
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvatomorph and polymorph screening of clopamide drug and its copper(II) complex crystals†
G. Tamás
Gál
a,
Nóra V.
May
 a,
László
Trif
b,
Judith
Mihály
b and
Petra
Bombicz
*a
a,
László
Trif
b,
Judith
Mihály
b and
Petra
Bombicz
*a
aCentre for Structural Science, Research Centre for Natural Sciences, Magyar Tudósok Körútja 2, H-1117 Budapest, Hungary. E-mail: bombicz.petra@ttk.hu
bInstitute of Material and Environmental Chemistry, Research Centre for Natural Sciences, 1117 Magyar Tudósok Körútja 2, Budapest, Hungary
First published on 8th October 2021
Abstract
Clopamide ((4-chloro-N-2,6-dimethylpiperidin-1-yl)-3-sulfamoylbenzamide, LH) drug is used worldwide in the treatment of hypertension and oedema. Despite its medical application its structure has not yet been published. We report the crystal structure of clopamide anhydrate and hemihydrate and a structural landscape of different polymorphs and solvatomorphs of its copper(II) complexes. Our main challenge was the growth of single crystals for X-ray diffraction experiments and then the analysis of the molecular arrangements of the pure drug as well as the series of complexes organised by the coordination bond and by different secondary interactions. We present how the inclusion of water contributes to the crystal perfection of the drug crystals (1 and 2). The newly defined chalcogen bond is recognised in the clopamide anhydrate crystals being in competition with an intramolecular halogen bond. All Cu(II) complexes have a square-planar coordination geometry, in which copper(II) centres are surrounded by piperidine-N and carbonyl-O donor atoms in a five-membered chelate ring. The two ligands are in trans positions. The solvatomorph crystals, containing small homologous alcohols (3–6), have been found to be isostructural (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) with increasing size of void and unit cell volumes which terminates using n-butanol. In the solvent-free polymorph [CuL2] crystals (7–9), the complex molecules form parallel columns strengthened by N–H⋯O interactions between the ligand sulfonamide groups. The arrangement of these columns in 7 (P) is similar to that of the solvate 3 (P), while 8 (P) transitions to 9 (R
) with increasing size of void and unit cell volumes which terminates using n-butanol. In the solvent-free polymorph [CuL2] crystals (7–9), the complex molecules form parallel columns strengthened by N–H⋯O interactions between the ligand sulfonamide groups. The arrangement of these columns in 7 (P) is similar to that of the solvate 3 (P), while 8 (P) transitions to 9 (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) ), where perfect threefold symmetry is realised, although leaving large empty spaces in the lattice. The columnal arrangement is preserved in the crystal containing dichloromethane (10) but the columns are no longer parallel. The solvate content and thermal behaviour of the crystals were studied by IR and thermoanalytical methods. The solution structure of the [CuL2] complex was investigated in DMSO by EPR spectroscopy at 77 K. These results of the systematic comparison of several polymorphic and solvatomorphic crystal arrangements contribute to the better understanding of the structure-directing effects, which provide strategies to control the molecular self-assembly.
), where perfect threefold symmetry is realised, although leaving large empty spaces in the lattice. The columnal arrangement is preserved in the crystal containing dichloromethane (10) but the columns are no longer parallel. The solvate content and thermal behaviour of the crystals were studied by IR and thermoanalytical methods. The solution structure of the [CuL2] complex was investigated in DMSO by EPR spectroscopy at 77 K. These results of the systematic comparison of several polymorphic and solvatomorphic crystal arrangements contribute to the better understanding of the structure-directing effects, which provide strategies to control the molecular self-assembly.
1 Introduction
Understanding the molecular conformation and crystal structures (including polymorphs and solvatomorphs) is imperative in the use and development of active pharmaceutical ingredients (APIs). Polymorphism plays a very important role in the bioavailability of a drug1 as the physicochemical properties (e.g., solubility, stability) of polymorphs can differ significantly.2 In many cases, a serious limitation is that only drugs that have GRAS (generally recognised as safe) compounds in their crystal lattice can obtain a marketing authorisation. It has to be considered at the time of the selection of the solvent from which the API is crystallized.3,4 Special attention should be paid to the crystallization conditions and the crystal structures of the APIs.Clopamide ((4-chloro-N-2,6-dimethylpiperidin-1-yl)-3-sulfamoylbenzamide) is an API used currently in the treatment of hypertension and oedema. It is an oral thiazide-like diuretic agent with antihypertensive activity.5 It has two interconnected functional groups: a piperidine and a sulfamoylbenzamide group. Despite the medical application of clopamide, its crystal structure has not yet been published. It has been found that the most widely used sulfonamide-type antihypertensive agents cause significant urinary potassium losses (hypokalemia).6,7 This side effect also exists when applying clopamide as well.8 It has also been shown that thiazide diuretics may also cause urinary loss of certain trace elements or modify their levels in blood and may induce changes in the levels of copper in normal hypertensives.9,10 According to the molecular structure it is noticeable that the carbonyl oxygen and the piperidine nitrogen of the ligand are able to coordinate to metal centres; thus the complexation of clopamide with metal ions in the human body may be responsible for this side effect. On the other hand, various metal complexes can advantageously modify the biological activity of an API compared to the free ligand. There are references which report an increased bioactivity by forming metal complexes.11,12
To gain a better insight into the structure and interaction of this drug molecule, the structure of clopamide and its coordination compounds with copper(II) has been studied (Scheme 1) using different crystallization solvents under different conditions. Our aim was to obtain insights into the solvatomorph and polymorph crystal forms of clopamide and its copper(II) complexes using single-crystal X-ray diffraction (SXRD). The outcome of successful crystallization experiments from a homologous series of alcohols allowed us to harvest single crystals of a series of clopamide derivatives (Table 1). The crystal structures of the anhydrous LH (1) and hemihydrate LH·0.5H2O (2) forms of clopamide have been determined. The bis-ligand copper(II) complex crystals were synthesized from a homologous series of alcohols, with the inclusion of solvent molecules, resulting in [CuL2]·2(MeOH)·2(H2O) (3), [CuL2]·2(EtOH)·2(H2O) (4), [CuL2]·2(PrOH)·2(H2O) (5) and [CuL2]·2(iPrOH)·2(H2O) (6). From organic solvents, three solvent-free polymorphs of [CuL2] 7–9 and a crystal containing dichloromethane [CuL2]·CH2Cl2 (10) were prepared. It has opened up a way to determine how far the molecular conformation and the packing arrangement can preserve isostructurality.13,14 The thermodynamic and kinetic stability of these complexes was investigated by thermoanalytical studies. The solution structure of the clopamide bis-ligand copper(II) complex was tested using electron paramagnetic resonance (EPR) spectroscopy.
 | ||
| Scheme 1 General formula diagram of the clopamide (LH) molecule (a) and bis-ligand copper(II) complex [CuL2] of clopamide (b). The carbonyl oxygens and the piperidine ring nitrogen atoms are coordinating to the copper centre. | ||
| 1 | LH |
| 2 | LH·0.5H2O |
| 3 | [CuL2]·2(MeOH)·2(H2O) |
| 4 | [CuL2]·2(EtOH)·2(H2O) |
| 5 | [CuL2]·2(PrOH)·2(H2O) |
| 6 | [CuL2]·2(iPrOH)·2(H2O) |
| 7 | CuL2 form I |
| 8 | CuL2 form II |
| 9 | CuL2 form III |
| 10 | [CuL2]·CH2Cl2 |
A better understanding of the structure-directing effects in crystallization contributes to the development of strategies to control the process of molecular self-assembly.15,16 The obtained results contain valuable information with regard to synthon engineering,17 as we performed a systematic comparison of several solvatomorphic and polymorphic crystal arrangements. These investigations enrich the knowledge on the aspects which contribute to the development of materials with specific properties.18
2 Materials and methods
2.1 Reagents and materials
All chemicals were of reagent grade and used as purchased without any further purification. Clopamide was received from EGIS Pharmaceuticals Private Limited Company and used without further purification. All solvents were of analytical grade and used without further purification. CuCl2·2H2O, CuSO4·5H2O and NaOH were purchased from Sigma-Aldrich in analytical quality.2.2 Synthesis of complexes and single crystal growth
Crystallization experiments were performed in order to obtain single crystals suitable for SXRD measurements. Crystals were kept in their mother liquor to preserve their quality. A scheme of the determined copper(II) complex crystals obtained by solvatomorph and polymorph screening is depicted in Scheme 2.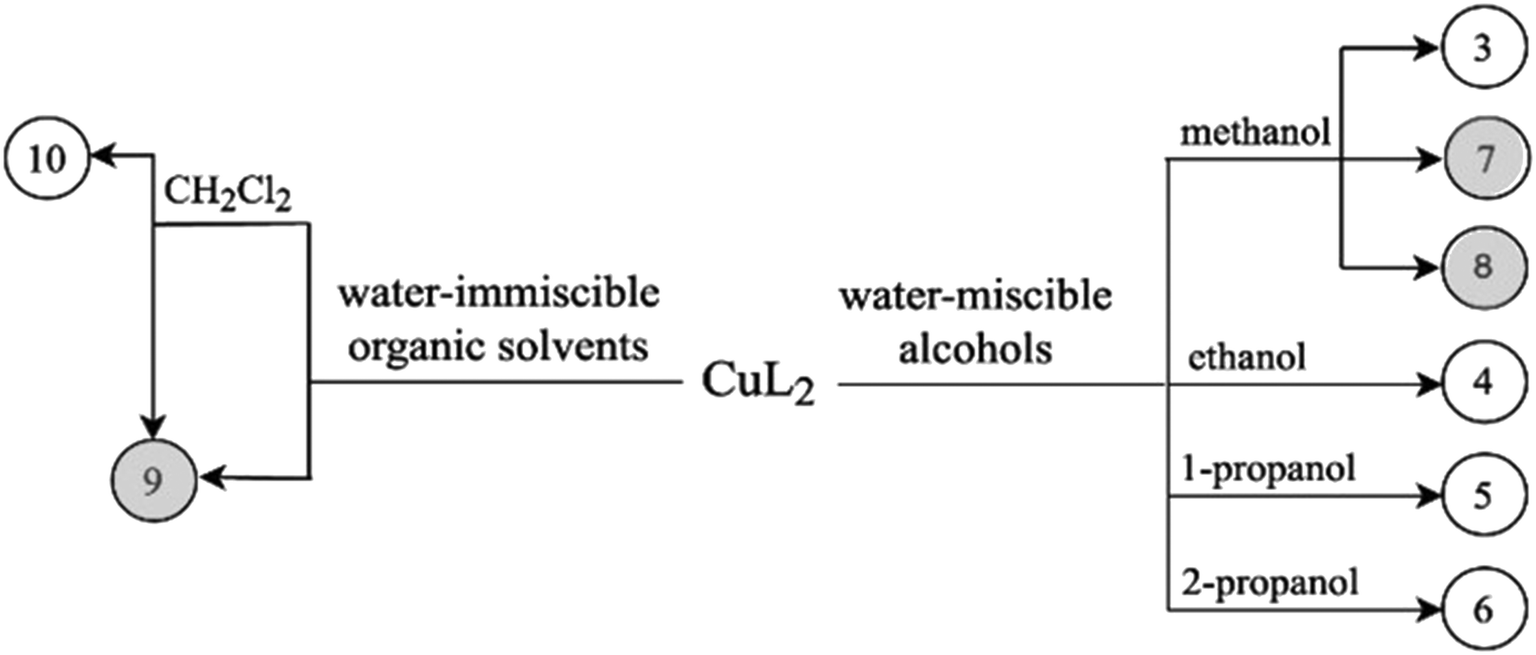 | ||
| Scheme 2 Crystallization experiments used to obtain crystals 3–10. Compound numbers with white background: crystals which enclathrate solvents; compound numbers with grey background: different polymorphic forms of CuL2. | ||
Clopamide in excess as ligand (23 mg, 0.07 mmol) and CuSO4·5H2O (5 mg, 0.02 mmol) were dissolved in 0.5 mL methanol. The pH of the reaction mixture was set to ∼9 with 0.02 mL 2 M NaOH solution. The mixture was kept in the fridge at 5 °C. Brown single crystals of 8 were obtained after one year.
Clopamide (10 mg, 0.03 mmol) and an aqueous solution of CuCl2 (0.1 mL, 0.1 M, 0.01 mmol) were added to water (0.3 mL). Clopamide is insoluble in water; thus a white suspension formed. After adding ethyl acetate (0.5 mL) two phases formed. Aqueous NaOH (0.02 mL, 2 M) was added dropwise to adjust the pH to approximately 9. The ampule was heated to 40 °C in order to increase the solubility of the complex in the organic solvent. The organic phase was separated and brown single crystals of 9 were obtained after one week by slow evaporation of the solvent in the fridge at 5 °C.
2.3 Single-crystal structure determination
The single crystals were mounted on loops and transferred to a goniometer. X-ray diffraction data were collected at 103 K (1–7), 123 K (8) 158 K (9) and 128 K (10) on a Rigaku RAXIS-RAPID II diffractometer. The diffraction measurements were performed using Mo-Kα radiation in all cases, except (1), (9) and (10), where a Cu-Kα source was used. Numerical absorption corrections were carried out using the software CrystalClear.19 Sir2018 (ref. 20) and SHELXL21 under WinGX22 software were used for structure solution and refinement, respectively. The structures were solved by direct methods. The models were refined using the full-matrix least squares method on F2. Refinement of the non-hydrogen atoms was carried out with anisotropic thermal parameters. Hydrogen atoms were placed into geometric positions (except alcohol and water hydrogens). The atomic positions of alcohol and water hydrogens were located in difference electron density maps, whereas all the other hydrogens were placed in geometric positions. Hydrogens were included in structure factor calculations, but they were not refined. The isotropic displacement parameters of the hydrogen atoms were approximated from the U(eq) value of the atom they were bonded to. In the case of crystal 7, several data collections have been performed but the high Rint value could not be lowered (Table S9†), which is most probably due to crystal defects. After the removal of the crystals of 10 from the mother liquor the enclathrated dichloromethane molecules evaporate rapidly causing partial degradation of the single crystal, resulting in a higher Rint value. The details of data collection and refinement parameters are collected in Table S1.† Selected bond lengths and angles were calculated using the software PLATON.23 The graphical presentations were prepared using Mercury24 and the editing of CIF files was done using the software EnCifer.252.4 NMR measurements and evaluation of the spectra
1H and 13C NMR spectra of clopamide in deuterated DMSO were recorded using a Bruker Avance 400 (400 MHz spectrometer) at 25 °C. The spectra were evaluated using the Mestrenova 9 NMR program.262.5 EPR measurements and evaluation of the spectra
X-band CW-EPR spectra were recorded on a BRUKER EleXsys E500 spectrometer (microwave frequency 9.4 GHz, microwave power 13 mW, modulation amplitude 5 G, modulation frequency 100 kHz). For the frozen solution measurement, a 0.2 mL sample of crystal 9 was prepared in 3.0 mM concentration, in pure DMSO, and placed into a quartz EPR tube. The EPR tubes were placed into a Dewar flask containing liquid nitrogen to perform the measurement at 77 K.The spectrum was simulated using the EPR program.27 Rhombic or axial g tensors and copper hyperfine A tensors were used for the simulation of the components. The splitting caused by the two coordinated nitrogen donor atoms is clearly visible in the spectrum and this superhyperfine coupling were taking into account by rhombic nitrogen tensor a. The linewidths were fitted with the help of orientation-dependent relaxation parameters (α, β, and γ) through the equation σMI = α + βMI + γMI2, where MI denotes the magnetic quantum number of the copper nucleus. Since natural CuCl2 was used for the measurements, the spectra were calculated as the sum of the spectra of 63Cu and 65Cu weighted by their natural abundances. The copper and nitrogen coupling constants and the relaxation parameters were obtained in field units (Gauss = 10−4 T).
2.6 Thermoanalytical methods
Thermal measurements were performed on a Setaram LabsysEvo (Lyon, France) TG–DSC system, in a flowing (90 mL min−1) helium gas (99.9999% purity) atmosphere. The sample was weighed directly into a 100 μL aluminium crucible (the reference cell was empty) and was heated from 20 °C to 500 °C at a heating rate of 20 °C min−1. After loading the sample into the thermal analyzer and prior to running the measurement, a 10 min purging step was run with a high helium flow rate (200 mL min−1). The obtained data were baseline corrected and further processed using the thermoanalyzer's processing software (Calisto Processing, ver. 2.092). The thermal analyzer (both the temperature scale and calorimetric sensitivity) was calibrated using a multipoint calibration method, in which seven different certified reference materials (CRMs) were used to cover the thermal analyzer's entire operating temperature range. In parallel with the thermal measurements, the analysis of evolved gases/volatiles was performed on a Pfeiffer Vacuum Omni Star™ mass spectrometric evolved gas analysis (MS-EGA) system, which was connected to the above-mentioned thermal analyzer. The gas splitter at the thermal analyzer's furnace exit was thermostated to 220 °C, while the transfer line to the mass spectrometer was thermostated to 200 °C in order to avoid condensation of some analytes. The temperature of the mass spectrometer gas inlet was programmed to 120 °C. In order to compensate for the dead time (the time elapsed from the release of any volatile until it reached the ion source of the MS) between the thermal analyzer and the mass spectrometer at a given gas flow rate, both instruments were electronically synchronized. The volume of gas entering the mass spectrometer is 1–2 standard cubic centimeters per minute. EGA measurements were done in SEM bar graph cycle acquisition mode, where the m/z interval of 10–170 was continuously scanned with a speed of 20 ms amu−1. The spectrometer was operated in electron impact mode.2.7 Infrared measurements
IR spectra were collected by means of a Varian 2000 (Scimitar Series) FT-IR spectrometer (Varian Inc., US) equipped with a broad-band mercury–cadmium–telluride (MCT) detector and with a ‘Golden Gate’ single reflection diamond attenuated total reflection (ATR) unit (Specac Ltd, UK). The ATR technique allows the measurement of the samples without any sample preparation; for comparability, all samples were pressed with a constant pressure of 70 cNm at the diamond surface with a sapphire anvil. For all spectra, 64 scans at a nominal spectral resolution of 4 cm−1 were applied. For evaluation, the GRAMS/AI software was used.3 Results and discussion
3.1 Characterisation of clopamide anhydrate LH (1) and clopamide hemihydrate LH·0.5H2O (2) crystals
Screening various crystallization conditions (polar and apolar solvents, temperature etc.), the clopamide (LH) molecule was found to form two different types of crystal structures: a solvent-free (1) and a hemihydrate form (2). Selected crystal data and parameters of structure refinement of crystals 1 and 2 are collected in Table 2, while more details can be found in Table S1.† Single crystals of 1 of acceptable quality suitable for SXRD measurements were obtained during a complex formation study. Although the colour of the brown copper complex could be observed in the methanol solution at room temperature, cooling to 5 °C and evaporation of the solvent resulted first in colourless needle-shaped crystals having the lowest solubility and identified as the crystals of clopamide anhydrate (1), then the blue CuSO4·5H2O crystallized. The colour changes of the solution during the crystallization processes can be followed in Fig. S1.† All other attempts, although they were numerous, to produce single crystals of 1 failed. 1 crystallizes in the monoclinic P21/n space group with two clopamide molecules in the asymmetric unit (Fig. 1a). The hemihydrate crystals of 2 were obtained from methanol, although clopamide dissolves poorly in methanol, even if it is heated. However, in the presence of some water the solubility increases. Clopamide hemihydrate crystallizes in the monoclinic C2/c space group. The asymmetric unit contains one clopamide molecule and one disordered water molecule (with site occupancy of 0.5, Fig. 1b). The void size is large enough (61.75 Å3) to allow two positions of the water molecule to be hydrogen bonded to two different clopamide molecules. The methyl groups of the 2,6-dimethylpiperidine moiety have a larger space available for motions due to the presence of solvent cavities in 2; this results in more elongated ellipsoids, shown on the ORTEP diagram in Fig. 1b. This hemihydrate form occurred during the different crystallization experiments using different non-absolute organic solvents such as methanol, ethanol, 1-propanol and acetone. Clopamide is poorly soluble in water, but it can be dissolved by the addition of some base (e.g., NaOH, diethylamine) and then it crystallizes as a hemihydrate. The hemihydrate form is present in medicaments, for example in Brinaldix tablets. The X-ray powder diffractogram of the clopamide sample used in the crystallization experiments agrees well with the calculated pattern based on the clopamide hemihydrate structure 2 (Fig. S2 and S3†).| (1) | (2) | |
|---|---|---|
| Formula | LH | LH·0.5H2O |
| Shape/colour | Needle/colourless | Prism/colourless |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | C2/c |
| a (Å) | 18.7914(4) | 14.5166(8) |
| b (Å) | 9.2921(4) | 9.3747(7) |
| c (Å) | 19.0621(4) | 24.7064(17) |
| β (°) | 99.322(7) | 100.965(2) |
| V (Å3) | 3284.51(18) | 3300.9(4) |
| Z/Z′ | 8/2 | 4/1 |
| D x (Mg m−3) | 1.399 | 1.428 |
| Temperature (K) | 128(2) | 103(2) |
| Data/restraints/parameters | 5936/0/401 | 3762/0/210 |
| R int | 0.0740 | 0.0564 |
| Final R indices [I > 2σ(I)] | 0.0998, 0.2229 | 0.0440, 0.1076 |
| R indices (all data) | 0.1199, 0.2315 | 0.0510, 0.1122 |
| Max. and mean shift/esd | 0.000; 0.000 | 0.000; 0.000 |
| Δρmax, Δρmin (e Å−3) | 0.617, −0.494 | 0.376, −0.412 |
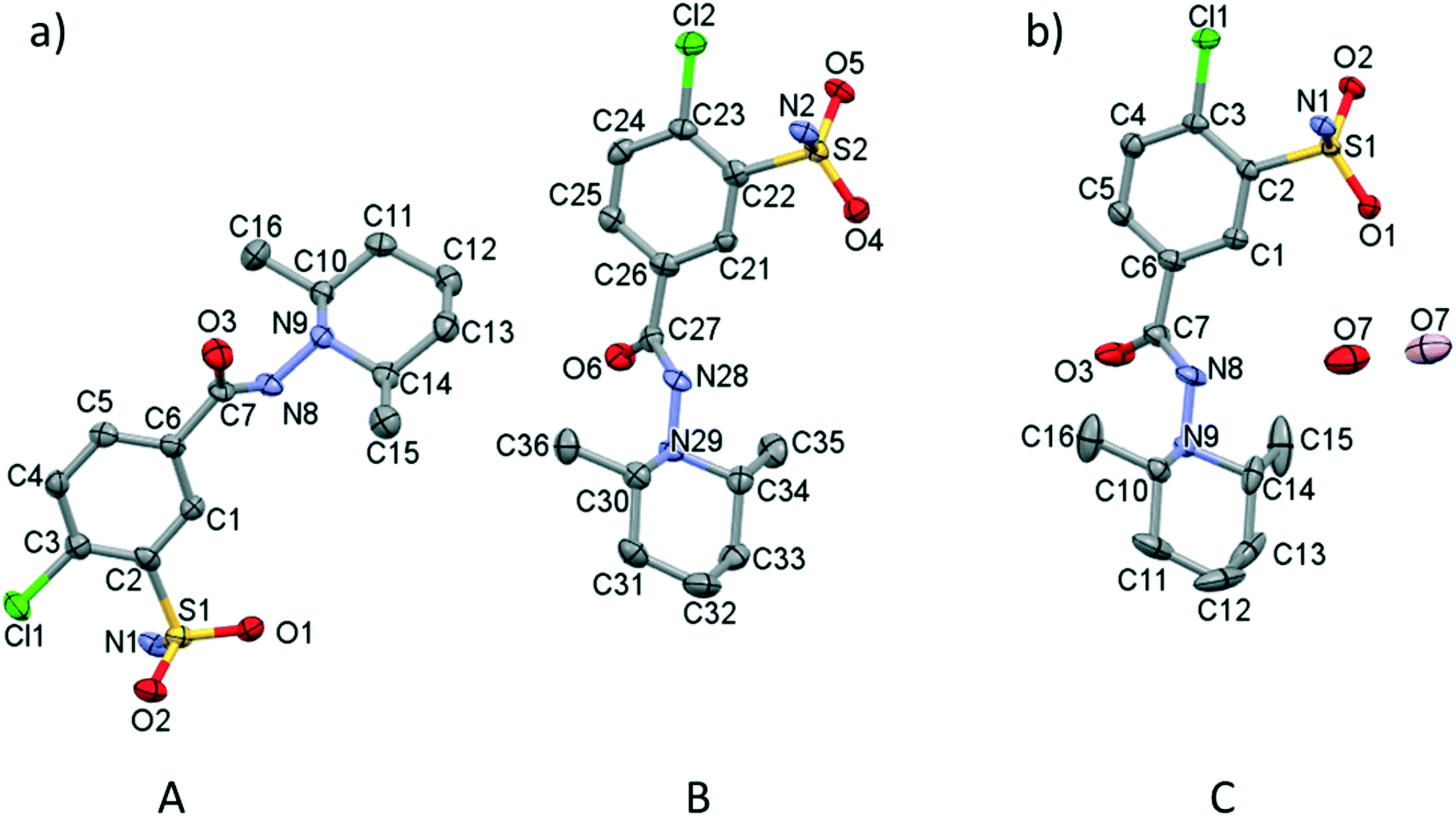 | ||
| Fig. 1 (a) ORTEP presentation of the asymmetric unit of crystal 1 (A and B molecules) and (b) 2 (C molecule) with atom labelling. The asymmetric unit of crystal 2 contains one water molecule (O7) in two disordered positions with 0.5 site occupancy (coloured light and dark red). Displacement ellipsoids are drawn at 50% probability level. Hydrogen atoms are omitted for clarity. | ||
In order to compare the two different conformations of molecules A and B in crystal 1 with that of molecule C determined in crystal 2, conformational analysis was performed. The overlaid molecules are shown in Fig. 2. The root-mean-square-deviation (rmsd) and maximum atomic distances (maxD) were calculated using Mercury and found to be 0.0728 Å and 0.1542 Å for A and B, 0.1137 Å and 0.2450 Å for A and C and 0.0936 Å and 0.2699 Å for B and C. Based on the calculated values the best fit was found between molecules A and B, the two molecules in the asymmetric unit of 1. The clopamide molecule C shows a slightly larger difference from both A and B molecules but still the conformation is very similar taking into account that the molecule contains three single bonds, N9–N8, N8–C7 and C6–C7, which allow high rotational freedom. A detailed comparison of the bond distances and angles can be found in Tables S2 and S3.†
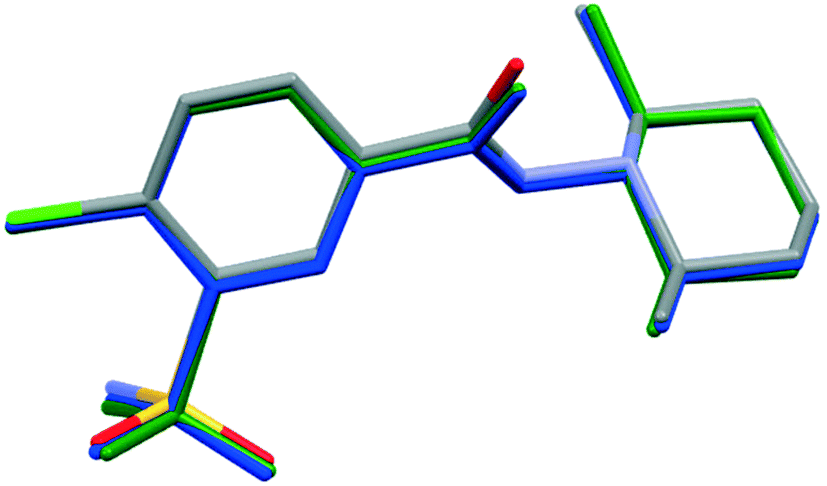 | ||
| Fig. 2 Overlaid structures of clopamide molecules A (blue) and B (green) from crystal 1 and molecule C (coloured by elements) from crystal 2. | ||
The very similar systems of secondary interaction formed among the molecules in the crystal lattices result in the very similar molecular conformations in 1 and 2. The sulfonamide group on the clopamide molecule contains two ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and an –NH2 group; consequently these molecules are able to form dimers via N–H⋯O type interactions. In both 1 and 2 crystal structures, isostructural planes of molecules are formed from these dimers by additional N–H⋯N, N–H⋯O and C–H⋯π interactions. Fig. 3a shows that the planes composed of molecules A (blue) and molecules B (green) are alternating in 1. There are no hydrogen bonds between layer A and layer B. The distance between the layers formed by molecule A is 14.503(4) Å in crystal 1, while the analogous distance is somewhat larger, 14.517(8) Å in crystal 2 (Fig. 3b). The water molecules in structure 2 act as spacers keeping the layers more distant than the layers are in structure 1. Thus a more favourable molecular conformation and higher symmetry can be realised in 2. The pseudo twofold axis in the P21/n, Z′ = 2 (1) structure becomes a real symmetry in 2, and the space group of C2/c is realised with Z′ = 1 (2), as C2/c is the minimal non-isomorphic supergroup of P21/n with a difference of a twofold axis.
O and an –NH2 group; consequently these molecules are able to form dimers via N–H⋯O type interactions. In both 1 and 2 crystal structures, isostructural planes of molecules are formed from these dimers by additional N–H⋯N, N–H⋯O and C–H⋯π interactions. Fig. 3a shows that the planes composed of molecules A (blue) and molecules B (green) are alternating in 1. There are no hydrogen bonds between layer A and layer B. The distance between the layers formed by molecule A is 14.503(4) Å in crystal 1, while the analogous distance is somewhat larger, 14.517(8) Å in crystal 2 (Fig. 3b). The water molecules in structure 2 act as spacers keeping the layers more distant than the layers are in structure 1. Thus a more favourable molecular conformation and higher symmetry can be realised in 2. The pseudo twofold axis in the P21/n, Z′ = 2 (1) structure becomes a real symmetry in 2, and the space group of C2/c is realised with Z′ = 1 (2), as C2/c is the minimal non-isomorphic supergroup of P21/n with a difference of a twofold axis.
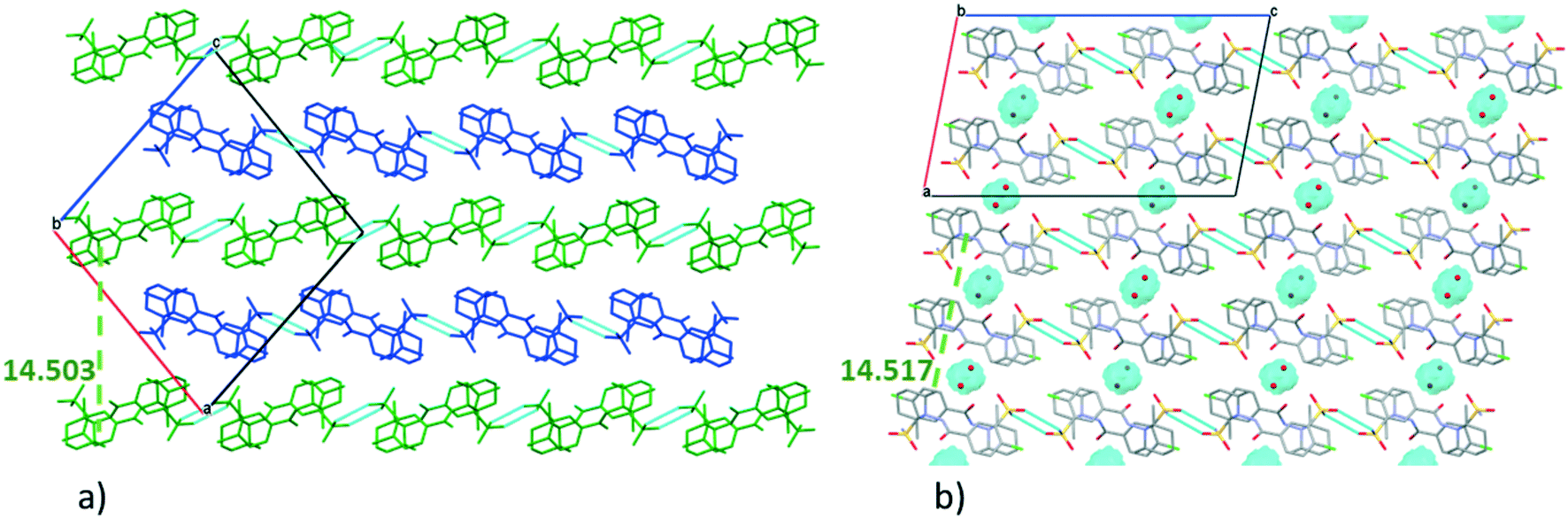 | ||
| Fig. 3 Packing arrangement of molecules in the crystal lattice of 1 (a) and 2 (b) viewed from the crystallographic b axis. Molecules are coloured A (blue) and B (green) in crystal 1. The cavities containing the water molecules are highlighted in blue in crystal 2. | ||
An intramolecular halogen bond can be observed between the chlorine and the oxygen of the chloro-sulfanoylbenzamide moiety (3.148(6) Å 1A, 3.096(4) Å 1B), while an intermolecular chalcogen bond is present between a sulfoxy oxygen as donor and the lone pair of the carbonyl group as acceptor ((3.036(7) Å 1A, 3.233(7) Å 1B)), showing a competition between the two types of supramolecular interactions in the two layers of the two crystallographically independent molecules in 1. Although the chlorine and the sulfonamide oxygen come closer (3.0581(16) Å C) in 2, there is no chance to form a chalcogen bond because of the presence of the water molecule, which forms a bridge between the neighbouring molecules. In the absence of the chalcogen bond in 2, the intramolecular halogen bond becomes the shortest here.
The distances and angles of selected interactions in crystals 1 and 2 are collected in Table S4.† Comparisons of the unit cells from different crystallographic views and the packing arrangements are shown in Fig. S4–S6.†
3.2 Characterization of solvatomorph [CuL2]·2(MeOH)·2(H2O) (3), [CuL2]·2(EtOH)·2(H2O) (4), [CuL2]·2(nPrOH)·2(H2O) (5) and [CuL2]·2(iPrOH)·2(H2O) (6) complex crystals
The outcome of crystallization experiments performed to obtain copper(II) complexes of clopamide is summarized in Scheme 2. At pH ∼9 the hydrazine N8 nitrogen deprotonates along with the increase in electron density on the N9 atom, and as a result the ligand is able to coordinate to the metal ion bidentately via N9 and the neighbouring carbonyl O3 atom. The bis-ligand copper(II) complexes [CuL2] could be obtained from different water-miscible and -immiscible solvents, producing solvatomorphic (3–6 and 10) and solvent-free polymorphic (7–9) crystals (Scheme 2).Solvatomorphic crystals of 3–6 were obtained by using a homologous series of alcohols: methanol, ethanol, 1-propanol and 2-propanol. All four solvatomorphs crystallize in the P space group with similar unit cell parameters and gradually increasing unit cell volume. Selected crystal data and parameters of structure refinement of crystals 3–6 are collected in Table 3, while more details can be found in Table S5.† All four crystals contain half of the [CuL2] complex, one alcohol and one water molecule in their asymmetric unit (Fig. 4). The copper(II) ion is coordinated by the N9 and O3 donor atoms of the ligand, forming a five-membered chelate ring in square planar geometry (Table S6†). The two ligands coordinate symmetrically in trans positions as the copper ion is located on an inversion centre. The complex formation reduces the strength of the intramolecular halogen bond, and the Cl⋯O distance increases to 3.208(2) Å in 3, 3.233(4) Å in 4, 3.189(5) Å in 5, and 3.204(3) Å in 6. The ORTEP diagrams show that the atomic displacement parameters presented by ellipsoids are growing from 3 to 6 with the increasing size of the solvent void, reflecting that the degree of freedom becomes higher especially for the piperidine rings of the hosts. In crystal 6 the piperidine ring could be split into two distinguishable disordered positions (occupancy: 2/3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1/3) (Fig. 4).
:1/3) (Fig. 4).
| 3 | 4 | 5 | 6 | |
|---|---|---|---|---|
| Moiety formula | [CuL2]·2MeOH·2H2O | [CuL2]·EtOH·2H2O | [CuL2]·2PrOH·2H2O | [CuL2]·2iPrOH·2H2O |
| Shape/colour | Prism/brown | Prism/brown | Prism/brown | Prism/brown |
| M r | 853.32 | 881.37 | 909.42 | 909.42 |
| Crystal system | Triclinic | Triclinic | Triclinic | Triclinic |
| Space group |
P |
P |
P |
P |
| Z, Z′ | 1, 0.5 | 1, 0.5 | 1, 0.5 | 1, 0.5 |
| a (Å) | 7.7518(3) | 8.2195(7) | 8.404(4) | 8.3107(14) |
| b (Å) | 8.0901(4) | 8.2439(7) | 8.497(5) | 8.7469(14) |
| c (Å) | 16.3683(7) | 16.4822(14) | 16.310(8) | 16.387(3) |
| α (°) | 83.1130(10) | 85.598(2) | 79.445(10) | 79.713(5) |
| β (°) | 78.1130(10) | 78.212(2) | 83.113(12) | 87.099(4) |
| γ (°) | 71.6590(10) | 68.273(2) | 67.366(10) | 65.629(4) |
| V (Å3) | 953.96(7) | 1015.62(15) | 1055.2(10) | 1067.3(3) |
| D x (Mg m−3) | 1.485 | 1.441 | 1.431 | 1.415 |
| Temperature (K) | 103(2) | 103(2) | 103(2) | 103(2) |
| Data/restraints/parameters | 6813/0/236 | 4634/0/244 | 3846/0/253 | 5523/0/368 |
| R int | 0.0600 | 0.0918 | 0.1536 | 0.0884 |
| Final R indices [I > 2σ(I)] R1, wR2 | 0.0472, 0.1021 | 0.0972, 0.2386 | 0.0849, 0.2092 | 0.0690, 0.1530 |
| R indices (all data), R1, wR2 | 0.0684, 0.1284 | 0.1179, 0.2561 | 0.1143, 0.2328 | 0.0816, 0.1599 |
| Max. and mean shift/esd | 0.000; 0.000 | 0.000; 0.000 | 0.000; 0.000 | 0.000; 0.000 |
| Δρmax, Δρmin (e Å−3) | 0.832, −0.888 | 1.573, −1.098 | 0.852, −0.963 | 0.644, −0.708 |
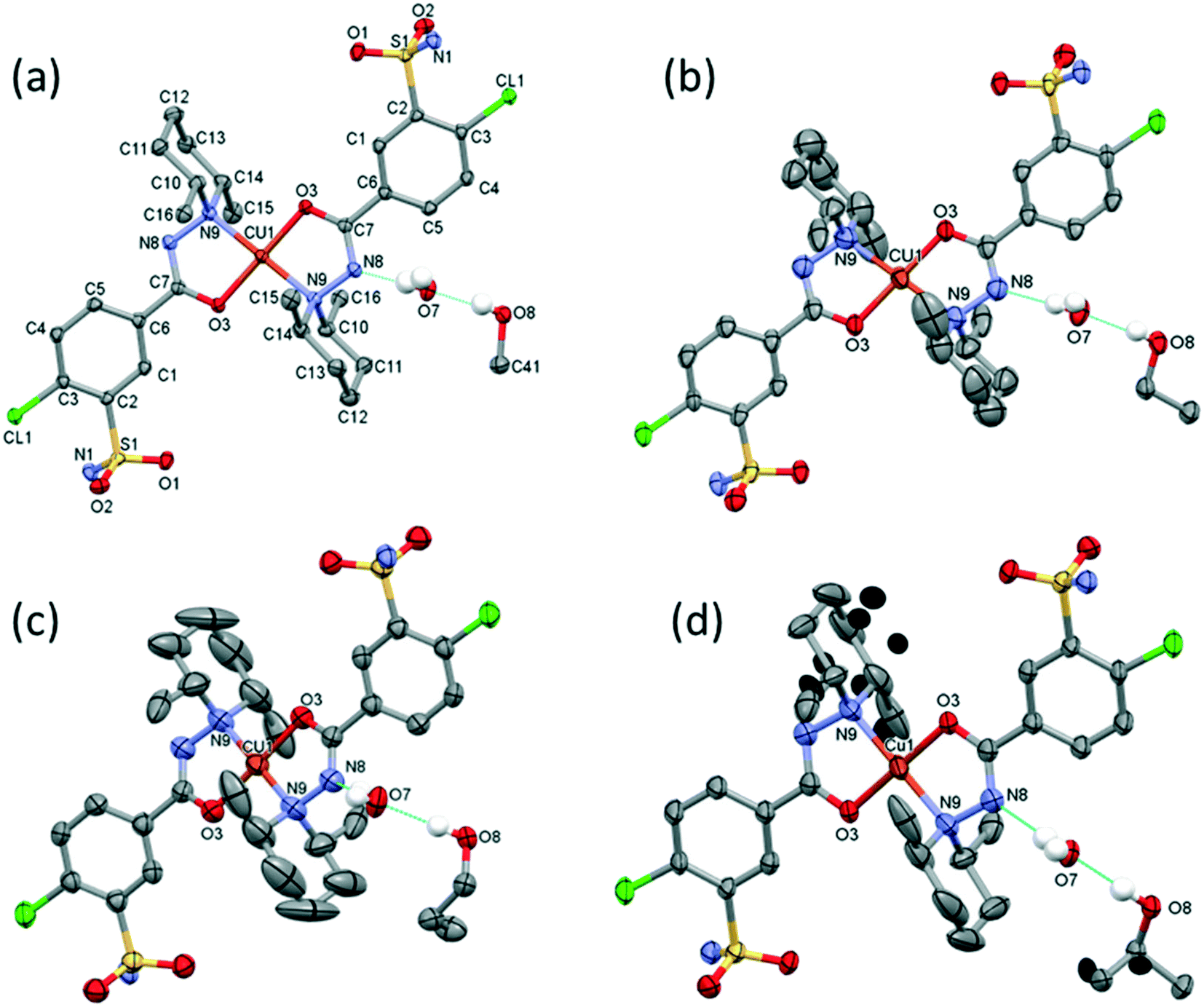 | ||
| Fig. 4 Molecular structure of the homologous series of solvatomorphic complexes of 3 (a), 4 (b), 5 (c) and 6 (d). Hydrogen atoms involved in H bonds are shown; all other ones are omitted for clarity. Atomic displacement parameters are presented at 50% probability level. One ligand is in the asymmetrical unit, the copper(II) ion is on the symmetry centre, and the other parts of the complexes are generated by inversion symmetry. Solvent molecules are shown on one side only for clarity. Labels of atoms are fully listed for crystal 3; the same was used for 4–6. The increasing displacements of the piperidines' carbon atoms can be observed with the increasing size of the solvent molecule. The disordered atoms of 4 are marked with black ellipsoids in crystal 6. | ||
The [CuL2] complexes connected by alcohol and water molecules simultaneously in similar positions resulted in isostructural crystal structures, all in the P space group. The cell similarity indexes (I = |(a + b + c)/(a′ + b′ + c′) − 1|) were found to be between 0.01 and 0.04 for the pairs of crystals (see Table S7†), which proves their very high similarity. The conformations of the molecules together with the similar solvent positions are compared in Fig. S7.† The comparison of packing arrangements is shown in Fig. 5 and S8.† The copper–copper distances between neighbouring complexes increase in the case of methanol 7.752(1) Å, ethanol 8.220(1) Å, 1-propanol 8.497(5) Å and 2-propanol clathrates to 8.747(1) Å. The complex molecules form an isostructural three-dimensional network connected by the solvent molecules in all structures from 3 to 6.
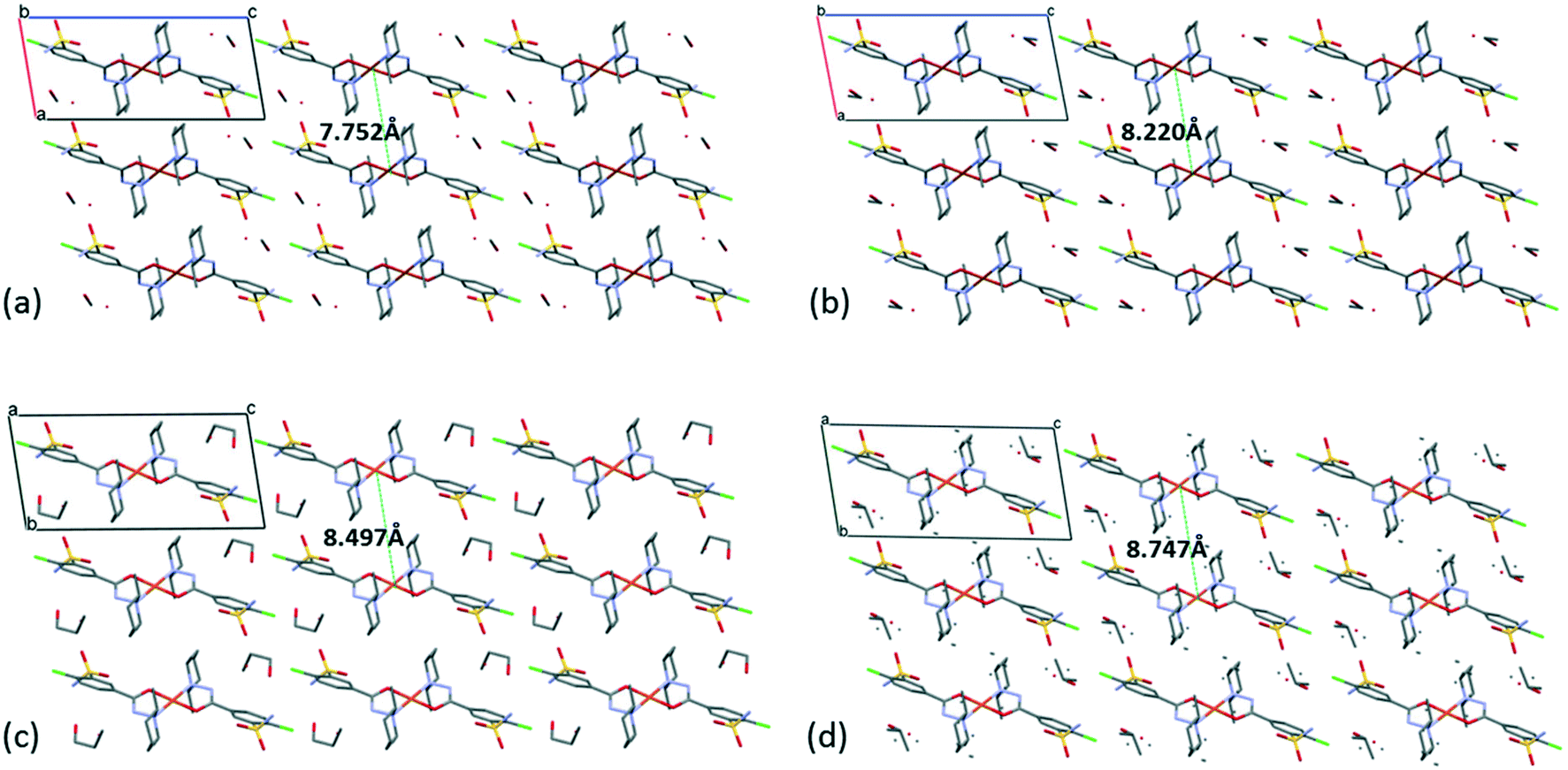 | ||
| Fig. 5 Packing arrangements in crystal 3 (a), 4 (b), 5 (c) and 6 (d) viewed from the crystallographic b axis, presenting also the increasing distances of the metal Cu(II) centres with the increasing size of the solvent guest molecules. | ||
The solvent molecules stabilize the crystal lattice similarly in all methanol (3), ethanol (4), 1-propanol (5), and 2-propanol (6) containing crystals, forming secondary interactions on both sides of the complex molecules: the alcohol oxygen atom O8 is an acceptor of adjacent sulfonamide NH2 protons with N1–H1NA⋯O8 and N1–H1NB⋯O8, and the alcohol proton connects to the neighbouring water oxygen atoms with O8–H1O8⋯O7. A water proton forms a H bond with the deprotonated ligand nitrogen O7–H2O7⋯N8, while the other one connects to sulfonamide oxygen O7–H1O7⋯O2 (Fig. 6). The details of H-bond interactions are collected in Table S8.† The distances between N8⋯O7 and O7⋯O8 atoms show no significant increase with the increasing size of the solvent molecules or with the increasing size of the available void volume (Table 4). As the volume of the unit cell and the volume of the void containing the solvent molecules increase in the order methanol, ethanol, 1-propanol and 2-propanol solvates from 3 to 6, the density of the crystals decreases, having an inverse relationship between the cell volume and the crystal density (Table 4 and Fig. 7).
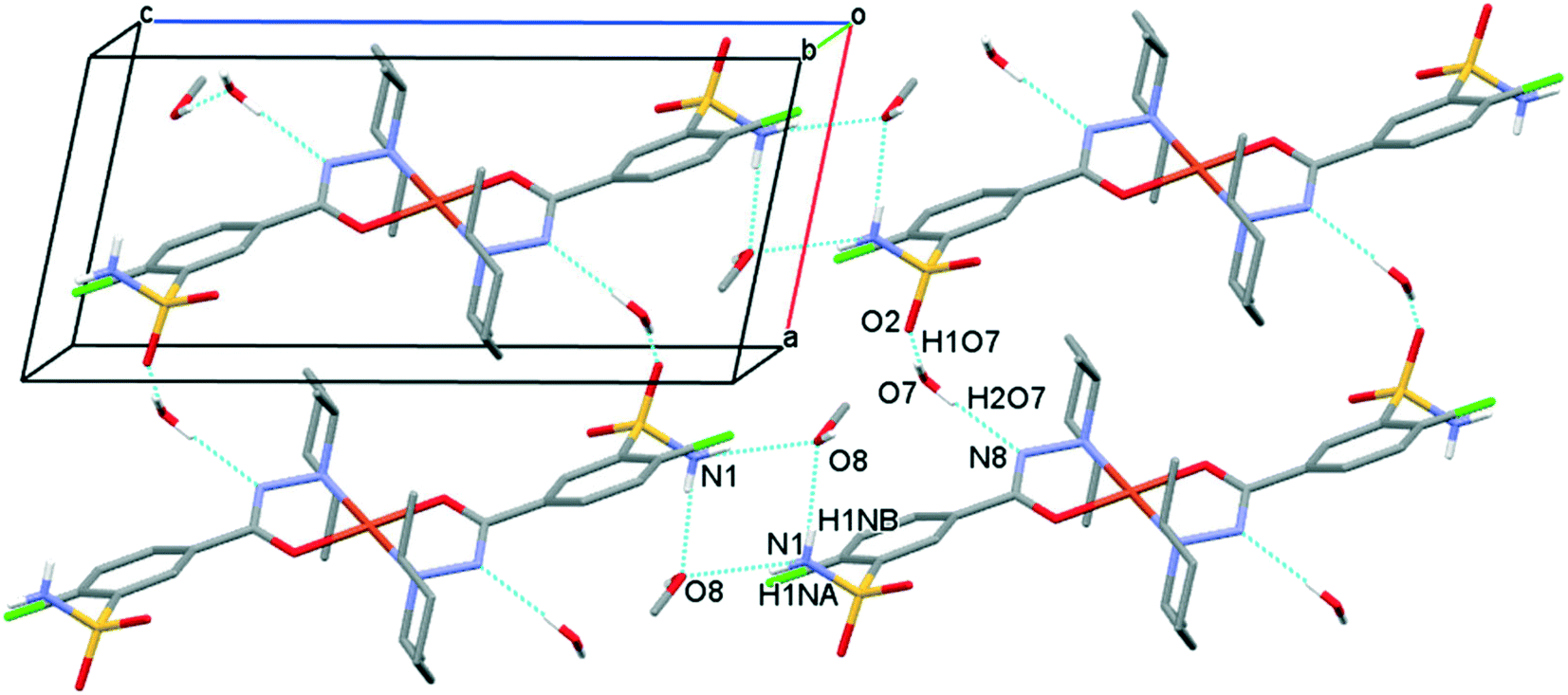 | ||
| Fig. 6 Packing arrangements in crystal 3 showing the most important H-bond interactions. Similar synthons can be found in crystals 4, 5 and 6. Hydrogen atoms not involved in the H-bond system are omitted for clarity. | ||
| Crystal number | V cell (Å3) | V void (Å3) | V void/Vcell (%) | Density (g cm−3) | d (N8⋯O7) (Å) | d (O7⋯O8) (Å) |
|---|---|---|---|---|---|---|
| 3 | 953.96(7) | 117.56 | 12.3 | 1.485 | 2.845(3) | 2.643(2) |
| 4 | 1015.62(15) | 158.00 | 15.6 | 1.441 | 2.857(7) | 2.673(6) |
| 5 | 1055.2(10) | 202.08 | 19.2 | 1.431 | 2.846(6) | 2.626(6) |
| 6 | 1067.3(3) | 193.73 | 18.2 | 1.415 | 2.882(4) | 2.707(4) |
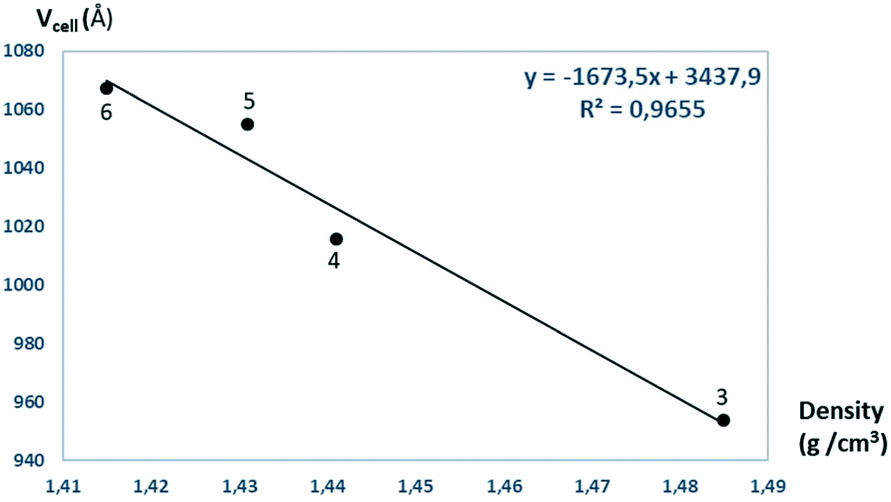 | ||
| Fig. 7 The unit cell volumes are plotted as a function of the calculated densities obtained for crystals 3–6. | ||
Crystallization experiments have been performed with longer chain homologous alcohols but crystals could not be obtained from n-butanol and longer chain alkylalcohols (n-pentane etc.), which shows that the above presented crystal lattice can no longer accommodate larger size molecules. As the length of the n-alcohol increases in its homologous order, the molecular size increases, and the strength of the intermolecular interaction to the bridging water decreases, and thus the isostructural crystal series of the Cu–clopamide complex could not be completed with more members.
3.3 Characterization of the polymorphic solvent-free [CuL2] (7–9) and [CuL2]·CH2Cl2 (10) complex crystals
Solvent-free [CuL2] crystals (Fig. 8a–c) were obtained from methanol solution using CuSO4 instead of CuCl2 salt. Use of CuSO4 in the complexation reaction with clopamide resulted in the brown colour of the methanol solution, unlike when using CuCl2, where the colour of the solution was blue. Two polymorphic solvent-free [CuL2] crystals were obtained from methanol solution: crystal 7 was harvested from a solution containing a slight ligand excess (10:9) and crystal 8 was harvested from a solution with a strong ligand excess (7:2), though the composition of the obtained complexes is the same. From water-immiscible organic solvents (e.g. ethyl acetate) another solvent-free polymorph, 9, was crystallized. Crystals 7 and 8 were found to crystallize in the triclinic crystal system in the P space group, while crystal 9 crystallizes in the trigonal crystal system R space group.
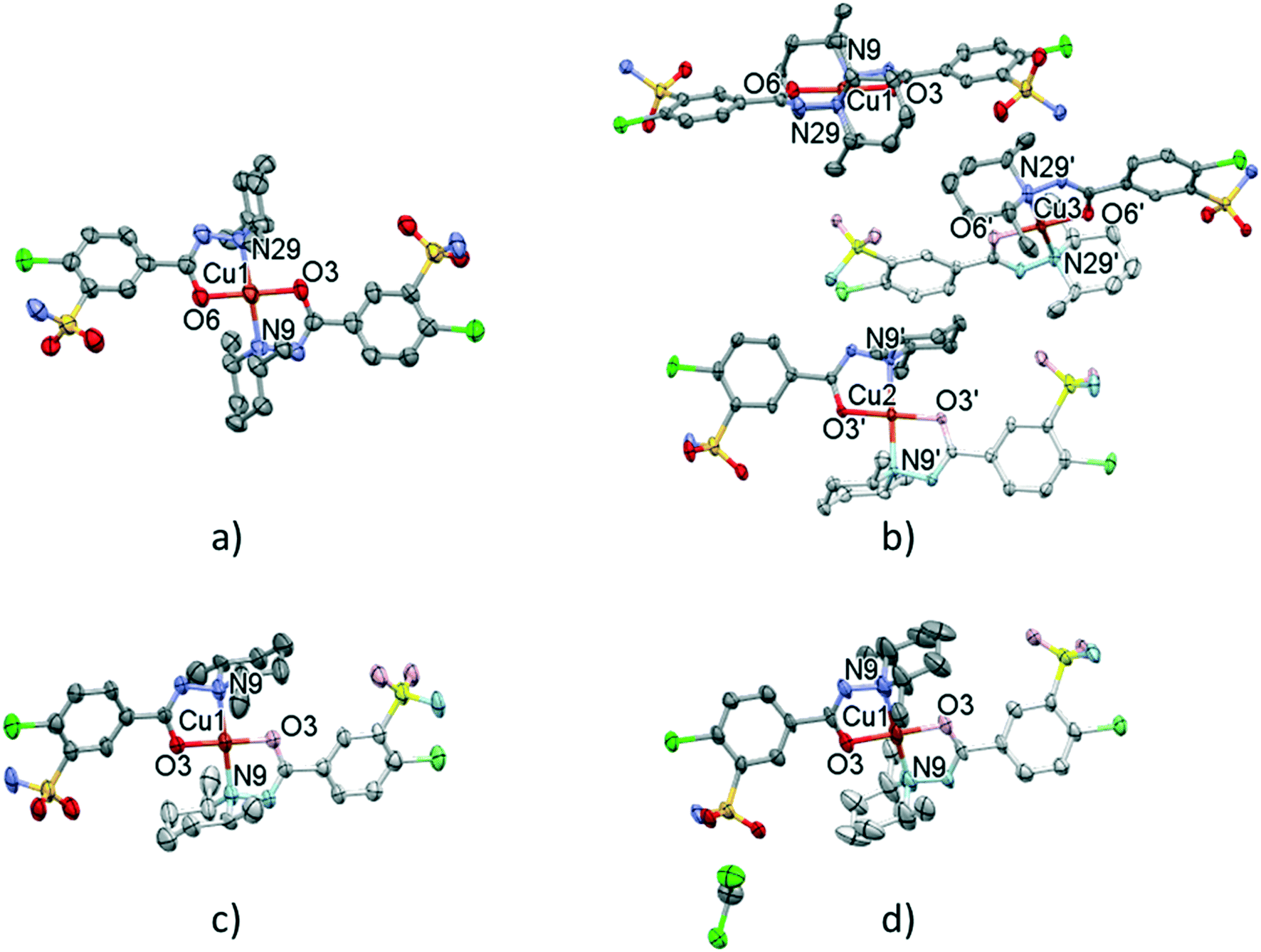 | ||
| Fig. 8 Molecular structure of the solvent-free polymorphic complexes of [CuL2] 7 (a), 8 (b), and 9 (c). Molecular structure of the solvate crystal [CuL2]·CH2Cl210 (d). Hydrogen atoms are omitted for clarity, atomic displacement parameters are presented at 50% probability level. The light-coloured atoms of the molecules are generated by inversion symmetry (full labelling can be found in Fig. S10 and S11†). | ||
Nevertheless, from dichloromethane solution crystal 10 (Fig. 8d) was obtained (besides 9) with the inclusion of a CH2Cl2 guest molecule in a 1:1 complex-to-solvent ratio. 10 crystallizes in the monoclinic crystal system in the C2/c space group. Selected crystallographic parameters are collected in Table 5, while detailed crystallographic data are given in Table S9.† Packing arrangements of crystals 7–10 are shown in Fig. S9.† In all cases a square planar coordination geometry with trans orientation of the two clopamide molecules was found, similarly to crystals 3–6; however, significant differences can be seen in the conformation of the piperidine ring and the packing arrangement of the complexes in the crystals.
| 7 | 8 | 9 | 10 | |
|---|---|---|---|---|
| Moiety formula | [CuL2] | [CuL2] | [CuL2] | [CuL2]·CH2Cl2 |
| Shape/colour | Chunk/brown | Chunk/brown | Chunk/brown | Chunk/prism |
| M r | 753.20 | 753.20 | 753.20 | 923.05 |
| Crystal system | Triclinic | Triclinic | Trigonal | Monoclinic |
| Space group |
P |
P |
R |
C2/c |
| a (Å) | 7.8247(14) | 13.4600(10) | 23.078(3) | 26.699(2) |
| b (Å) | 13.829(2) | 15.2424(11) | 23.078(3) | 9.8941(8) |
| c (Å) | 15.156(3) | 18.1855(16) | 18.3291(12) | 15.6876(14) |
| α (°) | 78.981(4) | 114.627(6) | 90 | 90 |
| β (°) | 81.309(4) | 93.362(8) | 90 | 104.750(7) |
| γ (°) | 85.041(4) | 90.021(6) | 120 | 90 |
| V (Å3) | 1588.5(5) | 3384.5(5) | 8454(2) | 4007.5(6) |
| Z, Z′ | 2, 1 | 4, 1 + 0.5 + 0.5 | 9, 0.5 | 4, 0.5 |
| D x (Mg m−3) | 1.575 | 1.478 | 1.331 | 1.530 |
| Temperature (K) | 103(2) | 123(2) | 153(2) | 128(2) |
| Data/restraints/parameters | 5798/0/410 | 12256/0/822 |
3429/0/207 | 3661/0/234 |
| R int | 0.1852 | 0.1301 | 0.1176 | 0.0901 |
| Final R indices [I > 2σ(I)], R1, wR2 | 0.1495, 0.3073 | 0.0777, 0.1198 | 0.1033, 0.1672 | 0.1231, 0.2351 |
| R indices (all data), R1, wR2 | 0.2415, 0.3583 | 0.1375, 0.1366 | 0.2142, 0.2052 | 0.1623, 0.2539 |
| Max. and mean shift/esd | 0.000; 0.000 | 0.001; 0.000 | 0.000; 0.000 | 0.000; 0.000 |
| Δρmax, Δρmin (e Å−3) | 1.205, −0.647 | 0.564, −0.576 | 0.553, −0.264 | 0.887, −0.619 |
The main conformational differences of the complexes in their different crystals 6–10 are shown in Fig. 9. The root-mean-square deviation (rmsd) and maximum atomic distance (maxD) values were calculated using Mercury and are compared in Tables S10 and S11.† The coordination spheres of the central Cu(II) cations are highly similar in all crystals; however, the conformation of the ligands may differ mainly because of two reasons: (1) the six-membered piperidine ring in chair conformation can be placed in two different ways, it can be parallel or perpendicular to the Cu–N9 bond and (2) the phenyl ring may be at different angles to the coordination plane defined by atoms Cu1, O3, C7, N8, and N9, according to different secondary interactions of the sulfonamide group in the different crystal lattices. These two characteristic values are compared in Table 6.
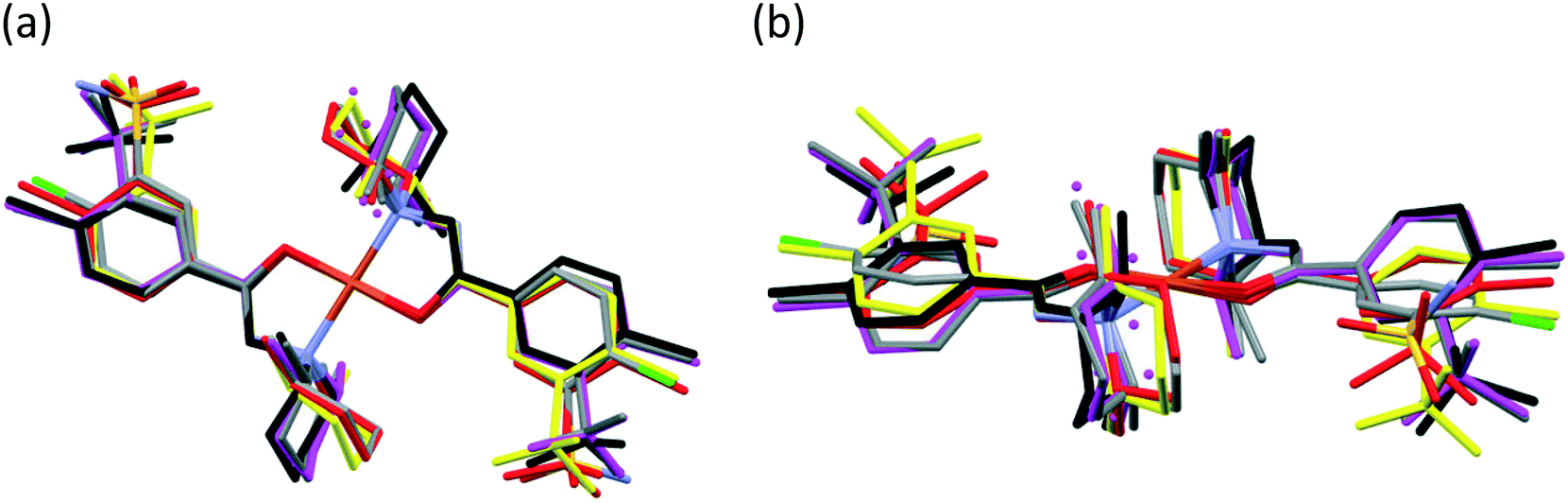 | ||
| Fig. 9 Top view (a) and side view (b) of selected [CuL2] complexes overlaid by the coordinating atoms of clopamide and the central metal ion. Colour code: 6 (magenta), 7 (black), molecule A in 8 (red), 9 (coloured by element) and 10 (yellow). Solvents and hydrogen atoms are omitted for clarity. | ||
| Crystal number | Angle between plane1 and plane2a (°) | Piperidine ring placement to Cu–N9b |
|---|---|---|
| a Plane1 is the coordination plane defined by atoms Cu1, O3, C7, N8, and N9 atoms. Plane2 is the plane of the phenyl ring. b The piperidine ring in chair conformation can be placed parallel or perpendicular to the Cu–N9 bond. | ||
| 3 | 31.27(10) | Parallel |
| 4 | 34.9(3) | Parallel |
| 5 | 34.6(3) | Parallel |
| 6 | 35.60 (18) | Parallel in major disordered positions |
| Perpendicular in minor disordered positions | ||
| 7 | 21.9(6) and 37.2 (6) | Parallel |
| 8A | 13.3(3) and 9.6 (3) | Perpendicular |
| 8B | 15.5(2) | Perpendicular |
| 8C | 3.8(3) | Perpendicular |
| 9 | 13.7(4) | Perpendicular |
| 10 | 23.9(4) | Perpendicular |
The conformations of the complexes in 3–6 are very similar; the two ligands coordinate symmetrically to the copper ion owing to an inversion centre. The piperidine ring is parallel to the Cu–N9 binding axes and the angle between the coordination plane and the phenyl ring was found to be between 31.27(10)° and 35.60(18)°. Interestingly, in crystal 6 the disordered positions of the piperidine ring appeared in both parallel (occupancy 2/3) and perpendicular placement (occupancy 1/3) to the Cu–N9 axis. In the case of 7 (Fig. 8a) the inversion symmetry is missing and the conformation of the two ligands is different. The angles between the phenyl rings and the coordination plane are 21.9(6)° and 37.2(6)°, respectively, for the two ligands, and the piperidine rings are parallel to the Cu–N bond (Table 6). In crystal 8 with one asymmetrical [CuL2] and two different symmetrical [CuL2] complexes (1 + 0.5 + 0.5) in the asymmetric unit (Fig. 8b), in all three complexes the piperidine ring has been found perpendicular to the Cu–N bond, and the difference between them is primarily originating from the conformation of the phenyl ring, which predicts that their sulfonamide groups are likely to form different types of secondary interactions in this crystal (see also the overlaid structures in Fig. S12;† H-bond interactions are presented in Fig. S13†). There is half of the complex [CuL2] in the asymmetric unit in 9 and 10, and an inversion centre is found where the metal ion is present; thus the two ligands are coordinated symmetrically. Similar to 8 the piperidine ring conformation is perpendicular to the Cu–N bond and differences in the phenyl ring angles have been observed. Comparing the distances and angles of the coordination spheres (Table S6†), it is noticeable that Cu–O coordination bonds are significantly shorter, while Cu–N bonds are longer in the complexes in crystals 8, 9 and 10 compared to the solvatomorphic crystals 3–6 or 7 which coincides with the perpendicular (shorter Cu–O) or parallel (longer Cu–O) conformation of the piperidine ring. The perpendicular position of the piperidine ring is stabilized by C–H⋯O intramolecular interactions between C13–H13A⋯O3 and C11–H11B⋯O3 (Table S12†). Although there are S–N–H⋯OS intermolecular interactions formed around the inversion centres in 7, 8 and 9, like in the pure clopamide crystals of 1 and 2, the N–H⋯Cl intramolecular hydrogen bonds become more important in the complexes and no halogen intramolecular interactions are observed.
Fig. 10 presents the similar arrangement of the molecules in the crystal lattices of 3 and 7. While the sulfonamide groups connect two columns through methanol solvent molecules (with N–H⋯O and O–H⋯S bonds) in 3, in the solvent-free form 7 the complexes are directly connected to each other through N–H⋯O bonds. An asymmetrical intermolecular interaction between a sulfonamide NH2 and the deprotonated N8 hydrazine nitrogen atom was found among neighbouring complexes, which causes the asymmetric coordination of the two ligands; therefore there is the whole complex molecule in the asymmetric unit in 7. Details of the H-bond interactions are collected in Table S12.† In crystals 8 and 9 the intermolecular sulfonamide N–H⋯O bonds generate linear columns of the complexes and adjacent columns then turn in a way that the angles between the columns of complexes are 62.7° in the case of 8 and 60° in the case of 9 (Fig. 11).
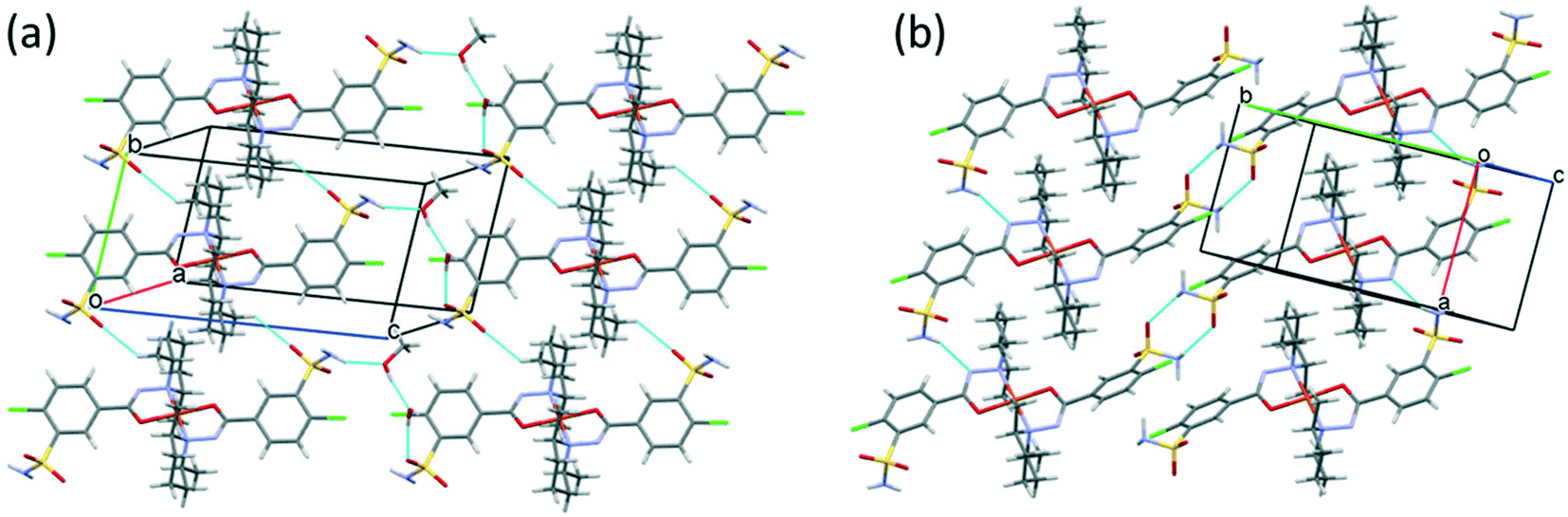 | ||
| Fig. 10 Analogous packing arrangements in [CuL2]·CH3OH·H2O crystal 3 (a) and in the solvent-free [CuL2] crystal 7 (b), showing the columns supported by the systems of secondary interactions. | ||
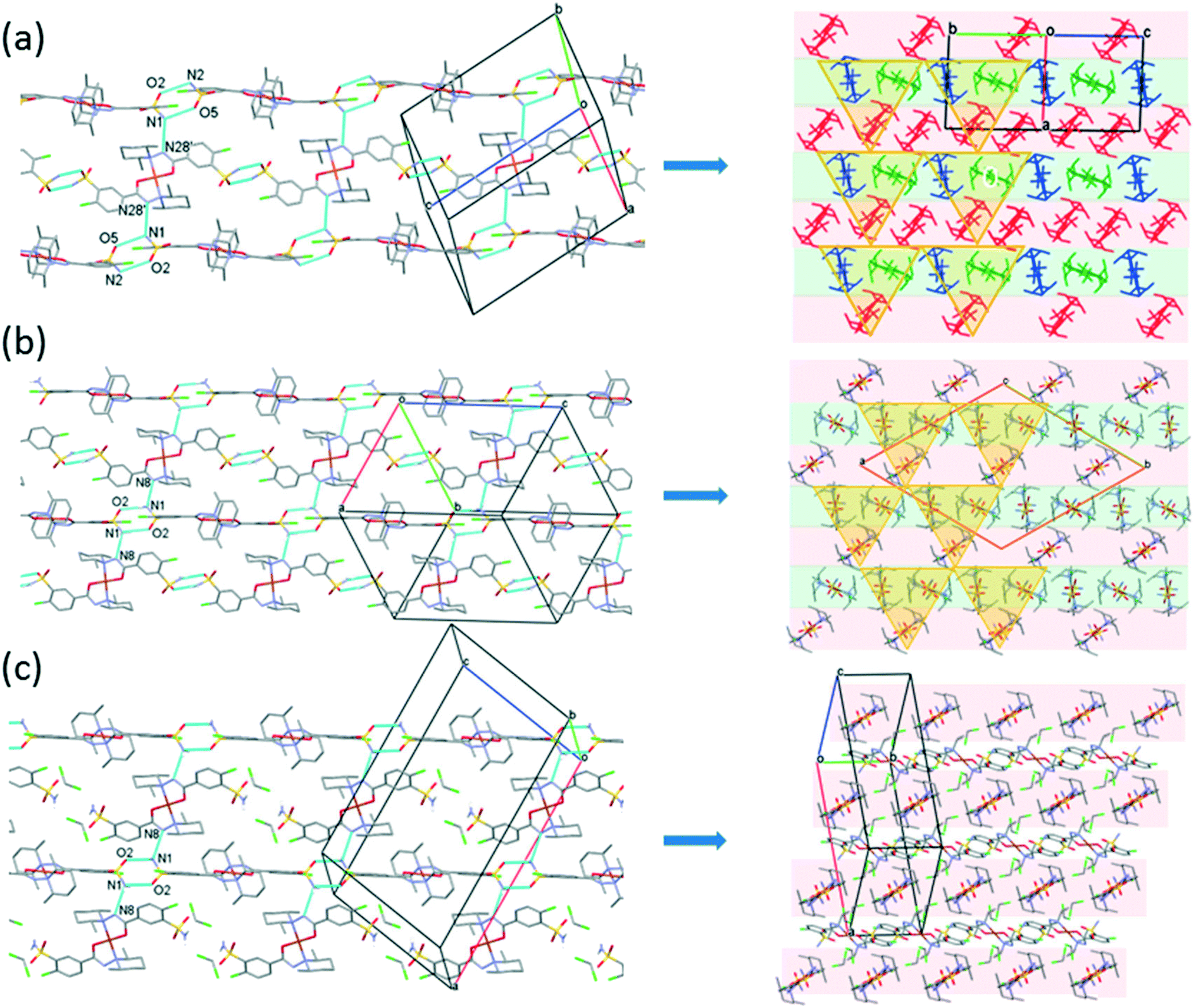 | ||
| Fig. 11 Packing arrangements in crystal 8 (a) (molecule A is red, B is green and C is blue), crystal 9 (b) and crystal 10 (c) showing the columns formed by N–H⋯O type hydrogen bonds viewed from the side and also from the top of the columns. The light pink and green colours show the different sheets; the yellow triangles highlight the triangular arrangements of the molecules in the different crystals. | ||
In crystal 8 the three different [CuL2] complexes from the asymmetric unit (coloured red, blue and green) are arranged in an approximate triangle (Fig. 11a). This becomes a perfect threefold symmetry in 9; there is one half complex molecule in the asymmetric unit in space group R (Fig. 11b). This molecular arrangement creates channels; the void size calculated by Mercury (probe radius 1.2 Å, approximate grid spacing 0.7 Å) is 1452.26 Å3 (16.8% of the unit cell volume). Solvent inclusion (residual electron density) could not be located during refinement. This type of complex crystal 9 crystallized from different water-immiscible organic solvents (e.g. ethyl acetate, 1-butanol, 1-pentanol, 1-hexanol, toluene, chloroform, etc.) and no solvent was found in either case in the channels. Small molecules like water would however fit into the channels by size, but the piperidine moieties create a hydrophobic surface of the channel. A similar Cu(II) complex, the structure (tetrakis(μ2-vanillato-O,O′)-bis(nicotinamide-N)-di-copper(II)), was found in the literature (Ref. code DOHWON) having even larger empty channels.28 TG–DSC–MSEGA coupled measurements have been performed on the crystals of 9 (Fig. 12), and around 3% solvent mixture (ethyl acetate and 1-butanol) loss was observed up to 224 °C without the collapse of the lattice. No structural change was observed by SXRD in the experiment when a single crystal of 9 was heated at 200 °C for an hour. The sample melts at 240.4 °C. Comparing the calculated crystal density of the three polymorphs it decreases in the order 7 (1.575 g cm−3), 8 (1.478 g cm−3) and 9 (1.331 g cm−3), with 9 being the lowest one.
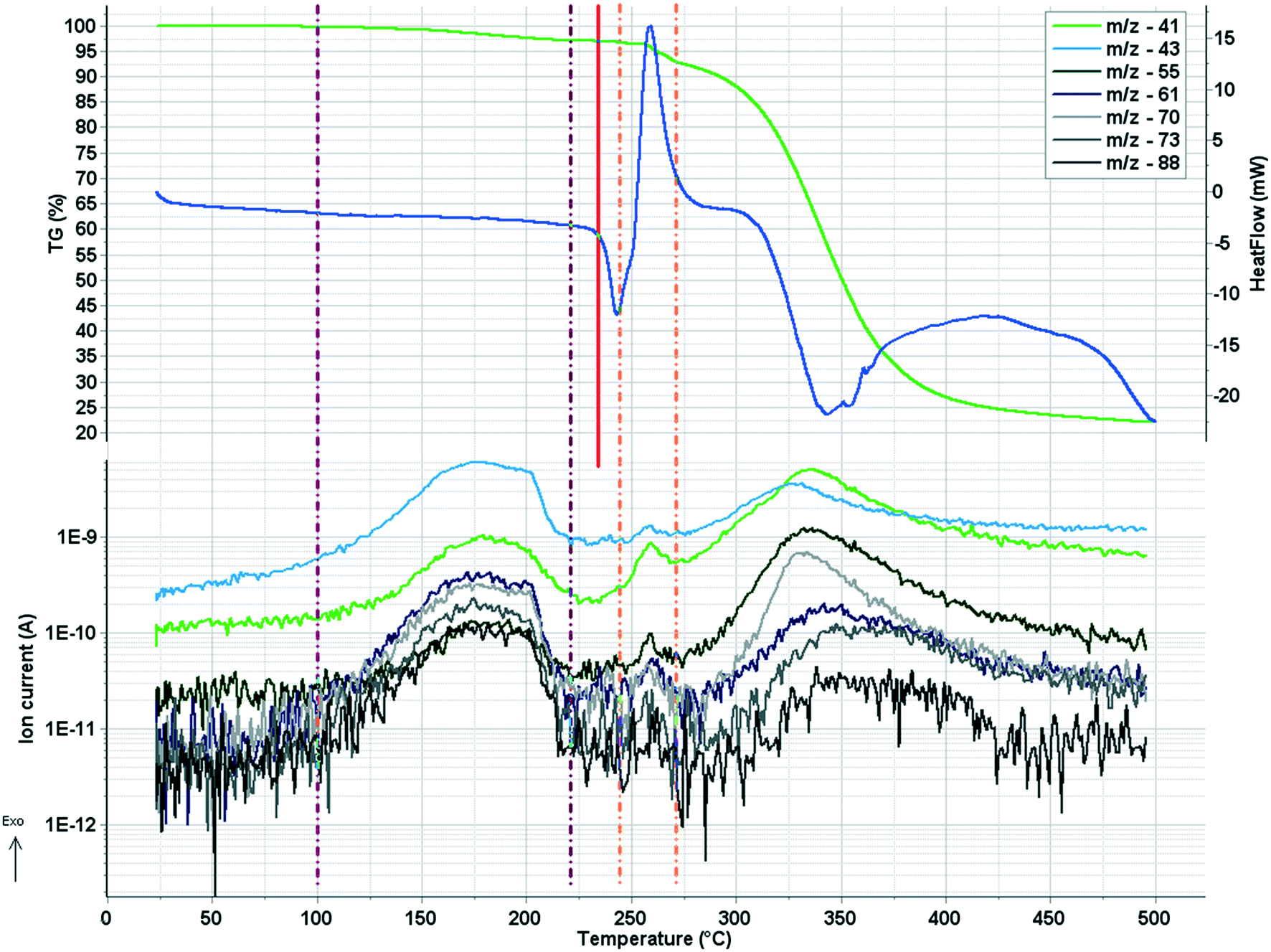 | ||
| Fig. 12 TG–DSC–MSEGA diagram of sample 9. On the upper part of the figure, the mass loss (TG – green curve) and heat flow (DSC – dark blue curve) signals are plotted, while on the lower part the ion current curves of some specific solvent fragments (ethyl acetate – m/z 88, 73, 70, 61, 43; 1-butanol – m/z 55 and 41) are plotted against temperature. The two vertical purple dash-dot lines mark the temperature interval, where the retained solvents (3%) are lost from the channels, while the red line shows the start of the melting of the sample. The orange dash-dot lines are marking a second small temperature interval, where traces of solvents are also lost and the decomposition starts. | ||
From the DCM solution, besides crystal 9 prismatic brown crystals of 10 could also be harvested in which the inclusion of one DCM molecule per asymmetric unit has been found (Fig. 8). We have seen that the conformation of the [CuL2] complex in this structure is very similar to that of 9 (Fig. 9 and S14†). The secondary interactions of the DCM molecules in the lattice are much weaker in 10 compared to the alcohol-containing complex crystals of 3–6. Crystal 10 decomposes rapidly when it is removed from its mother liquor. The packing of the molecules resembles that of crystal 9 as columns are formed by the sulfonamide connections; however, the angle between the Cu coordination planes of neighbouring complexes is much higher (80.7°) (Fig. 11c). The channels occurring in crystals 9 and 10 are compared in Fig. 13. The void volume occupied by the DCM molecules has grown from the empty 16.8% in 9 to 21.4% of the unit cell volume in 10.
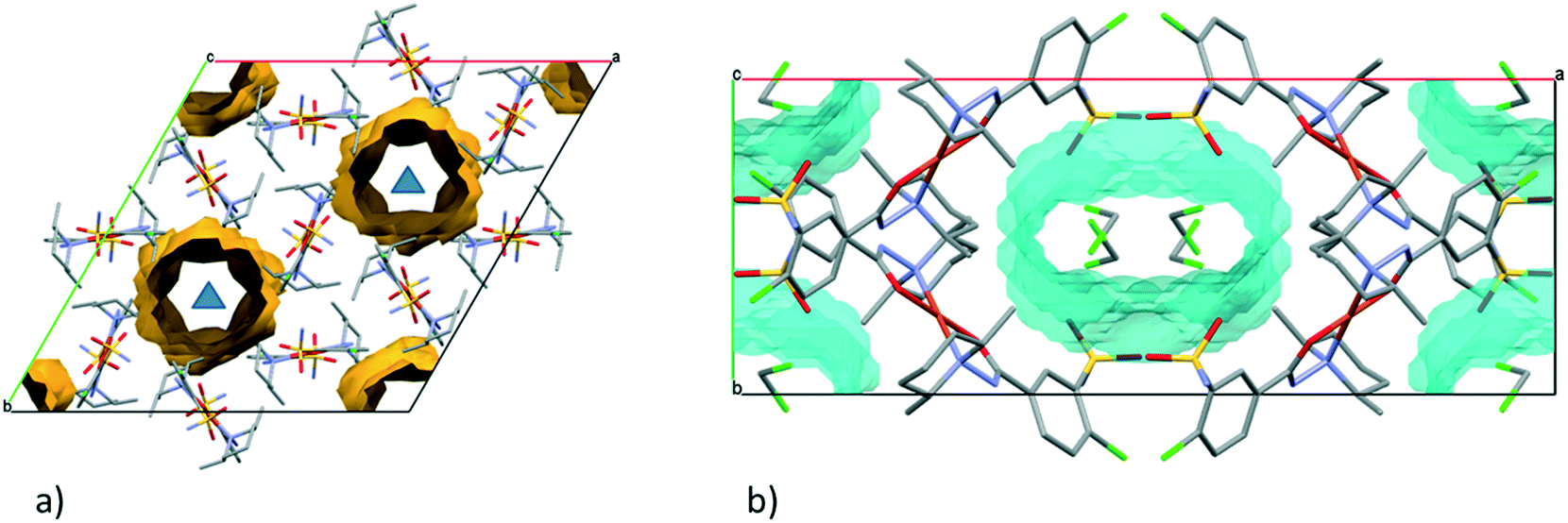 | ||
| Fig. 13 (a) Empty channels in the crystal structure of 9 viewed from the crystallographic c axis. The three-fold axes are shown as blue triangles. (b) Unit cell of crystal 10 viewed from the crystallographic c axis. The channels in the crystal lattice (highlighted by cyan) are filled by DCM molecules which are surrounded by four complex molecules. | ||
The difference in the conformation of clopamide in its pure crystal (2) and as a ligand in the complex crystal (3) is shown in Fig. 14. The sulfonamide group was found in the opposite direction in the complex as compared to the free ligand crystals, as the chloro-sulfamoylbenzamide group turns around the flexible C6–C7 single bond. Although in solution the molecular freedom is higher, comparing the molecular conformations may provide insights into the reaction mechanism of complex formation.
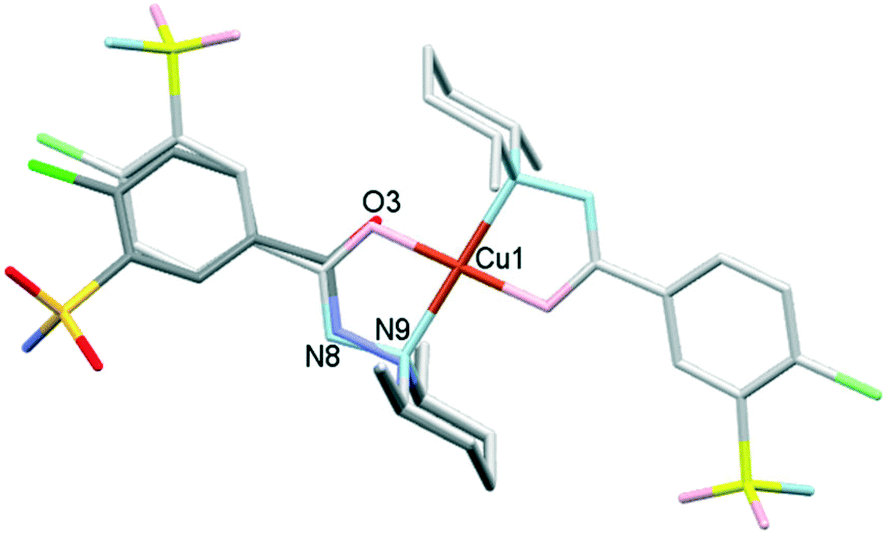 | ||
| Fig. 14 Comparison of the conformations of the clopamide molecule in its hemihydrate crystal 2 and in its complex molecule in MeOH containing crystal 3. The clopamide from 2 is shown in dark colours, while the complex from 3 is shown in light colours. | ||
3.4 Results of EPR spectroscopy measurements
In order to investigate the structure of the clopamide complex [CuL2] in solution, crystal 9 (a solvent-free polymorphic crystal form) was dissolved in pure DMSO in 3.0 mM concentration. The solution was poured in a quartz EPR tube and placed into a Dewar flask containing liquid nitrogen (77 K). The measured and simulated frozen solution spectra can be seen in Fig. 15. Depending on the stability of the complex when a crystal is dissolved, it may dissociate, thus the measured EPR spectrum is the superposition of the components appearing in the solution. When crystal 9 was dissolved in DMSO and EPR spectrum was measured for the frozen solution, the spectrum simulation showed the superposition of 80% bis-ligand and 20% mono-ligand complex. This means that in 20% of the complex, a ligand was released from the coordination sphere, resulting in the appearance of the mono-complex [CuL]+. The well-resolved superhyperfine line in the perpendicular range of the [CuL2] spectra originated from two nitrogen atoms coordinated to the copper ion. The trans position of the nitrogen atoms can be suggested based on the fact that magnetically equivalent couplings with the highest value in the y direction (referred to as the g tensor orientations) were obtained. The very low gz (2.185) value and strong copper hyperfine coupling Az (206 G) support the highly symmetrical square planar geometry of the complex with a high ligand field detected in the equatorial plane. In the case of the mono-complex, DMSO coordination beside one clopamide molecule in the equatorial plane can be presumed, and the higher gz and lower Az values reflect a lower ligand field compared to the bis-ligand complex. Nitrogen hyperfine couplings were not resolved for this complex.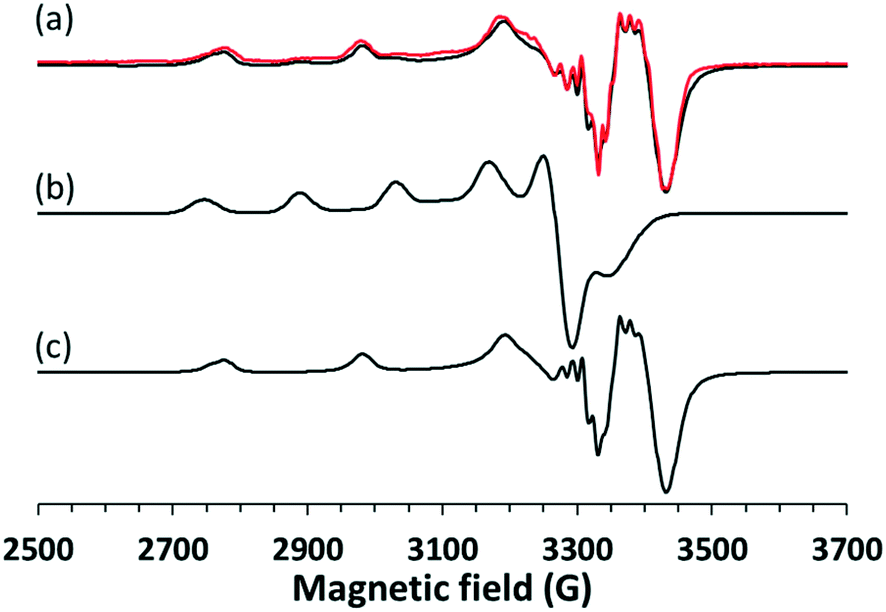 | ||
| Fig. 15 (a) Experimental (black) and simulated (red) EPR spectra recorded at 77 K for crystal 9 dissolved in pure DMSO. The spectra were fitted with the superposition of (b) [CuL] in 20% and (c) [CuL2] in 80%. The EPR parameters of [CuL2] are gx = 2.032, gy = 2.047, gz = 2.185, Ax = 33 G, Ay = 48 G, Az = 206 G and two nitrogens with ax = 5 G, ay = 15 G, az = 5 G, and of [CuL] are gx = 2.060, gy = 2.075, gz = 2.279, Ax = 16 G, Ay = 54 G, Az = 137 G. | ||
3.5 Results of the infrared spectroscopy measurements
Based on the IR spectra, the stretching vibration band of the –OH group is missing from crystal 1, while it appears at 3629 cm−1 in crystal 2 (Fig. S15 and Table S13†), which also shows a good agreement with the SXRD structures of the anhydrate and hemihydrate crystals of clopamide (Fig. S14†). In the case of the solvatomorph series, the shape (intensity and position) of the CO and NH vibrational bands change in the complexes (Fig. 16a). SO2 vibrations also alter; it is probably because of the violation of symmetry. In crystals 3, 4, 5 and 6, the bands assigned to C–O and –OH vibrations are amplified due to the presence of bound alcohol molecules. Interestingly, samples 3–6 and 4–5 can be paired based on their spectra. The spectra of solvent-free polymorphs exhibit sharper bands (Fig. 16b), suggesting a more ordered crystal structure. This is further supported by the splitting of the more intense SO2 stretching bands (Davydov splitting)29 compared with the spectra of the solvatomorph series. It cannot be determined from the IR spectra how many molecules are present in a given unit cell; however, the appearance of Davydov splits in the spectra indicates a good agreement with the SXRD structures. In the case of crystal 10, the bands of CH2Cl2 can be clearly seen (Fig. S16†). These extra bands do not appear in the other samples. A tentative assignment of the IR bands is presented in Tables S14 and S15.†30–33
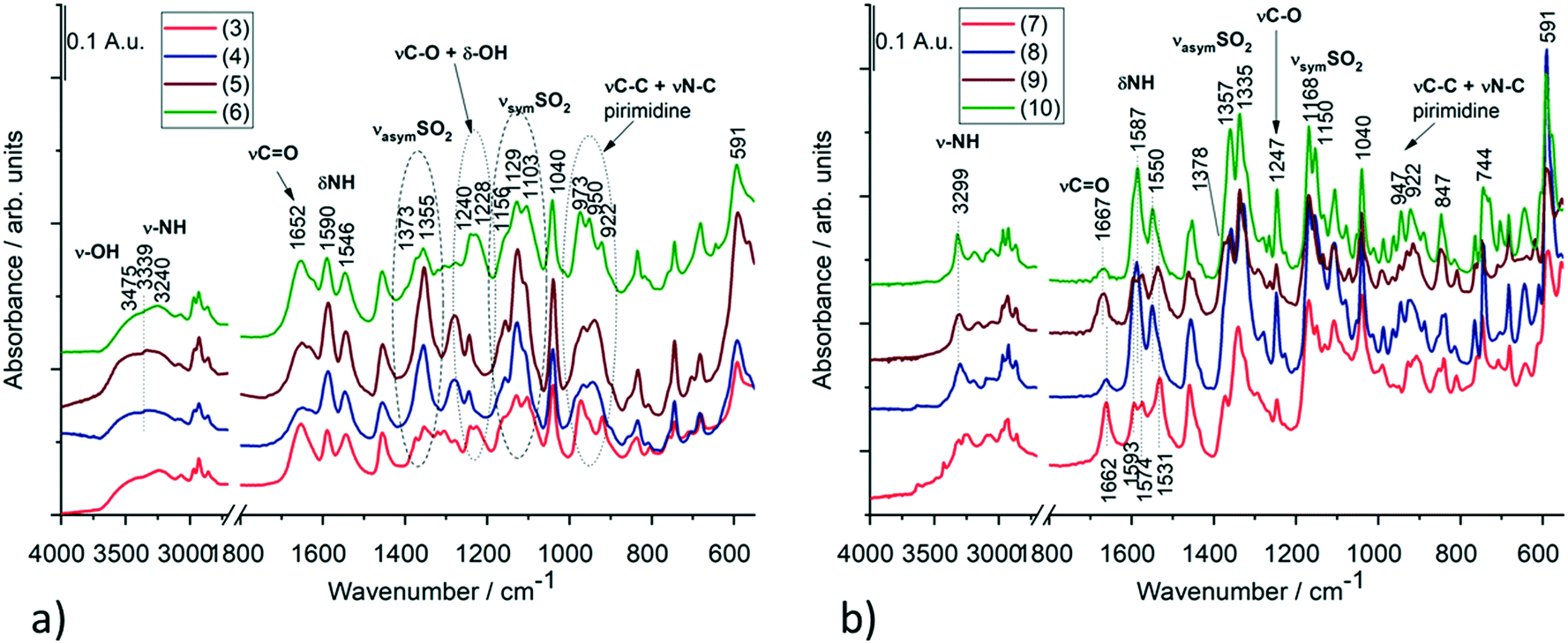 | ||
| Fig. 16 IR spectra of (a) the solvatomorph crystals of 3, 4, 5 and 6 and (b) the solvent-free polymorphs (7–9) as well as the DCM containing crystal 10 of the complexes. Selected IR bands of these compounds with tentative assignments are presented in Tables S14 and S15.† | ||
3.6 Results of the thermoanalytical measurements
The melting points of all crystalline samples were determined (Tables S1, S5 and S9†) by DSC. TG, DSC and DTG curves of 1 in inert atmosphere and in synthetic air atmosphere are presented in Fig. S17–S22.† The evolved sulfur dioxide is a very characteristic marker of the thermal degradation of clopamide, as the degradation of both the pure clopamide compound and its complexes starts at the –SO2NH2 group (Fig. S21†). The thermoanalytical results confirmed that 3% of water leaves crystal 2 up to 187.8 °C, followed by water leaving between 187.8 and 226.8 °C, with a weight loss of 26.7% accompanied by an endotherm peak. It means that a large amount of water is strongly bound in the crystal lattice (Fig. S23 and S24†). For samples 7 and 8, the mass spectrometric gas analysis results confirmed that no significant amounts of solvents were present in the crystals; these are solvent-free polymorphs (Fig. S25–S32†).4 Conclusions
Clopamide ((4-chloro-N-2,6-dimethylpiperidin-1-yl)-3-sulfamoylbenzamide, LH) drug is used worldwide in the treatment of hypertension and oedema. It has also been shown that thiazide diuretics may induce changes in the level of copper in blood. Despite its medical application its structure has not yet been published, most probably because of the difficulties in single-crystal growth. We have successfully crystallized and determined the crystal structures of clopamide anhydrate and hemihydrate and a structural landscape of different polymorphs and solvatomorphs of its copper(II) complexes, revealing the molecular arrangements, supramolecular interactions and coordination properties of the complexes.It is proved by PXRD that the drug Brinaldix is the hemihydrate form. As we were able to crystallize the anhydrate form of clopamide it is presented how the inclusion of water contributes to the crystal perfection of the drug (1 and 2). The water molecule acts as a spacer, keeping a distance between the molecular layers; thus a more favourable molecular conformation and higher symmetry of the space group can be realized.
The newly defined chalcogen bond is recognised in the clopamide anhydrate crystals being in competition with the intramolecular halogen bond present between the chlorine and the oxygen of the sulfonamide group. The intermolecular chalcogen bond is found between a sulfoxy oxygen as a donor and the lone pair of the carbonyl group as an acceptor. The longer the halogen bond, the shorter the chalcogen bond and vice versa. There is no chalcogen bond in 2 because the water molecule acts as a bridge between the neighbouring molecules. In the absence of the chalcogen bond in 2, the intramolecular halogen bond becomes the shortest.
All Cu(II) complexes have a square-planar coordination geometry, in which copper(II) centres are surrounded by piperidine-N and carbonyl-O donor atoms in a five-membered chelate ring. The two ligands are in trans positions. The 2,6-dimethylpiperidine units of the ligand molecules are found to be in parallel or in perpendicular positions to the Cu–N9 bond, which is in correspondence with the angle of the plane formed by the coordinating atoms to Cu(II) and the phenyl ring whose substituents form varying intermolecular interactions. The Cu–O coordination bonds are significantly shorter, while Cu–N bonds are longer in the complex crystals of 8, 9 and 10 with perpendicular piperidine conformation compared to the solvatomorph crystals 3–6 or solvent-free 7 with parallel conformation of the piperidine ring.
The solvatomorph crystals, containing small homologous alcohols (3–6), have been found to be isostructural (P) with increasing size of void and unit cell volumes which terminates using n-butanol. In the solvent-free polymorphic [CuL2] crystals (7–9) the complex molecules are forming parallel columns strengthened by N–H⋯O interactions between ligand sulfonamide groups. The arrangement of these columns in 7 (P) is similar to that of solvate 3 (P), while 8 (P) with its triangular arrangement transitions to 9 (R), where perfect threefold symmetry is realised, although leaving large empty spaces in the lattice. Thermal investigations prove that there is no more than about 3% remaining solvent in the voids. The columnal structure is preserved, although the columns become farther away in the complex crystal (10), notwithstanding that the columns are no longer parallel and enclathrate dichloromethane solvent molecules in the crystal lattice.
The solvent content and thermal behaviour of the crystals were studied by IR and thermoanalytical methods, whose results are in good agreement with the structural results. The solution structure of the [CuL2] complex was investigated in DMSO by EPR spectroscopy at 77 K. Upon dissolving the crystals they partly dissociate; 80% bis-ligand complex remained in the solution with a highly symmetrical square planar geometry but in 20% one ligand was released, resulting in the appearance of the mono complex [CuL]+ presuming the coordination of DMSO in the equatorial plane.
In addition to the structural description of a drug on the market, the systematic comparison of several polymorphic and solvatomorphic crystal arrangements provides valuable information to synthon and crystal engineering in order to develop new materials with specific properties.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The support of the National Research, Development and Innovation Office – NKFIH through OTKA KH129588 and K124544 is gratefully acknowledged.References
- R. Censi and P. Di Martino, Polymorph impact on the bioavailability and stability of poorly soluble drugs, Molecules, 2015, 20, 18759–18776 CrossRef CAS PubMed
.
- S. R. Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S. Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B. A. Narayanan, M. Soldani, D. Riley and K. McFarland, Dealing with the impact of ritonavir polymorphs on the late stages of bulk drug process development, Org. Process Res. Dev., 2000, 4, 413–417 CrossRef CAS
-
J. M. Miller, N. Rodríguez-Hornedo, A. C. Blackburn, D. Macikenas and B. M. Collman, Solvent Systems for Crystallization and Polymorph Selection, Part of the Biotechnology: Pharmaceutical Aspects book series, Springer Nature Switzerland AG, 2007, pp. 53–109 Search PubMed
- H. G. Brittain, Polymorphism and Solvatomorphism 2008, J. Pharm. Sci., 2010, 99, 3648–3664 CrossRef CAS PubMed
- J. J. McNeil, E. L. Conway, O. H. Drummer, L. G. Howes, N. Christophidis and W. J. Louis, Clopamide: Plasma concentrations and diuretic effect in humans, Clin. Pharmacol. Ther., 1987, 42, 299–304 CrossRef CAS PubMed
- R. Agarwal, Hypertension, hypokalemia, and thiazide-induced diabetes a 3-way connection, Hypertension, 2008, 52, 1012–1013 CrossRef CAS PubMed
- J. Zieg, L. Gonsorcikova and D. Landau, Current views on the diagnosis and management of hypokalaemia in children, Acta Paediatr., 2016, 105, 762–772 CrossRef PubMed
- F. K. Bauer, Body Potassium After Short-Term Diuretic Therapy With Clopamide, J. Clin. Pharmacol. J. New Drugs, 1969, 9, 16–23 CAS
- L. Reyes, A. J. Leary, W. P. Lockett and C. J. Alcocer, Diuretics and zinc, S. Afr. Med. J., 1982, 62, 373–375 Search PubMed
- M. J. du Preez and C. J. Lockett, Effect of clopamide, a thiazide diuretic, on copper and zinc levels in hypertensive patients, J. Am. Coll. Nutr., 1991, 10, 34–37 CrossRef CAS PubMed
- A. Kraker, S. Krezoski, J. Schneider, D. Minkel and H. Petering, Reaction of 3-Ethoxy-2-oxobutyraldehyde Cu ( I1 ) with Ehrlich Cells, J. Biol. Chem., 1985, 260, 13710–13718 CrossRef CAS
- D. Kovala-Demertzi, J. R. Miller, N. Kourkoumelis, S. K. Hadjikakou and M. A. Demertzis, Palladium(II) and platinum(II) complexes of pyridine-2-carbaldehyde thiosemicarbazone with potential biological activity. Synthesis, structure and spectral properties. Extended network via hydrogen bond linkages of Pd(PyTsc)Cl, Polyhedron, 1999, 18, 1005–1013 CrossRef CAS
- A. Kálmán, L. Párkányi and G. Argay, Classification of the isostructurality of organic molecules in the crystalline state, Acta Crystallogr., Sect. B: Struct. Sci., 1993, 49, 1039–1049 CrossRef
- P. Bombicz, The way from isostructurality to polymorphism. Where are the borders? The role of supramolecular interactions and crystal symmetries, Crystallogr. Rev., 2017, 23, 118–151 CrossRef
- K. Biradha, C. Y. Su and J. J. Vittal, Recent developments in crystal engineering, Cryst. Growth Des., 2011, 11, 875–886 CrossRef CAS
- G. R. Desiraju, Crystal engineering. From molecules to materials, J. Mol. Struct., 2003, 656, 5–15 CrossRef CAS
- P. Bombicz, T. Gruber, C. Fischer, E. Weber and A. Kálmán, Fine tuning of crystal architecture by intermolecular interactions: synthon engineering, CrystEngComm, 2014, 16, 3646–3654 RSC
- G. R. Desiraju, Crystal engineering: A holistic view, Angew. Chem., Int. Ed., 2007, 46, 8342–8356 CrossRef CAS PubMed
- Rigaku/MSC Inc., Rigaku/MSC, CrystalClear software, 2005.
- M. C. Burla, R. Caliandro, B. Carrozzini, G. L. Cascarano, C. Cuocci, C. Giacovazzo, M. Mallamo, A. Mazzone and G. Polidori, Crystal structure determination and refinement via SIR2014, J. Appl. Crystallogr., 2015, 48, 306–309 CrossRef CAS
- G. M. Sheldrick, Crystal structure refinement with SHELXL, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed
- L. J. Farrugia, WinGX and ORTEP for Windows: An update, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS
- A. L. Spek, Single-crystal structure validation with the program PLATON, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. Van De Streek, Mercury: Visualization and analysis of crystal structures, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS
- F. H. Allen, O. Johnson, G. P. Shields, B. R. Smith and M. Towler, CIF applications. XV. enCIFer: A program for viewing, editing and visualizing CIFs, J. Appl. Crystallogr., 2004, 37, 335–338 CrossRef CAS
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, NMR chemical shifts of common laboratory solvents as trace impurities, J. Org. Chem., 1997, 62, 7512–7515 CrossRef CAS PubMed
- A. Rockenbauer and L. Korecz, Automatic computer simulations of ESR spectra, Appl. Magn. Reson., 1996, 10, 29–43 CrossRef CAS
- B. Kozlevčar, P. Baškovič, A. Arko, A. Golobič, N. Kitanovski and P. Šegedin, Copper fixation to guaiacyl lignin units by nitrogen donor ligands, Z. Naturforsch., B: J. Chem. Sci., 2008, 63, 481–488 CrossRef
-
A. D. McNaught and A. Wilkinson, IUPAC, Compendium of Chemical Terminology, Oxford, 2nd edn, 1997 Search PubMed
-
G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, Wiley, 3rd edn, 2010 Search PubMed
- C. Topacli and A. Topacli, Infrared spectra simulation for some sulfonamides by using semi-empirical methods, Spectrosc. Lett., 2002, 35, 207–217 CrossRef CAS
-
L. J. Bellamy, The Infrared Spectra of Complex Molecules, Springer, 2013 Search PubMed
- S. Breda, I. D. Reva, L. Lapinski, M. J. Nowak and R. Fausto, Infrared spectra of pyrazine, pyrimidine and pyridazine in solid argon, J. Mol. Struct., 2006, 786, 193–206 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Detailed crystallographic data, results of IR and thermal investigations, additional figures. CCDC 2099748, 2099749 (1 and 2), 2099751–2099758 (3–10). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ce00995h |
| This journal is © The Royal Society of Chemistry 2021 |