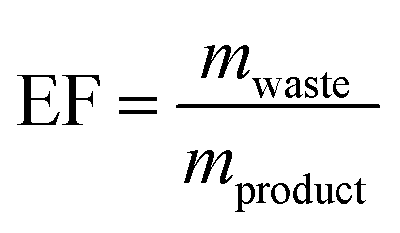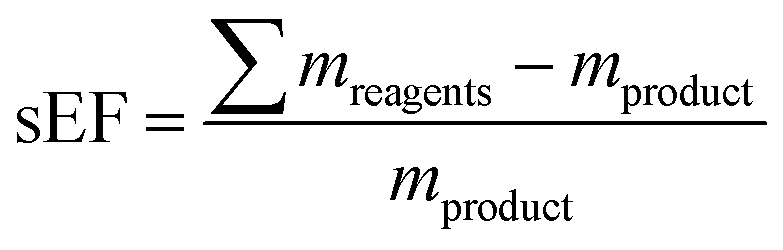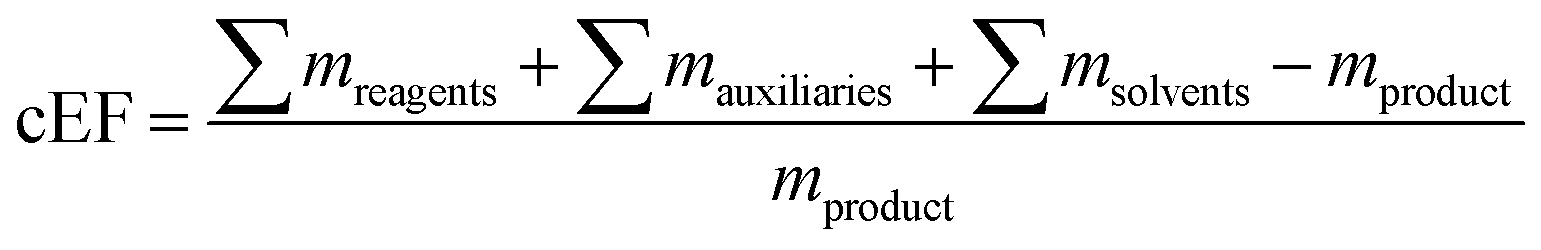DOI:
10.1039/D0GC02665D
(Critical Review)
Green Chem., 2021,
23, 37-50
Route efficiency assessment and review of the synthesis of β-nucleosides via N-glycosylation of nucleobases†
Received
3rd August 2020
, Accepted 16th November 2020
First published on 19th November 2020
Abstract
Nucleosides and their analogs are biomolecules central to nearly all areas of life science. Consequently, a variety of approaches have been developed to prepare these compounds. These methods typically employ N-glycosylation as a key step which installs a sugar moiety on a heterocyclic nucleobase. However, these methods vary drastically regarding their synthetic strategy, number of steps, yield, reagents, and conditions employed, making it difficult to compare and evaluate different approaches. Herein, we review the state of art for the synthesis of β-nucleosides by N-glycosylation and present a comprehensive sustainability assessment of these routes via an E-factor analysis. Our data reveal that the current methods and protocols are, in general, laborious and inefficient. Although impressive yields have been achieved in many cases, these typically came at the cost of long routes, leading to high overall E-factors (primarily composed of solvent contributions). Shorter routes using fewer protecting groups tended to perform equally well or better regarding their route E-factors, despite lower yields in many cases. Nearly all available approaches are currently hampered by a heavy reliance on chromatography, multiple protecting groups and bulky leaving groups. Biocatalytic methods bypass these limitations but suffer from poor substrate solubility and unfavorable reaction equilibria. To enable more efficient and sustainable nucleoside synthesis via N-glycosylation, future efforts should focus on using non-chromatographic purification steps, running shorter routes and higher substrate loading to minimize (solvent) waste accumulation.
 Felix Kaspar | Felix Kaspar is a Ph.D. candidate at the Institute for Biotechnology at the Technische Universität Berlin, Germany. He obtained a Bachelor's (Biotechnology, 2016) and Master's degree (Biochemistry, 2019) from the Technische Universität Braunschweig, Germany, and completed several research internships during his studies, leading him to Australia (Charles Sturt University, 2015; University of Queensland, 2017–2018) and the USA (University of Alabama, 2016). His research interests include all things biocatalysis, particularly biocatalytic nucleoside synthesis, as well as equilibrium thermodynamics, method development and environmentally acceptable chemistry. |
 M. Rhia L. Stone | Dr M. Rhia L. Stone completed her BSc and MSc (I) in Chemistry from Victoria University of Wellington in New Zealand, before moving to the University of Queensland, Australia for her PhD. Rhia completed her doctorate in July 2020, working on tagged antibiotics for the examination of bacterial resistance and infection. Rhia is currently a Postdoctoral Researcher at Rutgers University, the State University of New Jersey, where she is studying the Chemical Biology of nucleic acids. |
 Peter Neubauer | Peter Neubauer is Professor of Bioprocess Engineering at the Department of Biotechnology at the Technische Universität Berlin, Germany, since 2008. He holds a doctoral degree in Microbiology from Universität Greifswald, Germany, and completed postdocs at the Royal Institute of Technology (KTH), Sweden, and Martin-Luther-Universität of Halle, Germany, before he received a professorship in bioprocess engineering from the University of Oulu, Finland (2000–2008). His research is concerned with bioprocess scale up/scale down and microbial physiology in industrial scale bioprocesses, as well as the development of strategies for the production of difficult-to-express proteins and the mathematical modelling of bioprocesses. |
 Anke Kurreck | Anke Kurreck is a Postdoctoral Researcher in the Bioprocess Engineering group at the Technische Universität Berlin, Germany, and CEO of the Biotech company BioNukleo GmbH since 2014. She holds a doctoral degree in Microbiology from Martin-Luther-Universität of Halle (Germany) and completed a research internship at Wageningen University during her PhD. Subsequently she worked as a Postdoctoral Researcher in the Applied Biochemistry Group at the Technische Universität Berlin. Her research focusses on applied sciences with the aim to reduce the environmental burden and to bring (chemo)enzymatic synthesis routes increasingly into pharmaceutical application. |
Introduction
Nucleosides are highly functionalized biomolecules essential to life on earth and were among the first organic molecules on our planet.1 Now β-nucleosides serve as the building blocks of DNA and RNA, part of cellular energy transfer systems, and as enzymatic cofactors in all known organisms on earth. In recent years, nucleoside analogs have become indispensable as pharmaceutical agents against various cancers as well as viral infections and as molecular biology tools.2–4 For example, fluorinated nucleoside analogs such as floxuridine and islatravir are used for the treatment of colorectal cancer and HIV infections, respectively.5,6 Alkyne-containing nucleosides such as 5-ethynyluridine have been broadly applied for the labelling of nucleic acids, including the analysis of RNA synthesis and visualization of cellular localization.4 Consequently, the demand for these molecules in nearly all areas of life science has necessitated the development of chemical methods for their synthesis. More than six decades of research in the field have yielded a variety of robust methods to access these compounds.
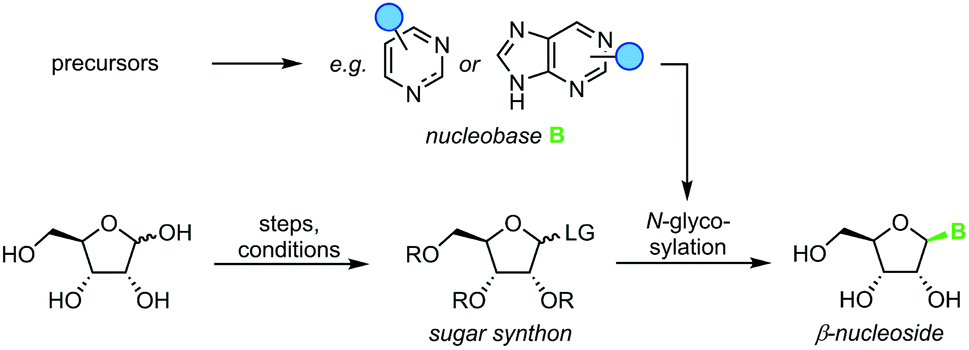
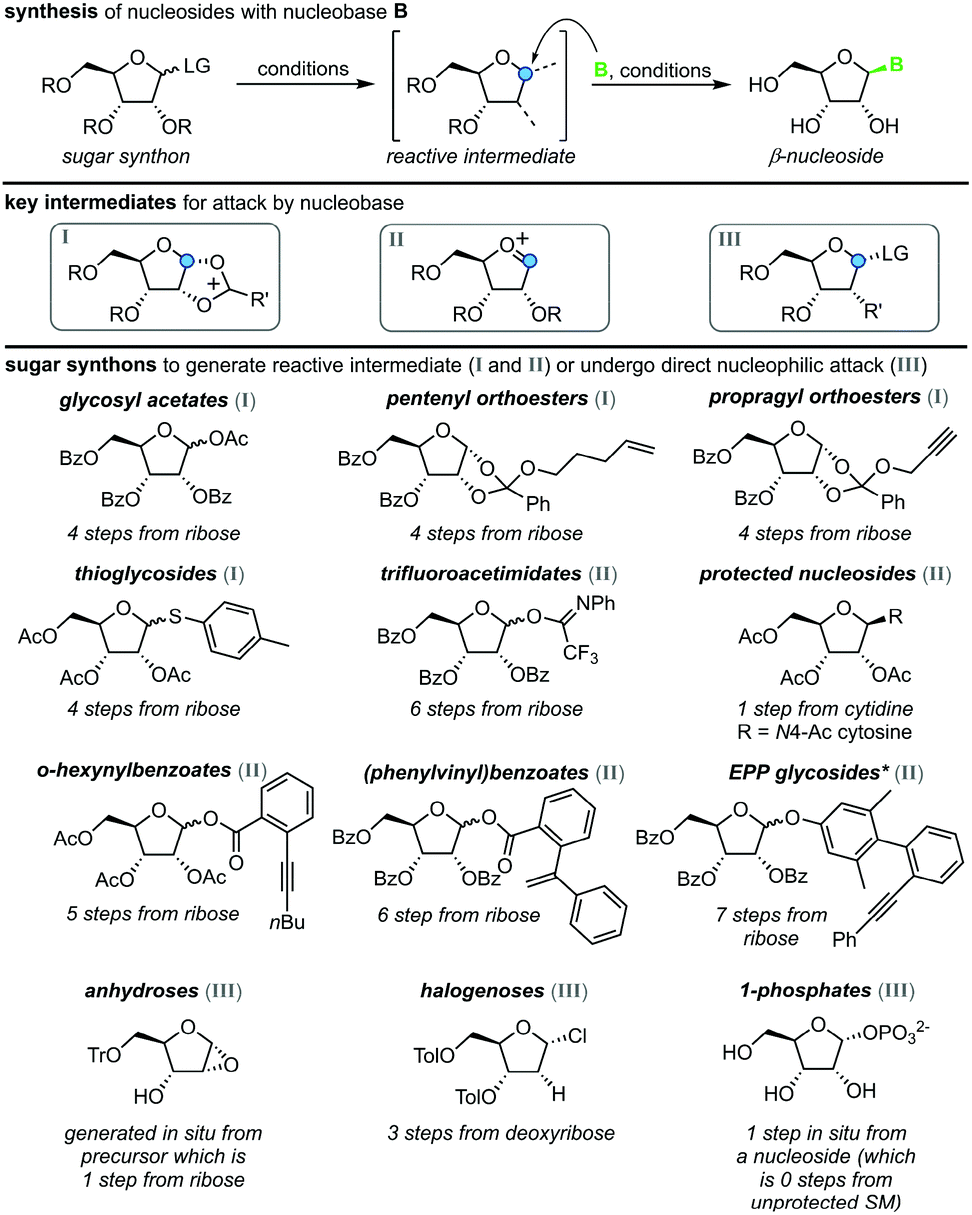
Nucleoside synthesis is generally performed in a convergent manner via N-glycosylation of a nucleobase, which installs a ribosyl moiety on a heterocyclic base (Scheme 1). Although the glycosylation of a nucleobase as a key step may appear rather simple at first glance, it is complicated by challenges in regio- and diastereoselectivity.7 These issues typically arise from the low nucleophilicity of nucleobases as well as the density of functional groups decorating the ribosyl moiety. Thus, the desired linkage of a nucleobase to the anomeric center of a ribosyl moiety to yield a β-nucleoside often competes with several side reactions, including unselective nucleophilic attack (forming the α-nucleoside) and attack of other nucleophilic functional groups, affording complex mixtures of products. To address these obstacles, a variety of creative approaches have been developed to prepare β-nucleosides in high yield and selectivity. However, these methods vary drastically regarding their strategy, number of steps, yield, reagents, and conditions employed, making it difficult to compare and evaluate different approaches.
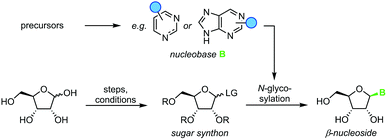 |
| | Scheme 1 Convergent synthesis of nucleosides with N-glycosylation as the key step. N-Heterocyclic nucleobases such as pyrimidines and purines with variable substitution patterns (blue circles) can be accessed directly from cheap precursors via condensation reactions while sugar synthons need to be prepared by multistep routes from unprotected sugars. | |
Driven by the increasing global effort to establish a sustainable economy, most branches of chemistry have questioned their practices and aimed to design “greener” processes and reagents.8,9 Among others, these efforts include the use of sustainably sourced solvents (and recycling thereof), the development of more concise and high-yielding routes to pharmaceuticals and the establishment of waste-minimizing cascade syntheses, to name just a few. The pharmaceutical industry (and related fields) has been quick to adopt green chemistry principles and environmental concerns are increasingly recognized in this area.10 Subsequent research has made a wealth of information publicly available to benchmark and predict various metrics of sustainability and efficiency of chemical syntheses.11–19 Several comprehensive assessments of different routes for the preparation of pharmaceutically relevant target molecules have been published, which have provided further insights into the pitfalls of some synthetic approaches and highlighted successful strategies from a sustainability perspective.20–25 Beyond that, assessments of individual newly published reactions or approaches versus established methods have become a common sight in the literature and continue to provide a valuable and critical evaluation of the state of the art.26–45 However, most of the above approaches focus either on (i) drug-like molecules and, consequently, the heterocycle chemistry commonly involved or (ii) single transformations. To the best of our knowledge, a comprehensive assessment of sustainability or efficiency for glycosylation-type chemistry is missing from the literature. Spurred by this lack of information, we were curious which of the available methods for nucleoside synthesis would be most compatible with a low-waste economy and be efficient in a sense of resource usage.
Since nucleosides will undoubtedly continue to be central to all areas of life science, we aimed to provide a transparent evaluation and specifically investigated which routes to nucleosides would yield the most efficient and sustainable synthesis. Rather than starting an assessment at a randomly chosen synthon, we opted to consider the entire route necessary for a given strategy. Since preparation of the sugar synthon is generally the most time- and material-intensive part of these routes,7 we assumed that all routes had to start from unprotected readily available materials. To this end, we surveyed the literature for glycosylation methods for the synthesis of β-nucleosides and extracted experimental data and protocols for several nucleoside examples to calculate representative environmental factors (E-factors, EF)9,46–50 for the entire synthetic routes. The E-factor is a mass-based metric to assess the amount of waste produced during a synthetic process or route, where
with
mproduct being the weight of the pure product and
mwaste being the mass of all materials involved in a synthesis that are not the product. In our analysis, we both considered the simple E-factor (sEF) which, as pioneered by Sheldon,
9 only considers the reagents used as
as well as the complete E-factor (cEF)
9 which considers all materials used as
where ∑
mreagents is the sum of the masses of all reagents (including starting materials), ∑
mauxillaries is the sum of the masses of all auxiliary materials (such as inorganic salts and silica gel, for example) and ∑
msolvents is the sum of the masses of all solvents, including organic solvents and water. Considering the number of reactions and routes we aimed to assess, as well as their heterogeneity, we herein opted for the use of the E-factor as a simple and accessible metric. A full life cycle assessment
51–53 would have far surpassed the scope of this work and, given the (sometimes) incomplete literature data, also proven unrealistic. Likewise, we did not consider energy consumption (
e.g. for inclusion in an E
+-factor)
54 as this data was not available from the literature. It should therefore also be recognized that our data only describe the amount and not the sort of waste generated, how harmful this waste is to the environment or how costly its treatment is (
i.e. a kilogram of NaCl or water had the same E-factor as a kilogram of fluorinated Lewis acids or methylation reagents).
This article reviews the state of the art for β-nucleoside synthesis via N-glycosylation and provides an assessment of the performance and efficiency of all available methods and their routes. Since the last comprehensive review of nucleoside synthesis by Vorbrüggen,7 there have been some notable additions to the toolbox, which we briefly introduce along with some general considerations. We further present a collection of 80 route E-factors (covering up to 11 total steps) which were extracted from over 30 papers using 12 different N-glycosylation methods. Our data highlight prominent sources of waste, reveal the inefficiency of some strategies and underscore that route and reaction design tremendously impact the overall E-factor of the synthesis. Based on these findings, we outline current obstacles and bottlenecks which future synthetic efforts should seek to address. Lastly, our freely available data allow straightforward benchmarking of future syntheses to evaluate their efficiency.
Nucleoside synthesis via N-glycosylation
All methods for N-glycosylation of nucleobases are united in employing a reactive (activated) glycosyl intermediate that is subjected to nucleophilic attack by a nucleobase (Scheme 2, top). All approaches published to date proceed via one of three key intermediates for attack by the nucleobase (Scheme 2, middle). Selective attack at the anomeric center is encouraged either by (i) generation of a 1,3-dioxolane cation through recruitment of the neighboring protecting group, (ii) formation of a reactive glycosyl cation with charge delocalization across the ring oxygen or (iii) employment of a good leaving group that sets the right configuration upon SN2-type substitution at the anomeric position. These intermediates can be accessed from various synthons, all of which typically bear one or more protecting groups and need to be prepared from (deoxy)ribose in 1–7 steps (Scheme 2, bottom). Despite these shared basic strategies, conditions and methodologies employed by the available methods differ significantly and are generally guided by the sugar synthon employed for N-glycosylation. It should be noted that the following overview only includes methods which predominantly yield the β-nucleoside in reasonable yield and selectivity. Therefore, nucleoside syntheses which favor α-nucleosides, afford minimal yields and/or poor selectivity, or do not employ an N-glycosylation step were not included. Furthermore, methods whose reports did not include sufficient detail to reconstruct the experimental procedures are also not included below or in our E-factor assessment.
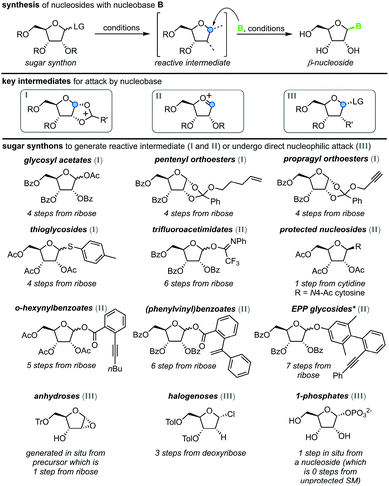 |
| | Scheme 2 Synthesis of nucleosides via N-glycosylation, including key intermediates and sugar synthons. Under varying conditions, a sugar synthon is transformed to a reactive intermediate primed for nucleophilic attack by an (activated) nucleobase, which furnishes a β-nucleoside. LG = leaving group, R = protecting group, B = nucleobase, SM = starting material, *EPP = (phenylethynylphenyl)phenyl. | |
Glycosyl acetates
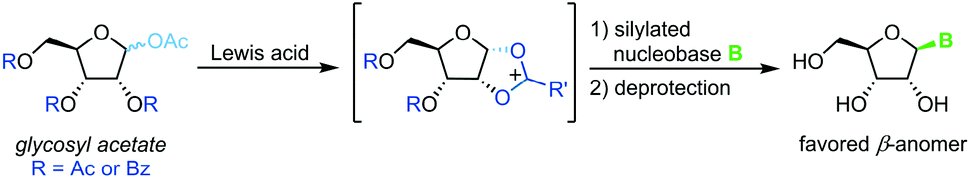
Vorbrüggen's classic synthesis of nucleosides built on silyl Hilbert–Johnson conditions and was originally only described for pyrimidine nucleosides.55 A fully protected glycosyl acetate is subjected to Lewis acid catalysis, yielding a glycosyl cation intermediate upon displacement of the anomeric acetate (Scheme 3). This labile species is then reacted with a silylated nucleobase to afford a nucleoside after global deprotection. Its exceptional substrate scope, easy adaptability and reliability have made it the most popular method for nucleoside synthesis in academia and industry. Nonetheless, downsides of this method include the need for silylated nucleobases, harsh reaction conditions and the stoichiometric amounts of Lewis acid generally used.56–59
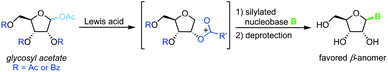 |
| | Scheme 3 Nucleobase glycosylation with glycosyl acetates. | |
Halogenoses
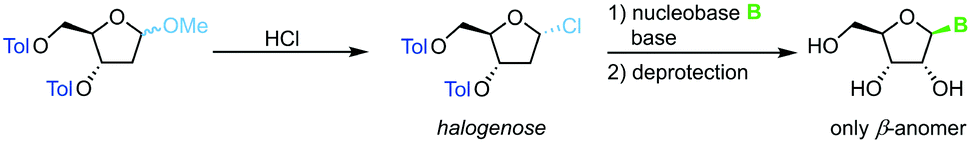
Direct glycosylation of nucleobases with halogenoses can be achieved through nucleophilic substitution at the anomeric center. This method is particularly popular for the synthesis of 2′-deoxy nucleosides that are otherwise difficult to access due to the lack of anchimeric assistance. Halogenoses can be accessed from the respective methoxyriboside and are subject to direct nucleophilic attack by the nucleobase (Scheme 4). To this end, different methods for nucleobase activation, including silylation and deprotonation by strong bases, have been employed.60–63 However, the functional group tolerance of this approach is hampered by the harsh conditions needed for this transformation. Further, the regioselectivity for purine nucleoside synthesis, as well as glycosylation yield, are generally limited. Despite its shortcomings, glycosylation with halogenoses offers a favorable atom economy compared to other methods.
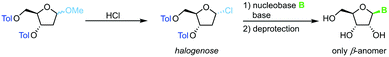 |
| | Scheme 4 Nucleobase glycosylation with halogenoses. | |
o-Hexynylbenzoates
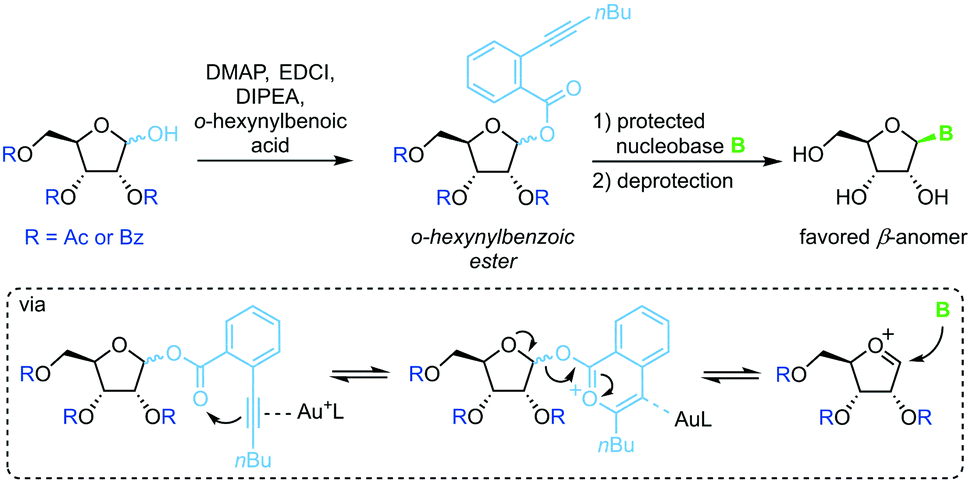
Spurred on by the difficulty to glycosylate purine bases via Vorbrüggen-type conditions, Yu and colleagues developed a glycosylation method based on ortho-hexynylbenzoic esters.64 Under gold catalysis, the benzoic ester at the anomeric position reversibly rearranges to an isocoumarin scaffold and yields a glycosyl cation (Scheme 5). This effectively minimizes competition of the leaving group with the nucleobase for attack at the anomeric center, enabling productive attack by weak nucleophiles such as purine nucleobases. Although this method allows glycosylation under mild conditions without the use of stoichiometric activating reagents, it demands a long reaction sequence and extensive use of protecting groups on both the sugar and the nucleobase.
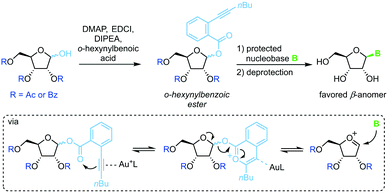 |
| | Scheme 5 Nucleobase glycosylation with o-hexynylbenozates. | |
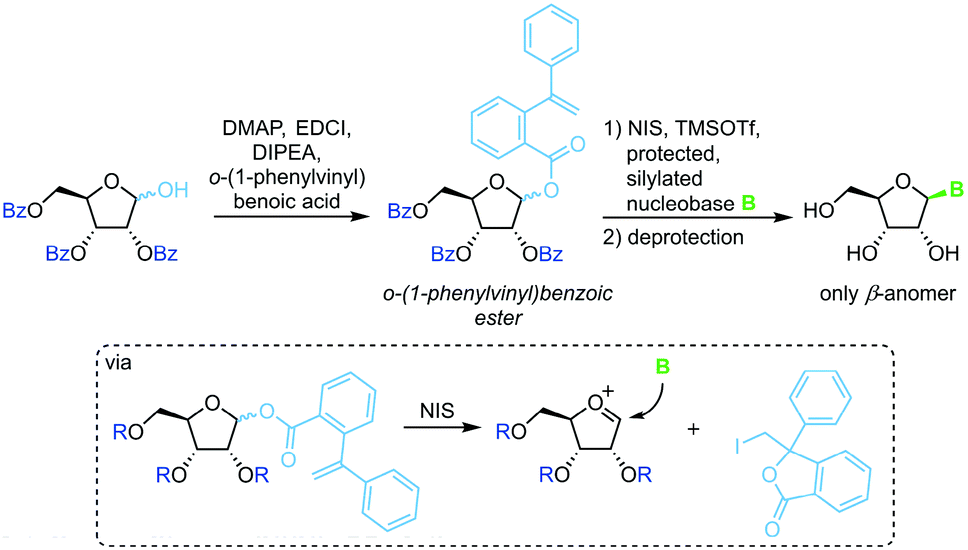
o-(1-Phenylvinyl)benzoates
A similar strategy that eliminates competition of the leaving group with the nucleobase relies on irreversible sequestration of a vinylbenzoic ester (Scheme 6).65 Esterification of tribenzoylated ribose with o-(1-phenylvinyl)benzoic acid accesses a stable sugar synthon. When subjected to an iodine source this synthon affords a glycosyl cation which can be intercepted by a silylated nucleobase. This method provides excellent glycosylation yields and regioselectivity under mild conditions. However, the need for a long reaction sequence with multiple protecting group manipulations make this approach rather laborious.
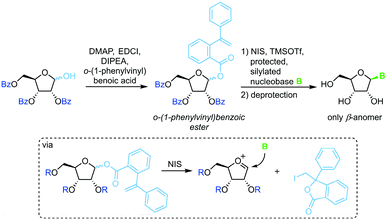 |
| | Scheme 6 Nucleobase glycosylation with o-(1-phenylvinyl)benzoates. | |
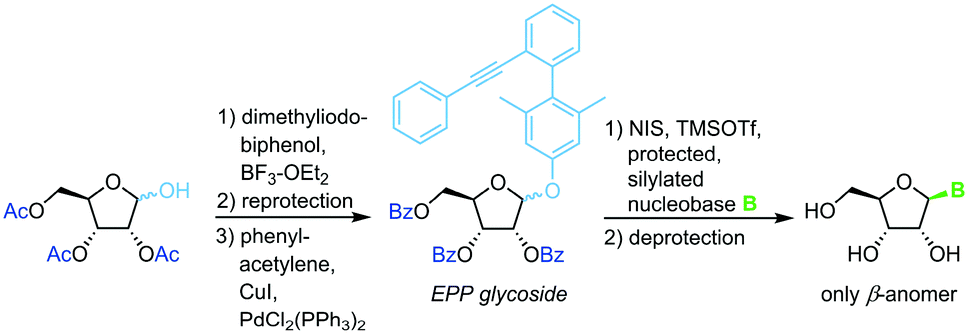
3,5-Dimethyl-4-(2′-phenylethynylphenyl)phenyl (EPP) glycosides
The same strategy and conditions can also be employed with (phenylethynylphenyl)phenyl glycosides as alternative leaving groups (Scheme 7).66
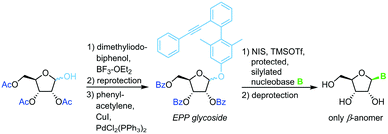 |
| | Scheme 7 Nucleobase glycosylation with EPP glycosides. | |
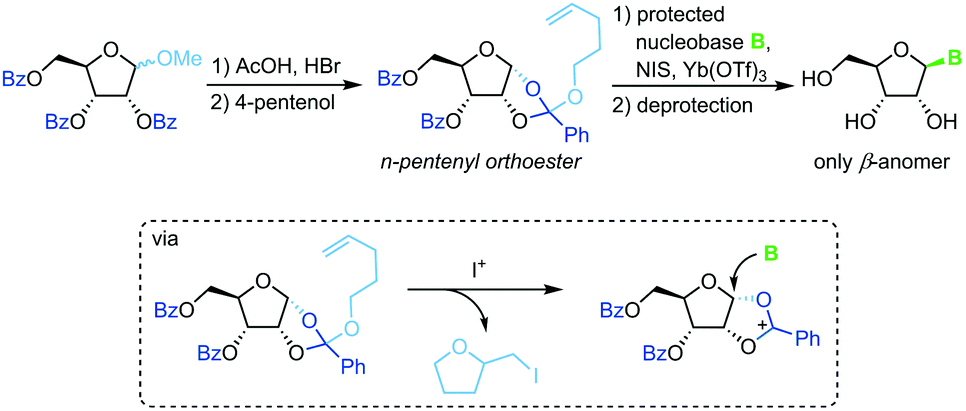
Pentenyl glycosides
Sequestration of a n-pentenyloxy group as an iodomethyl tetrahydrofuran to avoid competition of the leaving group with weakly nucleophilic nucleobases is a strategy developed recently by Fraser-Reid and colleagues.67 Starting from a perbenzoylated methyl riboside, a n-pentenyl orthoester is prepared through recruitment of the 2-benzoyl group. Similar to the above approaches, treatment of this ester with an iodonium source and a silylated nucleobase affords the corresponding β-nucleoside after global deprotection (Scheme 8). While this approach also suffers from a rather long reaction sequence (7 steps from unprotected starting materials) and a strict limitation to 2′-hydroxy nucleosides, it does provide excellent stereoselectivity and little problems with regioselectivity. Nonetheless, the substrate scope is limited and the need for the extensive use of protecting groups raises concerns from an efficiency perspective.
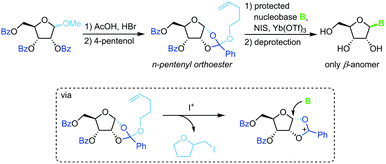 |
| | Scheme 8 Nucleobase glycosylation with pentenyl glycosides. | |
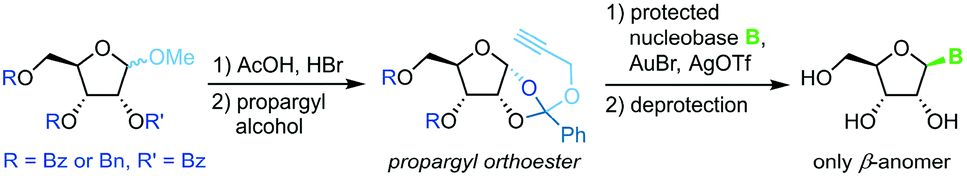
Propargyl-1,2-orthoesters
A similar methodology to Fraser-Reid et al. was developed by Rao and coworkers68 to facilitate high yields in the N-glycosylation of nucleobases. A propargyl-1,2-orthoester can be obtained from the respective perbenzoylated riboside through base-promoted attack of the alcohol to enable subsequent glycosylation under mild conditions. Addition of a silylated pyrimidine nucleobase under Lewis acid catalysis affords exclusively the β-anomer of the nucleoside (Scheme 9). Despite the method's promise of excellent stereo- and regioselectivity, it has only been applied to two nucleobases to date, presumably since the preparation of the sugar synthon is rather lengthy and labor-intensive.
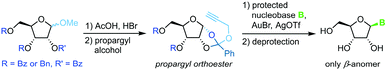 |
| | Scheme 9 Nucleobase glycosylation with propargyl-1,2-orthoesters. | |
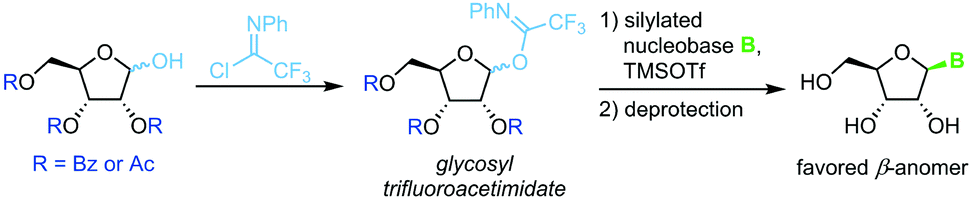
Trifluoroacetimidate glycosides
N-Glycosylation under mild conditions can also be achieved through activation of glycosyl donors as trifluoroacetimidates, which are excellent leaving groups.69,70 Nucleophilic attack of a silylated nucleobase provides near-quantitative yields of the thermodynamically favored β-nucleoside under Lewis acid catalysis (Scheme 10). However, this method has only been demonstrated for a small selection of pyrimidine nucleosides thus far. The extensive use of protecting groups, stoichiometric application of activating agents and unfavorable atom economy make the viability of this approach rather questionable, despite its impressive yields.
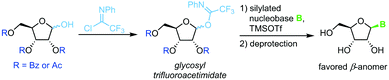 |
| | Scheme 10 Nucleobase glycosylation with trifluoroacetimidates. | |
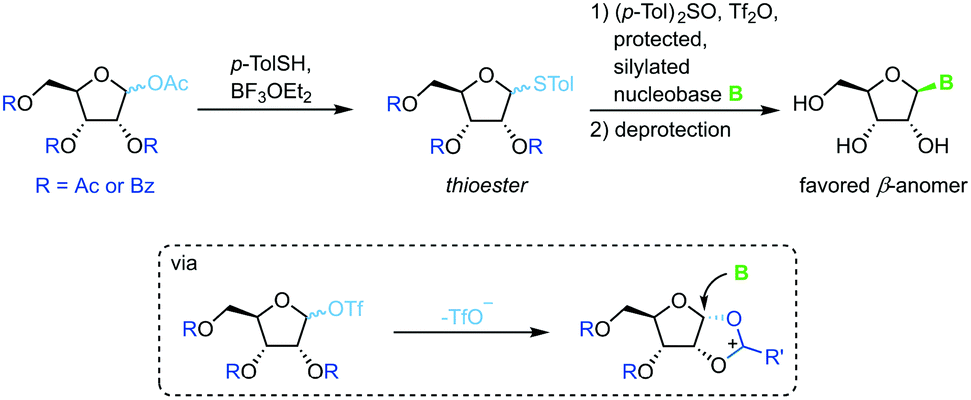
Thioglycosides
Thioglycosides have been employed in carbohydrate synthesis for various glycosylations and are notable for their versatility. Their use in nucleoside synthesis is rather rare, yet there are examples in the literature.71 Similar to other approaches, this method relies on in situ formation of a charged five-membered ring through recruitment of the protecting group at the 2-position after treatment of the thioglycoside with a triflate source (Scheme 11). This reactive intermediate is then intercepted by an activated nucleobase to generate the favored β-anomer. While yields of the glycosylation step are good to excellent, this approach requires a rather lengthy synthesis of the sugar synthons (4–5 steps) and suffers from similar drawbacks concerning protecting groups and silylating agents to analogous methods. It is noteworthy that even 2′-deoxynucleosides can be accessed with this approach, although not in high yield or anomeric selectivity.
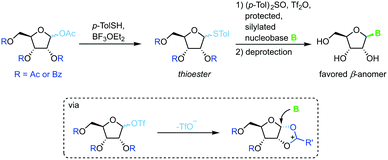 |
| | Scheme 11 Nucleobase glycosylation with thioglycosides. | |
Anhydroses
Building on Mitsunobu glycosylation conditions and pioneering work by Chow and Danishefsky,72 Hocek and colleagues developed73 and optimized74 a glycosylation strategy that relies on in situ tributylphosphine-mediated formation of a monoprotected anhydrose. Subsequent nucleophilic attack of a deprotonated nucleobase at the electronically favored 1-position exclusively provides the β-anomer of the nucleoside (Scheme 12). This concise route profits from employing only one protecting group (which can be installed in one high-yielding step from D-ribose), a respectable substrate scope and moderate to good yields of only the desired anomer, which significantly simplifies purification. Drawbacks of this method are few and mainly comprise the strict limitation to 2-hydroxy sugars.
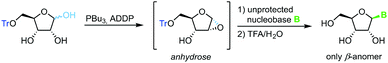 |
| | Scheme 12 Nucleobase glycosylation with anhydroses. | |
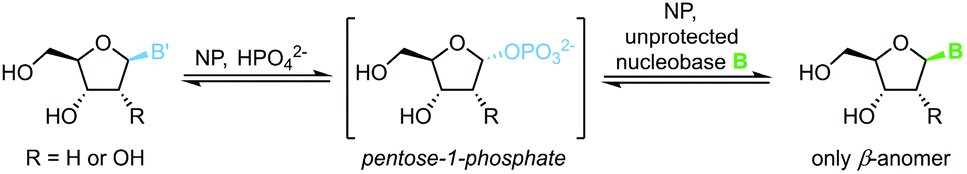
1-Phosphates
The biocatalytic synthesis of nucleosides via nucleoside phosphorylases (NPs) is well established and has recently attracted renewed interest.75 NP-catalyzed nucleoside transglycosylations employ an easily accessible nucleoside, such as natural uridine or thymidine, as the synthetic starting point.76 A pentose-1-phosphate is generated by phosphorolytic cleavage of the starting nucleoside and then serves as the glycosyl donor to a second nucleobase, which furnishes the nucleoside of interest (Scheme 13).77–83 These reactions capitalize on their promise of mild reaction conditions, perfect regio- and stereoselectivity of NPs, and overall excellent functional group tolerance. Both ribosyl and 2′-deoxyribosyl nucleosides with various nucleobases can be accessed easily with this methodology. While the substrate scope is somewhat limited by the capabilities of the available enzymes, further expansion of the substrate scope by enzyme engineering can be expected.84 Recent progress in the thermodynamic characterization of these reactions has further enabled robust optimization of reaction conditions by leveraging principles of thermodynamic reaction control.80,85 Thus, considerable progress in this field may be anticipated.
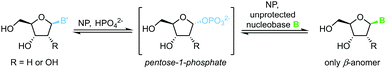 |
| | Scheme 13 Nucleobase glycosylation with 1-phosphates. | |
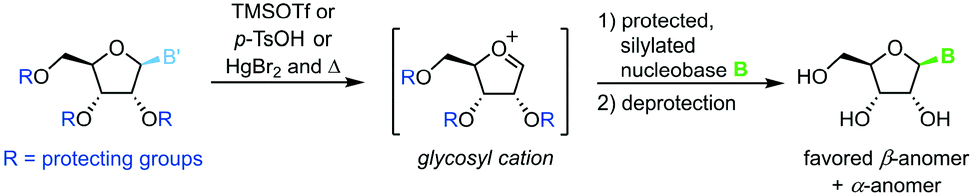
Protected nucleosides
Transglycosylation can also be achieved through heat- and Lewis acid-mediated dissociation of the nucleobase from a donor nucleoside (Scheme 14).86–88 This yields an unstable glycosyl cation which can be intercepted by a silylated nucleobase, affording the nucleoside of interest after global deprotection. Owing to several drawbacks of this approach, literature examples are rare and date back to the late 20th century. These include, among others, the need to protect every functional group, the limitation to purine nucleosides, poor control of the configuration at the anomeric center, as well as exceptionally harsh and hazardous reaction conditions.
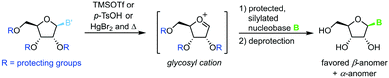 |
| | Scheme 14 Nucleobase glycosylation with protected nucleosides. | |
E-factor assessment of glycosylation methods for nucleobases
Considering the route length, methodology and yield of the individual steps of the available approaches, it is to be expected that these routes for nucleobase glycosylation would differ significantly regarding their efficiency and waste production.
Data collection
We sought to provide a fully transparent assessment of the efficiency and sustainability of the available methods for nucleobase glycosylation. Therefore, we surveyed the literature for examples of applications for these approaches and calculated the E-factor for the entire routes. In all cases, performing these calculations for every example in the literature (or even every example in a given publication) would have far surpassed the scope of this work. Thus, we selected representative examples that cover multiple pyrimidine and purine nucleosides each with various functional groups, as far as available from the literature (please see Chart S1† for an overview of all nucleosides considered herein).
To generate a level playing field for all methods, we opted to have all routes start from readily available unprotected starting materials. Consequently, we assumed that all routes started either from (deoxy)ribose or a natural nucleoside. Whenever possible and provided in the literature, we considered the E-factor for the synthesis of the sugar donor employed for glycosylation based on the route and procedures reported by the authors of that paper. However, this information was not available in most cases (i.e. the explicit methodology for synthon preparation was not always reported). Therefore, we assumed that whatever synthon these authors used for their synthesis was prepared according to classic literature procedures.89–102 Similarly, many nucleoside syntheses ended with the protected nucleoside and in those cases, we assumed that a suitable classic deprotection protocol from the literature was used.94 To avoid favoring one method over another, we applied these same assumptions equally across the board to all methods that built on a given synthon or required a given deprotection.
Calculations
To calculate the sEF and cEF of each route over all steps, we extracted experimental details and procedures from the reports of these methods. However, many reports across different journals did not provide a sufficient description of the experimental procedures to allow a precise reconstruction of the protocol. To still permit a calculation of cEF (which includes, for example, reaction solvents and solutions for quenching and extractions), several quantities had to be estimated. We based these estimates on previous reports of this kind,45 original papers on the matter44,103 as well as our own experience with typical laboratory procedures. A complete and transparent description of all calculations and estimates is given in the ESI.†
Route cEF and sEF
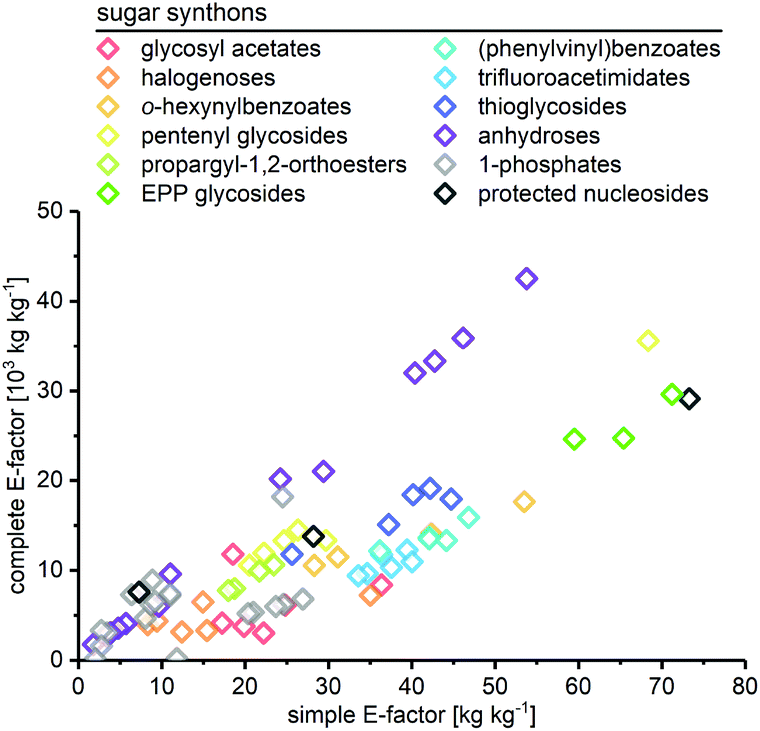
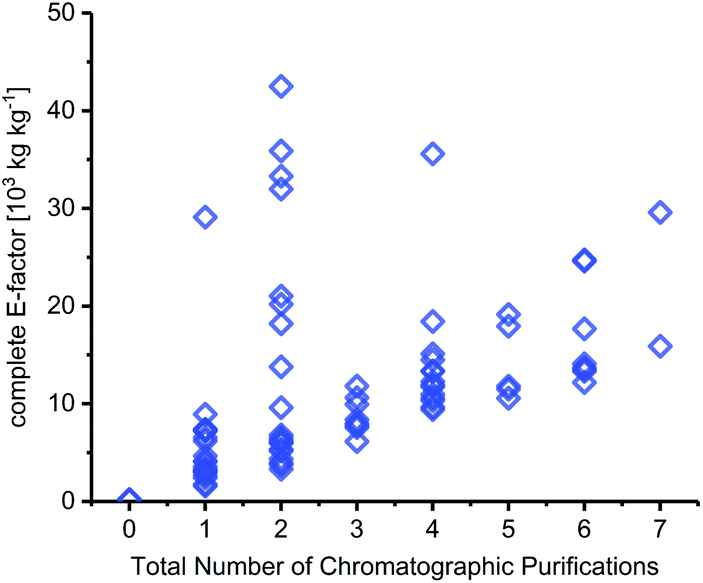
As expected, the sEF and cEF of the available routes differed significantly, both within and between methods. The sEFs in our dataset of 80 route E-factors (please see the ESI† for details) were as low as 1.8 and as high as 73.3, with most methods scoring between 10 and 30 (all kgwaste per kgproduct which is omitted from hereon for clarity). In contrast, the cEFs covered more than 2 orders of magnitude, with values from 165 to 42![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 499 (Fig. 1). Interestingly, some methods had E-factors in a rather narrow range, whereas others displayed significant variation between substrates/routes. Downey et al.'s anhydrose-based method is good example of the latter. While their originally reported procedure73 had quite unfavorable E-factors in a broad range (sEF = 11–53.8, cEF = 20195–42499, routes N1–N7 in the ESI†), the subsequently published improved protocol74 fared much better, but still displayed considerable variability (sEF = 1.8–9.6, cEF = 1760–6017, routes N8–N13). On the other hand, alternative methods such as Fraser-Reid and colleagues’ n-pentenyl orthoester-based procedure67 showed little E-factor variation (sEF = 22.2–29.7, cEF = 10590–14495), except for substrates where glycosylation yield suffered tremendously (N32–N37).
499 (Fig. 1). Interestingly, some methods had E-factors in a rather narrow range, whereas others displayed significant variation between substrates/routes. Downey et al.'s anhydrose-based method is good example of the latter. While their originally reported procedure73 had quite unfavorable E-factors in a broad range (sEF = 11–53.8, cEF = 20195–42499, routes N1–N7 in the ESI†), the subsequently published improved protocol74 fared much better, but still displayed considerable variability (sEF = 1.8–9.6, cEF = 1760–6017, routes N8–N13). On the other hand, alternative methods such as Fraser-Reid and colleagues’ n-pentenyl orthoester-based procedure67 showed little E-factor variation (sEF = 22.2–29.7, cEF = 10590–14495), except for substrates where glycosylation yield suffered tremendously (N32–N37).
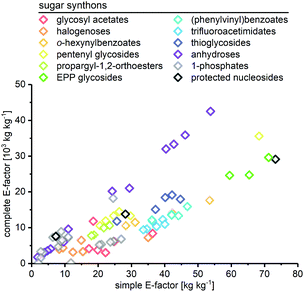 |
| | Fig. 1 Complete and simple E-factors of N-glycosylation methods for nucleobases cover an extensive range with great variability within and between methods. All E-factors represent route E-factors over all steps. Please see the ESI† for all details of the calculations. | |
In general, we were surprised to find how high most of these E-factors were. Both well-established and newly developed methods, and even biocatalytic approaches, typically had cEFs in the range of 5000–10000. This significantly surpasses many other types of transformations employed in industrial settings that typically have cEFs of less than 100 per step.15,16 While some of this may be ascribed to the fact that nucleosides are complex molecules with a high density of sensitive functional groups, these E-factors are still comparably high, considering the high demand and broad applicability of these compounds. Notably, none of the available methods performed well for all nucleobase substrates and/or delivered significantly lower E-factors than all other methods. Motivated by this lack of true efficiency104 in the sense of resource usage (and consequently high waste production), we were curious to find the sources of these high E-factors and identify areas where improvement is needed. At the same time, we sought to identify strategies that worked particularly well and may be employed by future “greener” nucleoside syntheses.
Yield
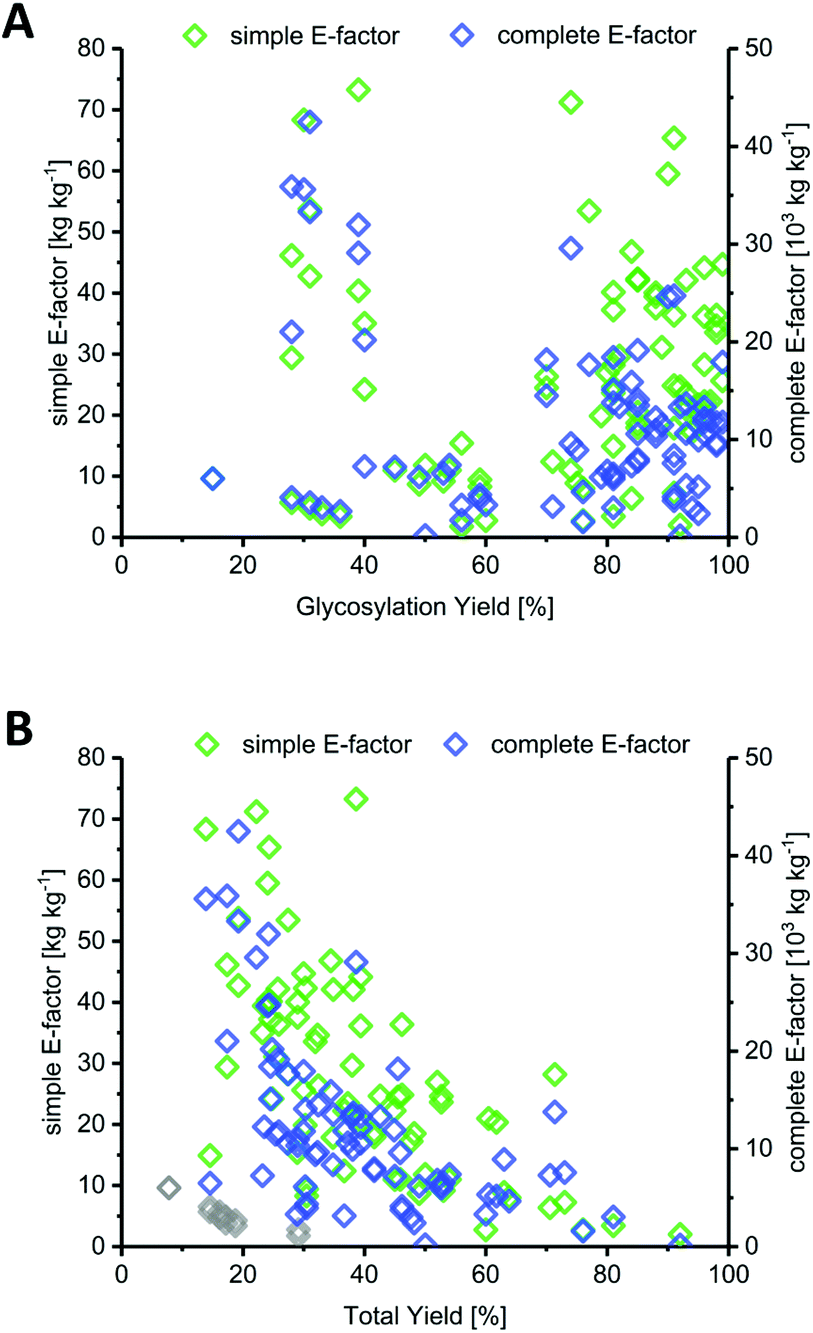
Most methods in the literature for nucleoside synthesis focused on optimization of glycosylation yield as the key metric. To this end, several strategies have been developed that employ highly reactive sugar synthons or disable competition of the leaving group with the nucleobase for (re)attack at the anomeric position. In many cases, these strategies succeeded in achieving glycosylation yields upwards of 90%. However, while the yield of an individual step is certainly a critical variable, it should always be viewed in light of the entire synthesis. Indeed, we found no correlation between glycosylation yield and sEF or cEF (Fig. 2A). Many of the strategies that sought to optimize glycosylation yield also performed quite lengthy routes and employed large leaving groups. Consequently, both the total yield and the atom economy of these routes suffered immensely, which is reflected by these E-factors.
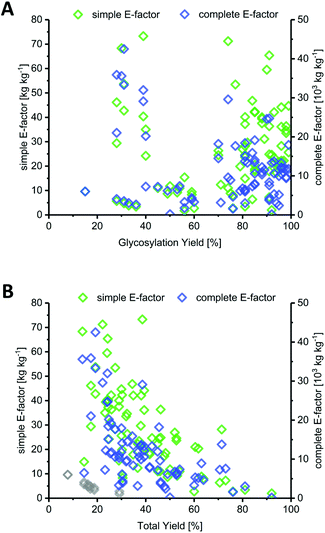 |
| | Fig. 2 Yield of the glycosylation step is a misleading predictor of method efficiency compared to total yield across the route. (A) Glycosylation yield shows no correlation with route sEF or cEF. (B) Total yield over the entire route correlates negatively with route sEF and cEF. Downey et al.'s improved anhydrose-based method (N8–N13) is shown as grey diamonds (sEF and cEF). | |
In contrast to glycosylation yield, we found that total yield (over the entire route) correlated negatively with route sEF and cEF, albeit only moderately (Fig. 2B). This comes as no surprise, as one would generally expect a higher efficiency for high-yielding routes versus those that barely generated any product. Nonetheless, there were some interesting outliers in the literature. The glycosylation and total yields of Downey's improved anhydrose-based approach were modest by most standards (in the range of 30% and 20%, respectively), yet this method displayed some of the lowest sEFs in our entire dataset (sEF = 1.8–9.6, see the grey diamonds in Fig. 2B).74 By employing a concise route (2 steps) and managing the atom economy by using only one protecting group that could be cleaved in situ, they were able to outweigh the rather moderate yields. Obviously, these sEFs could have been even lower with higher yields under the same conditions. However, these yields were still sufficient to achieve what could be considered an efficient (lower waste) synthesis. Other methods that had higher glycosylation and total yields, for example those employing trifluoroacetimidates (>88% glycosylation yield, N38–N43, please see the ESI† for details) or propargyl-orthoesters68 (>85% glycosylation yield, N44–N47), also necessitated longer routes and had more unfavorable atom economies, leading to much higher sEFs of 18–40. Clearly, yield is an important variable, but only to a certain extent. Even excellent yields are generally offset by cumulative reagent usage across a long route. Although yield of the key glycosylation step constitutes a bottleneck for some nucleobases, it appears that, from an efficiency standpoint, chasing maximum yields is not a fruitful strategy if it entails following longer routes.
Route length
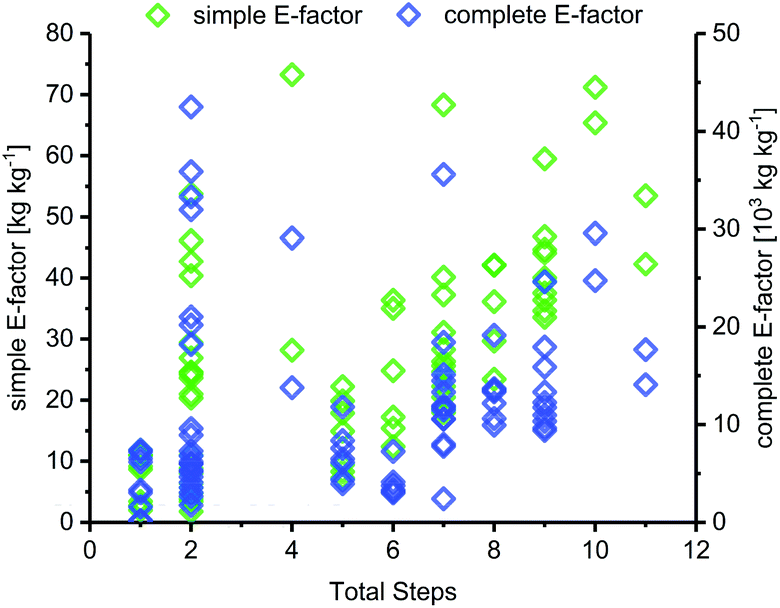
The length of a route, as given by the number of total steps,105 varied drastically among different glycosylation methods and appeared to dictate the lower bounds for possible route E-factors. The shortest available routes relied on nucleoside phosphorylases for biocatalytic glycosylation and had only one step, whereas the longest routes had around 9 to 11 steps, with several protecting group transformations. The cumulative sEFs and cEFs of all methods covered a broad range over all route lengths, and trended upwards with increasing route length (Fig. 3). This data highlights two important points for consideration. Routes employing only one step tended to perform rather favorably (sEF < 11), probably because the potential for waste accumulation in a one-step route is quite limited. On the other end of the spectrum, long routes with 9–11 steps all had sEFs higher than 40 and cEFs well above 10000. While these routes still displayed great heterogeneity, it appears that waste accumulation to a certain extent was a natural consequence of the number of transformations. It should be noted, however, that short routes don't guarantee lower E-factors, as our dataset featured 22 routes with 2 steps (all from Downey et al.'s method or biocatalytic) which mostly performed favorably regarding their E-factor, but also included a few outliers with cEFs above 15000 (Fig. 3). These data demonstrate that shorter routes do not necessarily translate to lower E-factors, but clearly have the potential to perform more efficiently than longer alternatives, especially if several protecting group transformations are involved.
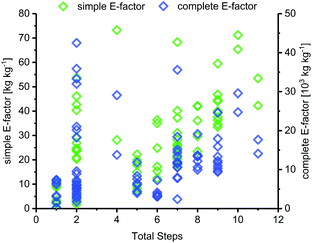 |
| | Fig. 3 sEF and cEF generally increase with route length. The total number of steps denotes the number of steps from an unprotected sugar starting material to an unprotected nucleoside. A step is considered as every transformation that was followed by a workup and/or purification of some sort. | |
E-factor contributions
Irrespective of yield and route length, the cEFs of all routes were mainly composed of solvent contributions (Fig. 4). The reagents used throughout a synthesis, as well as inorganics only added minorly to the route cEFs, as contributions from organic solvents and (for biocatalytic routes) water typically made up more than 95% of the cEF. This observation is somewhat intuitive as nucleobases and many nucleosides are generally poorly soluble in all solvents and solvents are known to be the main determiner for E-factors, if they are included in the calculation.11,36,44,45 Nonetheless, we were surprised to find that even routes which employed heterogenous steps were characterized by overshadowing solvent contributions. Biocatalytic routes were particularly plagued by the low water solubility of many nucleobases, which has so far largely restricted these syntheses to working concentrations in the low millimolar range. However, some routes which sought to prepare especially insoluble guanosine derivatives used this to their advantage to realize the lowest cEFs in our dataset. Zuffi et al.'s82 (cEF = 200, route N29) and Ubiali et al.'s83 (cEF = 165, route N31) one-step syntheses of 2′-deoxyguanosine from thymidine via transglycosylation employed a substrate loading which was an order of magnitude higher than other biocatalytic routes and profited from the target compound readily precipitating from the reaction mixture. Thus, the higher substrate loading in heterogenous reaction steps appears particularly attractive for sugar or nucleobase transformations. Based on our data it could be reasoned that (beyond heterogenous reactions) any strategy that allows higher substrate/reagent loading will result in lower E-factors. Yet even if one or multiple steps of a route can be realized heterogeneously, or with otherwise high substrate loading, solvent contributions from other steps may still be the main contributors to the cEF of that route. Again, this underscores that the demands and opportunities of a single step need to be considered as part of the entire route, and that shorter routes offer more potential for minimizing E-factors. It should also be noted that we did not consider recycling of any solvents in this analysis. Clearly, some solvents can be and are recycled in industrial settings to reduce the net waste arising from a reaction, especially in the case of low-boiling solvents such as dichloromethane or hexane. However, other solvents like pyridine, water or acetonitrile may be harder and much more energy-intensive to recover, purify and reuse. Thus, given the heterogeneity of solvents employed and their role in the respective syntheses, we opted to consider all solvents as waste without any recycling.
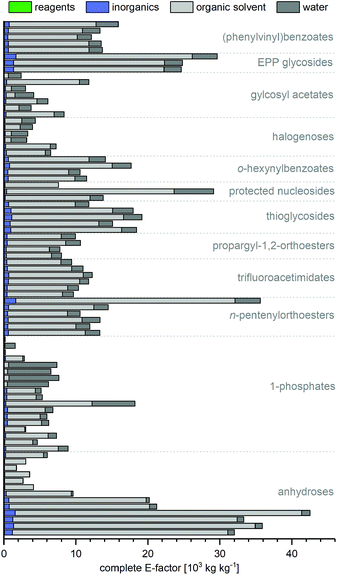 |
| | Fig. 4 Complete E-factors of nearly all routes are mainly composed of solvent contributions. All contributions are also given in the ESI† with reference to the procedure and details for calculation. | |
Chromatography
Solvent contributions for many steps originated to a large extent from chromatographic purification steps. In fact, the number of chromatography steps was equally good at setting the lower bounds for cEF as the total number of steps in a route – irrespective of the transformations, yields and types of workup performed (Fig. 5). It should be noted that very few syntheses in our dataset included quantities for their chromatography solvents, which required us to estimate these for most of the routes discussed herein. Like Hollmann and colleagues,45 we estimated 500 mL of solvent per gram of crude product and calculated the E-factor contributions via the density of the chromatography solvents employed (which are generally reported). Depending on the solvent used and the crude product, this equaled a cEF contribution of around 500–1500 per chromatography step in most cases. Considering published experimental data on this issue,44,103 and the fact that most chromatography steps in our dataset were done to isolate material from complex mixtures, this is a very conservative estimate for most syntheses analyzed here. Still, chromatography solvents dominated the cEFs of all routes that employed chromatographic purifications. Naturally, longer routes featured more chromatography steps, which is reflected in their cEFs (see e.g.N50–N52 or N56–N59). Conversely, the two routes that did not employ any chromatography steps (N29 and N31, see above) had the by far lowest cEFs. Admittedly, chromatography probably cannot be avoided altogether given the nature of the transformations required for nucleoside synthesis but limiting chromatography steps should be a primary goal to achieve more efficient and “greener” nucleoside synthesis.
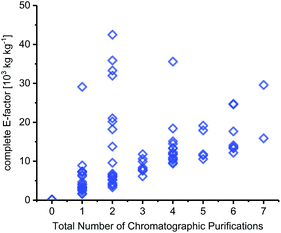 |
| | Fig. 5 The number of chromatography steps sets the lower bounds for route cEF. For all details regarding calculation of chromatography contributions to cEF, please see the ESI.† | |
Protecting groups
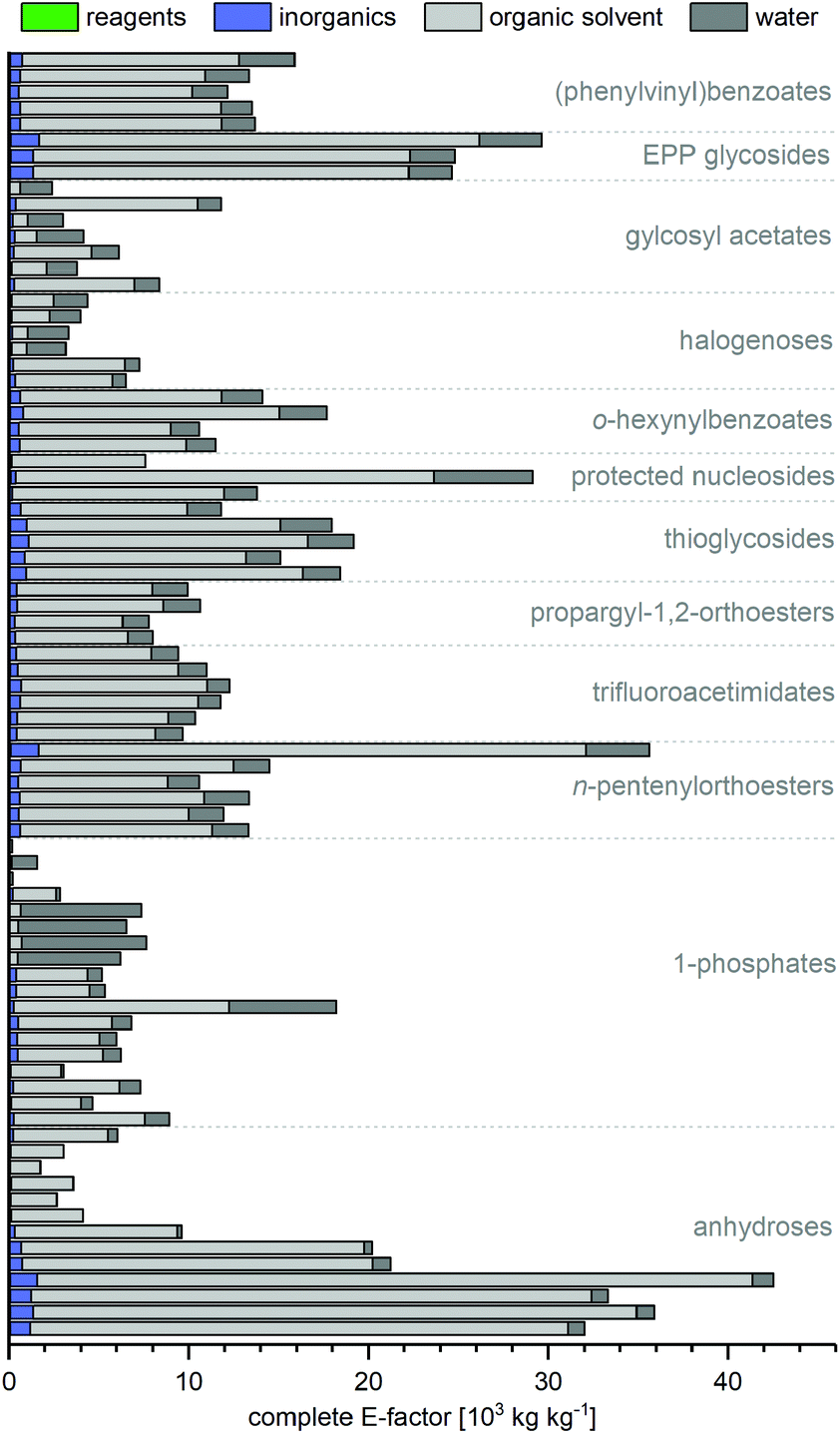
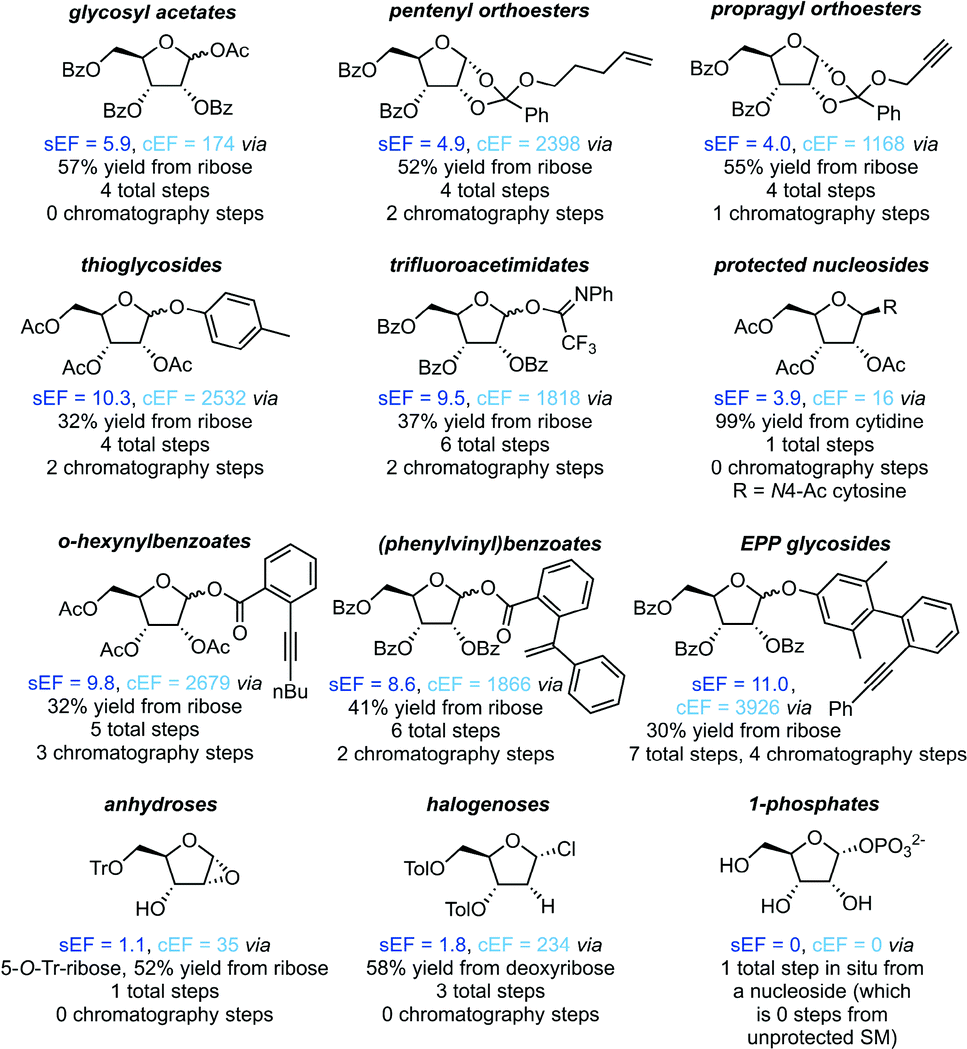
All non-biocatalytic syntheses considered herein employed protected sugar synthons, whose synthesis constitutes the most labor- and resource-intensive part of the route. Most sugar synthons need to be accessed in 3–7 steps from (deoxy)ribose through selective protection and introduction of the anomeric leaving group. Thus, the synthesis of these synthons accumulates a considerable E-factor even before the key glycosylation step. Even though yields for the required transformations are generally high to excellent, reagent usage and purification throughout these routes is reflected in the high sEFs and cEFs (Chart 1). Please note that these E-factors are somewhat skewed by the high molecular weights of the protected synthons and may not directly translate to full route E-factors since the E-factor is a mass-based metric, and the nucleoside products are generally a lot lighter than these synthons. Exceptions to these observations are presented by biocatalytic routes77–83 requiring no protecting groups (which considerably shortens these syntheses by all protecting and deprotecting steps) and Downey's method73,74 which only employs one (albeit large) protecting group, that can be installed in one step and cleaved in situ after the reaction. The only other method that consistently delivered E-factors close to these two approaches is halogenose-based glycosylation, which uses an easy to prepare synthon with a small leaving group. Clearly, the non-biocatalytic synthesis of nucleosides requires at least some protecting groups due to the complex arrangement of reactive functional groups in the target compounds. However, the choice of protecting groups and synthon for glycosylation should be made based on the most concise and efficient route to that synthon. Every protecting and deprotecting step that can be avoided in a synthesis typically results in lower waste production through a better atom economy and less purification effort.
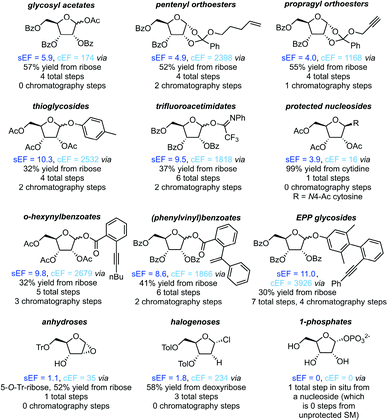 |
| | Chart 1 Please see the ESI† for procedures and references. | |
Transitioning to more efficient nucleoside synthesis
Benchmarks
Based on these observations, we propose some benchmarks for nucleoside synthesis to be termed “efficient”. Future synthetic efforts should seek to achieve sEFs below 10 and cEFs below 2000 in a route that takes 4 steps or less. We explicitly opt against inclusion of any recommendations regarding glycosylation or total yield, protecting groups, solvent usage or chromatographic purifications. However, a balance and improvement of all these metrics will be reflected in the E-factor. We chose to include route length as a relevant parameter, since the average step for nucleoside synthesis took roughly one day (Fig. S1†) and time investment in a synthesis is certainly a relevant factor. Selected routes in our dataset already meet these benchmarks (N11 and N29–N31), although only for some purines. We believe that there is potential for nucleoside synthesis to become more efficient in general by striving to meet these proposed benchmarks. To this end, there are some areas which require and deserve attention by researchers to effect immediate improvement.
Areas for improvement
The above data illustrate that nucleoside synthesis is currently hampered by several bottlenecks that manifest themselves in inefficient routes with high E-factors. Most notably, chromatographic purification steps present a significant source of waste in the form of solvent. Although some of this solvent may be recycled to reduce the net waste from these steps, they remain notoriously inefficient separation processes from a sustainability perspective. However, at least one chromatography step will probably be required for most target nucleosides to achieve sufficient purity, since N-glycosylation is a non-trivial transformation that (beyond the desired nucleoside) often yields several hard-to-separate byproducts. Thus, reduction of additional chromatography steps should be a central aim for all synthetic routes to the relevant sugar synthons. Whenever possible, precipitation or recrystallization steps allow tremendously lower resource investment and, therefore, lower E-factors. For some of the routes outlined above, it would also be worth considering if some of the steps required for either sugar donor synthesis or post-glycosylation deprotection could be performed in a one-pot manner to avoid intermediary purifications. This may also help to cut down the use of solvents like dichloromethane or hexane which serve as popular extraction and purification solvents but are recognized as environmentally concerning.106 To facilitate these aims, applied routes should be as short as possible, since additional steps such as protecting group manipulations on the sugar moiety have the potential to render the entire route inefficient – irrespective of metrics such as glycosylation or total yield. Therefore, glycosylation approaches which employ few protecting groups and do not rely on large leaving groups (which themselves necessitate prior installment) appear to have the most potential going forward. Future efforts to optimize existing methods or devise new methods may therefore focus on avoiding chromatography, shortening routes and/or doing multiple transformations in one pot. Furthermore, heterogenous reactions present an attractive strategy to cut down non-chromatography solvent waste, particularly since nucleosides are generally poorly soluble. This holds especially true for biocatalytic approaches which are currently severely hampered by the low water solubility of both the starting materials and products, as well as unfavorable reaction equilibria for some nucleosides. To overcome these obstacles, strategies to enable increased substrate loading and equilibrium shifts in favor of the target nucleosides should be key research goals.
Conclusions
Nucleosides and their analogs are indispensable biomolecules in nearly all areas of life science. However, the available methods to prepare these compounds via N-glycosylation of nucleobases suffer from severe drawbacks, which render these routes laborious and inefficient. Our comprehensive literature survey and E-factor analysis revealed that glycosylation methods for nucleoside synthesis cover an extended range of route E-factors and that glycosylation yield is an overrated metric of efficiency. Solvents, predominantly from chromatographic purification steps, are the main contributors to cEF and a heavy reliance on protecting groups tremendously increased both the sEF and cEF of most routes. Future syntheses should seek to address these bottlenecks to enable more efficient and sustainable nucleoside syntheses.
Conflicts of interest
A. K. is CEO of the biotech company BioNukleo GmbH. F. K. is a scientist at BioNukleo GmbH and P. N. is a member of the advisory board. These affiliations constitute no conflict of interest with the results presented and discussed in this report.
Acknowledgements
The authors thank Kerstin Heinecke (TU Berlin) for fruitful discussions.
Notes and references
- K. Ruiz-Mirazo, C. Briones and A. de la Escosura, Chem. Rev., 2014, 114, 285–366 CrossRef CAS
 .
.
- L. P. Jordheim, D. Durantel, F. Zoulim and C. Dumontet, Nat. Rev. Drug Discovery, 2013, 12, 447–464 CrossRef CAS .
- W. Xu, K. M. Chan and E. T. Kool, Nat. Chem., 2017, 9, 1043–1055 CrossRef CAS .
- C. Y. Jao and A. Salic, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15779–15784 CrossRef CAS .
- T. G. Allen-Mersh, S. Earlam, C. Fordy, K. Abrams and J. Houghton, Lancet, 1994, 344, 1255–1260 CrossRef CAS .
- D. Schürmann, D. J. Rudd, S. Zhang, I. De Lepeleire, M. Robberechts, E. Friedman, C. Keicher, A. Hüser, J. Hofmann and J. A. Grobler,
et al.
, Lancet HIV, 2020, 7, 164–172 CrossRef .
- H. Vorbrüggen and C. Ruh-Pohlenz, Org. React., 2004, 1–630 Search PubMed .
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC .
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC .
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. Leazer, L. Johnnie, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman and A. Wells,
et al.
, Green Chem., 2007, 9, 411–420 RSC .
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS .
- H. Loureiro, M. Prem and G. Wuitschik, Chim. Int. J. Chem., 2019, 73, 724–729 CrossRef CAS .
- D. Kaiser, J. Yang and G. Wuitschik, Org. Process Res. Dev., 2018, 22, 1222–1235 CrossRef CAS .
- T. Li and X. Li, Green Chem., 2014, 16, 4241–4256 RSC .
- J. Li, J. Albrecht, A. Borovika and M. D. Eastgate, ACS Sustainable Chem. Eng., 2018, 6, 1121–1132 CrossRef CAS .
- J. Li, E. M. Simmons and M. D. Eastgate, Green Chem., 2017, 19, 127–139 RSC .
- J. Li and M. D. Eastgate, React. Chem. Eng., 2019, 4, 1595–1607 RSC .
- E. R. Monteith, P. Mampuys, L. Summerton, J. H. Clark, B. U. W. Maes and C. R. McElroy, Green Chem., 2020, 22, 123–135 RSC .
- A. G. Parvatker, H. Tunceroglu, J. D. Sherman, P. Coish, P. Anastas, J. B. Zimmerman and M. J. Eckelman, ACS Sustainable Chem. Eng., 2019, 7, 6580–6591 CrossRef CAS .
- J. Tian, H. Shi, X. Li, Y. Yin and L. Chen, Green Chem., 2012, 14, 1990–2000 RSC .
- G. Rosini, V. Borzatta, C. Paolucci and P. Righi, Green Chem., 2008, 10, 1146–1151 RSC .
- Z. Amara, J. F. B. Bellamy, R. Horvath, S. J. Miller, A. Beeby, A. Burgard, K. Rossen, M. Poliakoff and M. W. George, Nat. Chem., 2015, 7, 489–495 CrossRef CAS .
- J. Andraos, Org. Process Res. Dev., 2009, 13, 161–185 CrossRef CAS .
- J. Andraos, E. Ballerini and L. Vaccaro, Green Chem., 2015, 17, 913–925 RSC .
- S. K. Ma, J. Gruber, C. Davis, L. Newman, D. Gray, A. Wang, J. Grate, G. W. Huisman and R. A. Sheldon, Green Chem., 2010, 12, 81–86 RSC .
- K. C. Badgujar and B. M. Bhanage, Biomass Bioenergy, 2016, 84, 12–21 CrossRef CAS .
- E. Busto, R. C. Simon, B. Grischek, V. Gotor-Fernández and W. Kroutil, Adv. Synth. Catal., 2014, 356, 1937–1942 CrossRef CAS .
- A. Cavezza, C. Boulle, A. Guéguiniat, P. Pichaud, S. Trouille, L. Ricard and M. Dalko-Csiba, Bioorg. Med. Chem. Lett., 2009, 19, 845–849 CrossRef CAS .
- F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, Green Chem., 2010, 12, 1301–1305 RSC .
- Y. Gaber, U. Törnvall, C. Orellana-Coca, M. Ali Amin and R. Hatti-Kaul, Green Chem., 2010, 12, 1817–1825 RSC .
- S. M. Kelly and B. H. Lipshutz, Org. Lett., 2014, 16, 98–101 CrossRef CAS .
- D. Kuhn, M. K. Julsing, E. Heinzle and B. Bühler, Green Chem., 2012, 14, 645–653 RSC .
- D. Kuhn, M. A. Kholiq, E. Heinzle, B. Bühler and A. Schmid, Green Chem., 2010, 12, 815–827 RSC .
- A. Ledoux, L. Sandjong Kuigwa, E. Framery and B. Andrioletti, Green Chem., 2015, 17, 3251–3254 RSC .
- J. Liang, J. Lalonde, B. Borup, V. Mitchell, E. Mundorff, N. Trinh, D. A. Kochrekar, R. Nair Cherat and G. G. Pai, Org. Process Res. Dev., 2010, 14, 193–198 CrossRef CAS .
- B. H. Lipshutz, N. A. Isley, J. C. Fennewald and E. D. Slack, Angew. Chem., Int. Ed., 2013, 52, 10952–10958 CrossRef CAS .
- S. Handa, J. C. Fennewald and B. H. Lipshutz, Angew. Chem., Int. Ed., 2014, 53, 3432–3435 CrossRef CAS .
- B. H. Lipshutz and S. Ghorai, Green Chem., 2014, 16, 3660–3679 RSC .
- L. Martínez-Montero, V. Gotor, V. Gotor-Fernández and I. Lavandera, Green Chem., 2017, 19, 474–480 RSC .
- O. Maurin, P. Verdié, G. Subra, F. Lamaty, J. Martinez and T.-X. Métro, Beilstein J. Org. Chem., 2017, 13, 2087–2093 CrossRef CAS .
- T.-X. Métro, J. Bonnamour, T. Reidon, A. Duprez, J. Sarpoulet, J. Martinez and F. Lamaty, Chem. – Eur. J., 2015, 21, 12787–12796 CrossRef .
- A. R. C. Morais, S. Dworakowska, A. Reis, L. Gouveia, C. T. Matos, D. Bogdał and R. Bogel-Łukasik, Catal. Today, 2015, 239, 38–43 CrossRef CAS .
- A. Palmieri, S. Gabrielli and R. Ballini, Green Chem., 2013, 15, 2344–2348 RSC .
- M. L. Parisi, A. Dessì, L. Zani, S. Maranghi, S. Mohammadpourasl, M. Calamante, A. Mordini, R. Basosi, G. Reginato and A. Sinicropi, Front. Chem., 2020, 8, 214 CrossRef CAS .
- J. H. Schrittwieser, F. Coccia, S. Kara, B. Grischek, W. Kroutil, N. d'Alessandro and F. Hollmann, Green Chem., 2013, 15, 3318–3331 RSC .
- R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC .
- R. A. Sheldon, J. R. Soc., Interface, 2016, 13, 20160087 CrossRef .
- F. Roschangar, J. Colberg, P. J. Dunn, F. Gallou, J. D. Hayler, S. G. Koenig, M. E. Kopach, D. K. Leahy, I. Mergelsberg and J. L. Tucker,
et al.
, Green Chem., 2017, 19, 281–285 RSC .
- R. A. Sheldon, ACS Sustainable Chem. Eng., 2018, 6, 32–48 CrossRef CAS .
- R. A. Sheldon, Green Chem., 2016, 18, 3180–3183 RSC .
- M. A. F. Delgove, A.-B. Laurent, J. M. Woodley, S. M. A. De Wildeman, K. V. Bernaerts and Y. van der Meer, ChemSusChem, 2019, 12, 1349–1360 CrossRef CAS .
- M. C. McManus and C. M. Taylor, Biomass Bioenergy, 2015, 82, 13–26 CrossRef CAS .
- A. A. Burgess and D. J. Brennan, Chem. Eng. Sci., 2001, 56, 2589–2604 CrossRef CAS .
- F. Tieves, F. Tonin, E. Fernández-Fueyo, J. M. Robbins, B. Bommarius, A. S. Bommarius, M. Alcalde and F. Hollmann, Tetrahedron, 2019, 75, 1311–1314 CrossRef CAS .
- U. Niedballa and H. Vorbrüggen, Angew. Chem., Int. Ed. Engl., 1970, 9, 461–462 CrossRef CAS .
- H. O. Kim, Arch. Pharmacal Res., 2001, 24, 508–513 CrossRef CAS .
- V. Parmenopoulou, D. S. M. Chatzileontiadou, S. Manta, S. Bougiatioti, P. Maragozidis, D.-N. Gkaragkouni, E. Kaffesaki, A. L. Kantsadi, V. T. Skamnaki and S. E. Zographos,
et al.
, Bioorg. Med. Chem., 2012, 20, 7184–7193 CrossRef CAS .
- H. Shirouzu, H. Morita and M. Tsukamoto, Tetrahedron, 2014, 70, 3635–3639 CrossRef CAS .
- C. Moreau, T. Kirchberger, J. M. Swarbrick, S. J. Bartlett, R. Fliegert, T. Yorgan, A. Bauche, A. Harneit, A. H. Guse and B. V. L. Potter, J. Med. Chem., 2013, 56, 10079–10102 CrossRef CAS .
- D. Hocková, M. Hocek, H. Dvořáková and I. Votruba, Tetrahedron, 1999, 55, 11109–11118 CrossRef .
- D. Ackermann and M. Famulok, Nucleic Acids Res., 2013, 41, 4729–4739 CrossRef CAS .
- J. N. Freskos, Nucleosides Nucleotides, 1989, 8, 549–555 CrossRef CAS .
- Z. Kazimierczuk, H. B. Cottam, G. R. Revankar and R. K. Robins, J. Am. Chem. Soc., 1984, 106, 6379–6382 CrossRef CAS .
- Q. Zhang, J. Sun, Y. Zhu, F. Zhang and B. Yu, Angew. Chem., Int. Ed., 2011, 50, 4933–4936 CrossRef CAS .
- P. Li, H. He, Y. Zhang, R. Yang, L. Xu, Z. Chen, Y. Huang, L. Bao and G. Xiao, Nat. Commun., 2020, 11, 405–414 CrossRef CAS .
- Z. Hu, Y. Tang and B. Yu, J. Am. Chem. Soc., 2019, 141, 4806–4810 CrossRef CAS .
- B. Fraser-Reid, P. Ganney, C. V. S. Ramamurty, A. M. Gómez and J. C. López, Chem. Commun., 2013, 49, 3251–3253 RSC .
- B. V. Rao, S. Manmode and S. Hotha, J. Org. Chem., 2015, 80, 1499–1505 CrossRef CAS .
- J. Liao, J. Sun and B. Yu, Tetrahedron Lett., 2008, 49, 5036–5038 CrossRef CAS .
- J. Liao, J. Sun and B. Yu, Carbohydr. Res., 2009, 344, 1034–1038 CrossRef CAS .
- G. Liu, X. Zhang and G. Xing, Chem. Commun., 2015, 51, 12803–12806 RSC .
- K. Chow and S. Danishefsky, J. Org. Chem., 1990, 55, 4211–4214 CrossRef CAS .
- A. M. Downey, C. Richter, R. Pohl, R. Mahrwald and M. Hocek, Org. Lett., 2015, 17, 4604–4607 CrossRef CAS .
- A. M. Downey, R. Pohl, J. Roithová and M. Hocek, Chem. – Eur. J., 2017, 23, 3910–3917 CrossRef CAS .
-
S. Kamel, H. Yehia, P. Neubauer and A. Wagner, in Enzym. Chem. Synth. Nucleic Acid Deriv, 2019, pp. 1–28 Search PubMed .
- F. Kaspar, R. T. Giessmann, K. F. Hellendahl, P. Neubauer, A. Wagner and M. Gimpel, ChemBioChem, 2020, 21, 1428–1432 CrossRef CAS .
- M. S. Drenichev, C. S. Alexeev, N. N. Kurochkin and S. N. Mikhailov, Adv. Synth. Catal., 2018, 360, 305–312 CrossRef CAS .
- C. S. Alexeev, M. S. Drenichev, E. O. Dorinova, R. S. Esipov, I. V. Kulikova and S. N. Mikhailov, Biochim. Biophys. Acta, Proteins Proteomics, 2020, 1868, 140292 CrossRef CAS .
- V. N. Barai, A. I. Zinchenko, L. A. Eroshevskaya, E. N. Kalinichenko, T. I. Kulak and I. A. Mikhailopulo, Helv. Chim. Acta, 2002, 85, 1901–1908 CrossRef CAS .
- H. Yehia, S. Westarp, V. Röhrs, F. Kaspar, T. R. Giessmann, F. T. H. Klare, K. Paulick, P. Neubauer, J. Kurreck and A. Wagner, Molecules, 2020, 25, 934 CrossRef CAS .
- X. Zhou, K. Szeker, L.-Y. Jiao, M. Oestreich, I. A. Mikhailopulo and P. Neubauer, Adv. Synth. Catal., 2015, 357, 1237–1244 CrossRef CAS .
- G. Zuffi, D. Ghisotti, I. Oliva, E. Capra, G. Frascotti, G. Tonon and G. Orsini, Biocatal. Biotransform., 2004, 22, 25–33 CrossRef CAS .
- D. Ubiali, S. Rocchietti, F. Scaramozzino, M. Terreni, A. M. Albertini, R. Fernández-Lafuente, J. M. Guisán and M. Pregnolato, Adv. Synth. Catal., 2004, 346, 1361–1366 CrossRef CAS .
- M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater and S. T. Grosser,
et al.
, Science, 2019, 366, 1255–1259 CrossRef CAS .
- F. Kaspar, R. T. Giessmann, P. Neubauer, A. Wagner and M. Gimpel, Adv. Synth. Catal., 2020, 362, 867–876 CrossRef CAS .
- T. Azuma and K. Isono, Chem. Pharm. Bull., 1977, 25, 3347–3353 CrossRef CAS .
- M. Miyaki, A. Saito and B. Shimizu, Chem. Pharm. Bull., 1970, 18, 2459–2468 CrossRef CAS .
- J. Boryski, Nucleosides Nucleotides, 1998, 17, 1547–1556 CrossRef CAS .
- O. V. Andreeva, M. G. Belenok, L. F. Saifina, M. M. Shulaeva, A. B. Dobrynin, R. R. Sharipova, A. D. Voloshina, A. F. Saifina, A. T. Gubaidullin and B. I. Khairutdinov,
et al.
, Tetrahedron Lett., 2019, 60, 151276 CrossRef CAS .
- Y. Kawase, M. Koizumi, S. Iwai and E. Ohtsuka, Chem. Pharm. Bull., 1989, 37, 2313–2317 CrossRef CAS .
- H. M. Kissman, C. Pidacks and B. R. Baker, J. Am. Chem. Soc., 1955, 77, 18–24 CrossRef CAS .
- N.-S. Li and J. A. Piccirilli, Synthesis, 2005, 2005, 2865–2870 CrossRef .
- Q. Liu, X. Cai, D. Yang, Y. Chen, Y. Wang, L. Shao and M.-W. Wang, Bioorg. Med. Chem., 2017, 25, 4579–4594 CrossRef CAS .
- P. Nauš, O. Caletková, P. Konečný, P. Džubák, K. Bogdanová, M. Kolář, J. Vrbková, L. Slavětínská, E. Tloušt'ová and P. Perlíková,
et al.
, J. Med. Chem., 2014, 57, 1097–1110 CrossRef .
- R. K. Ness and H. G. Fletcher, J. Am. Chem. Soc., 1953, 75, 3289–3290 CrossRef CAS .
- I. Nowak, M. Conda-Sheridan and M. J. Robins, J. Org. Chem., 2005, 70, 7455–7458 CrossRef CAS .
- A. Nudelman, J. Herzig, H. E. Gottlieb, E. Keinan and J. Sterling, Carbohydr. Res., 1987, 162, 145–152 CrossRef CAS .
- V. Rolland, M. Kotera and J. Lhomme, Synth. Commun., 1997, 27, 3505–3511 CrossRef CAS .
- C. V. S. Ramamurty, P. Ganney, C. S. Rao and B. Fraser-Reid, J. Org. Chem., 2011, 76, 2245–2247 CrossRef CAS .
- M. Thomas, J.-P. Gesson and S. Papot, J. Org. Chem., 2007, 72, 4262–4264 CrossRef CAS .
- S. A. Thadke, B. Mishra and S. Hotha, J. Org. Chem., 2014, 79, 7358–7371 CrossRef CAS .
- M. Kurosu and K. Li, J. Org. Chem., 2008, 73, 9767–9770 CrossRef CAS .
- F. Pessel, J. Augé, I. Billault and M.-C. Scherrmann, Beilstein J. Org. Chem., 2016, 12, 2351–2357 CrossRef CAS .
- ‘Efficiency’ is typically defined as the ability to obtain a result or desired product in a way that does not waste resources in the form of materials, money or time. Herein, we focus on the aspect of resource efficiency as quantified by the E-factor.
- We herein considered a step as any kind or number of transformations which was followed by a workup and/or purification of some sort.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC .
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0gc02665d |
|
| This journal is © The Royal Society of Chemistry 2021 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
M. Rhia L.
Stone
*ab,
M. Rhia L.
Stone