
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence3D and 4D printable dual cross-linked polymers with high strength and humidity-triggered reversible actuation†
Zhen
Jiang
 ,
Peidong
Shen
,
Ming
Li Tan
,
Qiao
Yan
,
Jekaterina
Viktorova
,
Chiara
Cementon
,
Xiaotong
Peng
,
Pu
Xiao
and
Luke A.
Connal
*
,
Peidong
Shen
,
Ming
Li Tan
,
Qiao
Yan
,
Jekaterina
Viktorova
,
Chiara
Cementon
,
Xiaotong
Peng
,
Pu
Xiao
and
Luke A.
Connal
*
Research School of Chemistry, Australian National University Canberra, ACT 2601, Australia. E-mail: Luke.connal@anu.edu.au
First published on 24th June 2021
Abstract
It is highly desirable but challenging to develop humidity-responsive polymers with simultaneously improved mechanical properties and 3D printability, while still displaying fast, reversible and complex shape transformations. Herein, a facile and scalable supramolecular strategy of fabricating a new class of humidity-responsive polymers is proposed to address this issue. The multiple hydrogen-bond cross-linked network is used to provide high humidity sensitivity and shear-dependent rheological behavior. Further introduction of metal coordination bonds can not only improve mechanical strength and creep resistance, but also promote reversible humidity-driven actuation and generate viscoelastic hydrogel inks. This humidity-responsive polymer with these unique combined attributes enables the potential to fabricate diverse functional materials from artificial muscles, smart electronic and catalytic devices. Moreover, diverse arbitrary architectures with spatial thickness contrast exhibiting sophisticated biomimetic 4D printing process were manufactured by direct ink writing (DIW). This material and method not only provides a general route to tune versatile functionalities and intelligent responsiveness of polymeric actuators at the molecular level, but also provides new opportunities for building exceptional 4D printed products.
1. Introduction
Dynamic structure and shape change in response to local moisture content are features of many biological entities. For example, pine cones open their scales when the environment is dry to release their seeds, and awns of wild wheat (Triticum turgidum) repeatedly open and close in response to variation of the moisture content.1 To mimic these sophisticated functions in smart biological systems, substantial efforts have been devoted to fabricating diverse polymeric systems such as polypyrrole,2 cellulose,3,4 liquid crystalline polymers,5–7 poly(ethylene glycol),8 agarose9 and alginate10 with controllable shape transformations induced by water vapor. These humidity-responsive polymers are usually engineered with hydrophilic groups such as hydroxyl, amide, carboxyl, or pyrrolyl, which are very responsive to moisture and undergo reversible water-sorption-induced swelling for actuation. Such fascinating dynamic responsiveness enables them to be particularly attractive for a wide range of technological applications such as soft robots,11 artificial muscles,12,13 sensors14 and electronic devices.15To mimic the delicate structures found in natural plants, a large body of work has been focused on achieving sophisticated humidity-responsive shape-morphing behaviors such as bending,6,16,17 twisting,5,9,18 spiraling19 and locomotion.2,20 These programmable 3D shapes could be realized through selective chemical modification of specific areas on the polymer films’ surface using photochemistry patterning,21 TiO2 patterning,22 ion patterning23 and local base and acid treatment.5,10 Moreover, controlling molecular alignment of liquid crystalline polymers,6 hierarchical self-assembly structures3 as well as orientation of incorporated inorganic nanofillers3,9 have also been reported to program sophisticated motions. Recently, the fabrication of multiresponsive polymers which are triggered by multiple stimuli, such as combination of humidity, light and heat,24,25 have been developed.
A major challenge in the preparation of humidity responsive polymers is to instill the materials with high mechanical properties and 3D printing processability which are also quintessential to the design and implementation of these material platforms in real applications. The high mechanical strength could enable the polymers to withstand a significant amount of loading without being damaged while operating in diverse applications. Furthermore, 3D printing techniques could further contribute to the development of polymer actuation technologies, because they enable the fabrication of more complex and programmable actuation devices and result in new actuation behavior.26–29 Material designs that enhance the mechanical properties such as the use of chemical cross-linked systems usually sacrifice their additive manufacturing processability. The enhanced mechanical properties may result in compromised actuation degree and slow down the shape morphing process.30 Moreover, the humidity responsive polymeric systems based on natural polymers such as agarose,19 alginate10 and cellulose3 lack the ability to precisely control over their compositions and chemistries. Therefore, there remains a challenge but significant need for the development of a humidity-responsive polymers with simultaneously improved mechanical properties and 3D printing processability while still displaying fast, reversible and programmable complex shape transformations.
Incorporating dynamic chemical bonds31,32 has been important in the development of high performance polymeric actuators.33–36 Incorporating multiple hydrogen bonding systems37–39 is an excellent candidate for programmable actuators as these show high intrinsic moisture-sensitivity,2 a wide spectrum of bonding strength,37 and cleavability by thermal or shear force.40 Combining multiple hydrogen bonding with other dynamic bonds could supply even greater diversity of fascinating properties.33,34 Herein, we present a simple supramolecular strategy to achieve a new class of humidity responsive polymeric networks composed of hydrogen bonds and carboxyl-Fe3+ coordination bond. The high humidity sensitivity of acid–ether hydrogen bonds enables fast deformation in response to changes in humidity. The carboxyl-Fe3+ coordination bond has been reported as an effective approach to strengthen and toughen hydrogels.41–43 While what distinguishes this work from these studies is that the actuation performance and 3D printability of physically cross-linked network could be simultaneously enhanced through introduction of carboxyl-Fe3+ coordination bond into hydrogen bonded network, increasing material stiffness, tensile strength, and tensile toughness of several orders of magnitude. The elastic deformation provided by the stable coordination cross-linking sites facilitates the reversible actuation upon humidity variations. This double cross-linked polymer featuring these attributes could be used as artificial muscle, smart electronic and catalytic devices. The addition of Fe3+ strikingly transforms the homogeneous polymer aqueous solutions into hydrogels by coordinate interactions between COOH and Fe3+, displaying excellent shear thinning behavior and the ability to rapidly self-heal after extrusion, which make it suitable as inks for direct ink writing (DIW). Taking advantage of 3D printing free design, we demonstrate complex shape changing including a “flower” architecture with actuating petals and a “mimosa” that could close its leaf on demand. These materials and methods thus also create a new platform for 4D printed structures.
2. Results and discussion
2.1 Materials design and preparation
To date, various polymeric systems such as poly(N,N-(dimethylacrylamide)-co-methacrylic acid),39,44 2-ureido-4-pyrimidone (UPy) based polymers,38 poly(N-acryloyl glycinamide),45 poly(N-acryloylsemicarbazide),46 poly(ether-thioureas)47 and poly(vinyl alcohol) cross-linked by small organic molecules48 have been reported to possess unique combinations of high strength, great toughness and large extensibility due to the presence of multiple hydrogen bonds. While in this work the multiple acid–ether hydrogen bond was utilized for synthesizing H-bond cross-linked polymers because of its fast dynamics,33,34,49 ease of synthesis and strong humidity sensitivity of oligo(ethylene glycol) units.8,33,34,50–52 The incorporation of soft oligo(ethylene glycol) methacrylate (OEGMA) units into hard methacrylic acid (MAA) units enables great flexibility in tailoring mechanical properties and stimulus-responsiveness over a wide range.33 We chose methyl ether terminated OEGMA as this does not have any dimethacrylate impurity. The content of MAA unit was controlled to be 75% mol in the copolymer chain which is much higher than that of OEGMA units (25% mol), for the purpose of efficient acid–ether hydrogen-bonded complexes formation.53 The poly(MAA-co-OEGMA) was synthesized through one-pot free radical polymerization (Fig. 1A), this composition was confirmed by 1H nuclear magnetic resonance (NMR) spectroscopy (Fig. S1A, ESI†), GPC results of this type of polymer showed inaccurate results due to aggregation and interactions on the column.54 Formation of hydrogen bonds between the self-associated COOH dimer, and between MAA and OEGMA were observed by FTIR spectroscopy (Fig. S1B, ESI†). The resultant H-bond network shows medium tensile stress (1.21 MPa), high stretchability (∼820%), high toughness (9.02 MJ m−3), and a Young's modulus of 14 MPa (Fig. 1B). Dynamic mechanical analysis (DMA) shows the consistently lower storage modulus (G′) than loss modulus (G′′) over the frequency range (0.5–100 Hz) (Fig. S2, ESI†), suggesting liquid like properties. Both G′ and G′′ showed strong frequency dependence across the frequency range, indicating the weak physical network structure which could be ascribed to the highly dynamic nature of hydrogen bonds.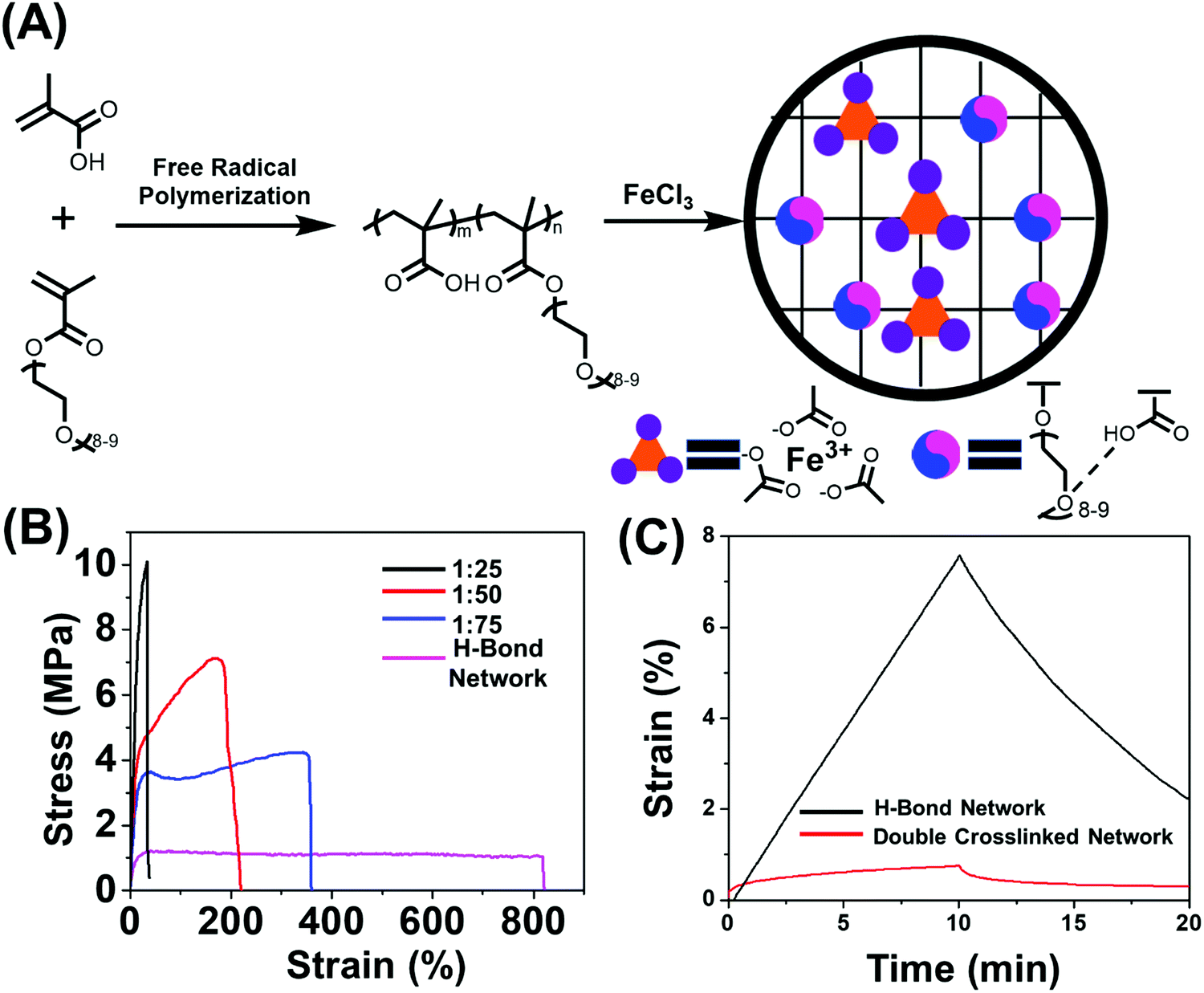 | ||
Fig. 1 Design and characterization of double cross-linked polymers. (A) Synthesis of copolymer, schematic representation of the molecular design and preparation of the doubly dynamic network. (B) Stress–strain curves, showing significant changes in mechanical properties with varying iron(III)-to-COOH molar ratios. (C) Creep and creep recovery behavior (load of 84 kPa) of double cross-linked (iron(III)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :COOH molar ratio of 1:75) and H-bonded networks, showing significant creep resistance in the iron(III) coordination system. :COOH molar ratio of 1:75) and H-bonded networks, showing significant creep resistance in the iron(III) coordination system. | ||
In order to further strengthen the network, stronger dynamic bonds should be incorporated as the secondary cross-linking sites. Metal-coordination bonds,55–57 are one kind of noncovalent interaction prevalent in biological systems, have been reported to substantially improve the mechanical properties of physically or chemically cross-linked polymers.41,43,58,59 These have also demonstrated fascinating properties such as shape memory41 and self-healing55 could be imparted through careful selection of metal ions. Another key feature of our macromolecular design is the presence of the COOH functional group in (poly(MAA-co-OEGMA)) which has the ability to form complexes with various metal ions such as Zn(II),60 Fe3+,41 Cu2+61 and Al3+.62 We selected the carboxyl-Fe3+ coordination complexes in this work due to its high coordination strength. Moreover, the commercially available ion chloride could be easily dissolved in water which is also a good solvent to dissolve poly(MAA-co-OEGMA). The ferric ions with different iron(III)-to-COOH molar ratios were directly added into aqueous solutions of poly(MAA-co-OEGMA) which were subsequently transferred to Teflon molds and dried at ambient conditions. The dried polymer films were immersed in a large amount of pure water for 1 day to remove excess Fe3+. However, it was observed that the films with low iron(III):COOH molar ratios of 1:100, 1:150 and 1:200 could be easily damaged after immersion in water for several hours due to low cross-linking densities. The samples with higher iron(III):COOH molar ratios of 1:75, 1:50, 1:25, 1:10 and 1:5 are swollen in water, indicating the successfully metal coordination cross-linking and were stable over a range of pH conditions (3–12, see Fig. S3, ESI†). The films were transparent suggesting the homogeneous nature of the resulting network. (Fig. S3, ESI†).
2.2 Mechanical properties
We further investigated the effect of metal coordination cross-linking on mechanical properties of the networks. Unfortunately, the sample with iron(III):COOH molar ratios of 1:10 and 1:05 were too brittle to be suitable for tensile testing because their ion cross-linking densities were too high. They were therefore excluded from further mechanical tests. Increasing the iron(III):COOH molar ratios from 0 to 1:25 caused a considerable increase in the tensile strength increased from 1.21 MPa to 10.2 MPa (Fig. 1B and Fig. S4A, ESI†). The corresponding Young's modulus also drastically increased from to 14 MPa to 58 MPa (Fig. S4B, ESI†). The increase in mechanical strength indicated that metal coordination bond effectively prohibits the chain movement by serving as cross-links to anchor the structure. Although the tensile strains were compromised after cross-linking with metal coordination bonds, the polymers with iron(III):COOH molar ratio of 1:50 and 1:75 still exhibit high stretchability with 355% and 183% respectively (Fig. S4C, ESI†). Meanwhile, the toughness of the polymers increased with increasing iron(III):COOH molar ratio from 0 to 1:25, while it decreased at higher iron(III) to COOH molar ratios (Fig. S4D, ESI†). It is likely that the double cross-linked polymer may be plasticized by moisture in the air, as confirmed by the presence of broad peak at 3400 cm−1 from interaction with water in Fig. S1B (ESI†). This may slightly affect the mechanical properties, however, the materials still have excellent mechanical properties.
Furthermore, the frequency sweep at room temperature revealed that the storage modulus (G′) is consistently higher than the loss modulus (G′′) over the frequency range for all of the double cross-linked samples, which is in contrast to the poly(MAA-co-OEGMA) with G′′ being higher than G′ (Fig. S2, ESI†). This result indicates that the metal coordination bonds play an indispensable role in stabilizing the networks (Fig. S5, ESI†) thus contributing to the enhanced mechanical properties.63 Higher iron(III):COOH molar ratios led to significantly increased storage modulus (G′). The formed network with iron(III):COOH molar ratio of 1:75 boasted the highest toughness and made it a prime candidate, and we use this throughout the rest of the study unless otherwise specified.
Creep is another important aspect of mechanical properties to be considered for polymeric actuators in real applications,42 as the rearrangement of weak physical cross-links may lead to undesirable creep during actuation that could permanently alter the material dimensions.64 We thus conducted creep-recovery experiments for both double cross-linked and H-bonded network samples. It was found that the double cross-linked polymer exhibited long-term network stability and that only a 0.7% strain was induced for the double cross-linked polymer after a loading applying a load for 10 min (Fig. 1C), while the hydrogen bonded network crept as high as 87% more than the ion cross-linked in under the same conditions. Such improved resistance to creep could be attributed to the restricted rearrangement of the polymer chain under external force as a result of strong coordination structures.42 Furthermore, the double cross-linked polymer recovered strain to a much greater extent during the creep recovery process. In fact, its recovery was found to be 88% more higher than that of the hydrogen bonded network at room temperature, indicating a much less permanent deformation. The double cross-linked sample also displayed excellent elasticity as shown by the complete and rapid recovery of the deformation after stretching to two times its initial length (Fig. S6A, ESI†), which is distinct from its counterpart H-bonded network undergoing obvious plastic and unrecoverable deformation (Fig. S6B, ESI†) after being mechanically stretched. It is because the hydrogen bond could be easily broken by mechanical stretching, resulting in rearrangement of network topology.65 The ionic cross-links, as “permanent” cross-linkers, hindered the rearrangement of the network to by dramatically restricting segment mobility, which improves the elasticity and protects the integrity of network.
2.3 Humidity responsiveness
To elucidate the internal changes at the molecular level of the doubly cross-linked polymer in response to water, one side of the polymer film was exposed to moisture from a humidifier, and then examined by Fourier-transform infrared spectroscopy (FTIR). The broadening and red-shifting of the absorption band at ≈3500 cm−1, ascribed to OH stretching vibrations, as well as the new strong peak at 1640 cm−1 due to stretching vibrations of the hydrogen bond between ester carbonyl groups and water (Fig. S7A and B, ESI†) suggesting the effective water adsorption of the polymer film. The shift of the carbonyl stretching bands shift to lower wavenumbers by ≈10 cm−1 after water diffuses diffusion into the film (Fig. S7A and B, ESI†) suggests the hydration of ester linkages. When this doubly cross-linked polymer was left in the dry conditions, the signals of these new peaks were found to gradually attenuate and eventually disappeared, suggesting the desorption of water (Fig. S7C and D, ESI†). Continuous absorption of water leads to the weight change of the polymer film (Fig. S8, ESI†), which could absorb water molecules up to 84% of the polymer film's weight from exposed humidity in 180 s and restored to the initial weight in a slightly slower rate after desorbing water in the dry environment.Exposing one side of a polymer film with the lower part being clamped between two glass slides generates rapid bending, with a bending angle of 91° in only 4 s (Fig. 2A and Movie 1, ESI†). The bending angle increased with an increase in exposure time (Fig. S9A, ESI†). To understand this humidity-induced bending mechanism, the FTIR spectra of exposed and unexposed surfaces were studied. The different chemical compositions were observed and only the exposed surface underwent the spectral changes induced by humidity (Fig. S9B, ESI†). This result suggests that only a thin surface layer absorbs a significant amount of water weakening the hydrogen bond network and thus creating a “more swellable” layer (Fig. 2B). The opposite face of the film constitutes a “less swellable” layer. Such mismatch in extent of swelling leads to the gradient expansion and subsequent exclusive bending to the back side. This bending deformation gradually recovered to the original unbending state within 30 s after the removal of water vapor (Fig. 2A and Fig. S9A, Movie 2, ESI†). The vibrating of the film was observed during the recovery process (Movie 2, ESI†), which could be attributed to the counteractive interaction between swelling force caused by absorbed moisture in the thin surface layer which gets weaker during drying, and the good elasticity provided by metal coordination bond which tend to unbend the film. The bending and unbending processes of the double cross-linked polymer were fully reversible and could be repeated at least five times upon alternating exposure to moisture and air without obvious fatigue (Fig. S9C, ESI†). The desorption of water in low humidity conditions was generally reported to be responsible for unbending behavior.2,6,19 However, in this work, the absorbed water in polymer film was still observed even after being left in dry ambient conditions for 3 min even with smaller volumes of water, demonstrated by the presence of peak at 1640 cm−1 and 3500 cm−1 as shown in Fig. S7C and D (ESI†). In order to understand the mechanism of the reversible bending behavior, the humidity responsiveness of the H-bonded cross-linked polymer was studied and found to remain in its unbent state even after switching to a dry environment for several hours (Fig. S10, ESI†). Therefore, we assumed that the carboxyl-Fe3+ coordination bond played an important role in such reversible actuation performance.
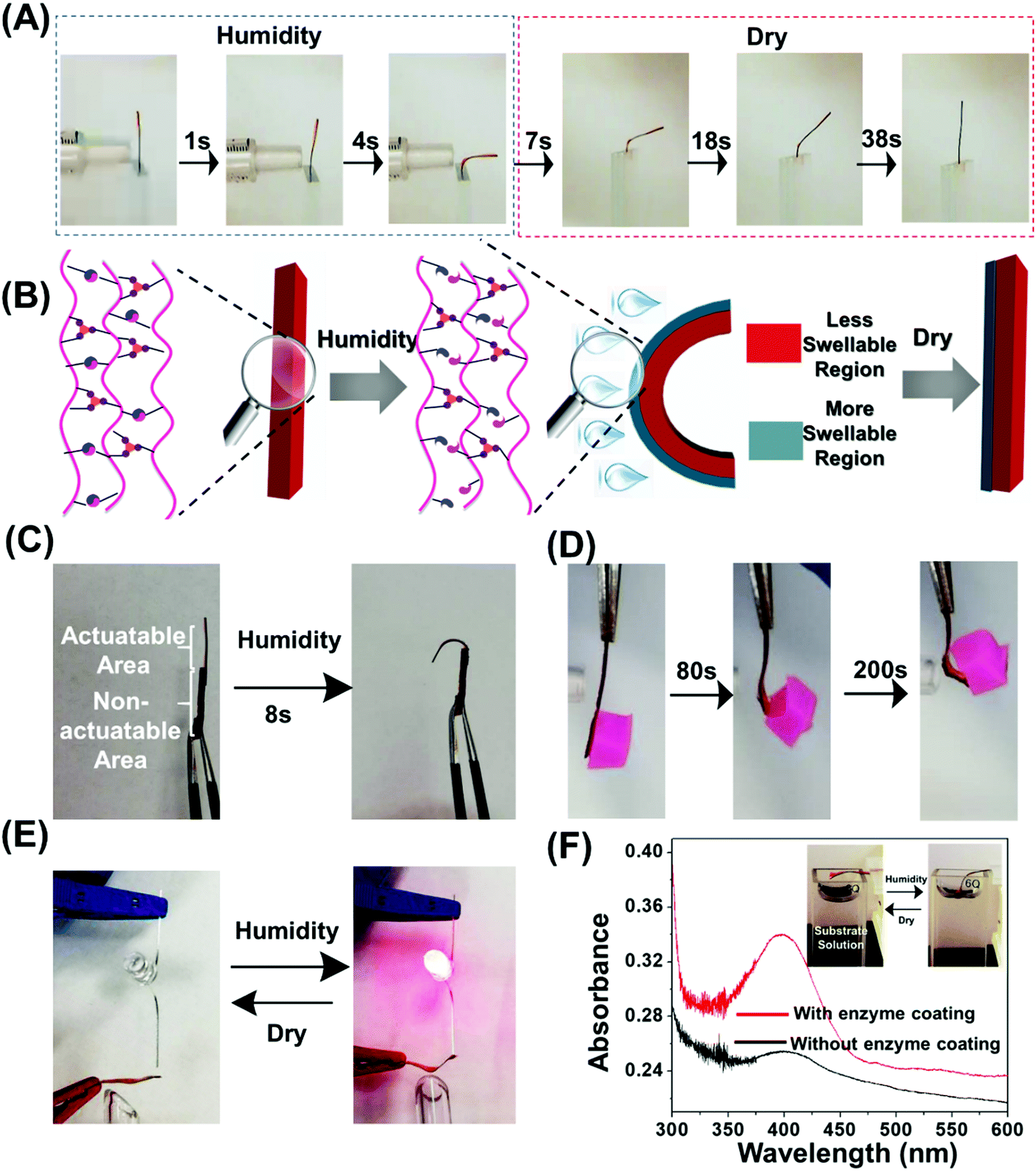 | ||
| Fig. 2 Humidity responsiveness of double cross-linked polymer. (A) Photographs showing the fast bending of double cross-linked polymer (thickness = 50 μm). Exposing film to humidity (RH change from 22% to 95%) causes the polymer to bend away from the humidity source. (B) Illustration showing the formation of the double cross-linked hydrogel with a gradient in the swelling ratio upon humidity, and stable and robust ion coordination cross-linkers could provide excellent elasticity and rigidity, contributing to the recovery of the bent film in dry condition (C) multi-material actuator generated from merging the double cross-linked polymer with different thickness in which only the thin part could be actuated upon humidity. (D) Optical images of double cross-linked polymer used as artificial muscles to lift a loaded silicone of twenty-fold weight. (E and F) Application of the double cross-linked polymer as smart electrical (E and F) catalysis devices. (E) Photographs of the polymer used as an electronical device which was assembled by a polymer film coated with conductive materials and a LED light. The left edge of the film was covered with silver ink. (F) Absorption spectra of the buffer solutions upon contacting polymers films with and without enzyme coating. Inset: Photographs of the polymer used to induce the enzyme catalyzed reaction. The left edge of the film was covered with enzyme. The humidity drove the film to bend and small part of it was then immersed into the buffer solution. | ||
Stress relaxation experiments were further carried out to test the rationality of our hypothesis. The wet H-bond cross-linked polymers were able to almost fully relax the exerted stress (Fig. S11, ESI†). After a 15 min relaxation, less than 10% of the original stress remained for the wet H-bonded cross-linked polymer. This could be ascribed to the formation of the hydrogen bonds between water and both the MAA and OEGMA units, resulting in the substantial reduction of intermolecular hydrogen bonds between the polymer chains and physical crosslinking density. In contrast, the wet doubly cross-linked polymer exhibited much slower and restricted stress relaxation behavior, with a much larger portion of the stress being retained after a similar time, indicating much better humidity-resistance of its coordination bond network than its hydrogen bond counterpart. Qu et al. observed the slightly hydration-induced dissociation of the carboxyl-Fe3+ coordination bond in the dynamic cross-linked network,43 but they pointed out that this coordination bond could be stabilized by the shielding effect of the hydrophobic units, and that this effect gets stronger with increasing iron cross-linking densities. Therefore, in this study, we interpret such enhanced moisture-stability of the coordination bond network to the presence of the hydrophobic methyl group in the MAA unit and high-density coordination cross-linking. Combined with the results in Fig. S6 (ESI†), we conclude that the stable and robust ion coordination cross-linkers in both “more swellable” and “less swellable” regions provide excellent elasticity and rigidity (Fig. 2B), contributing to the recovery of the bent film in dry conditions.
We further observed that increasing the film thickness could slow the bending speed and reduce the bending angle (Fig. S12A, ESI†). The bending deformation is essentially non-existent when the film with thickness was 500 μm even after being exposed to moisture for 2 min (Fig. S12B, ESI†). These results suggest that the humidity-driven actuation behaviors could be readily tunable by controlling the film thickness, this creating a unique method to control the actuation of objects made from the same material. We will harness this phenomenon for further intricate shape programming, which will be discussed in the following section.
Furthermore, we studied the self-healing properties of our double cross-linked polymer which could enable material wielding for sophisticated functions. However, self-healing of the dry sample could hardly occur in mild conditions due to the very high glass transition temperature (92 °C) which prohibited chain mobility (Fig. S13A, ESI†). This result also indicates that the double cross-linked polymer is in a glassy state at room temperature, which is consistent with the DMA result in Fig. S5 (ESI†) showing storage modulus on the order of 1 GPa. While the Young's modulus was measured to be ∼30 MPa, similar observation has also been reported for other glassy supramolecular polymers with Young's modulus of 35 MPa.66
The chain mobility could be promoted in wet conditions as demonstrated by the frequency sweep showing a crossover where G′ = G′′ at frequency of 2.8 Hz (Fig. S13B, ESI†). The dynamic bond lifetime was calculated to be 0.5 s using the reciprocal of the crossover angular frequency indicating favorable network mobility for self-healing. As shown in Fig. S14A (ESI†), the wet sample exhibits self-healing properties confirmed by optical microscopy images which attest to the gradual healing and disappearance of a scratch over time. The healed polymer sustained a large strain and held a weight that was 526 times greater than the film weight (Fig. S14B, ESI†), 68% recovery of the original fracture stress was observed after 8 h revealed by tensile testing (Fig. S14C, ESI†). The good self-healing efficiency under water might be attributed to the presence of metal coordination bonds at the interfaces.67 This self-healing ability offers the opportunity to achieve unique 3D geometries by integrating different components with distinct actuation performance into single object without other additives such as tapes and glues. As a simple demonstration, we molded two polymer films with thickness of 130 μm and 800 μm into one continuous rectangular sample. A hook like shape could be afforded when exposed to humidity by actuating only the thinnest segment (Fig. 2C).
Taking advantage of the excellent mechanical properties as well as fast and reversible shape changing capability, this doubly cross-linked polymeric system could serve as a smart device in several potential applications. We first studied whether the double cross-linked polymer could be used as an artificial muscle to lift weight in response to humidity. We used a film with thickness of 110 μm to perform the task which showed both fast and large deformation and good mechanical properties (Fig. 2D). Firstly, the polymer film was fixed using tweezers on one side with the other side hanging with an object (423 mg) with 20-fold the weight of the polymer film attached. Upon humidity exposure, the bottom of polymer film started to bend like an arm. The object was lifted up to a height of 2.0 cm within 200 s. The produced mechanical work (W) which is determined by W = mgx (m: mass of the weight; g: acceleration of gravity; x: length of the weight that is lifted), was 8.3 μJ. We further demonstrated an electrical device assembled by the polymer film covered with silver glue and a light-emitting diode (LED) light with red color. By controlling the humidity and hence actuation of a conductive network, we could reversibly open and close an electric circuit and thus power a LED light reversibly depending on the humidity (Fig. 2E). Moreover, the reversible, fast actuation could also be utilized to switch an enzyme catalyzed reaction (Fig. 2F). An enzyme (α-chymotrypsin) was physically immobilized on the top and left edge of the film. To demonstrate the efficacy of the actuation induced on–off switch of the enzyme, we monitored the hydrolysis of 4-nitrophenyl benzoate to 4-nitrophenol. The humidity drove the bending of the film, resulting in a small part of the network to be immersed into the substrate solution. The unbending of the film leads the immersed part to move out of the substrate solution within 15 min after removing moisture. The UV-vis spectroscopy was used to follow the catalytic reaction. Interestingly, the absorbance at 400 nm, attributed to 4-nitrophenolate ion increased significantly after the substrate solution was contacted with enzyme coated film during shape changing process (Fig. S15, ESI†). However, in contrast, the buffer solution still had low absorbance at 400 nm after being contacted with the polymer film without enzyme immobilization. These results indicate that we can turn an immobilized enzyme “on” and “off” by the reversible humidity-driven bending-unbending process driven by actuation into a solution with substrate.
2.4 3D/4D printing
In addition to having excellent mechanical properties and fast, reversible actuation, the ability for this humidity responsive polymer to be 3D printed opens further opportunities in advanced applications. Most of the previously reported humidity responsive systems contained covalent bonds5–8 which could prevent plastic deformation during the shape changing process. However, it may result in compromised polymer processabilities. In this work, engineering humidity responsive polymers with noncovalent bonds is expected to solve this issue. DIW, an extensively used 3D printing technology was utilized for manufacturing due to its flexibility of both hardware and software combined with inexpensive nozzles.40,68,69 The poly(MAA-co-OEGMA) concentrated aqueous solution (20 wt%) lacked the ability to retain structural integrity after extrusion due to its sol state (Fig. S16A, ESI†). It indicates that the multiple acid–ether hydrogen bond alone could not enable the stable hydrogel network formation in aqueous conditions.The addition of iron chloride induces the gelation of poly(MAA-co-OEGMA) solution and produces a stable homogenous hydrogel showing obvious viscoelastic properties (Fig. S16B, ESI†). The much lower modulus of the hydrogel compared to that of dry polymer network (Fig. S5, ESI†) could be ascribed to the presence of water which significantly reduces the intermolecular hydrogen bonding and the cross-linking densities. The produced hydrogel and high viscosity of 238 Pa s at low shear rate of 0.01 s−1 (Fig. 3A) which perfectly falls into an empirical ink viscosity range (0.1–1000 Pa s) reported as suitable for DIW.35 The viscosity of this hydrogel reduced rapidly with increased shear rate and dropped as low as 0.055 Pa s at a shear rate of 100 s−1. Such a reduction in viscosity facilitates flow through a syringe needle. A robust, stable and continuous hydrogel filament was formed without any fragmentation or forming of droplets when it came out of the nozzle (Fig. S16C, ESI†). Our previous study suggested that the intermolecular hydrogen bonding still presents even in the aqueous condition33 due to the stabilization effect of hydrophobic methyl motifs.39 David et al. observed the presence of a particular association of PEGMA and MAA in the water medium through light scattering measurements.53 Therefore, such shear thinning behavior could be ascribed to the multiple acid–ether hydrogen bonds which could display shear dependent behavior.70 Furthermore, higher iron(III)-to-COOH molar ratios of 1:05 and 1:10 were found to result in stiff hydrogels which clogged the nozzle (Fig. S17, ESI†). While hydrogels with iron(III)-to-COOH molar ratios of 1:50 and 1:75 could not be fabricated or were too soft and spreadable lacking sufficient self-support. (Fig. S17, ESI†).
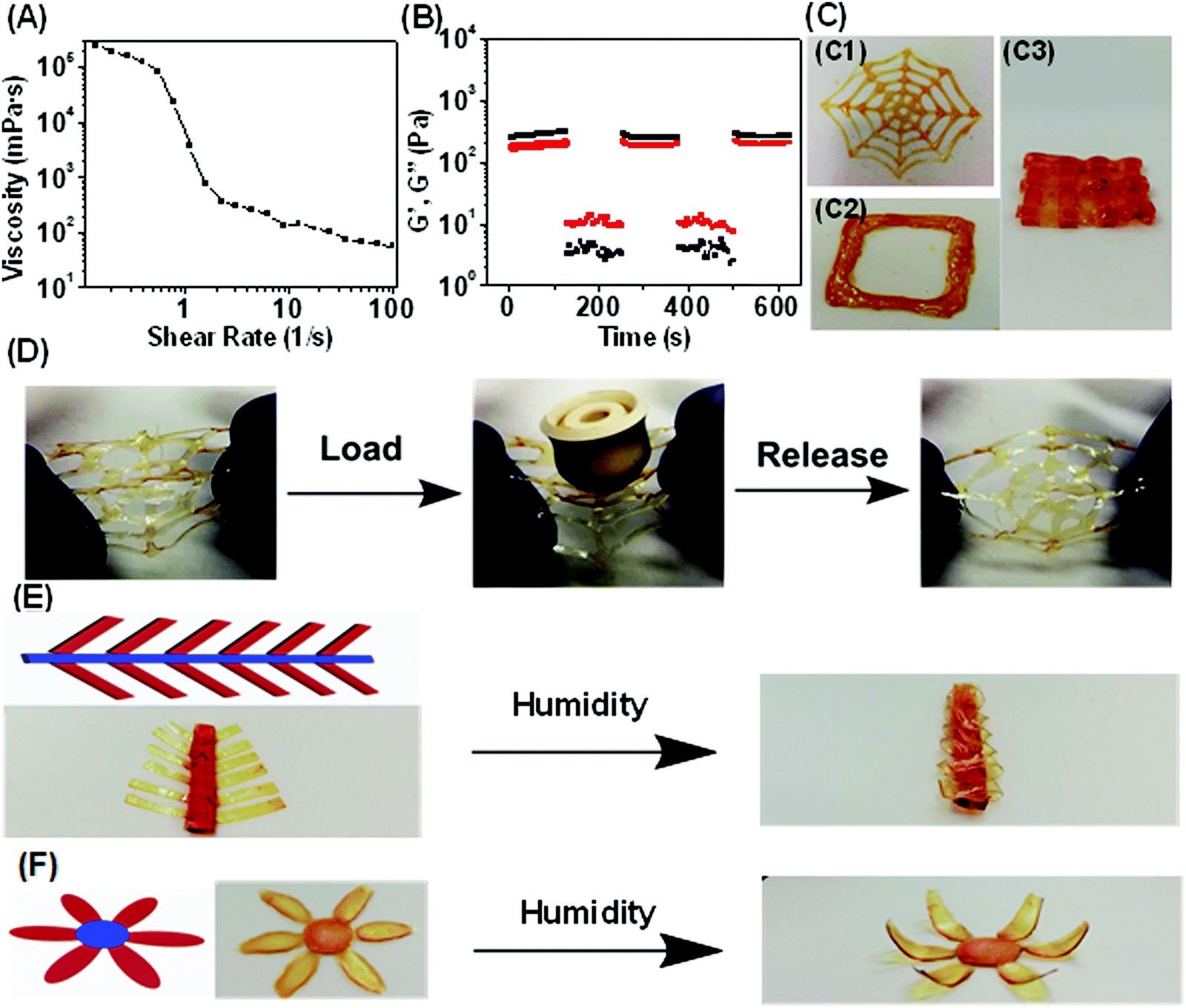 | ||
| Fig. 3 3D/4D printing of double cross-linked polymers. (A) Viscosity versus shear rate profile of hydrogel showing shear-thinning properties. (B) Reversible gel–sol transition and self-healing of hydrogels upon cyclic shearing at 1% and 600% strains. (C) Photographs of printed spider web with one printed layer (C1), square with five printed stacking layers (C2) (C3) stacking layer–layer mesh structures. (D) A printed spider web (average thickness = 30 μm) could sustain a heavy object with 60-fold of its own weight without breaking. (E) The printed mimosa where the thickness of the “leave” was controlled to be 80 μm while the “stem” was 600 μm. Eight and two stacking layers of inks printed on the thick and thin areas, respectively. (F) Printed flower where the thickness of petal and sepal was 110 μm and 640 μm, respectively. Nine and three layers of inks printed on the thick and thin areas, respectively. Blue represents the printed thick areas that are not actuatable while red represents the thin actuatable areas. | ||
In addition to shear thinning, a second important criterion to assess a polymers’ printability is the ability to retain structural fidelity after extrusion from the printhead. Oscillatory stress sweep testing suggested that G′ is maintained higher than G′′ up to crossover points of 521 Pa (Fig. S18, ESI†), suggesting that the hydrogel could bear a large deformation under external stress due to the existence of strong metal coordination bonds. Such high yield strength could be important for the printed structures to withstand their own weight against gravity without collapsing.71 While the G′′ is higher than G′ for higher shear rates the polymer shows a shear-induced solid–liquid transition.
To investigate the reversibility of such solid–liquid transition upon changing shear strains, continuous step strain measurements were further performed (Fig. 3B). The ink was subjected to three cycles of low (1%) and high (600%) strains. At low strain, the hydrogel was solid state indicated by G′ > G′′, while at high strain it turned into a viscous liquid indicated by G′ < G′′. This solid–liquid transformation of the polymeric ink was highly repeatable and could be attributed to the reversible shear stress-induced dissociation of hydrogen bonds,70 as well as the presence of strong metal coordination bond which facilitates the recovery to solid-like state after the shear force is removed. Enabled by suitable rheological properties, 3D printing of the polymeric ink can offer a promising route for facile fabrication of arbitrary customer-defined structures and devices. The simple preparation of the ink could enable the industrial scale up which is especially desirable for extrusion 3D printing technologies. The spider web, as well as layer-by-layer square and mesh structures could be successfully printed in high fidelity (Fig. 3C and Movie 3, ESI†). Notably, the printed spider web could resist the external force mimicking its natural counterparts demonstrated by the ability to withstand large load with 60 times of its own weight without damage (Fig. 3D).
Combining shape programmable polymers with 3D printing techniques resulted in 4D printing process,26,27 where static 3D printed objects evolve their configurations or functions with time in response to external stimuli. However, the majority of reported 4D printed objects evolved into complex biomimetic structures in which different printed parts with well-controlled size, shape, and organization of features can perform different functions.26,27 Whole areas of printed devices would generally be actuated in the presence of an external stimulus even though with spatially distinct actuation behaviors. Taking advantage of the thickness controllable humidity-driven actuation behaviors demonstrated above (Fig. S12, ESI†), our material system could enable a new approach of programming 4D printed shape transformations by allowing only certain regions of printed structures to be actuatable. We demonstrate this concept by printing a series of different shapes with controlled thickness in regions. We printed a “mimosa” with well controlled geometries in which the thickness of the “leaf” was controlled to be 80 μm such that they were actuatable regions while the non-actuable “stem” was made to be 600 μm. The flower geometry was constructed using similar process with only “petals” being actuatable. As shown in Fig. 3E and F, such a rationally designed 4D printed flower can exhibit motions that mimick petal closure. Additionally, the gradual closing of the leaves of the Mimosa was achieved from the gradual absorption of water vapor.
3. Conclusion
In summary, we presented a simple and innovative approach for engineering the properties of humidity responsive polymers, by introducing metal coordination complexes into hydrogen bond cross-linked network to form secondary cross-linking network. The hydrogen bonding was utilized to provide high humidity sensitivity and shear-dependent rheological behavior. The fascinating characteristics of the metal coordination bond brings several rather notable benefits. It enables the simultaneous improvement of diverse and independent material properties: the resultant doubly cross-linked polymer is orders of magnitude stiffer, stronger, and tougher than its iron-free counterpart due to considerably higher bonding energies than hydrogen bonding. Its intrinsic high humidity stability also enables the reversible humidity-driven actuations. Diverse smart devices including artificial muscle, smart electronic and immobilized catalytic devices were fabricated using the resultant doubly cross-linked polymer. The metal–ligand coordination bond could also modify the rheological properties by producing the hydrogels with excellent shear thinning and highly reversible shear-sensitive solid–liquid transition, which made them ideal inks for DIW. As a result, various sophisticated patterns and architectures were manufactured showing 4D printing process mimicking biomimetic movements of living organisms in nature. The polymer design concept proposed here is simple and should be applicable to other humidity-responsive polymeric systems toward robust mechanical properties and versatile functionalities. The unique combination of high mechanical properties and excellent 3D printability, in addition to fast and reversible shape changing ability, open avenues to smart devices such as soft robots, microfluid and biomedical devices. This study not only offers a strategy for efficient fabrication of humidity responsive polymeric actuators with superior performance which could play a prominent role in soft robots and artificial muscle applications, but also drastically expand the scale and scope of stimulus-responsive polymeric inks developed for 4D printing technologies.4. Experimental section
4.1 Materials
All chemicals were obtained from Sigma Aldrich and used as received unless otherwise specified. Deionized water was produced by an ELGA Laboratory water station and had a resistivity of 18.2 m cm−1. To remove inhibitors, oligoethyleneglycol methyl ether methacrylate [500 g mol−1,OEGMA] and methacrylic acid were passed through a basic alumina column immediately before use. (1,1′-AzobisIJcyclohexanecarbonitrile) (ACHN, Sigma-Aldrich, 98%) was recrystallized from ethanol and dried overnight prior to use.4.2 Synthesis of poly(MAA-co-OEGMA)
The procedure for preparation of well-defined poly(MAA-co-OEGMA) copolymers via free-radical polymerization is illustrated in Fig. 1A. As a representative example, OEGMA 500 (20 mL), MAA (10.98 mL), ACHN (56 mg) were dissolved in anhydrous DMSO (60.0 mL). The flask was sealed with a rubber septum and the solution was deoxygenated by sparging with nitrogen for 10 min. After polymerizing for 24 h at 65 °C, the reaction was cooled to 0 °C in an ice batch and exposed to air. The viscous polymer solution was diluted with DMSO and precipitated into diethyl ether three times.4.3 Preparation of double cross-linked polymer and hydrogel inks
Appropriate amounts of FeCl3 was dissolved into pure water to form an aqueous solution with concentration fixed at 2 mol L−1. Aqueous solution of poly(MAA-co-OEGMA) (5 wt%) and certain volume of Fe(III) aqueous solution (5–100 μL) were mixed in a small vial and stirred for 5 min. Then the solutions were poured into a PTFE mold and placed at room temperature for 24 h to dry and form double cross-linked polymer. At last, the samples were immersed in deionized water for 48 h to remove superfluous Fe3+. The similar procedure was used for preparing hydrogel inks follow except with higher poly(MAA-co-OEGMA) aqueous concentration (20 wt%).4.4 Catalyzed reduction of 4-nitrophenyl benzoate
4-Nitrophenyl benzoate solution (50 μL, 0.002 M) was mixed with aqueous buffer solution (16.0 mL, 0.06 M) and deionized water (3 mL). UV-Vis spectroscopy was used to follow the catalytic activity.4.5 3D printing
The ZMorph Multitool 3D Printer was used for direct ink writing experiment with thick paste extruder. The viscous polymer solutions were extruded out of nozzles onto a silicon substrate at room temperature and were immediately transferred into water for 8 h. Printing parameters: ambient temperature printing with 0.3 mm nozzle diameter, print head speed: 7.0 mm s−1.4.6 Material characterizations
1H NMR (500 MHz) spectra was recorded using a Bruker Avance BB 500. Chemical shifts were recorded in ppm (δ). Infrared spectra of various hydrogels were acquired using a Nicolet 5700 Fourier transform infrared (FTIR) spectrometer fitted with an attenuated total reflectance (ATR) accessory that used a diamond internal reflection element (IRE) and the angle of incidence of the beam was fixed at 45° relative to the IRE. Mechanical properties of the hydrogels were measured by tensile tests using a commercial tensile tester. The samples were cut from the polymer sheets into a rectangular shape with length of 12 mm, width of 2 mm and thickness of 0.17 mm. The stretching rate is 60 mm min−1. The nominal stress–strain curves were recorded, and the Young's modulus was calculated from the initial slope of the curve with a strain below 8%. Dynamic mechanical analysis (DMA) was performed to the hydrogels using a DMA 242E (NETZSCH). A rectangular sample (dimensions: 22.5 mm × 11.7 mm × 1 mm) was cut from the bulk polymer. The test was run in a tension mode with a frequency of 1 Hz when the temperature increased from 25 to 100 °C (heating rate: 2 °C min−1). Frequency sweeps were performed in the range of 0.5–100 Hz. The normalized stress-relaxation experiments were performed in a strain control (10% strain) mode. Rheological behaviors of the hydrogels were investigated using an rheometer (MCR 702). The Fe3+ crosslinked hydrogels were directly and evenly introduced to the plate. Amplitude sweeps, frequency sweeps, shear rate sweeps were performed at 25 °C. The amplitude sweep was carried out on hydrogels at 10 rad s−1 in a strain range from 0.1% to 1000%. The dynamic strain step tests were carried out by using 600% and 1% strain for 150 s, respectively for three cycles. Shear rate sweeps were carried out sequentially to measure viscosity. Shear rates from 0.01 to 100 s−1 were tested. Frequency sweeps were performed from 0.5 to 100 rad s−1 by using 1% strain. Shape changing properties of polymers were recorded using a digital camera in a cell phone. The water moisture source was a commercial humidifier.Author contributions
Z. J. and L. A. C. conceived the concept. Z. J. designed and performed the experiments, analyzed the data and wrote the manuscript with input from all authors. P. S. helped with materials preparation process and the preparation of Scheme and Figures. M. T. helped with 3D printing experiments. Q. Y. helped with the preparation of enzyme and substrate solutions. J. V. helped with the preparation of smart electrical devices. C. C helped with polymer synthesis and correction of language. X. P. and P. X. helped with rheological tests. L. A. C. supervised and funded the project.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Funding is gratefully acknowledged from the Australian Research Council (DP180103918), and the ANU Futures Scheme. The authors thank Associate Professor Zbigniew Stachurski for assistance with tensile testing.References
- C. Dawson, J. F. Vincent and A.-M. Rocca, Nature, 1997, 390, 668 CrossRef CAS.
- M. Ma, L. Guo, D. G. Anderson and R. Langer, Science, 2013, 339, 186–189 CrossRef CAS PubMed.
- R. M. Erb, J. S. Sander, R. Grisch and A. R. Studart, Nat. Commun., 2013, 4, 1712 CrossRef PubMed.
- Y. Kuang, C. Chen, J. Cheng, G. Pastel, T. Li, J. Song, F. Jiang, Y. Li, Y. Zhang, S.-H. Jang, G. Chen, T. Li and L. Hu, Extreme Mech. Lett., 2019, 29, 100463 CrossRef.
- L. T. de Haan, J. M. Verjans, D. J. Broer, C. W. Bastiaansen and A. P. Schenning, J. Am. Chem. Soc., 2014, 136, 10585–10588 CrossRef CAS PubMed.
- Y. Liu, B. Xu, S. Sun, J. Wei, L. Wu and Y. Yu, Adv. Mater., 2017, 29, 1604792 CrossRef PubMed.
- R. Lan, J. Sun, C. Shen, R. Huang, L. Zhang and H. Yang, Adv. Funct. Mater., 2019, 29, 1900013 CrossRef.
- C. Lv, H. Xia, Q. Shi, G. Wang, Y.-S. Wang, Q.-D. Chen, Y.-L. Zhang, L.-Q. Liu and H.-B. Sun, Adv. Mater. Interfaces, 2017, 4, 1601002 CrossRef.
- L. Zhang, S. Chizhik, Y. Wen and P. Naumov, Adv. Funct. Mater., 2016, 26, 1040–1053 CrossRef CAS.
- X. Qiu, S. Liang, X. Huang and L. Zhang, Chem. Commun., 2019, 55, 15049–15052 RSC.
- Y. Yang, Y. Wu, C. Li, X. Yang and W. Chen, Adv. Intell., 2019, 2, 1900077 CrossRef.
- G. M. Spinks, Adv. Mater., 2019, e1904093, DOI:10.1002/adma.201904093.
- S. Aziz and G. M. Spinks, Mater. Horiz., 2020, 7, 667–693 RSC.
- F. Muralter, F. Greco and A. M. Coclite, ACS Appl. Polym. Mater., 2020, 2, 1160–1168 CrossRef CAS PubMed.
- S. Taccola, F. Greco, E. Sinibaldi, A. Mondini, B. Mazzolai and V. Mattoli, Adv. Mater., 2015, 27, 1668–1675 CrossRef CAS PubMed.
- J. Cao, C. Zhou, G. Su, X. Zhang, T. Zhou, Z. Zhou and Y. Yang, Adv. Mater., 2019, 31, e1900042 CrossRef PubMed.
- D. Lei, Y. Yang, Z. Liu, S. Chen, B. Song, A. Shen, B. Yang, S. Li, Z. Yuan, Q. Qi, L. Sun, Y. Guo, H. Zuo, S. Huang, Q. Yang, X. Mo, C. He, B. Zhu, E. M. Jeffries, F.-L. Qing, X. Ye, Q. Zhao and Z. You, Mater. Horiz., 2019, 6, 394–404 RSC.
- Q. Chen, X. Yan, H. Lu, N. Zhang and M. Ma, ACS Appl. Mater. Interfaces, 2019, 11, 20473–20481 CrossRef CAS PubMed.
- L. Zhang and P. Naumov, Angew. Chem., Int. Ed., 2015, 54, 8642–8647 CrossRef CAS PubMed.
- Z. Zhao, Y. Hwang, Y. Yang, T. Fan, J. Song, S. Suresh and N. J. Cho, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 8711–8718 CrossRef CAS PubMed.
- L. Zhang, P. Naumov, X. Du, Z. Hu and J. Wang, Adv. Mater., 2017, 29, 1702231 CrossRef PubMed.
- Y. Tu, J. Yuan, D. Lei, H. Tan, J. Wei, W. Huang and L. Zhang, J. Mater. Chem. A, 2018, 6, 8238–8243 RSC.
- L. Zhang, X. Qiu, Y. Yuan and T. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 41599–41606 CrossRef CAS PubMed.
- Y. Dong, J. Wang, X. Guo, S. Yang, M. O. Ozen, P. Chen, X. Liu, W. Du, F. Xiao, U. Demirci and B. F. Liu, Nat. Commun., 2019, 10, 4087 CrossRef PubMed.
- G. Cai, J.-H. Ciou, Y. Liu, Y. Jiang and P. S. Lee, Sci. Adv., 2019, 5, eaaw7956 CrossRef CAS PubMed.
- X. Kuang, D. J. Roach, J. Wu, C. M. Hamel, Z. Ding, T. Wang, M. L. Dunn and H. J. Qi, Adv. Funct. Mater., 2019, 29, 1805290 CrossRef.
- A. S. Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413–418 CrossRef PubMed.
- S. Y. Zheng, Y. Shen, F. Zhu, J. Yin, J. Qian, J. Fu, Z. L. Wu and Q. Zheng, Adv. Funct. Mater., 2018, 28, 1803366 CrossRef.
- Y. Kim, H. Yuk, R. Zhao, S. A. Chester and X. Zhao, Nature, 2018, 558, 274–279 CrossRef CAS PubMed.
- Y. Tan, D. Wang, H. Xu, Y. Yang, X. L. Wang, F. Tian, P. Xu, W. An, X. Zhao and S. Xu, ACS Appl. Mater. Interfaces, 2018, 10, 40125–40131 CrossRef CAS PubMed.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS PubMed.
- Z. Jiang, A. Bhaskaran, H. M. Aitken, I. C. Shackleford and L. A. Connal, Macromol. Rapid Commun., 2019, 1900038 CrossRef PubMed.
- Z. Jiang, B. Diggle, I. C. Shackleford and L. A. Connal, Adv. Mater., 2019, 31, 1904956 CrossRef CAS PubMed.
- Z. Jiang, M. L. Tan, M. Taheri, Q. Yan, T. Tsuzuki, M. G. Gardiner, B. Diggle and L. A. Connal, Angew. Chem., Int. Ed., 2020, 132, 7115–7122 CrossRef.
- Z. Pei, Y. Yang, Q. Chen, E. M. Terentjev, Y. Wei and Y. Ji, Nat. Mater., 2014, 13, 36–41 CrossRef CAS PubMed.
- B. Jin, H. Song, R. Jiang, J. Song, Q. Zhao and T. Xie, Sci. Adv., 2018, 4, eaao3865 CrossRef PubMed.
- P. Song and H. Wang, Adv. Mater., 2019, e1901244, DOI:10.1002/adma.201901244.
- X. Yan, Z. Liu, Q. Zhang, J. Lopez, H. Wang, H. C. Wu, S. Niu, H. Yan, S. Wang, T. Lei, J. Li, D. Qi, P. Huang, J. Huang, Y. Zhang, Y. Wang, G. Li, J. B. Tok, X. Chen and Z. Bao, J. Am. Chem. Soc., 2018, 140, 5280–5289 CrossRef CAS PubMed.
- Y. J. Wang, X. N. Zhang, Y. Song, Y. Zhao, L. Chen, F. Su, L. Li, Z. L. Wu and Q. Zheng, Chem. Mater., 2019, 31, 1430–1440 CrossRef CAS.
- L. Li, Q. Lin, M. Tang, A. J. E. Duncan and C. Ke, Chem. – Asian J., 2019, 25, 10768–10781 CAS.
- S. Y. Zheng, H. Ding, J. Qian, J. Yin, Z. L. Wu, Y. Song and Q. Zheng, Macromolecules, 2016, 49, 9637–9646 CrossRef CAS.
- S. Wang, S. Ma, Q. Li, X. Xu, B. Wang, K. Huang, Y. Liu and J. Zhu, Macromolecules, 2020, 52, 2919–2931 CrossRef.
- Y. Deng, Q. Zhang, B. L. Feringa, H. Tian and D. H. Qu, Angew. Chem., Int. Ed., 2020, 132, 5316–5321 CrossRef.
- X. Hu, M. Vatankhah-Varnoosfaderani, J. Zhou, Q. Li and S. S. Sheiko, Adv. Mater., 2015, 27, 6899–6905 CrossRef CAS PubMed.
- X. Dai, Y. Zhang, L. Gao, T. Bai, W. Wang, Y. Cui and W. Liu, Adv. Mater., 2015, 27, 3566–3571 CrossRef CAS PubMed.
- C. Fan, B. Liu, Z. Xu, C. Cui, T. Wu, Y. Yang, D. Zhang, M. Xiao, Z. Zhang and W. Liu, Mater. Horiz., 2020, 7, 1160–1170 RSC.
- Y. Yanagisawa, Y. Nan, K. Okuro and T. Aida, Science, 2018, 359, 72–76 CrossRef CAS PubMed.
- P. Song, Z. Xu, Y. Lu and Q. Guo, Macromolecules, 2015, 48, 3957–3964 CrossRef CAS.
- P.-N. Song and J.-L. Hong, J. Mater. Chem. C, 2019, 7, 13161–13175 RSC.
- Z. Jiang, I. Blakey and A. K. Whittaker, Poly. Chem., 2017, 8, 4114–4123 RSC.
- Z. Jiang, H.-H. Cheng, I. Blakey and A. K. Whittaker, Mol. Syst. Des. Eng., 2018, 3, 627–635 RSC.
- Z. Jiang, R. J. P. Sanchez, I. Blakey and A. K. Whittaker, Chem. Commun., 2018, 54, 10909–10912 RSC.
- D. Rinaldi, T. Hamaide, C. Graillat, F. D'Agosto, R. Spitz, S. Georges, M. Mosquet and P. Maitrasse, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3045–3055 CrossRef CAS.
- B. Diggle, Z. Jiang, R. W. Li and L. A. Connal, Chem. Mater., 2021, 33, 3712 CrossRef CAS.
- C. H. Li and J. L. Zuo, Adv. Mater., 2019, 1903762 CrossRef PubMed.
- L. Zhang, Z. Liu, X. Wu, Q. Guan, S. Chen, L. Sun, Y. Guo, S. Wang, J. Song, E. M. Jeffries, C. He, F. L. Qing, X. Bao and Z. You, Adv. Mater., 2019, e1901402, DOI:10.1002/adma.201901402.
- E. R. L. Brisson, J. C. Griffith, A. Bhaskaran, G. V. Franks and L. A. Connal, J. Polym. Sci., Part A: Polym. Chem., 2019, 57, 1964–1973 CrossRef CAS.
- E. Filippidi, T. R. Cristiani, C. D. Eisenbach, J. H. Waite, J. N. Israelachvili, B. K. Ahn and M. T. Valentine, Science, 2017, 358, 502–505 CrossRef CAS PubMed.
- P. Lin, S. Ma, X. Wang and F. Zhou, Adv. Mater., 2015, 27, 2054–2059 CrossRef CAS PubMed.
- J.-C. Lai, L. Li, D.-P. Wang, M.-H. Zhang, S.-R. Mo, X. Wang, K.-Y. Zeng, C.-H. Li, Q. Jiang and X.-Z. You, Nat. Commun., 2018, 9, 1–9 CrossRef CAS PubMed.
- J. Cao, J. Li, Y. Chen, L. Zhang and J. Zhou, Adv. Funct. Mater., 2018, 28, 1800739 CrossRef.
- Y. Lei, W. Huang, Q. Huang and A. Zhang, New J. Chem., 2019, 43, 261–268 RSC.
- S. Chen, L. Sun, X. Zhou, Y. Guo, J. Song, S. Qian, Z. Liu, Q. Guan, E. Meade Jeffries, W. Liu, Y. Wang, C. He and Z. You, Nat. Commun., 2020, 11, 1107 CrossRef CAS PubMed.
- M. O. Saed, A. Gablier and E. M. Terentejv, Adv. Funct. Mater., 2019, 30, 1906458 CrossRef.
- Q. Zhao, W. Zou, Y. Luo and T. Xie, Sci. Adv., 2016, 2, e1501297 CrossRef PubMed.
- D. K. Hohl, A.-C. Ferahian, L. Montero de Espinosa and C. Weder, ACS Macro Lett., 2019, 8, 1484–1490 CrossRef CAS.
- Z. Wei, J. He, T. Liang, H. Oh, J. Athas, Z. Tong, C. Wang and Z. Nie, Poly. Chem., 2013, 4, 4601–4605 RSC.
- Z. Jiang, B. Diggle, M. L. Tan, J. Viktorova, C. W. Bennett and L. A. Connal, Adv. Sci., 2020, 7, 2001379 CrossRef CAS PubMed.
- M. Nadgorny, J. Collins, Z. Xiao, P. J. Scales and L. A. Connal, Polym. Chem., 2018, 9, 1684–1692 RSC.
- H. Sun, Y. Kim, Y. C. Kim, I. K. Park, J. Suhr, D. Byun, H. R. Choi, K. Kuk, O. H. Baek, Y. K. Jung, H. J. Choi, K. J. Kim and J. D. Nam, J. Mater. Chem. C, 2018, 6, 2996–3003 RSC.
- M. Nadgorny, Z. Xiao and L. A. Connal, Mol. Syst. Des. Eng., 2017, 2, 283–292 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and FTIR spectra of poly(MAA-co-OEGMA); frequency dependency of storage (solid dots, G′) and loss (hollow dots, G′′) moduli of H-bonded network poly(MAA-co-OEGMA); photographs of the double crosslinked film [with iron(III):COOH molar ratio of 1:75]; tensile strength, Young's modulus, tensile strain and toughness of the double crosslinked polymers with different iron(III)-to-COOH molar ratios; frequency dependency of storage (solid dots, G′) and loss (hollow dots, G′′) moduli of double cross-linked polymers with varying iron(III)-to-COOH molar ratios; photographs of the double crosslinked and H-bonded network in initial, stretching and unloading status, respectively; FTIR spectra of double cross-linked polymer after absorption of moisture and subsequent left in the dry condition; the weight change of the double cross-linked polymer during the humidification and dry process; humidity-induced shape transformations of polymers; humidity response of the H-bonded network; stress relaxation behaviors of wet H-bonded and double crosslinked networks; dependence of the bending curvature change of double cross-linked polymer films with different thickness; temperature and frequency dependence of the storage modulus G′, loss modulus G′′ and the loss factor tanδ of double crosslinked polymer; self-healing behavior of wet double cross-linked polymer; absorption spectra of the buffer solutions showing enzyme catalyzed reaction; shear thinning behavior of polymeric inks; photographs of concentrated aqueous solution of poly(MAA-co-OEGMA) after addition of ferric ions with different iron(III)-to-COOH molar ratios; storage (red) and loss modulus (black) of prepared ink. See DOI: 10.1039/d1ma00223f |
| This journal is © The Royal Society of Chemistry 2021 |