
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePoly-anthraquinone sulfide isomers as electrode materials for extended operating temperature organic batteries†
Benjamin
Flamme
,
Badr
Jismy
,
Mohamed
Abarbri
 and
Mérièm
Anouti‡
*
and
Mérièm
Anouti‡
*
Laboratoire PCM2E, EA 6299, Université de Tours, Faculté des Sciences et Techniques Parc de Grandmont, 37200 Tours, France. E-mail: meriem.anouti@univ-tours.fr
First published on 17th November 2020
Abstract
Three polyanthraquinone sulfide (PAQS) isomers were synthesized, characterized and included in a comparative study of electrochemical performances as organic material cathodes. The performances of the 1,5-PAQS, 2,6-PAQS, and 1,8-PAQS isomers were compared as a function of charge density, long-term stability and operating temperature. The results obtained showed superior performances for polymers that offer the best spatial flexibility, thus allowing the carbonyl groups to be more accessible to Li cations. The performances (200 mA h g−1 at 4C) of 1,5 and 2,6-PAQS isomers were preserved for up to 1000 cycles. The more flexible 2,6-PAQS isomer demonstrated a remarkable ability to endure fast cycling from 60 °C to −30 °C without a significant decrease in capacity retention (which decreased from 98.1% to 93.4%) thanks to its flexible structure. The reduced effect of the temperature on long term cycling results obtained with 2,6-PAQS allows it to deliver an energy density of 275 W h kg−1 at −30 °C. It therefore appears that, thanks to their low cost, environmental benignity and high sustainability, the selected PAQS isomers are promising candidates for alternative electrode materials in full-organic batteries dedicated to low temperature operation.
Introduction
Nowadays, lithium-ion batteries are already widely used in our daily lives and will continue to conquer new sectors, but their development lags behind the global demand for this type of energy storage device.1,2 The principal difficulties remain their low power density and their cost, but also the environmental impact linked to their manufacture.3,4 The intensive use of transition metal resources such as cobalt and nickel in LiMxOy (M = Co, Ni, Mn…) cathode electrodes is spurring the search for alternative greener electrode materials.5 The attention of many groups has therefore turned to different systems such as redox-flow batteries (RFB), lithium–sulfur (Li–S) or organic batteries (OBs). Organic electrode materials (OEMs) are attracting more attention because of their multiple properties,4,6,7 their durability, eco-compatibility,8–11 and their ability to operate with multiple alkali ions, such as Li+,12,13 Na+,14,15 or K+.16,17 OEMs could be used in a wide variety of systems,18i.e. supercapacitors,19 aqueous batteries20 and hybrid systems.21–23 Among the different families of OEMs,6 quinones and their derivatives appear to be the most promising type,24,25 thanks to their high availability and the good electrochemical reversibility of their redox system.26,27 However, the main drawback of quinone derivatives is their solubility in organic solvents that are commonly used in lithium-ion batteries.4 Small molecules of benzo-, naphtho- or anthraquinone homologues contaminate the electrolyte and reduce its electrochemical due to slow and continuous dissolution.28 One approach involves working with monomers and limiting the dissolution via the formulation as the Chen and Li groups reported recently.29–31 Another approach to avoid dissolution is to polymerize these small units in order to obtain an insoluble polymer. This approach is still currently in use and effective as shown recently by Liang et al. and Wang et al.32,33 The macromolecules thus obtained are almost insoluble in most organic solvents, and this was particularly the case for poly-anthraquinone sulfides (PAQS).28,34,35 Despite the reduced nominal capacity of PAQS (Cth = 225 mA h g−1) because of the higher molar mass of the anthraquinone monomer, and the introduction of a C–S bond by “Philipp's” polycondensation,36PAQS offer a greater degree of freedom and a wider diversity of spatial arrangement thanks to position isomers. It turns out that these two parameters influence the accessibility to carbonyl moieties and the diffusion of cations and consequently the performances. We report herein the comparative performances of three PAQS position isomers used as OEMs and the position effect on electrochemical performance and stability in a wide temperature range. The objective was to demonstrate that the spatial arrangements of PAQS isomers influence the charge distribution and the carbonyl group accessibility, and how much it impacts the electrochemical performances.Experimental section
Materials and synthesis
2,6-Dichloroanthraquinone (2,6-DCAQ) was prepared based on the procedure reported by Zhang et al.37 The reaction was carried out in two steps as described in Scheme 1. First, sodium nitrite (5.8 g, 84 mmol) was dissolved in ice-cold water (20 mL) and added slowly in a 1000 mL flask containing a solution of 2,6-diaminoanthraquinone (10 g, 42 mmol) and HCl (37%, 60 mL). The reaction mixture was maintained at 0 °C for 30 minutes to form the diazonium intermediate. Then, CuCl (8.32 g, 84 mmol) was then added slowly and the mixture was heated at 55 °C for 5 hours. The progress of the reaction was monitored by TLC (PE/EtOAc, 9/1).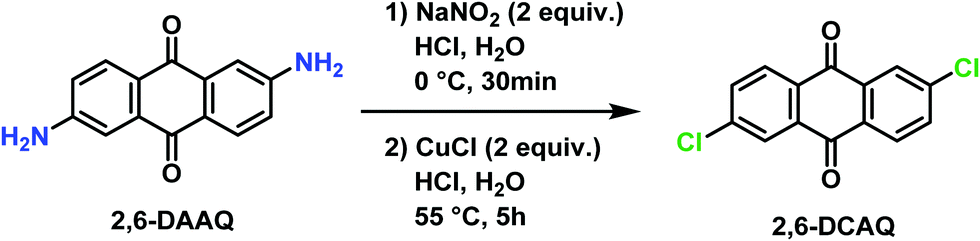 | ||
| Scheme 1 Synthesis of 2,6-dichloroanthraquinone (2,6-DCAQ). | ||
After completion of the reaction, the mixture was filtered, and the obtained solid was washed several times with HCl (37%, 3 × 20 mL) and H2O (3 × 200 mL) successively, and then dried under vacuum at 50 °C. The desired product was obtained after several extractions using CH2Cl2 (10 × 200 mL) followed by removal of the solvent. The crude product was purified by chromatography on silica gel using (PE/EtOAc, 9/1) as an eluent, to afford the pure 2,6-DCAQ as a beige solid in 52% yield (6.02 g). M.p. 292–294 °C (Lit.37 M.p.: 291 °C, Lit. M.p.: 295–297 °C38). 1H NMR (300 MHz, CDCl3) δ 8.35–8.21 (m, 4H), 7.77 (dd, J = 8.4, 2.1 Hz, 2H). HRMS (ESI) m/z [M + H]+ calcd for C14H7Cl2O2: 275.9703; found: 275.9708.
1,5-PAQS, 1,8-PAQS and 2,6-PAQS were synthesized by polycondensation using the corresponding dichloroanthraquinone (DCAQ) and anhydrous sodium sulfide as reagents according to methods described previously,6,16,28 The chemical reaction is shown in Scheme 2. All PAQS were prepared using anhydrous Na2S (1.41 g, 18 mmol) obtained by removing the hydrated water through azeotropic dehydration in toluene (10 mL) and NMP (40 mL). After evaporating toluene from the mixture, a stoichiometric quantity of corresponding DCAQ (5 g, 18 mmol) was added and the mixture was heated to reflux at 200 °C. The reaction mixture was maintained at the aforementioned temperature for 48 h with constant magnetic stirring. It was then allowed to cool down to room temperature and the polymer was obtained by vacuum filtration and washed with water (3 × 200 mL) and acetone (3 × 100 mL) several times to remove any unreacted Na2S and dichloroanthraquinone (DCAQ). The filtered polymer was then dried at 60 °C under vacuum for 12 h after which dark brown colored powders were obtained. The yield was about 81% (3.5 g) for 1,5-PAQS, 70% (3.0 g) for 1,8-PAQS and 89% (3.8 g) for 1,8-PAQS.
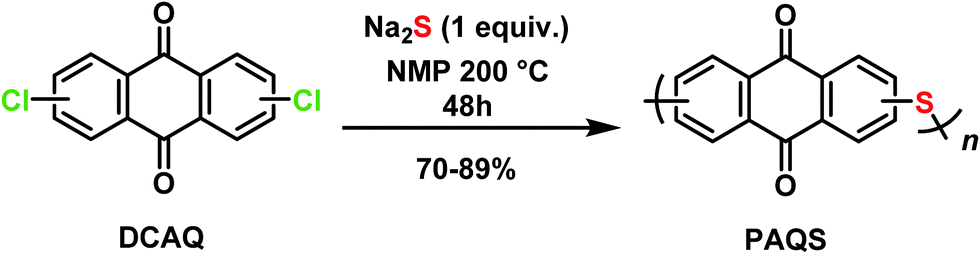 | ||
| Scheme 2 DCAQ polymerization affording the desired PAQS isomers. | ||
1H liquid NMR spectra were recorded at 300 MHz on a Bruker Avance 300 spectrometer, and elemental analysis of C, H, N, O, S and Cl was conducted by the “Institut des Sciences Analytiques” (ISA, UMR CNRS 5280, Lyon, France) on various equipment.39 XRD spectra were recorded at the “Institut de Recherche de Chimie Paris” (IRCP, UMR CNRS 8247, Paris, France) on a D8 Endeavor X-ray diffractometer (Bruker) equipped with a Cu source (Kα, λ = 1.54 Å). Raman spectra were recorded on a Horiba HR Evolution (532 nm) at 5% of the maximum power. Thermogravimetric analysis (TGA) was performed on a STA 6000 (PerkinElmer) within the temperature range of 30–700 °C at a heating rate of 5 °C min−1 under a N2 atmosphere.
Cell preparation and electrochemical measurements
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) mixture (both 99.5%, Sigma-Aldrich). The entire cell assembly process was performed inside a glove box in which the moisture and oxygen levels were below 5 ppm. Coin cells were placed in CR2032 coin cell holders (Origalys Electrochem) and electrochemical characterization was performed with a Biologic BCS-805 battery cycler and/or a Biologic VMP-3. For low temperature measurements, channel cables were entered in a dynamic climate chamber (Binder MK056) through a dedicated access and subsequently isolated by glass wool. The climate chamber was set at the targeted temperature (±0.1 °C) and a stabilization time of 2 h was applied before running each experiment in order to ensure that all the systems (coin cell, electrolyte, etc.) reached the operating temperature. For cycling at 60 °C, the same procedure was employed in an oven (Memmert UN30). 225 mA h g−1 was assumed as the theoretical capacity for calculating currents. The cutoff voltages of the cyclic voltammetry and galvanostatic cycling tests were 1.5–3.2 V. Coupled galvanostatic-EIS tests were performed on CR-2032 half-cells, with a voltage amplitude of 10 mV and a frequency range of 1 MHz to 1 mHz.
:1 v/v) mixture (both 99.5%, Sigma-Aldrich). The entire cell assembly process was performed inside a glove box in which the moisture and oxygen levels were below 5 ppm. Coin cells were placed in CR2032 coin cell holders (Origalys Electrochem) and electrochemical characterization was performed with a Biologic BCS-805 battery cycler and/or a Biologic VMP-3. For low temperature measurements, channel cables were entered in a dynamic climate chamber (Binder MK056) through a dedicated access and subsequently isolated by glass wool. The climate chamber was set at the targeted temperature (±0.1 °C) and a stabilization time of 2 h was applied before running each experiment in order to ensure that all the systems (coin cell, electrolyte, etc.) reached the operating temperature. For cycling at 60 °C, the same procedure was employed in an oven (Memmert UN30). 225 mA h g−1 was assumed as the theoretical capacity for calculating currents. The cutoff voltages of the cyclic voltammetry and galvanostatic cycling tests were 1.5–3.2 V. Coupled galvanostatic-EIS tests were performed on CR-2032 half-cells, with a voltage amplitude of 10 mV and a frequency range of 1 MHz to 1 mHz.
Results and discussion
1,5-PAQS, 1,8-PAQS and 2,6-PAQS (Fig. 1) were easily prepared by refluxing a solution of corresponding dichloroanthraquinone (DCAQ) with anhydrous sodium sulfide (Na2S) in N-methyl 2-pyrrolidone (NMP) at 200 °C according to methods described previously6,16,28 (Scheme 2). The reaction mixture was kept at the aforementioned temperature for 48 h with constant magnetic stirring. The purified 1,5-PAQS, 1,8-PAQS and 2,6-PAQS were isolated with a yield of 81%, 70% and 89%, respectively. The chemical structures of 1,5-PAQS, 1,8-PAQS and 2,6-PAQS obtained were checked by elemental analysis and polymerization degrees (DPn) were deduced from the data.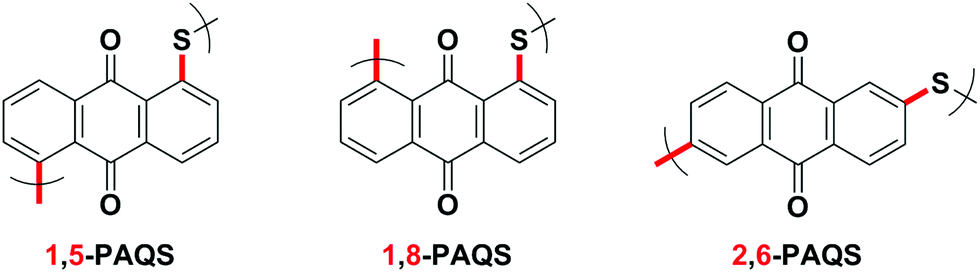 | ||
| Fig. 1 The chemical structure of 1,5-PAQS, 1,8-PAQS and 2,6-PAQS. | ||
Table 1 summarizes the data from elemental analysis and the evaluation of the degree of polymerization which is extracted from it as described by Wang et al.26 As an example, the reduction of Cl% from 25.59 in 2,6-DCAQ to 0.67 in 2,6-PAQS indicates that almost all of the Cl atoms were eliminated during the polymerization reaction. In this case the final C/Cl molar ratio is equal to 287 and the initial is 7. All the terminal groups of the polymer are assumed to be Cl atoms, as a consequence, the polymerization degree is about 287/7, DPn = 41.
| Carbon (%) | Hydrogen (%) | Oxygen (%) | Sulfur (%) | Chlorine (%) | DPn | |
|---|---|---|---|---|---|---|
| Theoretical | 70.57 | 2.54 | 13.43 | 13.46 | 0 | — |
| 1,5-PAQS | 61.48 | 2.48 | 12.17 | 10.92 | 2.10 | 12 |
| 1,8-PAQS | 66.51 | 2.80 | 12.51 | 13.35 | 3.63 | 8 |
| 2,6-PAQS | 65.13 | 2.72 | 12.34 | 9.84 | 0.67 | 41 |
The materials obtained were characterized by XRD and Raman spectroscopy (Fig. 2) and subjected to thermogravimetric analysis (Fig. S2, ESI†). XRD for the studied PAQS isomers shows low crystallinity to amorphous patterns depending on the isomer (Fig. 2a), which is common for polyquinones and consistent with the literature data for 1,5-PAQS.40 For 2,6-PAQS, peaks were less well-defined but still in the same 2θ angles, while 1,8-PAQS displayed only a very broad region without any defined peaks.
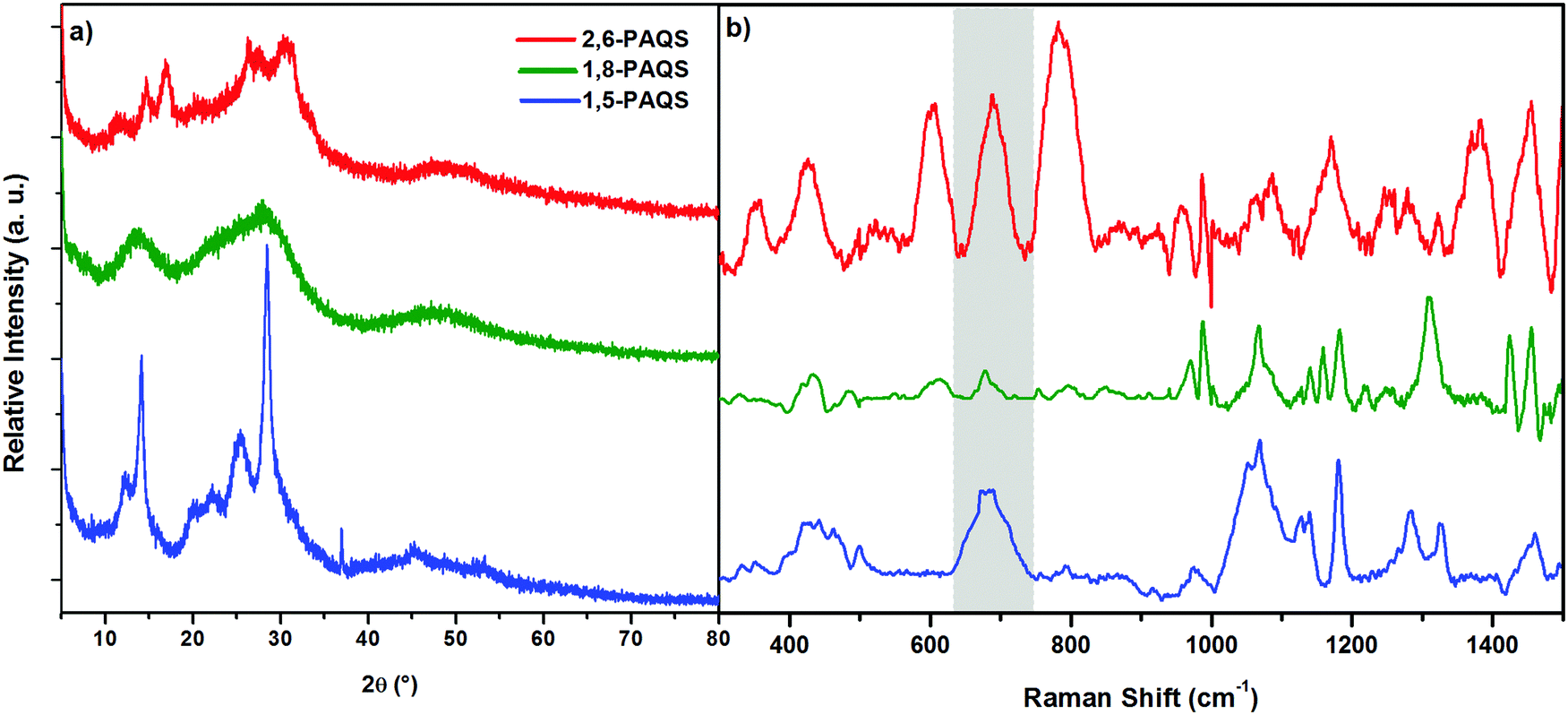 | ||
| Fig. 2 (a) XRD patterns of the synthesized PAQS isomers and (b) Raman spectra of PAQS isomer powders. | ||
The formation of C–S bonds was investigated by Raman spectroscopy and they were found to appear between 650 and 750 cm−1 (highlighted band) which is consistent with the range of the C–S bond stretching vibrations.41,42 As depicted in Fig. 2b, only a small peak can be observed for 1,8-PAQS, whereas a band can be seen for 1,5-PAQS and an intense one for 2,6-PAQS. These different intensities are correlated with the related degree of polymerization (DPn): the lower the DPn, the weaker the intensity of the C–S band (Table 1).
TGA analysis (Fig. S2, ESI†) for PAQS showed no variations up to 270 °C for 2,6-PAQS and 360 °C for 1,5-PAQS. The 1,8-PAQS curve seems to show a loss of water equal to 15% of the total mass which may correspond to residual water trapped in the polymer. The second weight loss appeared after 330 °C and corresponds to thermal degradation of 1,8-PAQS.
The main drawback of quinone as a monomer or oligomers is their solubility in organic solvents that are commonly used in lithium-ion batteries. As shown by visual solubility tests in Fig. 3, only the vial containing the 1,8-PAQS electrode exhibited a pale-yellow color after 24 h. This coloration did not evolve over a week, and the other two were still uncolored after seven days (Fig. S3, ESI†). 2,6-PAQS, thanks to its high polymerization degree, was not soluble in our electrolyte. As revealed by the pictures, 1,8-PAQS can be dissolved by the electrolyte; this was no doubt caused by its low degree of polymerization. Lastly, despite a very similar polymerization degree to that of 1,8-PAQS, 1,5-PAQS was not soluble in the 1 M LiTFSI DOL/DME mixture.
 | ||
| Fig. 3 Pictures showing the dissolution behavior of PAQS electrode materials in 1 M LiTFSI DOL/DME (1:1 v/v). | ||
Fig. 4a and b shows the comparative cyclic voltammograms (CV) for the three PAQS isomers at two scan rates v (mV s−1) with 1 M LiTFSI in 1,3-dioxolane/dimethoxyethane (DOL/DME: 1/1, v/v) as electrolyte. At a low scan rate v = 0.05 mV s−1, the two-step bielectronic redox mechanism for the reversible electrochemical reaction between lithium ions and carbonyl groups in the conjugated organic framework is visible and appear as a pair of peaks centered in the oxidation step at 2.3 V for 1,5-PAQS and 1,8-PAQS and 2.35 V for 2,6-PAQS. In the reduction step, a broad peak is observed at 2.25 V vs. Li+/Li. In all cases, the potential hysteresis is downsized to 0.10 V, indicating a low polarization during the redox process at this slow scan rate. At 1 mV s−1, the peaks are less clearly separated and the observed dissymmetry is attributed to the different diffusion rates into electrode materials between the oxidation and reduction steps. For 2,6-PAQS, electrochemical reversibility (electron transfer/Li+ compensation) of the concerted process was checked (Fig. 4c) by the analysis of Ipc and Ipa as a function of the square root of the scan rate, v1/2 for linearity.43
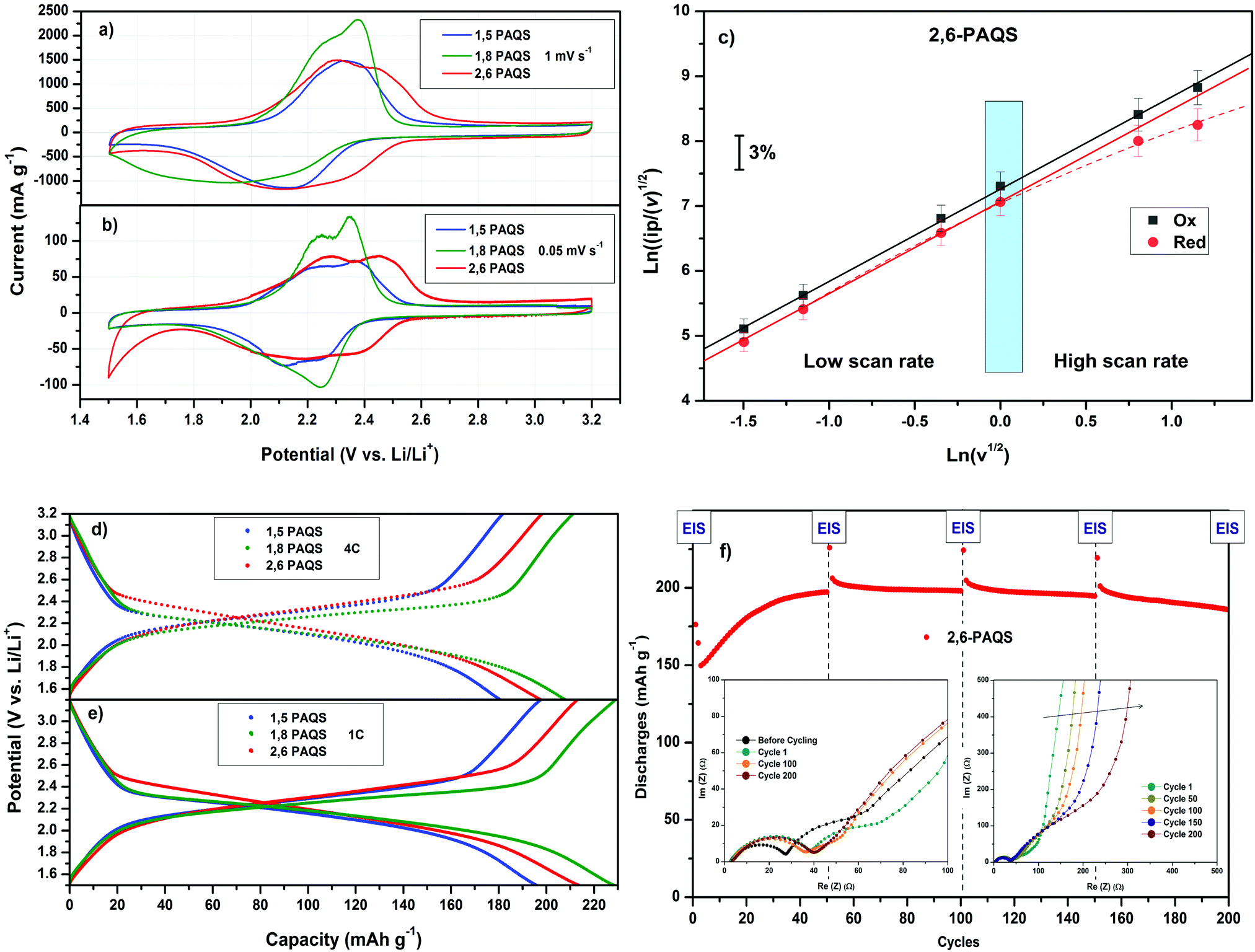 | ||
| Fig. 4 (a) and (b) Cyclic voltammetry of PAQS isomers with 1 M LiTFSI in DOL/DME at 1.00 and 0.05 mV s−1. (c) Evolution of normalized current (ip) as a function of the square root of the scan rate v. (d) and (e) Galvanostatic profiles of each PAQS isomer at 4C and 1C. (f) 2,6-PAQS impedance evolution during galvanostatic cycling at 4C. | ||
For both oxidation and reduction, the investigated systems show the expected behavior for a diffusion limited process. This can be explained by a fast electron transfer and Li+ charge compensation, which does not influence the electrode response substantially.44 At a high scan rate v > 1 mV s−1, ipc showed a slight deviation from linearity, which means that at this rate, the approach of the Li+ cation to compensate the charge is the limiting step.
Fig. 4d and e present the galvanostatic profiles of each PAQS isomer at two different C-rates. These profiles show very high capacities at 1C for each isomer, starting from 192 mA h g−1 for 1,5-PAQS, 212 mA h g−1 for 2,6-PAQS and ending at 224 mA h g−1 for 1,8-PAQS. At a faster C-rate (4C), the same ranking is found, with only slight decreases in capacities (only 10 to 15 mA h g−1). These capacities at a fast C-rate are higher than most conventional anodes and cathodes, such as LTO, LFP or LCO and also a competitive alternative of other related organic redox materials (Table 2).
As mentioned above, the capacities are in the following order: 1,8-PAQS > 2,6-PAQS > 1,5-PAQS. Unlike with a low C-rate (1C), with a faster cycling (4C) the half charge hysteresis is inverted and the order is changed as follows: 2,6-PAQS < 1,5-PAQS < 1,8-PAQS which is consistent with the previous observation for cyclic voltammetry. PAQS isomers seem to be sensitive to the C-rate, depending on the mechanical rigidity of the polymer backbone and its degree of polymerization. This hypothesis is corroborated by Fig. 4f and 5. During galvanostatic cycling at 4C, 2,6-PAQS shows progressive adaptation by gradual increases of its initial capacities due to the shaping of the polymer resulting from the repetitive redox processes. During the galvanostatic sequence, impedance measurements showed that the Warburg increases only for 2,6-PAQS, which indicates a slower diffusion in the material and consequently a greater resistance. These results confirm the effect of the isomerism on the polymer chain on the electrochemical response and consequently on the stability of PAQS during cycling. Thus 1,8-PAQS, which is more rigid, seems disfavored for a prolonged reversible redox process by comparison of the two other isomers, independently to their polymerization degree.
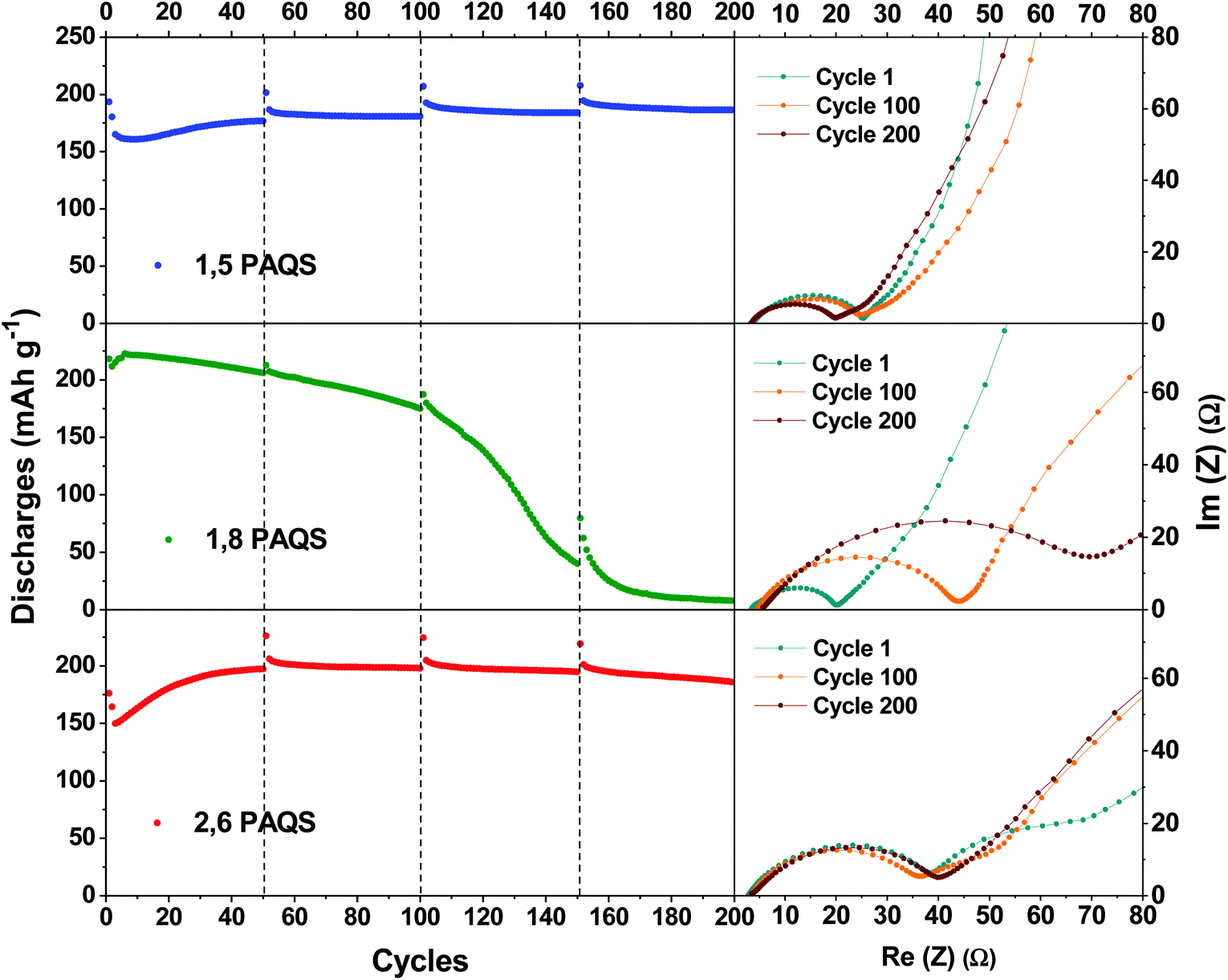 | ||
| Fig. 5 Nyquist plots of a Li-PAQS coin cell after the 1st, 100th, and 200th cycle under a current rate of 4C and the corresponding evolution of discharge capacities. | ||
Fig. 6a compares the rate capability for PAQS isomers at a current rate (from C/5 to 50C). PAQS delivers a reversible capacity above 220 mA h g−1 at C/5 for all isomers corresponding to the quasi-theoretical capacity, and it still delivers 120 mA h g−1 (0.6 A g−1) and 60 mA h g−1 (0.3 A g−1) for 2,6-PAQS and 1,5-PAQS respectively at 50C. In contrast, 1,8-PAQS does not support high current density and the capacity collapses for C-rates above 5C.
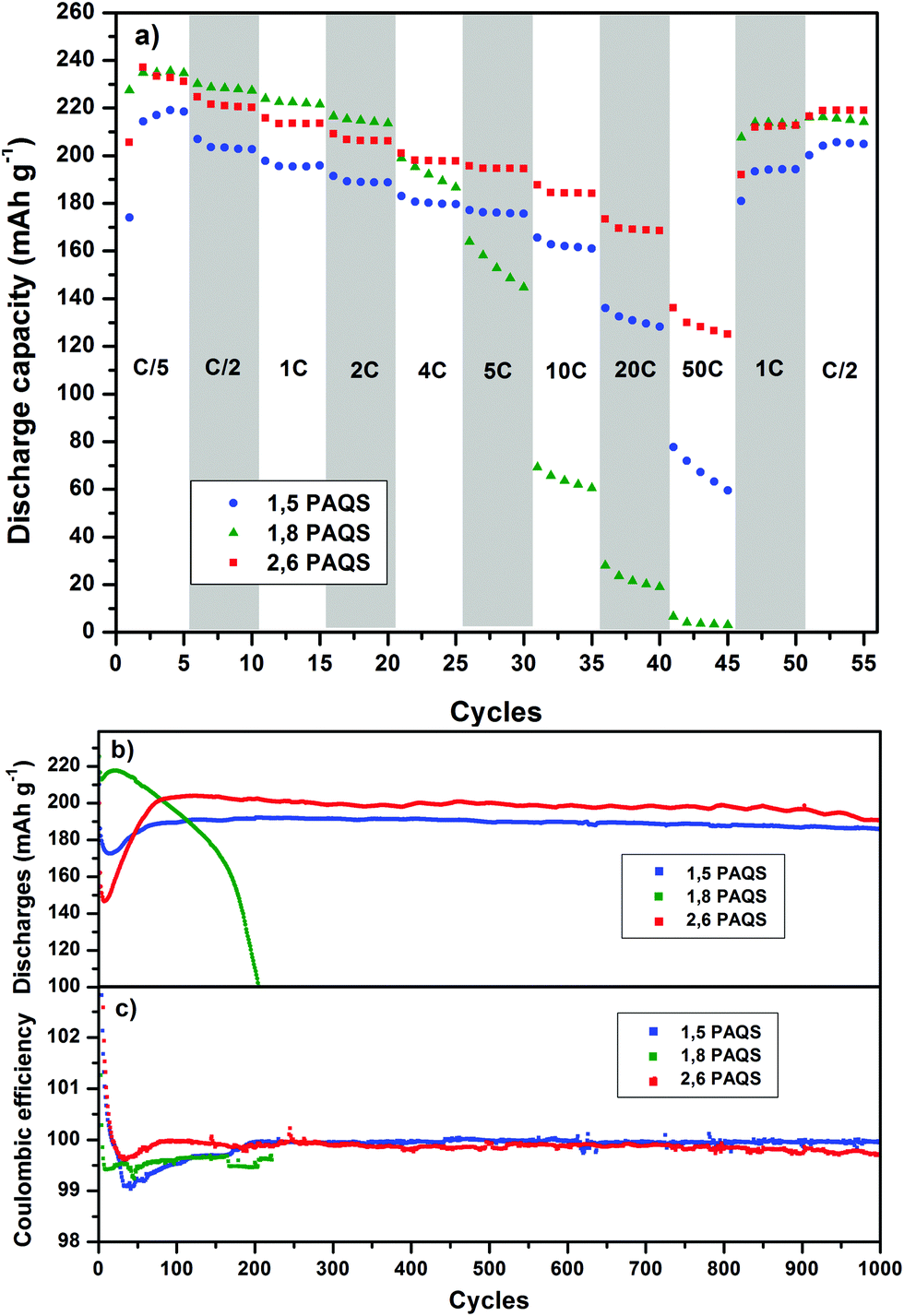 | ||
| Fig. 6 (a) Rate capability at room temperature for each PAQS isomer at a current rate from C/5 to 50C. (b) Long-term cycling profiles of PAQS isomers at 4C and (c) their corresponding coulombic efficiencies. | ||
Performances during long-term cycling up to 1000 cycles (Fig. 6b and c) show that 1,8-PAQS still suffers from fast cycling (4C) as its discharge capacities do not stabilize but fade continuously and end up collapsing after 200 cycles. In contrast, 1,5-PAQS and 2,6-PAQS, after stabilization, can be cycled over a thousand times with capacities close to 200 mA h g−1 (88% of the theoretical PAQS capacity). At the end of the 1000 cycles, the capacity retention is over 94–97% of the initial capacity for 2,6-PAQS and 1,5-PAQS, respectively.
The good performances of 1,5-PAQS demonstrate the secondary role of the polymerization degree (DPn = 12) in achieving long-term cycling, in contradiction with what has been previously described.26 By presenting at the same time a high polymerization degree, the best capacities, and a rate capability at very high current density, 2,6-PAQS seems to be the most promising PAQS isomer for an organic battery electrode.
The temperature effect on stability during cycling was evaluated by galvanostatic cycling (4C) over 200 cycles at temperatures ranging from 60 °C to −30 °C for all PAQS isomers. The results are presented in Fig. 7.
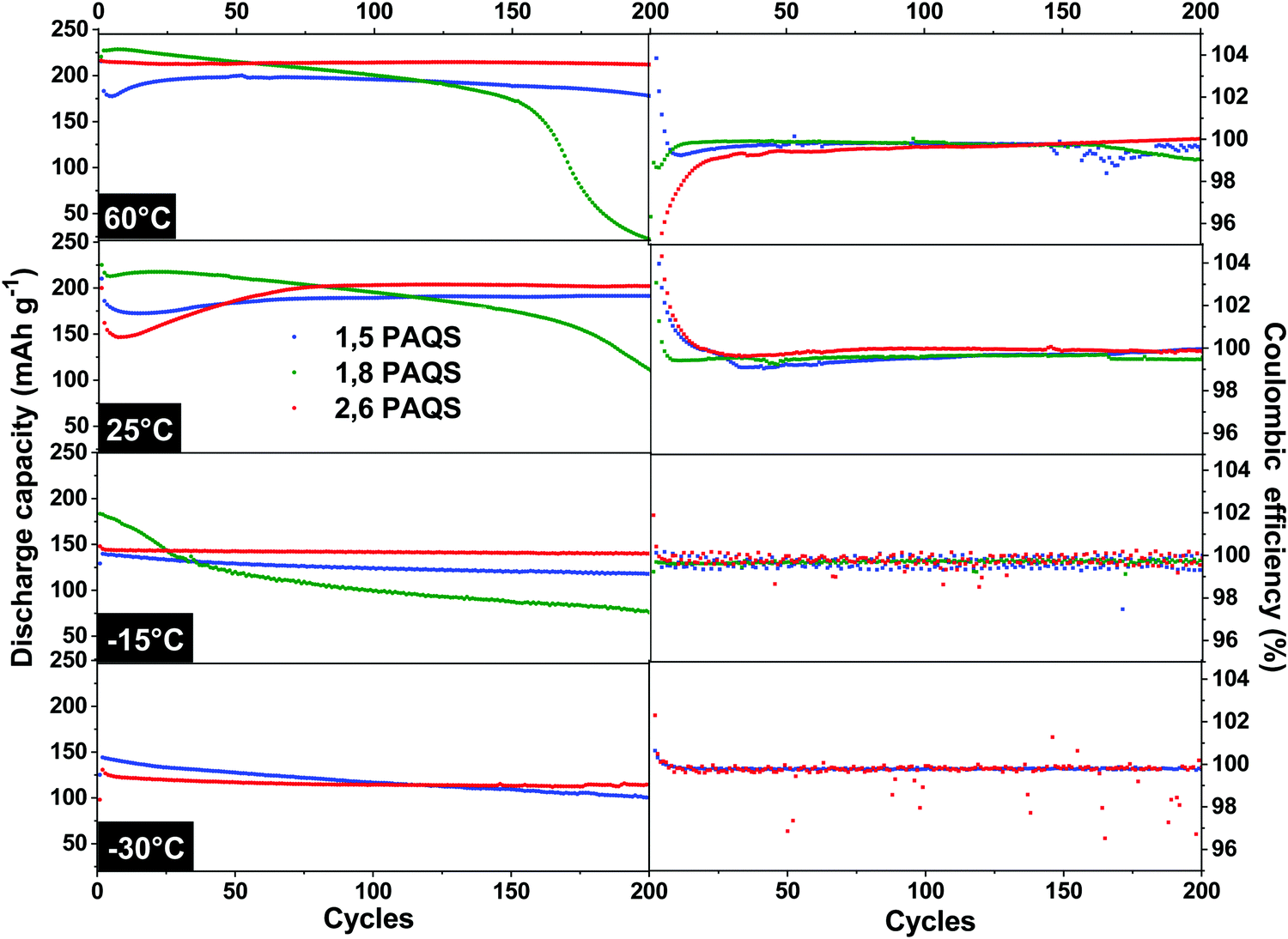 | ||
| Fig. 7 Discharge capacities and corresponding coulombic efficiencies of PAQS isomers from 60 °C to −30 °C at 4C. | ||
The 1,8-PAQS discharge capacities were even more unstable at 60 °C than at 25 °C, with faster fading. At −15 °C even if the starting capacities were better (185 mA h g−1) and the capacity fading more contained, performances still did not match the stability requirements. Lastly, unlike the other two isomers, 1,8-PAQS was not able to cycle at −30 °C. At 60 °C, 2,6-PAQS exhibited a better capacity and stability (211 mA h g−1, 200th cycle) than 1,5-PAQS with a capacity retention of 98.1% vs. 89.4% for 2,6-PAQS and 1,5-PAQS respectively, associated with the excellent coulombic efficiencies for both. At low temperatures, −15 °C and −30 °C, the capacity of the two PAQS showed an expected decrease due to the slowdown in electron transfers and diffusion. For 2,6-PAQS, a capacity retention of 95.2% was observed with 138 mA h g−1 after the 200th cycle at −15 °C, and 120 mA h g−1 after the 200th cycle at −30 °C. The 1,5-PAQS isomer exhibited comparable but slightly weaker results. It is clear that the 2,6-PAQS isomer adapts better to temperature variation and provides a better performance. At −30 °C, 2,6-PAQS delivered a remarkable discharge energy density, ED, of 275 W h kg−1 (ED = UD·C; with C = 120 mA h g−1 and UD = 2,29 V vs. Li+/Li) without any adaptation of the baseline electrolyte to low temperature operations.
Conclusions
In summary, three PAQS isomers were synthesized, and characterized in a comparative study of their electrochemical performances as organic electrode materials. Slightly higher charging and discharging voltages were observed during cyclic voltammetry and are clearly due to the C–S position of the isomer. The galvanostatic tests showed that only the 1,5 and the 2,6-PAQS isomers were able to tolerate rates higher than 4C and long-term cycling (1000 cycles). In addition, the 2,6-PAQS isomer demonstrated a remarkable ability to endure fast cycling from 60 °C to −30 °C without a significant decrease in capacity retention (which decreased from 98.1% to 93.4%) thanks to its flexible structure. These results allow 2,6-PAQS to deliver an energy density of 275 W h kg−1 at −30 °C. It therefore appears to be a promising candidate as an alternative cathode for primary batteries with extended operating temperatures as well as a negative electrode in full-organic batteries dedicated to low temperature operation.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors would like to thank “La Région Centre Val de Loire” for financial support through the Obama project under the Lavoisier II regional program.References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- D. Larcher and J. M. Tarascon, Nat. Chem., 2015, 7, 19–29 CrossRef CAS.
- C. P. Grey and J. M. Tarascon, Nat. Mater., 2017, 16, 45–56 CrossRef.
- A. Mauger, C. Julien, A. Paolella, M. Armand and K. Zaghib, Materials, 2019, 12, 1–86 Search PubMed.
- J. Xu, H. R. Thomas, R. W. Francis, K. R. Lum, J. Wang and B. Liang, J. Power Sources, 2008, 177, 512–527 CrossRef CAS.
- S. Muench, A. Wild, C. Friebe, B. Häupler, T. Janoschka and U. S. Schubert, Chem. Rev., 2016, 116, 9438–9484 CrossRef CAS.
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737–738 CrossRef CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS.
- H. Chen, M. Armand, M. Courty, M. Jiang, C. P. Grey, F. Dolhem, J.-M. Tarascon and P. Poizot, J. Am. Chem. Soc., 2009, 131, 8984–8988 CrossRef CAS.
- Y. Liang, Z. Tao and J. Chen, Adv. Energy Mater., 2012, 2, 702 CrossRef.
- Z. Song and H. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC.
- W. Huang, Z. Zhu, L. Wang, S. Wang, H. Li, Z. Tao, J. Shi, L. Guan and J. Chen, Angew. Chem., Int. Ed., 2013, 52, 9162–9166 CrossRef CAS.
- Z. Song, Y. Qian, X. Liu, T. Zhang, Y. Zhu, H. Yu, M. Otani and H. Zhou, Energy Environ. Sci., 2014, 7, 4077–4086 RSC.
- S. Wang, L. Wang, Z. Zhu, Z. Hu, Q. Zhao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 5892–5896 CrossRef CAS.
- L. Zhao, J. Zhao, Y.-S. Hu, H. Li, Z. Zhou, M. Armand and L. Chen, Adv. Energy Mater., 2012, 2, 962–965 CrossRef CAS.
- Y. Chen, W. Luo, M. Carter, L. Zhou, J. Dai, K. Fu, S. Lacey, T. Li, J. Wan, X. Han, Y. Bao and L. Hu, Nano Energy, 2015, 18, 205–211 CrossRef CAS.
- Z. Jian, Y. Liang, I. A. Rodríguez-Pérez, Y. Yao and X. Ji, Electrochem. Commun., 2016, 71, 5–8 CrossRef CAS.
- T. B. Schon, B. T. McAllister, P.-F. Li and D. S. Seferos, Chem. Soc. Rev., 2016, 45, 6345–6404 RSC.
- G. A. Snook, P. Kao and A. S. Best, J. Power Sources, 2011, 196, 1–12 CrossRef CAS.
- W. Choi, D. Harada, K. Oyaizu and H. Nishide, J. Am. Chem. Soc., 2011, 133, 19839–19843 CrossRef CAS.
- J. Cao and A. Emadi, IEEE Trans. Power Syst., 2012, 27, 122–132 Search PubMed.
- L. Kouchachvili, W. Yaïci and E. Entchev, J. Power Sources, 2018, 374, 237–248 CrossRef CAS.
- R. Thangavel, K. Kaliyappan, D.-U. Kim, X. Sun and Y.-S. Lee, Chem. Mater., 2017, 29, 7122–7130 CrossRef CAS.
- K. Oka, S. Furukawa, S. Murao, T. Oka, H. Nishide and K. Oyaizu, Chem. Commun., 2020, 56, 4055–4058 RSC.
- C. Han, H. Li, R. Shi, T. Zhang, J. Tong, J. Li and B. Li, J. Mater. Chem. A, 2019, 7, 23378–23415 RSC.
- Z. Song, Y. Qian, M. L. Gordin, D. Tang, T. Xu, M. Otani, H. Zhan, H. Zhou and D. Wang, Angew. Chem., Int. Ed., 2015, 54, 13947–13951 CrossRef CAS.
- Z. Song, H. Zhan and Y. Zhou, Chem. Commun., 2009, 448–450 RSC.
- S. Phadke, M. Cao and M. Anouti, ChemSusChem, 2018, 11, 965–974 CrossRef CAS.
- J. Tong, C. Han, X. Hao, X. Qin and B. Li, ACS Appl. Mater. Interfaces, 2020, 12, 39630–39638 CrossRef CAS.
- Y. Lu, X. Hou, L. Miao, L. Li, R. Shi, L. Liu and J. Chen, Angew. Chem., Int. Ed., 2019, 58, 7020–7024 CrossRef CAS.
- Y. Zou, V. Raveendran and J. Chen, Nano Energy, 2020, 77, 105303 CrossRef CAS.
- C. Liu, T. Ma, K. Xia, X. Hou, Q. Nian, Y. Cai and J. Liang, Sustainable Energy Fuels, 2020, 4, 132–137 RSC.
- Z. Man, P. Li, D. Zhou, R. Zang, S. Wang, P. Li, S. Liu, X. Li, Y. Wu, X. Liang and G. Wang, J. Mater. Chem. A, 2019, 7, 2368–2375 RSC.
- W. Deng, X. Liang, X. Wu, J. Qian, Y. Cao, X. Ai, J. Feng and H. Yang, Sci. Rep., 2013, 3, 2671 CrossRef.
- T. Kawai, K. Oyaizu and H. Nishide, Macromolecules, 2015, 48, 2429–2434 CrossRef CAS.
- J. T. Edmonds andH. W. Hill, Production of polymers from aromatic compounds, US Pat., US3354129A, 1967 Search PubMed.
- Y. Zhang, I. Murtaza, D. Liu, R. Tan, Y. Zhu and H. Meng, Electrochim. Acta, 2017, 224, 622–627 CrossRef CAS.
- K. W. Bair, Anthracene derivatives, US Pat., US4719049A, 1988 Search PubMed.
- https://isa-lyon.fr/?page_id=2908 .
- A. Ahmad, A. Imani, L. Mao, R. Iqbal, H. Zhang, Z. A. Ghazi, R. Ahmad, A. A. Khan, L. Xie, C.-M. Chen, Z. Zhang and Z. Wei, Adv. Mater. Technol., 2019, 4, 1900617 CrossRef CAS.
- D. Lin-Vien, N. B. Colthup, W. G. Fateley and J. G. Grasselli, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, 1991, pp. 225–250, ch. 14 Search PubMed.
- J. H. S. Green, Spectrochim. Acta, 1962, 18, 39–50 CrossRef CAS.
- N. Parveen and R. Kant, J. Phys. Chem. C, 2014, 118, 26599–26612 CrossRef.
- S. A. Petrova, M. V. Kolodyazhny and O. S. Ksenzhek, J. Electroanal. Chem. Interfacial Electrochem., 1990, 277, 189–196 CrossRef CAS.
- J. Kim, J. Kim, J. Lee, H.-K. Song and C. Yang, ChemElectroChem, 2014, 1, 1618–1622 CrossRef CAS.
- B. Häupler, T. Hagemann, C. Friebe, A. Wild and U. S. Schubert, ACS Appl. Mater. Interfaces, 2015, 7, 3473–3479 CrossRef.
- Z. Song, Y. Qian, T. Zhang, M. Otani and H. Zhou, Adv. Sci., 2015, 2, 1500124 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00497a |
| ‡ Present address: Faculté des Sciences et Techniques, Parc de Grandmont, 37200 Tours, France. |
| This journal is © The Royal Society of Chemistry 2021 |