
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChoosing the right strategy: cryogrinding vs. ball milling – comparing apples to apples†
Julia L.
Shamshina
 *a,
Robin S.
Stein
b and
Noureddine
Abidi
a
*a,
Robin S.
Stein
b and
Noureddine
Abidi
a
aFiber and Biopolymer Research Institute, Department of Plant and Soil Science, Texas Tech University, Lubbock, Texas 79409, USA. E-mail: jshamshi@ttu.edu
bMcGill University, Department of Chemistry, Montreal, QC H3A0B8, Canada
First published on 11th November 2021
Abstract
Despite many reports on ball milling (BM) of chitin, the effect of cryomilling of chitin has not yet been reported, even though it is a solventless and environmentally-friendly method for the processing of chitin polymer. In this work, commercially available chitin has been cryoground for up to 96 min and the resulting morphological and chemical changes after cryomilling were monitored and compared with those induced by BM. FE-SEM analysis (1000×) revealed that cryogrinding resulted in size reduction of chitin particles and the size reduction was proportional to cryogrinding time. FTIR followed by PCA analysis revealed the breaking of intra- and intermolecular hydrogen bonds, partial amorphization, and insignificant breaking of glycosidic bonds after up to 48 min of cryogrinding, whereas an increase of cryogrinding time to 96 min resulted in a slightly higher number of broken bonds and highly amorphous material. The crystalline structure analysis demonstrated a decrease of crystallinity from 68 to 58% after 48 min of cryogrinding, and to 51% after 96 min of cryogrinding. No change in the crystallite size was found in the (110) plane, while a ∼15% decrease in crystallite size was determined in plane (020). No changes in the molecular weight were detected, although deacetylation to some extent was determined, contrarily to BM. The thermal stability of the polymer was higher than that of the starting material. Overall, the study highlighted similarities and differences between BM at room temperature and cryomilling, and set a foundation for co-processing of chitin with temperature-labile compounds.
Introduction
Extensive research has been focused on biopolymers as potential renewable sources for the preparation of high-value materials as alternatives to synthetic plastics. Among those, chitin (Fig. 1) is the second most abundant natural polymer, and is made of β-(1–4)-linked 2-deoxy-2-acetamido-D-glucose units. It is obtained from crustacean shells, skeletons of insects, fungi, etc.,1 and is applicable in various fields including biomedicine.2–4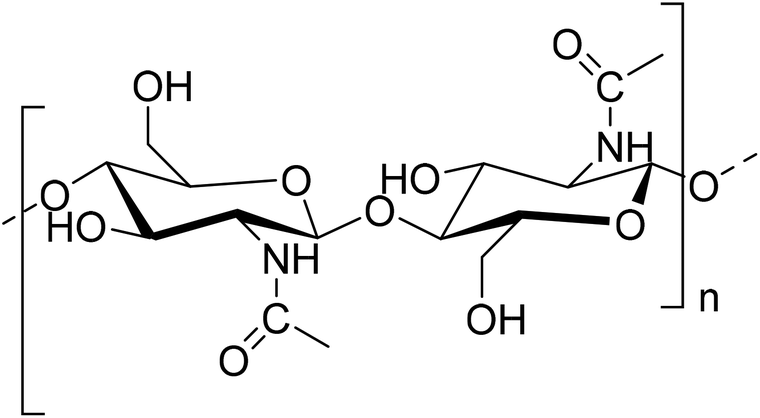 | ||
| Fig. 1 Structure of chitin. | ||
Successful replacements of synthetic plastics must preserve the low cost, versatility, and ease of processing in order to compete as biopolymeric alternatives. From the perspective of the field of Green Chemistry, the expansion of which has resulted in the development and implementation of new technologies, low chemical intensity solid-state processing methods are suitable complements to conventional solvent-assisted chemical methods.5 These include mechanochemistry, reaction milling, liquid- and electric discharge-assisted milling, etc.6–8 Mechanochemistry of insoluble (in traditional organic solvents) chitin polymer9 is an efficient route to its particle size reduction and amorphization; however, the terms “milling” and “grinding” thus far have been applied for relatively high-energy ball-milling (BM). The high number of large mechanical perturbations generated during BM often results in significant changes in morphology, crystallite size, shape, and crystallinity decrease, causing the desired partial or complete amorphization and also inducing structural changes.
BM of chitin breaks the intermolecular hydrogen bonds, often cleaving C–O–C glycosidic bonds, and, in the presence of a base, breaking amide C–NH bonds, causing deacetylation.10–15 Thus, recently, a solid-state single step mechanochemical method was developed for the synthesis of low molecular weight chitosan (LMWC) from shrimp shell powder or purified chitin in the presence of a sodium hydroxide.10 Similarly, a method based on the combination of mechanochemistry and aging was developed to yield high molecular weight chitosan (HMWC).11,12
At the same time, reducing the particle size of chitin with or without minimal structural changes would be desirable, since chitin itself is a valuable polymer for material preparation.16–21 In recent years, efforts have been put into the development of cryogrinding (cryogenic grinding, cryogenic milling, or cryomilling), that, as the name implies, is the process of grinding a material at low temperature (<123 K), achieved by cooling with, for instance, liquid nitrogen (LN2). It is an environmentally-friendly process that minimizes the energy expenditure while making the amorphization process relatively fast and effective. Depending on the conditions, the cryogrinding can be “wet” (milling materials mixed with cryogenic liquid) or “dry” (milling chamber is cooled externally).
The main advantage of cryogrinding over ball milling (BM) is the suppression of side processes at low temperature. A number of recent studies report that the processing of polymeric materials via cryogrinding facilitates particle size reduction,22 the preparation of powder coatings,23 microfibrillated substances24 and/or blending of polymers.25 For instance, cryogrinding was employed to generate blends of chitosan (CS) with poly(lactic acid) (PLA) and tricalcium phosphate (TCP), that were later electrospun into fibrous membranes for bone tissue engineering applications.26 The cryogrinding process is often applied towards the grinding of temperature-sensitive active pharmaceutical ingredients (APIs),27–30 producing amorphous systems without their thermal degradation and/or oxidation at cryogenic temperature. Cryogrinding of cellulose with a poorly water-soluble drug piroxicam resulted in the formation of a stabilized solid dispersion.31
However, despite the fact that cryogrinding of biopolymers could provide green, scalable access to biopolymers functionalization and composites, it has been overlooked in favor of BM. Applied to biopolymers, the cryogrinding of cellulose to induce changes in morphology, size, shape, and crystallinity was studied vastly more often than that of other biopolymers. Different types of celluloses (pure cellulose,32 natural sisal fibers,33 Norway spruce,34 orange tree,35 soy,35 sugarcane35) underwent cryogrinding following the study of morphological changes on the fiber surface, crystallinity, and stability.36 Cryogrinding studies of chitin, at the same time, are conspicuously lacking,37 and its mechanochemistry is restricted to BM, with or without10,11,15 the addition of base. We were able to find a single study of chitin cryogrinding where the discussion was dedicated to the design and construction of a cryogenic grinding system.38 Below we present a study of chitin cryogrinding with an emphasis on investigating chemical aspects, such as the evolution of structure, crystal size, thermal properties or molecular weight, and comparing the results with known BM studies.
Experimental
Materials
Chitin (powder, practical grade) was obtained from Fischer Scientific (Alfa Aesar™, chitin from shrimp shells, catalog number AAJ6120622) and dried overnight in a 50 °C-oven prior to processing. Ionic liquid 1-ethyl-3-methylimidazolium acetate ([C2mim][OAc], Lot 25PI281_4.3, 0.21 wt% water), was obtained from ProIonic, Gmbh (Grambach, Austria) and used without additional drying. N,N-Dimethylacetamide (DMAc), HPLC grade, manufactured by Alfa Aesar™, was obtained from VWR, transferred into an amber-glass bottle under nitrogen, and stored over 3 Å molecular sieves that were activated at 300 °C. Anhydrous lithium chloride (99%, for analysis) manufactured by Acros Organics was obtained from Fischer Scientific and used without additional purification.General procedures
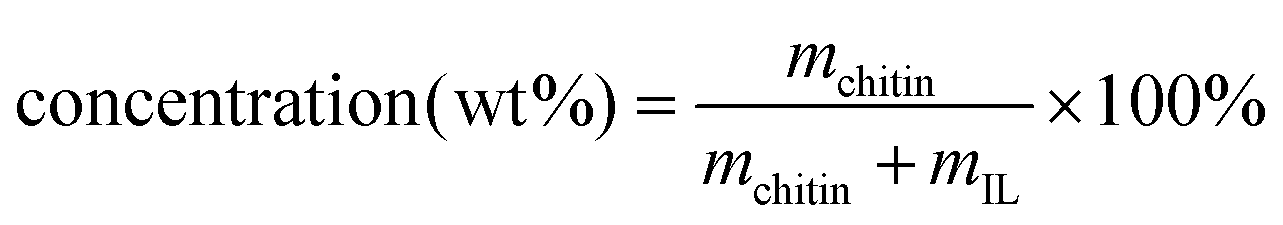 , see ESI, Table S2.† The vials were then placed into an oil bath (100 °C, 760 rpm) and heated overnight to ensure complete dissolution. After the dissolution, the vials were taken out of the heating bath, and the solutions were cooled to RT and subjected to rheological measurements.
, see ESI, Table S2.† The vials were then placed into an oil bath (100 °C, 760 rpm) and heated overnight to ensure complete dissolution. After the dissolution, the vials were taken out of the heating bath, and the solutions were cooled to RT and subjected to rheological measurements.
Methods
| CrI110 = (I110 − Iam) × 100%/I110 | (1) |
Method 2. After obtaining the diffraction intensity profiles of the samples in the same manner as described in Method 1, followed by smoothing and baseline correction, individual crystalline peaks were extracted by a curve-fitting process using a peak fitting feature in Origin (https://www.originlab.com/), assuming Gaussian functions for each peak. CrI was calculated using eqn (2)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 59,60 from the ratio of the sum of the areas under the crystalline diffraction peaks and the total area under the diffraction pattern, respectively, according to the eqn (2):
59,60 from the ratio of the sum of the areas under the crystalline diffraction peaks and the total area under the diffraction pattern, respectively, according to the eqn (2):
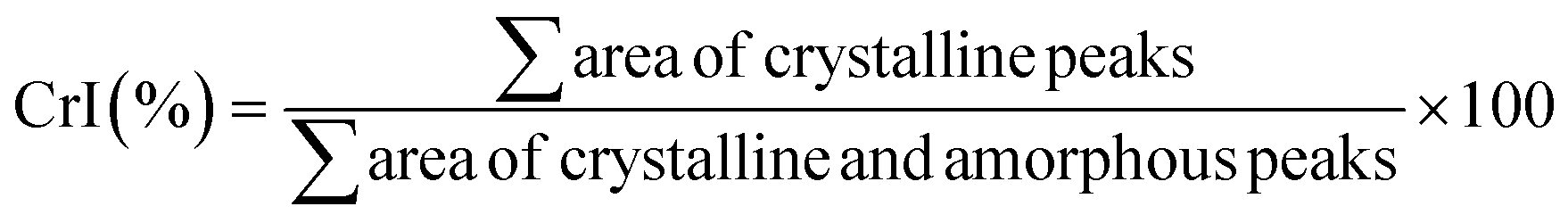 | (2) |
| β = (k × λ)/(L × cos(θ)) | (3) |
The FTIR spectra recorded at a spectra resolution of 4 cm−1 with 64 co-added scans over the range from 650 to 4000 cm−1 were also subjected to Principal Component Analysis (PCA) with leverage correction and mean-center cross validation boxes checked using Unscrambler V. 9.6 Camo Software AS (CAMO Software AS, Norway, http://www.camo.com/unscrambler/).
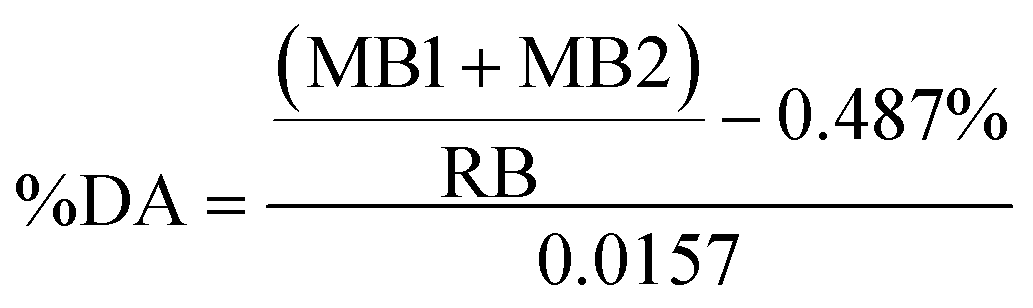 | (4) |
Method 2: solid-state cross-polarization magic angle spinning carbon-13 nuclear magnetic resonance (CP/MAS-NMR) spectroscopy
Solid-state 13C NMR spectra were acquired on a 400 MHz Varian VNMRS spectrometer using a 4 mm double-resonance Varian Chemagnetics T3 probe (now Agilent, Santa Clara, CA, USA). Approximately 35 mg of sample was center-packed into rotors and spun at a spinning rate of 8 kHz (C0, C3) and 8.3 kHz (C6, C12 and C24), to minimize spinning sidebands on the carbonyl carbon atom. Basic CP parameters were based on those suggested by Duarte et al.,43 with a recycle delay of 5 s and a contact time of 2000 μs using a spin-lock field of approximately 60 kHz (CP standard). 2048 scans were acquired for each sample, for a total of nearly 3 h per sample.
The spectra were processed using MestReNova software (MestReNova, Version 12.0.0-20080, Mestrelab Research, S.L., http://www.mestrelab.com). The spectra processing included a backwards linear prediction of the beginning of the FID, phase correction, and integration of the CH3 signal region and the polysaccharidic backbone region (C1–C5). The %DA was determined by measuring relative integrals of methyl group (CH3) compared to the backbone carbons.
Results and discussion
Mechanical processing of chitin is a known process, but it has been limited to BM. In order to effectively utilize chitin cryogrinding, information on the effect of cryogrinding conditions on the final properties of the polymer is essential. The critical properties include structural transformation, crystallinity, crystal size, thermal properties, and polymer quality, which is measured by its molecular weight (Mw) and degree of acetylation (%DA).44 In this context, a comparison of BM and cryogrinding is also of interest.Effect of cryogrinding conditions on particle morphology and microstructure of chitin
Commercial practical grade chitin powder, pre-dried in 50 °C-oven overnight, was cryoground using a cryogenic freezer mill (see Experimental). For this, polycarbonate vessels equipped with magnetically-driven 440C high carbon martensitic steel impactors were filled with chitin, chilled in liquid nitrogen, and then ground (in dry state) at cryogenic temperature for 3, 6, 12 and 24 cycles (12, 24, 48 and 96 min grinding time, respectively). These samples were abbreviated as C0 (starting material), C3, C6, C12 and C24.FE-SEM analysis revealed that raw chitin particles exhibited a flake-like shape (Fig. 2, 3 and ESI, Fig. S1 and S2†) and were reduced to smaller particles after the cryogrinding; a small number of large chitin particles also remained. The size reduction was proportional to cryogrinding time. Although the cryogrinding reduced particle sizes, no chitin fibrillation was observed, even after 24 cycles (96 min), consistent with the particle size analysis as discussed in crystallography section.
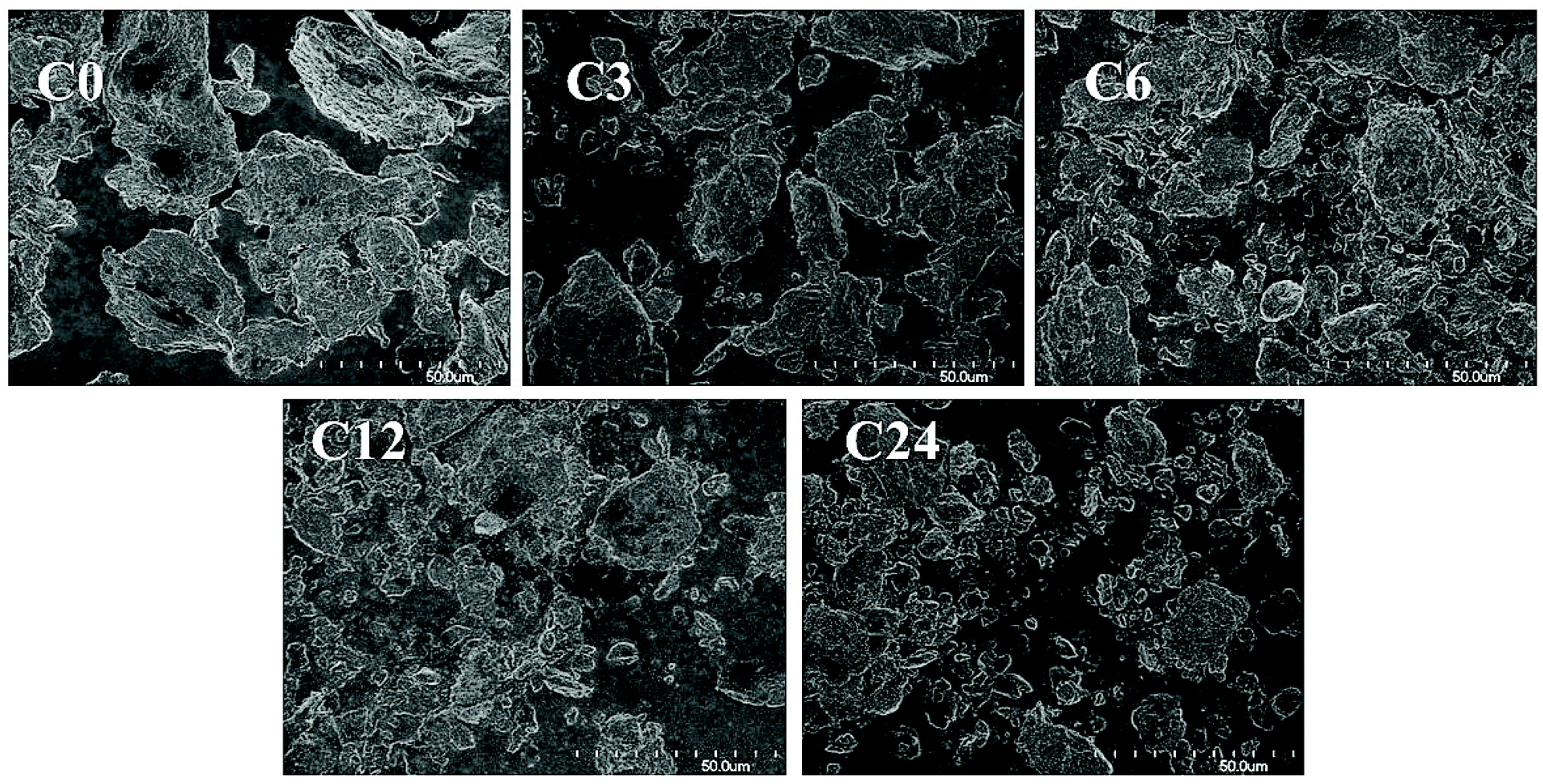 | ||
| Fig. 2 FE-SEM images (1000×) of raw chitin (C0) and the cryoground chitin particles (C3, C6, C12, C24). | ||
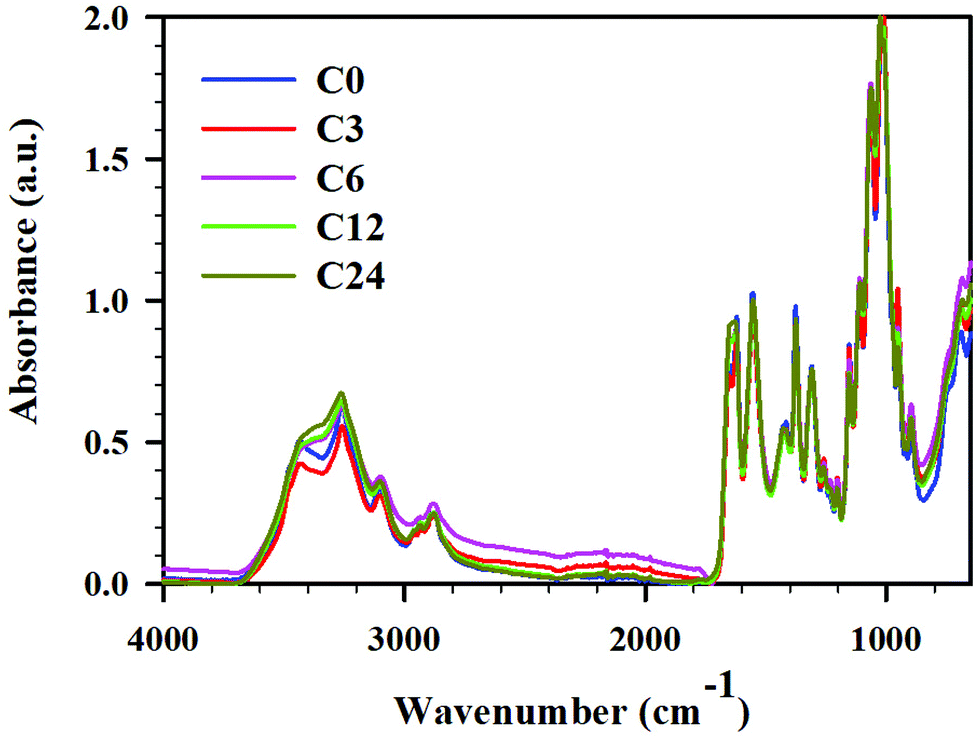 | ||
| Fig. 3 ATR FTIR spectra for cryoground chitin: C0 (blue), C3 (red), C6 (pink), C12 (green), and C24 (mustard), full spectra in the range 4000–600 cm−1. | ||
The observations were similar to those obtained for BM by Zhong et al.11 and Osada et al.12 who observed particle size reduction and claimed that BM-products exhibited a close-packed texture similar to that of raw chitin. The Moores group has recently shown a dependency of milling materials on results of BM and indicated that effectiveness of BM depended on both Vickers hardness (VH) of the milling balls and their mass.15 VH of 440C steel is ∼760 MPa at low temperatures, significantly lower than that of the stainless-steel (SS) balls that were utilized in BM (VH ∼ 2000 MPa).11,12 Even though the total mass of the SS balls in BM was 3- and 20-times larger than the mass of impactors in cryomilling (90 g (ref. 11) and 704 g (ref. 12) in BM vs. 32 g in cryogrinding), the impact and shearing forces were not sufficient to separate chitin into microfibril bundles.
Structural changes of cryomilled chitin detected by Fourier-transform infrared spectroscopy (FTIR) analysis
Zhang et al. claimed that the BM under neutral conditions rendered characteristic bands typical for raw chitin, and indicated that the chemical structure of the polymer was not altered under BM treatment.11 Contrarily, others claimed significant disassembly of hydrogen bonding network, amorphization, and decrease in glycosidic linkage content.12,13 To elucidate the effect of cryogrinding on chitin structure, FTIR ATR spectra of starting material and cryoground samples were recorded (Fig. 4, and ESI Fig. S3–S6† for expansion of regions of interest and complete analysis). After that, peaks were assigned by comparing the experimental spectra with those in literature reports (see ESI, Table S3† for a list of chitin peaks).45,46,47,48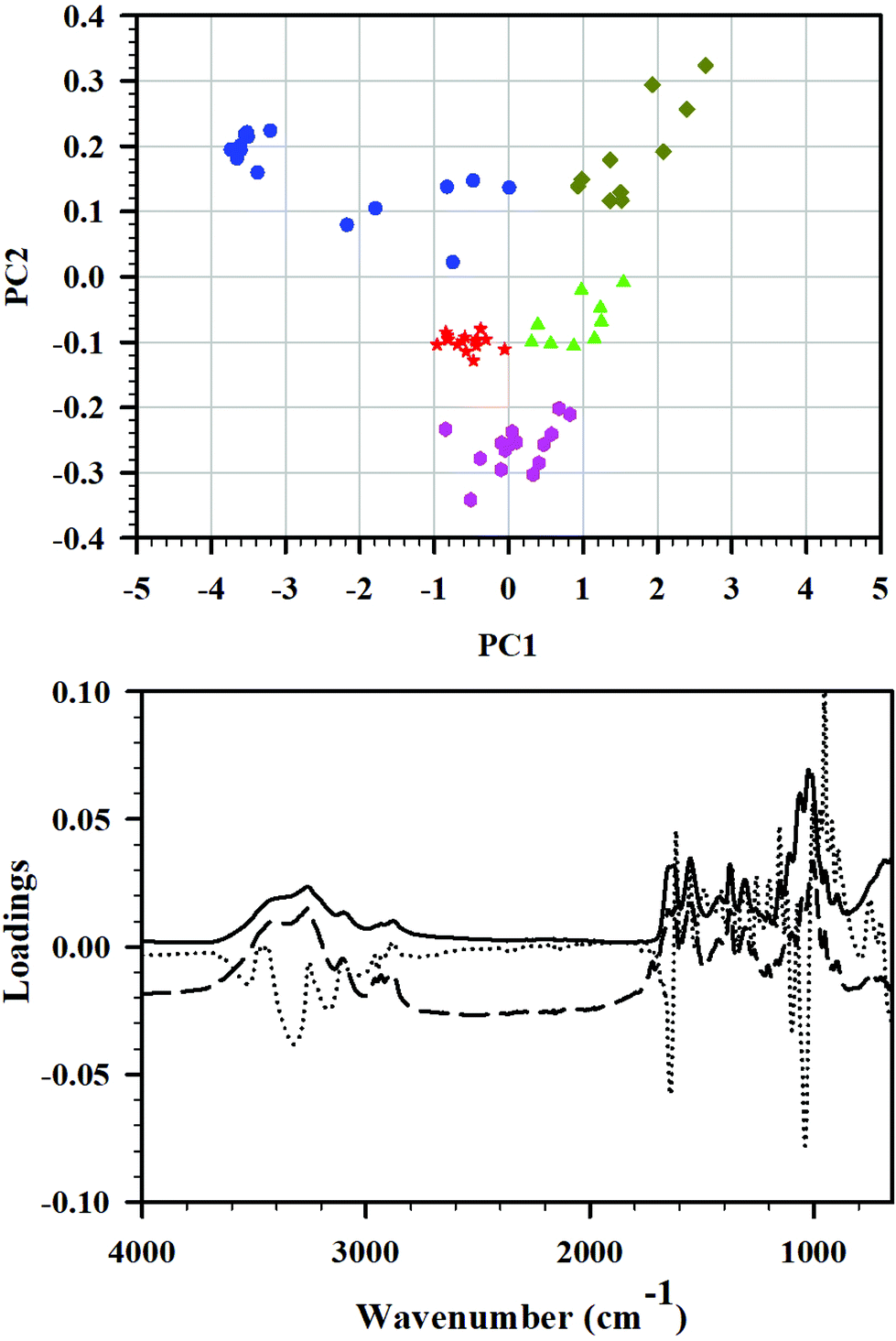 | ||
| Fig. 4 Top: PCA score plot, PC1 versus PC2, of the FTIR spectra of the total set of C0–C24 samples and all the wavenumbers included. The model was centered, and the axes cross each other at the origin. Color coding: C0 (blue), C3 (red), C6 (pink), C12 (green), and C24 (mustard), baseline corrected and normalized. Bottom: PC1, PC2, and PC3 loadings plots. | ||
While all spectra showed distinct peaks typical for chitin in all cryoground samples, there were a few differences noted in absorbances between raw chitin and cryoground chitin. There were no cryogrinding-induced changes in C–H stretches of CH, CH2 and CH3 groups of chitin at 2960 cm−1, 2922 cm−1 and 2877 cm−1, respectively, and the values were in agreement with the literature values (2965 cm−1,46 2927 cm−1,46 and 2883 cm−1,46 respectively). No obvious changes were found in location and intensity of C–N + N–H (Amide III) at 1306 cm−1 (literature value 1312 cm−1 (ref. 46)), as well as in asymmetric N–H at 3258 cm−1 (literature values 3265–3268 cm−1 (ref. 45 and 46)).
However, the O–H stretching initially located at 3436 cm−1 (literature values 3440–3448 cm−1 (ref. 47 and 48)) shifted slightly to lower wavenumbers and became much less pronounced as cryogrinding time increased (ESI, Fig. S4†). Specifically, this peak appeared pronounced in the starting material, slightly weakened in sample C3 (12 min of grinding), and significantly weakened in the C6 and C12 samples (24 and 48 min of cryogrinding, respectively). In sample C24 (96 min of cryogrinding) the peak largely vanished, appearing more like a shoulder. This analysis implies that the intra- and intermolecular hydrogen bonds between chitin chains were destroyed, slightly increasing the number of free hydroxyl groups and decreasing the number of bound hydroxyl groups. This observation agreed with BM results noted by others. Thus, Osada et al.12 noted weakening of hydrogen bonds (O–H stretching) between chitin interchains (3450 and 3486 cm−1) induced by BM, although hydrogen bonds at 3268 cm−1 were retained.
The most apparent spectral difference observed was the frequency of the vibration modes of amide I in the region 1660–1620 cm−1 (ESI, Fig. S5†). It is now an accepted explanation that two types of amides exist in chitin: half of the carbonyl groups that are bonded through hydrogen bonds to the amino group inside the same chain (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N) are responsible for the vibration located at 1660 cm−1, and another half of carbonyl groups additionally hydrogen-bond to C(6)OH from the side chain (–CH2OH⋯OC), producing a band at 1627 cm−1.46,49 In non-cryoground chitin (C0), we observed these peaks at 1654 cm−1 and 1623 cm−1. Upon grinding, we detected a decrease in the amide I band at 1623 cm−1, indicating the disruption of hydrogen-bonding between carbonyl groups and –C(6)OH from the side chain. Thus, samples C0 and C3 demonstrated two separate, clearly distinguishable peaks at 1654 cm−1 and 1623 cm−1, implying a higher degree of crystallinity, while samples C6 and C12 revealed a slight converging of these peaks and respective change in intensity, implying partial amorphization.50 Sample C24 exhibited a single band at 1656 cm−1. This observation is similar to BM. Thus, Margoutidis et al.13 reported the change in the ratio between the absorbance intensity at 1623 and 1654 cm−1 for BM-chitin and noted that the ratio and intensity of these peaks depended on milling time. A significant drop in this ratio was observed after 75 min of BM, and was indicative of hydrogen-bonding network weakening. Osada et al.12 also reported partial amorphization under BM.
O⋯H–N) are responsible for the vibration located at 1660 cm−1, and another half of carbonyl groups additionally hydrogen-bond to C(6)OH from the side chain (–CH2OH⋯OC), producing a band at 1627 cm−1.46,49 In non-cryoground chitin (C0), we observed these peaks at 1654 cm−1 and 1623 cm−1. Upon grinding, we detected a decrease in the amide I band at 1623 cm−1, indicating the disruption of hydrogen-bonding between carbonyl groups and –C(6)OH from the side chain. Thus, samples C0 and C3 demonstrated two separate, clearly distinguishable peaks at 1654 cm−1 and 1623 cm−1, implying a higher degree of crystallinity, while samples C6 and C12 revealed a slight converging of these peaks and respective change in intensity, implying partial amorphization.50 Sample C24 exhibited a single band at 1656 cm−1. This observation is similar to BM. Thus, Margoutidis et al.13 reported the change in the ratio between the absorbance intensity at 1623 and 1654 cm−1 for BM-chitin and noted that the ratio and intensity of these peaks depended on milling time. A significant drop in this ratio was observed after 75 min of BM, and was indicative of hydrogen-bonding network weakening. Osada et al.12 also reported partial amorphization under BM.
Multiple peaks were seen within the region 1020–1160 cm−1 and were associated with vibration modes of C–OH, C–O–C and C–C bonds (ESI, Fig. S6†).50 Thus, C–O–C ring asymmetric bridge stretching was located at 1154 cm−1 (literature value 1156–1157 cm−1 (ref. 46 and 50)). The intensity of this band decreased insignificantly upon cryomilling (C0 > C3 > C6 > C12 ∼ C24), indicating that cryomilling caused the breaking of glycosidic bonds in chitin chains to some extent. Importantly, the intensity of this peak was similar in C12 and C24. Interestingly, C–H deformation of the β-glycosidic bond at 895 cm−1 (literature 895–896 cm−1 (ref. 46 and 47)) demonstrated no identifiable trend.
The peaks at 1113 cm−1, 1025 cm−1 and 1068 cm−1 belong to various C–OH stretching of hydroxyl groups.51,52 The absorption band at 1025 cm−1 (C5–OH stretching) intensified and became more pronounced with an increase of grinding time while a peak at 1068 cm−1 (C3–OH stretching) slightly shifted to a lower wavenumber with an increase of grinding time. The same scenario was observed for a band located at 1113 cm−1 (asymmetric in phase ring stretching53) where C6, C12 and C24 demonstrated a more prominent peak than C0 and C3 did. Contrarily, the peak at 1010 cm−1 known to be C–O asymmetric stretch in phase ring,53 decreased in intensity with increase of time of cryogrinding; the same was true for CH3 wagging at 952 cm−1.47 All of this was attributed to environmental changes around these bonds due to glycosidic bond breaking. Overall, the results were consistent with those reported for BM: hydrogen bonding network breakage, partial amorphization, and glycosidic linkage breakage.12,13
To conclude, the most evident spectral features used to analyze the extent of amorphization in chitin is the change in the ratio and the intensity of amide I CO vibration modes at ∼1660 and ∼1623 cm−1, respectively while the decrease of C–O–C ring asymmetric bridge stretching (1154 cm−1) is typically indicative of the glycosidic bonds breaking in chitin chains. Zhong et al.11 showed no difference in the ratio or intensity of ∼1660/∼1623 cm−1 peaks between raw chitin and BM chitin, nor it demonstrated a decrease of the intensity of the peak at 1154 cm−1. However, no expansion of these specific regions, absence of spectra normalization, and somewhat different technique (spectra recorded using pellets made of potassium bromide (KBr) and chitin) made it difficult to either agree with or argue against the conclusion made by the authors. Contrarily, the spectra published by Osada et al. clearly indicated that BM resulted in amorphization while the study did not look at the glycosidic bond breaking. Data obtained by Margoutidis et al.13 demonstrated both amorphization and breaking of glycosidic bonds.
Principal component analysis
Principal component analysis (PCA) is frequently used for data reduction and analysis of high-dimensional data sets. During this analysis, a smaller set of principal components (PC) is obtained. It explains the variability on the data through creation of a new sub-space defined by PCs, and is easier to interpret than original data. In this study, PCA was applied to the FTIR spectra of the set of cryoground chitin samples, and PCA was applied to reveal differences between the samples and identify the main sources of variation in the spectra. Three principal components were enough to explain 98% differences among the samples.In the PCA score plot, the complete data matrix was modeled and included all the samples (10–15 FTIRs of C0–C24 each) and all the wavenumbers. The score plot of PC1 versus PC2 (Fig. 4, top) showed a distinct clustering of the samples that was related to cryogrinding time. Group A which consisted exclusively of starting material C0 before cryogrinding (n = 15) was located toward the negative side of PC1 and the positive side of PC2 and clearly had spectra deviating considerably from all other groups. Cluster B, which contained solely C3-points, was situated near the origin of the model, slightly toward the negative side of both PC1 and PC2 (group B, n = 14). Similarly, cluster C (n = 15) was comprised entirely of C6 and was located toward the more negative side of PC2. Cluster D (n = 10) that entailed C12 points exclusively was found near origin toward the positive side of PC1 and the negative side of PC2. Finally, cluster E, consisted exclusively of C24, was distinctively positioned on the positive sides of PC1 and PC2 (n = 10), away from the other samples. Clusters B, C, and D were relatively close to each other, while clusters A and D were distinctively set apart. Such difference was attributed to strong intramolecular O–H bonding, its weakening in C3, C6, and C12, and the full breaking of this bonding in C24. It was also dependent on the extent of amorphization and the extent of breaking of glycosidic bonds in chitin chains.
This is in line with the observation that the PC1, PC2, and PC3 loadings plots showed particularly high loadings for several wavenumbers in the regions from 3500 to 3200 cm−1 (O–H stretching), 1650 to 1600 cm−1 (CO stretching of amide I), and 1200 to 900 cm−1 (C–O–C and C–O stretching) as illustrated in Fig. 4, bottom, confirming the interpretation of FTIR results.
Changes in crystalline structure of cryomilled chitin
To obtain the crystallinity index (CrI) of the samples, powder X-ray diffractograms were recorded and are presented in Fig. 5. All diffractograms showed six peaks prominent for α-chitin, at 2θ 9.24, 12.69, 19.23, 20.58, 23.18, and 26.27°.39,54,55 These peaks corresponded to the crystalline planes of (020), (021), (110), (120), (130), and (013), suggesting a typical α-chitin crystalline structure.11,56,57 Less pronounced peaks were located at 2θ 27.86 and 39.20°.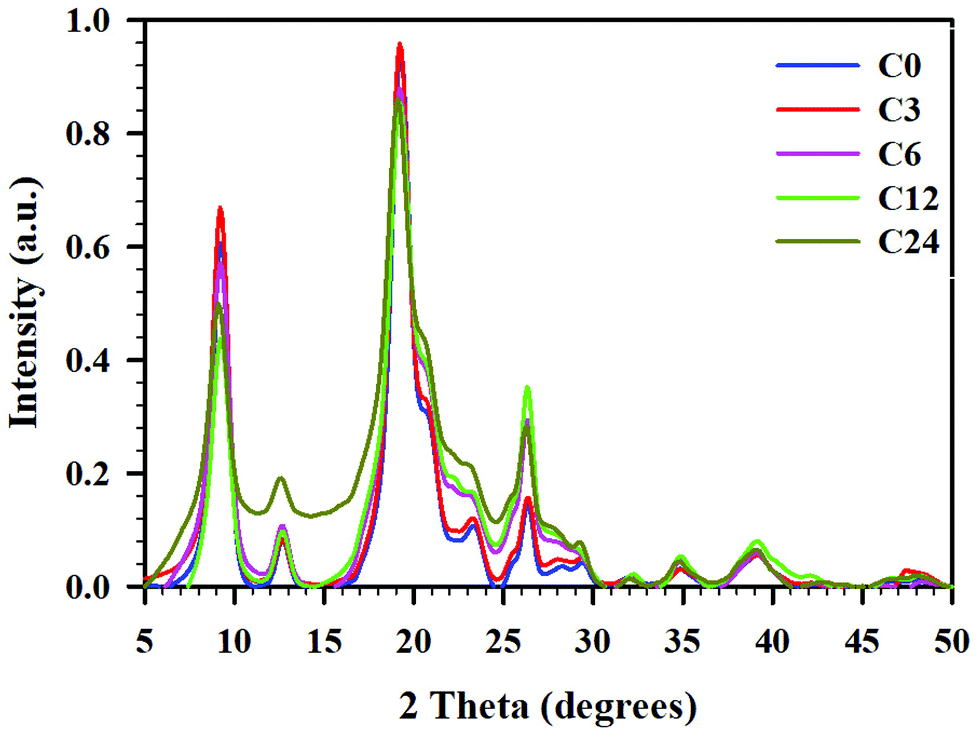 | ||
| Fig. 5 pXRD difractogramms for cryoground chitin: C0 (blue), C3 (red), C6 (pink), C12 (green), and C24 (mustard), baseline corrected and normalized. | ||
As seen in the pXRD patterns of cryomilled chitin, there were noticeable changes in crystallinity (crystallinity index, CrI) after chitin processing; these changes depended on processing time. First, we conducted an evaluation of CrI by the peak height method, comparing the relative intensities of crystalline (2θ 19.2°) vs. amorphous peak of the same unit (2θ 12.6°), as reported by Cardenas et al.,47 or with a baseline height (2θ 16.0°) as reported by Kaya et al.39 However, since intensities of the amorphous peak (or baseline height) were similar for all samples but C24, crystallinity indices calculated by this method were nearly identical for C0–C12 (see ESI, Table S6†).
Yet, as it could be seen from Fig. 5, the total area under the diffractograms noticeably increased with cryogrinding time clearly indicating the reduction in crystallinity and following the order C0 ∼ C3 < C6 ∼ C12 < C24. This discrepancy could be a result of the fact that the single highest peak (110) at 19.2° was used in the calculation of CrI, while contributions from the other five crystalline peaks were excluded. Besides, peaks in the diffractogram were relatively broad and varied considerably in their width, resulting in comparison of intensities to be an inaccurate measure of the polymer's crystallinity.58
Hence, we turned our attention to the second method, peak deconvolution,59,60 for a crystallinity evaluation. After peak fitting (ESI, Fig. S7–S11†), CrI was calculated as the ratio of sum of the areas under the crystalline diffraction peaks and the total area under the diffraction pattern, respectively (see ESI, Table S6† for peak areas). Thus, samples C0 and C3 had similar crystallinity values (CrI 68 ± 0.2%), samples C6 and C12 were slightly less crystalline (CrI 58 ± 2%), and the sample C24 had the lowest crystallinity (CrI 51 ± 1%). The diffractogram of C24 was distinctive from the rest in that the peak at 12.6° had a significantly larger area than the same peak in all other samples. The method resulted in significantly lower CrI values (∼20–30%) than peak height method, as was already shown for cellulose.58
Such peak broadening and significant decrease of crystallinity during cryogrinding was previously noted by Liang et al.;23 however, diffractograms reported in this study were recorded in the narrow range of 2θ 20–50° and excluded three of the most prominent crystalline peaks in (020), (021), and (110) planes from consideration. Comparing the crystallinity of chitin after cryogrinding with that after BM, we noticed a high dependency on size and mass of the grinding balls.11,15 Thus, Margoutidis et al. reported the CrI decrease from 91% to 51% by BM with 2 × ∅ 12.7 mm (17 g) balls and to 37% by BM with 16 × ∅ 6.35 mm (16 g) balls, during a 90-min time period.13 Osada et al. reported a gradual decrease of the crystallinity with increasing BM-time: from 91 to 68% after BM with 1400 × ∅ 5 mm (704 g) chromium steel balls for 10 min, and to 40% after BM with the same number and type of balls for 30 min. DiNardo et al. demonstrated a crystallinity decrease from 67 to 27–34%, after BM with 1 × ∅ 5.53–12.64 mm (0.7–8.4 g) SS balls.15 Zhong et al. who used BM with 14 × ∅ 10 mm and 38 × ∅ 6.25 mm (total mass 90 g) SS balls reported complete amorphization of chitin.11
The crystallite size values were calculated using Scherrer's equation.56 Cryogrinding had no impact on the destruction of the plane (110) and slightly impacted plane (020). Thus, no decrease in the average sizes of the crystalline regions (∼84 nm) was detected in (110) direction, while there was a ∼15% decrease in crystallite size in the (020) direction, from 73 to 63 nm (Table S7†). Osada et al.12 reported a ∼20% decrease in crystallite size (110), from ∼11 to 9 nm, after 10 min of BM time, and to ∼3 nm after 30 min of BM time.
Degree of acetylation (%DA) and molecular weight
The most important properties of chitin are the Mw and %DA. Chitin is made of repeated β-(1→4)-linked N-acetyl-D-glucosamine units. When more than 50% of N-acetyl residues in a polymer are cleaved with the formation of β-(1→4)-linked D-glucosamine, the polymer is called chitosan.61 Quantitatively, the %DA represents the proportion of chitosan units with respect to the total number of units. Extent of deacetylation brought about by polymer processing conditions depends on variations in chitin sources, its Mw, crystallinity, types of crystal morphologies, etc.For the determination of %DA FTIR has been used by many through the application of empirical equations,62 frequently combined with titration.63 According to Alvarenga, all FTIR methods are only useful in the 45–100 %DA range.61 We have used the method of Beil et al. for %DA determination based on first derivative ATR FTIR spectroscopy (ESI, Fig. S12–S16 and Table S9†).42 The %DA was determined to be ∼82 ± 3% for both the starting material and all the cryoground samples, suggesting that no deacetylation took place during processing. This agrees with the results of BM reported by Margoutidis et al.13 who did not observe any deacetylation.
Because ATR equipped instruments do not deliver precise values in too low or high a DA range, and a non-uniform contact with the ATR crystal, that might result in poor resolution,64 we have also evaluated %DA using Solid-State Cross-Polarization Magic Angle Spinning Carbon-13 Nuclear Magnetic Resonance CP/MAS-NMR, to be consistent with Experimental Spectroscopy. This was done by measuring relative integrals of methyl group (CH3) compared to the carbon integrals of the polysaccharidic backbone (Fig. 6).43
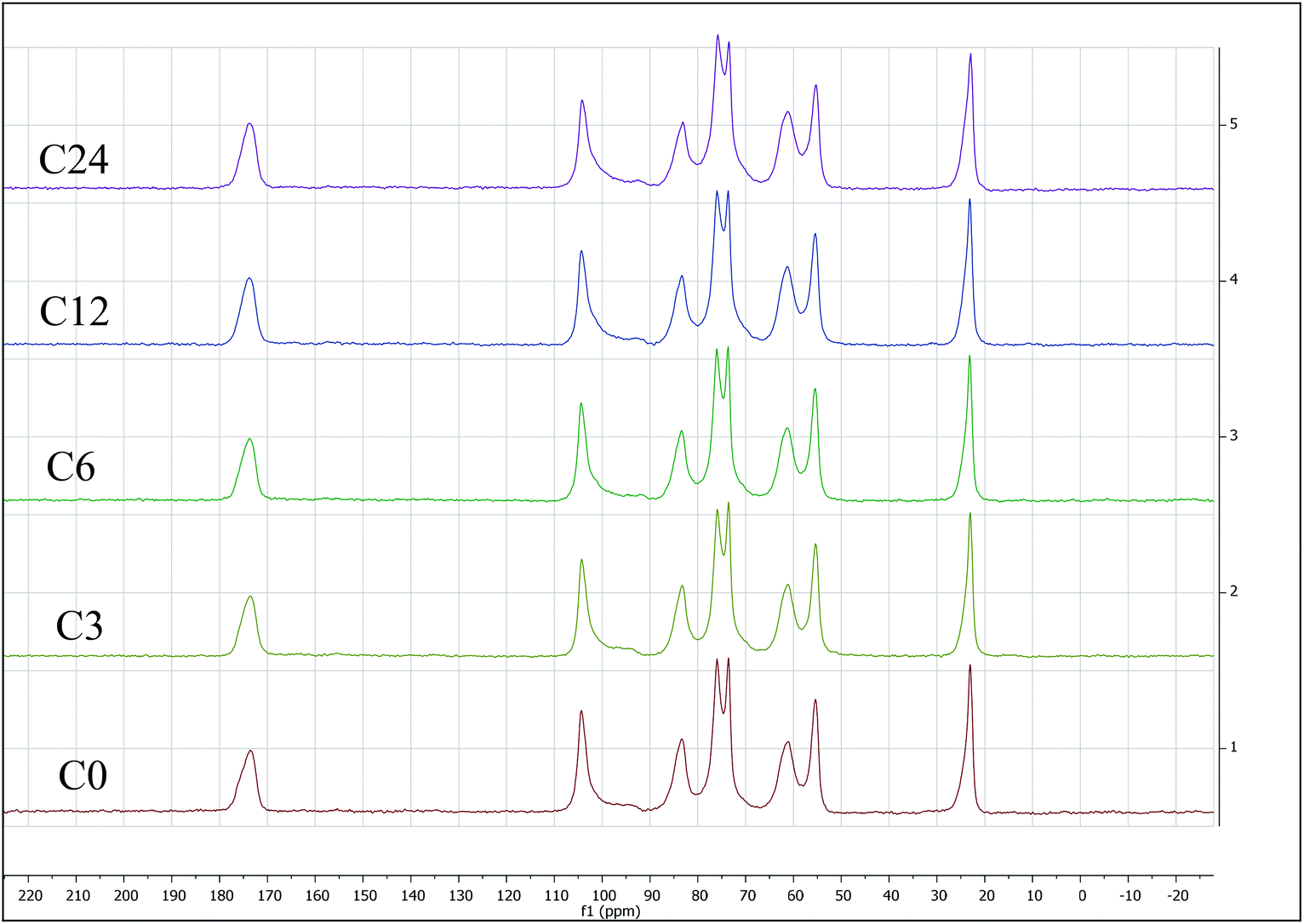 | ||
| Fig. 6 Solid-state cross-polarization magic angle spinning carbon-13 nuclear magnetic resonance (CP/MAS-NMR) for starting material C0 (bottom) through C24 sample (top). All integrated CP/MAS-NMR spectra are presented in the ESI.† | ||
The %DA determined via CP/MAS-NMR was 94% for C0, and gradually decreased with increasing of cryogrinding time. Specifically, the samples C3 and C6 exhibited 85–87 %DA, just slightly lower than that for C0. Contrarily, %DA of the sample C12 was found to be 76%, while %DA of the sample C24 was determined to be as little as 61 %DA (ESI, Fig. S17–S21 and Table S10†). This observation is contrarily to unchanged %DA in BM reports,13,14 that claimed that mechanical force alone was not able to induce deacetylation. It appears that extended cryogrinding results in breaking of NH–C(O) bond.
High energy milling methods, such as the use of a BM process, were previously reported to significantly (2–3 times) decrease molecular weight (Mw) of chitin.12,15 Thus, we set to evaluate whether cryogrinding decreased the Mw and to what extent. While synthetic polymers are soluble in volatile organic solvents (VOCs), chitin is not, and various solvent systems are employed for its dissolution. A typical solvent system for chitin is N,N-dimethylacetamide/lithium chloride (DMAc/LiCl),65–67 which is a classic solvent system used in the literature for Mw determination. However, despite numerous attempts, DMAc/LiCl (8% w/v) did not allow complete dissolution of chitin samples, an issue that was previously reported.12,16 While for material preparation incomplete dissolution of the polymer does not present an issue, for Mw determination it is critical that all chitin is dissolved; otherwise any measurement would result in an erroneous value.
Considering that the value of Mw was not a purpose in this work, we set to compare relative viscosities of the samples, to elucidate any differences in Mw. For that, we measured the rheological properties of 1 wt% chitin solutions in ionic liquid 1-ethyl-3-methylimidazolium acetate [C2mim][OAc]. The flow curves were plotted (3–5 replicates for each sample, ESI, Fig. S22†), and statistical analysis showed no significant differences between them, indicating no decrease in Mw value.
Thermal properties
Thermal properties were studied using Thermal Gravimetric Analysis (TGA). TGA curves and differential thermogravimetry (DTG) thermograms for chitin are shown in ESI, Fig. S23 and S24.† It could be seen that there were three weight loss intervals in TGA curves of C0–C24 samples. The first characteristic weight loss, at 49–51 °C, corresponded to the evaporation of physically adsorbed and strongly hydrogen-bonded water to chitin.68 The samples contained 9–11% water (Table 1) and the ability of cryoground samples to absorb water of the samples increased with amount of grinding time.| % water | T onset, °C | T 1 50%, °C | T transition, °C | T 2 50%, °C | Ref. | |
|---|---|---|---|---|---|---|
| a Individual peaks were extracted by a curve-fitting process from the thermogram profiles using a peak fitting feature in Origin (https://www.originlab.com/), assuming Gaussian functions for each peak. b Not reported. c Elucidated from reported thermogram. | ||||||
| C0 | 7.6 | 292.5 | 326.2a | 342.9 | 370.8 | This work |
| C3 | 8.5 | 298.1 | 348.9a | 366.2 | 391.6 | This work |
| C6 | 9.0 | 302.5 | 353.4a | 366.2 | 393.2 | This work |
| C12 | 9.4 | 297.9 | 346.4 | 366.2 | 390.2 | This work |
| C24 | 11.0 | 303.1 | 351.6 | 371.4 | 388.6 | This work |
| α-Chitin | —b | —b | —b | —b | 396.6 | 70 |
| α-Chitin | —b | —b | ∼340.0c | ∼350.0c | ∼380.0c | 69 |
| BM α-chitin | —b | —b | 270.0 | 390.6 | 11 | |
The onset temperature (Tonset) for C0 decomposition was observed at 292.5 °C (Table 1). All other samples exhibited Tonset at temperature slightly higher than that for C0. Thus, Tonset for C3, C12, C24, and C6 was observed at 298.1, 302.5, 298.0, and 303.1 °C, respectively. The decomposition of the chitin polymer was a two-step decomposition in all cases. The first step was attributed to the depolymerization/decomposition of polymer chains through deacetylation and cleavage of glycosidic linkage, and the second step was attributed to the thermal destruction of the pyranose ring.69 Because these peaks were close to each other in the DTG plots, individual peaks were extracted by a curve-fitting process from the thermogram profiles using a peak fitting feature in Origin (https://www.originlab.com/), assuming Gaussian functions for each peak (ESI, Fig. S25–S29†).
Thus, T1 50% was significantly higher for cryoground samples C3–C24 (∼349–352 °C) than for the starting material C0 (∼326 °C). Specifically, the value of T1 50% was approximately the same for all cryoground samples 348.9 (C3), 353.4 (C6), 346.4 (C12), and 351.6 °C (C24). The second step decomposition (T2 50%) took place at 370.8 °C for the starting material and increased to ∼390 °C for the cryoground samples. Namely, T2 50% was found to be 391.6, 393.2, 390.2, and 388.6 for C12–C24, respectively. The amount of ash was very similar between all samples and varied from 13% in C0 to 16.5% in C3.
In the case of α-chitin, the decomposition temperature at 10 °C min−1 decomposition rate was reported to be 396.6 °C,70 with the sample degrading in two steps, but it was lower in the case of our starting material. It has also been reported by Zhong et al.11 that chitin ball-milled with SS balls (14 × ∅ 10 mm + 38 × ∅ 6.25 mm, 90 g) displayed two stages of weight loss, at ∼270 °C and 390.6 °C. Other studies did not report the thermal decomposition data.
Conclusions
The work reported here suggests that the cryogrinding method is suitable for chitin polymer processing. With this study, we demonstrated the morphological, crystalline, and structural changes in chitin after cryomilling, and compared it with BM-grinding (Table 2). It was found that similarly to BM, a size reduction takes place during cryogrinding. FTIR analysis showed a significant breaking of intra- and intermolecular hydrogen bonds and partial amorphization, with the extent of amorphization proportional to cryogrinding time. The crystallinity, determined from the ratio of the sum of the areas under the crystalline diffraction peaks and the total area under the diffraction pattern, was found to be 51%, somewhat higher than that typically obtained through BM (27–51%), which might be explained by the relatively low mass of the 440C steel impactor. The crystallite size in plane (110) was determined to be 84.5 nm and was not affected by cryogrinding while the crystal size in plane (020) decreased ∼15%, from 73 to 63 nm.| Analysis | Cryogrinding | Ball milling | Ref. | |
|---|---|---|---|---|
| Conditions | Results | |||
| Morphology | No chitin morphological changes other than size reduction | 14 × ∅ 10 mm + 38 × ∅ 6.25 mm (90 g) | Close-packed texture similar to that of raw chitin | 11 |
| 1400 × ∅ 5 mm (704 g) | Size reduction observed after 10 minutes | 12 | ||
| FTIR-ATR spectroscopy | 1. Breaking intra- and intermolecular hydrogen bonds | 14 × ∅ 10 mm + 38 × ∅ 6.25 mm (90 g) | Chemical structure of the polymer was not altered | 11 |
| 2. Extent of amorphization proportional to cryogrinding time | 16 × ∅ 6.35 mm (16 g), 90 min | 1. Weakening of the hydrogen bonding | 13 | |
| 3. Insignificant breaking of glycosidic bonds after 48 minutes of cryogrinding | 2. Partial amorphization | |||
| 3. Glycosidic linkage breaking | ||||
| 1400 × ∅ 5 mm (704 g) | 1. Weakening of hydrogen bonds between chitin interchains | 12 | ||
| 2. Partial amorphization | ||||
| Crystalline structure | Decreased from 68 to 51% | 14 × ∅ 10 mm + 38 × ∅ 6.25 mm (90 g) | Complete amorphization | 11 |
| 1400 × ∅ 5 mm (704 g), 10 min | Decreased from 91 to 68% | 12 | ||
| 1400 × ∅ 5 mm (704 g), 30 min | Decreased from 91 to 40% | 12 | ||
| 2 × ∅ 12.7 mm (17 g), 90 min | Decreased from 91 to 51% | 13 | ||
| 16 × ∅ 6.35 mm (16 g), 90 min | Decreased from 91 to 37% | 13 | ||
| 1 × ∅ 5.53–12.64 mm (0.7–8.4 g) | Decreased from 66 to 34% | 15 | ||
| Crystallite size (110) | Size of 84.5 nm did not change | 1400 × ∅ 5 mm (704 g), 30 min | Size decrease from 10.7 to 8.8 nm | 12 |
| Crystallite size (020) | A slight decrease in crystallite size in (020) direction, from 73 to 63 nm | — | — | |
| Deacetyl. degree | Gradual decrease in %DA, from 91% (C0) to 61% (C24) | 16 × ∅ 6.35 mm (16 g), 90 min | No change in %DA | 13 |
| MW | No change in MW | 1 × ∅ 5.53–12.64 mm (0.7–8.4 g) | Significant decrease of Mw | 15 |
| 1400 × ∅ 5 mm (704 g), 30 min | Significant decrease of Mw | 12 | ||
| Thermal properties | T 1 50% ∼ 350, T2 50% ∼ 390 °C | 14 × ∅ 10 mm + 38 × ∅ 6.25 mm (90 g) | T 1 50% ∼ 270, T2 50% ∼ 390 °C | 11 |
The study also demonstrated that the method of chitin grinding dramatically affected two main characteristics of chitin, which are its Mw and DA. From this viewpoint, Kim et al.71 and Mao et al.72 have extensively reviewed existing information on the influence of chitin and/or chitosan properties, specifically, Mw and DA, on the performance of the delivery systems. Our study showed that while DA was not affected by BM, it continuously reduced during cryogrinding. Likewise, chitin of high Mw could not be obtained from BM, but only from cryogrinding, greatly expanding chitin's potential applications.
In relation to Green Chemistry process, relatively recent article73 evaluated technological aspects of cryomilling such as its environmental impact, scale-up potential, and potential applications. It has been shown that from the point of Green Chemistry principles, the cryomilling is an eco-friendly process, and that the large-scale cryogrinding can be utilized in different applications, especially those that involve temperature-sensitive materials (i.e., drug delivery vehicles, matrices loaded with pesticides, etc.). The process utilizes liquid nitrogen, economical and efficient coolant, which is eco-friendly as it turns into non-toxic and abundant N2 gas. The major input raw materials in cryomilling are coolant, and milling materials. The processing cost of this technique consists of the electricity cost and the cost of liquid coolant. As research into this area continues to progress, cryogenic grinding is gaining recognition as an efficient alternative for biopolymer processing that meets the demands of today's industry.
When we know how chitin is behaving under this process, and what changes in its properties we can expect – we now have enough information and are at starting point for the preparation of respective composites with temperature-labile materials (volatiles, pharmaceutical actives, DNA and siRNA, etc.). We believe that these findings will be useful in designing further cryogrinding strategies in the production of chitin composites and blends. In this context, the application of cryogrinding can be advantageous in establishing strategies for chitin processing aligned with the Green Chemistry principles.
Conflicts of interest
The authors declare the following interest(s): Dr J. L. Shamshina is a former employee of 525 Solutions, Inc., and a former employee of Mari Signum, Ltd.References
- S.-K. Kim, Chitin, chitosan, oligosaccharides and their derivatives: biological activities and applications, CRC Press, 2010 Search PubMed.
- B. K. Park and M. M. Kim, Int. J. Mol. Sci., 2010, 11, 5152–5164 CrossRef CAS PubMed.
- R. Jayakumar, D. Menon, K. Manzoor, S. V. Nair and H. Tamura, Carbohydr. Polym., 2010, 82, 227–232 CrossRef CAS.
- J. L. Shamshina, P. Berton and R. D. Rogers, ACS Sustainable Chem. Eng., 2019, 7, 6444–6457 CrossRef CAS.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef CAS PubMed.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., 2020, 59, 1018–1029 CrossRef PubMed.
- A. Calka and D. Wexler, Nature, 2002, 419, 147–151 CrossRef CAS PubMed.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- J. C. Roy, F. Salaün, S. Giraud and A. Ferri, Solubility of Chitin: Solvents, Solution Behaviors and Their Related Mechanisms, InTechOpen, 2017, pp. 109–127 Search PubMed.
- X. Chen, H. Yang, Z. Zhong and N. Yan, Green Chem., 2017, 19, 2783–2792 RSC.
- T. Zhong, M. P. Wolcott, H. Liu, N. Glandon and J. Wang, Cellulose, 2020, 27, 6205–6216 CrossRef CAS.
- M. Osada, C. Miura, Y. S. Nakagawa, M. Kaihara, M. Nikaido and K. Totani, Carbohydr. Polym., 2013, 92, 1573–1578 CrossRef CAS PubMed.
- G. Margoutidis, V. H. Parsons, C. S. Bottaro, N. Yan and F. M. Kerton, ACS Sustainable Chem. Eng., 2018, 6, 1662–1669 CrossRef CAS.
- T. Di Nardo, C. Hadad, V. N. A. Nguyen and A. Moores, Green Chem., 2019, 21, 3276–3285 RSC.
- T. Di Nardo and A. Moores, Beilstein J. Org. Chem., 2019, 15, 1217–1225 CrossRef CAS PubMed.
- H. Wineinger, J. L. Shamshina, A. Kelly, C. King and R. D. Rogers, Green Chem., 2020, 22, 3734–3741 RSC.
- J. L. Shamshina and P. Berton, Front. Bioeng. Biotechnol., 2020, 8, 1–14 CrossRef PubMed.
- P. Berton, X. Shen, R. Rogers and J. L. Shamshina, Ind. Eng. Chem. Res., 2019, 58, 19862–19876 CrossRef CAS.
- X. Shen, Y. Xie, Q. Wang, X. Yi, J. L. Shamshina and R. D. Rogers, Cellulose, 2019, 26, 4005–4019 CrossRef CAS.
- J. L. Shamshina, P. Berton and R. D. Rogers, ACS Sustainable Chem. Eng., 2019, 7, 6444–6457 CrossRef CAS.
- J. L. Shamshina, O. Zavgorodnya, H. Choudhary, B. Frye, N. Newbury and R. D. Rogers, ACS Sustainable Chem. Eng., 2018, 14713–14722 CrossRef CAS.
- J. Schmidt, M. Plata, S. Tröger and W. Peukert, Powder Technol., 2012, 228, 84–90 CrossRef CAS.
- S. B. Liang, D. P. Hu, C. Zhu and A. B. Yu, Chem. Eng. Technol., 2002, 25, 401–405 CrossRef CAS.
- A. Chakraborty, M. Sain and M. Kortschot, Holzforschung, 2005, 59, 102–107 CAS.
- A. M. Poulose, S. Piccarolo, D. Carbone and S. M. Al-Zahrani, J. Appl. Polym. Sci., 2016, 133, APP.43083 CrossRef.
- S. Ramesh, L. Lungaro, D. Tsikritsis, E. Weflen, I. V. Rivero and A. P. D. Elfick, J. Appl. Polym. Sci., 2018, 135, APP.46692 CrossRef.
- K. Kaminski, K. Adrjanowicz, Z. Wojnarowska, K. Grzybowska, L. Hawelek, M. Paluch, D. Zakowiecki and J. Mazgalski, J. Pharm. Sci., 2010, 7, 2642–2657 Search PubMed.
- K. Adrjanowicz, K. Grzybowska, K. Kaminski, L. Hawelek, M. Paluch and D. Zakowiecki, Philos. Mag., 2011, 91, 1926–1948 CrossRef CAS.
- T. P. Shakhtshneider, F. Danede, F. Capet, J. F. Willart, M. Decamps, S. A. Myz, E. V. Boldyreva and V. V. Boldyrev, J. Therm. Anal. Calorim., 2007, 89, 699–707 CrossRef CAS.
- T. P. Shakhtshneider, F. Danede, F. Capet, J. F. Willart, M. Decamps, L. Paccou, E. V. Surov, E. V. Boldyreva and V. V. Boldyrev, J. Therm. Anal. Calorim., 2007, 89, 709–715 CrossRef CAS.
- A. Penkina, K. Semjonov, M. Hakola, S. Vuorinen, T. Repo, J. Yliruusi, J. Aruvali, K. Kogermann, P. Veski and J. Heinamaki, Drug Dev. Ind. Pharm., 2016, 42, 378–388 CrossRef CAS PubMed.
- M. Pouzar, A. Krejcova, T. Cernohorsky and K. Peskova, Talanta, 2008, 76, 254–258 CrossRef CAS PubMed.
- M. Benitez-Guerrero, L. A. Perez-Maqueda, R. Artiaga, P. E. Sanchez-Jimenez and J. Pascual-Cosp, J. Nat. Fibers, 2017, 14, 26–39 CrossRef CAS.
- P. Karinkanta, M. Illikainen and J. Niinimaeki, Holzforschung, 2013, 67, 277–283 CAS.
- M. S. Gomes, D. Santos Jr., L. C. Nunes, G. G. Arantes de Carvalho, F. O. Leme and F. J. Krug, Talanta, 2011, 85, 1744–1750 CrossRef CAS PubMed.
- A. Penkina, K. Semjonov, M. Hakola, S. Vuorinen, T. Repo, J. Yliruusi, J. Aruvali, K. Kogermann, P. Veski and J. Heinamaki, Drug Dev. Ind. Pharm., 2016, 42, 378–388 CrossRef CAS PubMed.
- S. Gui, Faming Zhuanli Shenqing, CN106519075A20170322 Search PubMed, 2017.
- S. B. Liang, D. P. Hu, C. Zhu and A. B. Yu, Chem. Eng. Technol., 2002, 25, 401–405 CrossRef CAS.
- M. Kaya, T. Baran, I. Saman, M. A. Ozusaglam, Y. S. Cakmak and A. Menteşet, J. Crustacean Biol., 2014, 34, 283–288 CrossRef.
- P. Scherrer, Nachr. Ges. Wiss. Goettingen, Math.-Phys. Kl., 1918, 26, 98–100 Search PubMed.
- J. I. Langford and A. J. C. Wilson, J. Appl. Crystallogr., 1978, 11, 102–113 CrossRef CAS.
- S. Beil, A. Schamberger, W. Naumann, S. Mauchill and K. van Pee, Carbohydr. Polym., 2012, 87, 117–122 CrossRef CAS PubMed.
- M. L. Duarte, M. C. Ferreira, M. R. Marvão and J. Rocha, Int. J. Biol. Macromol., 2001, 28, 359–363 CrossRef CAS PubMed.
- E. Khor, Chitin and Chitosan: Making the Grade, in Fulfilling the biomaterial promise, ed. E. Khor, Elsevier, Oxford, UK, 2014, ch. 7, pp. 101–111 Search PubMed.
- Y. Yamaguchi, T. T. Nge, A. Takemura, N. Hori and H. Ono, Biomacromolecules, 2005, 6, 1941–1947 CrossRef CAS PubMed.
- S. Darmon and K. M. Rudall, Discuss. Faraday Soc., 1950, 9, 251–260 RSC.
- J. Kumirska, M. Czerwicka, Z. Kaczyński, A. Bychowska, K. Brzozowski, J. Thöming and P. Stepnowski, Mar. Drugs, 2010, 8, 1567–1636 CrossRef CAS PubMed.
- G. Cárdenas, G. Cabrera, E. Taboada and S. P. Miranda, J. Appl. Polym. Sci., 2004, 1876–1885 CrossRef.
- S. Tanner, H. Chanzy, M. Vincendon, J. Roux and F. Gaill, Macromolecules, 1990, 23, 3576–3583 CrossRef CAS.
- I. Akpan, O. P. Gbenebor and S. O. Adeosun, Int. J. Biol. Macromol., 2018, 106, 1080–1088 CrossRef PubMed.
- P. Kolhe and R. M. Kannan, Biomacromolecules, 2003, 4, 173–180 CrossRef CAS PubMed.
- Z. Zakaria, Z. Izzah, M. Jawaid and A. Hassan, BioResources, 2012, 7, 5568–5580 CrossRef.
- M. Kaya, M. Mujtaba, H. Ehrlich, A. M. Salaberria, T. Baran, C. T. Amemiya, R. Galli, L. Akyuzh, I. Sargin and J. Labidi, Carbohydr. Polym., 2017, 176, 177–186 CrossRef CAS PubMed.
- E. S. Abdou, K. S. A. Nagy and M. Z. Elsabee, Bioresour. Technol., 2008, 99, 1359–1367 CrossRef CAS PubMed.
- F. A. Sagheer, M. A. Al-Sughayer, S. Muslim and M. Z. Elsabee, Carbohydr. Polym., 2009, 77, 410–419 CrossRef.
- Y. Fan, T. Saito and A. Isogai, Biomacromolecules, 2008, 9, 192–198 CrossRef CAS PubMed.
- R. Minke and J. Blackwell, The structure of α-chitin, J. Mol. Biol., 1978, 120, 167–181 CrossRef CAS PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef PubMed.
- A. K. Gupta and R. P. Singhal, J. Polym. Sci., Polym. Phys. Ed., 1983, 21, 2243–2262 CrossRef CAS.
- G. Nansé, E. Papirer, P. Fioux, F. Moguet and A. Tressaud, Carbon, 1997, 35, 175–194 CrossRef.
- E. Alvarenga, Characterization and Properties of Chitosan, in Biotechnology of Biopolymers, ed. M. Elnashar, InTechOpen, 2011, pp. 91–110 Search PubMed.
- M. Kasaai, Carbohydr. Polym., 2008, 71, 497 CrossRef CAS.
- N. Varan, J. Text. Sci. Eng., 2017, 6, 288 Search PubMed.
- J. Brugnerotto, J. Lizardi, F. M. Goycoolea, W. Argüelles-Monal, J. Desbrières and M. Rinaudo, Polymer, 2001, 42, 3569–3580 CrossRef CAS.
- N. L. B. M. Yusof, L. Y. Lim and E. Khor, J. Biomed. Mater. Res., 2001, 54, 59–68 CrossRef CAS PubMed.
- P. R. Austin, Chitin solution, US4059457A, 1977 Search PubMed.
- K. D. Nguyen and T. Kobayashi, J. Chem., 2020, 6645351 CAS.
- H. Moussout, H. Ahlafi, M. Aazza and M. Bourakhouadar, Polym. Degrad. Stab., 2016, 130, 1–9 CrossRef CAS.
- S. S. Kim, S. J. Kim, Y. D. Moon and Y. M. Lee, Polymer, 1994, 35, 3212 CrossRef CAS.
- M. Jang, B. Kong, Y. Jeong, C. H. Lee and J. Nah, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3423–3432 CrossRef CAS.
- T.-H. Kim, H. L. Jiang, D. Jere, I.-K. Park, M.-H. Cho, J.-W. Nah, Y.-J. Choi, T. Akaike and C.-S. Cho, Prog. Polym. Sci., 2007, 32, 726–753 CrossRef CAS.
- S. Mao, W. Sun and T. Kissel, Chitosan-based formulations for delivery of DNA and siRNA, Adv. Drug Delivery Rev., 2010, 62, 12–27 CrossRef CAS PubMed.
- N. K. Katiyar, K. Biswas and C. S. Tiwary, Int. Mater. Rev., 2021, 66, 493–532 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FE-SEM images, ATR FTIR spectra, crystallinity indices (CrI), pXRD diffractograms (include peak fitting and related parameters, crystallite size (110) and (020), the first derivative of the digitally filtered FTIRs, solid-state cross-polarization magic angle spinning carbon-13 nuclear magnetic resonance (CP MAS NMR), flow curves for 1 wt% chitin solutions in ionic liquid 1-ethyl-3-methylimidazolium acetate [C2mim][OAc], TGA curves, DTG curves and related peak fitting. See DOI: 10.1039/d1gc03128g |
| This journal is © The Royal Society of Chemistry 2021 |