
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeparanized chitosans: towards the third generation of chitinous biomaterials
Julia
Revuelta
 *,
Isabel
Fraile
,
Dianelis T.
Monterrey
,
Nerea
Peña
,
Raúl
Benito-Arenas
,
Agatha
Bastida
,
Alfonso
Fernández-Mayoralas
and
Eduardo
García-Junceda
*
*,
Isabel
Fraile
,
Dianelis T.
Monterrey
,
Nerea
Peña
,
Raúl
Benito-Arenas
,
Agatha
Bastida
,
Alfonso
Fernández-Mayoralas
and
Eduardo
García-Junceda
*
BioGlycoChem Group, Departamento de Química Bio-Orgánica, Instituto de Química Orgánica General, CSIC (IQOG-CSIC), Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: julia.revuelta@iqog.csic.es; eduardo.junceda@csic.es
First published on 22nd June 2021
Abstract
The functionalization of chitosans is an emerging research area in the design of solutions for a wide range of biomedical applications. In particular, the modification of chitosans to incorporate sulfate groups has generated great interest since they show structural similarity to heparin and heparan sulfates. Most of the biomedical applications of heparan sulfates are derived from their ability to bind different growth factors and other proteins, as through these interactions they can modulate the cellular response. This review aims to summarize the most recent advances in the synthesis, and structural and physicochemical characterization of heparanized chitosan, a remarkably interesting family of polysaccharides that have demonstrated the ability to mimic heparan sulfates as ligands for different proteins, thereby exerting their biological activity by mimicking the function of these glycosaminoglycans.
 Julia Revuelta, Eduardo García-Junceda, Agatha Bastida, Alfonso Fernández-Mayoralas, Isabel Fraile, Dianelis T. Monterrey, Nerea Peña and Raúl Benito-Arenas | In the BioGlycoChem group, we seek the revaluation of polysaccharides of marine origin, obtaining from them molecules and materials of biological interest and with biomedical applications, increasing the sustainability of the production processes within the framework of the blue and circular economy. From a methodological perspective, our purpose is the development of sustainable and selective protocols for the synthesis, modification and chemical and biocatalytic transformation of carbohydrates and analogues, based on the principles of Green Chemistry. Besides, we seek to obtain a basic understanding of the structure/function/activity relationship at the molecular level of those polysaccharides in order to develop, validate and apply the necessary tools to exploit the potential of chitosan derivatives in the development of novel applications in the field of therapeutic polymers and smart materials. |
1. Introduction
The 2030 Agenda for Sustainable Development, adopted by all United Nations Member States in 2015, provides the so-called 17 Sustainable Development Goals (SDG's), which are an urgent call for action by all countries, focusing on improving human and planetary wellbeing.1 This includes “doing more and better with less”, reducing resource use, degradation and pollution and, therefore, increasing sustainability. Agro-food by-products are abundant and are easily accessible renewable resources and their use with innovative approaches can lead to a successful paradigm shift towards sustainability.2 In this context, the revaluation of these by-products is currently a great challenge that seeks to make these a source of raw materials, able to obtain products with high added value.3 Bio-polymers, in particular, have emerged as a potent solution for replacing petroleum-based polymeric materials, with polysaccharides being the most interesting class of functional bio-polymers. In this sense, sulfated polysaccharides, natural or synthetic, have attracted enormous interest due to their multiple biomedical applications.4Chitins are the second-most abundant polysaccharides after celluloses and are widely distributed in marine invertebrates, insects, fungi and yeast (Fig. 1). These are linear homo-polysaccharides, consisting of N-acetyl-D-glucosamine monomeric units, linked by β-1,4-glycosidic bonds. The removal of most of the acetyl groups of chitins via treatment with strong alkali yields chitosans, a family of binary co-polymers of D-glucosamine and N-acetyl-D-glucosamine residues with superb properties and versatile biological functionalities. For this reason, the sustainable production and employment of chitinous biomass for various applications in many fields and for biomedical usage have become part of many global development strategies (Fig. 1).5
 | ||
| Fig. 1 Chemical structure, natural sources, properties, activities and applications of chitosan. | ||
The hatching of chitosan as a sustainable polymer derived from abundant renewable feedstocks has propitiated the discovery of the most varied biological activities (analgesic, antitumor, anti-inflammatory, antimicrobial, etc.) of these polysaccharides and their derivatives, making chitosan an exciting and promising biopolymer for biomedical applications.6–10 However, it should be taken into account that the term “chitosan” represents a large group of structurally different chemicals that may not only demonstrate various biological activities, but also different functionalities (i.e. non-toxicity, biodegradability, up-take, etc.).11 All these diverse bioactive attributes as well as other chitosan functional properties (i.e. their material-forming capacity and their biodegradability) depend on the different structural variables that define chitosan, since it is a random biopolymer.
The ‘first generation’ chitosans of the past were rather poorly defined mixtures of polymers of varying purity and compositions to be used solely as a biomaterial. However, today's ‘second generation’ chitosans are polysaccharides that are better defined in terms of their degrees of polymerization and acetylation, and more sustainable for the development of reliable products due to the increasingly known molecular structure–function relationship. Future ‘third generation’ chitosans will be even more closely defined in terms of their properties and functionalities, with defined biological activities, and known cellular modes of action allowing a further refinement of products and creating new opportunities.12
Additionally, and given its capability to undergo versatile structural modifications and functionalization, chitosan and its derivatives present a great opportunity to design solutions for a wide range of biomedical and technological applications.13,14 In this sense, the modification of chitosan with sulfate groups provides new and attractive physicochemical properties compared to the starting chitosan, as well as interesting pharmacological properties and biological activities, such as immunomodulation, antioxidant, antiviral, anti-radiation, anti-inflammatory, neuroprotective, anti-proliferative and anticoagulant effects.15–18
This review aims to address the so-called heparanized chitosans, a very interesting family of polysaccharides that have demonstrated the ability to mimic heparan sulfates and heparin as ligands of different proteins, thereby exerting their biological activity mimicking the function of these glycosaminoglycans (Fig. 2).
 | ||
| Fig. 2 Sulfated chitosans as heparan sulfate mimics: (a) general chemical structure of heparin/heparan sulfates; (b) general structure of sulfated chitosans and biomedical applications of these as heparan sulfate mimics. | ||
The development of heparan sulfate-based drugs has been widely limited, despite their interesting therapeutic activities, as a result of the limited availability of homogeneous heparan sulfates from natural sources and due to the expensive and time-consuming procedures for their preparation.19,20 Nevertheless, the availability and low cost of chitosan and the straightforward synthesis of its sulfonyl derivatives compensate for this inconvenience.
In this context, it should be pointed out that the development of heparanized chitosans encompasses several broad fields of research.
Thus, this approximation involves not only the specific sulfonation of chitosan, but also the control of its macromolecular architecture. For this reason, rather than attempting an exhaustive review of the biological literature pertaining to chitosan sulfate, which has been undertaken successfully elsewhere,21–23 the aim of this review is to focus on the findings detailing how the chemical and structural properties of heparanized chitosans are intertwined with their functions as heparan sulfate and heparin mimics (Fig. 3). Finally, the outlooks regarding future research opportunities in this field will be discussed.
 | ||
| Fig. 3 Fields of research encompassed in heparanized chitosan development. | ||
2. Relevant chemistry and functional properties of heparan sulfate and heparin
Heparan sulfates are linear sulfated, heterogeneous polysaccharides composed of repeating units of uronic acid [β-D-glucuronic or α-L-iduronic acid, in the salt form – a urinate] linked to 2-amino-2-deoxy D-glucopyranose by (1,4)-glycosidic bonds. The anticoagulant heparin, which was first discovered in 1916 and derived its name from its abundance in hepatic tissue,24 can be considered a highly sulfated variant of heparan sulfate. In fact, heparan sulfate was originally known as heparatin sulfate since it was initially identified as an impurity of heparin.25However, under this conventional definition, heparan sulfates and heparin display several structural differences. Firstly, in heparin, the urinates are predominantly α-L-iduronate, whereas in heparan sulfates, the urinates are mainly its C-5 epimer, β-D-glucuronate. Secondly, in heparan sulfates, the D-glucosamine residues are predominantly N-acetylated, whereas in heparin, they are N-sulfonated. Finally, although at least 70–80% of heparin is composed of the disaccharide L-iduronate 2-sulfate α (1 → 4) D-glucosamine N,6-sulfate, in heparan sulfates around 40–60% of the disaccharides consist of D-glucuronate β (1 → 4) D-glucosamine, that can be either N-acetylated or N-sulfonated and O-sulfonated at various positions, including C2 of the uronic acid and C6 of the glucosamine units. Sulfation at the C3 position of glucosamine is a relatively rare modification, present in only a limited number of chains, which occurs when D-glucosamine is previously N-sulfated.26,27 These types of modifications create a pattern along each heparan sulfate chain with stretches of unmodified N-acetylated disaccharide units (denoted NA domains), consecutive sequences of N-sulfonated disaccharide units (NS domains), and interspacing domains composed of alternating N-sulfonated and N-acetylated disaccharides (mixed or NA/NS domains) (Fig. 4a).28 Heparin, however, is more uniformly sulfated and resembles a continuous NS domain. Together, these structural characteristics make heparin more sulfated and, hence, more charged than heparan sulfates (2.3–2.8 sulfates/disaccharide in heparin vs. 0.6–1.5).29–31 Furthermore, heparan sulfates also have a much higher maximum average molecular weight (ca. 50 kDa) than heparin (ca. 20 kDa).32
 | ||
| Fig. 4 (a) Domain structure of heparan sulfates; (b) biological activities modulated by the interaction of proteins with heparan sulfates; and (c) regulation of cell signalling pathways by heparan sulfate proteoglycans (adapted with permission from ref. 37). | ||
Heparan sulfates are ubiquitously expressed on cell surfaces and in the extracellular matrix and basement membrane and are covalently attached to a range of core proteins to form heparan sulfate proteoglycans. Although the core proteins can function independently of the heparan sulfate chains they carry,33 these predominantly dictate the ligand-binding capability and therefore the biological roles of heparan sulfate proteoglycans.34 In particular, heparan sulfate chains due to their vast structural diversity are able to bind and interact with a wide variety of proteins (growth factors, chemokines, morphogens, extracellular matrix components, and enzymes, among others), that modulate different biological processes through their interaction.35 Similar interactions are also characteristic of various pathophysiological settings, including cancer, amyloid diseases, infectious diseases, inflammatory conditions and certain developmental disorders (Fig. 4b).36 In general, the most important role of heparan sulfates is in cell signalling, regulating the signalling pathways in many different ways (Fig. 4c).37 Thus, they can act cell-autonomously as receptors or co-receptors and recruiters (increasing the ligand or receptor concentration at the cell surface), by regulating receptor membrane trafficking (during endocytosis) or by controlling ligand secretion. They can also act non-cell-autonomously as direct cues, or by controlling the distribution of signalling gradients as well as the composition of the extracellular matrix.38
Meanwhile, although heparan sulfates are produced by virtually all types of cells, heparin is present in only a limited type of cells, notably connective tissue-type mast cells.39 The major biological role of heparin is the regulation of the coagulation system, and it has been used as a clinical anticoagulant for over 90 years.40 Heparin serves as a molecular scaffold in the antithrombin/thrombin and antithrombin/factor Xa interactions, which results in the inhibition of the blood coagulation cascade. Antithrombin alone is not an efficient inhibitor of thrombin and factor Xa. However, its inhibitory activity increases up to several thousand-fold by the binding of heparin. More recently, attention has been drawn to the non-anticoagulant activities of heparin. Most of its potential applications seem to be associated with its anti-inflammatory effects,41 as well as its interactions with a multitude of proteins.42 It inhibits different enzymes involved in pathological processes, such as heparanase43 and metalloproteases,44 and also acts as a heparan sulfate mimic.
3. Chitosan sulfate synthesis and characterization: chemical tools for heparanized chitosan development
Given that the chemical and structural properties of chitosan and its derivatives are intertwined and related to their possible biological activities and, therefore, to their specific applications, it is necessary to prepare the most homogeneous and well-defined molecular structures possible. Thus, the chemo- and regioselective modifications are key for the development of novel chitosan derivatives with new and improved functional properties.45 This review is not intended to be a comprehensive account of all the chemical transformations that can be performed on chitosan;46 rather, the case studies selected will focus on systematically analysing the key and most recent methods described for the selective sulfonation of chitosan (Fig. 5).47 | ||
| Fig. 5 (a) Selective sulfonation strategies from original chitosan (see Table 1). (b) Sulfonation methods employing O- or N-protected derivatives. Reagents and conditions: [a] phthalic anhydride, ethyleneglycol, DMF, and 130 °C; [b] (i) trityl chloride, DMAP, py, 90 °C and (ii) H2N–NH2·H2O, 100 °C; [c] (i) SO3·py, py, 80 °C; and (ii) dichloroacetic acid, 20 °C; [d] (i) SO3·py, py, 80 °C and (ii) H2N–NH2·H2O, 100 °C; [e] Me3SiH, Me3SiCl, py, 100 °C; and [f] (i) SO3·py, DMSO, 40 °C. See ref. 66 and 67. | ||
Chitosan sulfonation can occur at three key positions in the glucosamine and acetyl glucosamine residues: The C-2, C-3 and C-6 positions carrying the amino, secondary and primary hydroxyl groups, respectively.48,49 This gives rise to N-, O- or N,O-sulfated chitosans and, depending on the type of sulfonating reagent and reaction conditions, the reaction can lead to a selective or non-selective derivative. Traditionally, chitosan sulfates have been prepared by using different sulfonating agents such as chlorosulfonic acid (HClSO3), 1-piperidinesulfonic, sulfuryl chloride, sulfuric acid, SO3, or sulfamic acid.50–52 The common difficulty of these procedures is that the reaction is performed in a heterogeneous medium, because most of the polysaccharides are insoluble or only slightly soluble in organic solvents used as the reaction medium in the conventional sulfonation procedures. Consequently, these conditions lead to heterogeneous products and mono-, di-sulfated compounds or copolymers containing both block-types, which are randomly distributed on the polymer chain and can be obtained with poor reproducibility.53–56 To solve these drawbacks, improved methodologies have been developed. These include optimization of the standard conditions for sulfonation from the original chitosan (Fig. 5a) or the use of protecting groups to achieve fully regioselective chitosan sulfonations (Fig. 5b).
| Entry | Regioselectivity | Selected conditionsa | Entry | Regioselectivity | Selected conditionsa |
|---|---|---|---|---|---|
| a The conditions presented are a selection of those most frequently employed. b FA: formic acid. c DCAA: dichloroacetic acid. d N-Methylpyrrolidinone. | |||||
| 1 | 6-O-Sulfated | HClSO3/H2SO457 | 4 | 2-N,6-O-Disulfated | (1) HClSO3/H2SO4; (2) SO3py/Na2CO3/H2O57,62 |
| HClSO3·DMF/DMF59 | |||||
| SO3·DMF/Cu2SO4·5H2O/DMF61 | |||||
| (1) HCONH2; (2) HClSO3 | |||||
| 2 | 3,6-O-Disulfated | HClSO3/DMF/FAb (or DCAAc)50,60 | 5 | 3-O-Sulfated | (1) HClSO3/DMF/FA; (2) NMPd/H2O50,61 |
| (1) HCONH2; (2) HClSO3/DCCA58 | |||||
| 3 | 2-N-Sulfated | SO3·py/Na2CO3/H2O60 | 6 | 2-N,3,6-O-Trisulfated | (1) HClSO3/DMF/FA (2) SO3·py/Na2CO3/H2O50,62 |
| SO3·DMF/DMF/FA/MW irradiation61 | |||||
3.1. Direct sulfonation reaction of chitosan
The direct sulfonation of chitosan has allowed the chemo- and regioselective synthesis of various sulfonated derivatives (Fig. 5a). 6-O-Sulfated chitosan can be selectively obtained by using a chlorosulfonic acid/sulfuric acid system.57 This is a convenient way for obtaining 6-O-sulfated chitosans of different degrees of sulfation (DS), by changing the H2SO4/HClSO3 ratio.50 However, the presence of a strong sulfuric acid solution leads to a high degree of depolymerization and, as a consequence, to derivatives having larger polydispersities compared to the original chitosan.58 To avoid this, the complex HClSO3·DMF has been widely used to add sulfonate functions on the hydroxyl groups of chitosan. In this case, 6-O-sulfated or 3,6-O-disulfated derivatives are obtained by tuning the reaction conditions (time, temperature and molecular ratio of the sulfonating reagent). When the reaction is performed in aprotic organic solvents, such as DMF, this is executed under non-homogeneous conditions due to the very poor solubility of chitosan in these solvents, and a previous activation of chitosan seems to be essential for a dominant substitution of primary hydroxyl groups (HClSO3/DMF).59 Moreover, sulfonation under these conditions provides a higher sulfur content when the medium contains formic acid or dichloroacetic acid, yielding preferentially 3,6-O-disulfated chitosan.50,60 A complete substitution of the primary hydroxyl groups can be performed by sulfonating chitosan formate with HClSO3, either under homogeneous or heterogeneous conditions.59 Additionally, it is commonly assumed that the 3,6-O-disulfated chitosan can be prepared using HClSO3 in the presence of a formamide/dichloroacetic acid system.50,60 Furthermore, it is possible to selectively synthesize 6-O-sulfated chitosan in the presence of Cu2SO4·5H2O with SO3·DMF. Under these conditions, the copper salts seem to act as a temporary protective group, preventing the reaction at O-3.61Chemoselective 2-N-sulfonation can be accomplished by employing SO3·pyridine as the sulfonating agent in a basic medium,62 whereas 3-O-sulfated derivative can be obtained by the regioselective 6-O-desulfonation of 3,6-O-disulfated chitosan.61 On the other hand, 2-N,6-O-disulfated and 2-N,3,6-O-trisulfated chitosan can be prepared using a combination of the aforementioned procedures. The fully sulfated derivative (2-N,3,6-O-trisulfated chitosan) has also been obtained by using SO3·DMF complex in DMF/formic acid mixtures under microwave irradiation.61
Finally, during the last few years, significant effort has been dedicated to the development of sustainable processes for chitosan modification because the re-evaluation chitosan processes cannot be sustainable if their modification does not imply chemical safety, recyclability and a low environmental impact. To solve this issue, the dissolution of natural polymers in ionic liquids has been pointed out as a promising strategy that combines two green chemistry principles, namely the use of environmental solvents and bio-renewable feedstocks.63 Additionally, and since ionic liquids allow the dissolution of both organic and inorganic reagents, they are considered excellent interfaces for promoting chitosan derivatization.64 In particular, the hydrophilic ionic liquid BMImCl (1-butyl-3-methylimidazolium chloride) has demonstrated great efficiency in disrupting the inter- and intramolecular hydrogen bonds in chitosan, promoting homogeneous media and thus enhancing the efficiency of the reactions. In particular, the sulfonation of polysaccharides employing BMImCl as the reaction medium and SO3·py as the sulfonating agent, affords the sulfated derivatives with good reproducibility, capable of modulating the sulfur content by varying the reaction conditions (time or temperature). Finally, the observed increase in molecular weight (Mw) indicates that these conditions allow for efficient polysaccharide modification, avoiding side effects such as depolymerisation or degradation.65
3.2. Chitosan sulfonation employing protecting groups
Although the sulfonation of original chitosan allows the synthesis of derivatives with great regioselectivity as previously described, the methods that ensure selective modification are those that employ suitably protected polysaccharides as the starting material (Fig. 5b). For example, certain authors have used SO3·py with N-phthaloylated chitosan to assure selective O-sulfonation. Thus, this treatment affords 3,6-O-disulfated chitosan favouring an almost complete reaction at C-3, in contrast to the aforementioned procedures, whereas C-6 is only partially sulfonated.66 On the other hand, the use of protecting groups has allowed the synthesis of derivatives with new regioselectivities. Thus, 2,3-O-disulfated derivatives can be selectively prepared from 6-O-trityl chitosan.67Finally, highly sulfated chitosans (DS > 2.0) have been prepared under homogenous conditions via trimethylsilylation, which significantly enhanced the reactivity and solubility of chitosan in organic solvents. In particular, silylated chitosans have been easily sulfonated with SO3·py complex under homogeneous conditions in DMSO and at low temperatures (20–40 °C). It is important to note that the use of certain protecting groups, for example trimethylsilyl, protects the chitosan from degradation during sulfonation, suggesting that depolymerisation could be associated with the free hydroxyl groups on the polysaccharide.68
3.3. Physico-chemical characterization of chitosan sulfates
Analyzing and determining the different structural variables that define chitosan is a complex task encountering significant difficulties, given that it is a random biopolymer. Hence, the structural characterization of chitosan sulfates covers a broad range of chemical problems and structural hierarchies within the molecules that, in general, addresses several aspects. In this context, it is noteworthy that, although the primary structure is well known, chitosan sulfates exhibit polyfunctionality and a typical polydispersity that lead to innumerable structural possibilities. For this reason, the characterization of chitosan sulfates must address several aspects and requires the use of a wide range of analytical techniques as summarized in Fig. 6. Therefore, despite their relevance, studies on the structure ↔ functionality ↔ biological activity relationship are few and detailed data relating to findings at the molecular level are scarce and often the results obtained are contradictory.69,70 | ||
| Fig. 6 Overview on different approaches for the analysis of various structural features in chitosan sulfates. | ||
Regarding the molecular weight, it should be remembered that chitin (the starting material for chitosan derivatives) by itself is a polydispersed polymer. Besides, the depolymerisation processes that the polymer can undergo in the different extractions, the deacetylation and sulfonation processes could increase the polydispersity. To know the Mw and the polydispersity index (PDI) of chitosan sulfates to suit the application is crucial and key to tackling targets. The techniques most employed to determine the molecular weight—or size—of chitosan sulfates are viscosimetry and gel permeation chromatography (GPC).71 Viscosimetry is the most widely used method due to its simplicity and low cost. The intrinsic viscosity, [η], is directly proportional to the average molecular weight of the polymer. This method has the disadvantage of not being absolute. It depends on the hydrodynamic volume and the degree of polymerization of the chain and on inter and intramolecular interactions.72 Thus, chitosan sulfate, which is a polyanion in alkaline media, under neutral and acidic conditions contains anionic and cationic groups that can neutralize one another and form zwitterion bonds. These zwitterion bonds are formed at pH values of 5–6, obtaining a minimum in the intrinsic viscosity of the polymer.
In molecular separation techniques, such as GPC, the separation of solutes is actually carried out based on their hydrodynamic volume and not on their Mw, in a strict sense. For the results obtained with these separation techniques to be reliable, it is essential that neither the charge of the molecules nor the effects of ionic exclusion or adsorption on the column are determinative in the separation process. This is particularly relevant in the case of chitosan sulfate, which is a polyampholyte. Besides, certain detectors that may be coupled to the GPC, as the refractive index detector, require the use of standards to determine the Mw. Although there are no specific standards for chitosan, standards for other polysaccharides, such as dextrans or pullulans, have been used with good correlation, although with some overestimation due to their greater flexibility. Furthermore, these polysaccharides are uncharged and therefore, are not always useful standards for determining the Mw of the highly charged chitosan sulfate polymer.72 On the other hand, light scattering detectors, both multi-angle light scattering (MALS) and right-angle and low-angle light scattering (RALS/LALS), provide the absolute molecular weight of the polymer. The combined use of refractive index and multi-angle light scattering (MALS) detectors makes it possible to determine the molecular weight and radius of the gyration of the individual fractions as they elute out of the column, thereby obtaining their distribution as a function of concentration. In this way, we obtain the weight-average molecular weight (Mw), number-average molecular weight (Mn) and, by dividing Mw and Mn, the polydispersity index (PI) which clarifies the width of the Gaussian distribution of sizes in the sample.73
In the context of chemical composition, it should be noted that chitosan sulfates are polyfunctionalised polysaccharides since, in addition to the sulfate groups, a variable proportion of amine groups are acetylated. The determination of the number of sulfate groups in chitosan sulfates is usually carried out via elemental analysis.74 Other analytical methods including FT-IR and FT-Raman spectroscopies can also be applied to analyse chitosan sulfates (Fig. 7). Both methods are simple and do not require any excessive pre-treatment of the sample; therefore, they are sometimes referred to as “green analytical methods”.75 Chitosan sulfate presents characteristic signals in FT-Raman spectroscopy.76,77 Generally, bands of around 1000 cm-1 are characteristic of the stretching vibrations of the O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) SO groups, whilst bands of around 750 cm−1 are characteristic of stretching vibrations of the C–O–S groups (Fig. 7a).78 These characteristic vibrations derived from sulfate substituents can be used to determine the total DS attributed to these substituents.79 On the other hand, the FT-IR spectra have two bands that are representative of an O-sulfate at approximately 1234 cm−1 (νsym OSO) and 802.06 cm−1 (ν C–O–S). In N-sulfated chitosan, this last peak is not observed, allowing for the differentiation between O- and N-sulfated chitosan (Fig. 7b).74
SO groups, whilst bands of around 750 cm−1 are characteristic of stretching vibrations of the C–O–S groups (Fig. 7a).78 These characteristic vibrations derived from sulfate substituents can be used to determine the total DS attributed to these substituents.79 On the other hand, the FT-IR spectra have two bands that are representative of an O-sulfate at approximately 1234 cm−1 (νsym OSO) and 802.06 cm−1 (ν C–O–S). In N-sulfated chitosan, this last peak is not observed, allowing for the differentiation between O- and N-sulfated chitosan (Fig. 7b).74
 | ||
| Fig. 7 (a) FT Raman spectra of chitosan (1), and chitosan sulfate with a total DS = 0.82 (2), 1.09 (3) and 1.67 (4). (b) FT-IR spectra of O-sulfated chitosan (blue) and N-sulfonated chitosan (red). (c) Key regions of 15NCP/MAS NMR spectra of 3,6-O-disulfated chitosan (brown) and 2-N-sulfated chitosan (blue) (d) 1H-NMR of 3,6-O-disulfated chitosan. Signals enclosed in the colour boxes are employed for DDA (deacetylation degree) determination. (e) Essential region of heteronuclear single quantum coherence (HSQC) spectra of 3,6-O-disulfated chitosan. Densities enclosed in the colour boxes were integrated for sulfation degree estimation: 6-position (dashed red line) and 3-position (solid green line). (a) Adapted from ref. 79, with permission from Elsevier. (b–e) Adapted with permission from ref. 74; copyright 2020 American Chemical Society. | ||
This selectivity can also be determined also by 15NCP/MAS NMR spectroscopy. In this case, the spectra of N-sulfonated chitosan indicate the presence of a signal around ∼283 ppm, which corresponds to the sulfamic acid (NHSO3) that is not detected in O-sulfated chitosans (Fig. 7c). It should be noted that, although there is a relationship between DS and the intensity of the corresponding band, FT-IR is not the method of choice for its quantifications.
UV spectrophotometry using glucosamine and N-acetyl-glucosamine as standards is a simple, convenient and accurate method to determine the deacetylation degree (DDA) of chitosan.80 However, in the case of sulfated samples, the difficulty in accessing pure samples of sulfated standards reduces the usefulness of the technique.
Other methods for measuring the DDA of chitosan include titration,81 IR-spectroscopy,82 elemental analysis, circular dichroism,83N-acetyl group hydrolysis84 and gel permeation chromatography (GPC).85 Unfortunately, these techniques often show considerable discrepancies in the obtained DDA values. In addition, many of these techniques are inaccurate, time consuming or complicated to perform. An alternative method for solving these discrepancies and drawbacks is NMR, a fast, precise, reproducible and accurate method that allows DDA determination by a simple integration of peak around δ 1.92 ppm assigned to CH3 of N-acetylated monomer and a peak around δ 2.95 ppm assigned to H2 of deacetylated monomer in the 1H-NMR spectra (Fig. 7d).86 NMR is also a useful technique for determining the sulfation distribution along the chain. In particular, 13C allows the determination of the sulfation profile, by observing significantly different characteristic signals in the spectra depending on which position is sulfated (Table 2).
| Compound | Chemical shifts (ppm) | ||
|---|---|---|---|
| C2 | C3 | C6 | |
| Chitosan | 57.1 | 71.6 | 61.7 |
| 2-N-Sulfated chitosan | 63.5 | ||
| 3-O-Sulfated chitosan | 77.2 | ||
| 6-O-Sulfated chitosan | 68.2 | ||
| 3,6-O-Disulfated chitosan | 77.3 | 68.2 | |
Finally, 2D-NMR experiments have also been employed for the characterization of chitosan sulfate (HSQC-DEPT, COSY, ROESY, etc.). In particular, HSQC-DEPT spectra provide an estimation of the DS by integration of representative signals. For example, the HSQC–DEPT spectrum of a 3,6-O-disulfated chitosan (Fig. 7e) displays antiphase signals at position 6 due to sulfated (δH,C = 4.25/66.5) and non-sulfated (δH,C = 3.83/60.0 ppm) CH2 groups. Furthermore, two-phase signals at δH,C = 4.3/78.8 ppm (minor) and at δH,C = 3.8/72.8 ppm (major) have been attributed to the sulfated and non sulfated CH at position 3. In both cases, the integration of each array/body of signals with respect to the CH-2 density allows for an estimation of the degree of sulfation.
4. Factors for consideration in heparanized chitosan development
The progress made in terms of chemo- and/or regioselective chitosan sulfonation and physico-chemical characterization has paved the way for the development of sulfated chitosan-based entities with a wide range of choices. However, to deliver therapeutic solutions based on heparanized chitosans, certain other factors need to be taken into consideration. Firstly, it is very important to note that all the aforementioned procedures can have a huge impact on various chitosan parameters such as molecular weight (Mw), degree of sulfation, solubility, etc. In addition, it is noteworthy that heparanized chitosan bioactive attributes as well as other functional properties (i.e. bioavailability, biodegradability, etc.) are very dependent upon these parameters. In general, the development of heparanized chitosan should involve two complementary approaches, one which covers the analysis from a physico-chemical point of view and another one that employs biochemical approaches. The former includes not only a compositional analysis of the polysaccharide, but also a deeper understanding of the mechanism involved in their functionalities and biological activities together with the possibility of controlling the architecture to achieve greater control over the final biomedical properties. On the other hand, the biochemical approach includes protein assays, in vitro cell assays, and in vivo and ex vivo experiments. Although both are importantly correlated, rarely does one incorporate the studies of the other.The following sections will provide an overview of the most significant advances that emerged from both approaches, with a focus on establishing the relationship between both, where possible.
5. Understanding the interdependence between chemical structure and properties: a key task in the development of heparanized chitosans
Successful process optimization and development of tailored heparanized chitosan are currently only possible by understanding and controlling the details of how the specific properties of polysaccharides determine their interaction with proteins which are ultimately responsible for their biological activities. However, this field is still in its infancy, and the unsolved question of how the specific structural properties of polysaccharides determine their biological activities continues to frustrate many research efforts today.69,70 In this context, key challenges continue to include identifying the roles of chemistry, structure, and understanding and harnessing these roles in biomedical applications. Recent advances in this field are centred primarily in deciphering the structural determinants in heparanized chitosan responsible for binding to growth factors and signalling proteins that play relevant functions in many normal and abnormal processes.Traditionally it was though that, since the interactions between the sulfated polysaccharides and growth factors take place between the negative charges of the chain and the positive charges of the protein surface, the binding affinity could increase with the number of sulfate groups. However, very recently, it has been observed that, as in natural polysaccharides, heparanized chitosan has the capacity to organize sulfation patterns that adhere to a glycosaminoglycan-like helical periodic format.74 For this reason, the interaction should be governed not by the total charge of the chain, but by the superficial charge of the adopted helical structure. Interestingly, recent studies have demonstrated that the 3D-organization of the sulfates is modulated at the same time by the particular sulfate distribution within the repeating-units.72,74,87 For example, it has been observed that the 6-O-sulfated motifs seem to induce a disposition of the sulfate groups pointing outside the 3D-helical structure, while the presence of the 3,6-O-disulfated motifs induces a disposition of the sulfate groups inside the helix, as can be deduced by the increase in the zeta potential values (ζ-potential) (a physico-chemical indicative parameter of the superficial net charge) when the proportion of these di-sulfated units is increased (Fig. 8). Interestingly, this has a significant influence on the binding activity of these with several growth factors.72 The positive band observed in circular dichroism around 245 nm for these polysaccharides is indicative of a helical (right-handed) secondary structure. A similar effect has been observed in chondroitin sulfates, natural polysaccharides that regulate important neural processes by interacting with growth factors in a sulfation dependent manner.88,89
 | ||
| Fig. 8 Schematic representation of the way in which the 3D structure of 3,6-O-disulfated chitosan can determine that a lower sulfation degree provides a higher net charge on the surface and, consequently, a great affinity with FGF-2 (see ref. 72). | ||
The identification of this structure–function relationship strengthens the hypothesis that the sulfation pattern of heparanized chitosan modulates the 3D-polysaccharide structure which, as in natural glycosaminoglycans, has a significant influence on their capacities to bind growth factors. However, it is worth noting that, in accordance with recent studies, the strong and specific binding between heparan sulfates and growth factors is not simply regulated by the sulfate distribution along the chain. Indeed, the microheterogeneities resulting from the variation in sulfation and epimerization patterns represent only the first level of molecular diversity in heparan sulfates. In addition to this, these polysaccharides present a second level of diversity due to the presence of regions or domains throughout the polymer of defined size, spacing, and general composition known as NS domains (NS), NA domains (NA) and transition zones (NS/NA) (Fig. 4a). These provide numerous docking sites for protein ligands, enabling selective interactions in a topologically and temporally controlled manner.
The primary interaction between heparan sulfate and a protein is the attraction between the highly negatively charged NS domains and the clusters of basic residues at the protein surface. In certain cases, for example with AT-III or FGFs, a single NS-domain is sufficient to allow a high affinity interaction,90,91 while with other proteins such as IFN-γ or MIP-1α a single NS-domain is too short for high affinity binding92,93 and a longer fragment, including an NA-domain “spacer”, is needed for an efficient interaction.94,95 In this organization, the flexibility of an NA-domain may allow the heparan sulfate chains to adapt their shapes and facilitate protein interactions with the sulfate residues of the relatively rigid NS-domains.95 Indeed, many interactions appear to depend more on the overall organization of heparan sulfate domains than on their fine structure. In this regard, it has been proposed that the heparan sulfate chain may adapt its conformation in order to meet the requirements for the recognition of a protein, namely, the flexibility or rigidity of such domain determinants on the binding processes.96 For this reason, we considerer that in heparanized chitosan mimics, as in natural polysaccharides, selective recognition properties could reside at the domain topology. As an answer to this question, we have, very recently, analysed unprecedented chitosan sulfates decorated with different domain structural motifs of natural polysaccharides (Fig. 9a).
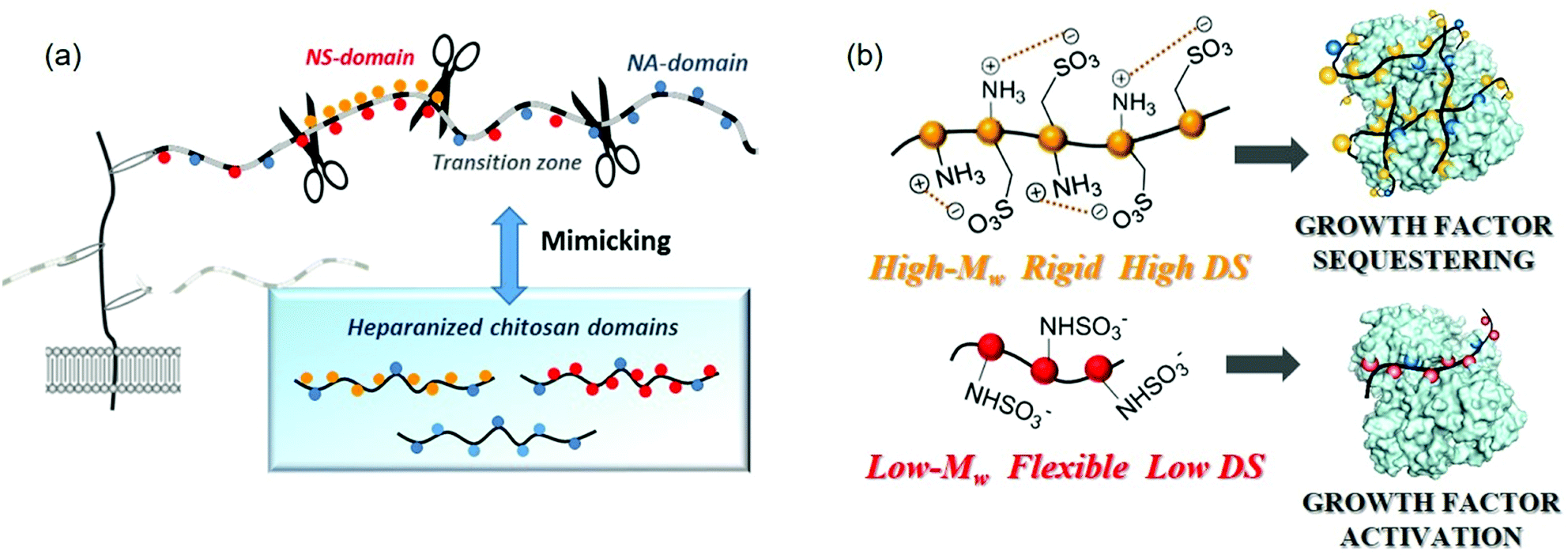 | ||
| Fig. 9 (a) Heparan sulfate-domains of chitosan mimics. (b) Proposed modes of binding of heparanized chitosans to proteins, according to their structure. Adapted with permission from ref. 74; Copyright 2020 American Chemical Society. | ||
The intrinsic structures of these polysaccharides were estimated by analyzing the 3D-structure of the chains, employing CD and hydrodynamic volume measurements, and ζ-potential determinations for a simple estimation of the superficial net charge. The “degree of contraction”—expressed as the ratio between the intrinsic viscosity [η] in water ([η]H2O) and 0.1 M NaCl ([η]NaCl)—was employed as an estimation of the intrinsic chain flexibility. Previous studies have proposed that although the electrical charge density drives the interaction between polyanionic polysaccharides and proteins, the unique properties of each protein–polysaccharide complex are determined by other polysaccharide characteristics such as chain flexibility.97 In particularly, studies by our group indicate that heparanized chitosans must adopt a completely different 3D-structure depending on chitosan functionalization.74 Thus, O-sulfonated derivatives appear to adopt a rigid linear conformation stabilized by cooperative intrapolymer electrostatic interactions between the sulfate groups at C-6 and the protonated amino groups on adjacent residues (Fig. 9b). By contrast, an increase in acetylation degree to mimic the transition zones of heparan sulfates produces a relevant conformational change that corresponds to a more expanded and less rigid polymeric structure.
Finally, in N-sulfonated polysaccharides with high Mw, a charge-driven self-association between chains takes place, giving rise to rigid polyelectrolytic complexes. In this case, the structure has been confirmed through the use of additional physico-chemical techniques such as isotermal titration calorimetry (ITC) and difussion ordered spectroscopy (DOSY); the results of which are consistent with the presence of self-assembling structures. Otherwise, N-sulfonated chitosans with low Mw adopt fewer compact structures (Fig. 9b), which is in agreement with the existence of a critical chain length, above which chitosan and its derivatives tend to form stable self-assembled structures. These conformational changes were associated with different behaviours in the binding of polysaccharides with FGF-2, giving rise to distinct biological responses. Thus, we observed that the combination of O-sulfates and N-acetyl groups (mimicking the transition zones of heparan sulfates) favours cell proliferation. This result can be explained by the fact that these heparanized chitosan could enhance growth factor signaling activity, as the bound protein is still able to bind to its cognate receptor, promoting an effect on cell proliferation as we demonstrated for PC12 cells. A similar effect is produced by low molecular weight N-sulfated derivatives. On the contrary, highly sulfated polysaccharides (mimicking NS-domains) display a tendency to sequester the growth factor, through the binding of the rigid structures to the protein, inhibiting cell division (Fig. 9b).
These studies point out the relevance of the 3D-structure of heparanized chitosan in its binding to proteins. However, much interesting research lies ahead in the efforts to solve the paradigm regarding the way in which the physical–chemical properties relate to its protein binding, leading the to enhanced predictability of its biological functions.
6. Biomedical applications of heparanized chitosans
The modification of chitosan with sulfate groups can provide new or improved properties while retaining the original physicochemical and biochemical properties of chitosan, e.g. its low immunogenicity, biodegradability and wound-healing activity. Herein, we will focus on the biological activities of chitosan sulfate that are more characteristic of the so-called “heparanized chitosans”. These are derived from their ability to mimic heparan sulfate and heparins and are exerted through their ability to bind with different growth factors and other proteins, enhancing or inhibiting their binding to their receptors,74 whilst as a heparin mimic, anticoagulant activities have been reviewed.6.1. Heparanized chitosan and cancer
Growth factors are involved in tumor development and progression, playing a significant role in cancer pathobiology.98 In particular, FGF-2 affects the proliferation, drug sensitivity and apoptosis of cancer cells.99 The roles that heparanized chitosans can play in fighting the carcinogenic processes, such as cell proliferation, survival, adhesion, migration, and angiogenesis, are several, as are the roles that heparan sulfate plays in the development of these processes.100,101Thus, it has been described how sulfated chitosan and benzylidenimine sulfated chitosan significantly inhibit the proliferation and induce the apoptosis of breast cancer MCF-7 cells in a dose-dependent manner.102 The results of this study indicated that the inhibition of cell proliferation was due to the interference of signaling mediated by FGF-2 since, although they did not have direct evidence for the interaction of chitosan derivatives with the growth factor, the pretreatment of MCF-7 cell cultures with these compounds significantly reduced ERK phosphorylation induced by FGF-2 and thereby interrupting its downstream signaling pathway. Interestingly, none of these effects was elicited by the unmodified chitosan. Analysing the structure of these derivatives, and considering the significant increase in the anti-proliferative and apoptotic effects of benzylidenimine sulfated chitosan compared to sulfated chitosan, illustrates that the phenyl groups could demonstrate a significant effect, due to van der Waals interactions between the phenyls and hydrophobic residues of FGF-2 away from the allosteric zone, inhibiting the FGF-2 signalling activity.
Suppressing the angiogenesis in tumours has become a valuable approach in anticancer treatment.103 Perhaps, the most crucial regulators of all known angiogenic factors are vascular endothelial growth factors (VEGF).104 In this sense, various studies have described that heparin is capable of inhibiting the VEGF/VEGFR2 signalling pathway.105 Given the strong anticoagulant activity of heparin, which precludes its clinical use, it is necessary to develop analogues in which this activity has been eliminated. Thus, it has recently been shown that low molecular weight chitosan sulfates can inhibit angiogenesis in combination with VEGF and by blocking the VEGF/VEGFR signalling pathway (Fig. 10a).106 It is noteworthy, that this inhibitory effect depends largely on the polysaccharide structure. On the one hand, the inhibitory effect is reduced when position 6 is partially sulfated (DS < 1), while, when DS is higher than 1.00, the effect was no longer significantly modified (Fig. 10b). On the other hand, the polymer size (Mw) showed the opposite effect; the larger the size, the smaller the inhibitory effect (Fig. 10c). The authors justified this effect because the shorter chitosan can directly bind to VEGF and block the VEGF/VEGFR signalling pathway. In the same study, it was found that these sulfated chitosans effectively inhibited tumour growth in vivo without the common side effects of heparin, such as bleeding.106
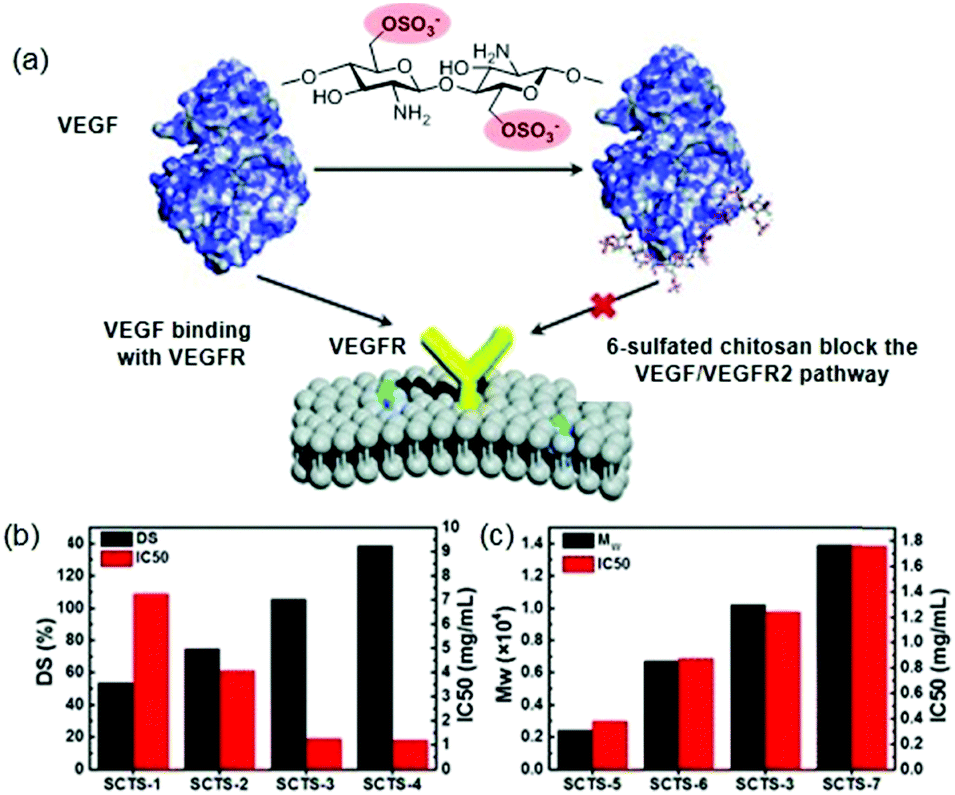 | ||
| Fig. 10 (a) Schematic effect of sulfated chitosans in VEGF/VEGRF2 pathway inhibition. (b) Effect of DS in inhibitory effect. (c) Effect of Mw in inhibitory effect. Reproduced by permission of The Royal Society of Chemistry from ref. 106 | ||
On the other hand, it is accepted that the P-selectin-mediated initial adhesion of tumour cells to platelets or endothelial cells plays a critical role in haematogeneous metastasis, constituting an adhesion process based mostly on mucin- and glycosaminoglycan-type selectin ligands.107 For this reason, heparanized chitosans have been proposed as heparan sulfate-like ligands to prevent P-selectin from binding to its native carbohydrate ligands. In particular, it has been observed that 6-O-sulfonation of chitosan is indispensable for inhibition and that additional N-sulfonation or 3-O-sulfonation dramatically enhanced the inhibitory activity.108 Interestingly, the authors highlight the possibility that the interaction of P-selectin with these polysaccharides does not involve a linear defined sequence but a clustered saccharide patch that can be generated by the appropriate spatial arrangement of sulfate esters along the chain. Similar results were observed when oleic acid sulfated chitosans were analysed.109 In this case, these derivatives were designed as sulfatide (a generic denomination for sulfated glycolipids presented in different tissues) mimics. Since sulfatide mediates metastatic progression through binding to P-selectin,107 the oleic acid sulfated chitosan derivatives were designed to act as “decoy” ligands for selectins, thus inhibiting the metastatic process.110 These studies only represent preliminary approximations for the development of anticancer agents; however, these promising results render heparanized chitosan as a promising candidate for drug development, especially as we move into an era of precision and personalized cancer therapy.
6.2. Heparanized chitosans for tissue engineering
At the end of the last century, tissue engineering emerged as a new field in medicine.111 The primary goal of all approaches in tissue engineering is the restoration of function through the delivery of living elements, which become integrated into the patient. From the very beginning, the standard approach was to seed cells on a three-dimensional biomaterial scaffold combined with suitable biochemical signals.111,112Given their native-like biological properties, high growth factor retention capacity and porous nature, scaffolds based on sulfated polysaccharides hold great promise for a number of tissue engineering applications.113 Focusing on chitosan sulfates, these polymers combine a number of properties of great interest in tissue engineering. Their structural and functional similarities with heparan sulfate allow them to influence and modulate both the morphology and the function of cells, thus directing their proliferation and differentiation. Moreover, as these polymers mimic the important properties of tissues such as bone and cartilage, they are ideal for orthopaedic tissue engineering.114
Bone morphogenetic protein-2 (BMP-2) has demonstrated remarkable ability to induce bone formation and bone tissue reconstruction, playing critical roles in osteogenesis and bone metabolism.115
Interestingly, it has been reported that heparanized chitosans not only stimulate the osteoblast differentiation induced by BMP-2 in vitro, but also ectopic bone formation in vivo.116 In particular, from a structural point of view, the enhanced bioactivity of BMP-2 has been attributed primarily to the stimulation from 6-O-sulfated chitosan, while 2-O-sulfate gives rise to less activation. However, when both functionalizations are conjugated to obtain 2-N,6-O-disulfated chitosan, a large increase in stimulation takes place. The synergistic mechanism between 2-N,6-O-disulfated chitosan and BMP-2 has been further investigated.117 CD studies have shown that disulfated chitosan produces a significant change in the BMP-2 secondary structure, mainly due to the reduction of the antiparallel conformation of the β-sheet. Interestingly, it was found that at a low concentration of disulfated chitosan the BMP-2 induced osteogenic differentiation was greater than at a higher concentration of the chitosan sulfate. Other studies have revealed the applicability of these results in the preparation of various scaffolds based on these polysaccharides (Fig. 11). On the one hand, BMP-2 loaded 2-N,6-O-disulfated chitosan nanoparticles incorporated into gelatin (G)-based scaffolds have been prepared (Fig. 11a). This composite delivery system not only allows a sustained release of bioactive BMP-2, but also produces relevant osteoconductive and osteoinductive effects.116 On the other hand, the use of 2-N,6-O-disulfated chitosan in combination with poly(lactide-co-glycolide) (PLGA) has allowed the development of efficient scaffolds in which the release profiles of BMP-2 are 30% slower than that in non-functionalized PLGA.118,119 Moreover, cell adhesion and proliferation were improved, probably due to the higher hydrophilicity of the surface, and the levels of growth factors immobilized on the scaffold were higher while their release rate was slowed (Fig. 11b).120 In addition, systematic in vivo studies on the ability of the dual-modular scaffolds functionalized with 2-N,6-O-disulfated chitosan to induce bone and vascular regeneration showed that these two processes were well coordinated and an acceleration in regeneration, induced in terms of rapid blood reperfusion, significantly increased the expression of type H vessels and tissue ingrowth.121,122 2-N,6-O-Disulfated chitosan not only improved the bioactivity of growth factors by giving the environment a greater bio-similarity to the extracellular matrix, but may also have been involved in the induction of a favourable immune microenvironment that improves the crosstalk between immune cells and stem cells, undergoing osteogenic differentiation. 2-N,6-O-Disulfated chitosan is also responsible for the observed improved development of bone tissue.122,123
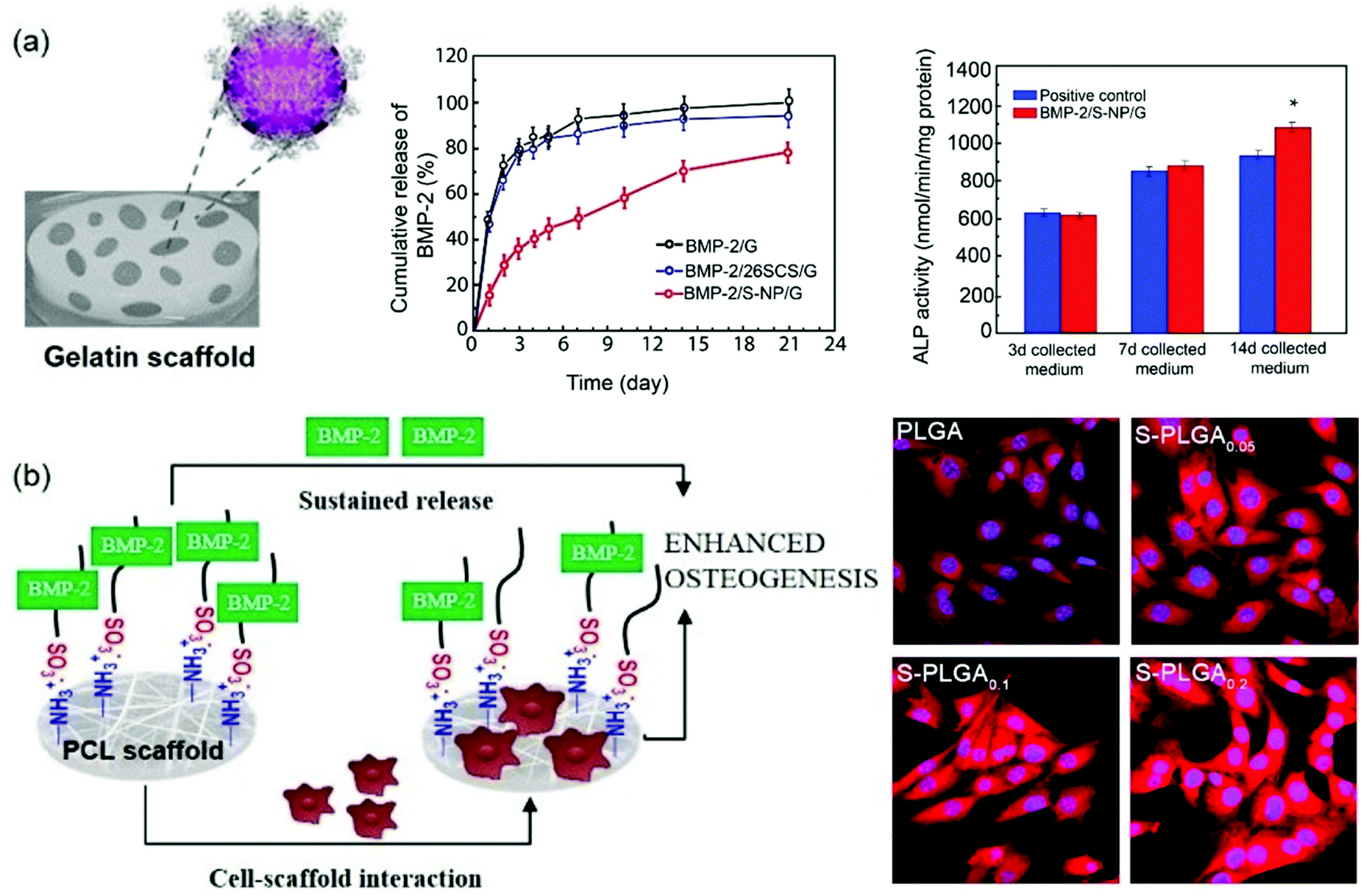 | ||
| Fig. 11 2-N,6-O-Disulfated chitosan-based scaffolds for orthopaedic tissue engineering. (a) Schematic showing the design principle behind 2-N,6-O-disulfated chitosan nanoparticles incorporating gelatin scaffolds (left). The sustained release of BMP-2 from the scaffold and in vitro alkaline phosphatase activity are also shown (right). (b) Schematic diagram of the S-PLGA/rhBMP-2 scaffolds on bone regeneration (left) and effect of S-PLGA scaffolds on the binding efficiency between rhBMP-2 and its receptor (right). Panel (a) reprinted from ref. 114 Copyright 2019, with permission from Elsevier and panel (b) adapted/reproduced from ref. 120 with permission from Elsevier. | ||
On the other hand, the angiogenic factor VEGF is associated with stimulating endothelial cell proliferation, migration and sprouting to enhance new blood vessel formation. However, its high cost and short half-life are significant drawbacks for its therapeutic applications. Recent studies have demonstrated the synergic effect between 2-N,6-O-disulfated chitosan and VEGF, prolonging the life-span in vitro and in vivo and enhancing bioactivity through the activation of the receptor phosphorylation and pro-angiogenic related genes expression.18 In a recent structure–activity analysis, the efficiency of this polysaccharide in contrast with other sulfated derivatives, such as 3,6-O-disulfated- and 6-O-sulfated-derivatives, is explained again as the basis to the spatial structure of the chain.124 By taking into account the fact that 3,6-O- and 2-N,6-O-disulfated derivatives showed similar zeta potential values and sulfur contents, it is plausible to hypothesize that, as in the binding to BMP-2, the presence of sulfate groups in position 2 should allow the conformation adaptation of the polysaccharide during the binding process. Based on these results, scaffolds that served as cytokine reservoirs for capturing VEGF in situ to facilitate angiogenesis in order to accelerate tissue regeneration have been prepared (Fig. 12a).125
 | ||
| Fig. 12 (a) Scheme of 2-N,6-O-disulfated chitosan-coated scaffolds capturing VEGF in situ. (b) Different sequential releases of BMP-2 and VEGF obtained in 2-N,6-O-disulfated chitosan-based dual-loading systems. Fast releasing of BMP-2 made for rapid initiation of osteogenesis, while through VEGF release guaranteed persistent angiogenesis. Panel (a) reproduced by permission of The Royal Society of Chemistry from ref. 125 and panel (b) reprinted with permission from ref. 121 copyright 2019 American Chemical Society. | ||
In accordance with these results, 2-N,6-O-disulfated chitosan has demonstrated, on the one hand, the capability of improving the osteogenic and angiogenic activities of BMP-2 during bone formation and, on the other hand, the ability to enhance the VEFG-mediated angiogenesis. In this context, very recently, dual-loading systems constructed with hydrogels and microspheres have been developed. These systems have been designed to achieve the different releasing patterns of BMP-2 and VEFG. It has been observed that the introduction of 2-N,6-O-disulfated chitosan in the system accelerates endochondral ossification and promotes angiogenesis (Fig. 12b).121
In another approach, a dual-modular growth factor delivery scaffold has been developed based on an organic–inorganic modular system functionalized with 2-N,6-O-disulfated chitosan.122 Systematic in vitro and in vivo studies have proven that the two coupled processes of osteogenesis and angiogenesis are well-orchestrated and both are enhanced and ascribed to the specific BMP-2 and VEGF delivery modes and 2-N,6-O-disulfated chitosan decoration. These studies highlight the importance of differentiating between the delivery pattern of different GFs and sheds light on the future design of growth factor-based bone grafts.
The 2-N,6-O-disulfated chitosan has also been employed to fabricate delivery vehicles of epithelial growth factors (EGFs) through encapsulation of the chitosan–GF complex in poly(lactic-co-glycolic acid) (PLGA) nanofibers.126 In this case, the sulfated polysaccharide may cooperate with EGF not only for binding and controlled release, buy may also have a synergistic effect on promoting wound healing through migration of the Hacat cells as well as facilitating the maturation of the vascular system as a cofactor for VEGF and FGF-2 that contribute to angiogenesis.
Finally, this polysaccharide has also been used to modify the surface of polycaprolactone stent (PCL) in order to improve the biocompatibility of the original PCL stents. Moreover, the coating with 2-N,6-O-disulfated chitosan produces a good surface which is suitable for endothelial cell attachment and growth, maintaining the mechanical properties similar to those of the existing bioresorbable polymeric stents.127
3,6-O-Disulfated chitosans have also demonstrated great utility in tissue engineering applications. In particular, the employment of these polysaccharides in microsphere scaffolds, allows a long-term release profile of transform growth factor-β1 (TGF-β1), a power protein to induce the chondrogenesis of mesenchymal stem cells that has been widely applied in studies of cartilage restoration. Under the protection of the sulfated chitosan, around 13% TGF-β1 was preserved even after being stored for 14 days.128 On the other hand, it has been observed that the combination of 3,6-O-disulfated chitooligosaccharides with acidic fibroblast growth factor (FGF-2) in thermo-sensitive hydrogels protects against peripheral nerve injuries, promoting the repair of the injured rat sciatic nerve.129 These results are explained in relation to the ability of these polysaccharides to improve the bioactivity of the growth factor.
Additionally, the capacity of heparanized chitosans on defining the cell phenotype has been investigated. To this end, chitosan-N-arginine derivatives have been employed. Interestingly, it has been observed that a soluble arginine functionalized chitosan promoted an osteogenic phenotype in primary human foetal chondroblasts for a period of seven days in the absence of an osteogenic medium, while its sulfated derivative promoted a chondrogenic phenotype in the same cells. These results demonstrate the fine control that can be exerted on the phenotype of progenitor cells by the sulfation of chitosan, which can be attributed to the greater structural similarity of sulfated-derivatives with glycosaminoglycans, the natural ligands of growth factors.130 These derivatives are also good candidates for wound dressing, having demonstrated on the one hand that they bind FGF-2 with a higher affinity than heparan sulfates (Fig. 13a). On the other hand, they also promote epithelial cell migration and support the formation of an expanded epidermis in an organotypic skin model. Furthermore, sulfated chitosan-N-arginine promotes the expression of the heparan sulfate proteoglycan, perlecan, by both epithelial and fibroblast cells (Fig. 13b).131 Interestingly, in this study, an important dependence of the pattern and DS on biological effects has been observed.
 | ||
| Fig. 13 (a) Level of FGF-2 bound to chitosan-N-arginine and their sulfated derivatives, perlecan, and perlecan without heparan sulfate chains measured by SPR. (b) Analysis by quantitative real-time PCR (qPCR) of the expression of the perlecan gene induced by chitosan-N-arginine and its derivatives in keratinocytes and fibroblasts. Reproduced from ref. 131 with permission from Wiley, the owner of publishing rights. | ||
In a similar way, the ability to induce neural differentiation of the embryonic stem cells of heparanized chitosans is also controlled by these parameters.50 Compared with 2-N,6-O- and 3,6-O-disulfated chitosans, 6-O-sulfated chitosan demonstrated the most optimal effects in promoting neural differentiation. Furthermore, this effect correlated with the DS of the sulfated chitosan; at a higher DS an increase in the efficiency of neural differentiation was observed.50
6.3. Heparanized chitosans as anticoagulant agents
Chitosan sulfates have also demonstrated their ability to mimic heparin for decades.132 As has been highlighted above (Section 2), heparin is a highly sulfated variant of heparan sulfate and its main biological activity is to act as an anticoagulant.38 Evidence from different studies shows that the anti-coagulant activity of heparanized chitosan is strongly dependent on its structure, being influenced by the degree of sulfation, the degree of acetylation, the molecular weight of the polymer, and its sulfation pattern. It has been reported that the anticoagulation activity of heparanized chitosans is closely related to the DS. Thus, clotting assays have showed that highly sulfated chitosans (DS > 2.1) significantly prolong activated partial thromboplastin time (APTT) and thrombin time (TT), but not prothrombin time (PT).68 The lack of PT activity of heparanized chitosan is similar to heparin and other sulfated polysaccharides, which is indicative that highly sulfated derivatives mainly affect the intrinsic pathway and have little effect on the extrinsic pathway of the clotting cascade.133Many authors have suggested that the high anticoagulant activity observed in samples with a high DS value could be related to their increased negative surface charge density, which provides them with a greater capacity to neutralize the positively charged protein amino acid residues. However, certain studies have showed that the anticoagulant activities (APTT and TT values) do not show a regular increase with an increase in DS. Such differences have been attributed to the possible differences in sulfation pattern; it is possible that polysaccharides with a lower DS possess specific sequences of saccharide residues that have a high-affinity binding to the plasma serine protease inhibitors such as antithrombin III (ATIII) and heparin cofactor II (HCII). In this context, it has been proposed that to act as anticoagulant, heparanized chitosan must possess at least 36 consecutive sulfate groups along the polymer backbone.134 On the other hand, different studies have proposed that anticoagulant activity of heparanized chitosans is influenced by its Mw. Several authors have associated this influence with the mechanism of action, as is the case with heparin. Thus, this is the main mechanism by which unfractionated heparin (UFH) equally inhibits serine proteases, such as thrombin (IIa factor) and Xa factor, while preparations of low molecular weight heparin exhibit a higher anti-factor Xa activity (aXa).135 A second mechanism by which UFH can specifically inactivate thrombin is through its binding to heparin cofactor II (HCII), a serine protease inhibitor that binds many glycosaminoglycans to enhance its inhibition of thrombin.136 In this way, certain authors observed that decreasing the Mw of heparanized chitosans could result in a higher anti-factor Xa activity.137 Subsequent studies suggested that the main mechanics of the anticoagulant activity of low Mw heparanized chitosans could be mediated by HCII,138,139 while the binding of these derivatives to antithrombin III fails to produce a conformational change critical for the action of this serpin on Factor Xa and thrombin. However, the few reports available in the literature, and their inconsistent results require additional studies for a clear establishment of the relationship between Mw and anticoagulant activity.
Finally, different studies reveal that the N-acetyl groups also have a relevant influence on anticoagulant activities.140 In particular, it has been proposed that the introduction of acyl groups into chitosan sulfate chains could improve their hydrophobicity, which has been confirmed to enhance anticoagulant activity in other polysaccharides, such as dextran sulfate.141 To prove this hypothesis, different hydrophobic groups have been introduced onto the amino groups of chitosan sulfate. For example, N-succinylation of chitosan sulfates produces an important increase in activated partial thromboplastin time, with only minimal effects having been observed in terms of prothrombin and thrombin times.142 A similar effect has been observed with the introduction of hexanoyl and propanoyl groups.54 More recently, the mechanism of action of N-acylated chitosan sulfates has been analysed. The interaction of these with ATIII was able to inhibit the proteolytic activation of FX by the intrinsic FXase complex, as well as the formation of FIIa via the prothrombinase complex in a dose-dependent manner (Fig. 14).143 The analysis of the binding affinities revealed that the values of the equilibrium dissociation constant (KD) of N-acylated polysaccharides for FXa and FIIa in the presence of ATIII were 67.4 nM and 112.6 nM, respectively, indicating that the ATIII/polysaccharide complex had a greater inhibition effect on FXa, while both FXa and FIIa in the common pathway were inhibited by the ATIII/polysaccharide complex.
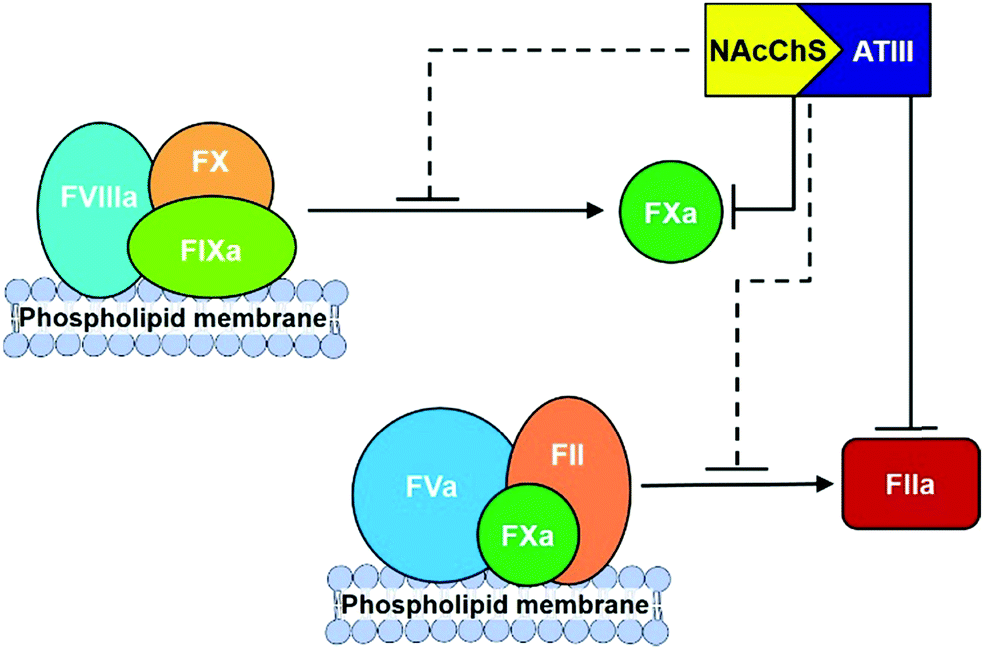 | ||
| Fig. 14 Schematic diagram for anticoagulant mechanism of N-acylated chitosans sulfate. Adapted from ref. 143. | ||
Interesting effects have been also showed when sulfated groups are introduced directly in the hydrophobic residue, such as for example N-arylsulfonates.144 In this case, a double effect has been observed, increasing both activated partial thromboplastin time and prothrombin time. These results have been applied in the development of anticoagulant scaffolds. For example, low-molecular-weight sulfoethyl chitosan has been used as a model template for the generation of silver core–shell nanoparticles with high potential as anticoagulants for medical applications.145 The main interaction mechanism of these nanoparticles lies in the interference with factor Xa, an important target in the heparin dosage therapy. These results may lead to completely new anticoagulants on the basis of capped nanoparticles. Finally, it has been proposed that heparanized chitosan-based films, prepared from 2-N,3,6-O-trisulfated chitosan, are suitable candidates for coating blood-contacting medical devices, due to their excellent haemo-compatibility.146
7. Conclusions and outlook
Chitosan is a natural polymer that has attracted a great deal of attention due to its great abundance in nature and its many favourable properties, such as its biocompatibility and biodegradability, which make it an attractive biomaterial for multiple technological applications. In addition, its structure presents many reactive primary and secondary hydroxyl and amino functional groups, susceptible to being modified, consequently providing it with new functionalities. In this sense, the sulfation of chitosan makes this polymer a closer mimic of heparins and heparan sulfates, giving rise to the “third generation” of chitosans, more sustainable and with multiple new and groundbreaking biological activities and technological applications (Fig. 15).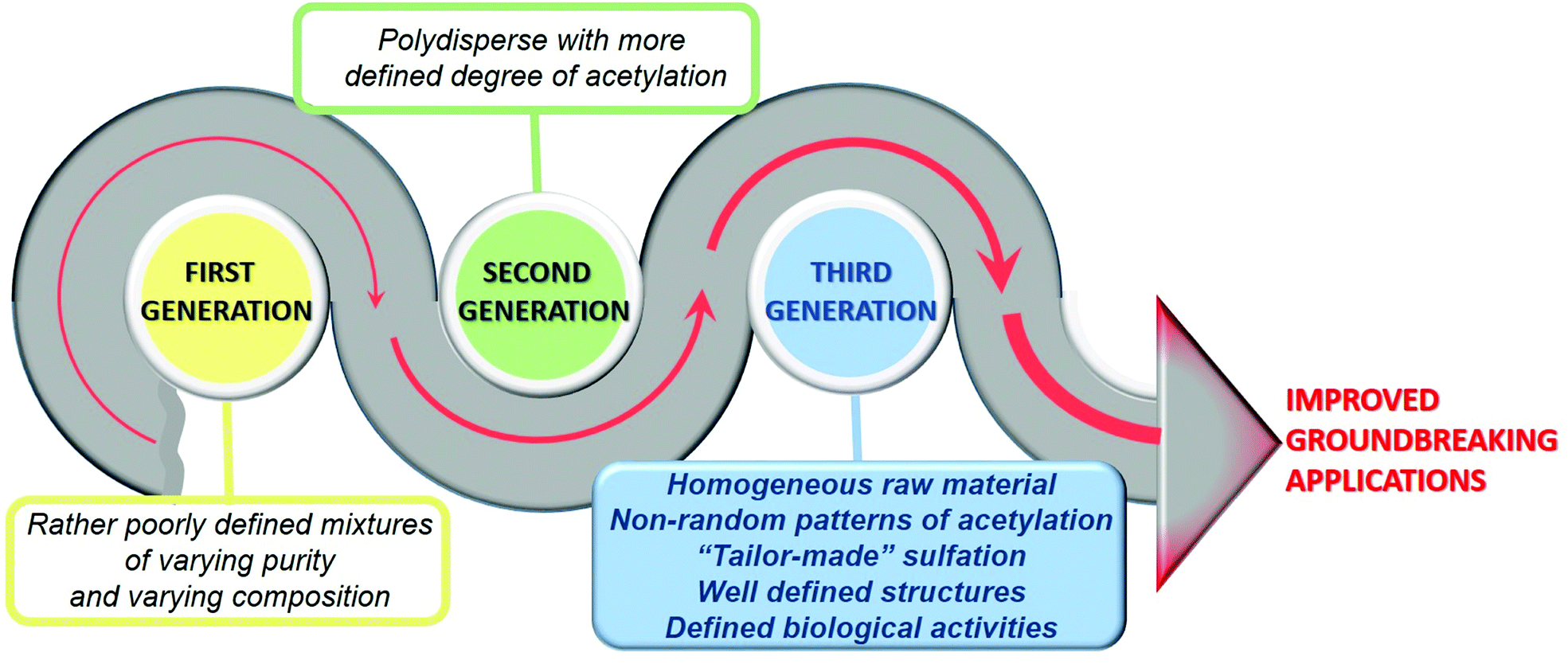 | ||
| Fig. 15 The three generations of chitosan biopolymers. To answer these issues, multi- and interdisciplinary research is necessary, involving complementary knowledge of various scientific fields and generating synergies between research groups with different scientific-technical “know-how”. | ||
In this review, we have tried to highlight certain studies, which demonstrate the enormous development and progress that both the chemical sulfation methods of chitosan and the techniques for its structural determination have undergone in recent years. These methods and technologies presented and discussed herein share great potential in terms of improving our understanding of how the specific structural properties of chitosans sulfate determine their biological activities.
In addition, we have tried to provide an overview of the main applications of third generation of heparanized chitosans, especially in the field of biomedicine. Despite the tremendous advances made in recent years in this field, key challenges must still be resolved in relation to facilitating and generalizing the practical applications of chitosan sulfate.
Thus, the origin of chitosans has been considered problematic at times. In this case, it will be important to answer the following question: which biological organisms are able to produce the materials required for the specific applications? The use of crustacean exoskeletons (from shrimps, crabs, lobsters or prawns), the main source of chitosan nowadays, may become unsustainable, due to the continuous harvesting of these without replacement, coupled with the fact that they are limited seasonally, necessitating the search for alternative sources. Furthermore, the destructive and environmentally unfriendly nature of the isolation process from this crustaceous source (strong alkalis at high temperatures for long time periods are required) leads to random mixtures of raw materials, the properties and functionalities of which are difficult to predict and even more difficult to reproduce.147 Chitins from algae, fungi and insects will probably attract increasing attention as it is easier to isolate chitin from these sources than from crustacean waste.148 In addition, the development of new processes adapted to the source is necessary, to produce a high quality chitin, and subsequently (after partial de-acetylation) a pure and “homogeneous” raw material.
In addition, it is important to underline the emphasis on the sustainability. For, what is the purpose of sustainability in terms of revaluating agro-food by-products, if the revaluation process implies greater damage to the natural environment? To answer this question, it will be necessary to develop methodologies in accordance with the recommendations of the sustainable production trend. Sustainable chemistry offers a wide range of controlled synthesis processes, specific chemical modification reactions or new assembly techniques that, nowadays, can be applied to obtain new “tailor-made” chitosan biopolymers, offering great versatility in relation to their structure and functionality. Additionally, the production of well-defined chitosans with known structures and functionalities through biotechnological approaches has acquired great significance in recent years.149 These tailor-made polysaccharides will allow not only their effective binding to known targets, but also the development of new potential biomedical applications based on their binding to new targets such as, for example, lipoproteins, a recently discovered target of HSs.150
Author contributions
J. R.: conceptualization; writing, reviewing and editing. I. F.: writing original draft. D. T. M.: writing original draft. N. P.: writing original draft. R. B.-A.: writing original draft. A. B.: writing original draft. A. F.-M.: writing original and reviewing. E. G.-J.: conceptualization; writing, reviewing and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financial support provided by the grants PID2019-105337RB-C21 (MICINN/FEDER) and IND2019/IND-17121 (Consejería de Ciencia, Universidades e Innovación, CAM). I. F. acknowledges the Comunidad de Madrid for a Doctorado Industrial contract.References
- Sustainable Development Goals, https://www.un.org/sustainabledevelopment/sustainable-development-goals/.
- Q. V. Vuong, Utilisation of Bioactive Compounds from Agricultural and Food Waste, CRC Press, Boca Raton, FL, 1st edn, 2017 Search PubMed.
- C. Xu, M. Nasrollahzadeh, M. Selva, Z. Issaabadi and R. Luque, Chem. Soc. Rev., 2019, 48, 4791–4822 RSC.
- Ø. Arlov, D. Rütsche, M. Asadi Korayem, E. Öztürk and M. Zenobi-Wong, Adv. Funct. Mater., 2021, 2010732 CrossRef.
- Chitin and Chitosan Derivatives – Global Market Trajectory & Analytics, https://www.researchandmarkets.com/research/vwfnch/2018_global?w=4.
- M. S. Riaz Rajoka, L. Zhao, H. M. Mehwish, Y. Wu and S. Mahmood, Appl. Microbiol. Biotechnol., 2019, 103, 1557–1571 CrossRef CAS PubMed.
- A. Zhu and J. Shen, in Chitosan-Based Hydrogels, ed. K. Yao, J. Li, F. Yao and Y. Yin, CRC Press, 2011, pp. 109–177 Search PubMed.
- D. Araújo, I. C. Ferreira, C. A. V. Torres, L. Neves and F. Freitas, J. Chem. Technol. Biotechnol., 2020, 95, 1277–1289 CrossRef.
- D. A. Aguilera, N. Tanchoux, M. Fochi and L. Bernardi, Eur. J. Org. Chem., 2020, 3779–3795 CrossRef CAS.
- S. Takeshita, S. Zhao, W. J. Malfait and M. M. Koebel, Angew. Chem., Int. Ed., 2020, 59, 2–26 CrossRef.
- T. Kean and M. Thanou, Adv. Drug Delivery Rev., 2010, 62, 3–11 CrossRef CAS PubMed.
- B. Bellich, I. D’Agostino, S. Semeraro, A. Gamini and A. Cesàro, Mar. Drugs, 2016, 14, 99 CrossRef PubMed.
- H. Mittal, S. S. Ray, B. S. Kaith, J. K. Bhatia, Sukriti, J. Sharma and S. M. Alhassan, Eur. Polym. J., 2018, 109, 402–434 CrossRef CAS.
- S. Pokhrel and P. N. Yadav, J. Macromol. Sci., Part A: Pure Appl.Chem., 2019, 56, 450–475 CrossRef CAS.
- F. Krichen, Z. Ghlissi, I. Ben Amor, N. Sayari, R. Kallel, J. Gargouri, Z. Sahnoun, T. Boudawara and A. Bougatef, Exp. Toxicol. Pathol., 2017, 69, 45–53 CrossRef CAS PubMed.
- V. H. Pomin, Carbohydr. Res., 2015, 413, 41–50 CrossRef CAS PubMed.
- N. Sayari, R. Balti, M. Ben Mansour, I. Ben Amor, I. Graiet, J. Gargouri and A. Bougatef, Biomed. Pharmacother., 2016, 80, 322–330 CrossRef CAS PubMed.
- Y. Yu, R. Chen, Y. Sun, Y. Pan, W. Tang, S. Zhang, L. Cao, Y. Yuan, J. Wang and C. Liu, Acta Biomater., 2018, 71, 510–521 CrossRef CAS PubMed.
- M. Mende, C. Bednarek, M. Wawryszyn, P. Sauter, M. B. Biskup, U. Schepers and S. Bräse, Chem. Rev., 2016, 116, 8193–8255 CrossRef CAS PubMed.
- J. Li, C. Cai, L. Wang, C. Yang, H. Jiang, M. Li, D. Xu, G. Li, C. Li and G. Yu, ACS Macro Lett., 2019, 8, 1570–1574 CrossRef CAS.
- S. Dimassi, N. Tabary, F. Chai, N. Blanchemain and B. Martel, Carbohydr. Polym., 2018, 202, 382–396 CrossRef CAS PubMed.
- N. M. Alves and J. F. Mano, Int. J. Biol. Macromol., 2008, 43, 401–414 CrossRef CAS PubMed.
- R. Jayakumar, N. Nwe, S. Tokura and H. Tamura, Int. J. Biol. Macromol., 2007, 40, 175–181 CrossRef CAS PubMed.
- W. H. Howell and E. Holt, Am. J. Physiol. Content, 1918, 47, 328–341 CrossRef CAS.
- A. Linker, P. Hoffman, P. Sampson and K. Meyer, Biochim. Biophys. Acta, 1958, 29, 443–444 CrossRef CAS.
- B. E. Thacker, D. Xu, R. Lawrence and J. D. Esko, Matrix Biol., 2014, 35, 60–72 CrossRef CAS PubMed.
- P. Chopra, A. Joshi, J. Wu, W. Lu, T. Yadavalli, M. A. Wolfert, D. Shukla, J. Zaia and G.-J. Boons, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2012935118 CrossRef CAS PubMed.
- J. D. Esko and U. Lindahl, J. Clin. Invest., 2001, 108, 169–173 CrossRef CAS.
- J. T. Gallagher and A. Walker, Biochem. J., 1985, 230, 665–674 CrossRef CAS PubMed.
- B. Casu, Ann. N. Y. Acad. Sci., 1989, 556, 1–17 CrossRef CAS PubMed.
- R. D. Rosenberg and L. Lam, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 1218–1222 CrossRef CAS PubMed.
- B. Mulloy, E. Gray and T. W. Barrowcliffe, Thromb. Haemostasis, 2000, 84, 1052–1056 CrossRef CAS.
- K. L. Kramer and H. J. Yost, Annu. Rev. Genet., 2003, 37, 461–484 CrossRef CAS PubMed.
- S. Sarrazin, W. C. Lamanna and J. D. Esko, Cold Spring Harbor Perspect. Biol., 2011, 3, a004952 Search PubMed.
- S. D. Vallet, O. Clerc and S. Ricard-Blum, J. Histochem. Cytochem., 2021, 69, 93–104 CrossRef CAS PubMed.
- U. Lindahl and L. Kjellén, J. Intern. Med., 2013, 273, 555–571 CrossRef CAS PubMed.
- F. E. Poulain and H. J. Yost, Development, 2015, 142, 3456–3467 CrossRef CAS PubMed.
- E. Bedini, M. M. Corsaro, A. Fernández-Mayoralas and A. Iadonisi, in Extracellular Sugar-Based Biopolymers Matrices, ed. E. Cohen and H. Merzendorfer, Springer, Cham, 2019 Search PubMed.
- U. Lindahl, J. Couchman, K. Kimata and J. D. Esko, in Essentials of Glycobiology, 3rd edn, ed. A. Varki, R. D. Cummings, J. D. Esko, P. Stanley, G. W. Hart, M. Aebi, A. G. Darvill, T. Kinoshita, N. H. Packer, J. H. Prestegard, R. L. Schnaar and P. H. Seeberger, Cold Spring Harbor Laboratory Press, NY, 2015, pp. 207–221 Search PubMed.
- M. Petitou, Biochimie, 2003, 85, 83–89 CrossRef CAS.
- S. Mousavi, M. Moradi, T. Khorshidahmad and M. Motamedi, Adv. Pharmacol. Sci., 2015, 2015, 1–14 Search PubMed.
- M. C. Z. Meneghetti, A. J. Hughes, T. R. Rudd, H. B. Nader, A. K. Powell, E. A. Yates and M. A. Lima, J. R. Soc., Interface, 2015, 12, 20150589 CrossRef PubMed.
- G. Cassinelli, G. Torri and A. Naggi, in Heparanase. Advances in Experimental Medicine and Biology, ed. I. Vlodavsky, R. Sanderson and N. Ilan, Springer, Cham, 2020, pp. 493–522 Search PubMed.
- D. Zhao, R. Ding, Y. Liu, X. Yin, Z. Zhang and X. Ma, Exp. Ther. Med., 2017, 14, 5515–5522 CrossRef CAS PubMed.
- V. K. Mourya and N. N. Inamdar, React. Funct. Polym., 2008, 68, 1013–1051 CrossRef CAS.
- L. C. R. Carvalho, F. Queda, C. V. A. Santos and M. M. B. Marques, Chem. – Asian J., 2016, 11, 3468–3481 CrossRef CAS PubMed.
- E. Bedini, A. Laezza, M. Parrilli and A. Iadonisi, Carbohydr. Polym., 2017, 174, 1224–1239 CrossRef CAS PubMed.
- P. Vongchan, W. Sajomsang, D. Subyen and P. Kongtawelert, Carbohydr. Res., 2002, 337, 1239–1242 CrossRef CAS.
- S. Ahmed, Annu, A. Ali and J. Sheikh, Int. J. Biol. Macromol., 2018, 116, 849–862 CrossRef CAS PubMed.
- K. Ding, Y. Wang, H. Wang, L. Yuan, M. Tan, X. Shi, Z. Lyu, Y. Liu and H. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 20043–20050 CrossRef CAS PubMed.
- A. Gamzazade, A. Sklyar, S. Nasibov, I. Sushkov, A. Shashkovb and Y. Knirelb, Structural features of sulfated chitosans, 1997, vol. 34 Search PubMed.
- K. Zhang, A. Weltrowski, D. Peschel, S. Fischer and T. Groth, in Functional Materials from Renewable Sources, ACS Symposium Series, ed. F. Liebner and T. Rosenau, American Chemical Society, Washington, DC, 2012, vol. 1107, pp. 297–314 Search PubMed.
- S. Hirano and J. Kinugawa, Carbohydr. Res., 1986, 50, 295–299 CrossRef.
- R. Huang, Y. Du, J. Yang and L. Fan, Carbohydr. Res., 2003, 338, 483–489 CrossRef CAS PubMed.
- K. Nagasawa, Y. Tohira, Y. Inoue and N. Tanoura, Carbohydr. Res., 1971, 18, 95–102 CrossRef CAS.
- N. Nishi, A. Ebina, S. Nishimura, A. Tsutsumi, O. Hasegawa and S. Tokura, Int. J. Biol. Macromol., 1986, 8, 311–317 CrossRef CAS.
- A. M. Naggi, G. Torri, T. Compagnoni and B. Casu, in Chitin in Nature and Technology, ed. R. Muzzarelli, C. Jeuniaux and G. W. Gooday, Springer US, Boston, MA, 1986, pp. 371–377 Search PubMed.
- N. R. Pires, P. L. R. Cunha, J. S. Maciel, A. L. Angelim, V. M. M. Melo, R. C. M. de Paula and J. P. A. Feitosa, Carbohydr. Polym., 2013, 91, 92–99 CrossRef CAS PubMed.
- K. Zhang, J. Helm, D. Peschel, M. Gruner, T. Groth and S. Fischer, Polymer, 2010, 51, 4698–4705 CrossRef CAS.
- M. Tan, H. Wang, Y. Wang, G. Chen, L. Yuan and H. Chen, J. Mater. Chem. B, 2014, 2, 569–576 RSC.
- R. Xing, X. He, S. Liu, H. Yu, Y. Qin, X. Chen, K. Li, R. Li and P. Li, Mar. Drugs, 2015, 13, 3072–3090 CrossRef CAS PubMed.
- K. R. Holme and A. S. Perlin, Carbohydr. Res., 1997, 302, 7–12 CrossRef CAS PubMed.
- A. Farrán, C. Cai, M. Sandoval, Y. Xu, J. Liu, M. J. Hernáiz and R. J. Linhardt, Chem. Rev., 2015, 115, 6811–6853 CrossRef PubMed.
- S. S. Silva, J. F. Mano and R. L. Reis, Green Chem., 2017, 19, 1208–1220 RSC.
- N. Chopin, C. Sinquin, J. Ratiskol, A. Zykwinska, P. Weiss, S. Cérantola, J. Le Bideau and S. Colliec-Jouault, Biomed Res. Int., 2015, 2015, 1–9 CrossRef PubMed.
- H. Baumann and V. Faust, Carbohydr. Res., 2001, 331, 43–57 CrossRef CAS PubMed.
- S.-I. Nishimura, H. Kai, K. Shinada, T. Yoshida, S. Tokura, K. Kurita, H. Nakashima, N. Yamamoto and T. Uryu, Carbohydr. Res., 1998, 306, 427–433 CrossRef CAS.
- J. Yang, K. Luo, D. Li, S. Yu, J. Cai, L. Chen and Y. Du, Int. J. Biol. Macromol., 2013, 52, 25–31 CrossRef CAS PubMed.
- P. Sahariah and M. Másson, Biomacromolecules, 2017, 18, 3846–3868 CrossRef CAS PubMed.
- M. Collado-González, Y. González Espinosa and F. M. Goycoolea, Biomimetics, 2019, 4, 32 CrossRef PubMed.
- C. Peniche, W. Argüelles-Monal and F. M. Goycoolea, in Monomers, Polymers and Composites from Renewable Resources, ed. M. N. Belgacem and A. Gandini, Elsevier, 2008, vol. 1, pp. 517–542 Search PubMed.
- E. Doncel-Pérez, I. Aranaz, A. Bastida, J. Revuelta, C. Camacho, N. Acosta, L. Garrido, C. Civera, E. García-Junceda, A. Heras and A. Fernández-Mayoralas, Carbohydr. Polym., 2018, 191, 225–233 CrossRef PubMed.
- J. Lizardi-Mendoza, W. M. Argüelles Monal and F. M. Goycoolea Valencia, in Chitosan in the Preservation of Agricultural Commodities, Elsevier, 2016, pp. 3–31 Search PubMed.
- J. Revuelta, I. Aranaz, N. Acosta, C. Civera, A. Bastida, N. Peña, D. T. Monterrey, E. Doncel-Pérez, L. Garrido, Á. Heras, E. García-Junceda and A. Fernández-Mayoralas, ACS Appl. Mater. Interfaces, 2020, 12, 25534–25545 CrossRef CAS PubMed.
- A. Rohman, A. Windarsih, E. Lukitaningsih, M. Rafi, K. Betania and N. A. Fadzillah, Biomed. Spectrosc. Imaging, 2020, 8, 55–71 Search PubMed.
- K. Zhang, J. Helm, D. Peschel, M. Gruner, T. Groth and S. Fischer, Polymer, 2010, 51, 4698–4705 CrossRef CAS.
- H. Zhou, J. Qian, J. Wang, W. Yao, C. Liu, J. Chen and X. Cao, Biomaterials, 2009, 30, 1715–1724 CrossRef CAS PubMed.
- K. R. Holme and A. S. Perlin, Carbohydr. Res., 1997, 302, 7–12 CrossRef CAS PubMed.
- K. Zhang, D. Peschel, J. Helm, T. Groth and S. Fischer, Carbohydr. Polym., 2011, 83, 60–65 CrossRef CAS.
- D. Liu, Y. Wei, P. Yao and L. Jiang, Carbohydr. Res., 2006, 341, 782–785 CrossRef CAS PubMed.
- P. Broussignac, Chim. Ind., Genie Chim., 1968, 99, 1241–1247 CAS.
- S. Sabnis and L. H. Block, Polym. Bull., 1997, 39, 67–71 CrossRef CAS.
- A. Domard, Int. J. Biol. Macromol., 1987, 9, 333–336 CrossRef CAS.
- F. Niola, N. Basora, E. Chornet and P. F. Vidal, Carbohydr. Res., 1993, 238, 1–9 CrossRef CAS.
- S. Aiba, Int. J. Biol. Macromol., 1986, 8, 173–176 CrossRef CAS.
- M. Lavertu, Z. Xia, A. N. Serreqi, M. Berrada, A. Rodrigues, D. Wang, M. D. Buschmann and A. Gupta, J. Pharm. Biomed. Anal., 2003, 32, 1149–1158 CrossRef CAS PubMed.
- L. Yuan, Z. Yue, H. Chen, H. Huang and T. Zhao, Colloids Surf., B, 2009, 73, 346–350 CrossRef CAS PubMed.
- Q. Long, Z. Zhang, G. Qi, Z. Wang, Y. Chen and Z.-Q. Liu, ACS Sustainable Chem. Eng., 2020, 8, 2512–2522 CrossRef CAS.
- G. Vessella, J. A. Vázquez, J. Valcárcel, L. Lagartera, D. T. Monterrey, A. Bastida, E. García-Junceda, E. Bedini, A. Fernández-Mayoralas and J. Revuelta, Polymers, 2021, 13, 313 CrossRef CAS PubMed.
- C. A. A. van Boeckel and M. Petitou, Angew. Chem., Int. Ed. Engl., 1993, 32, 1671–1690 CrossRef.
- J. Angulo, R. Ojeda, J.-L. de Paz, R. Lucas, P. M. Nieto, R. M. Lozano, M. Redondo-Horcajo, G. Giménez-Gallego and M. Martín-Lomas, ChemBioChem, 2004, 5, 55–61 CrossRef CAS PubMed.
- H. Lortat-Jacob, J. E. Turnbull and J. A. Grimaud, Biochem. J., 1995, 310, 497–505 CrossRef CAS PubMed.
- S. E. Stringer, M. J. Forster, B. Mulloy, C. R. Bishop, G. J. Graham and J. T. Gallagher, Blood, 2002, 100, 1543–1550 CrossRef CAS PubMed.
- P. W. Park, O. Reizes and M. Bernfield, J. Biol. Chem., 2000, 275, 29923–29926 CrossRef CAS PubMed.
- M. Mobli, M. Nilsson and A. Almond, Glycoconjugate J., 2008, 25, 401–414 CrossRef CAS PubMed.
- A. Lubineau, H. Lortat-Jacob, O. Gavard, S. Sarrazin and D. Bonnaffé, Chem. – Eur. J., 2004, 10, 4265–4282 CrossRef CAS PubMed.
- B. Menchicchi, J. P. Fuenzalida, A. Hensel, M. J. Swamy, L. David, C. Rochas and F. M. Goycoolea, Biomacromolecules, 2015, 16, 924–935 CrossRef CAS PubMed.
- M. R. Akl, P. Nagpal, N. M. Ayoub, B. Tai, S. A. Prabhu, C. M. Capac, M. Gliksman, A. Goy and K. S. Suh, Oncotarget, 2016, 7, 44735–44762 CrossRef PubMed.
- P. Chiodelli, A. Bugatti, C. Urbinati and M. Rusnati, Molecules, 2015, 20, 6342–6388 CrossRef CAS PubMed.
- E. H. Knelson, J. C. Nee and G. C. Blobe, Trends Biochem. Sci., 2014, 39, 277–288 CrossRef CAS PubMed.
- N. Afratis, C. Gialeli, D. Nikitovic, T. Tsegenidis, E. Karousou, A. D. Theocharis, M. S. Pavão, G. N. Tzanakakis and N. K. Karamanos, FEBS J., 2012, 279, 1177–1197 CrossRef CAS PubMed.
- M. Jiang, H. Ouyang, P. Ruan, H. Zhao, Z. Pi, S. Huang, P. Yi and M. Crepin, Anticancer Res., 2011, 31, 1321–1328 CAS.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS PubMed.
- G. D. Yancopoulos, S. Davis, N. W. Gale, J. S. Rudge, S. J. Wiegand and J. Holash, Nature, 2000, 407, 242–248 CrossRef CAS PubMed.
- M. Presta, D. Leali, H. Stabile, R. Ronca, M. Camozzi, L. Coco, E. Moroni, S. Liekens and M. Rusnati, Curr. Pharm. Des., 2003, 9, 553–566 CrossRef CAS PubMed.
- Y. Li, W. Wang, Y. Zhang, X. Wang, X. Gao, Z. Yuan and Y. Li, Biomater. Sci., 2019, 7, 1584–1597 RSC.
- J. Garcia, N. Callewaert and L. Borsig, Glycobiology, 2007, 17, 185–196 CrossRef CAS PubMed.
- R. Wang, J. Huang, M. Wei and X. Zeng, Biosci., Biotechnol., Biochem., 2010, 74, 1697–1700 CrossRef CAS PubMed.
- I. Ishizuka, Prog. Lipid Res., 1997, 36, 245–319 CrossRef CAS PubMed.
- S. Kocabay and B. Akkaya, Int. J. Biol. Macromol., 2020, 147, 792–798 CrossRef CAS PubMed.
- J. P. Vacanti and R. Langer, Lancet, 1999, 354, 32–34 CrossRef.
- J. Elisseeff, C. Puleo, F. Yang and B. Sharma, Orthod. Craniofac. Res., 2005, 8, 150–161 CrossRef CAS PubMed.
- K. Zeng, T. Groth and K. Zhang, ChemBioChem, 2019, 20, 737–746 CrossRef CAS PubMed.
- J. Dinoro, M. Maher, S. Talebian, M. Jafarkhani, M. Mehrali, G. Orive, J. Foroughi, M. S. Lord and A. Dolatshahi-Pirouz, Biomaterials, 2019, 214, 119214 CrossRef CAS PubMed.
- E. Gazzerro and E. Canalis, Rev. Endocr. Metab. Disord., 2007, 7, 51–65 CrossRef PubMed.
- H. Zhou, J. Qian, J. Wang, W. Yao, C. Liu, J. Chen and X. Cao, Biomaterials, 2009, 30, 1715–1724 CrossRef CAS PubMed.
- A. Zheng, X. Wang, J. Wang, X. Xin, Y. Yu, Y. Liu, J. Wang, K. Lv and L. Cao, Carbohydr. Polym., 2021, 117888 CrossRef CAS PubMed.
- L. Cao, Y. Yu, J. Wang, J. A. Werkmeister, K. M. McLean and C. Liu, Mater. Sci. Eng., C, 2017, 74, 298–306 CrossRef CAS PubMed.
- L. Cao, X. Kong, S. Lin, S. Zhang, J. Wang, C. Liu and X. Jiang, Artif. Cells, Nanomed., Biotechnol., 2018, 46, S1–S17 CrossRef CAS PubMed.
- X. Kong, J. Wang, L. Cao, Y. Yu and C. Liu, Colloids Surf., B, 2014, 122, 359–367 CrossRef CAS PubMed.
- S. Zhang, J. Chen, Y. Yu, K. Dai, J. Wang and C. Liu, ACS Biomater. Sci. Eng., 2019, 5, 1944–1955 CrossRef CAS PubMed.
- W. Tang, Y. Yu, J. Wang, H. Liu, H. Pan, G. Wang and C. Liu, Biomaterials, 2020, 232, 119645 CrossRef CAS PubMed.
- Y. Shu, Y. Yu, S. Zhang, J. Wang, Y. Xiao and C. Liu, Biomater. Sci., 2018, 6, 2496–2507 RSC.
- G. Han, X. Xia, Z. Pan, Y. Lin, L. Li, Y. Jiao, C. Zhou and S. Ding, J. Biomater. Sci., Polym. Ed., 2020, 31, 1237–1253 CrossRef CAS PubMed.
- C. Wang, Y. Yu, H. Chen, S. Zhang, J. Wang and C. Liu, J. Mater. Chem. B, 2019, 7, 1882–1892 RSC.
- X. Peng, Y. Yu, Z. Wang, X. Zhang, J. Wang and C. Liu, RSC Adv., 2017, 7, 43161–43171 RSC.
- T. Qiu, W. Jiang, P. Yan, L. Jiao and X. Wang, Front. Bioeng. Biotechnol., 2020, 8, 1–12 CrossRef PubMed.
- F. Li, L. Ma, B. Li and C. Gao, Pure Appl. Chem., 2014, 86, 1885–1895 CAS.
- Y. Liu, F. Yu, B. Zhang, M. Zhou, Y. Bei, Y. Zhang, J. Tang, Y. Yang, Y. Huang, Q. Xiang, Y. Zhao, Q. Liang and Y. Liu, Asian J. Pharm. Sci., 2019, 14, 511–520 CrossRef PubMed.
- M. S. Lord, B. M. Tsoi, B. L. Farrugia, S. R. Simon Ting, S. Baker, W. P. Wiesmann and J. M. Whitelock, J. Mater. Chem. B, 2014, 2, 6517–6526 RSC.
- B. L. Farrugia, Y. Mi, H. N. Kim, J. M. Whitelock, S. M. Baker, W. P. Wiesmann, Z. Li, P. Maitz and M. S. Lord, Adv. Funct. Mater., 2018, 28, 1802818 CrossRef.
- W. G. Malette, H. J. Quigley, R. D. Gaines, N. D. Johnson and W. G. Rainer, Ann. Thorac. Surg., 1983, 36, 55–58 CrossRef CAS PubMed.
- J. Yang, Y. Du, R. Huang, Y. Wan and T. Li, Int. J. Biol. Macromol., 2002, 31, 55–62 CrossRef CAS PubMed.
- Y. Zou and E. Khor, Carbohydr. Polym., 2009, 77, 516–525 CrossRef CAS.
- T. C. Laurent, A. Tengblad, L. Thunberg, M. Höök and U. Lindahl, Biochem. J., 1978, 175, 691–701 CrossRef CAS PubMed.
- J. Rau, J. Mitchell, Y. Fortenberry and F. Church, Semin. Thromb. Hemostasis, 2011, 37, 339–348 CrossRef CAS PubMed.
- G. Vikhoreva, G. Bannikova, P. Stolbushkina, A. Panov, N. Drozd, V. Makarov, V. Varlamov and L. Gal’braikh, Carbohydr. Polym., 2005, 62, 327–332 CrossRef CAS.
- J. Suwan, Z. Zhang, B. Li, P. Vongchan, P. Meepowpan, F. Zhang, S. A. Mousa, S. Mousa, B. Premanode, P. Kongtawelert and R. J. Linhardt, Carbohydr. Res., 2009, 344, 1190–1196 CrossRef CAS PubMed.
- P. Vongchan, W. Sajomsang, D. Subyen and P. Kongtawelert, Carbohydr. Res., 2002, 337, 1239–1242 CrossRef CAS PubMed.
- S. Hirano, Y. Tanaka, M. Hasegawa, K. Tobetto and A. Nishioka, Carbohydr. Res., 1985, 137, 205–215 CrossRef CAS PubMed.
- R. Huynh, F. Chaubet and J. Jozefonvicz, Carbohydr. Res., 2001, 332, 75–83 CrossRef CAS PubMed.
- T. Wang, Y. Zhou, W. Xie, L. Chen, H. Zheng and L. Fan, Int. J. Biol. Macromol., 2012, 51, 808–814 CrossRef CAS PubMed.
- P. Chandika, S.-Y. Heo, G.-W. Oh, I.-W. Choi, W. S. Park and W.-K. Jung, Int. J. Biol. Macromol., 2020, 161, 1552–1558 CrossRef CAS PubMed.
- S. Ouerghemmi, S. Dimassi, N. Tabary, L. Leclercq, S. Degoutin, F. Chai, C. Pierlot, F. Cazaux, A. Ung, J.-N. Staelens, N. Blanchemain and B. Martel, Carbohydr. Polym., 2018, 196, 8–17 CrossRef CAS PubMed.
- K. Heise, M. Hobisch, L. Sacarescu, U. Maver, J. Hobisch, T. Reichelt, M. Sega, S. Fischer and S. Spirk, Int. J. Nanomed., 2018, 13, 4881–4894 CrossRef CAS PubMed.
- A. F. Moraes, R. N. F. Moreira Filho, C. C. O. Passos, A. P. Cunha, L. M. A. e Silva, L. B. N. Freitas, N. F. Vasconcelos, N. M. P. S. Ricardo, K. M. Canuto, M. F. Rosa, L. K. A. M. Leal and R. S. Vieira, J. Appl. Polym. Sci., 2019, 136, 47128 CrossRef.
- I. Aranaz, M. Mengibar, R. Harris, I. Panos, B. Miralles, N. Acosta, G. Galed and A. Heras, Curr. Chem. Biol., 2009, 3, 203–230 CAS.
- V. Ghormade, E. K. Pathan and M. V. Deshpande, Int. J. Biol. Macromol., 2017, 104, 1415–1421 CrossRef CAS PubMed.
- M. B. Kaczmarek, K. Struszczyk-Swita, X. Li, M. Szczęsna-Antczak and M. Daroch, Front. Bioeng. Biotechnol., 2019, 7, 243 CrossRef PubMed.
- H. Kang, J. Lu, J. Yang, Y. Fan and X. Deng, Med. Nov. Technol. Devices, 2019, 3, 100016 CrossRef.
| This journal is © The Royal Society of Chemistry 2021 |