
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel 18F-labeled clickable substrate for targeted imaging of SNAP-tag expressing cells by PET in vivo†
Dominic Alexej
Depke‡
a,
Christian Paul
Konken‡
 ab,
Lukas
Rösner‡
c,
Sven
Hermann
a,
Michael
Schäfers
*ab and
Andrea
Rentmeister
*c
ab,
Lukas
Rösner‡
c,
Sven
Hermann
a,
Michael
Schäfers
*ab and
Andrea
Rentmeister
*c
aEuropean Institute for Molecular Imaging (EIMI), University of Münster, Germany. E-mail: michael.schaefers@uni-muenster.de
bDepartment of Nuclear Medicine, University Hospital Münster, Germany
cInstitute of Biochemistry, University of Münster, Germany. E-mail: a.rentmeister@uni-muenster.de
First published on 31st August 2021
Abstract
Bioorthogonal covalent labeling with self-labeling enzymes like SNAP-tag bears a high potential for specific targeting of cells for imaging in vitro and also in vivo. To this end, fluorescent SNAP substrates have been established and used in microscopy and fluorescence imaging while radioactive substrates for the highly sensitive and whole-body positron emission tomography (PET) have been lacking. Here, we show for the first time successful and high-contrast PET imaging of subcutaneous SNAP-tag expressing tumor xenografts by bioorthogonal covalent targeting with a novel 18F-based radioligand in vivo.
A number of chemical and biochemical strategies for labeling specific types of cells have been developed in the past years. Several of these strategies have been used for targeted labeling of mammalian cells, e.g. in tumors.1 While labeling of cells in vitro often can be realized straightforwardly using cell-specific physiologically or pathophysiologically expressed cellular targets, cell-specific targeted labeling for imaging in vivo is more intricate and often unsuccessful due to the lack of exclusivity of naturally expressed targets, unfavorable biodistribution of the chemical targeting unit, and its metabolism and excretion. Another important limitation of imaging cells in vivo is the physical detection principle of the various imaging strategies. Fluorescence-based targeting allows for in vivo targeting; however, the imaging readout by microscopy is typically done ex vivo – preventing dynamic and serial imaging – or intravitally with restricted surgical access to very confined tissue volumes of single organs. Fluorescence reflection imaging (FRI) provides whole-body imaging but is limited by penetration depth and scattering of light in organisms. In contrast, scintigraphic imaging methods such as SPECT or PET provide high sensitivity and quantitative, fully non-invasive whole-body imaging of organisms, which has also the unique capability of clinical translation. Scintigraphic imaging has been successfully used to target individual cells in vivo, e.g. using non-cell-specific metabolic markers such as [18F]fluorodeoxyglucose ([18F]FDG) or gene reporters such as thymidine kinase genetically introduced into cells or mouse models and targeted by [18F]fluoro-guanine ([18F]FHBG).2
The so-called SNAP-tag stands out as a versatile genetic approach for irreversible covalent labeling of cells with, in principle, any type of reporter in a chemo-enzymatic strategy. It is a variant of the O6-alkylguanine-DNA alkyltransferase suitable for labeling with O6-benzylguanine (BG) derivatives which can be coupled with reporters at the para-position of the benzyl moiety.3 This self-labeling tag exhibits high specificity and kinetics for in vivo labeling of fusion proteins on cells with synthetic fluorophores and it has been used for in vivo imaging in mice using far-red emitting fluorescent dyes, such as BG-547,4 BG-8005 or novel charge-free fluorescence-switchable near-infrared (NIR) dyes.6 Non-invasive imaging of tumors in nude mice was successful after subcutaneous implantation of murine breast carcinoma cells engineered to express SNAP-GPI using intravenous injection of the infrared probe BG-782.4 Furthermore, the Cre-dependent expression of SNAP-tag in mice has been shown to be a powerful method for the genetic targeting of chemical indicators in vivo, as demonstrated by labeling in complex tissues in vivo with fluorophores from green to near infrared emission.7
However, as discussed above, the application of SNAP-tag-based biomedical imaging in vivo using fluorescent dyes might be restricted by post-mortem approaches (microscopy), invasivity (intravital microscopy), limited sensitivity due to tissue penetration and lack of absolute quantification due to scattering of light (FRI). Such challenges would be ideally addressed by a radioactive SNAP-tag substrate combined with scintigraphic, non-invasive and dynamic whole-body imaging by PET; however, radioactive SNAP-tag substrates have not been reported so far. Therefore, we aimed at the development of a novel 18F-labeled SNAP-tag substrate, its characterization in vitro, and a proof-of-principle PET study in SNAP-tag expressing subcutaneous tumors in vivo (Fig. 1A).
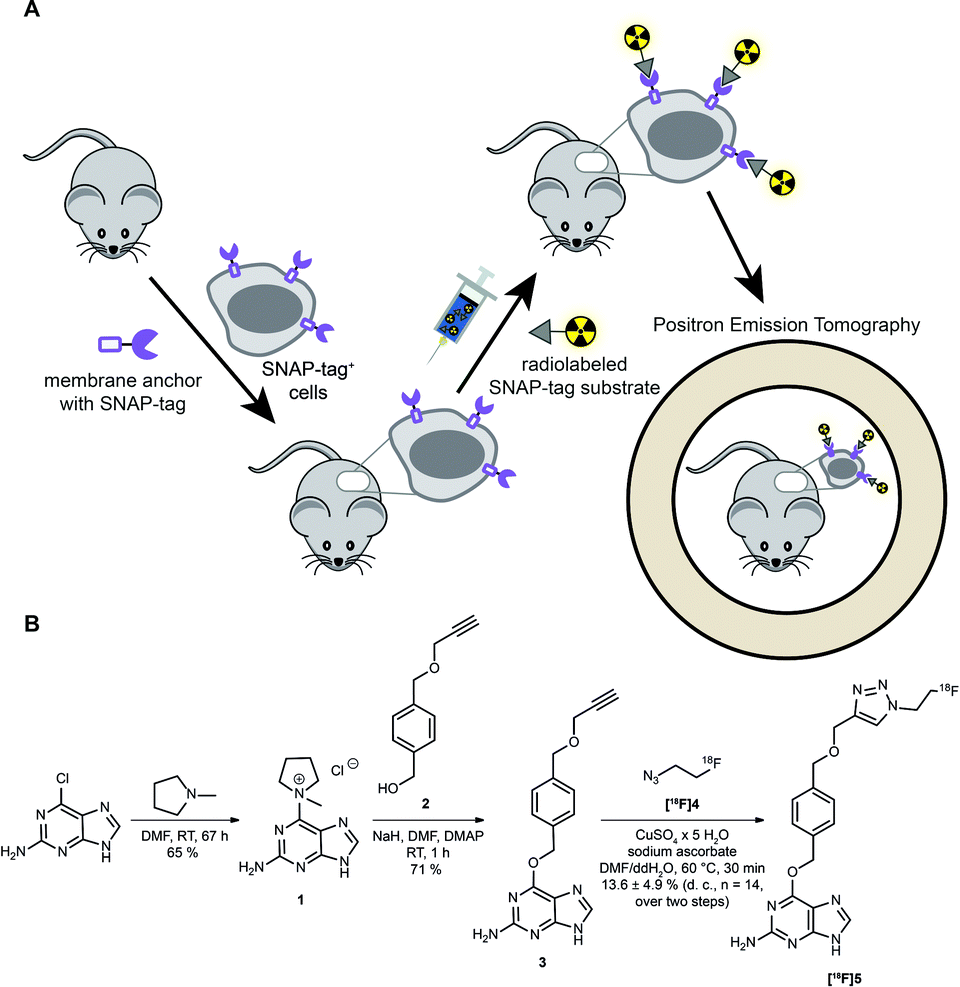 | ||
| Fig. 1 Scheme for cell-specific labeling and synthesis of the 18F-PET sensor. (A) Schematic illustration of specific labeling and imaging strategy in vivo with a newly synthesized SNAP-tag substrate. (B) Synthetic route to O6-((4-(((1-(2-[18F]fluoroethyl)-1H-1,2,3-triazol-4-yl)methoxy)methyl)benzyl)guanine) [18F]5, termed [18F]FTBG, a radioactive SNAP-tag substrate for PET imaging. | ||
Synthesis and labeling of [ 18 F]FTBG: For radioactive labeling of the SNAP-tag substrate we favored a copper-catalyzed azide–alkyne cycloaddition (CuAAC) which provides the required specificity, yield and velocity for introduction of [18F]fluorine. Accordingly, we synthesized an O6-benzylguanine with a terminal alkyne and 1-azido-2-[18F]fluorethane (Fig. 1B).
To this end, the activated guanine 1 was prepared by reacting 6-chloroguanine with 1-methylpyrrolidine.3,8 A straightforward approach to introduce a terminal alkyne to the para-position of the benzylic moiety was to start from the symmetric 1,4-benzenedimethanol and react one of the benzylic alcohol functions by reaction with propargyl bromide and sodium hydride to from 2.8,9 Purified 1 and 2 were then reacted in NaH and catalytic amounts of DMAP to yield the clickable SNAP-tag substrate O6-(4-((prop-2-yn-1-yloxy)methyl)benzyl)guanine (PYBG) (3), termed PYBG, which can be stored for months.8
To obtain click reagent [18F]4, a three step synthesis was necessary. First, the reaction of 2-bromoethanol with sodium azide was performed to obtain 2-azidoethanol (6),10,11 which was subsequently tosylated to give 2-azidoethyl-4-methylbenzosulfonate (7) in 66% yield.12 Compound [18F]4 was obtained via fluorination of 7 using [18F]fluoride, Kryptofix®2.2.2 and potassium carbonate 71 ± 6% (d. c., n = 14). It was used for CuAAC with the PYBG (3) after distillation to yield the desired 18F-labeled SNAP-tag substrate [18F]5, termed [18F]FTBG. The reaction mixture was first purified by C18-solid phase extraction after ligand exchange of copper-complexes with [18F]FTBG ([18F]5) using ammonia buffer. A second purification by semi-preparative HPLC equipped with a gamma-detector was necessary to obtain pure [18F]5 13.6 ± 4.9% (d. c., n = 14, total yield over two steps of radiosynthesis). Furthermore, we synthesized BG-Dy549P1, a cell-impermeable fluorescent SNAP-tag ligand, to facilitate characterization of [18F]FTBG on cells in vitro (see ESI†).
In vitro characterization of [ 18 F]FTBG: We first performed in vitro uptake experiments to study selectivity and binding characteristics of [18F]FTBG to SNAP-tag epitopes expressed on the cell surface of transduced Gli36 glioblastoma cells (SNAP-tag+). We found that [18F]FTBG binds to epitopes on SNAP-tag+ cells with high selectivity and binding was saturated at high ligand concentrations (Fig. 2A, Bmax = 170.6 ± 7.8 Bq, Kd = 7784 ± 1605 Bq, R2 = 0.8903, parameters ± standard error (SE)). In contrast, non-significant low binding of the ligand to non-transduced WT cells (SNAP-tag−) was observed. In line with these findings, pre-dosing of SNAP-tag+ cells with non-radioactive cell-permeable SNAP-Cell® or impermeable SNAP-Surface® blocking substrates (to study cell permeability) prior to incubation with [18F]FTBG led to a significantly reduced accumulation of the radioligand in the cells (Fig. 2B).
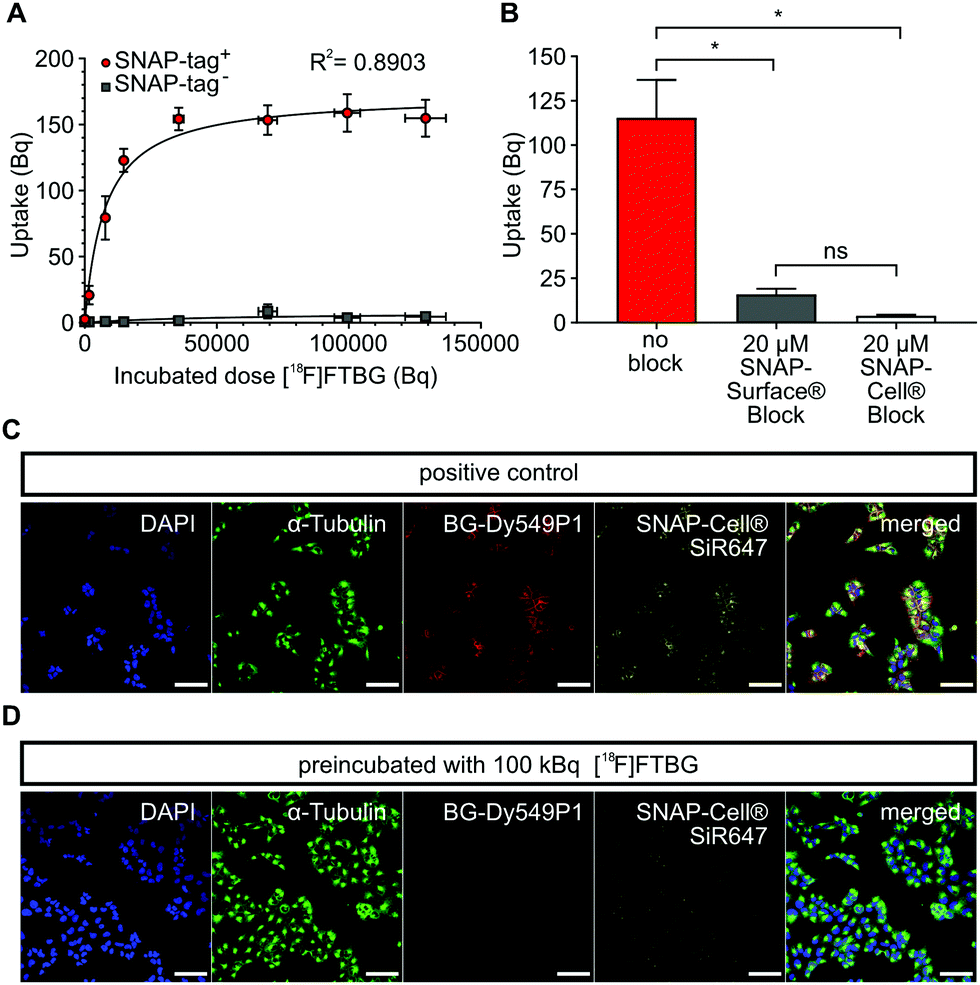 | ||
| Fig. 2 In vitro binding study of [18F]FTBG to SNAP-tag+ Gli36 cells. (A) 1 × 105 SNAP-tag+ or SNAP-tag− cells were incubated with a dilution series of [18F]FTBG for 30 min. Cells were washed, harvested and radioactivity bound to cells was measured with a gamma-counter. SNAP-tag+ cells accumulate much higher amounts of radioactivity and approach a plateau-phase at higher concentrations of the ligand (n = 4, R2 = 0.89, Bmax = 170.6 ± 7.8 Bq, Kd = 7784 ± 1605 Bq, non-linear regression analysis with one-side fit with total and non-specific binding) as compared to SNAP-tag− cells. (B) 1× 105 SNAP-tag+ cells were incubated with cell-permeable SNAP-Cell® or non-permeable SNAP-Surface® block substrates or were left untreated before 100 kBq [18F]FTBG was added. Blocking resulted in significantly lower accumulation of radioactivity in the cells when compared to control cells (n = 4, RM One-way ANOVA with Tukey's test for multiple comparisons, *p < 0.05). (C) Labeling of SNAP-tag+ Gli36 cells with fluorescent O6-benzylguanines. Labeling with cell-impermeable BG-Dy549P1 shows bright staining at the plasma membrane, incubation with cell-permeable SNAP-Cell® 647-SiR stains intracellular and extracellular epitopes. (D) Fluorescent signals are strongly reduced when cells were first incubated with 100 kBq [18F]FTBG. Scale bars 100 μm. All data is shown as mean ± SEM. | ||
In addition, we studied cell permeability of [18F]FTBG by confocal imaging of SNAP-tag+ cells. Cells were either pre-dosed with [18F]FTBG for 30 min or left untreated followed by staining with the cell-impermeable SNAP-tag dye BG-Dy549P1and the highly cell-permeable dye SNAP-Cell® 647-SiR. Pre-dosing of cells with [18F]FTBG largely abolished fluorescent labeling of both extra- and intracellular epitopes (Fig. 2D) while we observed bright staining of these structures in untreated SNAP-tag+ cells (Fig. 2C). This together with the above radiotracer uptake experiments suggests that [18F]FTBG is cell-permeable.
In vivo biodistribution: Following successful evaluation of SNAP-tag-specific binding in vitro we aimed at studying biodistribution of [18F]FTBG in vivo. In vivo biodistribution was determined by dynamic PET in 4 adult female C57/BL6 mice after intravenous injection. [18F]FTBG showed a fast clearance from the blood and from non-targeted tissues such as muscle following the perfusion phase (Fig. 3A and B). In contrast, [18F]FTBG accumulated in the liver and the kidneys (Fig. 3C), followed by a highly efficient and almost balanced net excretion to the gallbladder and the urinary bladder (Fig. 3D). We calculated a total clearance of 79.77 ± 4.48% Injected Dose (%ID) at 60 min, of which 49.78 ± 5.75% ID were excreted through liver and 50.22 ± 5.75% ID through kidney (mean ± standard deviation (SD)). The in vivo biodistribution data was corroborated by ex vivo counting of tissues and fluids harvested 90 min p.i. (ESI† Table S2).
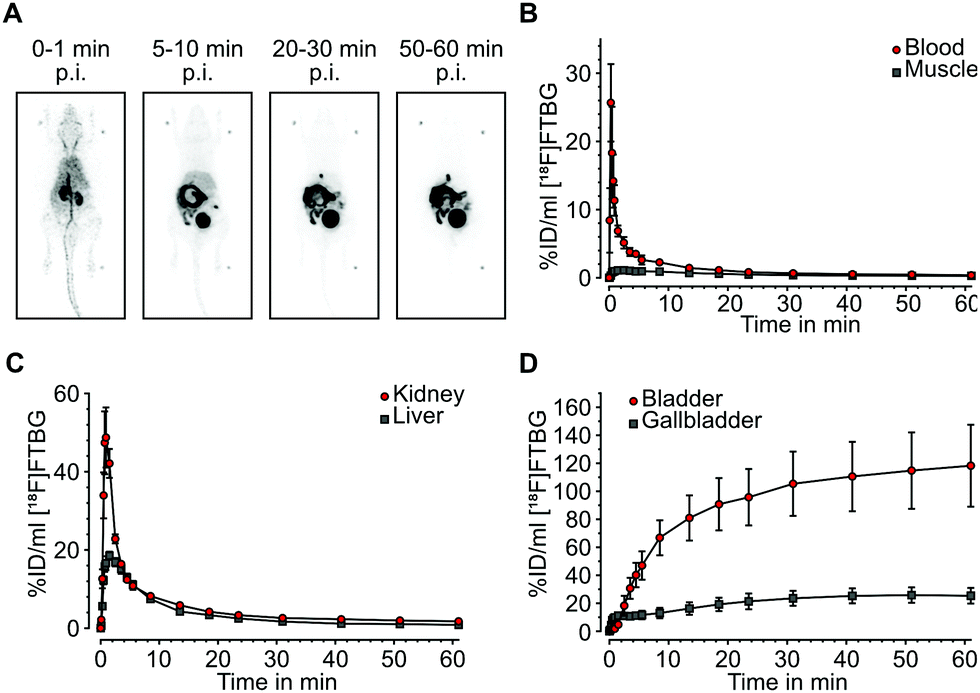 | ||
| Fig. 3 In vivo biodistribution and excretion analysis of [18F]FTBG in adult female C57/Bl6 mice after intravenous injection. (A) Maximum intensity projections of the biodistribution of radiotracer at different time points post injection. (B) In vivo radiotracer dynamics (time-activity-curves) in arterial blood and muscle tissue, (C) in kidneys and the liver and (D) in bladder and gallbladder showing a fast clearance of the tracer from blood and tissues through hepatic and renal excretion. All data is shown as mean ± SEM. %Injected Dose per mL (%ID per mL) gives the concentration of radioactivity in a tissue volume of 1 cm3 as the percentage of the total injected dose. | ||
SNAP-tag targeted imaging in tumor xenografts using [ 18 F]FTBG: Feasibility of targeting SNAP-tag by [18F]FTBG in vivo was studied in Gli36 tumor cells transduced with a SNAP-tag fusion construct (SNAP-tag+) versus wild type cells (SNAP-tag−) (see ESI†). Tumor cells were injected subcutaneously into the left (SNAP-tag+) and right (SNAP-tag−) shoulders/flanks of female C57/BL6 mice (n = 6) and PET imaging was performed 7–10 days after tumor cell implantations. Upon intravenous injection of [18F]FTBG a 60 min dynamic imaging acquisition was initiated. Following initial perfusion-related uptake of [18F]FTBG in all tumors, [18F]FTBG was quickly washed out from SNAP-tag− tumors with a remaining mean activity concentration of 0.15 ± 0.08%ID per mL (Fig. 4B) at 60 min p.i. However, in contrast, [18F]FTBG was retained in all SNAP-tag+ tumors with 0.38 ± 0.14%ID per mL (Fig. 4B and C) and a SNAP-tag+/SNAP-tag− tumor ratio of 2.5 at 60 min p.i. Fig. 4A shows PET images of tumors from individual animals 60 min p.i. with a significant uptake of [18F]FTBG only in SNAP-tag+ tumors.
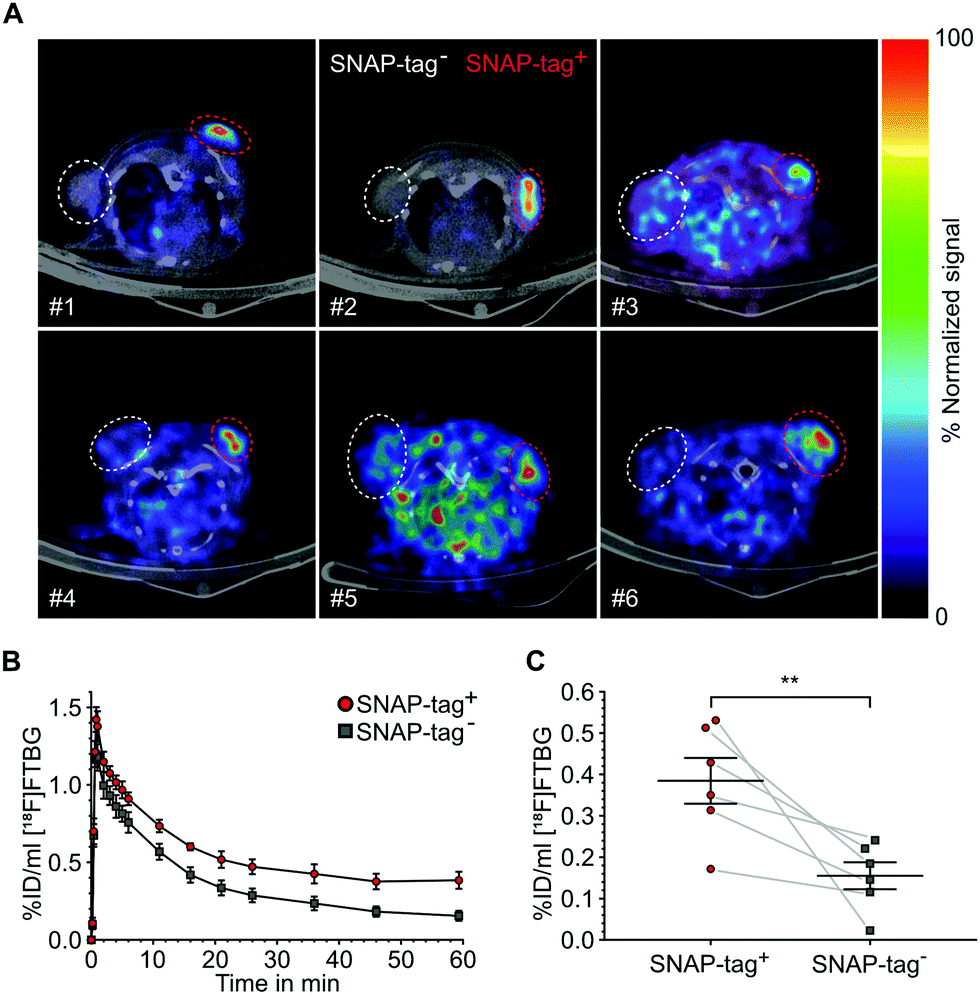 | ||
| Fig. 4 In vivo imaging of [18F]FTBG accumulation in a subcutaneous xenograft tumor model. (A) Transaxial PET images of 6 individual animals (#1–#6) coregistered to computer tomography 60 min post radiotracer injection showing tumor uptake only in SNAP-tag+ tumors (red circles: SNAP-tag+ tumors, white circles: SNAP-tag− tumors). Signals were normalized to the percentage of the maximum value of the SNAP-tag+ tumor. (B) Time-activity-curves of the volume-of-interest (VOI) analysis of SNAP-tag+ and SNAP-tag− tumors (mean ± SEM). Signals in SNAP-tag+ tumors reach a stable plateau at 40–60 min while signals from SNAP-tag− tumors show a steady decrease over time. (C) Comparison of radiotracer accumulation in tumors at 60 min. SNAP-tag+ tumors show a significantly higher amount of radioactivity as compared to SNAP-tag− tumors. Gray lines connect data points from individual animals (n = 6, statistical significance was calculated using an unpaired t-test (two-tailed): **p < 0.01). | ||
At the end of the PET scans, tumors were harvested, weighed and counted. Even more striking as compared to the in vivo VOI analysis, uptake in SNAP-tag+ tumors ex vivo (%ID per g) was 7.94-fold higher than that of SNAP-tag− tumors (Fig. 5D). In addition, all tumors were cryo-fixated and sliced for histology (Fig. 5B) and autoradiography. Again, only SNAP-tag+ tumor tissues showed a high uptake whereas SNAP-tag− tumor tissues did only show background activity (Fig. 5A and C).
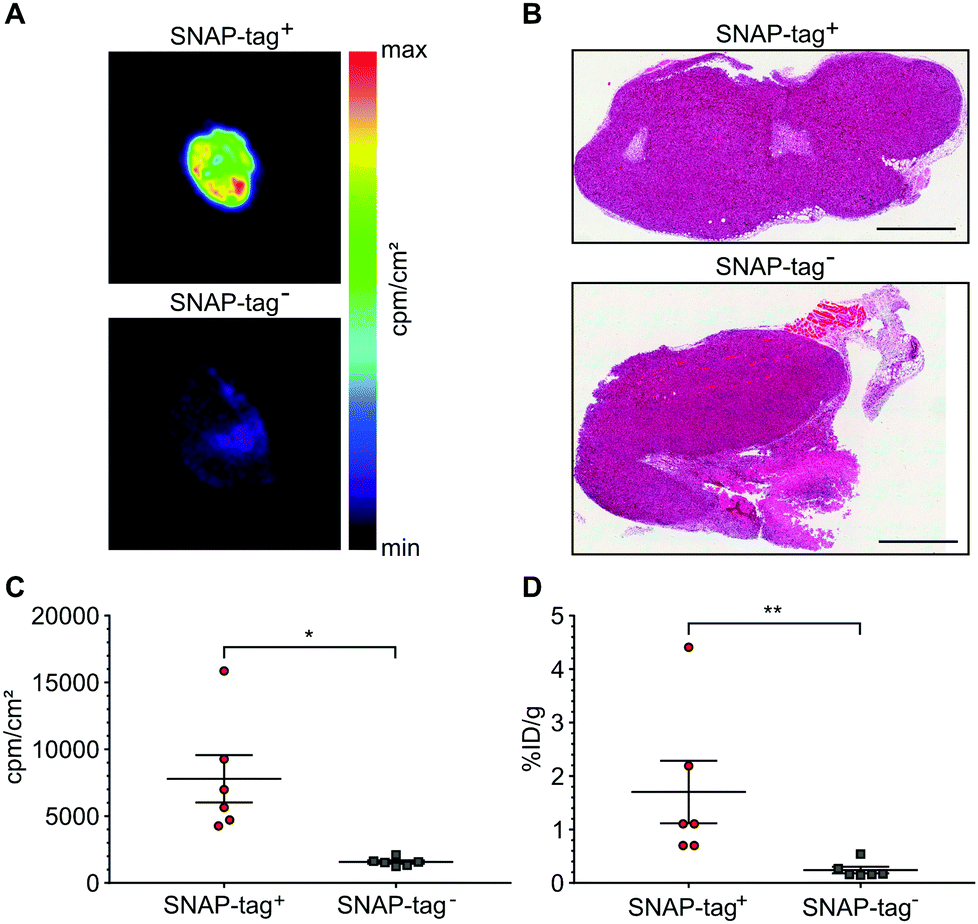 | ||
| Fig. 5 Ex vivo validation of tumor radiotracer accumulation. (A) Representative autoradiographic images of axial tumor sections harvested 90 min post radiotracer injection. (B) Representative hematoxylin and eosin staining of axial tumor sections. SNAP-tag+ and SNAP-tag− tumors appear as solid tumors with a high amount of mitotic nuclei and a low degree of necrosis. Scalebars 1 mm. (C) Quantification of the autoradiographic images. SNAP-tag+ tumors show a significantly higher accumulation of radioactivity as compared to SNAP-tag− tumors (n = 6, statistical significance was calculated using an unpaired t-test (two-tailed) with Welch's correction, *p < 0.05). (D) Quantification of ex vivo tumor signals by scintillation counting. Statistical significance was calculated using an unpaired Mann–Whitney-U test (two-tailed) with **p < 0.01. All data is shown as mean ± SEM. | ||
In conclusion, this study shows for the first time a radiochemical strategy for labeling of a SNAP-tag substrate by [18F]fluorine, its favorable binding characteristics and specificity in vitro, and successful bioorthogonal covalent targeting and imaging of SNAP-tag expressing tumor cells by PET in mice in vivo. This approach uniquely establishes SNAP-tag-based targeting for PET and thus also opens new avenues for multiscale imaging, i.e. the transfer and exchange of biological information from high-resolution microscopy to whole-body quantitative PET imaging. Future studies will define the overall sensitivity and specificity of this new approach compared to other SNAP-tag directed imaging strategies as well as to metabolic and gene reporter-based strategies and its application to various cell types including immune cells and stem cells. Besides preclinical applications in disease models, translational attempts are foreseen. E.g., therapeutic CAR T cells could be genetically equipped by SNAP-tags to enable tracking of their distribution in the individual patient. Further, fusion of the SNAP-tag to antibody fragments has been employed to introduce a variety of labels or theranostic agents such as photosensitizers for future biomedical applications.13
The authors would like to thank Sandra Höppner, Dirk Reinhardt, Nina Kreienkamp, Roman Priebe, Christine Bätza, Irmgard Hoppe, Sabine Hüwel and Christa Möllmann for technical support, as well as Prof. Uwe Karst and Michael Holtkamp for ICP-OES measurements. This work was supported in part by funding from the Deutsche Forschungsgemeinschaft (DFG) CRC 1450 inSight – 431460824 (projects A01, C01 and C03), the EU/EFPIA/Innovative Medicines Initiative 2 Joint Undertaking Immune-Image n° 831514, and the IZKF Münster, Germany, core unit PIX.
Conflicts of interest
There are no conflicts do declare.References
- J. Perrin, M. Capitao, M. Mougin-Degraef, F. Guerard, A. Faivre-Chauvet, L. Rbah-Vidal, J. Gaschet, Y. Guilloux, F. Kraeber-Bodere, M. Cherel and J. Barbet, Front. Med., 2020, 7, 34 CrossRef PubMed.
- M. Thunemann, B. F. Schorg, S. Feil, Y. Lin, J. Voelkl, M. Golla, A. Vachaviolos, U. Kohlhofer, L. Quintanilla-Martinez, M. Olbrich, W. Ehrlichmann, G. Reischl, C. M. Griessinger, H. F. Langer, M. Gawaz, F. Lang, M. Schäfers, M. Kneilling, B. J. Pichler and R. Feil, Nat. Commun., 2017, 8, 444 CrossRef PubMed.
- A. Keppler, S. Gendreizig, T. Gronemeyer, H. Pick, H. Vogel and K. Johnsson, Nat. Biotechnol., 2003, 21, 86–89 CrossRef CAS PubMed.
- K. Bojkowska, F. Santoni de Sio, I. Barde, S. Offner, S. Verp, C. Heinis, K. Johnsson and D. Trono, Chem. Biol., 2011, 18, 805–815 CrossRef CAS PubMed.
- H. Gong, J. L. Kovar, B. Baker, A. Zhang, L. Cheung, D. R. Draney, I. R. Correa, Jr., M. Q. Xu and D. M. Olive, PLoS One, 2012, 7, e34003 CrossRef CAS PubMed.
- H. J. Chen, C. Y. Chew, E. H. Chang, Y. W. Tu, L. Y. Wei, B. H. Wu, C. H. Chen, Y. T. Yang, S. C. Huang, J. K. Chen, I. C. Chen and K. T. Tan, J. Am. Chem. Soc., 2018, 140, 5224–5234 CrossRef CAS PubMed.
- G. Yang, F. de Castro Reis, M. Sundukova, S. Pimpinella, A. Asaro, L. Castaldi, L. Batti, D. Bilbao, L. Reymond, K. Johnsson and P. A. Heppenstall, Nat. Methods, 2015, 12, 137–139 CrossRef CAS PubMed.
- X. Song, C. Wang, Z. Han, Y. Xu and Y. Xiao, RSC Adv., 2015, 5, 23646–23649 RSC.
- G. Lemercier, S. Gendreizig, M. Kindermann and K. Johnsson, Angew. Chem., 2007, 46, 4281–4284 CrossRef CAS PubMed.
- B. K. H. Chan, Seeking new understanding and applications of 1,1'-bisisoquinolines, PhD thesis, University of New South Wales, 2007 Search PubMed.
- J. McConathy, D. Zhou, S. E. Shockley, L. A. Jones, E. A. Griffin, H. Lee, S. J. Adams and R. H. Mach, Mol. Imaging, 2010, 9, 329–342 CrossRef CAS PubMed.
- V. Hugenberg, B. Riemann, S. Hermann, O. Schober, M. Schäfers, K. Szardenings, A. Lebedev, U. Gangadharmath, H. Kolb, J. Walsh, W. Zhang, K. Kopka and S. Wagner, J. Med. Chem., 2013, 56, 6858–6870 CrossRef CAS PubMed.
- A. F. Hussain, P. A. Heppenstall and F. Kampmeier, et al., One-step site-specific antibody fragment auto-conjugation using SNAP-tag technology, Nat. Protoc., 2019, 14, 3101–3125 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1cc03871k |
| ‡ Shared first authors. |
| This journal is © The Royal Society of Chemistry 2021 |