. L. Wu, Y. C. Yin, W. B. Chen, F. Xin, Y. T. Lu, K. Qin, L. Zhang, Y. X. Song and M. F. Li, Chem. Eng. Sci., 2020, 214, 115446 Search PubMed.
- B. Wang, C. K. Xiao, P. F. Li, Z. S. Zhao, C. M. Xu, Z. Zhao, Q. Meng, J. M. Li, A. J. Duan and Z. T. Chen, Ind. Eng. Chem. Res., 2018, 57, 11868–11882 Search PubMed.
- C. Peng, R. Guo, X. Feng and X. C. Fang, Chem. Eng. J., 2019, 377, 119706 Search PubMed.
- S. Dasgupta, P. Gupta, Aarti, A. Nanoti, A. N. Goswami, M. O. Garg, E. Tangstad, O. B. Vistad, A. Karlsson and M. Stocker, Fuel, 2013, 108, 184–189 Search PubMed.
- X. He and C. Zhang, J. Mater. Sci., 2019, 54, 8831–8851 Search PubMed.
- D. Ravelli, D. Dondi, M. Fagnoni and A. Albini, Chem. Soc. Rev., 2009, 38, 1999–2011 Search PubMed.
- X. Chen, N. Li, Z. Kong, W.-J. Ong and X. Zhao, Mater. Horiz., 2018, 5, 9–27 Search PubMed.
- W.-J. Ong, L. K. Putri and A. R. Mohamed, Chem. – Eur. J., 2020, 26, 9710–9748 Search PubMed.
- W. Zhang, A. R. Mohamed and W.-J. Ong, Angew. Chem., Int. Ed., 2020, 59, 22894–22915 Search PubMed.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 Search PubMed.
- A. Samokhvalov, ChemPhysChem, 2011, 12, 2870–2885 Search PubMed.
- M. Zarrabi, M. H. Entezari and E. K. Goharshadi, RSC Adv., 2015, 5, 34652–34662 Search PubMed.
- W.-J. Ong and K. P. Y. Shak, Sol. RRL, 2020, 4, 2000132 Search PubMed.
- J. Peng, X. Chen, W.-J. Ong, X. Zhao and N. Li, Chem, 2019, 5, 18–50 Search PubMed.
- C. N. C. Hitam, A. A. Jalil and A. A. Abdulrasheed, J. Ind. Eng. Chem., 2019, 74, 172–186 Search PubMed.
- A. Samokhvalov, Catal. Rev.: Sci. Eng., 2012, 54, 281–343 Search PubMed.
- A. Bourane, O. Koseoglu, A. Al-Hajji, F. Adam and H. Muller, React. Kinet., Mech. Catal., 2019, 126, 365–382 Search PubMed.
- M. W. Wan, M. D. G. de Luna, L. R. Golosinda, C. M. Futalan, P. Phatai and M. C. Lu, Clean Technol. Environ. Policy, 2019, 21, 1459–1469 Search PubMed.
- D. Julião, F. Mirante, S. O. Ribeiro, A. C. Gomes, R. Valenca, J. C. Ribeiro, M. Pillinger, B. de Castro, I. S. Gonçalves and S. S. Balula, Fuel, 2019, 241, 616–624 Search PubMed.
- J. C. Vedrine, ChemSusChem, 2019, 12, 577–588 Search PubMed.
- S.-F. Ng, M. Y. L. Lau and W.-J. Ong, Adv. Mater., 2021, 2008654, DOI:10.1002/adma.202008654.
- A. A. Olajire, A. Kareem and A. Olaleke, J. Nanostruct. Chem., 2017, 7, 159–170 Search PubMed.
- M. Liu, J. He, P. Wu, L. Lu, C. Wang, L. Chen, M. Hua, W. Zhu and H. Li, Inorg. Chem. Front., 2020, 7, 1212–1219 Search PubMed.
- W. A. W. Abu Bakar, R. Ali, A. A. A. Kadir and W. N. A. W. Mokhtar, Fuel Process. Technol., 2012, 101, 78–84 Search PubMed.
- A. Akbari, M. Omidkhah and J. T. Darian, Ultrason. Sonochem., 2014, 21, 692–705 Search PubMed.
- L. Cedeno-Caero, H. Gomez-Bernal, A. Fraustro-Cuevas, H. D. Guerra-Gomez and R. Cuevas-Garcia, Catal. Today, 2008, 133, 244–254 Search PubMed.
- C. Deng, P. Wu, L. Zhu, J. He, D. Tao, L. Lu, M. He, M. Hua, H. Li and W. Zhu, Appl. Mater. Today, 2020, 20, 100680 Search PubMed.
- J. He, P. Wu, Y. Wu, H. Li, W. Jiang, S. Xun, M. Zhang, W. Zhu and H. Li, ACS Sustainable Chem. Eng., 2017, 5, 8930–8938 Search PubMed.
- W. Jin, Y. Tian, G. Wang, D. Zeng, Q. Xu and J. Cui, RSC Adv., 2017, 7, 48208–48213 Search PubMed.
- A. Liu, M. Zhu and B. Dai, Appl. Catal., A, 2019, 583, 117134 Search PubMed.
- J. Luis Garcia-Gutierrez, G. C. Laredo, P. Garcia-Gutierrez and F. Jimenez-Cruz, Fuel, 2014, 138, 118–125 Search PubMed.
- S. Xun, C. Hou, H. Li, M. He, R. Ma, M. Zhang, W. Zhu and H. Li, J. Mater. Sci., 2018, 53, 15927–15938 Search PubMed.
- J. T. Sampanthar, H. Xiao, H. Dou, T. Y. Nah, X. Rong and W. P. Kwan, Appl. Catal., B, 2006, 63, 85–93 Search PubMed.
- X. Carrier, S. Royer and E. Marceau, Metal Oxides, Metal Oxides in Heterogeneous Catalysis, 2018, ch. 43, p. 61 Search PubMed.
- W. R. K. Thalgaspitiya, T. Kankanam Kapuge, J. He, D. Rathnayake and S. L. Suib, Appl. Mater. Today, 2020, 19, 100616 Search PubMed.
- M. Zhang, W. Liao, Y. Wei, C. Wang, Y. Fu, Y. Gao, L. Zhu, W. Zhu and H. Li, ACS Appl. Nano Mater., 2021, 4, 1085–1093 Search PubMed.
- Q. Zhang, J. Zhang, H. Yang, Y. Dong, Y. Liu, L. Yang, D. Wei, W. Wang, L. Bai and H. Chen, Catal. Sci. Technol., 2019, 9, 2915–2922 Search PubMed.
- Y. Liu, L. Han, J. Zhang, R. Yao, H. Zhan, H. Yang, L. Bai, L. Yang, D. Wei and W. Wang, Ind. Eng. Chem. Res., 2020, 59, 6488–6496 Search PubMed.
- Y. Dong, J. Zhang, Z. Ma, H. Xu, H. Yang, L. Yang, L. Bai, D. Wei, W. Wang and H. Chen, Chem. Commun., 2019, 55, 13995–13998 Search PubMed.
- Y. Jia, G. Li and G. Ning, Fuel Process. Technol., 2011, 92, 106–111 Search PubMed.
- W. N. A. W. Mokhtar, W. A. W. Abu Bakar, R. Ali and A. A. A. Kadir, Arabian J. Chem., 2018, 11, 1201–1208 Search PubMed.
- Z. E. A. Abdalla, B. Li and A. Tufail, J. Ind. Eng. Chem., 2009, 15, 780–783 Search PubMed.
- D. Wang, E. W. Qian, H. Amano, K. Okata, A. Ishihara and T. Kabe, Appl. Catal., A, 2003, 253, 91–99 Search PubMed.
- R. Sheldon, J. Mol. Catal., 1980, 7, 107–126 Search PubMed.
- O. Gonzalez-Garcia and L. Cedeno-Caero, Catal. Today, 2009, 148, 42–48 Search PubMed.
- M. Muñoz, L. J. Mendoza-Herrera, G. P. Romanelli, D. Gazzoli and C. I. Cabello, Catal. Today, 2020 DOI:10.1016/j.cattod.2020.10.011.
- X. W. Lv, C. C. Weng, Y. P. Zhu and Z. Y. Yuan, Small, 2021, 2005304, DOI:10.1002/smll.202005304.
- L. Chen, Z.-P. Hu, J.-T. Ren, Z. Wang and Z.-Y. Yuan, Microporous Mesoporous Mater., 2021, 315, 110921 Search PubMed.
- S. Xun, Q. Ti, Z. Jiao, L. Wu, M. He, L. Chen, L. Zhu, W. Zhu and H. Li, Ind. Eng. Chem. Res., 2020, 59, 18471–18479 Search PubMed.
- R. Zhao, X. Li, J. Su and X. Gao, Appl. Surf. Sci., 2017, 392, 810–816 Search PubMed.
- R. Ma, J. Guo, D. Wang, M. He, S. Xun, J. Gu, W. Zhu and H. Li, Colloids Surf., A, 2019, 572, 250–258 Search PubMed.
- C. Wang, Y. Qiu, H. Wu, W. Yang, Q. Zhu, Z. Chen, S. Xun, W. Zhu and H. Li, Fuel, 2020, 270, 117498 Search PubMed.
- S. Said and A. A. Abdelrahman, J. Sol–Gel Sci. Technol., 2020, 95, 308–320 Search PubMed.
- Y. Qin, S. Xun, L. Zhan, Q. Lu, M. He, W. Jiang, H. Li, M. Zhang, W. Zhu and H. Li, New J. Chem., 2017, 41, 569–578 Search PubMed.
- Y. Cao, H. Wang, R. Ding, L. Wang, Z. Liu and B. Lv, Appl. Catal., A, 2020, 589, 117308 Search PubMed.
- C. Wang, W. Jiang, H. Chen, L. Zhu, J. Luo, W. Yang, G. Chen, Z. Chen, W. Zhu and H. Li, Chin. J. Catal., 2021, 42, 557–562 Search PubMed.
- C. Wang, A. Li, J. Xu, J. Wen, H. Zhang and L. Zhang, J. Chem. Technol. Biotechnol., 2019, 94, 3403–3412 Search PubMed.
- S.-W. Li, W. Wang and J.-S. Zhao, Appl. Catal., A, 2020, 602, 117671 Search PubMed.
- Q. Luo, Q. Zhou, Y. Lin, S. Wu, H. Liu, C. Du, Y. Zhong and C. Yang, Catal. Sci. Technol., 2019, 9, 6166–6179 Search PubMed.
- L. Seifikar Gomi and M. Afsharpour, Appl. Organomet. Chem., 2019, 33, e4830 Search PubMed.
- S.-W. Li, W. Wang and J.-S. Zhao, Appl. Catal., B, 2020, 277, 119224 Search PubMed.
- S. K. Singh and A. W. Savoy, J. Mol. Liq., 2020, 297, 112038 Search PubMed.
- A. Bösmann, L. Datsevich, A. Jess, A. Lauter, C. Schmitz and P. Wasserscheid, Chem. Commun., 2001, 2494–2495, 10.1039/B108411A.
- C. Dai, J. Zhang, C. Huang and Z. Lei, Chem. Rev., 2017, 117, 6929–6983 Search PubMed.
- X. Chen, H. Guo, A. A. Abdeltawab, Y. Guan, S. S. Al-Deyab, G. Yu and L. Yu, Energy Fuels, 2015, 29, 2998–3003 Search PubMed.
- Y.-F. Guo, C.-Y. Gao, K.-G. Yang, Z.-J. Wang, Y.-H. Duan, S.-X. Feng and H. Xu, Energy Fuels, 2019, 33, 10728–10733 Search PubMed.
- H. A. Elwan, M. T. Zaky, A. S. Farag, F. S. Soliman and M. Ezel Dean Hassan, J. Mol. Liq., 2017, 248, 549–555 Search PubMed.
- H. A. Elwan, M. T. Zaky, A. S. Farag, F. S. Soliman and M. E. D. Hassan, J. Mol. Liq., 2020, 310, 113146 Search PubMed.
- M. Zhang, J. Liu, H. Li, Y. Wei, Y. Fu, W. Liao, L. Zhu, G. Chen, W. Zhu and H. Li, Appl. Catal., B, 2020, 271, 118936 Search PubMed.
- M. Safa, B. Mokhtarani, H. R. Mortaheb and K. Tabar Heidar, Energy Fuels, 2016, 30, 10909–10916 Search PubMed.
- Y. Chen, H.-y. Song, Y.-z. Lu, H. Meng, C.-x. Li, Z.-g. Lei and B.-h. Chen, Ind. Eng. Chem. Res., 2016, 55, 10394–10403 Search PubMed.
- S. Xun, W. Jiang, T. Guo, M. He, R. Ma, M. Zhang, W. Zhu and H. Li, J. Colloid Interface Sci., 2019, 534, 239–247 Search PubMed.
- W. Jiang, H. Jia, X. Fan, L. Dong, T. Guo, L. Zhu, W. Zhu and H. Li, Appl. Surf. Sci., 2019, 484, 1027–1034 Search PubMed.
- L. Zhang, J. Wang, Y. Sun, B. Jiang and H. Yang, Chem. Eng. J., 2017, 328, 445–453 Search PubMed.
- A. P. Abbott, G. Capper, D. L. Davies, R. K. Rasheed and V. Tambyrajah, Chem. Commun., 2003, 70–71, 10.1039/B210714G.
- D. Chandran, M. Khalid, R. Walvekar, N. M. Mubarak, S. Dharaskar, W. Y. Wong and T. C. S. M. Gupta, J. Mol. Liq., 2019, 275, 312–322 Search PubMed.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 Search PubMed.
- F. Lima, L. C. Branco, A. J. D. Silvestre and I. M. Marrucho, Fuel, 2021, 293, 120297 Search PubMed.
- Z. Zhu, H. Lü, M. Zhang and H. Yang, Phys. Chem. Chem. Phys., 2021, 23, 785–805 Search PubMed.
- J. Yin, J. Wang, Z. Li, D. Li, G. Yang, Y. Cui, A. Wang and C. Li, Green Chem., 2015, 17, 4552–4559 Search PubMed.
- L. Hao, M. Wang, W. Shan, C. Deng, W. Ren, Z. Shi and H. Lü, J. Hazard. Mater., 2017, 339, 216–222 Search PubMed.
- C.-f. Mao, R.-x. Zhao and X.-p. Li, RSC Adv., 2017, 7, 42590–42596 Search PubMed.
- L. Hao, T. Su, D. Hao, C. Deng, W. Ren and H. Lü, Chin. J. Catal., 2018, 39, 1552–1559 Search PubMed.
- H. Lü, P. Li, C. Deng, W. Ren, S. Wang, P. Liu and H. Zhang, Chem. Commun., 2015, 51, 10703–10706 Search PubMed.
- W. Jiang, K. Zhu, H. Li, L. Zhu, M. Hua, J. Xiao, C. Wang, Z. Yang, G. Chen, W. Zhu, H. Li and S. Dai, Chem. Eng. J., 2020, 394, 124831 Search PubMed.
- W. Jiang, H. Jia, H. Li, L. Zhu, R. Tao, W. Zhu, H. Li and S. Dai, Green Chem., 2019, 21, 3074–3080 Search PubMed.
- W. Liu, T. Li, G. Yu, J. Wang, Z. Zhou and Z. Ren, Fuel, 2020, 265, 116967 Search PubMed.
- L. Sun, Z. Zhu, T. Su, W. Liao, D. Hao, Y. Chen, Y. Zhao, W. Ren, H. Ge and H. Lü, Appl. Catal., B, 2019, 255, 117747 Search PubMed.
- H. Xu, D. Zhang, F. Wu, X. Wei and J. Zhang, Fuel, 2018, 225, 104–110 Search PubMed.
- W. Zhu, P. Wu, Y. Chao, H. Li, F. Zou, S. Xun, F. Zhu and Z. Zhao, Ind. Eng. Chem. Res., 2013, 52, 17399–17406 Search PubMed.
- W. Jiang, L. Dong, W. Liu, T. Guo, H. Li, M. Zhang, W. Zhu and H. Li, RSC Adv., 2017, 7, 55318–55325 Search PubMed.
- C. M. Granadeiro, P. M. C. Ferreira, D. Juliao, L. A. Ribeiro, R. Valenca, J. C. Ribeiro, I. S. Goncalves, B. de Castro, M. Pillinger, L. Cunha-Silva and S. S. Balula, Energies, 2018, 11, 1696 Search PubMed.
- B. Bertleff, J. Claussnitzer, W. Korth, P. Wasserscheid, A. Jess and J. Albert, ACS Sustainable Chem. Eng., 2017, 5, 4110–4118 Search PubMed.
- R. Wang, G. Zhang and H. Zhao, Catal. Today, 2010, 149, 117–121 Search PubMed.
- S. O. Ribeiro, C. M. Granadeiro, P. L. Almeida, J. Pires, M. C. Capel-Sanchez, J. M. Campos-Martin, S. Gago, B. de Castro and S. S. Balula, Catal. Today, 2019, 333, 226–236 Search PubMed.
- J. Xu, Z. Zhu, T. Su, W. Liao, C. Deng, D. Hao, Y. Zhao, W. Ren and H. Lu, Chin. J. Catal., 2020, 41, 868–876 Search PubMed.
- H. Li, C. Lian, L. Chen, J. Zhao and G.-Y. Yang, Nanoscale, 2020, 12, 16091–16101 Search PubMed.
- P. Deshlahra, Encyclopedia of Interfacial Chemistry, 2018, pp. 553–559 DOI:10.1016/B978-0-12-409547-2.14126-5.
- A. E. S. Choi, S. Roces, N. Dugos and M.-W. Wan, Fuel, 2016, 180, 127–136 Search PubMed.
- A. E. S. Choi, S. Roces, N. Dugos and M.-W. Wan, Sustainable Environ. Res., 2016, 26, 184–190 Search PubMed.
- W. Trakarnpruk and K. Rujiraworawut, Fuel Process. Technol., 2009, 90, 411–414 Search PubMed.
- M. Ammam, J. Mater. Chem. A, 2013, 1, 6291–6312 Search PubMed.
- A. Blazevic and A. Rompel, Coord. Chem. Rev., 2016, 307, 42–64 Search PubMed.
- C. Komintarachat and W. Trakarnpruk, Ind. Eng. Chem. Res., 2006, 45, 1853–1856 Search PubMed.
- M. Te, C. Fairbridge and Z. Ring, Appl. Catal., A, 2001, 219, 267–280 Search PubMed.
- N. Mizuno, K. Kamata and K. Yamaguchi, in Bifunctional Molecular Catalysis, ed. T. Ikariya and M. Shibasaki, 2011, vol. 37, pp. 127–160 Search PubMed.
- A. Akbari, M. Chamack and M. Omidkhah, J. Mater. Sci., 2020, 55, 6513–6524 Search PubMed.
- X. Li, P. Li, J. Liu, Z. Lin and C. Hu, RSC Adv., 2020, 10, 22515–22521 Search PubMed.
- F. Mirante, A. C. Alves, D. Juliao, P. L. Almeida, S. Gago, R. Valenca, J. C. Ribeiro, B. de Castro, C. M. Granadeiro and S. S. Balula, Fuel, 2020, 259, 116213 Search PubMed.
- A. Mojaverian Kermani, V. Mahmoodi, M. Ghahramaninezhad and A. Ahmadpour, Sep. Purif. Technol., 2021, 258, 117960 Search PubMed.
- J. Xiao, L. Wu, Y. Wu, B. Liu, L. Dai, Z. Li, Q. Xia and H. Xi, Appl. Energy, 2014, 113, 78–85 Search PubMed.
- X. Liao, Y. Huang, Y. Zhou, H. Liu, Y. Cai, S. Lu and Y. Yao, Appl. Surf. Sci., 2019, 484, 917–924 Search PubMed.
- A. K. Dizaji, B. Mokhtarani and H. R. Mortaheb, Fuel, 2019, 236, 717–729 Search PubMed.
- Y. Zhu, M. Zhu, L. Kang, F. Yu and B. Dai, Ind. Eng. Chem. Res., 2015, 54, 2040–2047 Search PubMed.
- Z. Shokri Aghbolagh, M. R. Khanmohammadi Khorrami and M. S. Rahmatyan, Energy Fuels, 2020, 34, 16366–16380 Search PubMed.
- Z. Yu, X. Huang, S. Xun, M. He, L. Zhu, L. Wu, M. Yuan, W. Zhu and H. Li, J. Mol. Liq., 2020, 308, 113059 Search PubMed.
- Z. Ma, J. Zhang, H. Zhan, M. Xu, H. Yang, L. Yang, L. Bai, H. Chen, D. Wei and W. Wang, Process Saf. Environ. Prot., 2020, 140, 26–33 Search PubMed.
- Y. L. Peng, J. Y. Liu, H. F. Zhang, D. Luo and D. Li, Inorg. Chem. Front., 2018, 5, 1563–1569 Search PubMed.
- R. Abazari, L. Esrafili, A. Morsali, Y. Wu and J. Gao, Appl. Catal., B, 2021, 283, 119582 Search PubMed.
- C. Wang, A.-R. Li and Y.-L. Ma, Fuel Process. Technol., 2021, 212, 106629 Search PubMed.
- P. P. Wei, Y. Yang, W. Z. Li and G. M. Li, Fuel, 2020, 274, 117834 Search PubMed.
- Y. Lu, C. Yue, B. Liu, M. Zhang, Y. Li, W. Yang, Y. Lin, Y. Pan, D. Sun and Y. Liu, Microporous Mesoporous Mater., 2021, 311, 110694 Search PubMed.
- L. Xu, Y. Wang, T. Xu, S. Liu, J. Tong, R. Chu, X. Hou and B. Liu, ChemCatChem, 2018, 10, 5386–5390 Search PubMed.
- S. W. Li, R. M. Gao, R. L. Zhang and J. S. Zhao, Fuel, 2016, 184, 18–27 Search PubMed.
- S. W. Li, Z. Yang, R. M. Gao, G. Zhang and J. S. Zhao, Appl. Catal., B, 2018, 221, 574–583 Search PubMed.
- S. W. Li, J. R. Li, Q. P. Jin, Z. Yang, R. L. Zhang, R. M. Gao and J. S. Zhao, J. Hazard. Mater., 2017, 337, 208–216 Search PubMed.
- H. W. Yang, B. Jiang, Y. L. Sun, L. H. Zhang, Z. H. Huang, Z. N. Sun and N. Yang, J. Hazard. Mater., 2017, 333, 63–72 Search PubMed.
- H. Liu, Z. Li, J. Dong, D. Liu, C. Liu, Y. Chi and C. Hu, Nanoscale, 2020, 12, 16586–16595 Search PubMed.
- H. Zhang, X. Xu, H. Lin, M. A. Ud Din, H. Wang and X. Wang, Nanoscale, 2017, 9, 13334–13340 Search PubMed.
- G. Ye, L. Hu, Y. Gu, C. Lancelot, A. Rives, C. Lamonier, N. Nuns, M. Marinova, W. Xu and Y. Sun, J. Mater. Chem. A, 2020, 8, 19396–19404 Search PubMed.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastré, J. Mater. Chem., 2006, 16, 626–636 Search PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 974 Search PubMed.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 Search PubMed.
- B. N. Bhadra and S. H. Jhung, Nanoscale, 2018, 10, 15035–15047 Search PubMed.
- M. Safaei, M. M. Foroughi, N. Ebrahimpoor, S. Jahani, A. Omidi and M. Khatami, TrAC, Trends Anal. Chem., 2019, 118, 401–425 Search PubMed.
- O. K. Farha and J. T. Hupp, Acc. Chem. Res., 2010, 43, 1166–1175 Search PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 Search PubMed.
- S.-N. Kim, J. Kim, H.-Y. Kim, H.-Y. Cho and W.-S. Ahn, Catal. Today, 2013, 204, 85–93 Search PubMed.
- C. Valles-Garcia, A. Santiago-Portillo, M. Alvaro, S. Navalon and H. Garcia, Appl. Catal., A, 2020, 590, 117340 Search PubMed.
- X. Liao, X. Wang, F. Wang, Y. Yao and S. Lu, J. Inorg. Organomet. Polym. Mater., 2020, 31, 756–762, DOI:10.1007/s10904-020-01808-y.
- Y. Zhang, G. Li, L. Kong and H. Lu, Fuel, 2018, 219, 103–110 Search PubMed.
- P. Yang, Y. Huang, Z.-W. Zhang, N. Li and Y. Fan, Dalton Trans., 2020, 49, 10052–10057 Search PubMed.
- G. Ye, Y. Gu, W. Zhou, W. Xu and Y. Sun, ACS Catal., 2020, 10, 2384–2394 Search PubMed.
- M. Y. Masoomi, M. Bagheri and A. Morsali, Inorg. Chem., 2015, 54, 11269–11275 Search PubMed.
- H. Ma, Z. Zhu, P. Tang, T. Su, P. Wu and H. Lü, Microporous Mesoporous Mater., 2021, 311, 110702 Search PubMed.
- A. M. Viana, S. O. Ribeiro, B. de Castro, S. S. Balula and L. Cunha-Silva, Materials, 2019, 12, 3009 Search PubMed.
- X. Zhang, P. Huang, A. Liu and M. Zhu, Fuel, 2017, 209, 417–423 Search PubMed.
- W. Xiao, Q. Dong, Y. Wang, Y. Li, S. Deng and N. Zhang, CrystEngComm, 2018, 20, 5658–5662 Search PubMed.
- A. M. Viana, D. Juliao, F. Mirante, R. G. Faria, B. de Castro, S. S. Balula and L. Cunha-Silva, Catal. Today, 2021, 362, 28–34 Search PubMed.
- G. Fu, B. Bueken and D. De Vos, Small Methods, 2018, 2, 1800203 Search PubMed.
- S. Lee and W. Choe, Elaboration and Applications of Metal-Organic Frameworks, 2018, pp. 143–182 DOI:10.1142/9789813226739_0003.
- Y. Gu, G. Ye, W. Xu, W. Zhou and Y. Sun, ChemistrySelect, 2020, 5, 244–251 Search PubMed.
- X. Li, L. Zhang and Y. Sun, Catalysts, 2020, 10, 1091 Search PubMed.
- Z. Zhu, H. Ma, H. Xu, B. Wang, P. Wu and H. Lü, J. Hazard. Mater., 2020, 393, 122458 Search PubMed.
- J. R. Ludwig and C. S. Schindler, Chem, 2017, 2, 313–316 Search PubMed.
- L. Dai, Y. Xue, L. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 Search PubMed.
- Q. Zhao, Q. Mao, Y. Zhou, J. Wei, X. Liu, J. Yang, L. Luo, J. Zhang, H. Chen, H. Chen and L. Tang, Chemosphere, 2017, 189, 224–238 Search PubMed.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 Search PubMed.
- W. S. Hummers Jr and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 Search PubMed.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 Search PubMed.
- J. He, P. Wu, L. Lu, H. Sun, Q. Jia, M. Hua, M. He, C. Xu, W. Zhu and H. Li, Energy Fuels, 2019, 33, 8302–8311 Search PubMed.
- C. Xu, J. Chen, S. Li, Q. Gu, D. Wang, C. Jiang and Y. Liu, J. Hazard. Mater., 2021, 403, 123806 Search PubMed.
- S. Navalón, W.-J. Ong and X. Duan, Processes, 2020, 8, 672 Search PubMed.
- G. Abdi, A. Alizadeh and M. M. Khodaei, Mater. Chem. Phys., 2017, 201, 323–330 Search PubMed.
- G. Abdi, M. Ashokkumar and A. Alizadeh, Fuel, 2017, 210, 639–645 Search PubMed.
- Y. Zhang and R. Wang, Diamond Relat. Mater., 2017, 73, 161–168 Search PubMed.
- Q. Gu, G. Wen, Y. Ding, K.-H. Wu, C. Chen and D. Su, Green Chem., 2017, 19, 1175–1181 Search PubMed.
- Q. Gu, Y. Ding, Z. Liu, Y. Lin, R. Schloegl, S. Heumann and D. Su, J. Energy Chem., 2019, 32, 131–137 Search PubMed.
- Z. C. Kampouraki, D. A. Giannakoudakis, K. S. Triantafyllidis and E. A. Deliyanni, Green Chem., 2019, 21, 6685–6698 Search PubMed.
- T. S. Miller, A. B. Jorge, T. M. Suter, A. Sella, F. Cora and P. F. McMillan, Phys. Chem. Chem. Phys., 2017, 19, 15613–15638 Search PubMed.
- W.-J. Ong, L.-L. Tan, Y. H. Ng, S.-T. Yong and S.-P. Chai, Chem. Rev., 2016, 116, 7159–7329 Search PubMed.
- L. Shen, G. Lei, Y. Fang, Y. Cao, X. Wang and L. Jiang, Chem. Commun., 2018, 54, 2475–2478 Search PubMed.
- G. Lei, Y. Cao, W. Zhao, Z. Dai, L. Shen, Y. Xiao and L. Jiang, ACS Sustainable Chem. Eng., 2019, 7, 4941–4950 Search PubMed.
- Q. Jia, J. He, P. Wu, J. Luo, Y. Wei, H. Li, S. Xun, W. Zhu and H. Li, Mol. Catal., 2019, 468, 100–108 Search PubMed.
- Y. Wei, P. Wu, J. Luo, L. Dai, H. Li, M. Zhang, L. Chen, L. Wang, W. Zhu and H. Li, Microporous Mesoporous Mater., 2020, 293, 109788 Search PubMed.
- L. Lu, J. He, P. Wu, Y. Wu, Y. Chao, H. Li, D. Tao, L. Fan, H. Li and W. Zhu, Green Chem., 2018, 20, 4453–4460 Search PubMed.
- P. Wu, W. Zhu, Y. Chao, J. Zhang, P. Zhang, H. Zhu, C. Li, Z. Chen, H. Li and S. Dai, Chem. Commun., 2016, 52, 144–147 Search PubMed.
- Z. Quan, Y. Wang and J. Fang, Acc. Chem. Res., 2013, 46, 191–202 Search PubMed.
- P. Wu, L. Lu, J. He, L. Chen, Y. Chao, M. He, F. Zhu, X. Chu, H. Li and W. Zhu, Green Energy Environ., 2020, 5, 166–172 Search PubMed.
- N. Lv, L. Sun, L. Chen, Y. Li, J. Zhang, P. Wu, H. Li, W. Zhu and H. Li, Phys. Chem. Chem. Phys., 2019, 21, 21867–21874 Search PubMed.
- L. Dai, Y. Wei, X. Xu, P. Wu, M. Zhang, C. Wang, H. Li, Q. Zhang, H. Li and W. Zhu, ChemCatChem, 2020, 12, 1734–1742 Search PubMed.
- P. Wu, Y. Wu, L. Chen, J. He, M. Hua, F. Zhu, X. Chu, J. Xiong, M. He, W. Zhu and H. Li, Chem. Eng. J., 2020, 380, 122526 Search PubMed.
- J. He, Y. Wu, P. Wu, L. Lu, C. Deng, H. Ji, M. He, W. Zhu and H.-m. Li, ACS Sustainable Chem. Eng., 2020, 8, 2032–2039 Search PubMed.
- P. Wu, Q. Jia, J. He, L. Lu, L. Chen, J. Zhu, C. Peng, M. He, J. Xiong, W. Zhu and H. Li, J. Hazard. Mater., 2020, 391, 122183 Search PubMed.
- Y. Gao, Z. Y. Lv, R. M. Gao, G. Zhang, Y. Zheng and J. S. Zhao, J. Hazard. Mater., 2018, 359, 258–265 Search PubMed.
- H. Song, N. Zhu, B. Chen, F. Wang, M. Bai and X. Wang, Russ. J. Appl. Chem., 2016, 89, 2076–2083 Search PubMed.
- S. Matsuzawa, J. Tanaka, S. Sato and T. Ibusuki, J. Photochem. Photobiol., A, 2002, 149, 183–189 Search PubMed.
- H. Dong, G. Zeng, L. Tang, C. Fan, C. Zhang, X. He and Y. He, Water Res., 2015, 79, 128–146 Search PubMed.
- K. Kalantari, M. Kalbasi, M. Sohrabi and S. J. Royaee, Ceram. Int., 2016, 42, 14834–14842 Search PubMed.
- C. Wang, W. Zhu, Y. Xu, H. Xu, M. Zhang, Y. Chao, S. Yin, H. Li and J. Wang, Ceram. Int., 2014, 40, 11627–11635 Search PubMed.
- G. Zhang, M. Gao, M. Tian and W. Zhao, J. Solid State Chem., 2019, 279, 120953 Search PubMed.
- G. Guo, H. Guo, F. Wang, L. J. France, W. Yang, Z. Mei and Y. Yu, Green Energy Environ., 2020, 5, 114–120 Search PubMed.
- C. N. C. Hitam, A. A. Jalil and Y. O. Raji, Top. Catal., 2020, 63, 1169–1181 Search PubMed.
- K. Chen, X.-M. Zhang, X.-F. Yang, M.-G. Jiao, Z. Zhou, M.-H. Zhang, D.-H. Wang and X.-H. Bu, Appl. Catal., B, 2018, 238, 263–273 Search PubMed.
- S. H. Ammar, Y. S. Kareem and A. D. Ali, J. Environ. Chem. Eng., 2018, 6, 6780–6786 Search PubMed.
- R. Ma, S. Zhang, T. Wen, P. Gu, L. Li, G. Zhao, F. Niu, Q. Huang, Z. Tang and X. Wang, Catal. Today, 2019, 335, 20–30 Search PubMed.
- X. Li, Z. Zhang, C. Yao, X. Lu, X. Zhao and C. Ni, Appl. Surf. Sci., 2016, 364, 589–596 Search PubMed.
- W. L. Haden and I. A. Schwint, Ind. Eng. Chem., 1967, 59, 58–69 Search PubMed.
- X. Li, W. Zhu, X. Lu, S. Zuo, C. Yao and C. Ni, Chem. Eng. J., 2017, 326, 87–98 Search PubMed.
- J. Cui, G. Wang, W. Liu, P. Ke, Q. Tian, X. Li and Y. Tian, Fuel, 2021, 290, 120066 Search PubMed.
- M. Mousavi-Kamazani and S. Ashrafi, Ultrason. Sonochem., 2020, 63, 104948 Search PubMed.
- Y. Yang, M. Wu, X. Zhu, H. Xu, S. Ma, Y. Zhi, H. Xia, X. Liu, J. Pan, J.-Y. Tang, S.-P. Chai, L. Palmisano, F. Parrino, J. Liu, J. Ma, Z.-L. Wang, L. Tan, Y.-F. Zhao, Y.-F. Song, P. Singh, P. Raizada, D. Jiang, D. Li, R. A. Geioushy, J. Ma, J. Zhang, S. Hu, R. Feng, G. Liu, M. Liu, Z. Li, M. Shao, N. Li, J. Peng, W.-J. Ong, N. Kornienko, Z. Xing, X. Fan and J. Ma, Chin. Chem. Lett., 2019, 30, 2065–2088 Search PubMed.
- Y. Ren, D. Zeng and W.-J. Ong, Chin. J. Catal., 2019, 40, 289–319 Search PubMed.
- Y. Li, X. Li, H. Zhang and Q. Xiang, Nanoscale Horiz., 2020, 5, 765–786 Search PubMed.
- X. Yu, S.-F. Ng, L. K. Putri, L.-L. Tan, A. R. Mohamed and W.-J. Ong, Small, 2021, 2006851, DOI:10.1002/smll.202006851.
- L. K. Putri, L.-L. Tan, W.-J. Ong, W. S. Chang and S.-P. Chai, Appl. Mater. Today, 2016, 4, 9–16 Search PubMed.
- Y. Zhu, X. Li and M. Zhu, Catal. Commun., 2016, 85, 5–8 Search PubMed.
- X. Zhang, H. Song, C. Sun, C. Chen, F. Han and X. Li, Mater. Chem. Phys., 2019, 226, 34–43 Search PubMed.
- X. Zeng, X. Xiao, Y. Li, J. Chen and H. Wang, Appl. Catal., B, 2017, 209, 98–109 Search PubMed.
- S.-F. Ng, M. Y. L. Lau and W.-J. Ong, Sol. RRL, 2021, 2000535, DOI:10.1002/solr.202000535.
- Z. Huang, H. Song, A. Li, Z. An, K. Zhang, X. Xiang, X. Shu and J. He, Energy Fuels, 2020, 34, 14676–14687 Search PubMed.
- Y. Cai, H. Song, Z. An, X. Xiang, X. Shu and J. He, Green Chem., 2018, 20, 5509–5519 Search PubMed.
- S. Masoumi and S. A. Hosseini, Fuel, 2020, 277, 118137 Search PubMed.
- B. Li, H. Song, F. Han and L. Wei, Appl. Catal., B, 2020, 269, 118845 Search PubMed.
- X. Y. Zeng, X. Y. Xiao, J. Y. Chen, Y. Wang and H. L. Wang, J. Hazard. Mater., 2020, 391, 122198 Search PubMed.
- M. Ja'fari, S. L. Ebrahimi and M. R. Khosravi-Nikou, Ultrason. Sonochem., 2018, 40, 955–968 Search PubMed.
| This journal is © The Royal Society of Chemistry 2021 |