
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProgress and challenges in self-healing composite materials
Shafiqul
Islam
 ab and
Gajanan
Bhat
*a
ab and
Gajanan
Bhat
*a
aDepartment of Textiles, Merchandising and Interiors, University of Georgia, Athens, GA, USA. E-mail: si59291@uga.edu; gbhat@uga.edu
bDepartment of Textile Engineering, Dhaka University of Engineering and Technology, Gazipur 1700, Bangladesh. E-mail: shafiqul.islam@duet.ac.bd
First published on 2nd February 2021
Abstract
Synthetic materials used in a wide range of applications are prone to damage in the form of cracking/microcracking. Catastrophic failure of the materials may occur due to the growth and merging of the microcracks, which results in the reduction of the service life of the materials. To avoid these problems and increase the service life, early detection and mending of the microcracks are extremely important. Self-healing materials can be utilized in these cases, which have the capability not only to detect cracks early but also to repair cracks automatically. These materials will also have the potential to improve material reliability, extend the service life, reduce replacement costs, and improve product safety. Due to these attractive features, numerous research studies are conducted every year on the development of self-healing systems. This paper summarizes the latest progress in the design and fabrication techniques of self-healing materials through a wide range of materials, including metals, ceramics, concrete, and polymer composites. Based on recent research, this article provides an overview of different chemistries and approaches involved in preparing self-healing composites. Comparative healing efficiency and related fabrication methods are tabulated. Finally, existing problems, gaps, and challenges, and future research directions and opportunities for commercial applications are highlighted.
 Shafiqul Islam | Shafiqul Islam is a PhD student and researcher at the department of TMI of the University of Georgia. He pursued his undergraduate and master’s degrees in textile engineering from the Bangladesh University of Textiles. After completing the undergraduate degree, Shafiqul worked in research and developed sections of an export oriented industry. Later he joined at Dhaka University of Engineering and Technology as a faculty member. Currently, he is on study leave and pursuing his PhD degree at UGA. His main research focus is on the design and development of composite materials and production of environmentally friendly, sustainable insulation materials. He is also working on lifecycle assessment (LCA) to check the true sustainability of produced composite materials and comparing them with those of commercially available products. Another desirable property of such composites is that of self-repairing. At present, Shafiqul is investigating the possibility of incorporating the right ingredients to make self-healing composite materials. |
 Gajanan Bhat | Gajanan Bhat is the Georgia athletic association professor of fibers and textiles and department head of Textiles, Merchandising and Interiors of the University of Georgia. After earning his PhD from Georgia Tech in textile and polymer engineering, Dr Bhat joined the University of Tennessee, Knoxville (UTK) in August 1990, where his research covered nonwovens – melt blown, spunbonded and biodegradable, plastics recycling, nanotechnology, sustainable materials, and high performance fibers. Recently he joined University of Georgia as the Head of the TMI department. He has served as the president of the Fiber Society and is also an active member of INDA, TAPPI and the Textile Institute. His current research projects include: Recycling of Textile Waste into Value-added Products; Cotton-based nonwovens and composites; Stretchable cotton nonwovens; Biodegradable/Compostable Meltbown Nonwovens for Filter media and other Applications; Flushable Nonwovens; LightWeight Ballistic Materials with Improved Performance; Processing and Characterization of Carbon Fibers from Alternative Precursors. |
1. Introduction
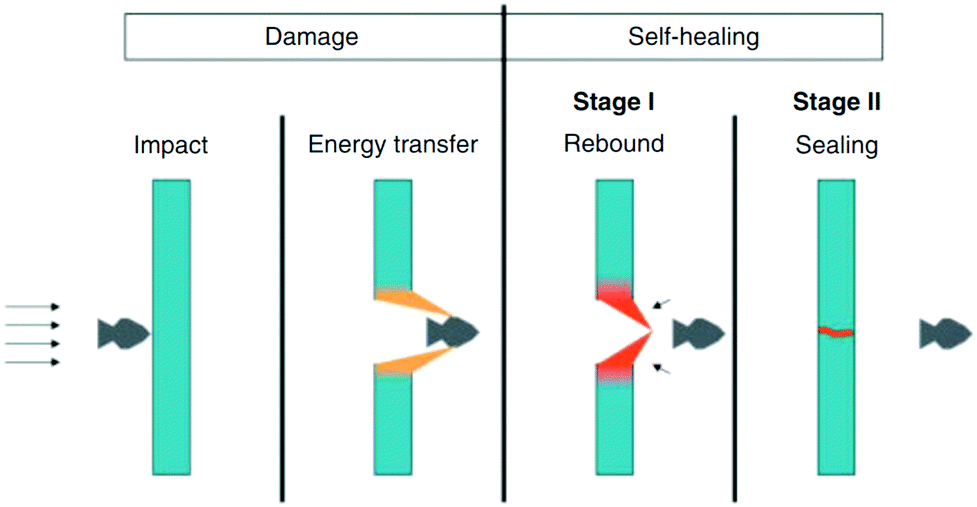
Synthetic materials have been widely used in a variety of applications, but these materials are susceptible to damage. A small imperfection or microcracks of the materials may lead to premature failure if it is not detected or repaired in time.1 Some of these microcracks are formed within the material structures, which are invisible, difficult to repair manually, and result in catastrophic failure before the service life of the products.1 On the other hand, biological materials have the ability to recover their damage and resume their original function automatically, which contributes to their longevity.2 Inspired by nature, researchers are trying to develop a new class of synthetic materials known as self-healing materials that can automatically detect and repair damage. This increases the durability and reliability and reduces the maintenance cost of the material.3 As a result of these exciting features, several researchers are trying to incorporate self-healing properties into a wide variety of materials, including polymers,4–11 ceramics,12,13 concretes,14,15 metals, and composites.16These different types of materials have their own self-healing mechanisms which can be categorized into several groups, including intrinsic and extrinsic self-healing approaches.17 Intrinsic self-healing normally occurs by reversible covalent bond formation with supramolecular and physical interactions of polymers.3 On the other hand, extrinsic self-healing occurs typically with the help of healing agents, which are usually stored in reservoirs like capsules,18 hollow fibers,19,20 or vascular networks,21,22 which are inserted in polymers or other suitable materials. When cracks occur within the materials, the capsules or vascular networks also rupture, releasing the healing agent that will close the gap and repair the material.23 Similar to the repairing process of fractured bones, three steps are involved in the extrinsic healing process.24 The first step is the triggering action that occurs soon after the damage/crack happens, the second step is the transportation of the healing agent to the damage/crack area, and the third step is the chemical interactions that heal the polymer materials.25 In both intrinsic and extrinsic self-healing, one step that is required is the initiation of self-healing in response to a stimulus/trigger.26 Along with the damage/crack, which is the main stimulus for autonomous healing, there are several other external stimuli, including optical,4,5,27,28 thermal,6,29 electrical,30 ballistic,31 or chemical,32 which can initiate a self-healing process. In all cases, microcracks in the materials will be repaired before propagation and catastrophic failure, resulting in increased service life of the materials.
The potential for reducing catastrophic failure and increasing the service life in self-healing materials has drawn huge attention from researchers. Every year, a significant amount of research is reported on the development of self-healing materials and use of these techniques in practical applications. The number of published papers on the topic of self-healing materials has been growing rapidly (Fig. 1).
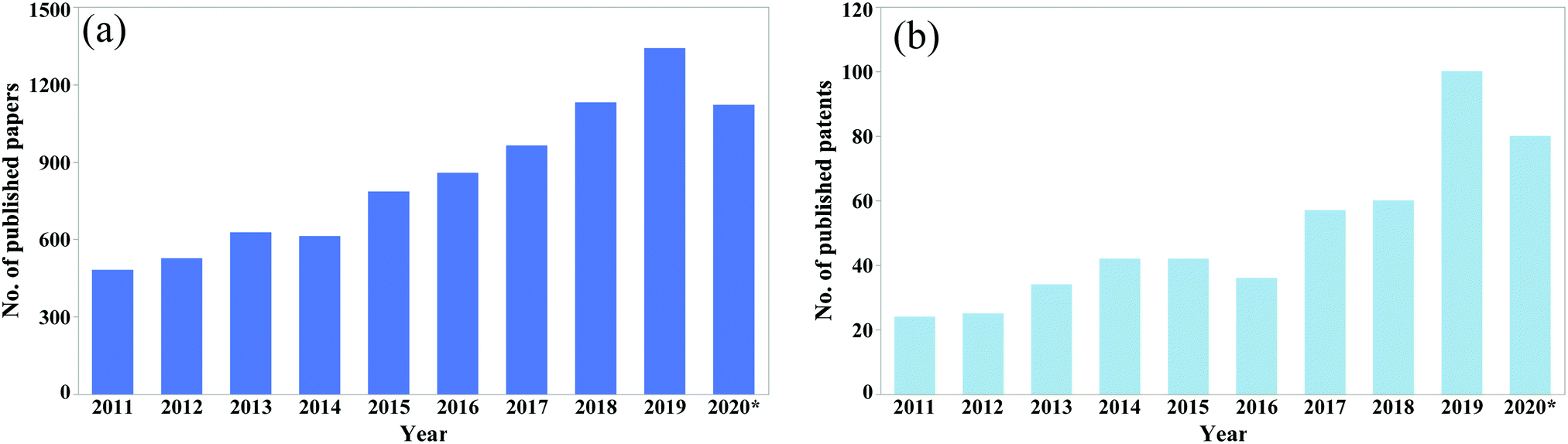 | ||
| Fig. 1 No. of published (a) papers and (b) patents from 2011 to August 2020 (according to google scholar, searched by self-healing in the title of the article). | ||
From the data shown in Fig. 1, it is clear that the number of publications has been increasing rapidly over the last decade. Hence a comprehensive review article regarding self-healing materials is essential. To the best of our knowledge, previous reviews mainly focus on a general process of producing self-healing materials conducted by several researchers. A few of them focus on comparison of self-healing performance conducted by the researchers. Again, as the number of publications is increasing rapidly, a new comprehensive review paper is essential to summarize the recent advances. Hence the main objectives of this review paper are to summarize recent progresses and current challenges in self-healing materials and compare the healing performance achieved by different materials.
This paper summarizes the design and fabrication techniques of self-healing materials through a wide range of material classes, including metals, ceramics, concrete, and polymer composites. Based on recent research, this article provides an overview of different chemistries and approaches involved in the preparation of self-healing polymer composites. Comparative healing efficiencies obtained by researchers and related fabrication methods involved to obtain those efficiencies are tabulated. Finally, existing problems, gaps, and challenges of the present research are identified. The future direction of research and opportunities for commercial applications are also highlighted.
2. Approaches of self-healing
Self-healing methods in living organisms are mainly autonomous. For synthetic artificial materials, the process of self-healing is diverse. The first self-healing polymeric material demonstrated by Chen et al. was based on the mechanism of dynamic covalent bonds with the help of external heating.33 The first room-temperature self-healing elastomer was explained by Cordier et al., which was fabricated based on hydrogen bonding.34 The first microcapsule-based self-healing method was reported by White et al.18 Gosh and Urban illustrated the UV-triggered self-healing polymers utilizing oxetane-substituted chitosan polyurethane networks.28 With the passage of time, several other researchers developed new approaches to self-healing. In general, most of these self-healing methods can be categorized as intrinsic and extrinsic self-healing processes.3. Intrinsic self-healing
Intrinsic self-healing materials have the ability to restore molecular or micro-scale damage by means of inherent reversibility of bonding of the polymer matrix.17 The inherent reversibility of bonding provides some excellent benefits to intrinsic self-healing materials, including multiple healing cycles, and it is possible to achieve healing without using any external healing materials.35 However, there are some limitations as well. Self-healing can be achieved only in micro-scale or small damaged areas, which restricts its range of applications.36 As healing takes place at the molecular level, the reversibility of bonding occurs when the chemicals’ distances are in the nanometer range.37 Thus, for healing to occur, it is necessary to bring the two damaged parts together either manually or by other mechanisms.37 In the case of hard materials, heating is also required for making full contact with the two damaged parts.37 For repairing any damage, the healing functionality needs to be triggered/initiated by a stimulus.Normally, healing functionalities of intrinsic self-healing materials remain in latent conditions in the polymeric structure. These latent functionalities are triggered by damage and driven by surface tension and elastic energy of the stress source or by an external stimulus such as heat, light, solvent, and others.35 The initiation of self-healing also relies on several other factors, including chain mobility and entanglement, reversible polymerizations, melting of thermoplastic phases, hydrogen bonding, or ionic interactions.17
3.1. Chemical interactions
Chemical interactions at the damaged site are mainly responsible for the re-bonding and intrinsic self-healing capability of fracture surfaces.3 Healing efficiency of a material depends on the extent of the reaction. However, all types of chemical reactions do not have the capability of incorporating self-healing properties. So far, several reactive groups (free radicals, hydrogen bonds, ionomeric interactions –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–, –COOH, –NH2, –OH, –SH, –Si–O, S–S, –CO, and/or formation of cyclic structures) have been investigated that have re-bonding and self-healing capability.38 A significant number of self-healing materials have been developed by incorporating these reactive groups into a polymer network and are typically categorized into reversible covalent bonds and supramolecular chemistry.2
C–, –COOH, –NH2, –OH, –SH, –Si–O, S–S, –CO, and/or formation of cyclic structures) have been investigated that have re-bonding and self-healing capability.38 A significant number of self-healing materials have been developed by incorporating these reactive groups into a polymer network and are typically categorized into reversible covalent bonds and supramolecular chemistry.2
Several types of reversible covalent bonds like cycloaddition reactions, exchange reactions, and transesterification reactions can be utilized for self-healing purposes.
3.1.1.1. Cycloaddition reactions. Cycloaddition is a reversible covalent reaction where unsaturated molecules join together and form a ring.42 Diels–Alder (DA) thermoreversible reaction is a common cycloaddition reaction where reversible cross-linking occurs through a [4+2] cycloaddition involving a diene and a dienophile as precursors (Fig. 2).42 The notation within the brackets denotes the number of electrons each molecule provides.42 Similar to other addition reactions, the DA reaction is useful for polymer preparation where a multifunctional diene and dienophile are used as monomers.43,44 The DA reaction does not require any additional chemical like catalysts, and it is ideal for the synthesis of repairable polymers.41 In addition to that, a reversible DA reaction does not involve free radicals, which is helpful for the limitation of the side reaction and avoiding reconversion.45
 | ||
| Fig. 2 Diels–Alder reaction. | ||
A number of self-healing materials like epoxies, polyacrylates, and polyamides can be developed utilizing DA reactions.46 At elevated temperatures, the bond between diene and dienophile breaks. After that lowering, the temperature causes the bonds to reform and heal the cracks.47 Fortunato et al. obtained reversible networks by reacting a series of linear copolymers of furfuryl methacrylate with aliphatic bismaleimides through the DA reaction between furan and maleimide moieties and developed highly transparent and colorless self-healing polyacrylate coatings.48
One of the drawbacks of DA reactions is that it requires higher temperatures to activate the self-repair process.2 The activation temperature of the retro-DA reaction is above its glass transition temperature (100 °C).2
In order to reduce the healing temperature, researchers employed photochemical cycloadditions ([2+2] cycloaddition of 1,1,1-tris-(cinnamoyloxy-methyl)ethane (TCE)49 or coumarin,50 and [4+4] cycloaddition of anthracene derivatives)27 which provide an optically induced self-healing process at lower temperature.2 Self-healing chemistry of these materials is similar to DA reactions where light at different wavelengths is utilized for ring opening and closing the bond within a polymer network (Fig. 3).49 For example, polymerization of 1,1,1-tris-(cinnamoyloxymethyl)ethane (TCE) monomers via photo-stimulated [2+2] cycloaddition gives a rigid polymer.49 Coumarin [2+2] is photo-dimerized at 350 nm, and the reaction is reversed at 254 nm.50
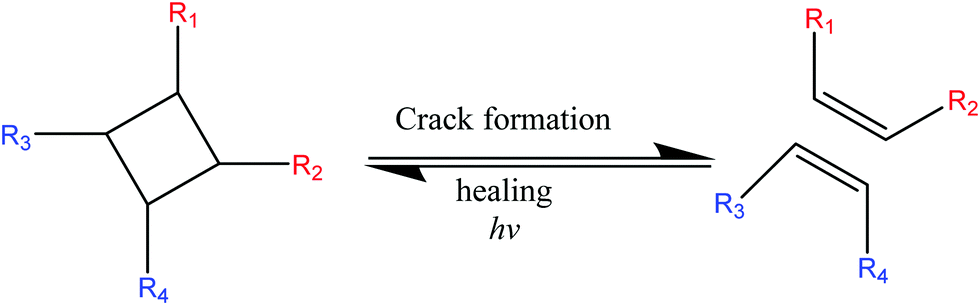 | ||
| Fig. 3 Schematic [2+2] cycloaddition upon light exposure [reproduced from ref. 35 with permission from Elsevier]. | ||
Cycloaddition reactions have been extensively investigated by the researchers due to their good adaptability to various polymers and obtaining good self-healing properties.2 Many of the most commonly used reactions based on several recent research studies are summarized in Table 1.
| Healing mechanisms | Materials | Healing conditions | Mechanical properties | Healing efficiency | Ref. |
|---|---|---|---|---|---|
| Healing efficiency (%) = recovered properties/original properties.a Tensile strength. | |||||
| Diels–Alder reactions | Furan and bismaleimide are grafted to an epoxy matrix system | 150 °C for 120 min then 60 °C for 120 min | 0.82 kJ m−2 (fracture toughness) | >100% | 63 |
| 41.6 MPa (shear strength) | |||||
| Recyclable polyurethane nanocomposites | 90 °C for 5 min, then 65 °C for 48 h | 40.07 ± 2.86 MPaa | >90% | 64 | |
| Recyclable polyurethane composites | NIR laser for 30 s, 120 °C for 2 h, then 80 °C for 24 h. | 24 MPaa | 93.1% | 65 | |
| Polyacrylate coating produced by reaction of furfuryl methacrylate and bismaleimides | 150 °C (90 min) then cooling at RT | — | >90% scratches | 48 | |
| Non-isocyanate polyurethanes | 120 °C for 30 min, then 60 °C for 24 h | 2.2 MPaa | 100% | 66 | |
| Bio-based dendritic methacrylates | 150 °C, 35 h | 12–20 MPa | 89.04% (razor blade cut) | 67 | |
| Photochemical [2+2] cycloaddition | Poly(TCE-co-DCE) capsules prepared by 1,1,1-tris(cinnamoyloxymethyl)ethane (TCE) and 1,1-di(cinnamoyloxymethyl)ethane (DCE) | 15 min to a light of λ > 280 nm UV light | — | Reduced crack sized | 68 |
| Cinnamoyl mechanophore | 48 h UV irradiation | — | 24% | 69 | |
| Dihydroxyl coumarin based polyurethane | 254 nm UV then 350 nm UV, >2 h | 3.5 MPa | 64.4% (tensile strength) | 50 | |
| Disulfide (S–S) exchange reactions | Polyurethane nanocomposite (polyurethane and carbon nanotubes) | 80 °C for 24 h, Irradiation of NIR laser for 1 min | 2.99 MPaa | 90.1% | 70 |
| Waterborne polyurethane | RT, 24 h | 12–25 MPaa | 95.18% | 71 | |
| Superamphiphobic coatings, fabricated by deposition of Al2O3 nanoparticles into polyurethane acrylic resin | 80 °C for 90 min | 12.8–16.5 MPaa | Complete (almost 100%) removal of scratch | 72 | |
| Polydimethylsiloxane–polyurethane | 120 °C, 12 h | 3.31 MPaa | 97% | 73 | |
| Disulfide–thiol exchange reactions | Thermoset rubber from dipropyl disulfide, dibutyl disulfide, and 1-penthanethiol | 24 h, RT | 320 KPaa | Up to 99% | 74 |
| S-Aromatic thiourethane exchange reaction | Polythiourethane networks | 160 °C, 20 min | — | Complete removal of cracks | 75 |
| Alkoxyamine | Polyurethane elastomer | 80 °C in argon, 2.5 h | — | 70% | 76 |
| Transesterification/ester exchange reactions | Blends of thermoplastic polyurethane and phenoxy resin | 150 °C | 48.4 MPaa | 100% | 77 |
| Isocyanate-based crosslinked star-shaped polyglycolide network | 120–160 °C for 10 min | 10 MPaa | 70% | 78 | |
| Anhydride modified polybenzoxazines | 140 °C for 40 min | 2.8 GPa (storage modulus) | 99% | 79 | |
| Bentonite/natural rubber | 150 °C, 3 h | 4.5 MPaa | 96% | 80 | |
| CNC-containing gel | Visible light | — | 100% | 81 | |
3.1.1.2. Exchange reactions. Dynamic exchange reactions, another type of reversible covalent bond, can also be utilized for the development of self-healing polymers.2 One of the major advantages of an exchange reaction is that it requires lower temperature than DA reactions for the activation of the healing mechanism.2 Disulfide bonds (Fig. 4(a)) have the ability to give exchange reactions between two adjacent S–S bonds via free radical or ionic intermediates where healing of cracks or damage can be activated at low temperatures using photolysis, heating, or oxidation.51 Amamoto et al. incorporated thiuram disulfide units in a low Tg polyurethane and found good self-healing properties at room temperature under visible light.52 The concentration of disulfide groups determined the percent recovery of strength, and healing efficiency increases with the increases in concentration (Fig. 5).
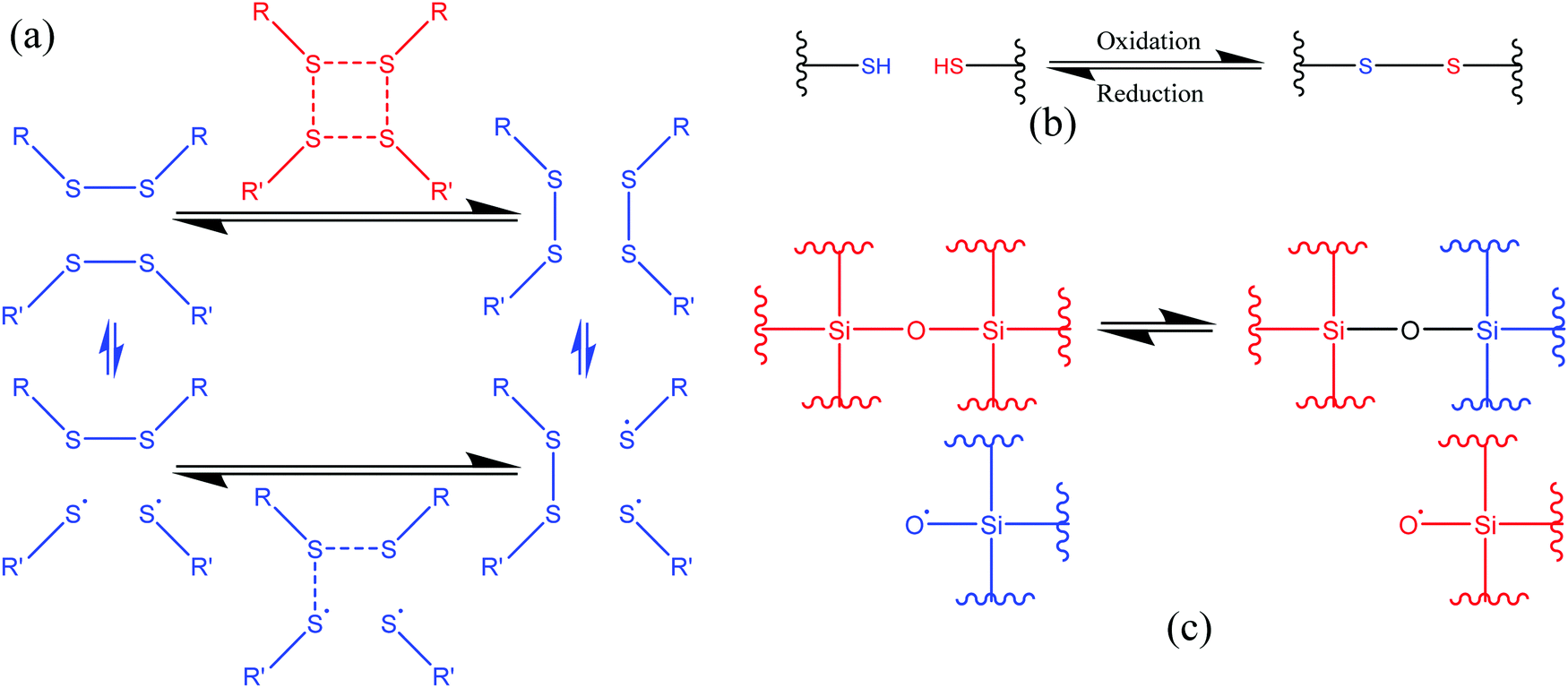 | ||
| Fig. 4 Schematic representation of the exchange reaction. (a) [2+2] metathesis (red) and [2+1] radical-mediated (blue) mechanisms [reproduced from ref. 53 with permission from The Royal Society of Chemistry]. (b) Oxidative disulfide formation from thiols [reproduced from ref. 35 with permission from the Elsevier]. (c) Siloxane exchange reaction (used only top part)54 [reproduced from ref. 54 with permission from the ACS]. | ||
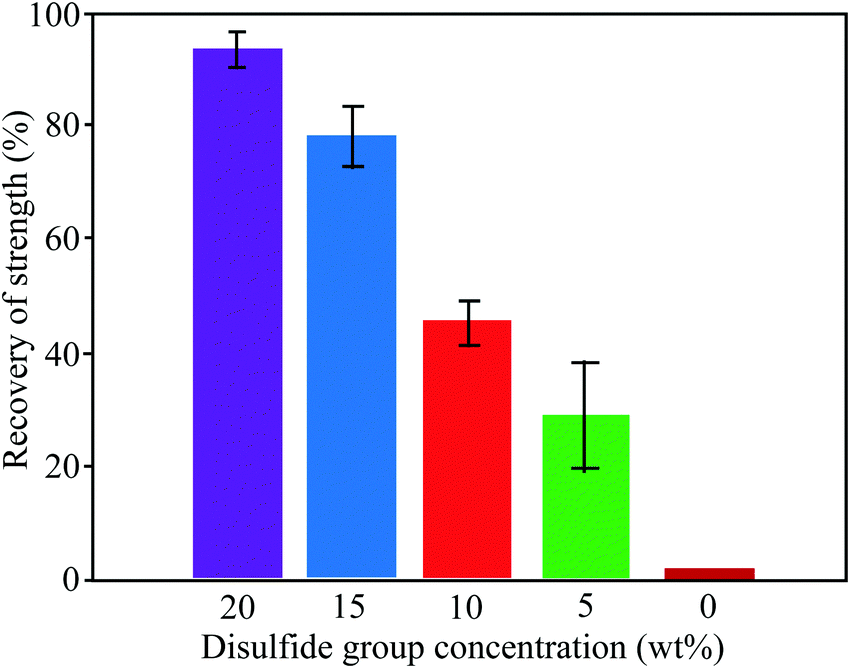 | ||
| Fig. 5 Recovery of strength (%) of healed samples with different concentrations of disulfide groups [reproduced from ref. 55 with permission from the ACS]. | ||
Similar to disulfide bonds, thiol–disulfide (Fig. 4(b)) and silicone-based (Fig. 4(c)) exchange reactions also give self-healing properties. Tsarevsky and Matyjaszewski presented thiol–disulfide reactions based on a polystyrene copolymer, which provides good healing properties through subsequent oxidation of the thiols regenerating the disulfide linkers.56 Zheng and McCarthy showed the self-healing mechanism of crosslinked silicone-based polymers containing ethylene bridges and active silanolate end groups synthesized from tetramethylammonium silanolate-initiated ring-opening copolymerization of octamethylcyclotetrasiloxane (D4) and bis(heptamethyl cyclotetrasiloxanyl)ethane (bis-D4).57 Several of the exchange-based self-healing materials with self-healing conditions and efficiency are listed in Table 1.
In some cases, exchange reactions occur through free radical intermediates.42 When cracks or damage occur in polymer matrix, free radicals are generated due to the splitting of covalent bonds.2 Healing occurs by restoring covalent linkages when recoupling of free radicals occurs in opposite fractured surfaces.2 Higaki et al. first incorporated thermoreversible alkoxyamine reaction into a crosslinked reversible network where crack healing occurs when free radicals are generated after cracking.58 The self-healing capacity of free-radical based materials depends on the mobility of free-radicals and avoidance of radical–oxygen interactions.42 For effective self-healing, split chain ends with reactive groups have to move to contact each other and complete all the reactions before other reactions, like radical–oxygen interactions, and capture of free radicals.42,59 Healing of the damaged part will not occur if the free radicals interact with oxygen instead of interacting with the broken end.60
3.1.1.3. Transesterification reactions. Transesterification reactions that normally occur between an ester group and alcohol/carboxylic acid groups can be utilized for developing self-healing materials with good healing and mechanical properties (Table 1). Self-healing properties have been shown when transesterification occurs between pendant ester and hydroxy groups and forms a network with fatty acids (Fig. 6).61 Piermattei et al. investigated transesterification reactions and found that silver(I) complexes of polymer-functionalized N-heterocyclic carbenes were able to effectively catalyze transesterification reactions when exposed to ultrasound in solution.62
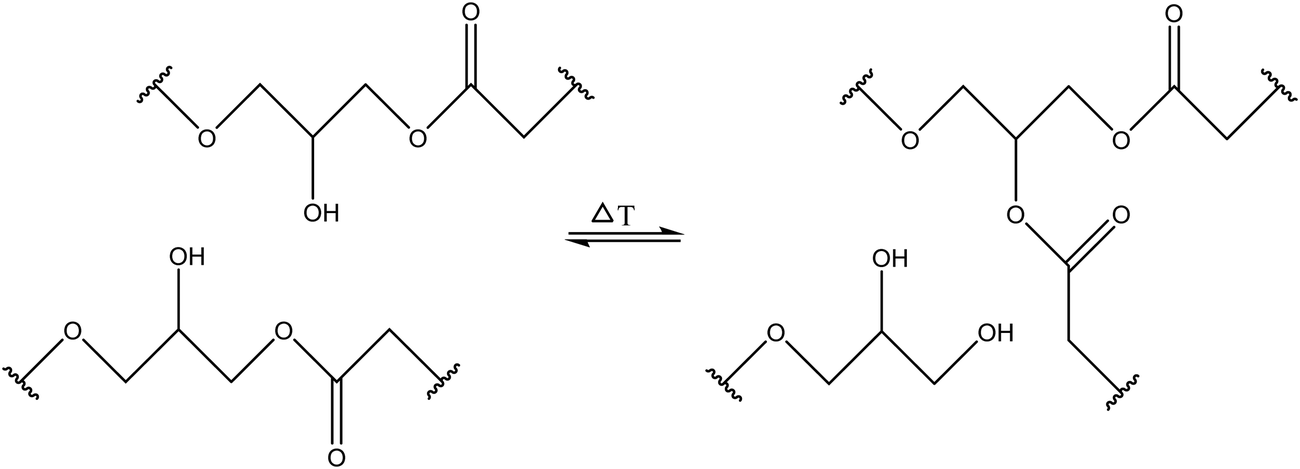 | ||
| Fig. 6 Hydroxy and ester groups undergo transesterification reactions [reproduced from ref. 35 with permission from Elsevier]. | ||
The self-healing of supramolecular materials are generally obtained by the formation of non-covalent interactions that include hydrogen bonds, metal coordination, π–π stacking, ionic interactions, and host–guest interactions.2,84,85 Upon applying mechanical force, the weaker supramolecular bonds dissociate, and then due to their dynamic character of this chemistry, subsequent regeneration and self-healing take place.2
3.1.2.1. Hydrogen bonds. Hydrogen bonds can be utilized to prepare a wide range of supramolecular polymers with diverse mechanical properties.2 In general, hydrogen bonds are weaker than common covalent bonds, but it is possible to produce strong rubber-like materials using hydrogen bonds (Table 2) as they have some excellent features like directionality and affinity.2 A number of studies have revealed that combining multiple H-bonds into a functional unit of urea isopyrimidone (Upy) (Fig. 7) resulted in increased association strengths between Upy units and polysiloxane, polyethers, and polyesters.86,87 For example, Yamauchi et al. showed that enhanced mechanical strength could be obtained if Upy was incorporated into flexible poly(ethylene-co-butylene) and PDMS.88 Meijer and other co-authors conducted several studies on the preparation of stable DDAA (donor–donor–acceptor–acceptor) dimers from ureidopyrimidinone (Upy) utilizing hydrogen bonds.89–91 Guadagno and other coauthors utilized reversible hydrogen bonding for developing carbon-based nanocomposites, which has potential to be used in structural applications.92,93 Along with reasonable mechanical strength, hydrogen bonds can be utilized to produce materials with multiple healing properties at the same damaged sites.41 However, it is required that the damaged ends come in contact immediately after breakage. Otherwise, hydrogen bonding units may react with the closest ones resulting in reduction of healing capability.41
| Healing mechanisms | Materials | Healing conditions | Mechanical properties | Healing efficiency | Ref. |
|---|---|---|---|---|---|
| Healing efficiency (%) = recovered properties/original properties.a Tensile strength, RT = room temperature. | |||||
| Hydrogen bonding | Actuators, manufactured by embedding carbon nanotubes into elastomer | NIR light, 66 °C, for 2 s | 12.52 MPaa | 87.68% (toughness); 99.50% (actuating) | 106 |
| Ureido-pyrimidinone functionalized PLA grafted with PEG | 24 h, RT | — | 99.4% (Coulombic efficiency after 350 cycle) | 107 | |
| Polycarbonate hydrogel from 2-methyl-2-benzyloxy carbonyl propylene carbonate and methoxy PEG | 4 h, RT | 0.5–1.5 MPa (storage modulus) | Almost completely heal the scratch | 108 | |
| Epoxidized natural rubber embedded with nanochitin, carboxymethyl chitosan, and carbon nanotubes | 24 h, RT | 3.5 MPaa | 91% | 109 | |
| Polyurethane elastomer/polypropylene glycol | 45 °C, for 120 min | Up to 33.6 MPaa | 79.85% | 110 | |
| Metal–ligand coordination | Hydrogels from acrylamide and hydrophobic acrylic monomer | 10 min at RT | 51.34 KPaa | 100% (stress) | 111 |
| 90% (energy) | |||||
| Methyl vinyl silicone rubber, coated by carbon nanotubes and silver film | 120 °C for 84 h | 130 KPaa | 84–91% | 112 | |
| Ni2+ coordination of PEG with bistriazole pyridine ligands | 2 min at ambient temperature | — | Completely heal, no scratch | 113 | |
| Polydimethylsiloxane-based soft polymer, imine-based ligands | 2 h, RT | 0.23–10.2 MPa (elastic modulus) | 61–88% | 114 | |
| Metal–ligand domains into the carboxylated nitrile rubber | 80 °C for 24 h | 4.3–10.3 MPa | 88–100% | 115 | |
| π–π interactions | Cyclometalated platinum(II) complex/polydimethylsiloxane | 12 h, RT | 300 KPaa | Almost 100% | 116 |
| Chitosan/graphene oxide | RT | — | 91% (compressive stress) | 117 | |
| Pyrene-containing poly(ε-caprolactone) network and single-walled carbon nanotubes | NIR light for 120 min | 21–32 MPaa | Scratch completely removed | 118 | |
| Azobenzene-containing liquid crystalline polyester | 60 °C for 5 h | 12.8 MPaa | 73.5% | 119 | |
| Ionomeric interactions | Poly(styrene-co-methacrylate) and poly(styrene-co-itaconate) ionomers | 60 °C for 4 h | — | 97% | 120 |
| Natural rubber-based carboxylate ionomers | 80 °C for 2 h | 2.7 MPaa | 75% | 121 | |
| Zwitterionic copolymers containing sulfobetaine moieties | 120 °C for 2 h | — | 98% | 122 | |
| Ionomeric carboxylated styrene–butadiene polymer | 100 °C for 3 h | 5.2 MPaa | 68% | 123 | |
| 1-Butyl-3-methylimidazolium hydrogen sulfate | RT, 24 h | 15.8 KPaa | 93.8% | 124 | |
| Host–guest interactions | Cyclodextrin and adamantane | 60 °C for 10 min | 2.5–8 MPaa | 70–72% | 125 |
| Thermoplastic polyurethane, prepared by hexamethylene diisocyanate and tetraethylene glycol | 80 °C for 7 min | 11 MPaa | 87% | 126 | |
| Polymer hydrogel of poly(N-isopropylacrylamide) | 37 °C for 20 min | 250 kPa (storage modlus) | Fully recover of mechanical properties | 127 | |
| Silk fibroin-based hydrogels modified with cholesterol (guest) or β-cyclodextrin (host) | 2 h at RT | 3.16 MPa | 93.78% | 128 | |
| p(HEMA-co-BA)–Fe3O4 | RT | — | 94.98% (electromagnetic absorption) | 129 | |
| 40% (tensile) | |||||
| Diels–Alder and hydrogen bonds | Multi-arm UV-cured polyurethane, N,N′-(4,4′-methylenediphenyl)bismaleimide, and furfuryl alcohol | 120 °C, 10 min | — | 100% | 130 |
| Disulfide and hydrogen bonding | Poly(vinyl alcohol)-graft-(ε-caprolactone) | 12 h at 90 °C | 20 MPaa | 94% | 131 |
| π–π stacking and hydrogen bonding | PDA/Fe3O4/carbon black/PAM | RT | — | 100% | 132 |
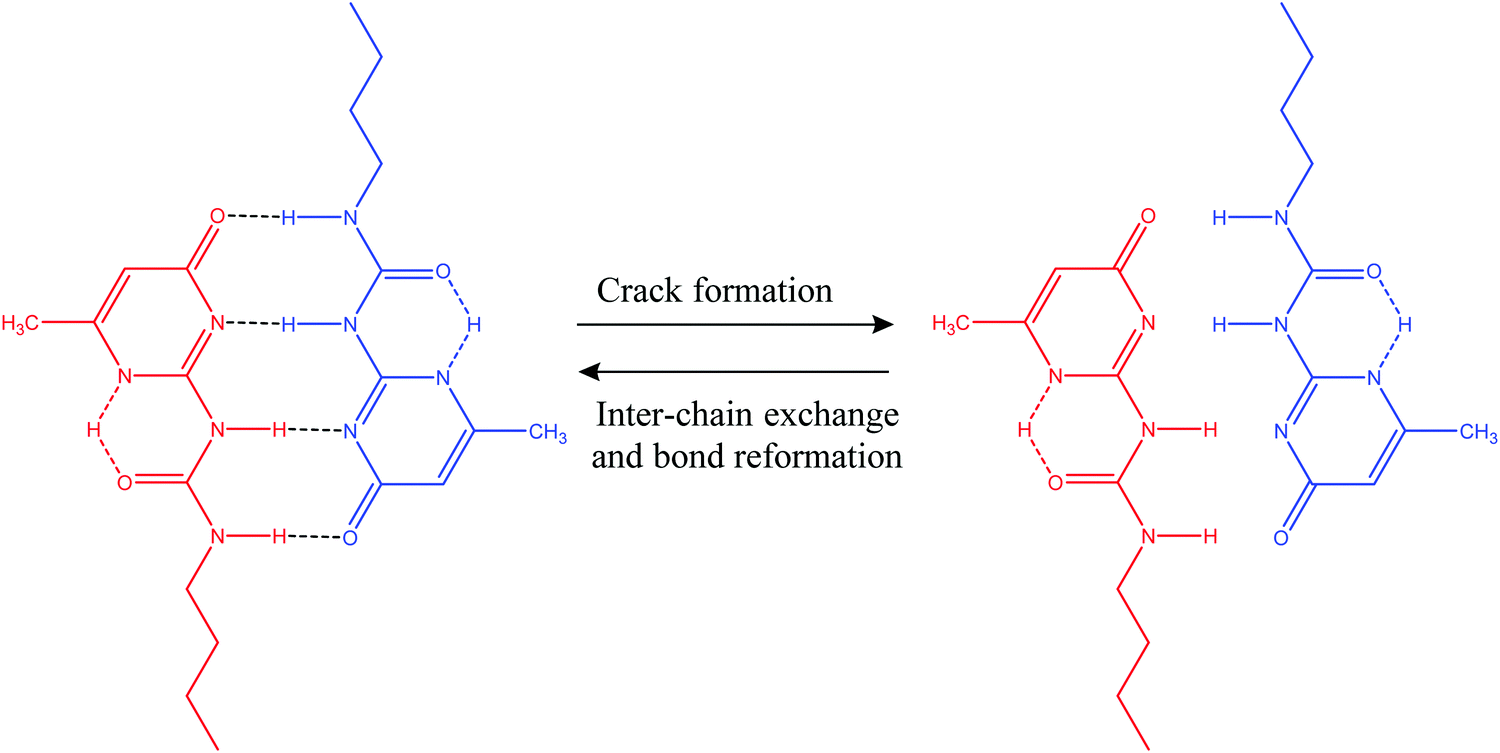 | ||
| Fig. 7 Self-healing utilizing H-bond formation in a functional unit of urea isopyrimidone (Upy) [reproduced from ref. 38 with permission from the Royal Society of Chemistry]. | ||
3.1.2.2. Metal–ligand coordination. Metal–ligand coordination bond formation, another noncovalent supramolecular interaction, occurs between a metal ion (known as coordination center) and its surrounding array of organic molecules (known as a ligand).94 These coordination bonds have very good potentiality for developing self-healing polymers due to their special properties: spontaneous metal bond formation ability and highly tunable bond energies.95 In some cases, the bond can be thermodynamically stable but kinetically liable to change.95 The strength of coordination bonds lies between covalent bonds and van der Waals interactions, but it is possible to alter the bond strength with a precise combination of ligand and metal ion.95 The capability of altering the bond strength offers reversibility similar to that of H-bonds and is very much favorable for developing self-healing materials.2 However, the healing temperature is relatively high because of high coordination bond strength, and there is a dearth of soft portions (Table 2).2
Theoretically, it is possible to produce self-healable materials using all types of ligands due to the tunability of bond strength by changing the metal ions. But, in practice, several factors need to be considered before choosing ligands, including availability of the starting materials, the level of complexity in synthesizing the ligand, and the degree of difficulty in incorporating the ligand into a polymer matrix.95 Some ligands are soluble in water, which may not be desirable under some circumstances.38 Until now, several ligands have been shown to be successful in coordinating with metal ions and produce self-healing polymers, including catechol (Fig. 8), histidine, pyridinyl, and carboxylate.95
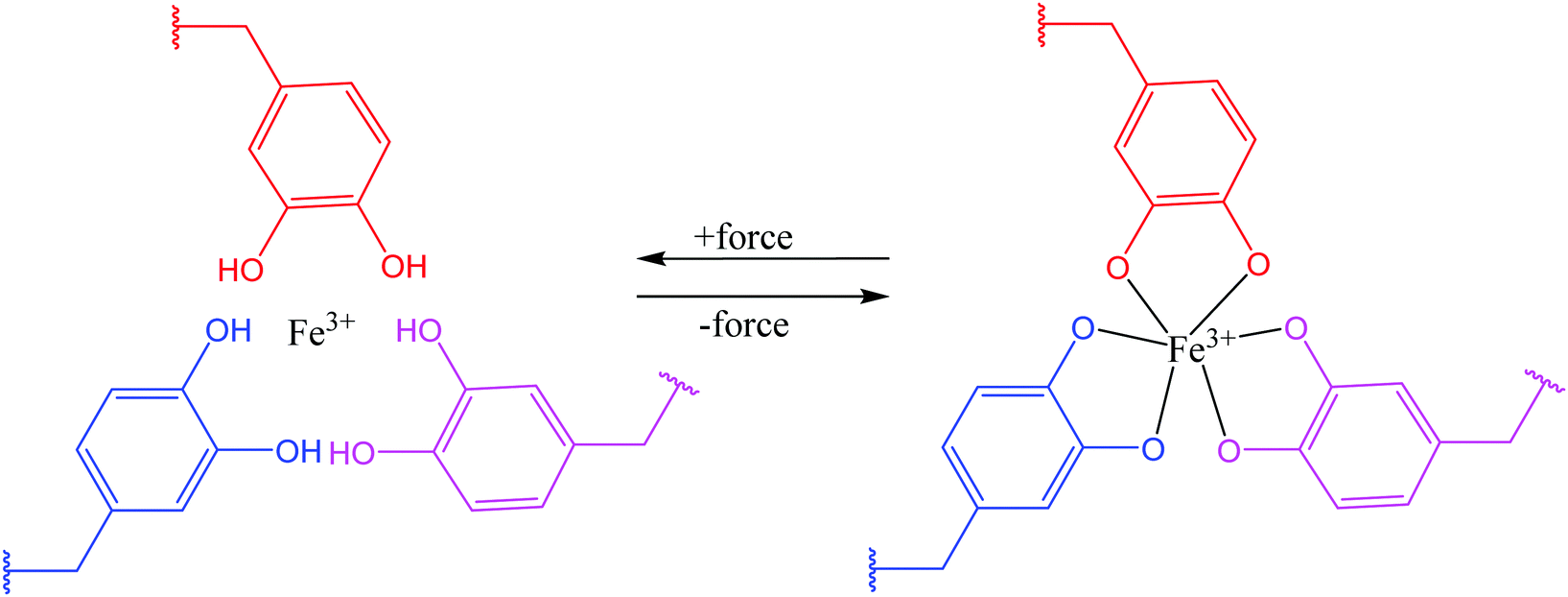 | ||
| Fig. 8 Coordination bond between catechol ligands and Fe3+ [reproduced from ref. 38 with permission from the Royal Society of Chemistry]. | ||
3.1.2.3. π–π stacking interactions. π–π stacking interaction, another non-covalent interaction, that occurs between end-capped π electron-deficient groups and other π electron-rich aromatic backbone molecules,38 can be utilized for the development of thermal-triggered reversible self-healing materials (Table 2). Burattini and other co-authors utilized aromatic π–π stacking between the π electron-deficient diimide groups and the π electron-rich pyrenyl units and developed an elastomeric, self-healable, supramolecular polymer blend consisting of a chain-folding polyimide and a telechelic polyurethane with pyrenyl end groups (Fig. 9).11 The tested results showed good self-healing efficiency after damage with 95% regains of tensile modulus, 91% regain of the elongation at break, and 77% regain of toughness.11
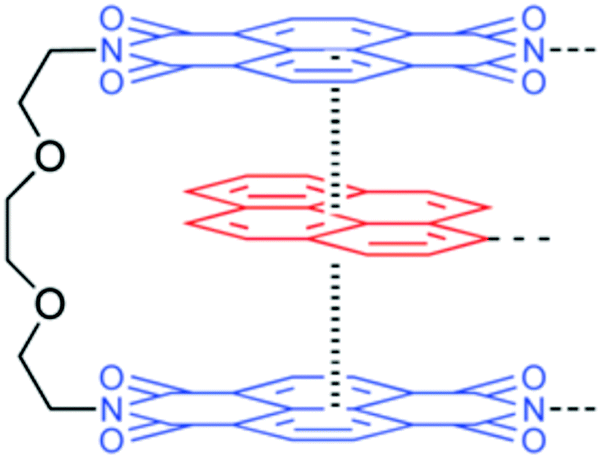 | ||
| Fig. 9 Structure of the electronically complementary π–π stacking interaction between a bis(diimide) chain fold in polyimide [reproduced from ref. 11 with permission from the ACS]. | ||
One of the advantages of π–π stacking interaction is that self-healing can be achieved at a wide range of temperatures (∼50–100 °C) as Tg of the polymeric materials can be altered by adjusting the spacer and the composition of the blend.38 For example, a blend of folding “tweezer-type” polyimide and linear polysiloxane as a backbone heals at 100 °C temperature,96 whereas the healing temperature of a polyimide–polyamide network is 50 °C.97 The limitation of this mechanism is that the bonded strength within the network is relatively weak.42
3.1.2.4. Ionomeric interactions. Ionomeric interactions, another process of producing self-healing materials, mainly occur when ionomers form in the polymer matrix.98 Ionomers comprise up to 15% of ionic groups that can form clusters and act as reversible cross-links.17 Healing of materials, initiated by high impact, occurs immediately and autonomously in the absence of healing agents, additional heat, stimuli, or any other external species (Fig. 10).41,99 When a projectile impact disrupts the ionomeric network, heat generates due to the friction between the projectile and the material. The produced heat is passed to nearby damaged areas of the polymer network resulting in localized melting. After that, the molten surfaces merged together as polymer chains with high mobility.42 Several other researchers developed self-healing materials based on ionomeric interactions, which are summarized in Table 2. Kalista et al. investigated a self-healing mechanism of a number of poly(ethylene-co-methacrylic acid) copolymers following projectile puncture and found that ionic interactions are responsible for puncture reversal.31,100 Their experiment revealed that instead of external heat or any other external stimuli, heat was generated during projectile puncture, which stimulates polymer diffusion in a damaged area, and repairs occur at low temperature, even at −30 °C.31,100 However, the application areas of this mechanism are still limited because healing happens only within a narrow temperature range and only when the damage produced by projectiles is in specific shapes.101
 | ||
| Fig. 10 Schematic presentation of self-healing ionomers [reproduced from ref. 102 with permission from the Wiley]. | ||
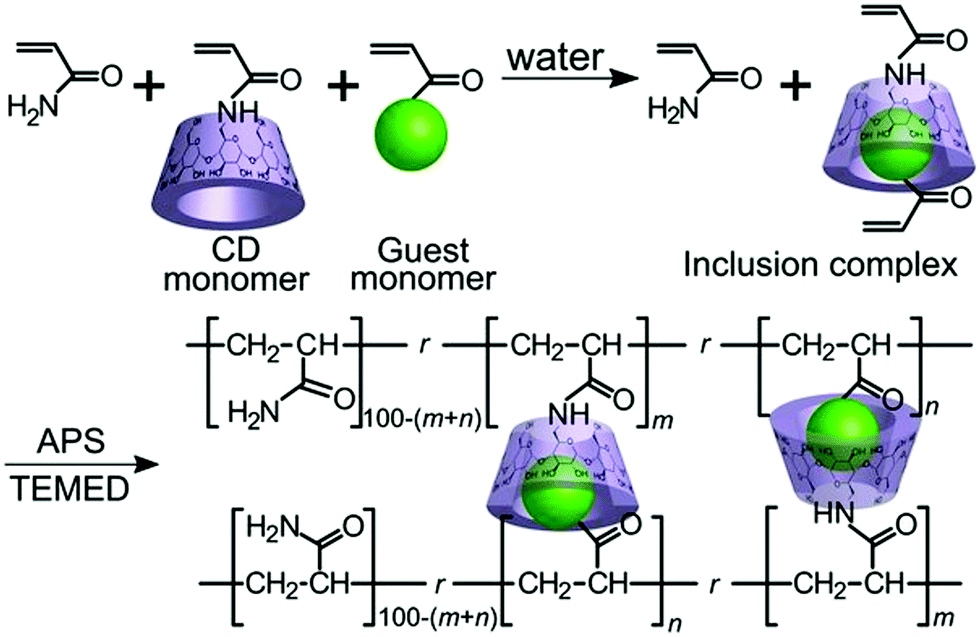
3.1.2.5. Host–guest interactions. Host–guest chemistry, a branch of supramolecular chemistry where a host molecule interacts with a guest molecule or ion, has become important in many biological and chemical processes. A host molecule normally consists of structures with large cavity volumes such as cyclodextrins (CD), cucurbiturils (CB), or calixarenes. Guests usually have matching shapes with host molecules and undergo different noncovalent interactions with host molecules, including hydrogen bonding, electrostatics, and van der Waals, and hydrophobic interactions.103 Synthetically produced host–guest pairs have the capability to provide reversible guest exchanges, which may be utilized for the fabrication of self-repairable supramolecular gels.2,104
Kakuta et al. utilized host–guest interaction and developed self-healable supramolecular materials composed of cyclodextrins (CD) – guest gel crosslinked between poly(acrylamide) chains with inclusion complexes (n-butyl acrylate and N-adamantane-1-yl-acrylamide dissolved in aqueous solutions of acrylamide-CDs to form inclusion complexes) (Fig. 11).105 The produced self-healable material was capable of recovering its initial strength even after being sectioned in the middle.105 Several other researchers also found similar results, which are summarized in Table 2.
 | ||
| Fig. 11 Host–guest interaction of the αCD–nBu gel(m, n) and the βCD–Ad gel(m, n) [reproduced from ref. 105 with permission from the Wiley]. | ||
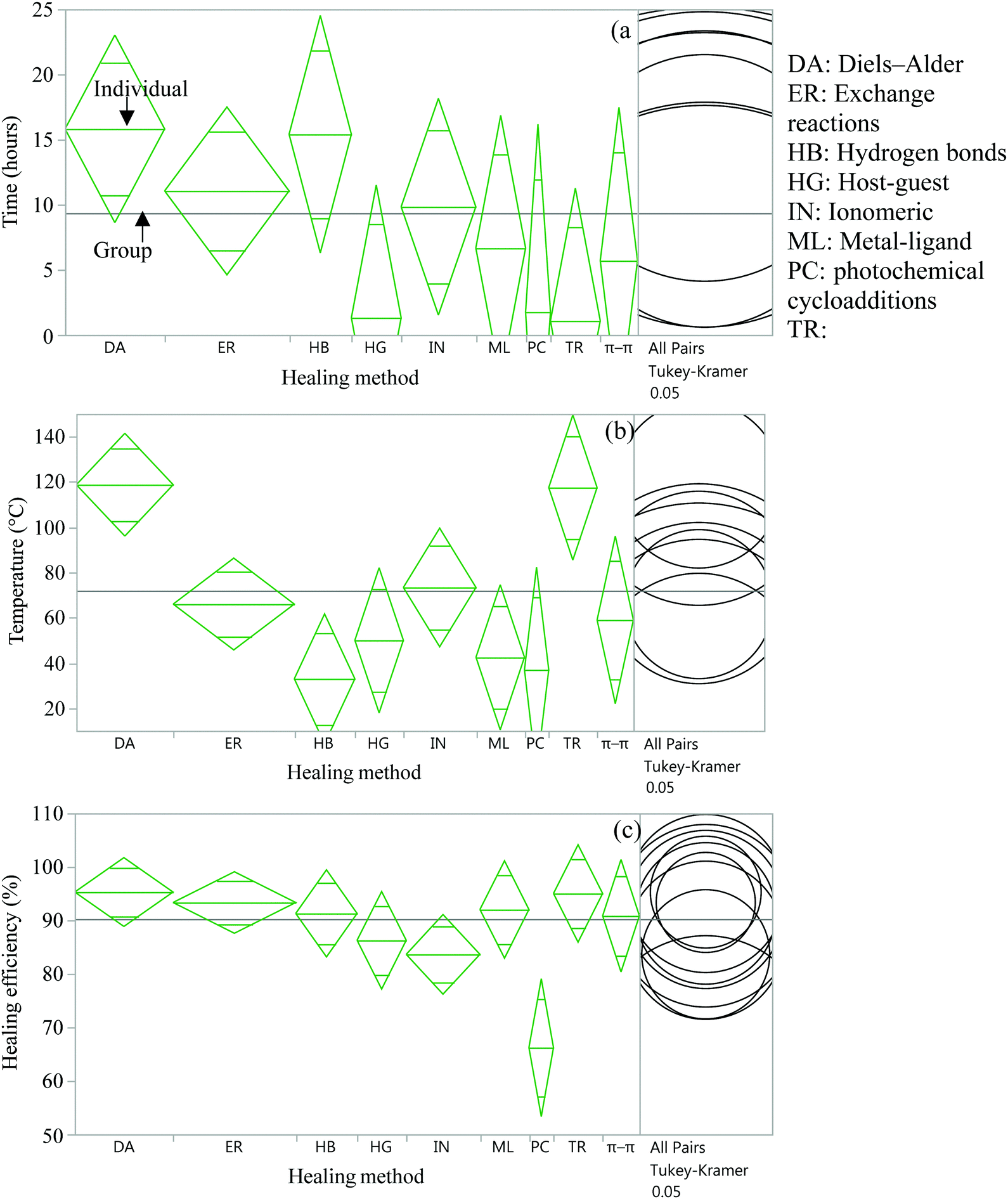 | ||
| Fig. 12 Analysis of variance and Tukey HSD test to compare time (a), temperature (b), and efficiency (c) of several healing approaches. | ||
From the statistical analysis of several self-healing reactions in terms of healing time, temperature and efficiency, it was found that there is a significant statistical difference among several intrinsic approaches in terms of healing temperature and healing efficiency (p < 0.05) (Fig. 12(b and c)) but no significant difference in healing time (Fig. 12(a)). It is found that DA and transesterification reactions require significantly higher temperatures (p < 0.05), whereas hydrogen bonds require significantly lower temperatures (p < 0.05) for self-healing compared to other intrinsic healing approaches. In the case of healing efficiency, photochemical cycloaddition provides significantly lower healing efficiency (p < 0.05) compared to other self-healing methods.
Since the data for these analyses were obtained by different researchers under different conditions, one has to be careful with this interpretation. However, the data were randomly selected for analysis to minimize the effect of intrinsic variables.
3.2. Physical interactions
Polymeric materials can recover their properties through physical interaction.133 The healing process, after damage, can be initiated simply by entropy of molecular diffusion (Fig. 13) caused by an external stimulus like heating.35 When polymeric materials are heated, diffusion of the polymer chain occurs and results in the formation of new entanglements and closure of the crack.134,135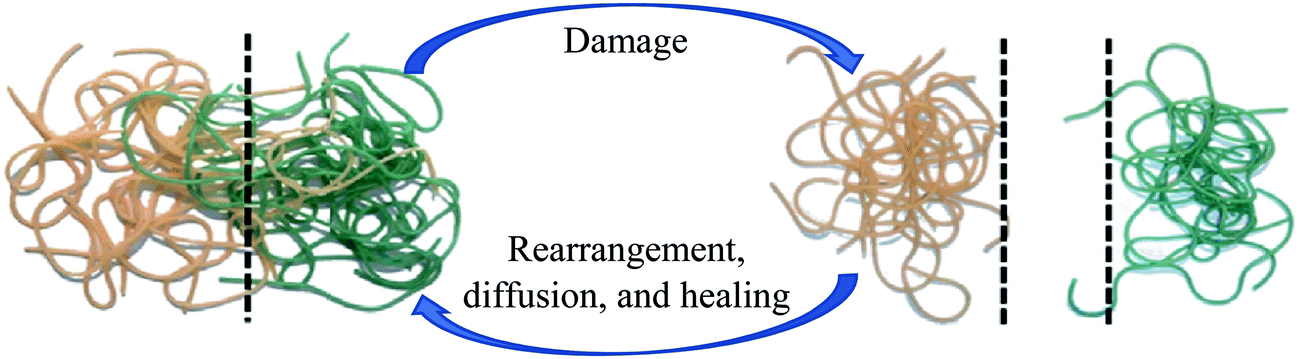 | ||
| Fig. 13 Schematic of physical self-healing process [reproduced from ref. 136 with permission from the Wiley]. | ||
Several researchers utilized this mechanism to develop self-healing materials (Table 3). Yamaguchi et al. developed self-healing materials utilizing molecular diffusion and entanglement of dangling chains in a weak PU gel.137,138 Rahmathullah et al. demonstrated that epoxy-amine thermoset materials showed multiple self-healing properties when the materials were heated at 185 °C temperature as molecular diffusion occurred due to heating.139 Both thermoplastic and thermosetting polymers have the capability of self-healing by molecular diffusion.
| Healing mechanisms | Materials | Healing conditions | Tensile strength (MPa) | Healing efficiency | Ref. |
|---|---|---|---|---|---|
| Healing efficiency (%) = recovered properties/original properties. | |||||
| Diffusion of polymer chains | PVAc/graphene | 60 °C | — | 89% | 162 |
| Lignin-modified graphene and waterborne PU | Infrared | — | 171% (elastic modulus) | 163 | |
| n-Butyl acrylate based zinc ionomer with magnetic nanoparticles | 70 °C for 30 min or magnetic field for 15 min | — | 90% | 164 | |
| Polymer blends | Epoxy resin and cyclic olefin copolymer | 190 °C for 1 h | 20–35 | 89% | 165 |
| Thermosetting vitrimer with thermoplastic polyurethane | RT, 3 min | 0.46 | Fracture surface joined | 166 | |
| Polymer blends and shape memory | PU elastomer and a low melting temperature Polycaprolactone | 90 °C for 30 min | 37.2 | 100% | 167 |
| Shape memory assisted self-healing | Polycaprolactone and polyfurfuryl methacrylate | 120 °C, 15 min | 13 | 75.1% | 168 |
| Graphene oxide incorporated into a thermoset PU | NIR (2.5 W cm−2) light for 30 min | 16.6 | 85% | 169 | |
| Epoxy vitrimer, fabricated by epoxy soybean oil and fumaropimaric acid | 180 °C for 60 min | 16 | Reducing crack size | 170 | |
| Epoxy/polycaprolactone copolymers cured with cashew nut shell | 80 °C for 4 h | 12.52 | 93.7% | 171 | |
| Boron nitride/epoxy composites | 140 °C for 2 h | 55 | Almost 100% | 172 | |
| Shame memory and remote self-healing | Poly ethylene glycol (PEG), poly(ε-caprolactone), and 2-ureido-4-pyrimidone supramolecular moieties | 70 °C for 10 min and irradiating with light (365 nm) for 1 min | 7.2 | 86% | 173 |
| Remote self-healing | Fluorescence-responsive hydrogel with a triple network structure, prepared from PVA, agarose, chitosan, and glycerin | RT, 1 min | 10.21 | 100% | 174 |
| Polyurethane nanocomposite and carbon nanotubes | 80 °C for 24 hours or NIR irradiation for 1 min | 3.64 | 91.1% (heat), 84.9% (NIR) | 175 | |
| Azobenzene-functionalized multiwalled carbon nanotubes | NIR (808 nm) irradiation for 15 min or UV (365 nm) irradiation for 25 min | — | Almost recover the scratch to original | 176 | |
Thermoplastic polymers are very much suitable to introduce self-healing properties by molecular interfusion as molecules within this polymer become mobile at temperatures above their glass transition (Tg) temperature.140,141 When two parts of the damaged or cracked polymer are brought into contact at a temperature above the glass transition (Tg), the molecular diffusion occurs between two parts resulting in healing of the cracks and recovery of mechanical strength of those polymers.141 Experimental results regarding healing conditions also revealed that polymers generally heal at atmospheric pressure or in a vacuum, with a range of temperature from −50 to +100 °C, and time from minutes to years.141 However, in some thermoplastic polymers like PMMA, the Tg is relatively high (above 100 °C) and requires high temperature for the initiation of self-healing. This limitation can be overcome by mixing the polymer with a substance that has a plasticizing effect on the polymer. For example, Lin et al. and Wang et al. treated PMMA with methanol and ethanol, respectively, and reduced the effective glass transition temperature of PMMA to a range of 40 to 60 °C.134,142 Sirajuddin and Jamil demonstrated self-healable hydrogel of poly(2-hydroxyethyl methacrylate), where 92% healing efficiency was achieved due to the intermolecular diffusion of dangling chains and the chain slippage when the damaged hydrogel was heated to 48.5 °C.143
Similar to thermoplastic, thermoset polymers can also achieve healing properties via physical interactions. At ambient or increased temperatures, healing of cracks or damage occurs in the polymeric network through interdiffusion of dangling chains or chain slippage.138 However, thermosetting materials have more limitations than thermoplastics as they cross-link together during the manufacturing process and form an irreversible chemical bond.144 Hence, the glass transition temperature of the polymer is very high. For obtaining mobility, it is necessary to heat to very high temperatures where molecular degradation may occur, and materials can lose their stability.35 These limitations can be minimized by blending thermoplastic additives into a thermoset polymer matrix (Table 3).
Self-healing materials can also be obtained by incorporating low melting thermoplastic additives into a thermoset matrix. Upon heating, the thermoplastic material would mobilize and diffuse through the thermosetting matrix resulting in crack healing.17 Hence the original thermoset matrix will remain stable. Healing will occur due to the flow of low melting thermoplastic additives, which will solidify again upon removal of heat. This technology was first reported by Zako and Takano in 1999, where thermoplastic additives were embedded in a glass/epoxy composite.145 About 100% healing of stiffness was obtained when damaged materials were heated at 120 °C for 10 min.145 Hayes et al. blended a thermoplastic material of linear poly(bisphenol-A-coepichlorohydrin), into thermosetting resin composites and obtained a self-healable material that can recover its 70% pre-fractured strength after healing at 130 °C for 1 h.146 Luo et al. developed a thermally re-mendable thermoset epoxy resin by dispersing phase-separated poly(caprolactone) in an epoxy matrix.147 When the material is heated at 190 °C for eight min, the PCL melts and undergoes a volumetric thermal expansion to fill the damage with thermal-mending efficiencies of about 100%. Several other researchers also introduce healing properties to thermoplastic, thermosetting and blended materials using a physical interaction method, which is summarized in Table 3.
3.3. Shape memory assisted self-healing
Shape memory materials are characterized by the ability to recover their original shape from a significant and apparently plastic deformation upon application of a specific stimulus like temperature, electrical or magnetic fields, solution, or light.38,148 Shape memory assisted self-healing (SMASH) materials have attracted increasing attention of researchers, as both self-healing and shape memory properties are intelligent and have promising applications.149One of the biggest concerns of developing self-healing materials is that, in some cases, it is necessary to manually close or bring in contact the two damaged parts for the initiation of self-healing. SMASH materials have excellent potential to overcome this limitation. In this process, the cracks or damaged parts will close automatically as shape-memory materials have a shorter predefined memorized shape, which generates a shrinking force on split parts and forces them to return back to the original position (Fig. 14).150 Due to this excellent feature, a number of research projects to fabricate self-healing materials utilizing shape-memory properties have been growing over the last few decades (Table 3). Cheng et al. patented Self-healing tribological surfaces in 2006, where complete recovery of scratched surfaces has occurred upon heating to 150 °C and then cooling the materials.151 Luo and Mather reported SMASH coating fabricated by electro-spun thermoplastic poly(ε-caprolactone) fibers, which were randomly distributed in a shape-memory epoxy matrix.152 The obtained SMASH coatings not only heal structural damage but also functionally restore nearly total corrosion resistance.152 Rodriguez et al. demonstrated SMASH, fabricated by blending a cross-linked poly(ε-caprolactone) network with linear poly(ε-caprolactone) and obtained excellent self-healing of films with nearly complete healing properties.153
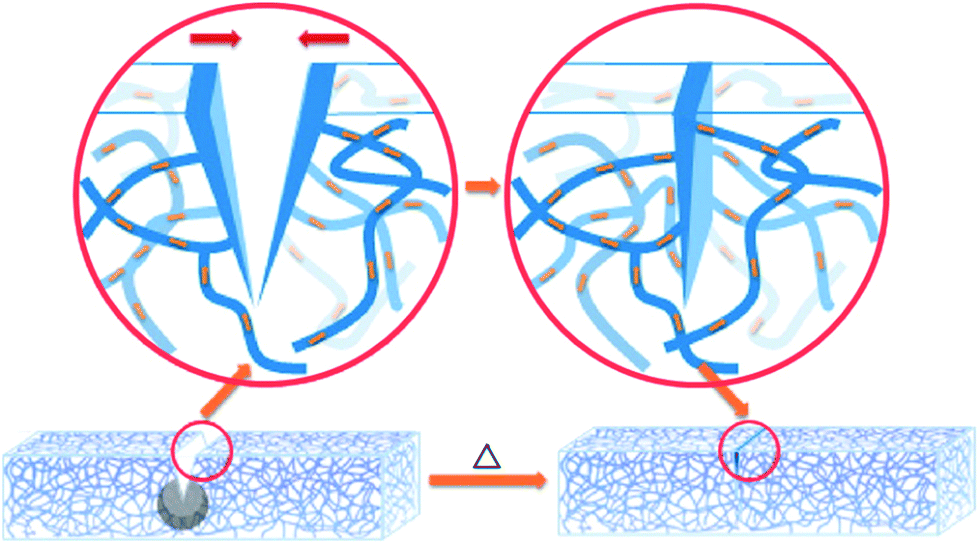 | ||
| Fig. 14 Schematic illustration of the shape memory assisted self-healing [reproduced from ref. 152 with permission from the ACS]. | ||
3.4. Remote self-healing
Remote self-healing is a process of using energy (like electric and magnetic fields) from remote sources and converting this energy into localized heat in damaged areas.2 Due to this localized heat, chain mobility of polymers in damaged areas increases, and interfacial diffusion occurs, which causes self-repairs.38 One of the examples of remote self-healing is using superparamagnetic γ-Fe2O3 into a thermoplastic polymer network (Fig. 15).154 When damage occurs, a remotely oscillating magnetic field is applied, which causes oscillation of γ-Fe2O3 nanoparticles, and as a result, generates heat, and interfacial diffusion occurs. Finally, permanent self-healing (multiple times) on the polymer network has been observed with a healing efficiency of up to 98% in terms of Young's modulus and strain at break.154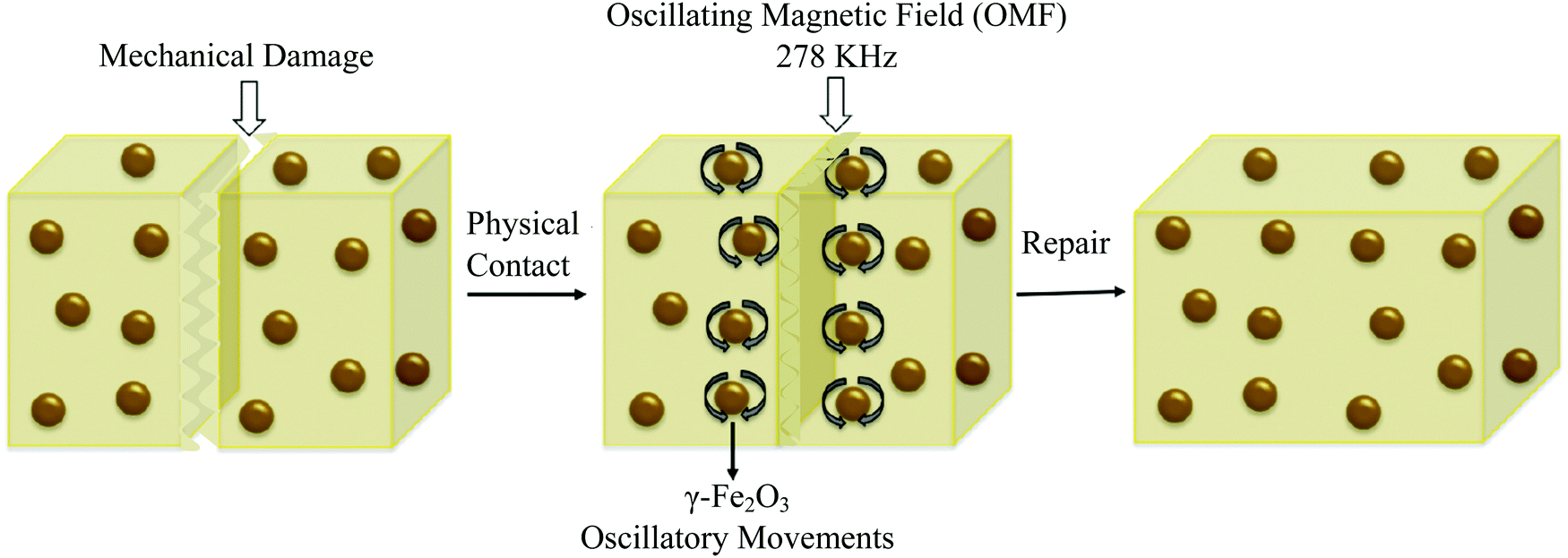 | ||
| Fig. 15 Remote self-healing based on magnetic nanoparticle incorporated thermo-plastic polymers [reproduced from ref. 2 with permission from Elsevier]. | ||
Another method of remote self-healing is the use of selective wavelengths that have the ability to transform electromagnetic radiation into other forms of energy.38 Some metal nanoparticles like AuNPs and AgNPs can transform incident light (usually near-infrared spectrum) into local heat by generating surface plasmon resonance, resulting in initiation of localized healing.155–160 Similarly, graphene layers within a polymer matrix cause localized heating upon application of infrared light, electricity, or electromagnetic waves.42 Huang et al. integrated few graphene layers with a thermoplastic polyurethane matrix. It was found that the polymer composite repeatedly healed (up to 20 cycles) with excellent healing efficiencies, higher than 98%, when infrared light, electricity, or electromagnetic waves were applied to the composite.161
4. Extrinsic self-healing
The mechanism of extrinsic self-healing is completely different from the intrinsic one. Here, the material itself does not give any reversible or healing reaction. A healing agent, which is stored in a container (capsules, vascular networks, or nanoparticles), has to be inserted into the materials during the production process. As soon as cracks occur within the materials, the capsules or vascular networks rupture, release the healing agent into the crack planes due to the capillary effect, fill the damaged surface, and restore surface integrity by curing.35,177 Catalysts are embedded, which trigger the polymerization of healing agents and restore the crack surface.178 Materials used as a liquid healing agent must contain certain properties, including good stability (to store in the capsule/vascular network for long periods without reaction), suitable flowability (to fill the damage easily by capillary action), and fast reactivity with the matrix for quick healing.3,17,179 The mechanical properties of the healing agent should be similar to the matrix where it was incorporated.3,17,179So far, a number of materials have been developed using several healing agents like dicyclopentadiene (DCPD), epoxy and amine, mercaptan, phenylacetate, ethyl phenyl acetate, chlorobenzene, linseed oil, tung oil, and alkyd resins.180 White et al. first reported a microcapsule-based self-healing material incorporating DCPD healing agent triggered by a ruthenium-based Grubbs' catalyst that initiates ring-opening metathesis polymerization (ROMP) resulting in bonding of the crack surface.18 Ruthenium-based catalysts exhibit excellent functional group tolerance and higher air and water stability, but thermolytic decomposition limits the usefulness of these catalysts.181,182 In order to overcome this limitation, Guadagno et al. used a Hoveyda–Grubbs’ second-generation catalyst to polymerize healing agents at high temperatures.181
In some structural applications, like in the field of aeronautic applications, self-healing mechanisms should be carried out at very low temperatures. Raimondo et al. developed self-healing materials which have healing functionality as low as −50 °C and have very good potentiality to be used in aeronautic applications.178 The material was developed based on the metathesis polymerization of 5-ethylidene-2-norbornene/dicyclopentadiene (ENB/DCPD) blend activated by a Hoveyda–Grubbs’ 1st generation (HG1) catalyst.178 The experiment revealed that the lower healing efficiency (72%) is related to the deactivation of some catalyst particles which are in contact with the oxirane rings (during the curing reactions), reducing the amount of active catalyst in the ROMP reactions.178 When a Hoveyda–Grubbs' 1st generation catalyst is solubilized at a molecular level, an equimolecular reaction occurs between the epoxide ring and alkylidene of the ruthenium resulting in deactivation of the catalyst, lower healing efficiency, and increase of the cost of the catalyst based self-healing system.178 This catalyst deactivation was overcome by the authors in their subsequent research and increased healing efficiency by about 95%.183 In subsequent research, Guadagno et al. prepared a protected Ru-catalyst, immobilized on graphene sheets that causes the hindrance of catalyst deactivation because of equimolecular reactions occurring between the catalyst complex and the oxirane ring.184 Longo et al. synthesized a new stable ruthenium initiator suitably designed for self-repairing applications which preserves the catalytic activity at high temperatures (180–200 °C), rapidly polymerizes cyclic olefins in reactive environments, reduces the amount of initiator and cost of structural self-healing materials, and provides healing efficiency of 103%.185
4.1. Chemistry of extrinsic healing
Chemical reactions occur in extrinsic healing approaches among healing components and matrix materials to form permanent bonds and repair the materials. Most of these reactions occur by forming covalent bonds, some of which are discussed in the previous section (reversible covalent bonds). Along with reversible covalent bonds, several other nonreversible bonds, including living polymerization, ring-opening, and polycondensation reaction, have been utilized to form bonds among healing components and matrix materials.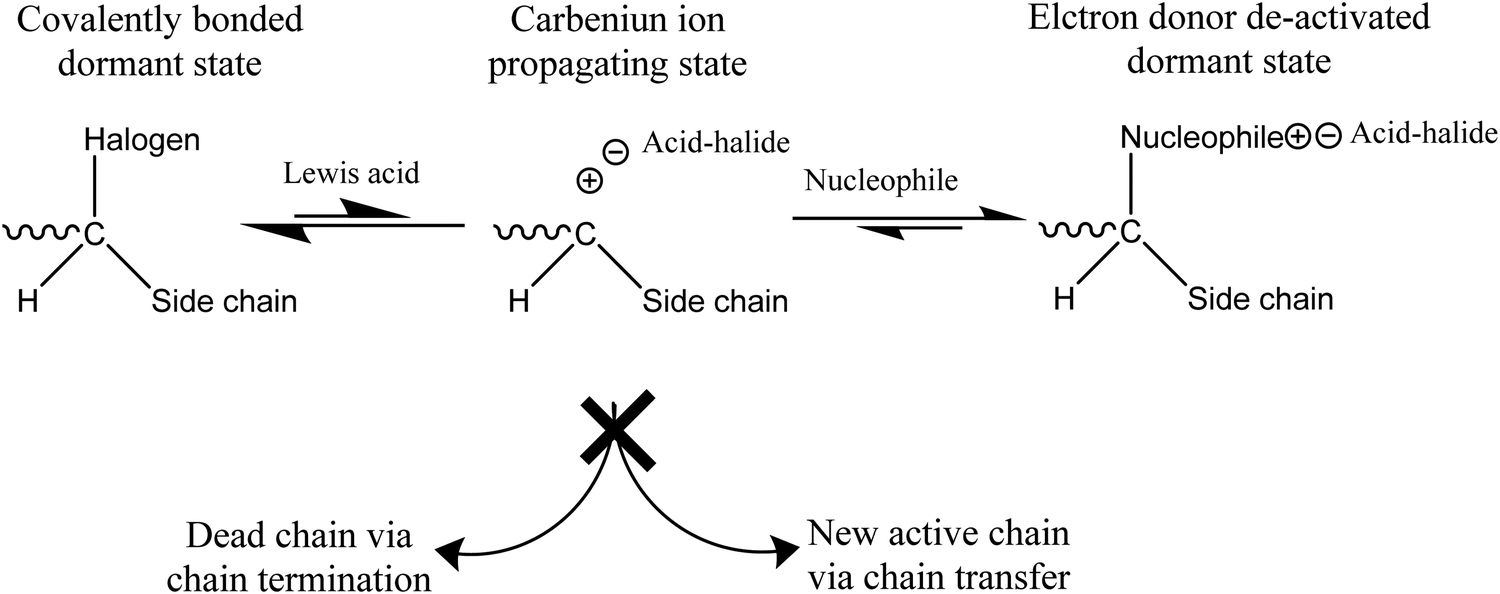 | ||
| Fig. 16 Mechanism of a living polymerization [reproduced from ref. 141 with permission from the Elsevier]. | ||
The manufacturing process of living polymerization can be accomplished by both ionic polymerization and free-radical polymerization. For designing self-healing materials, a free radical living polymerization process is likely more suitable due to its high reactivity.141,187,188
One of the commonly used living polymerizations in self-healing processes is atom transfer radical polymerization (ATRP). ATRP is incorporated as a healing component in the matrix in the form of capsules or a vascular system. Whenever crack occurs in the matrix, monomer is released, and cracks re-bond, continuously healing the matrix at ambient temperature.3 So far, several methacrylate monomers, including methyl methacrylate, benzyl methacrylate, and glycidyl methacrylate, have been effectively homopolymerized in ATRP systems.3
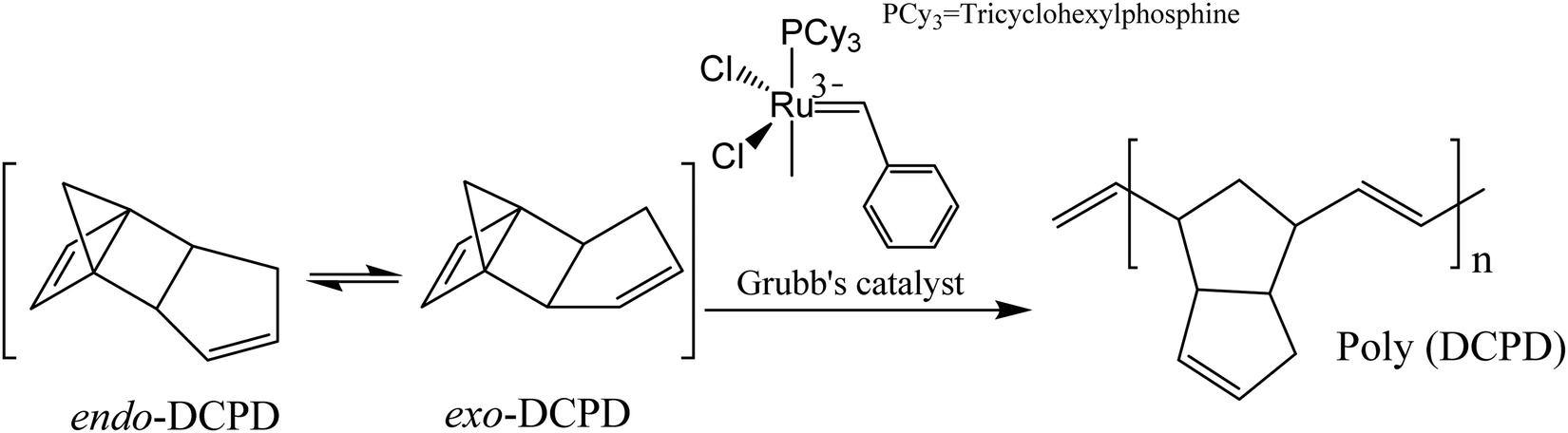 | ||
| Fig. 17 ROMP of endo- and exo-DCPD with first-generation Grubbs’ catalyst. | ||
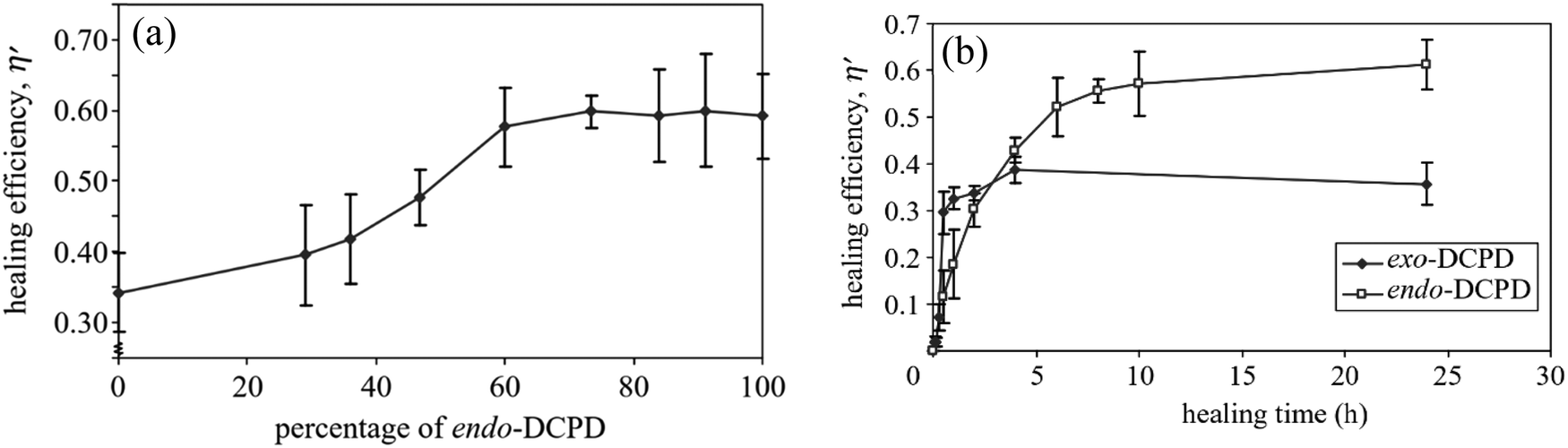 | ||
| Fig. 18 Effect of DCPD on self-healing materials. (a) Effect of exo/endo-DCPD blends on healing efficiency. (b) Progress of healing efficiency for endo- and exo-DCPD with time [reproduced from ref. 189 with permission from the Royal Society of Chemistry]. | ||
Although DCPD and Grubbs’ catalyst are the most explored healing chemistry of ROMP, they have some drawbacks as well. The melting temperature of Grubb's catalyst is low (153 °C), and its reactivity alters and decreases with prolonged exposure to oxygen and moisture.190 To avoid these limitations, Kamphaus et al. investigated tungsten chloride (WCl6), which has relatively high melting point (275 °C),191 and Lee et al. proposed 5-ethylidene-2-norbornene (ENB), which is known for much faster ROMP reaction.192 Several other researchers also suggested ENB as a prospective material for the polymerizable monomer of a two-component healing agent based on the ROMP mechanism.193–198 However, Aïssa et al. mentioned that the formed cross-link structure of ENB is relatively weaker than the polymerized linear chain structure created using DCPD.199 Hence some others suggested combination of DCPD and ENB to obtain a balance of reactivity and structural strength.200
 | ||
| Fig. 19 Schematic illustration of polycondensation between HOPDMS and PDES [reproduced from ref. 3 with permission from Elsevier]. | ||
4.2. Capsule-based self-healing
One of the most common extrinsic self-healing methods is capsule-based healing, where chemicals are encapsulated within a suitable material. Originally, the encapsulation techniques within the materials were evolved from nature. For example, natural latex is encapsulated in elongated plants’ cells called laticifers.37 In some trees, like Hevea brasiliensis, the function of latex is to heal physical damage.204 Inspired from nature, synthetic materials are also designed by incorporating capsules, ranging from macroscale to nanoscale, in order to obtain healing properties.Among different ranges of capsule size, microcapsule-based systems have drawn much interest during the last decade where micron-sized particles, normally liquid healant or liquid crosslinking hardeners (e.g., catalyst), are encapsulated in an inert shell.150 The role of the inert shell is to isolate and protect the encapsulated substance from unwanted or undesirable reactions in the external environment.150,205 These microcapsules are then embedded in the matrix, either a single or double or a dual-microcapsule archetype. In single microcapsule-based systems, the catalysts are co-distributed throughout the matrix material to initiate curing.83 In double or dual-microcapsule-based systems, liquid healant and crosslinking hardeners are encapsulated in separate capsules.133 In double-walled microcapsules, two healing components are incorporated in the same microcapsule, separated by the shell wall. Fig. 20 illustrates the single capsule-based self-healing concept.
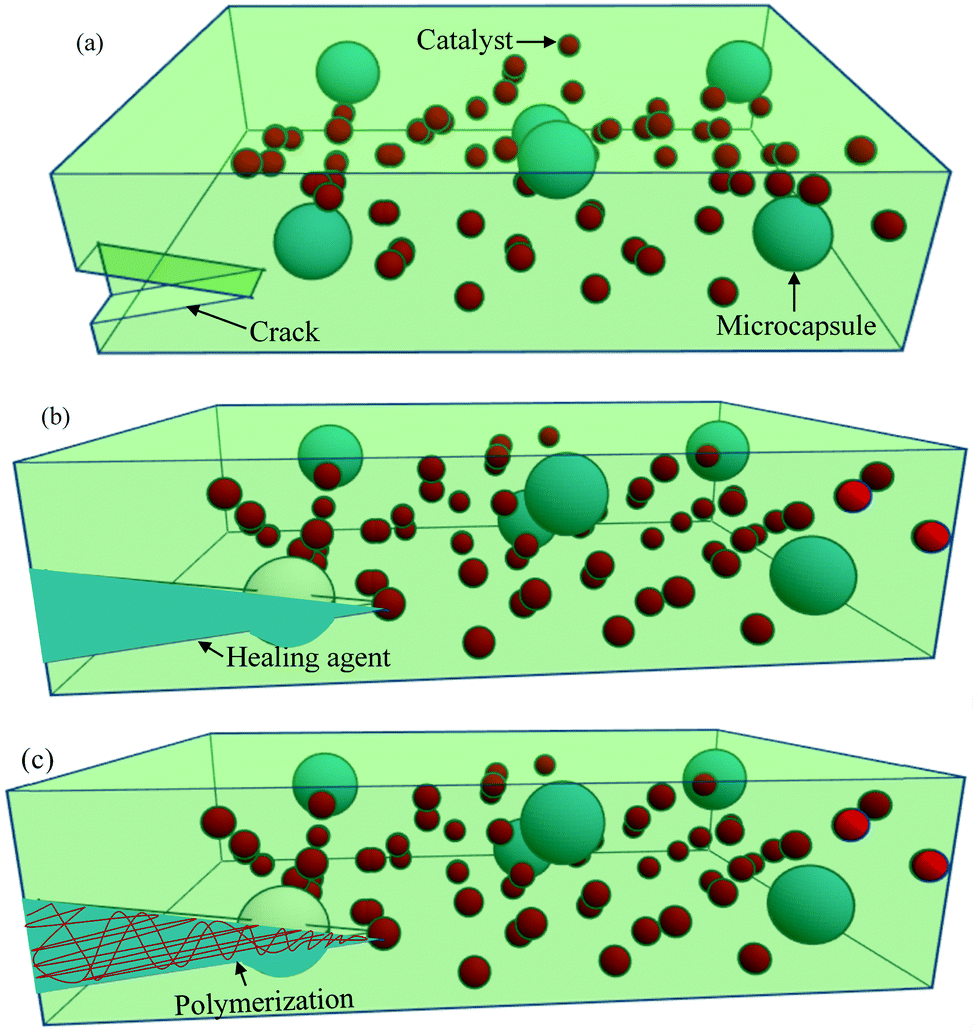 | ||
| Fig. 20 Microencapsulated based self-healing process: (a) crack forms in the matrix, (b) crack ruptures the microcapsules, releasing the healing agent into the crack through capillary action, (c) healing agent contacts the catalyst, resulting in polymerization and repair of the matrix. | ||
Researchers have developed a number of self-healing materials based on the capsule-based approach (Table 4). White et al. developed the first successful autonomic self-healing technology based on this approach, where 75% of toughness was recovered after damage.18 Brown et al. reported a 90% recovery of fracture toughness after healing and demonstrated the effects of size and concentration of microcapsules on healing performance.206
| Healing type | Healing components | Material to be healed | Healing conditions | Healing efficiency | Ref. |
|---|---|---|---|---|---|
| Healing efficiency (%) = recovered properties/original properties; RT = room temperature.a Tensile strength. | |||||
| Single microcapsule | Epoxy resin | Composite of fluorescent latent curing agents | 60 °C for 4 h | 85.6% | 247 |
| Epoxy resin | Polymer composite | 120 °C for 24 h | 79.1%a | 248 | |
| 5-Ethylidene-2-norbornene mixed with carbon nanotubes and Grubbs catalyst | Polymer composite | RT, 24 h | 80%a, 82% (conductivity) | 249 | |
| Double or dual microcapsule | Epoxy + ethylenediamine | Polymer composite | RT, 24 h then 50 °C for 6 h | 81.1%a | 250 |
| Isophorone diisocyanate + polyaspartic acid ester | Epoxy coatings | RT, 12 h | 93% | 251 | |
| Epoxy and hardener | Glass carbon fiber reinforced epoxy matrix | RT, 24 h | >100%a | 252 | |
| Encapsulated epoxy + encapsulated polyetheramine | Epoxy resin | RT, 24 h | 84.5%. | 253 | |
| Double wall microcapsule | 1,6-Diaminohexane (inner core), isophorone diisocyanate (outer core) | GO-modified double-walled polyurea microcapsules/epoxy composites | RT, 72 h | 80.43%a | 254 |
| All-in-one microcapsules | Encapsulated GMA–(PMMA–Br + CuBr/PMDETA)j | PS | RT, 48 h | 100%. | 255 |
| Microcapsules and hollow fiber | Diluted epoxy | Glass fibers–epoxy composites | 130 °C for 60 min | 97.6%a | 256 |
| >100% (shear) | |||||
| Hollow fiber/tube | Grubbs catalyst and DCPD | Glass fiber-reinforced epoxy composite | RT, 24 h | 60% | 257 |
| Epoxy–polyacrylonitrile (PAN) and mercaptan–PAN | Elctrospun nanofiber composite | 10 °C, 200 min | 87% | 258 | |
| Epoxy resin and hardener | Epoxy/glass composite | — | 42.2%a | 259 | |
| Epoxy resin and hardener | Glass fibers/epoxy composites | RT, 7 days | 55% | 260 | |
| Oily rejuvenator | Bituminous composites | 30 °C for 24 h | 78% | 261 | |
| Microvascular network | Sodium silicate | Cementitious structure | RT, 28 days | 81% (crack closing) | 262 |
| Epoxy monomer and curing agent | Waterborne polyurethane matrix | — | 99.34% (corrosion protection) | 263 | |
| Epoxy resin epichlorohydrin hardener | Glass fiber reinforced epoxy composites | 25 °C for 7 days | 89%a | 264 | |
| Grubbs catalyst and DCPD | Epoxy coating | 25 °C for 12 h | 70%a | 241 | |
4.2.1.1. In situ polymerization. In situ polymerization was the first encapsulation technique utilized by Brown et al. to produce microcapsule containing DCPD with poly(urea-formaldehyde) as the shell material.208 The shell material, produced by in situ polymerization, is hydrophilic35 and the core healing agent is hydrophobic,35 which acts as the dispersed phase. The shell was produced by dissolving monomers and initiators, either the dispersed or the continuous phase.3 Polymerization, initiated by heat and catalyst, normally occur or accumulate on the surface of the core material droplets resulting in microcapsules with the desired core material.3,35 The produced microcapsules are normally spherical in shape with an average diameter from 10 to 1000 μm.3 So far, this technique has been successfully used to produce urea-formaldehyde, melamine-formaldehyde, melamine-urea-formaldehyde (UF), phenol-formaldehyde, and several other microcapsules.37
4.2.1.2. Interfacial polymerization. Interfacial polymerization is another encapsulation technique that has been successfully utilized to produce several microcapsules, including polyurethane and polyacrylate.37 For example, Liu et al. developed a self-healing coating where they utilized interfacial polymerization of epoxy resin droplets with ethylenediamine to produce microcapsules.209
In this technique, microcapsules are produced by dispersing one phase containing a reactive monomer (normally core material) into a second immiscible phase.210 After that, another monomer that is soluble in the dispersed phase is added.3 Both monomers react at the droplet surface (interface), forming a polymeric membrane around the core material.210 This technique can also be utilized for producing double-shell wall microcapsules.37 For example, Caruso et al. produced robust double-walled polyurethane/poly(urea-formaldehyde) microcapsules for developing self-healing materials.211 A double shell wall is also used when two reactive healing agents are utilized to develop self-healing materials.35
Interfacial polymerization is very much suitable for producing microcapsules with high encapsulation efficiency. However, in certain cases, microcapsules contain unreacted shell monomers, which may react and deactivate the core materials.3
4.2.1.3. Miniemulsion polymerization. Minieemulsion polymerization has been used to produce nano size capsules. In some applications, especially in thin coating and biomedical, nano-size capsules between 50 and 300 nm are highly suitable for developing self-healing materials. A miniemulsion technique can be utilized in these cases to produce stable and narrowly distributed capsules with sizes ranging from 50 nm to 500 nm.207 For example, Van den Dungen and Klumperman synthesized liquid-filled nanocapsules with a size range of 150 nm to 350 nm for self-healing through the miniemulsion technique.212 However, this technique is not suitable for healing of large size cracks as produced nanocapsules cannot store a sufficient amount of healing materials.3
Temperature is one of the key factors that influences the size of the capsules and production rate. At lower temperatures, capsule size decreases, but the production rate also decreases. Hence, an optimum temperature of about 55–70 °C has been used for the synthesis of microcapsules in self-healing applications.213 Another important parameter that influences the size of microcapsules is the agitation rate. It was observed that capsule size decreases with the increase of agitation rates and vice versa (Fig. 21).208,215,216 Higher agitation rates favor synthesis of microcapsules with smoother surfaces.217 The role of pH is also important for the synthesis of microcapsules with smoother surfaces. For example, during the synthesis of UF microcapsules, a soother surface has been obtained when the pH value was kept at 3.213 Surfactant/emulsifier is another important factor that has an influence on capsule size and the shell wall of microcapsules.218,219 For example, Ting et al. used different concentrations of emulsifiers (0.5 wt%, 3 wt%, and 5 wt%) and obtained capsules with the smallest size at 5 wt% of emulsifiers with uneven size distribution.220
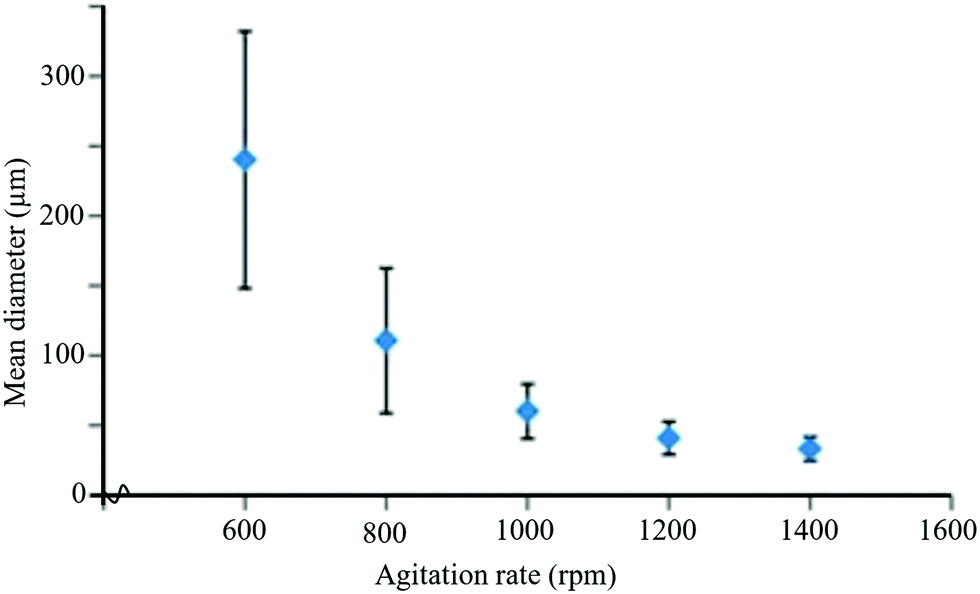 | ||
| Fig. 21 Change in mean diameter of microcapsules with agitation rate during the encapsulation process [reproduced from ref. 221 with permission from the Royal Society of Chemistry]. | ||
4.3. Hollow-fiber systems
Hollow fiber systems significantly contribute to the development of polymer and fiber-reinforced self-healing materials. Discrete microcapsules, with their inherent geometric incompatibility to fiber-reinforced composite materials, lead to the development of hollow fiber-based self-healing systems.83 The main principle of this system is to store some functional components inside of the hollow fiber, which should remain fluid at least at the healing temperature.150,177 The functional components have been filled through the open ends of hollow fibers either by capillary action or vacuum assistance or through surface pores.224 After filling, the open end was sealed. When damage or rupture occurs, the functional component or filling agent flows out to the damage site and heals the rupture.177 So far, three different types of self-healing designs have been developed based on hollow fiber systems (Fig. 22).19,20,225–228 These are single-part adhesives where hollow fibers are filled with the same type of functional unit, two-part adhesive where two different kinds of functional units are filled in adjacent hollow fibers, and two-part adhesive where one type of functional unit is incorporated in hollow fibers, and another type is into microcapsules.177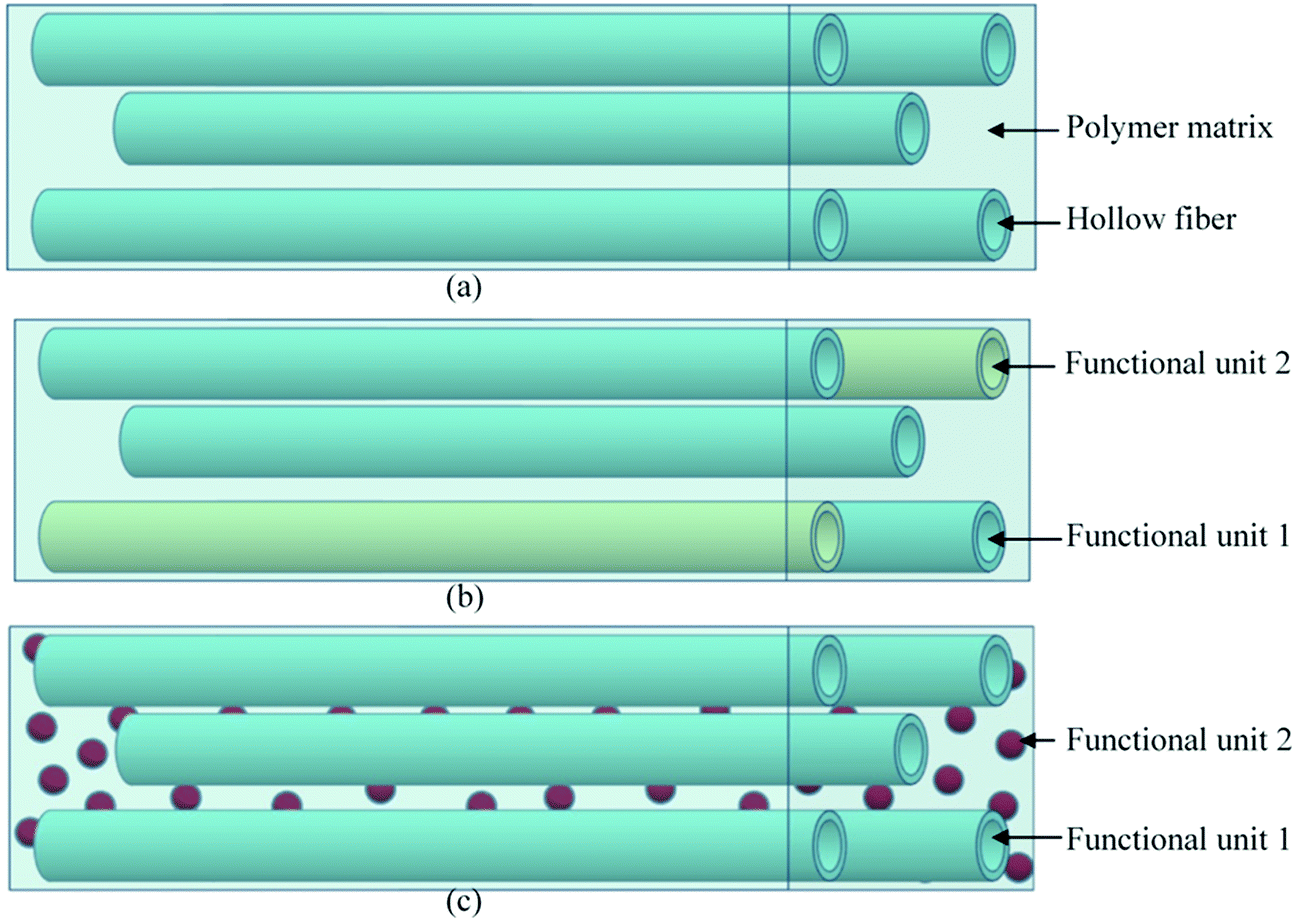 | ||
| Fig. 22 Schematic diagram of the hollow fiber-based self-healing system; (a) single-part adhesive, (b) two-part adhesive and (c) two-part adhesive (one in a hollow tube and another in microcapsules). | ||
This concept of hollow fiber-based self-healing was originally introduced by Dry for repairing concrete where hollow glass fibers (HGF) were filled with a liquid epoxy resin.229 However, one of the limitations of the HGF is that it is comparatively larger in diameter (on millimeter-scale) than those of the reinforcing fibers in composites, which may initiate the failure of composite materials.230 To minimize the limitation related to large fiber diameter, Bleay et al. developed HGF with smaller diameters (external diameter of 15 μm and an internal diameter of 5 μm).230 However, fibers with smaller diameters have a problem in delivering the resin into the crack due to the high-viscous epoxy resin.99 To overcome this problem, Bond and co-workers optimized the production process to obtain HGF with a diameter of 30–100 μm and hollowness of about 65 percent.19,226,227 Along with HGF, recently, researchers have focused on hollow nanofibers and several other polymeric fibers.231 Compared to HGF, hollow polymer fiber is more efficient in controlling the diameter from nanometer to micrometer range.232 The production process and filling of functional materials have been claimed to be more effective.133 Liu et al. utilized water degradable poly(lactic acid) (PLA) fibers to develop self-healing materials.233 The PLA fibers were filled with a metal oxide precursor healing agent (TiCl4) and then incorporated into subsurface polymer layers of multilayer films. When cracks occurred in the multilayer films, atmospheric moisture degraded the PLA fibers, releasing TiCl4 into the damaged region resulting in repair of the multilayer film.233 Lanzara et al. proposed tougher and automated self-healing materials using carbon nanofiber, which also acts as a reinforcing material for the composites. The study shows that when the fiber wall suffers the formation of a crack, a certain amount of organic molecules stored inside the fibers escape into space in a few picoseconds, which indicates the potential of carbon nanofibers to be successfully used in the next generation of stronger, lighter and self-healing materials.234 Some other researchers also developed self-healing materials based on the hollow fiber system which are summarized in Table 4.
Numerous benefits have been obtained when self-healing materials have been developed using hollow fiber systems. One of the major advantages of this system is that hollow fiber can be used both as a structural reinforcement and as large storage of functional or healing materials.224 In this process, it is possible to obtain multiple healing after repetitive damage in the same place of materials. Healing of large cracks is also possible through this method as large volumes of functional materials can be transported to longer distances and refilled from outside. However, there have been some challenges as well in this technique, including uniform distribution of fibers throughout the materials, including accurate stoichiometric mixing (where the agent is multi-component), and proper viscosity of the healing agent to ensure flowability.83,177
4.4. Vascular self-healing
The vascular self-healing method is one of the most promising extrinsic healing approaches that mimics vascular networks of both animals and plants. Inspired by the transport networks of the human circulatory system235 and plant vasculature system,22,236 this method utilizes microchannels or vascules to mimic those networks and deliver healing agents to the damaged areas (Fig. 23).83,133 This method has lots of similarities with hollow fiber systems. The main difference is that instead of discrete tubes, interconnected vascular networks transport the healing agents.83 Due to this interconnected network, it is possible to continuously deliver the healing agent to the damaged area by attaching larger storage vessels at one end of the vascular network.23 Hence, this approach has excellent potential to give multiple healing events of one damage site, which is one of the major advantages of this approach.224 | ||
| Fig. 23 A schematic of the microvascular healing concept. | ||
The manufacturing stage of developing a microvascular network is very important. There are several potential techniques for the production of microvascular networks, including laser micromachining, soft lithography, electrostatic discharge, and fugitive inks.99 Some researchers have utilized the technique of fugitive ink, which is a mixture of high- and low-molecular-weight hydrocarbons.237 At first, fugitive inks are deposited according to the design through a cylindrical nozzle that becomes solid upon cooling.238 Then the scaffold is infiltrated with epoxy resin. After consolidating the resin, the fugitive ink is removed by applying heat under a light vacuum resulting in a 3D vascular interconnected network.177 The fabrication process can also be accomplished precisely with the help of 3D printers where structural design is programmed in the computer, and fugitive ink is printed by a robotic arm using a micronozzle of the desired channel size (Fig. 24).239 After fabrication of interconnected vascular networks, healing agents have been injected into the networks with the help of a syringe or a pump.177 Hence, this process allows continuous refilling of the healing agent, which gives opportunity for multiple healing of damage at the same location. Now researchers are trying to integrate pumps and reservoirs that can automatically sense and supply healing agents based on necessity.
 | ||
| Fig. 24 Fabrication process of 3D printed microvascular scaffolds. (a) Fugitive ink deposition; (b) infiltration of matrix resin, (c) resin solidification and (d) removal of fugitive ink and formation of a vascular network [reproduced from ref. 37 with permission from the Wiley]. | ||
Toohey et al. developed a 3D vascular self-healing epoxy system using direct-ink writing where epoxy was coated with Grubbs’ catalyst, and DCPD monomer was injected into the vascular network as a healing agent.240,241 Multiple healing up to 7 cycles was obtained by refilling healing agents; after that, the healing property was diminished due to the consumption of the embedded Grubbs’ catalyst that cannot be refilled similar to DCPD.240 To increase the number of healing cycles, Toohey et al. and Hansen et al. modified the design, and instead of using a solid catalyst, both healing agents and catalysts are transported by two isolated vascular networks.21,242 Toohey et al. achieved 16 repeated healing cycles with a 60% recovery of fracture toughness,21 and Hansen et al. obtained more than 30 repeated healing cycles with a 50–100% recovery of fracture toughness.242 Patrick et al. further modified the design of vascular networks and obtained full healing efficiency of 100%.243 Several other researchers also developed self-healing materials based on vascular network systems, which are summarized in Table 4.
Although vascular self-healing is the most effective technique of biomimetic healing approaches, still a lot of challenges need to be overcome for successful applications. The design structure of vascular networks is very complex, and it is difficult to achieve practically at large scales in synthetic materials.244 Similar to microcapsule and hollow fibers, two healing components require certain stoichiometry and good mixing and need to remain inactive until damage occurs.37 In microcapsules and hollow fiber systems, the healing agent is covered by a shell that prevents this reactive material from any direct contact with the host matrix. But the design of interconnected vascular networks allows direct contact of reactive healing materials with the host matrix, which is the major challenge of using this approach in several applications.133 Suitable release of healing materials to the damaged site is also very challenging. It is necessary to perfectly balance tube diameter, capillary forces, and flow properties of healing materials.35 Injecting healing components into vascular networks is another big task. At present, manual pumping is used to refill the healing component, but for autonomous healing, a sensing and automatic refilling system are required.83 Another challenge is to minimize the loss of healing components during repair of large or wide damage. Some healing components may lose or may not fully cover the damaged parts if two forces that act on healing agents, intrinsic forces (such as surface tension) and extrinsic forces (such as gravity), are not equal.133,245,246
4.5. Shape memory alloy-based self-healing
Shape memory alloy (SMA) wires are also used for self-healing purposes as SMA wires and particles can remember their shape and return back to the original shape after damage.150,265 In this technique, micron-size SMA wires were used to bring the two damaged or cracked parts together and then repair the damaged parts using partial melting techniques.266 When SMA wires are incorporated along with capsule or vascular based healing system, good healing efficiency is obtained with a minimal amount of healing agents due to the reduction of crack size (Table 5).267,268 Kirkby et al. embedded SMA wires with polymer composites and obtained 49% to 77% higher healing efficiency compared to a self-healable polymer composite without SMA wires.268 Similarly, Cohades et al. stitched SMA wires with fiber-reinforced polymer composites (woven glass fiber–epoxy–polycaprolactone matrix) and obtained 55% higher healing efficiency over the same polymer composites without SMA wires.269 However, there are some limitations to SMA wires as well. Integration of SMA wires alters the overall physical properties of host materials.267 Suitable incorporation of SMA wires with host materials is also very challenging. Improper placement of SMA wires in polymer matrixes or other materials may widen the crack size and decrease the healing performance.38| Healing method | Mechanism/healing components | Material to be healed | Healing conditions | Healing efficiency | Ref. |
|---|---|---|---|---|---|
| Healing efficiency (%) = recovered properties/original properties; RT = room temperature.a Tensile strength. | |||||
| Shape memory alloy (SMA) | SMA wires (Flexinol) as z-pins and epoxy resin | Glass fiber reinforced vitrimer-based shape memory polymer | 150 °C, 2 h | 100%a | 273 |
| Flexinol SMA wires and polycaprolactone-diol | Vitrimer based self-healing composite | 150 °C, 2 h | 58.6%a | 274 | |
| SMA wires, Araldite epoxy resin, and Aradur hardener | Graphene–glass fiber–reinforced polymer nanocomposites | — | 64% (bending strength) | 275 | |
| SMA wires, epoxy resin Araldite, hardener | Thermally reversible polymer matrix and SMA | 110 °C for 24 h | 92%a | 276 | |
| SMA strips, epoxy resin and hardener | Glass fibers/epoxy composites | RT, 7 days | 55% | 260 | |
| Nanoparticle | Diels–Alder reaction | Polydopamine nanoparticles based polyurethane elastomer | NIR (0.5 W cm−2), 30 s, or 130 °C, 10 min then 80 °C, 6 h | >90% | 277 |
| Metal–ligand (zinc–imidazole) interactions | Imidazole-containing silsesquioxane nanoparticles | 50 °C for 24 h | 80%a | 278 | |
| Reversible S–S, hydrogen, and iron(III)-carboxylate coordinate bonds | Magnetic gel based on thioctic acid (TA), FeCl3 and (Fe3O4) | Air temperature, 10 min | 100%a | 279 | |
| Dodecylamine (DDA) and n-methylthiourea (NMTU) | Epoxy coating | 5 days | >95% (corrosion inhibition) | 280 | |
| Ionic interaction between –COOH of poly(acrylic) acid and Al3+ ions. | Poly(acrylic) acid and aluminium (Al3+) ions, AgCl | 37 °C for 12 h | 80.2%a | 281 | |
4.6. Self-healing by nanoparticles
Self-healing by nanoparticles is another emerging and potential approach where cracks of polymers are repaired utilizing nanoparticles.141 In this process, nanoparticles are dispersed throughout the materials during fabrication. When a material breaks, the dispersed nanoparticles can migrate to cracks and repairs them.270 Lee et al. first demonstrated the self-healing potentiality of nanoparticles in polymers by using computer simulations.271 By integrating computer simulations, they showed that when a crack was generated in a multilayer composite, nanoparticles were driven to fill the cracks, and heal the composite repeatedly (average healing efficiency of 75 to 100%) until the depletion of nanoparticle concentration within the composites.271 Gupta et al. also confirmed some aspects of this computer simulation.272Chen et al. utilized core–shell nanoparticles and developed a new self-healing material that could almost completely heal a cut sample. In this process, the core–shell nanoparticles were fabricated by grafting dynamic self-healing polymer brushes of poly(acrylate amide) shell onto cross-linked polystyrene (PS) nanoparticles (NPs) via atom transfer radical polymerization (ATRP).12 The secondary amide group of poly(acrylate amide) brushes gives a simple reversible hydrogen-bonding motif, which fulfills the purpose of self-healing.12 Recently, Nan and Yan fabricated quasi-one-dimensional silver-nanoparticle-based metamolecules, which has fascinating self-healing ability in a linearly polarized optical field. It was found that the source of self-healing is the heterogeneous interparticle electrodynamic interactions, which were enhanced by the formation of unusual nanoparticle dimers.13 Several other researchers also developed self-healing materials based on nanoparticle systems, which are summarized in Table 5.
5. Self-healing ceramics
Ceramics have very good compression strength but are brittle in nature and susceptible to cracks that limit their area of application.140 Incorporating self-healing properties in ceramics may increase its potential application area. However, the healing process of ceramics is not similar to that of polymer matrix. To initiate self-healing in ceramics using diffusion processes, very high energy is required to overcome the strong directional chemical bond of ceramics. In general, temperature needs to increase to more than 1000 °C to obtain this high energy and initiate a self-healing process in ceramics.282 This limitation can be minimized utilizing oxidation induced healing of ceramic surface cracks.283Oxidation induced healing of ceramics can be achieved by dispersing suitable healing agents in ceramic composites during fabrication. When cracks occur, the environmental oxygen reacts with the dispersed healing components, rapidly fills the volume of a surface crack, and recovers the strength of ceramic composites.283 Nakao and Abe developed self-healing ceramics by dispersing SiC nanoparticles with alumina. It was found that the use of nanometer-sized dispersed SiC particles as a healing agent significantly reduced the activation energy required for the initiation of self-healing.284
While the crack healing temperature of solid-state Al2O3 is higher than 1400 °C, using SiC and Ti2SnC nanoparticles as healing agents, the temperature can be reduced to 870 °C and 500 °C, respectively.283 Some other researchers also achieved self-healing properties on ceramics, which are summarized in Table 6.
| Healing method | Mechanism/healing components | Material to be healed | Healing conditions | Healing efficiency | Ref. |
|---|---|---|---|---|---|
| Healing efficiency (%) = recovered properties/original properties. | |||||
| Ceramics | Oxidation | Al2O3/TiC/TiB2 ceramic | 700 °C for 60 min | 91.35% (strength) | 314 |
| Oxidation | Laminated Al2O3 cutting tool composites | 900 °C for 30 min | Almost completely filled the cracks | 315 | |
| Oxidation, submicron-SiB6 | MoSi2 coatings | 1200 °C for 40 h | Completely filled the cracks | 316 | |
| Oxidation | Yttrium silicate-composites dispersed with SiC particles | 1300 °C for 1 h | Cracks completely disappeared | 317 | |
| Concrete | Bacterial spores | Green cement from carbide slag, fly ash, and desulfurized gypsum | Soaked in DM (16 h), exposed in air (8 h), and cured (20 °C at N60% RH, 28 d) | 96.67–100% (150–550 μm wide cracks) | 318 |
| Cement, superabsorbent polymers, PEG, epoxy resin, and sand | Concrete | 28 days | 60% (sealed of 100–200 μm wide cracks) | 319 | |
| 18% (flexural strength) | |||||
| Magnesia expansive agents | Strain-hardening cement-based composites | 90 days | 82% (sealed area) | 320 | |
| Microbial | Alkali sulpho-aluminate cement | 28 days | 99.2% (sealed area) | 321 | |
| Artificial aggregates (CaO, SiO2, Al2O3, and others) | Concrete in marine environments | 7 days | 62% (sealed area) | 322 | |
| Metal matrix composites | Zinc ZA-8/NiTi shape memory alloy | Metal matrix composite | 400 °C for 17 h | 30% (tensile strength) | 323 |
| Low melting point alloy (60Sn–40Pb) | Aluminum hybrid composite | Solution heat-treatment (500 °C, 1 h), thermally aged (160 °C, 10 min, then 50 °C, 1 h) | 61% | 324 | |
| NiTi SMA | Aluminum Alloy A380 (Al–8Si–3.5Cu) | 600 °C for 3 h | Crack closed but no bonding | 325 | |
| NiTi shape memory alloy (SMA) | Metal matrix composites (Sn-13 wt% Bi alloy) | 190 °C for 24 h | 66.08% (tensile strength) | 326 | |
When cracks occur in the ceramics, the healing components (like SiC) come in contact with the atmospheric oxygen that would cause oxidation at high temperatures (eqn (1)). The oxidation reaction causes the expansion of the volume by almost two times, resulting in complete filling of cracks with the formed oxide (Fig. 25). Strong bonding between the ceramic matrix and formed oxides also occurs, which leads to complete recovery of the lost mechanical strength.285 Similar to carbide-based particles, nitride-based particles (like Si3N4) also have the ability of autonomous self-healing by oxidation.140
| SiC + O2 → SiO2 + CO | (1) |
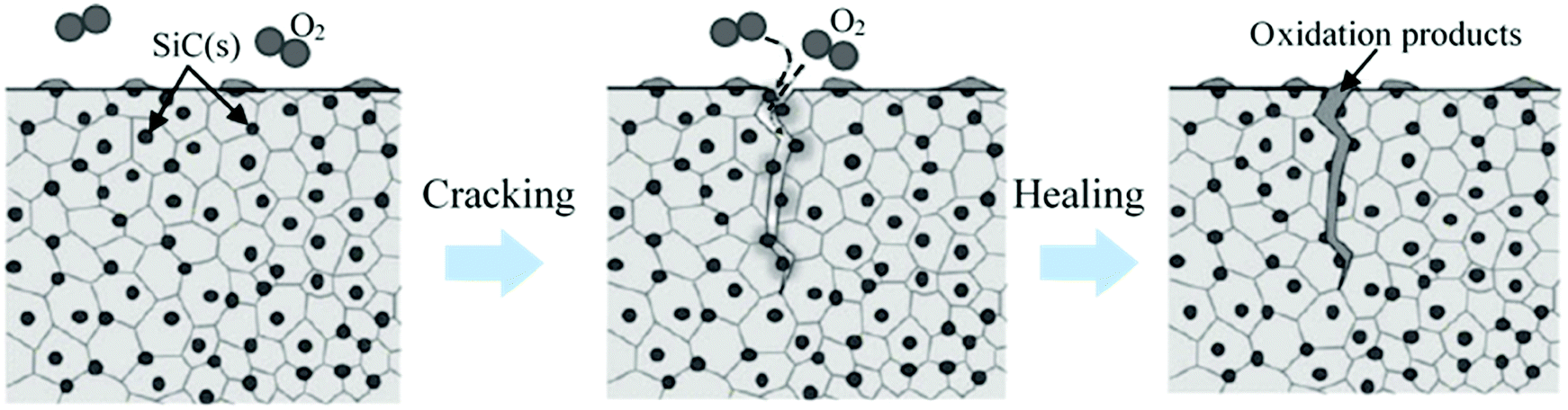 | ||
| Fig. 25 Schematic diagram of the self-healing mechanism of ceramics based oxidation reaction of SiC nanoparticles [reproduced from ref. 286 with permission from the Woodhead Publication]. | ||
6. Self-healing concrete
Concrete, the most widely used construction material in the world, yielded from aggregates, cement, and water, is vulnerable to cracking.15,33,287 Introducing self-healing properties in concrete can help repair the cracks automatically once they appear and can prevent catastrophic failure. However, the self-healing mechanism of concrete is not completely similar to polymer or ceramics. So far, several self-healing approaches have been developed, including autogenous, mineral-admixture-based, and bacteria-based methods (Table 6).288Autogenous self-healing of concrete occurs by two processes, hydration and carbonation.289 Of them, hydration of unhydrated cement particles is the major healing process of concrete in its initial stage, and at a later stage, precipitation of CaCO3 is the key healing process.290 For the hydration healing process, the presence of water is an essential part. When a crack occurs in concrete, water penetrates through the cracks, dissolves the unhydrated cement particles on the crack surfaces resulting in diffusion of Ca2+ and other ions on the cracks, and finally repair of the cracks.291 In this process, narrower cracks can be healed completely but there is only limited healing of wider cracks.292 Based on research, it was found that cracks with more than 300 μm width cannot be healed through this process.292 To minimize this problem, Li and coworkers utilize fiber reinforced cementitious composites that have capabilities to control the crack width and thus promote autogenous healing.293–297 Kuang and Ou proposed SMA wires which can close the cracks to their original position, resulting in complete healing performance.298,299 Another limitation of autogenous healing is the availability of water in the concrete materials, which is essential for the initiation of a repair.
Researchers have been trying to overcome this limitation by providing additional water into concrete materials. Some researchers blended super absorbent polymers or hydrogel with cementitious materials that can retain lots of water.300–302 When water is released from the superabsorbent, it promotes further hydration of the cementitious materials.150
Another potential approach to self-healing is the utilization of calcium carbonate-producing bacteria as a healing agent in concrete.140 Bacteria based solutions can be externally applied by spraying onto cracked surfaces or by direct injection into cracks, which then heal the cracks by precipitating minerals.303 Several researchers have investigated the prospect of this biological healing approach by the introduction of bacteria into concrete.304–306 Tittelboom used ureolytic bacteria such as Bacillus sphaericus to develop an environment-friendly biological repair system of concrete.304Bacillus sphaericus can convert urea into ammonium and carbonate, thus CaCO3 precipitates in their micro-environment. These precipitated CaCO3 crystals can fill the cracks when bacteria are protected in silica gel.304 The negatively charged cell wall of bacteria can draw cations, including Ca2+-ions, which then react with the CO32− ions leading to the precipitation of CaCO3 and filling of the cracked surface.150 Jonkers and coworkers demonstrated that the emitted CO2 from the bacteria's metabolic conversion reacts with Ca(OH)2 (leached from the cementitious matrix) and forms further CaCO3 crystals.305,307
The potential for a bacteria-based self-healing process is very high as comparatively wider cracks can be healed through this process. However, the challenges are that the bacteria cannot survive inside the harsh environment of concrete.288 In order to ensure bacterial growth and repair of cracks, extra cares need to be included, like providing enough nutrients and space to grow and protection from squeezing and the high alkaline medium of the surrounding environment.308
7. Metal and metal matrix self-healing
Introducing self-healing properties in metallic materials has great potential to recover the damage in metals at an early-stage, and thus improve the lifetime of metallic components. However, the goal of producing successful self-healing metals, unfortunately, is still far from being reached because introducing self-healing properties in metals is inherently more complicated than other material classes.309 High bond strengths, small size of metal atoms, and low diffusion rates of metallic atoms compared to that of polymers make it very difficult to repair after damage.265 Even at temperatures close to the melting point, the diffusion rate is not good enough to carry an adequate amount of healing component to the damaged parts. Under these circumstances, currently available processes of healing in metals are only related to healing of damage with a rather small volume.135So far, researchers have demonstrated several techniques to produce self-healing metals, including precipitation healing, incorporation of SMA wires and particles, inclusion of low-melting temperature alloys, electroplating on metal surface, and solder techniques (Table 6).135 Among these techniques, precipitation healing is the most intensively studied method where healing of a damaged metal occurs by the formation of precipitates at the defect sites that immobilizes further growth of damage.140 In order to obtain precipitation healing, the original microstructure must contain a supersaturated amount of solute atoms, which can be obtained by a conventional metallurgical treatment.310 Precipitation does not occur all over the place of metal. Precipitates tend to form at high stress/high energy regions such as nanovoids or cracks, which normally occur by damage or defect resulting in reversal of crack growth and some degree of healing.266 He et al. investigated the influence of defects on the isothermal precipitation at 550 °C of Fe–Cu and Fe–Cu–B–N alloys and found similar tendency where Cu precipitate at the voids of defects results in reduction of the size of defects.311
A self-healing metal composite can also be manufactured by utilizing a low-melting temperature alloy to the defected sites.266 In this process, the low melting temperature alloy acts as a healing agent within the matrix of a high melting temperature metal alloy composite.266 A micro-tube or micro balloon containing a low melting alloy is incorporated in a high-melting temperature alloy metal matrix composite (Fig. 26). When cracks occur in the metal composite, bursting the micro-tubes or micro balloons, the cracks are filled with healing components upon heating and finally the cracks are welded upon cooling.265 Based on this process, Lucci et al. developed a self-healing metal composite by incorporating hollow ceramic tubes filled with low melting alloy (Sn–40Pb solder) as a healing agent then embedded into a high melting metal matrix composite (Al alloy).312,313
 | ||
| Fig. 26 Self-healing process of a metal matrix composite. | ||
8. Gaps and challenges
Over the last few decades, numerous studies have been conducted to improve the healing performance of self-healing composites. However, lots of challenges still need to be overcome for the successful utilization of this technology in practical applications. Some of the challenges related to specific approaches have already been discussed in the above sections.One of the main objectives of producing synthetic self-healing materials is to mimic biological healing, but in reality, we need to go a long way before even mimicking the simplest biological healing process. Biological systems use multiple healing mechanisms simultaneously to completely recover its original properties, but in synthetic systems, these healing characteristics are still unimaginable.
Another limitation of presently available synthetic self-healing materials is that this research and development is still in theoretical stages. Products produced with this technology are not yet suitable for practical applications due to uncertainties in their healing performances, which varies with the surrounding environments and size of damage.23 But for practically useful self-healing products, healing efficiency (healing strength), healing rate, and healing capability should efficiently carry on irrespective of environmental conditions, such as pressure, increased temperature, and mechanical mixing, the size of the damage, and aging of materials.23 Healing rate should be carried out in a controlled and speedy way. Normally, healing time ranges from a couple of hours to several days, which is not sufficient enough to avoid catastrophic failure of a material.3 Aging is another concern that gradually decreases the healing properties with time. Healing components lose their reactivity with the passage of time. But there is a lack of time series analysis regarding aging.
In several approaches, composites need to be brought into contact to initiate healing, which is one of the biggest hindrances for using these approaches in real-world applications, especially materials used in load-bearing engineering structures. Again in some processes, introduction of self-healing properties into a material significantly changes the physicomechanical properties of that material due to incorporation of extra healing components, fibers, microcapsules, or microvascular networks, which significantly change the structure of the original material.26
Hence, although substantial progress has been made in self-healing materials, a lot more challenges need to be overcome before implementing them in practical applications.
9. Conclusions
Self-healing composites have excellent prospects in the field of innovative product research. Inspired by nature, numerous researchers are putting their efforts into mimicking the healing processes of biological materials within non-living materials to substantially improve the working life of the materials. Regardless of some limitations, lots of technological developments have taken place in the last decade, which has been reviewed in this paper. This paper summarized and analyzed most of the available healing approaches in polymers, ceramics, concretes, metals, and their composites with the corresponding healing mechanisms, efficiency, and conditions.Intrinsic healing approaches provide excellent healing efficiency within small damaged areas. Several intrinsic approaches like reversible Diels–Alder, hydrogen, and other supramolecular chemistry are very favorable for the synthesis of highly oriented self-healable polymers. Polymers produced with these types of interactions are capable of fully recovering their original properties (high healing efficiency) at the molecular level multiple times without any additional chemicals. But the problems of these approaches are that they are not suitable for large damage areas, as there is a need to contact the damaged parts manually, and external stimuli (heat, light, and pH) are required for the initiation of healing, and healing efficiency is reduced with the number of healing repetitions due to side reactions.
Similar to intrinsic healing, self-healing materials can also be fabricated utilizing extrinsic healing mechanisms like capsule-based, hollow fibers, and vascular based healing approaches. Capsule-based healing materials can be easily incorporated within the polymer matrix, but they are only suitable for small size damage, and their healing capability in a specific location is diminished after a single repairing cycle. Whereas vascular based healing approaches provide multiple healing to comparatively larger damaged areas as healing components can be repetitively delivered to the damaged areas. However, the challenge is in the integration of a vascular network to the composite materials without affecting the properties of the composite materials and optimizing the tube diameter, capillary forces, and flowability of the healing components.
Besides these approaches, some other methods, including physical interactions, remote self-healing, shape-memory assisted self-healing (SMASH), and nanoparticle-based self-healing, are also very promising. Among them, SMASH can be incorporated with other healing approaches that have the capability to reduce the crack diameter and increase the healing efficiency.
Along with polymers and their composites, self-healing properties can also be incorporated in several other materials, including concretes, ceramics, and metals. Among these, self-healable concrete materials can be fabricated by several approaches where bacteria-based healing proved to be very effective for relatively wider size cracks. The self-healable metal matrix composites have good potential to be used in cylinder liners, pistons, CV joints, gears, and sliding surfaces, which are very susceptible to damage due to friction, creep, and wear between parts. Incorporating healing properties in ceramics can increase their structural integrity and reduce the machining and non-destructive inspection costs. However, incorporation of self-healing components in metals and ceramics is much more complex, and current progress is still at the rudimentary level.
In summary, it can be said that although there are some limitations, substantial progress has been made in laboratory-based healing materials. Now it's time to expand this technology to be used in real applications. In addition to fundamental science, future research should concentrate on the practical challenges and issues of employing self-healing materials in industrial applications.
Conflicts of interest
There are no conflicts to declare.Notes and references
- P. S. Tan, Characterisation of Self-Healing Polymer Composites, The University of Auckland, 2013, pp. 1–187. https://researchspace.auckland.ac.nz/handle/2292/22733 Search PubMed.
- Y. Yang, X. Ding and M. W. Urban, Prog. Polym. Sci., 2015, 49–50, DOI:10.1016/j.progpolymsci.2015.06.001.
- D. Y. Zhu, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2015, 49–50, 175–220, DOI:10.1016/j.progpolymsci.2015.07.002.
- H. Otsuka, S. Nagano, Y. Kobashi, T. Maeda and A. Takahara, Chem. Commun., 2010, 46, 1150–1152, 10.1039/B916128G.
- Y. Amamoto, J. Kamada, H. Otsuka, A. Takahara and K. Matyjaszewski, Angew. Chem., 2011, 123, 1698–1701, DOI:10.1002/ange.201003888.
- A. J. Inglis, L. Nebhani, O. Altintas, F. G. Schmidt and C. Barner-Kowollik, Macromolecules, 2010, 43, 5515–5520, DOI:10.1021/ma100945b.
- S. Burattini, B. W. Greenland, D. Chappell, H. M. Colquhoun and W. Hayes, Chem. Soc. Rev., 2010, 39, 1973–1985, 10.1039/B904502N.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27, DOI:10.1038/nmat2891.
- N. Holten-Andersen, et al. , Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2651–2655, DOI:10.1073/pnas.1015862108.
- M. Burnworth, et al. , Nature, 2011, 472, 334–337, DOI:10.1038/nature09963.
- S. Burattini, et al. , J. Am. Chem. Soc., 2010, 132, 12051–12058, DOI:10.1021/ja104446r.
- Y. Chen and Z. Guan, Polym. Chem., 2013, 4, 4885–4889, 10.1039/C3PY00078H.
- F. Nan and Z. Yan, Angew. Chem., Int. Ed., 2019, 58, 4917–4922, DOI:10.1002/anie.201814060.
- Z. Yang, J. Hollar, X. He and X. Shi, Cem. Concr. Compos., 2011, 33, 506–512, DOI:10.1016/j.cemconcomp.2011.01.010.
- V. Wiktor and H. M. Jonkers, Cem. Concr. Compos., 2011, 33, 763–770, DOI:10.1016/j.cemconcomp.2011.03.012.
- L. Yao, M. Z. Rong, M. Q. Zhang and Y. C. Yuan, J. Mater. Chem., 2011, 21, 9060–9065, 10.1039/C1JM10655D.
- B. J. Blaiszik, S. L. B. Kramer, S. C. Olugebefola, J. S. Moore, N. R. Sottos and S. R. White, Annu. Rev. Mater. Res., 2010, 40, 179–211, DOI:10.1146/annurev-matsci-070909-104532.
- S. R. White, et al. , Nature, 2001, 409, 794–797, DOI:10.1038/35057232.
- J. W. C. Pang and I. P. Bond, Compos. Sci. Technol., 2005, 65, 1791–1799, DOI:10.1016/j.compscitech.2005.03.008.
- G. Williams, R. Trask and I. Bond, Composites, Part A, 2007, 38, 1525–1532, DOI:10.1016/j.compositesa.2007.01.013.
- K. S. Toohey, C. J. Hansen, J. A. Lewis, S. R. White and N. R. Sottos, Adv. Funct. Mater., 2009, 19, 1399–1405, DOI:10.1002/adfm.200801824.
- R. S. Trask and I. P. Bond, J. R. Soc., Interface, 2010, 7, 921–931, DOI:10.1098/rsif.2009.0420.
- Y. Wang, D. T. Pham and C. Ji, Cogent Eng., 2015, 2, 1–28, DOI:10.1080/23311916.2015.1075686.
- K. Mphahlele, S. S. Ray and A. Kolesnikov, Polymer, 2017, 9, 1–22, DOI:10.3390/polym9100535.
- S. Sangadji and E. Schlangen, Procedia Eng., 2013, 54, 315–326, DOI:10.1016/j.proeng.2013.03.029.
- N. K. Guimard, K. K. Oehlenschlaeger, J. Zhou, S. Hilf, F. G. Schmidt and C. Barner-Kowollik, Macromol. Chem. Phys., 2012, 213, 131–143, DOI:10.1002/macp.201100442.
- P. Froimowicz, H. Frey and K. Landfester, Macromol. Rapid Commun., 2011, 32, 468–473, DOI:10.1002/marc.201000643.
- B. Ghosh and M. W. Urban, Science, 2009, 323, 1458–1460, DOI:10.1126/science.1167391.
- Y. Zhang, A. A. Broekhuis and F. Picchioni, Macromolecules, 2009, 42, 1906–1912, DOI:10.1021/ma8027672.
- D. Kowalski, M. Ueda and T. Ohtsuka, J. Mater. Chem., 2010, 20, 7630–7633, 10.1039/C0JM00866D.
- S. J. Kalista Jr, T. C. Ward and Z. Oyetunji, Mech. Adv. Mater. Struct., 2007, 14, 391–397, DOI:10.1080/15376490701298819.
- T. Ono, T. Nobori and J.-M. Lehn, Chem. Commun., 2005, 1522–1524, 10.1039/B418967A.
- X. Chen, et al. , Science, 2002, 295, 1698–1702, DOI:10.1126/science.1065879.
- P. Cordier, F. Tournilhac, C. Soulié-Ziakovic and L. Leibler, Nature, 2008, 451, 977–980, DOI:10.1038/nature06669.
- K. Urdl, A. Kandelbauer, W. Kern, U. Müller, M. Thebault and E. Zikulnig-Rusch, Prog. Org. Coat., 2016, 104, 232–249, DOI:10.1016/j.porgcoat.2016.11.010.
- N. Zhong and W. Post, Composites, Part A, 2015, 69, 226–239, DOI:10.1016/j.compositesa.2014.11.028.
- C. E. Diesendruck, N. R. Sottos, J. S. Moore and S. R. White, Angew. Chem., Int. Ed., 2015, 54, 10428–10447, DOI:10.1002/anie.201500484.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467, 10.1039/c3cs60109a.
- J. Dahlke, S. Zechel, M. D. Hager and U. S. Schubert, Adv. Mater. Interfaces, 2018, 5, 1–14, DOI:10.1002/admi.201800051.
- Z. P. Zhang, M. Z. Rong and M. Q. Zhang, Prog. Polym. Sci., 2018, 80, 39–93, DOI:10.1016/j.progpolymsci.2018.03.002.
- J. A. Syrett, C. R. Becer and D. M. Haddleton, Polym. Chem., 2010, 1, 978–987, 10.1039/c0py00104j.
- M. Scheiner, T. J. Dickens and O. Okoli, Polymer, 2016, 83, 260–282, DOI:10.1016/j.polymer.2015.11.008.
- A. Sanyal, Macromol. Chem. Phys., 2010, 211, 1417–1425, DOI:10.1002/macp.201000108.
- M. A. Tasdelen, Polym. Chem., 2011, 2, 2133–2145, 10.1039/C1PY00041A.
- F. R. Kersey, D. M. Loveless and S. L. Craig, J. R. Soc., Interface, 2007, 4, 373–380, DOI:10.1098/rsif.2006.0187.
- Y. L. Liu and T. W. Chuo, Polym. Chem., 2013, 4, 2194–2205, 10.1039/c2py20957h.
- C. Goussé, A. Gandini and P. Hodge, Macromolecules, 1998, 31, 314–321, DOI:10.1021/ma9710141.
- G. Fortunato, E. Tatsi, B. Rigatelli, S. Turri and G. Griffini, Macromol. Mater. Eng., 2020, 305, 1–6, DOI:10.1002/mame.201900652.
- C. M. Chung, Y.-S. Roh, S.-Y. Cho and J.-G. Kim, Chem. Mater., 2004, 16, 3982–3984, DOI:10.1021/cm049394.
- J. Ling, M. Z. Rong and M. Q. Zhang, Polymer, 2012, 53, 2691–2698, DOI:10.1016/j.polymer.2012.04.016.
- B. Adhikari, D. De and S. Maiti, Prog. Polym. Sci., 2000, 25, 909–948, DOI:10.1016/S0079-6700(00)00020-4.
- Y. Amamoto, H. Otsuka, A. Takahara and K. Matyjaszewski, Adv. Mater., 2012, 24, 3975–3980, DOI:10.1002/adma.201201928.
- S. Nevejans, N. Ballard, J. I. Miranda, B. Reck and J. M. Asua, Phys. Chem. Chem. Phys., 2016, 18, 27577–27583, 10.1039/C6CP04028D.
- D. J. Fortman, J. P. Brutman, G. X. De Hoe, R. L. Snyder, W. R. Dichtel and M. A. Hillmyer, ACS Sustainable Chem. Eng., 2018, 6, 11145–11159, DOI:10.1021/acssuschemeng.8b02355.
- J. Canadell, H. Goossens and B. Klumperman, Macromolecules, 2011, 44, 2536–2541, DOI:10.1021/ma2001492.
- N. V. Tsarevsky and K. Matyjaszewski, Macromolecules, 2002, 35, 9009–9014, DOI:10.1021/ma021061f.
- P. Zheng and T. J. McCarthy, J. Am. Chem. Soc., 2012, 134, 2024–2027, DOI:10.1021/ja2113257.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121–2125, DOI:10.1021/ma052093g.
- T. Kawasaki and T. Iwamoto, US Pat. 6,890,978 B2, 2005 Search PubMed.
- M. W. Urban, Nat. Chem., 2012, 4, 80–82, DOI:10.1038/nchem.1249.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968, DOI:10.1126/science.1212648.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137, DOI:10.1038/nchem.167.
- P. Wu, L. Liu and Z. Wu, Macromol. Mater. Eng., 2020, 305, 2000359 CrossRef CAS.
- C. Lin, et al. , Polymer, 2020, 206, 122894 CrossRef CAS.
- S. Yang, et al. , Chem. Eng. J., 2020, 398, 125654 CrossRef CAS.
- D. Wang, S. Chen, J. Zhao and Z. Zhang, Mater. Today Commun., 2020, 23, 1–7, DOI:10.1016/j.mtcomm.2020.101138.
- J. Handique, J. Gogoi, J. Nath and S. K. Dolui, Polym. Eng. Sci., 2020, 60, 140–150, DOI:10.1002/pen.25267.
- S. Kim, B.-H. Kim, M. Oh, D. H. Park and S. Lee, Polymer, 2019, 11, 104, DOI:10.3390/polym11010104.
- R. Gunckel, et al. , ACS Appl. Polym. Mater., 2020, 2, 3916–3928, DOI:10.1021/acsapm.0c00599.
- H. Jia and S.-Y. Gu, J. Polym. Res., 2020, 27, 1–13, DOI:10.1007/s10965-020-02186-2.
- M. Zhang, F. Zhao, W. Xin and Y. Luo, ChemistrySelect, 2020, 5, 4608–4618, DOI:10.1002/slct.201904316.
- D. Zhao, et al. , ACS Appl. Polym. Mater., 2019, 1, 2951–2960, DOI:10.1021/acsapm.9b00656.
- X. Wu, et al. , J. Appl. Polym. Sci., 2018, 135, 46532, DOI:10.1002/app.46532.
- M. Pepels, I. Filot, B. Klumperman and H. Goossens, Polym. Chem., 2013, 4, 4955–4965, 10.1039/C3PY00087G.
- A. Erice, et al. , Polymer, 2020, 196, 1–9, DOI:10.1016/j.polymer.2020.122461.
- C. Yuan, M. Z. Rong and M. Q. Zhang, Polymer, 2014, 55, 1782–1791, DOI:10.1016/j.polymer.2014.02.033.
- J. Y. Liang, S.-R. Shin, S.-H. Lee and D.-S. Lee, Polymer, 2020, 12, 1–16, DOI:10.3390/polym12051011.
- Q. Fu, Q. Yan, X. Jiang and H. Fu, React. Funct. Polym., 2020, 146, 104440, DOI:10.1016/j.reactfunctpolym.2019.104440.
- F. Fu, M. Huang, W. Zhang, Y. Zhao and X. Liu, Sci. Rep., 2018, 8, 1–6, DOI:10.1038/s41598-018-27942-9.
- C. Xu, R. Cui, L. Fu and B. Lin, Compos. Sci. Technol., 2018, 167, 421–430, DOI:10.1016/j.compscitech.2018.08.027.
- W. Li, et al. , Polymer, 2018, 10, 1392, DOI:10.3390/polym10121392.
- L. M. de Espinosa, G. L. Fiore, C. Weder, E. J. Foster and Y. C. Simon, Prog. Polym. Sci., 2015, 49, 60–78, DOI:10.1016/j.progpolymsci.2015.04.003.
- A. Cohades, C. Branfoot, S. Rae, I. Bond and V. Michaud, Adv. Mater. Interfaces, 2018, 5, 1–20, DOI:10.1002/admi.201800177.
- J. M. Lehn, Chem. Soc. Rev., 2007, 36, 151–160, 10.1039/B616752G.
- J. L. Atwood, Comprehensive supramolecular chemistry II, Elsevier, Amsterdam, Netherland, 2nd edn, 2017 Search PubMed.
- A. W. Bosman, R. P. Sijbesma and E. W. Meijer, Mater. Today, 2004, 7, 34–39, DOI:10.1016/S1369-7021(04)00187-7.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487–7493, DOI:10.1021/ja000435m.
- K. Yamauchi, J. R. Lizotte, D. M. Hercules, M. J. Vergne and T. E. Long, J. Am. Chem. Soc., 2002, 124, 8599–8604, DOI:10.1021/ja020123e.
- K. E. Feldman, M. J. Kade, T. F. A. de Greef, E. W. Meijer, E. J. Kramer and C. J. Hawker, Macromolecules, 2008, 41, 4694–4700, DOI:10.1021/ma800375r.
- T. F. A. de Greef and E. W. Meijer, Nature, 2008, 453, 171–173 CrossRef CAS.
- T. F. A. De Greef, M. M. J. Smulders, M. Wolffs, A. P. H. J. Schenning, R. P. Sijbesma and E. W. Meijer, Chem. Rev., 2009, 109, 5687–5754, DOI:10.1021/cr900181u.
- L. Guadagno, et al. , Composites, Part B, 2019, 157, 1–13, DOI:10.1016/j.compositesb.2018.08.082.
- L. Guadagno, et al. , Polymers, 2019, 11, 1–23, DOI:10.3390/polym11050903.
- G. Wilkinson, R. D. Gillard and J. A. McCleverty, Comprehensive Coordination Chemistry: The synthesis, reactions, properties applications of coordination compounds, Pergamon Press, Oxford, UK, 1987 Search PubMed.
- C. Li and J. Zuo, Adv. Mater., 2020, 32, 1–29, DOI:10.1002/adma.201903762.
- S. Burattini, H. M. Colquhoun, B. W. Greenland and W. Hayes, Faraday Discuss., 2009, 143, 251–264, 10.1039/B900859D.
- S. Burattini, et al. , Chem. Commun., 2009, 6717–6719, 10.1039/B910648K.
- A. Eisenberg, B. Hird and R. B. Moore, Macromolecules, 1990, 23, 4098–4107, DOI:10.1021/ma00220a012.
- J. Kumar Banshiwal and D. Nath Tripathi, Funct. Mater., 2019, 1–24, DOI:10.5772/intechopen.82420.
- S. J. Kalista Jr and T. C. Ward, J. R. Soc., Interface, 2007, 4, 405–411, DOI:10.1098/rsif.2006.0169.
- J. D. Rule, Polymer chemistry for improved self-healing composite materials, University of Illinois at Urbana-Champaign, 2005 Search PubMed.
- S. J. Kalista Jr., Self-Healing Ionomers, in Self-Healing Materials: Fundamentals, Design Strategies, and Applications, ed. S. K. Ghosh, Wiley-VCH, Weinheim, Germany, 2009, pp. 73–100 DOI:10.1002/9783527625376.ch3.
- H. Yang, B. Yuan, X. Zhang and O. A. Scherman, Acc. Chem. Res., 2014, 47, 2106–2115, DOI:10.1021/ar500105t.
- M. D. Pluth and K. N. Raymond, Chem. Soc. Rev., 2007, 36, 161–171, 10.1039/B603168B.
- T. Kakuta, Y. Takashima, M. Nakahata, M. Otsubo, H. Yamaguchi and A. Harada, Adv. Mater., 2013, 25, 2849–2853, DOI:10.1002/adma.201205321.
- X. Qiu, et al. , ACS Appl. Mater. Interfaces, 2020, 12, 41981–41990, DOI:10.1021/acsami.0c11708.
- J. Nam, E. Kim, K. K. Rajeev, Y. Kim and T.-H. Kim, Sci. Rep., 2020, 10, 1–12, DOI:10.1038/s41598-020-71625-3.
- C. Chen, et al. , J. Mol. Liq., 2020, 319, 114134, DOI:10.1016/j.molliq.2020.114134.
- J. Nie, J. Huang, J. Fan, L. Cao, C. Xu and Y. Chen, ACS Sustainable Chem. Eng., 2020, 8, 13724–13733, DOI:10.1021/acssuschemeng.0c04136.
- Z. Liang, et al. , J. Inorg. Organomet. Polym. Mater., 2020, 30, 1–12, DOI:10.1007/s10904-020-01697-1.
- P. Panda, A. Dutta, D. Ganguly, S. Chattopadhyay and R. K. Das, J. Appl. Polym. Sci., 2020, 137, 1–11, DOI:10.1002/app.49590.
- Z. Li, Y. Shan, X. Wang, H. Li, K. Yang and Y. Cui, Chem. Eng. J., 2020, 394, 1–9, DOI:10.1016/j.cej.2020.124932.
- X. Xu, V. V. Jerca and R. Hoogenboom, Macromol. Rapid Commun., 2020, 41, 1–6, DOI:10.1002/marc.201900457.
- J. Pignanelli, Z. Qian, X. Gu, M. J. Ahamed and S. Rondeau-Gagné, New J. Chem., 2020, 44, 8977–8985, 10.1039/D0NJ01119C.
- M. Das, S. Pal and K. Naskar, eXPRESS Polym. Lett., 2020, 14, 860–880, DOI:10.3144/expresspolymlett.2020.71.
- J. Mei, et al. , Macromol. Rapid Commun., 2016, 37, 1667–1675, DOI:10.1002/marc.201600428.
- X. Jing, H.-Y. Mi, B. N. Napiwocki, X.-F. Peng and L.-S. Turng, Carbon, 2017, 125, 557–570, DOI:10.1016/j.carbon.2017.09.071.
- W. X. Xiao, C.-J. Fan, B. Li, W.-X. Liu, K.-K. Yang and Y.-Z. Wang, Compos. Commun., 2019, 14, 48–54, DOI:10.1016/j.coco.2019.05.006.
- H. Y. Zhong, et al. , J. Mater. Chem. C, 2017, 5, 3306–3314, 10.1039/C6TC05493E.
- E. Kim, M. G. Jo and J. Kim, J. Appl. Polym. Sci., 2020, 137, 1–13, DOI:10.1002/app.49046.
- J. Liu, C. Xiao, J. Tang, Y. Liu and J. Hua, Ind. Eng. Chem. Res., 2020, 59, 12755–12765, DOI:10.1021/acs.iecr.0c01538.
- J. Dahlke, et al. , NPG Asia Mater., 2020, 12, 1–10, DOI:10.1038/s41427-019-0190-2.
- S. Mukhopadhyay, P. Sahu, H. Bhajiwala, S. Mohanty, V. Gupta and A. K. Bhowmick, J. Mater. Sci., 2019, 54, 14986–14999, DOI:10.1007/s10853-019-03921-x.
- M. Zhu, et al. , ACS Sustainable Chem. Eng., 2018, 6, 17087–17098, DOI:10.1021/acssuschemeng.8b04452.
- J. Park, et al. , Adv. Mater., 2020, 1–9, DOI:10.1002/adma.202002008.
- C. Jin, et al. , Polymer, 2020, 12, 1–13, DOI:10.3390/polym12061393.
- D. Y. Zhu, et al. , ACS Appl. Mater. Interfaces, 2020, 12, 22534–22542, DOI:10.1021/acsami.0c03523.
- X. Huang, M. Zhang, J. Ming, X. Ning and S. Bai, ACS Appl. Bio Mater., 2020, 3, 7103–7112 CrossRef CAS.
- Y. Wang, M. Pan, X. Liang, B. Li and S. Zhang, Macromol. Rapid Commun., 2017, 38, 1–8, DOI:10.1002/marc.201700447.
- J. Liu, Z. Zhou, X. Su, J. Cao, M. Chen and R. Liu, Prog. Org. Coat., 2020, 146, 1–7, DOI:10.1016/j.porgcoat.2020.105699.
- M. Liu, et al. , Eur. Polym. J., 2020, 124, 1–10, DOI:10.1016/j.eurpolymj.2020.109475.
- L. Han, et al. , NPG Asia Mater., 2017, 9, 1–12, DOI:10.1038/am.2017.33.
- P. Zhang and G. Li, Prog. Polym. Sci., 2016, 57, 32–63, DOI:10.1016/j.progpolymsci.2015.11.005.
- C. B. Lin, S. Lee and K. S. Liu, Polym. Eng. Sci., 1990, 30, 1399–1406, DOI:10.1002/pen.760302109.
- B. M. D. Hager, P. Greil, C. Leyens, S. Van Der Zwaag and U. S. Schubert, Adv. Mater., 2010, 22, 5424–5430, DOI:10.1002/adma.201003036.
- W. H. Binder, Self-Healing Polymers: From Principles to Applications, Wiley-VCH, Weinheim, Germany, 2013 DOI:10.1002/9783527670185.
- M. Yamaguchi, S. Ono and K. Okamoto, Mater. Sci. Eng., B, 2009, 162, 189–194, DOI:10.1016/j.mseb.2009.04.006.
- M. Yamaguchi, S. Ono and M. Terano, Mater. Lett., 2007, 61, 1396–1399, DOI:10.1016/j.matlet.2006.07.039.
- M. A. M. Rahmathullah and G. R. Palmese, J. Appl. Polym. Sci., 2009, 113, 2191–2201, DOI:10.1002/app.30152.
- S. Van Der Zwaag, N. H. Van Dijk, H. M. Jonkers, S. D. Mookhoek and W. G. Sloof, Philos. Trans. R. Soc., A, 2009, 367, 1689–1704, DOI:10.1098/rsta.2009.0020.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522, DOI:10.1016/j.progpolymsci.2008.02.001.
- P. Wang, S. Lee and J. P. Harmon, J. Polym. Sci., Part B: Polym. Phys., 1994, 32, 1217–1227, DOI:10.1002/polb.1994.090320709.
- N. A. Sirajuddin and M. S. M. Jamil, Sains Malays., 2015, 44, 811–818, DOI:10.17576/jsm-2015-4406-06.
- V. K. Stokes, Polym. Eng. Sci., 1989, 29, 1310–1324, DOI:10.1002/pen.760291903.
- M. Zako and N. Takano, J. Intell. Mater. Syst. Struct., 1999, 10, 836–841, DOI:10.1106/YEIH-QUDH-FC7W-4QFM.
- S. A. Hayes, F. R. Jones, K. Marshiya and W. Zhang, Composites, Part A, 2007, 38, 1116–1120, DOI:10.1016/j.compositesa.2006.06.008.
- X. Luo, R. Ou, D. E. Eberly, A. Singhal, W. Viratyaporn and P. T. Mather, ACS Appl. Mater. Interfaces, 2009, 1, 612–620, DOI:10.1021/am8001605.
- K. Otsuka and C. M. Wayman, Shape memory materials, Cambridge University Press, Madrid, Spain, 1998 Search PubMed.
- H. Y. Lai, H.-Q. Wang, J.-C. Lai and C.-H. Li, Molecules, 2019, 24, 1–12, DOI:10.3390/molecules24183224.
- M. Wu, B. Johannesson and M. Geiker, Constr. Build. Mater., 2012, 28, 571–583, DOI:10.1016/j.conbuildmat.2011.08.086.
- Y. T. Cheng, W. Ni, M. J. Lukitsch, A. M. Weiner and D. S. Grummon, Self-healing tribological surfaces, US Pat. 7,060,140, 2006 Search PubMed.
- X. Luo and P. T. Mather, ACS Macro Lett., 2013, 2, 152–156, DOI:10.1021/mz400017x.
- E. D. Rodriguez, X. Luo and P. T. Mather, ACS Appl. Mater. Interfaces, 2011, 3, 152–161, DOI:10.1021/am101012c.
- C. C. Corten and M. W. Urban, Adv. Mater., 2009, 21, 5011–5015, DOI:10.1002/adma.200901940.
- I. Alessandri, Small, 2010, 6, 1679–1685, DOI:10.1002/smll.201000638.
- M. B. Cortie and A. M. McDonagh, Chem. Rev., 2011, 111, 3713–3735, DOI:10.1021/cr1002529.
- V. Giannini, A. I. Fernández-Domínguez, S. C. Heck and S. A. Maier, Chem. Rev., 2011, 111, 3888–3912, DOI:10.1021/cr1002672.
- V. Amendola and M. Meneghetti, J. Mater. Chem., 2012, 22, 24501–24508, 10.1039/C2JM33464J.
- G. L. Fiore, S. J. Rowan and C. Weder, Chem. Soc. Rev., 2013, 42, 7278–7288, 10.1039/C3CS35471G.
- H. Zhang, H. Xia and Y. Zhao, J. Mater. Chem., 2012, 22, 845–849, 10.1039/C1JM14615G.
- L. Huang, et al. , Adv. Mater., 2013, 25, 2224–2228, DOI:10.1002/adma.201204768.
- M. Sabzi, M. Babaahmadi, N. Samadi, G. R. Mahdavinia, M. Keramati and N. Nikfarjam, Polym. Int., 2017, 66, 665–671, DOI:10.1002/pi.5303.
- S. I. Seyed Shahabadi, J. Kong and X. Lu, ACS Sustainable Chem. Eng., 2017, 5, 3148–3157, DOI:10.1021/acssuschemeng.6b02941.
- N. Hohlbein, A. Shaaban and A. M. Schmidt, Polymer, 2015, 69, 301–309, DOI:10.1016/j.polymer.2015.04.024.
- H. Mahmood, A. Dorigato and A. Pegoretti, eXPRESS Polym. Lett., 2020, 14, 368–383, DOI:10.3144/expresspolymlett.2020.31.
- Z. Chen, Y. C. Sun, J. Wang, H. J. Qi, T. Wang and H. E. Naguib, ACS Appl. Mater. Interfaces, 2020, 12, 8740–8750, DOI:10.1021/acsami.9b21411.
- S. Bhattacharya, R. K. Hailstone and C. L. Lewis, ACS Appl. Mater. Interfaces, 2020, 12, 46733–46742, DOI:10.1021/acsami.0c13645.
- P. Mondal, P. K. Behera, B. Voit, F. Böhme and N. K. Singha, Macromol. Mater. Eng., 2020, 305, 1–11, DOI:10.1002/mame.202000142.
- Y. Bai, et al. , Compos. Sci. Technol., 2020, 187, 1–8, DOI:10.1016/j.compscitech.2019.107940.
- X. Yang, L. Guo, X. Xu, S. Shang and H. Liu, Mater. Des., 2020, 186, 1–10, DOI:10.1016/j.matdes.2019.108248.
- N. Lorwanishpaisarn, et al. , Polym. Test., 2020, 81, 1–10, DOI:10.1016/j.polymertesting.2019.106159.
- Y. Cao, et al. , J. Mater. Sci., 2020, 55, 11325–11338, DOI:10.1007/s10853-020-04842-w.
- J. Yan, M. Li, Z. Wang, C. Chen, C. Ma and G. Yang, Chem. Eng. J., 2020, 389, 1–12, DOI:10.1016/j.cej.2019.123468.
- M. Li, et al. , Mater. Horiz., 2019, 6, 703–710, 10.1039/C8MH01441H.
- H. Jia and S.-Y. Gu, Eur. Polym. J., 2020, 126, 1–11, DOI:10.1016/j.eurpolymj.2020.109542.
- H. Liu, et al. , Compos. Commun., 2020, 19, 233–238, DOI:10.1016/j.coco.2020.03.014.
- Y. C. Yuan, T. Yin, M. Z. Rong and M. Q. Zhang, eXPRESS Polym. Lett., 2008, 2, 238–250, DOI:10.3144/expresspolymlett.2008.29.
- M. Raimondo, P. Longo, A. Mariconda and L. Guadagno, Adv. Compos. Mater., 2015, 24, 519–529, DOI:10.1080/09243046.2014.937135.
- G. O. Wilson, H. M. Andersson, S. R. White, N. R. Sottos, J. S. Moore and P. V. Braun, Encycl. Polym. Sci. Technol., Willey, New Jersey, 2002 DOI:10.1002/0471440264.pst469.
- S. Ataei, S. N. Khorasani and R. E. Neisiany, Prog. Org. Coat., 2019, 129, 77–95, DOI:10.1016/j.porgcoat.2019.01.012.
- L. Guadagno, et al. , Composites, Part B, 2011, 42, 296–301, DOI:10.1016/j.compositesb.2010.10.011.
- R. H. Grubbs and D. J. O’Leary, Handbook of Metathesis, Volume 2: Applications in Organic Synthesis, Wiley VCH, Weinheim, Germany, 2015 Search PubMed.
- L. Guadagno, M. Raimondo, C. Naddeo, P. Longo and A. Mariconda, Polym. Eng. Sci., 2014, 54, 777–784, DOI:10.1002/pen.23621.
- L. Guadagno, A. Mariconda, A. Agovino, M. Raimondo and P. Longo, Composites, Part B, 2017, 116, 352–360, DOI:10.1016/j.compositesb.2016.10.075.
- P. Longo, et al. , J. Ind. Eng. Chem., 2017, 54, 234–251, DOI:10.1016/j.jiec.2017.05.038.
- K. Matyjaszewski and P. Sigwalt, Polym. Int., 1994, 35, 1–26, DOI:10.1002/pi.1994.210350101.
- E. J. Goethals and F. Du Prez, Prog. Polym. Sci., 2007, 32, 220–246, DOI:10.1016/j.progpolymsci.2007.01.001.
- G. W. Coates, P. D. Hustad and S. Reinartz, Angew. Chem., Int. Ed., 2002, 41, 2236–2257, DOI:10.1002/1521-3773(20020703)41:13<2236::AID-ANIE2236>3.0.CO;2-3.
- T. C. Mauldin, J. D. Rule, N. R. Sottos, S. R. White and J. S. Moore, J. R. Soc., Interface, 2007, 4, 389–393, DOI:10.1098/rsif.2006.0200.
- T. S. Coope, U. F. J. Mayer, D. F. Wass, R. S. Trask and I. P. Bond, Adv. Funct. Mater., 2011, 21, 4624–4631, DOI:10.1002/adfm.201101660.
- J. M. Kamphaus, J. D. Rule, J. S. Moore, N. R. Sottos and S. R. White, J. R. Soc., Interface, 2008, 5, 95–103, DOI:10.1098/rsif.2007.1071.
- J. K. Lee, S. J. Hong, X. Liu and S. H. Yoon, Macromol. Res., 2004, 12, 478–483, DOI:10.1007/BF03218430.
- X. Liu, X. Sheng, J. K. Lee and M. R. Kessler, Macromol. Mater. Eng., 2009, 294, 389–395, DOI:10.1002/mame.200900015.
- Y. C. Yuan, X. J. Ye, M. Z. Rong, M. Q. Zhang, G. C. Yang and J. Q. Zhao, ACS Appl. Mater. Interfaces, 2011, 3, 4487–4495, DOI:10.1021/am201182j.
- R. Wang, et al. , J. Appl. Polym. Sci., 2011, 121, 2202–2212, DOI:10.1002/app.33829.
- D. Y. Zhu, M. Z. Rong and M. Q. Zhang, Polymer, 2013, 54, 4227–4236, DOI:10.1016/j.polymer.2013.06.014.
- H. Wang, Y. Yuan, M. Rong and M. Zhang, Colloid Polym. Sci., 2009, 287, 1089–1097, DOI:10.1007/s00396-009-2072-6.
- L. Yuan, et al. , Polym. Adv. Technol., 2013, 24, 81–89, DOI:10.1002/pat.3053.
- B. Aissa, et al. , Int. Scholarly Res. Not., 2012, 2012, 1–16, DOI:10.5402/2012/351205.
- X. Liu, J. K. Lee, S. H. Yoon and M. R. Kessler, J. Appl. Polym. Sci., 2006, 101, 1266–1272, DOI:10.1002/app.23245.
- S. H. Cho, H. M. Andersson, S. R. White, N. R. Sottos and P. V. Braun, Adv. Mater., 2006, 18, 997–1000, DOI:10.1002/adma.200501814.
- S. H. Cho, S. R. White and P. V. Braun, Adv. Mater., 2009, 21, 645–649, DOI:10.1002/adma.200802008.
- J. L. Moll, H. Jin, C. L. Mangun, S. R. White and N. R. Sottos, Compos. Sci. Technol., 2013, 79, 15–20, DOI:10.1016/j.compscitech.2013.02.006.
- A. Nellesen, M. Von Tapavicza, J. Bertling, A. M. Schmidt, G. Bauer and T. Speck, Polym. Renewable Resour., 2011, 2, 149–156, DOI:10.1177/204124791100200402.
- S. K. Ghosh, Functional Coatings: by Polymer Microencapsulation, Wiley Online Library, Weinheim, Germany, 2006 Search PubMed.
- E. N. Brown, N. R. Sottos and S. R. White, Exp. Mech., 2002, 42, 372–379, DOI:10.1007/BF02412141.
- D. Crespy and K. Landfester, Beilstein J. Org. Chem., 2010, 6, 1132–1148, DOI:10.3762/bjoc.6.130.
- E. N. Brown, M. R. Kessler, N. R. Sottos and S. R. White, J. Microencapsul., 2003, 20, 719–730, DOI:10.3109/02652040309178083.
- X. Liu, H. Zhang, J. Wang, Z. Wang and S. Wang, Surf. Coat. Technol., 2012, 206, 4976–4980, DOI:10.1016/j.surfcoat.2012.05.133.
- C. Perignon, G. Ongmayeb, R. Neufeld, Y. Frere and D. Poncelet, J. Microencapsul., 2015, 32, 1–15, DOI:10.3109/02652048.2014.950711.
- M. M. Caruso, et al. , ACS Appl. Mater. Interfaces, 2010, 2, 1195–1199, DOI:10.1021/am100084k.
- E. T. A. van den Dungen and B. Klumperman, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5215–5230, DOI:10.1002/pola.24322.
- H. Ullah, K. A. M. Azizli, Z. B. Man, M. B. Che Ismail and M. I. Khan, Polym. Rev., 2016, 56, 429–485, DOI:10.1080/15583724.2015.1107098.
- J. D. Rule, N. R. Sottos and S. R. White, Polymer, 2007, 48, 3520–3529, DOI:10.1016/j.polymer.2007.04.008.
- L. Yuan, G. Liang, J. Xie, L. Li and J. Guo, Polymer, 2006, 47, 5338–5349, DOI:10.1016/j.polymer.2006.05.051.
- D. S. Xiao, M. Z. Rong and M. Q. Zhang, Polymer, 2007, 48, 4765–4776, DOI:10.1016/j.polymer.2007.06.018.
- C. Fan and X. Zhou, Colloids Surf., A, 2010, 363, 49–55, DOI:10.1016/j.colsurfa.2010.04.012.
- H. Yoshizawa, E. Kamio, E. Kobayashi, J. Jacobson and Y. Kitamura, J. Microencapsul., 2007, 24, 349–357, DOI:10.1080/02652040601162699.
- H. Yoshizawa, E. Kamio, N. Hirabayashi, J. Jacobson and Y. Kitamura, J. Microencapsul., 2004, 21, 241–249, DOI:10.1080/02652040410001673946.
- Z. Ting, Z. Min, T. Xiao-Mei, C. Feng and Q. Jian-Hui, J. Appl. Polym. Sci., 2010, 115, 2162–2169, DOI:10.1002/app.31329.
- Y. K. Song and C. M. Chung, Polym. Chem., 2013, 4, 4940–4947, 10.1039/C3PY00102D.
- Y. Xue, et al. , Colloids Surf., A, 2020, 603, 1–10, DOI:10.1016/j.colsurfa.2020.125201.
- A. Ghaemi, A. Philipp, A. Bauer, K. Last, A. Fery and S. Gekle, Chem. Eng. Sci., 2016, 142, 236–243, DOI:10.1016/j.ces.2015.11.002.
- T. C. Mauldin and M. R. Kessler, Int. Mater. Rev., 2010, 55, 317–346, DOI:10.1179/095066010X12646898728408.
- C. M. Dry, Self-repairing, reinforced matrix materials, US Pat. 7,022,179 B1, 2006 Search PubMed.
- J. W. C. Pang and I. P. Bond, Composites, Part A, 2005, 36, 183–188, DOI:10.1016/j.compositesa.2004.06.016.
- R. S. Trask and I. P. Bond, Smart Mater. Struct., 2006, 15, 704, DOI:10.1088/0964-1726/15/3/005.
- R. S. Trask, G. J. Williams and I. P. Bond, J. R. Soc., Interface, 2007, 4, 363–371, DOI:10.1098/rsif.2006.0194.
- C. M. Dry, Cem. Concr. Res., 2000, 30, 1969–1977, DOI:10.1016/S0008-8846(00)00415-4.
- S. M. Bleay, C. B. Loader, V. J. Hawyes, L. Humberstone and P. T. Curtis, Composites, Part A, 2001, 32, 1767–1776, DOI:10.1016/S1359-835X(01)00020-3.
- H. Pulikkalparambil, M. R. Sanjay, S. Siengchin, A. Khan, M. Jawaid and C. I. Pruncu, Self-repairing hollow-fiber polymer composites, in Self-Healing Composite Materials, ed. M. Jawaid, S. N. Raveendran and A. M. Asiri, Elsevier, Duxford, United Kingdom, 2020, pp. 313–326 Search PubMed.
- J. Park and P. V. Braun, Adv. Mater., 2010, 22, 496–499, DOI:10.1002/adma.200902465.
- H. A. Liu, B. E. Gnade and K. J. Balkus Jr, Adv. Funct. Mater., 2008, 18, 3620–3629, DOI:10.1002/adfm.200701470.
- G. Lanzara, Y. Yoon, H. Liu, S. Peng and W. I. Lee, Nanotechnology, 2009, 20, 1–7, DOI:10.1088/0957-4484/20/33/335704.
- H. R. Williams, R. S. Trask and I. P. Bond, Compos. Sci. Technol., 2008, 68(15–16), 3171–3177, DOI:10.1016/j.compscitech.2008.07.016.
- H. R. Williams, R. S. Trask, A. C. Knights, E. R. Williams and I. P. Bond, J. R. Soc., Interface, 2008, 5, 735–747, DOI:10.1098/rsif.2007.1251.
- D. Therriault, S. R. White and J. A. Lewis, Appl. Rheol., 2007, 17, 1–8, DOI:10.1515/arh-2007-0001.
- D. Therriault, R. F. Shepherd, S. R. White and J. A. Lewis, Adv. Mater., 2005, 17, 395–399, DOI:10.1002/adma.200400481.
- D. Therriault, S. R. White and J. A. Lewis, Nat. Mater., 2003, 2, 265–271, DOI:10.1038/nmat863.
- K. S. Toohey, N. R. Sottos and S. R. White, Exp. Mech., 2009, 49, 707–717, DOI:10.1007/s11340-008-9176-7.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581–585 CrossRef CAS.
- C. J. Hansen, W. Wu, K. S. Toohey, N. R. Sottos, S. R. White and J. A. Lewis, Adv. Mater., 2009, 21, 4143–4147, DOI:10.1002/adma.200900588.
- J. F. Patrick, et al. , Adv. Mater., 2014, 26, 4302–4308, DOI:10.1002/adma.201400248.
- M. R. Kessler, Proc. Inst. Mech. Eng., Part G, 2007, 221, 479–495, DOI:10.1243/09544100JAERO172.
- S. R. White, J. S. Moore, N. R. Sottos, B. P. Krull, W. A. Santa Cruz and R. C. R. Gergely, Science, 2014, 344, 620–623, DOI:10.1126/science.1251135.
- Z. Zhao and E. M. Arruda, An internal cure for damaged polymers, Science, 2014, 344, 591–592, DOI:10.1126/science.1254317.
- H. Feng, F. Yu, Y. Zhou, M. Li, L. Xiao and Y. Ao, RSC Adv., 2020, 10, 33675–33682, 10.1039/D0RA03197F.
- R. Rodriguez, D. G. Bekas, S. Flórez, M. Kosarli and A. S. Paipetis, Polymer, 2020, 187, 1–7, DOI:10.1016/j.polymer.2019.122084.
- H. H. Zamal, D. Barba, B. Aïssa, E. Haddad and F. Rosei, Sci. Rep., 2020, 10, 1–13, DOI:10.1038/s41598-020-59725-6.
- H. Hu, et al. , Mater. Des., 2020, 189, 1–12, DOI:10.1016/j.matdes.2020.108535.
- M. Guo, Y. He, J. Wang, X. Zhang and W. Li, J. Appl. Polym. Sci., 2020, 137, 1–14, DOI:10.1002/app.48478.
- K. P. Jhanji, R. Asokan, R. A. Kumar and S. Sarkar, The flexural properties of self healing fiber reinforced polymer composites, in IOP Conference Series: Materials Science and Engineering, 2020, vol. 912, pp. 1–8, DOI:10.1088/1757-899X/912/5/052022.
- Q. Li, N. H. Kim, D. Hui and J. H. Lee, Composites, Part B, 2013, 55, 79–85, DOI:10.1016/j.compositesb.2013.06.006.
- Y. Ma, et al. , Mater. Des., 2020, 189, 1–16, DOI:10.1016/j.matdes.2020.108547.
- D. Y. Zhu, B. Wetzel, A. Noll, M. Z. Rong and M. Q. Zhang, J. Mater. Chem. A, 2013, 1, 7191–7198, 10.1039/C3TA11008G.
- H. Ebrahimnezhad-Khaljiri and R. Eslami-Farsani, Eng. Fract. Mech., 2020, 230, 1–10, DOI:10.1016/j.engfracmech.2020.106937.
- I. Radovic, et al. , Mater. Chem. Phys., 2020, 256, 1–9, DOI:10.1016/j.matchemphys.2020.123761.
- S. A. M. Sadeghi, S. Borhani, A. Zadhoush and M. Dinari, Polymer, 2020, 186, 1–12, DOI:10.1016/j.polymer.2019.122007.
- A. Adli, K. Shelesh-Nezhad, M. Khoshravan Azar and M. Mohammadi-Aghdam, Plast., Rubber Compos., 2020, 49, 79–90, DOI:10.1080/14658011.2019.1706278.
- G. R. Aghamirzadeh, S. M. R. Khalili, R. Eslami-Farsani and A. Saeedi, Polym. Compos., 2019, 40, 1883–1889, DOI:10.1002/pc.24953.
- Y. D. Guo, et al. , Constr. Build. Mater., 2019, 225, 1026–1035, DOI:10.1016/j.conbuildmat.2019.08.036.
- Z. Li, L. R. de Souza, C. Litina, A. E. Markaki and A. Al-Tabbaa, Mater. Des., 2020, 190, 1–14, DOI:10.1016/j.matdes.2020.108572.
- S. Xu, J. Li, H. Qiu, Y. Xue and J. Yang, Compos. Commun., 2020, 19, 220–225, DOI:10.1016/j.coco.2020.04.007.
- R. Eslami-Farsani, S. M. R. Khalili, A. Khademoltoliati and A. Saeedi, Mech. Adv. Mater. Struct., 2019, 1–7, DOI:10.1080/15376494.2019.1567882.
- A. D. Moghadam, B. F. Schultz, J. B. Ferguson, E. Omrani, P. K. Rohatgi and N. Gupta, JOM, 2014, 66, 872–881, DOI:10.1007/s11837-014-0948-5.
- J. B. Ferguson, B. F. Schultz and P. K. Rohatgi, JOM, 2014, 66, 866–871, DOI:10.1007/s11837-014-0912-4.
- E. L. Kirkby, V. J. Michaud, J.-A. Månson, N. R. Sottos and S. R. White, Polymer, 2009, 50, 5533–5538, DOI:10.1016/j.polymer.2009.05.014.
- E. L. Kirkby, J. D. Rule, V. J. Michaud, N. R. Sottos, S. R. White and J. E. Månson, Adv. Funct. Mater., 2008, 18, 2253–2260, DOI:10.1002/adfm.200701208.
- A. Cohades, N. Hostettler, M. Pauchard, C. J. G. Plummer and V. Michaud, Compos. Sci. Technol., 2018, 161, 22–31, DOI:10.1016/j.compscitech.2018.03.040.
- Anonymous, Nano World: Nano for self-healing material, 2006, https://phys.org/news/2006-02-nano-world-self-healing-material.html.
- J. Y. Lee, G. A. Buxton and A. C. Balazs, J. Chem. Phys., 2004, 121, 5531–5540, DOI:10.1063/1.1784432.
- S. Gupta, Q. Zhang, T. Emrick, A. C. Balazs and T. P. Russell, Nat. Mater., 2006, 5, 229–233, DOI:10.1038/nmat1582.
- J. Konlan, P. Mensah, S. Ibekwe, K. Crosby and G. Li, Composites, Part B, 2020, 200, 1–10, DOI:10.1016/j.compositesb.2020.108324.
- H. Suslu, J. Fan, S. Ibekwe, D. Jerro, P. Mensah and G. Li, Smart Mater. Struct., 2020, 29, 1–8, DOI:10.1088/1361-665X/ab85a7.
- R. Abedinzadeh and M. F. Nejad, Polym. Bull., 2020, 1–14, DOI:10.1007/s00289-020-03253-w.
- A. Saeedi and M. M. Shokrieh, J. Intell. Mater. Syst. Struct., 2019, 30, 1585–1593, DOI:10.1177/1045389X19844015.
- S. Yang, et al. , Polymer, 2020, 190, 1–11, DOI:10.1016/j.polymer.2020.122219.
- Y. Sasaki and H. Mori, Mater. Chem. Front., 2020, 4, 2655–2664, 10.1039/D0QM00225A.
- W. Zheng, Y. Li, H. Wei, G.-L. Gao, D. Zhang and Z. Jiang, New J. Chem., 2020, 44, 10586–10591, 10.1039/C9NJ05885K.
- S. Habib, et al. , Nanomaterials, 2020, 10, 1–20, DOI:10.3390/nano10040791.
- S. P. Pasaribu, M. Ginting, I. Masmur and J. Kaban, J. Mol. Liq., 2020, 310, 1–8, DOI:10.1016/j.molliq.2020.113263.
- S. S. Lucas, M. Von Tapavicza, A. M. Schmidt, J. Bertling and A. Nellesen, J. Intell. Mater. Syst. Struct., 2016, 27, 2577–2598, DOI:10.1177/1045389X16641205.
- P. Greil, Adv. Eng. Mater., 2019, 22, 1–8, DOI:10.1002/adem.201901121.
- W. Nakao and S. Abe, Smart Mater. Struct., 2012, 21, 1–7, DOI:10.1088/0964-1726/21/2/025002.
- S. J. Kalista Jr., Self-Healing Ionomers, in Self-Healing Materials: Fundamentals, Design Strategies, and Applications, ed. S. K. Ghosh, Wiley-VCH, Weinheim, Germany, 2009, pp. 73–100 DOI:10.1002/9783527625376.ch3.
- T. Osada, N. Wataru, K. Takahashi and K. Ando, Self-crack-healing behavior in ceramic matrix composites, in Advances in Ceramic Matrix Composites, ed. I. M. Low, Woodhead Publishing, Cambridge, MA, 2018, pp. 515–544 DOI:10.1016/B978-0-08-102166-8.00021-9.
- M. Luo, C. Qian and R. Li, Constr. Build. Mater., 2015, 87, 1–7, DOI:10.1016/j.conbuildmat.2015.03.117.
- H. Xu, J. Lian, M. Gao, D. Fu and Y. Yan, Materials, 2019, 12, 1–16, DOI:10.3390/ma12142313.
- T. G. Nijland, J. A. Larbi, R. P. J. van Hees, B. Lubelli and M. de Rooij, Proc. 1st Int. Conf. on Self-Healing Mater., 2007, vol. 1, pp. 1–9, https://www.researchgate.net/profile/Mario_De_Rooij/publication/267991706_SELF_HEALING_PHENOMENA_IN_CONCRETES_AND_MASONRY_MORTARS_A_MICROSCOPIC_STUDY/links/55ab6c1208aea9946724249f.pdf.
- A. Neville, Concr. Int., 2002, 24, 76–82 Search PubMed . https://www.concrete.org/publications/internationalconcreteabstractsportal/m/details/id/12356.
- H. Huang and G. Ye, Cem. Concr. Compos., 2012, 34, 460–467, DOI:10.1016/j.cemconcomp.2012.01.003.
- K. Van Tittelboom and N. De Belie, Materials, 2013, 6, 2182–2217, DOI:10.3390/ma6062182.
- M. Li and V. C. Li, ACI Mater. J., 2011, 108, 333–340 CAS . https://www.concrete.org/publications/internationalconcreteabstractsportal.aspx?m=details&id=51682499.
- V. C. Li and E.-H. Yang, Self healing in concrete materials, in Self healing materials, ed. S. van der Zwaag, Springer, Dordrecht, Netherlands, 2007, pp. 161–193 Search PubMed.
- M. Şahmaran and V. C. Li, Cem. Concr. Res., 2009, 39, 1033–1043, DOI:10.1016/j.cemconres.2009.07.009.
- Y. Yang, E.-H. Yang and V. C. Li, Cem. Concr. Res., 2011, 41, 176–183, DOI:10.1016/j.cemconres.2010.11.002.
- V. C. Li, C. Wu, S. Wang, A. Ogawa and T. Saito, ACI Mater. J., 2002, 99, 463–472 CAS . http://hdl.handle.net/2027.42/84693.
- Y. Kuang and J. Ou, Smart Mater. Struct., 2008, 17, 1–7, DOI:10.1088/0964-1726/17/2/025020.
- Y. Kuang and J. Ou, J. Cent. South Univ. Technol., 2008, 15, 411–417, DOI:10.1007/s11771-008-0077-9.
- H. X. D. Lee, H. S. Wong and N. R. Buenfeld, Adv. Appl. Ceram., 2010, 109, 296–302, DOI:10.1179/174367609X459559.
- D. Snoeck, S. Steuperaert, K. Van Tittelboom, P. Dubruel and N. De Belie, Cem. Concr. Res., 2012, 42, 1113–1121, DOI:10.1016/j.cemconres.2012.05.005.
- D. Snoeck, K. Van Tittelboom, S. Steuperaert, P. Dubruel and N. De Belie, J. Intell. Mater. Syst. Struct., 2014, 25, 13–24, DOI:10.1177/1045389X12438623.
- W. De Muynck, D. Debrouwer, N. De Belie and W. Verstraete, Cem. Concr. Res., 2008, 38, 1005–1014, DOI:10.1016/j.cemconres.2008.03.005.
- K. Van Tittelboom, N. De Belie, W. De Muynck and W. Verstraete, Cem. Concr. Res., 2010, 40, 157–166, DOI:10.1016/j.cemconres.2009.08.025.
- H. M. Jonkers, Self healing concrete: a biological approach, in Self healing materials, ed. S. van der Zwaag, Springer, Dordrecht, Netherlands, 2007, pp. 195–204 Search PubMed.
- H. M. Jonkers and E. Schlangen, Development of a bacteria-based self healing concrete, in Tailor made concrete structures—new solutions for our society, ed. J. C. Walraven and D. Stoelhorst, CRC Press, London, UK, 2008, pp. 425–430 Search PubMed.
- H. M. Jonkers, A. Thijssen and K. van Breugel, Bacteria mediated remediation of concrete structures, in Proceedings of the second international symposium on service life design for infrastructures, Delft, The Netherlands, 2010, pp. 833–840. https://www.rilem.net/publication/publication/74?id_papier=3745 Search PubMed.
- X. Chen, J. Yuan and M. Alazhari, Materials, 2019, 12, 1303, DOI:10.3390/ma12081303.
- S. van der Zwaag, Self-healing materials: an alternative approach to 20 centuries of material science, Springer, Dordrecht, Netherlands, 2007 Search PubMed.
- B. Grabowski and C. C. Tasan, Self-healing metals, in Self-healing materials, ed. M. D. Hager, S. van der Zwaag and U. S. Schubert, Springer, Switzerland, 2016, pp. 387–407 Search PubMed.
- S. M. He, N. H. Van Dijk, H. Schut, E. R. Peekstok and S. Van Der Zwaag, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 1–10, DOI:10.1103/PhysRevB.81.094103.
- J. M. Lucci, R. S. Amano and P. K. Rohatgi, Heat Mass Transfer, 2017, 53, 825–848, DOI:10.1007/s00231-016-1837-y.
- J. M. Lucci, R. S. Amano, P. K. Rohatgi and B. Schultz, Self-healing in an aluminum alloy reinforced with microtubes, in Proceedings of Energy Nano08 2008b ASME Turbo Expo, ENIC2008-53011, 2008, pp. 79–88, DOI: http://10.1115/ENIC2008-53011.
- Z. Chen, L. Ji, N. Guo, C. Xu and S. Zhang, Int. J. Refract. Met. Hard Mater., 2020, 87, 1–9, DOI:10.1016/j.ijrmhm.2019.105167.
- J. Sun, C. Liu, R. Zhang and T. Hussain, Ceram. Int., 2020, 46, 20463–20471, DOI:10.1016/j.ceramint.2020.05.145.
- L. Wang, W. Wang and Q. Fu, J. Eur. Ceram. Soc., 2020, 40, 2896–2906, DOI:10.1016/j.jeurceramsoc.2020.01.048.
- H. D. Vu and M. Nanko, J. Asian Ceram. Soc., 2020, 8, 298–308, DOI:10.1080/21870764.2020.1734153.
- M. Wu, X. Hu, Q. Zhang, W. Cheng, D. Xue and Y. Zhao, Cem. Concr. Compos., 2020, 113, 1–13, DOI:10.1016/j.cemconcomp.2020.103718.
- J. Feng, H. Dong, R. Wang and Y. Su, Cem. Concr. Res., 2020, 133, 1–13, DOI:10.1016/j.cemconres.2020.106053.
- P. Zhang, et al. , Mater. Today Commun., 2020, 25, 1–11, DOI:10.1016/j.mtcomm.2020.101421.
- T. Zheng, Y. Su, C. Qian and H. Zhou, Biochem. Eng. J., 2020, 163, 1–14, DOI:10.1016/j.bej.2020.107756.
- X. Wu, et al. , Composites, Part B, 2020, 182, 1–10, DOI:10.1016/j.compositesb.2019.107605.
- J. B. Ferguson, B. F. Schultz and P. K. Rohatgi, Mater. Sci. Eng., A, 2015, 620, 85–88, DOI:10.1016/j.msea.2014.10.002.
- O. P. Oladijo, M. O. Bodunrin, K. Sobiyi, N. B. Maledi and K. K. Alaneme, Thin Solid Films, 2016, 620, 201–205, DOI:10.1016/j.tsf.2016.08.071.
- P. K. Rohatgi, Mater. Sci. Eng., A, 2014, 619, 73–76, DOI:10.1016/j.msea.2014.09.050.
- M. A. Poormir, S. M. R. Khalili and R. Eslami-Farsani, J. Intell. Mater. Syst. Struct., 2018, 29, 3972–3982, DOI:10.1177/1045389X18803448.
| This journal is © The Royal Society of Chemistry 2021 |