
Prediction of room-temperature ferromagnetism and large perpendicular magnetic anisotropy in a planar hypercoordinate FeB3 monolayer†
Cheng
Tang
 ab,
Kostya (Ken)
Ostrikov
ab,
Stefano
Sanvito
c and
Aijun
Du
*ab
ab,
Kostya (Ken)
Ostrikov
ab,
Stefano
Sanvito
c and
Aijun
Du
*ab
aCentre for Materials Science, Queensland University of Technology, Brisbane, QLD 4000, Australia. E-mail: aijun.du@qut.edu.au
bSchool of Chemistry and Physics, Queensland University of Technology, Brisbane, QLD 4000, Australia
cSchool of Physics and CRANN Institute, Trinity College, Dublin 2, Ireland
First published on 13th November 2020
Abstract
Two-dimensional (2D) magnets simultaneously possessing a high transition temperature and large perpendicular magnetic anisotropy are extremely rare, but essential for highly efficient spintronic applications. By using ab initio and global minimization approaches, we for the first time report a completely planar hypercoordinate metalloborophene (α-FeB3) with high stability, unusual stoichiometry and exceptional magnetoelectronic properties. The α-FeB3 monolayer exhibits room-temperature ferromagnetism (Tc = 480 K), whose origin is first revealed by the B-mediated RKKY interaction in the 2D regime. Its perpendicular magnetic anisotropy is almost six times larger than that of the experimentally realized 2D CrI3 and Fe3GeTe2. Moreover, metallic α-FeB3 shows n- and p-type Dirac transport with a high Fermi velocity in both spin channels. Our results not only highlight a promising 2D ferromagnet for advanced spintronics, but also pave the way for exploring novel 2D magnetism in boron-based magnetic allotropes.
New conceptsThe discovery of two-dimensional (2D) magnetism opens the gate for advanced spintronic nanodevices. However, the practical application of 2D magnets is limited by their own disadvantages, such as low Cuire temperature and/or moderate magnetic anisotropy. In this work, by combining comprehensive PSO and first-principles approaches, we present a new type of metalloborophene (α-FeB3) monolayer with a planar hypercoordinate structure, high stability and novel magnetoelectronic properties. α-FeB3 exhibits out-of-plane ferromagnetism with a Curie temperature of 480 K. The magnetic anisotropy energy is almost six-times larger than that of 2D CrI3 and Fe3GeTe2. Such novel magnetism in α-FeB3 originates from the B-mediated RKKY interaction, which is first revealed in the 2D regime. Furthermore, metallic α-FeB3 shows n- and p-type Dirac transport with a high Fermi velocity in both spin channels. Our findings highlight the α-FeB3 monolayer with novel magnetoelectronic properties for highly efficient spintronic applications and pave the way for exploring new 2D magnets among boron-based allotropes. |
Introduction
The realization of two-dimensional (2D) magnetism provides unprecedented opportunities for future magnetoelectronic and magneto-optic applications.1–4 However, the long-range 2D ferromagnetic (FM) order is limited by its instability originating from short-range interactions and weak anisotropy, as established in the Mermin–Wagner theorem.5 After unremitting efforts, a few 2D magnets were experimentally fabricated.6–10 Herein, the sizable magnetocrystalline anisotropy plays the critical role in lifting the Mermin–Wagner restriction, thus stabilizing the 2D magnetic order. Among them, the CrI3 and Fe3GeTe2 monolayers exhibit persistent ferromagnetism and large perpendicular magnetic anisotropy (∼0.70 meV per magnetic atom).7,9 However, the Curie temperatures of these monolayers remain well below room temperature, imposing great limitations on practical applications.11 The exception is the room-temperature 2D magnet VSe2, which, however, can only be synthesized on metal substrates.12 Its wide application is then limited by the unstable magnetic order in the free-standing form.13 In addition, a very limited number of single-layer magnets have been theoretically predicted, all with a magnetic anisotropy significantly lower than that of the CrI3 monolayer.14–21 Hence, the exploration of new 2D magnets with high transition temperatures and large perpendicular magnetic anisotropy remains a huge challenge and is of great importance for both fundamental research and future applications.Boron, B, is electron deficient and can form diverse compounds with metals. These span a range of different stoichiometry and present rich electrochemical properties.22,23 Recently, 2D boron nanosheets, i.e. borophene, have been experimentally realized in abundant allotropes24,25 with novel Dirac dispersion, superconductivity and high thermal conductivity.26–28 Going beyond borophene, metalloborophene (MB) could be a promising platform for the exploration of a new family of 2D compounds with hypercoordinate metal–boron bonds.29 However, unlike the rich stoichiometries in the bulk compounds and planar structures in clusters, to date theoretically investigated MB nanosheets are limited to the MB2n composition, rarely possessing a completely planar geometry.30–34 Particularly, MB monolayers with transition metal centred boron rings promise great potential in stabilizing 2D magnetism since the intrinsic magnetic coupling is likely induced by the unpaired d or f electrons.35 Nevertheless, current studies have been mainly focused on their electronic and optical properties.30–34 Therefore, the study of the origin of the magnetism in 2D hypercoordinate MB is still urgent and significant. If it displays novel 2D magnetism, the MB monolayer might offer hope for applications toward high-speed, low dissipation spintronic nanodevices.
In this work, we investigate 2D MB allotropes by departing from the conventional MB2n configurations. Herein, Fe is chosen as the magnetic element, simply because its multiple oxidation states can facilitate the coordination with B. By combining a global structure search and density functional theory (DFT) calculations, we have discovered a completely planar α-FeB3 monolayer with an unusual hypercoordinate structure, high stability, and novel magnetic and electronic properties. In such a planar structure, hexacoordinate and pentacoordinate B coexist around the Fe centre. Electron transfer from Fe to B atoms in the Fe©B6 ring endows the 2D FeB3 structure with significant dynamical and thermal stability.31 Remarkably, the α-FeB3 sheet possesses perpendicular FM with a high Curie temperature of 480 K. Its high magnetic anisotropy energy (MAE) also suggests great potentials for applications in low energy spintronics. The origin of the 2D magnetism in the MB monolayer is first realized in terms of the B-mediated Ruderman–Kittel–Kasuya–Yosida (RKKY) interactions. Furthermore, n- and p-type Dirac dispersions with high Fermi velocities are discovered in both spin channels. This work provides new insights into the chemical bonding in 2D boron-based hypercoordinate materials and aims at stimulating experimental efforts towards magnetic α-FeB3 for novel spintronic devices.
Results and discussion
The geometric structures of 2D unconventional iron borides including FeB, FeB3 and FeB5 are first generated and then filtered by comprehensive particle swarm optimization (PSO) and DFT calculations. Among thousands of generated structures, iron triboride, FeB3, possesses the lower relative energy and the completely planar structure is expected to exhibit high stability and unique properties. Fig. S1 (ESI†) shows the convex hull diagram of the predicted FeBx monolayers. Compared to the discovered FeB2n monolayers, FeB3 nanosheets are more energetically favourable. Accordingly, five FeB3 monolayers with relatively lower energies are plotted in Fig. S2 (ESI†). The first two planar structures are named α- and β-FeB3. The α-FeB3 monolayer (space group Pmmm) possesses a rectangular lattice with parameters a = 2.90 and b = 4.49 Å, while β-FeB3 with a space group of P6/mmm presents a hexagonal lattice with a = 3.89 Å, and contains kagome B and trigonal Fe sublattices. Since a large total energy gap (−0.37 eV per atom) exists between these two structures, we mainly focus on the α-FeB3 monolayer, with the details for β-FeB3 in the ESI.† In Fig. 1(a), each iron atom is coordinated by 6 boron ones, forming planar iron-centred boron wheels (Fe©B6). Here, the Fe©B6 unit is composed of hexacoordinate (B1) and pentacoordinate (B2) boron atoms with bond lengths of 2.25 Å for Fe–B1 and 2.04 Å for Fe–B2. Notably, the bond angle of Fe–B2–Fe is near to 90° along the a direction, while the Fe–B1–Fe one is linear along the b direction. In addition, the B–B bond length (1.66 Å) is comparable to that in borophene (∼1.67 Å),24,36 suggesting the stability of α-FeB3. To understand the bonding nature, we calculate the electron localization function (ELF) of the FeB3 monolayer. As shown in Fig. 1(b), the electrons are mainly delocalized around Fe atoms, and an electron gas with an ELF value of around 0.5 is distributed between Fe and B. These indicate ionic Fe–B bonds and electron transfer from Fe to B. Bader charge analysis also confirms that each Fe atom transfers 0.87e electrons to the B framework.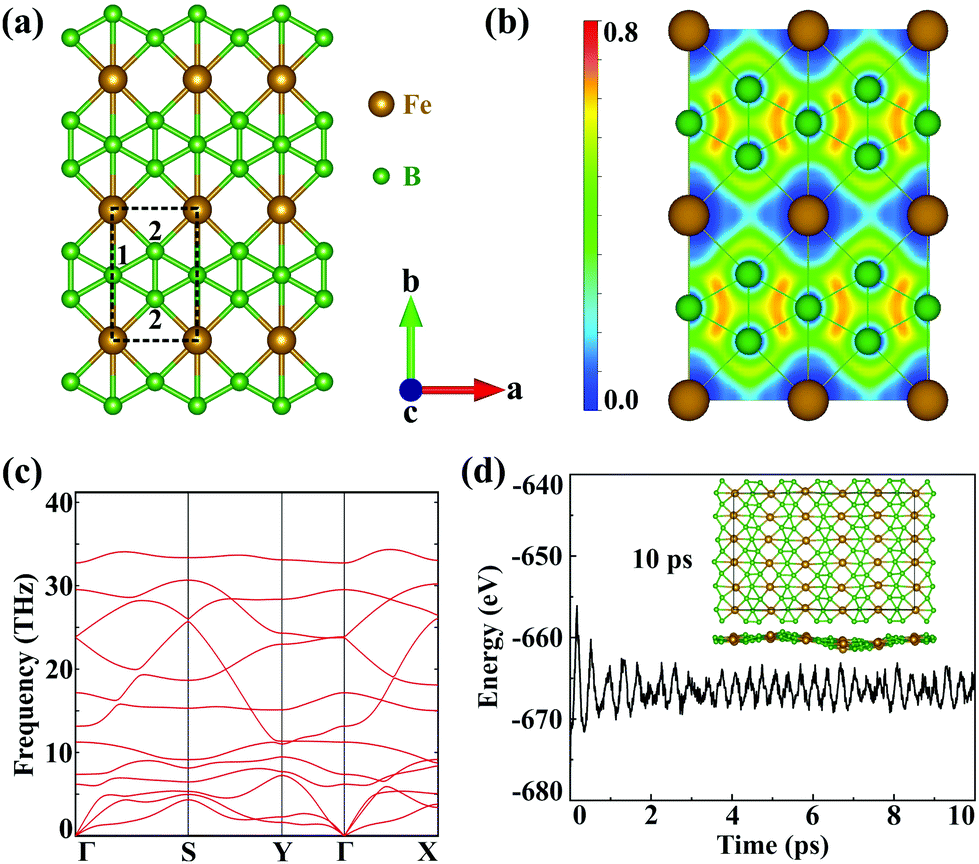 | ||
| Fig. 1 (a) Geometric structures, (b) ELF, (c) phonon spectrum and (d) AIMD simulations for the α-FeB3 monolayer. The brown and green spheres represent Fe and B atoms, respectively. The top and side views of α-FeB3 at the end of the AIMD simulation are shown in (d). | ||
Next, we evaluate the stability of the newly predicted FeB3 monolayer. The thermodynamic stability is examined by calculating the cohesive energies. The estimated energy is 5.93 eV per atom, which is larger than that of other stable monolayers, such as FeB2 (4.87 eV per atom) and FeB6 (5.56–5.79 eV per atom).30,31 Such large cohesive energy suggests that the FeB3 sheet is thermodynamically stable. Then, Fig. 1(c) shows that the phonon spectrum presents no imaginary frequency, meaning that the compound is also kinetically stable. In addition, we also evaluate the thermal stability of α-FeB3 by an ab initio molecular dynamics (AIMD) simulation at 800 K for 10 ps. In Fig. 1(d), the energy of the α-FeB3 monolayer fluctuates in a small range (∼0.1 eV per atom) during the entire simulation, and the final structure is well-preserved at the high temperature, indicating good thermal stability. To further investigate the mechanical stability, we calculate the elastic constants of the FeB3 monolayer. These are computed as C11 = 198.80, C12 = 58.42, C22 = 145.34 and C66 = 22.02 N m−1, namely they satisfy the Born criteria (C11, C22 and C66 > 0, and C11C22 – C122 > 0). This indicates that the FeB3 monolayer is mechanically stable. Moreover, the calculated Young's modulus shows a large difference between the a and b directions (175.04 and 128.17 N m−1), suggesting its strong structural anisotropy (see Fig. S3 in the ESI†).
Planar α-FeB3 with a highly stable structure is a promising candidate for 2D magnetism, given the presence of the partially occupied Fe 3d orbitals. Thus, to determine its magnetic ground state, we examine the 2 × 2 supercells having different magnetic configurations (see Fig. S4, ESI†). The calculated corresponding total energies are listed in Table S1 (ESI†). Accordingly, the magnetic ground state is found to be FM with the lowest energy among the examined configurations. As shown in Fig. 2(a), the spin density is localized around the Fe atoms, suggesting that the magnetism is mainly contributed by the Fe 3d electrons. The net magnetic moment of each Fe atom is 2.89 μB. We then calculate the magnetic coupling parameters of α-FeB3 based on the Ising model. Herein, J1, J2 and J3 describe the exchange interactions between nearest-neighbour (NN), second-NN and third-NN Fe atoms, respectively. The calculated parameters are J1 = 10.51, J2 = 58.03 and J3 =18.30 meV; thus, the estimated Curie temperature from Monte Carlo simulations is about 480 K [Fig. 2(b)], much higher than room temperature (details are in the ESI†). Finally, we calculate the MAE with respect to the out-of-plane direction by the equation of MAE = Ex−y − Ez (a positive value represents perpendicular magnetism). Fig. 2(c) shows that the α-FeB3 monolayer possesses perpendicular uniaxial anisotropy with a MAE reaching 4.13 meV per Fe, almost six times larger than that of the most-studied 2D magnets, such as CrI3 (∼0.70 meV per Cr)37 and Fe3GeTe2 (∼0.67 meV per Fe).38 The orbital-resolved MAE (see Fig. S5, ESI†) shows that the significant magnetic anisotropy in α-FeB3 is mainly contributed by the coupling between Fe dyz and dxz orbitals. The contribution of the coupling of dxy–dx2−y2, dyz–dz2 and dxz–dz2 orbitals is only half of that of dyz–dxz coupling.
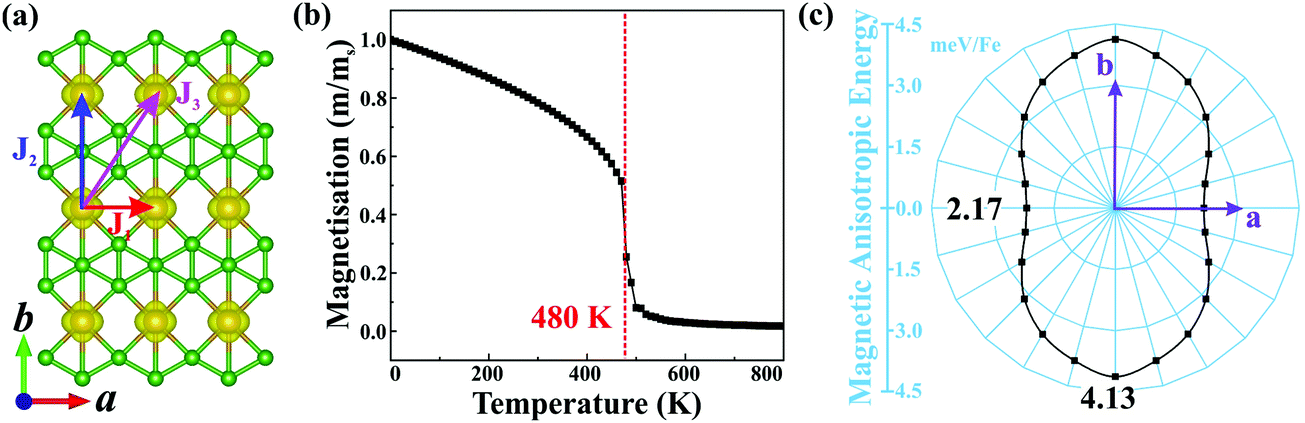 | ||
| Fig. 2 (a) Spin-dependent charge density of the α-FeB3 monolayer, (b) average magnetic moment as a function of temperature and (c) MAE in the x–y plane with respect to the out-of-plane direction. The isovalue of the spin charge density is set to be 0.01 e Å−3. | ||
In order to understand the origin of such 2D magnetism, we analyse the exchange interactions between the magnetic atoms. In α-FeB3, the distance between NN Fe atoms is 2.90 Å, suggesting weak Fe–Fe direct exchange.11 Thus, the dominant magnetic coupling is likely to be the indirect RKKY interaction, due to the existence of electron deficient orbitals in boron, as found in other rare earth borides.39,40 Importantly, the exchange parameters of the RKKY interaction decay in an oscillatory way with the distance between the magnetic atoms. To elucidate this point, we examine the relation between the exchange parameters and the Fe–Fe distance. Herein, biaxial strains from −5% to 5% per formula unit (f.u.) are adopted to change the distance between Fe atoms. The calculated total energies of α-FeB3 in Table S2 (ESI†) indicate that the magnetic ground state of α-FeB3 undergoes a FM to stripe antiferromagnetic (AFM-1 and AFM-2) transition under compressive and tensile strains of 4% per f.u., respectively. As shown in Fig. S6 (ESI†), the estimated magnetic exchange fluctuates between FM and AFM coupling with respect to the change in the Fe–Fe distance, consistent with an RKKY-type interaction.
Since magnetism controllable by an external stimulus may extend the range of application of a magnet, we explore two possible approaches, strain and charge-carrier doping, to externally modulate the magnetism. In brief, we find that the Fe magnetization increases under tensile strains, while it decreases upon compression [Fig. S7(a), ESI†]. Upon charge-carrier doping (≤0.5 e/h per f.u.), the Fe magnetization first slightly drops and then rises again with the hole concentration, whereas it remains monotonous for electron doping [Fig. S7(b), ESI†]. The transition temperatures of the FeB3 monolayer are then estimated through Monte Carlo simulations (Fig. S8 and S9, ESI†), whose results are summarised in Fig. 3(a) and (b). Clearly, biaxial strains remarkably decrease the transition temperature of FeB3 with the value dropping to 30 K (at 5% per f.u.), due to the large oscillations of the RKKY exchange. Interestingly, the Curie temperature of the α-FeB3 monolayer drops to 160 K after peaking at 610 K (at 0.2 h per f.u.) for hole doping, whereas it decreases monotonically under electron doping. In addition, Fig. 3(c) and (d) summarise the changes of the MAE under the external stimulus. Herein, the α-FeB3 monolayer keeps the perpendicular magnetic anisotropy under biaxial strains, whereas the MAE changes to in-plane (along the a and b directions) under electron and hole doping over 0.4 e/h per f.u, respectively. In Fig. 3(c), a decrease of the MAE is shown under biaxial strains. Besides, as shown in Fig. 3(d), the presence of additional electrons reduces the magnetic anisotropy, while a hole concentration of around 0.1 h per f.u. maximizes the MAE (4.55 meV per Fe). This then suggests that moderate hole-doping represents the ideal condition for magnetism by simultaneously promoting the magnetic exchange and the uniaxial MAE.
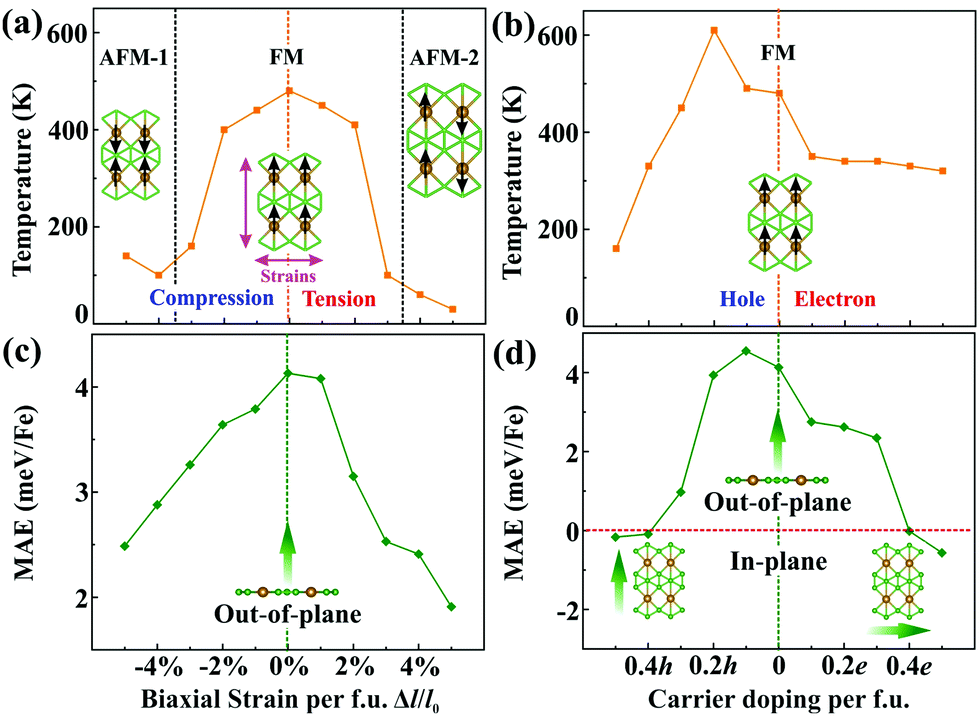 | ||
| Fig. 3 Transition temperature and magnetic anisotropy energy (MAE) of the FeB3 monolayer as a function of (a and c) external strains and (b and d) charge-carrier doping, respectively. | ||
Next, we turn our attention to the electronic properties of the planar α-FeB3. As shown in Fig. 4, the α-FeB3 monolayer is metallic with several Dirac points above and below the Fermi surface, having an n- and p-type dispersion, respectively. Due to the strong interaction between Fe and B, the contribution of these Dirac points involves orbitals from both atoms (Fig. S10, ESI†). For instance, the p-type Dirac point I in the spin-up channel is contributed by both B pz, px + py and Fe dyz, dx2−y2 orbitals [Fig. 4(c)], while the orbital components for the n-type one in the spin-down channel are the pz orbital of B1 and B2 and the Fe dyz, dxz orbitals [Fig. 4(d)]. Besides, the Fermi velocities of Dirac point I and II are calculated to be 20.84 and 8.39 × 105 m s−1, which are comparable to the value of graphene and borophene (∼106 m s−1).28,41 Since spin–orbit coupling (SOC) will affect the electronic properties, we then calculate the band structure of the α-FeB3 monolayer by considering the SOC effect, as shown in Fig. S11 (ESI†). We can see that the p-type Dirac point I keeps the linear dispersion with the SOC effect, whereas the n-type Dirac point II shows a small gap of 0.18 eV. Thus, the SOC effect truly affects some of the Dirac-type dispersions; however, some Dirac dispersions are still retained in α-FeB3 monolayers. The combination of the intriguing magnetic and electronic properties makes 2D FeB3 a candidate for the exploration of diverse spintronic effects (such as the anomalous Hall effect).42
 | ||
| Fig. 4 Band structures of the α-FeB3 monolayer for the (a) spin up and (b) spin down channels. The orbital-resolved band structures of Dirac point (c) I and (d) II. | ||
Finally, we investigate one possible route for experimental synthesis of the α-FeB3 monolayer. As we all know, atomically thin boron allotropes have been synthesised for a long time.43,44 Accordingly, the newly predicted α-FeB3 monolayer is expected to be experimentally realized by adsorbing the Fe atoms onto the δ4 boron nanosheet. Although a δ4 boron layer has not been synthesised yet, its lower formation energy than some of the fabricated ones suggests feasibility in experimental synthesis.45 The possible adsorption sites for Fe atoms on the δ4 boron sheet are marked in Fig. S12(a) (ESI†). The Fe adsorption energy is calculated by Eads = EFe–boron − Eboron − EFe, where EFe–boron, Eboron and EFe represent the total energies of the Fe adsorbed boron sheet, the pure boron sheet and a single Fe atom. Generally, the adsorption of Fe on the single-layer boron sheet is energetically favourable with negative Eads for all sites [see Fig. S12(b), ESI†]. Notably, the adsorption energy at site I is remarkably lower than the other examined sites, indicating that the Fe atoms prefer to adsorb on that site. As shown in Fig. S12(c) (ESI†), the Fe adsorbed δ4 boron sheet can change its phase to our predicted α-FeB3 monolayer under uniaxial strains along the b direction with a very small barrier energy of around 16 meV. Therefore, the α-FeB3 monolayer is likely to be realized in an experiment by adsorbing Fe atoms and then applying external strains on the δ4 boron sheets.
Conclusions
In conclusion, based on PSO global minimization and ab initio calculations, we have discovered an iron triboride polymorph (α-FeB3) with completely planar hypercoodination. This is also a member of a new family of magnetic MB allotropes with unusual B stoichiometry. The predicted α-FeB3 monolayer exhibits ferromagnetism at a high temperature of 480 K. The origin of its magnetic coupling is ascribed to the B-mediated RKKY interactions. Intriguingly, α-FeB3 possesses perpendicular magnetic anisotropy reaching a MAE value of 4.13 meV per Fe, which is several times larger than what is found in experimentally realized 2D magnets. Notably, both external strain and charge-carrier doping can be used to effectively tune its 2D magnetism. Finally, the α-FeB3 monolayer is metallic with Dirac dispersions in both spin channels for n- and p-type doping. Our findings pave the way for the discovery of new hypercoodinate MB allotropes as a promising 2D material platform for high-performance spin-enabled applications.Computational details
We have performed a swarm-intelligence structural search by mean of the CALYPSO code.46,47 This is powered by DFT using the Vienna ab initio simulation package (VASP)48,49 at the level of the Perdew–Burke–Erzenhof (PBE)50 exchange–correlation functional within the projector-augmented wave (PAW) method.51 In order to describe the strong interaction of d electrons, a Hubbard on-site Coulomb potential was set for Fe to U − J = 4.0 eV.52,53 This value is comparable to pioneering work.11,54 The Heyd–Scuseria–Ernzerhof functional was adopted for electronic calculations.55 The cut-off energy was set to 500 eV, and a 13 × 7 × 1 Monkhorst k-mesh was used to sample the first Brillouin zone. The interactions between neighbouring layers were shielded by a vacuum layer over 20 Å. Additionally, DFT-D3 corrections were included for interlayer van der Waals interactions.56 The structures were optimized until the energy and forces on each atom converged to 10−6 eV and 0.001 eV Å−1, respectively. Moreover, a 2 × 2 supercell of the α-FeB3 monolayer was adopted when considering the different magnetic configurations (more details can be found in the ESI†).Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge generous grants of high-performance computer resources provided by Queensland University of Technology, NCI National Facility, the Pawsey Supercomputing Centre through the National Computational Merit Allocation Scheme supported by the Australian Government, the Government of Western Australia. A. D. greatly appreciates the financial support from the Australian Research Council under Discovery Project (DP170103598). S. S. thanks the Irish Research Council Advanced Laureate Award (IRCLA/2019/127) for financial support.References
- C.-Z. Chang, J. Zhang, X. Feng, J. Shen, Z. Zhang, M. Guo, K. Li, Y. Ou, P. Wei and L.-L. Wang, Science, 2013, 340, 167–170 CrossRef CAS.
- W. Han, R. K. Kawakami, M. Gmitra and J. Fabian, Nat. Nanotechnol., 2014, 9, 794 CrossRef CAS.
- Z. Wang, C. Tang, R. Sachs, Y. Barlas and J. Shi, Phys. Rev. Lett., 2015, 114, 016603 CrossRef.
- S. Jiang, J. Shan and K. F. Mak, Nat. Mater., 2018, 17, 406–410 CrossRef CAS.
- N. D. Mermin and H. Wagner, Phys. Rev. Lett., 1966, 17, 1133 CrossRef CAS.
- C. Gong, L. Li, Z. Li, H. Ji, A. Stern, Y. Xia, T. Cao, W. Bao, C. Wang and Y. Wang, Nature, 2017, 546, 265–269 CrossRef CAS.
- B. Huang, G. Clark, E. Navarro-Moratalla, D. R. Klein, R. Cheng, K. L. Seyler, D. Zhong, E. Schmidgall, M. A. McGuire and D. H. Cobden, Nature, 2017, 546, 270–273 CrossRef CAS.
- M. Bonilla, S. Kolekar, Y. Ma, H. C. Diaz, V. Kalappattil, R. Das, T. Eggers, H. R. Gutierrez, M.-H. Phan and M. Batzill, Nat. Nanotechnol., 2018, 13, 289–293 CrossRef CAS.
- Y. Deng, Y. Yu, Y. Song, J. Zhang, N. Z. Wang, Z. Sun, Y. Yi, Y. Z. Wu, S. Wu and J. Zhu, Nature, 2018, 563, 94–99 CrossRef CAS.
- D. J. O’Hara, T. Zhu, A. H. Trout, A. S. Ahmed, Y. K. Luo, C. H. Lee, M. R. Brenner, S. Rajan, J. A. Gupta and D. W. McComb, Nano Lett., 2018, 18, 3125–3131 CrossRef.
- A. Bandyopadhyay, N. C. Frey, D. Jariwala and V. B. Shenoy, Nano Lett., 2019, 19, 7793–7800 CrossRef CAS.
- M. Bonilla, S. Kolekar, Y. Ma, H. C. Diaz, V. Kalappattil, R. Das, T. Eggers, H. R. Gutierrez, M.-H. Phan and M. Batzill, Nat. Nanotechnol., 2018, 13, 289 CrossRef CAS.
- S.-J. Gong, C. Gong, Y.-Y. Sun, W.-Y. Tong, C.-G. Duan, J.-H. Chu and X. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8511–8516 CrossRef CAS.
- C. Zhang, Y. Nie, S. Sanvito and A. Du, Nano Lett., 2019, 19, 1366–1370 CrossRef.
- B. Cui, X. Zheng, J. Wang, D. Liu, S. Xie and B. Huang, Nat. Commun., 2020, 11, 1–8 Search PubMed.
- M. Xu, C. Huang, Y. Li, S. Liu, X. Zhong, P. Jena, E. Kan and Y. Wang, Phys. Rev. Lett., 2020, 124, 067602 CrossRef CAS.
- X. Li and J. Yang, J. Am. Chem. Soc., 2018, 141, 109–112 CrossRef.
- C. Huang, J. Feng, F. Wu, D. Ahmed, B. Huang, H. Xiang, K. Deng and E. Kan, J. Am. Chem. Soc., 2018, 140, 11519–11525 CrossRef CAS.
- Y. Sun, Z. Zhuo, X. Wu and J. Yang, Nano Lett., 2017, 17, 2771–2777 CrossRef CAS.
- Y. Ma, Y. Dai, M. Guo, C. Niu, Y. Zhu and B. Huang, ACS Nano, 2012, 6, 1695–1701 CrossRef CAS.
- X. Zhang, B. Wang, Y. Guo, Y. Zhang, Y. Chen and J. Wang, Nanoscale Horiz., 2019, 4, 859–866 RSC.
- G. Akopov, M. T. Yeung and R. B. Kaner, Adv. Mater., 2017, 29, 1604506 CrossRef.
- S. Carenco, D. Portehault, C. Boissiere, N. Mezailles and C. Sanchez, Chem. Rev., 2013, 113, 7981–8065 CrossRef CAS.
- X. Wu, J. Dai, Y. Zhao, Z. Zhuo, J. Yang and X. C. Zeng, ACS Nano, 2012, 6, 7443–7453 CrossRef CAS.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563 CrossRef CAS.
- E. S. Penev, A. Kutana and B. I. Yakobson, Nano Lett., 2016, 16, 2522–2526 CrossRef CAS.
- D. Li, J. Gao, P. Cheng, J. He, Y. Yin, Y. Hu, L. Chen, Y. Cheng and J. Zhao, Adv. Funct. Mater., 2020, 30, 1904349 CrossRef CAS.
- F. Ma, Y. Jiao, G. Gao, Y. Gu, A. Bilic, Z. Chen and A. Du, Nano Lett., 2016, 16, 3022–3028 CrossRef CAS.
- W.-L. Li, X. Chen, T. Jian, T.-T. Chen, J. Li and L.-S. Wang, Nat. Rev. Chem., 2017, 1, 0071 CrossRef CAS.
- H. Zhang, Y. Li, J. Hou, A. Du and Z. Chen, Nano Lett., 2016, 16, 6124–6129 CrossRef CAS.
- H. Zhang, Y. Li, J. Hou, K. Tu and Z. Chen, J. Am. Chem. Soc., 2016, 138, 5644–5651 CrossRef CAS.
- X. Qu, J. Yang, Y. Wang, J. Lv, Z. Chen and Y. Ma, Nanoscale, 2017, 9, 17983–17990 RSC.
- C. Zhang, T. He, S. K. Matta, T. Liao, L. Kou, Z. Chen and A. Du, J. Phys. Chem. Lett., 2019, 10, 2567–2573 CrossRef CAS.
- B. Song, Y. Zhou, H.-M. Yang, J.-H. Liao, L.-M. Yang, X.-B. Yang and E. Ganz, J. Am. Chem. Soc., 2019, 141, 3630–3640 CrossRef CAS.
- B. Wang, X. Zhang, Y. Zhang, S. Yuan, Y. Guo, S. Dong and J. Wang, Mater. Horiz., 2020, 7, 1623–1630 RSC.
- H. Tang and S. Ismail-Beigi, Phys. Rev. Lett., 2007, 99, 115501 CrossRef.
- J. L. Lado and J. Fernández-Rossier, 2D Mater., 2017, 4, 035002 CrossRef.
- Y. J. Deng, Y. J. Yu, Y. C. Song, J. Z. Zhang, N. Z. Wang, Z. Y. Sun, Y. F. Yi, Y. Z. Wu, S. W. Wu, J. Y. Zhu, J. Wang, X. H. Chen and Y. B. Zhang, Nature, 2018, 563, 94 CrossRef CAS.
- S. Gabani, K. Flachbart, K. Siemensmeyer and T. Mori, J. Alloys Compd., 2020, 821, 153201 CrossRef CAS.
- T. Mori, T. Takimoto, A. Leithe-Jasper, R. Cardoso-Gil, W. Schnelle, G. Auffermann, H. Rosner and Y. Grin, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 104418 CrossRef.
- J. Scott, Science, 2007, 315, 954–959 CrossRef CAS.
- Z. Liu, G. Zhao, B. Liu, Z. Wang, J. Yang and F. Liu, Phys. Rev. Lett., 2018, 121, 246401 CrossRef.
- A. J. Mannix, X.-F. Zhou, B. Kiraly, J. D. Wood, D. Alducin, B. D. Myers, X. Liu, B. L. Fisher, U. Santiago and J. R. Guest, Science, 2015, 350, 1513–1516 CrossRef CAS.
- B. Feng, J. Zhang, Q. Zhong, W. Li, S. Li, H. Li, P. Cheng, S. Meng, L. Chen and K. Wu, Nat. Chem., 2016, 8, 563–568 CrossRef CAS.
- W. Li, L. Kong, C. Chen, J. Gou, S. Sheng, W. Zhang, H. Li, L. Chen, P. Cheng and K. Wu, Sci. Bull., 2018, 63, 282–286 CrossRef CAS.
- Y. Wang, J. Lv, L. Zhu and Y. Ma, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 094116 CrossRef.
- Y. Wang, M. Miao, J. Lv, L. Zhu, K. Yin, H. Liu and Y. Ma, J. Chem. Phys., 2012, 137, 224108 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- S. Dudarev, G. Botton, S. Savrasov, C. Humphreys and A. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505 CrossRef CAS.
- L. Wang, T. Maxisch and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 195107 CrossRef.
- Z. Fang, K. Terakura, H. Sawada, T. Miyazaki and I. Solovyev, Phys. Rev. Lett., 1998, 81, 1027 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nh00598c |
| This journal is © The Royal Society of Chemistry 2021 |