
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA boron–nitrogen transborylation enabled, borane-catalysed reductive cyanation of enones†
Kieran
Benn
a,
Kieran
Nicholson
 *a,
Thomas
Langer
b and
Stephen P.
Thomas
*a
*a,
Thomas
Langer
b and
Stephen P.
Thomas
*a
aEaStCHEM School of Chemistry, University of Edinburgh, Edinburgh EH9 3FJ, UK. E-mail: K.nicholson-3@sms.ed.ac.uk; stephen.thomas@ed.ac.uk
bPharmaceutical Technology & Development, Chemicals Development U.K., AstraZeneca, Macclesfield SK10 2NA, UK
First published on 24th August 2021
Abstract
Cyanation offers a simple method for the introduction of a nitrile group into organic molecules and an orthogonal route for the installation of a wide array of functional groups using simple transformations. Cyanation methods are dominated by transition metal catalysis and the use of hydrogen cyanide gas. Here, the electrophilic cyanation of enones was achieved using a main-group catalyst and a non-toxic, electrophilic cyanide source. This protocol was applied across a broad substrate scope including those containing reducible functional groups. Mechanistic studies indicated an amino-borane intermediate which underwent B–N transborylation (B–N/B–H exchange) to achieve catalytic turnover.
Nitriles are common synthetic intermediates which provide facile access to a wide array of functional groups including amines,1–3 ketones,4 amides,5 carboxylic acids6 and aldehydes,7 amungst others.8–10 Industrially, a nitrile group is most often introduced by metal-catalysed hydrocyanation, with the DuPont synthesis of adiponitrile being the seminal example.11 However, the acute toxicity of HCN (LD50 1.52 mg kg−1, b.p. 25.6 °C) has limited applications outside of specialist environments.12–14
Electrophilic cyanation offers an alternative strategy for the synthesis of nitriles, and one without the need for HCN gas. Main-group strategies12–15 for the cyanation of ketones have been developed which proceed by the generation of an enolate and reaction with an electrophilic cyanide source (Scheme 1a). These telescope, stoichiometric methods include the use of lithium,16 silicon17 and zinc enolates;18 in each case the use of stoichiometric base was required. Minakata used stoichiometric dialkylboranes (HBR2) for the 1,4-hydroboration and cyanation of enones without the requirement for stoichiometric base.19,20 Catalytic cyanation of α-bromo amides can be achieved with copper catalysis, however, this method is limited to sterically congested substrates.21
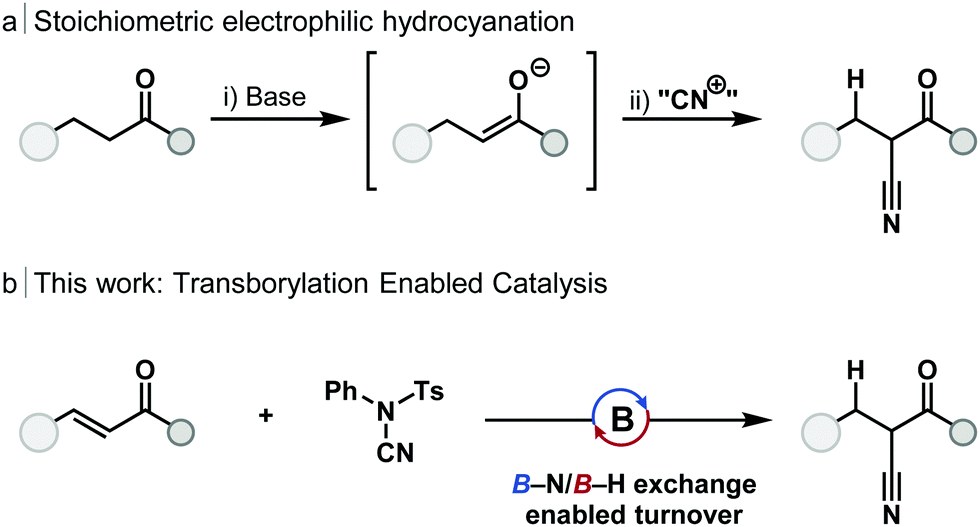 | ||
| Scheme 1 Electrophilic cyanation. | ||
Catalytic, non-metal methods for the synthesis of β-ketonitriles include the cyanation of silyl enol ethers22 catalysed by B(C6F5)3 and amine-catalysed23 reductive cyanation of β-keto esters and β-keto amides. However, a general strategy for main-group-catalysed cyanation without the requirement for pre-functionalised substrates would provide an advantage over existing methodologies.
Transborylation, a boron–boron exchange reaction akin to transmetallation, has emerged as a powerful mechanism to enable catalytic turnover using main-group species. Reaction between a heteroatom or carbo-dialkylborane and an appropriate dioxaborolane (e.g. HBpin, HBcat), through a σ-bond metathesis-type pathway, regenerates the borane catalyst and releases a boronic ester product.24 Transborylation has enabled borane-catalysed alkene25,26 and alkyne24–26 hydroboration, Midland reduction,27 chemoselective enone reduction,28 esterification, and Friedel–Crafts-type reactions.29 To date, transborylation has only been demonstrated at B–C and B–O bonds within catalysis. Application of transborylation to the catalytic, reductive cyanation of enones would require the development of, previously unreported, B–N/B–H exchange; B–N transborylation. In order to achieve catalytic turnover, a number of mechanistic challenges would need to be overcome: (1) control of regioselectivity of hydroboration and cyanation; (2) chemoselectivity for 1,4-hydroboration over 1,2-reduction of the alkene, or carbonyl group; (3) unwanted reaction of the electrophilic cyanation source with the borane catalyst must be avoided.
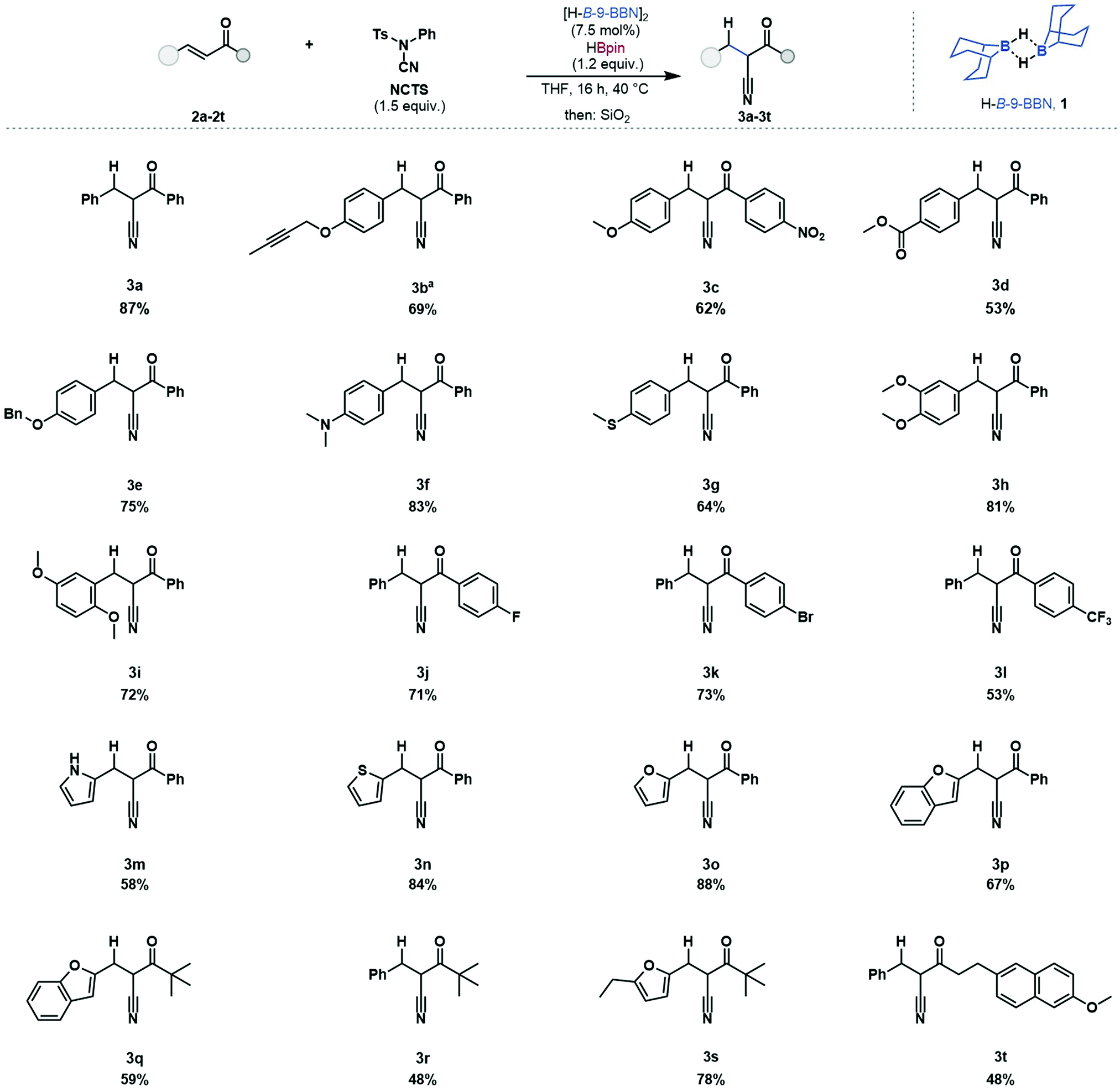
Investigations began with simple, dialkylboranes as potential catalysts and using HBpin as a turnover reagent. In line with stoichiometric reactivity,19N-cyano-N-phenyl-p-toluenesulfonamide (NCTS) was successfully applied as the cyanation reagent in catalysis, however, in contrast to the reported stoichiometric reactivity, tosylcyanide (TsCN)20 was not a suitable cyanation reagent. Commercially available [H-B-9-BBN]2 gave full conversion of chalcone to the β-ketonitrile product with full control of chemo- and regioselectivity. While catecholborane and Me2S·BH3 can undergo stoichiometric 1,4-hydroboration of enones to give an O–B enolate,30,31 these boranes were found to be catalytically incompetent, and dicyclohexylborane exhibited minimal catalytic activity (see ESI,† Table S1). This catalytic transformation was possible in several solvent systems but achieved the best results in THF, which is beneficial as [H-B-9-BBN]2 is commercially available as a 0.5 M solution in THF. Cyclic enones, α,β-unsaturated esters and amides were found to be unreactive in accordance with previous reports.28 The turnover reagent HBpin gave trace product without catalyst under reaction conditions (see ESI,† Table S1, entry 18).
The scope and limitations of this hydrocyanation protocol were investigated (Table 1). Chalcone 2a was reacted and 2-benzyl-3-(phenyl)-3-oxopropanenitrile was isolated in good yield (87%). Of particular focus were enones bearing reactive functionalities, incompatible with existing methods. This reaction protocol was applied to substrates bearing functionalities which react with metal hydrides, alkyne 2b (69%), nitro 2c (62%), ester 2d (53%) and benzyloxy 2e (75%) were successfully reacted without any observed side-reactivity such as hydroboration of the alkene or carbonyl moieties.32 The Lewis acidic catalyst was tolerant of substrates bearing Lewis basic and electron donating functionalities such as dimethylamino 2f (83%), thioether 2g (64%), and dimethoxy 2h (81%) 2i (72%) with no observed catalyst coordination or deactivation. Aryl halides were tolerated with substrates bearing fluoride 2j (71%) and bromide 2k (73%) substituents undergoing reductive cyanation. Aryl halide substituents can be problematic when using transition metal catalysts due to protodehalogenation.33 The inclusion of heteroaromatic structures was tolerated, including pyrrole 2m (58%), thiophene 2n (84%), furan 2o (88%) and benzofuran 2p (67%). The inclusion of alkyl-enones was successful, 2q (59%), 2r (48%) and 2s (78%). This reductive cyanation protocol was applied to a biologically active molecule, a derivative of Nabumetone (an anti-inflammatory drug) 2t (48%) which underwent reductive cyanation in good yield. Substrates 2b and 2m bear functional groups which have not been reported using borane-mediated cyanation and, in our hands, gave improved product yields when using the catalytic methodology (see ESI† for details).
| a Isolated as an inseparable mixture with starting material. |
|---|
|
O-Bpin enolate 4 did not react with NCTS (see ESI†) under reaction conditions, or in the presence of substoichiometric H-B-9-BBN, suggesting that nucleophilic attack occurs through the O-B-9-BBN enolate, and that boron–boron exchange at the enolate is limited (Scheme 2a). Minakata proposed an aminoborane side product, 5, from stoichiometric cyanation studies.19 The aminoborane 5 was prepared by reaction of tosylaniline with [H-B-9-BBN]2 (Scheme 2b) and used as a pre-catalyst for the hydrocyanation of chalcone 2a (Scheme 2c). This gave comparable yield to that using [H-B-9-BBN]2, which suggests aminoborane 5 is, or is in rapid equilibrium with, an on-cycle species. Thus, a mechanism for the borane-catalysed hydrocyanation of enones is proposed (Scheme 2d) (for alternative plausible mechanisms see ESI,† Scheme S4): the dissociation of [H-B-9-BBN]21, to a solvent coordinated monomer, followed by 1,4-hydroboration of the enone, 2 gives O-B-9-BBN enolate, 6. The nucleophilic boron enolate 6 reacts with the electrophilic cyanide-source, NCTS, resulting in borylated imine 7 which eliminates the cyanation product 3, and gives the amino-B-9-BBN, 5. This amino borane 5 then undergoes B–N/B–H transborylation with HBpin to regenerate the borane catalyst.
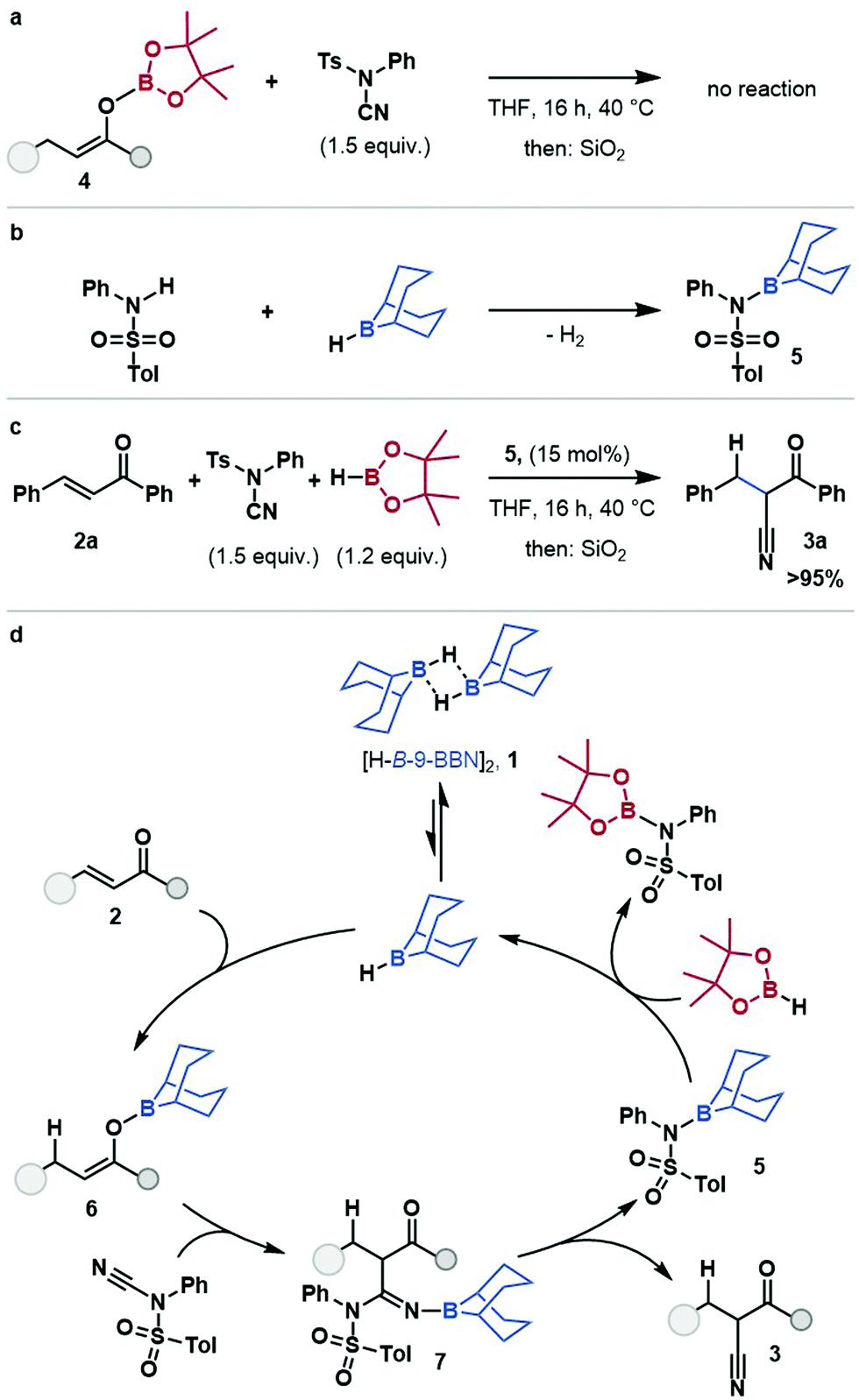 | ||
| Scheme 2 (a) Reaction of Bpin enolate, 4 with NCTS. (b) Synthesis of intermediate 5. (c) Aminoborane 5 as a pre-catalyst, yield determined by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard. (d) Proposed reaction mechanism. | ||
In summary, B–N/B–H transborylation has been used as a turnover strategy for the formal hydrocyanation of enones, transforming previously stoichiometric reagents into catalysts and provides a main-group-catalysed alternative to transition metal catalysis and stoichiometric strategies. The catalytic protocol showed excellent functional group tolerance and wide applicability with chemo- and regioselective enone hydrocyanation in all cases. Amino-B-9-BBN 5 was identified as a key intermediate and B–N/B–H transborylation with HBpin shown to provide catalytic turnover.
K. W. B. and K. N. completed all practical laboratory work. K. N. and S. P. T. conceived the reactions and wrote the manuscript. S. P. T. and T. L. advised investigations. S. P. T. thanks the Royal Society for a University Research Fellowship (URF/R/191015). S. P. T. and K. N. thank AstraZeneca and the EPSRC for an iCASE PhD studentship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. J. Ritter and J. Kalish, J. Am. Chem. Soc., 1948, 70, 4048–4050 CrossRef CAS PubMed.
- B. L. H. Amundsen and L. S. Nelson, J. Am. Chem. Soc., 1951, 73, 242–244 CrossRef.
- K. Lévay and L. Hegedűs, Curr. Org. Chem., 2019, 23, 1881–1900 CrossRef.
- C. G. Swain, J. Am. Chem. Soc., 1947, 69, 2306–2309 CrossRef CAS.
- J. J. Ritter and P. P. Minieri, J. Am. Chem. Soc., 1948, 70, 4045–4048 CrossRef CAS PubMed.
- V. K. Krieble and C. I. Noll, J. Am. Chem. Soc., 1939, 61, 560–563 CrossRef CAS.
- H. Stephen, J. Chem. Soc., Trans., 1925, 127, 1874–1877 RSC.
- L. Bosch and J. Vilarrasa, Angew. Chem., Int. Ed., 2007, 3926–3930 CrossRef CAS.
- V. Aureggi and G. Sedelmeier, Angew. Chem., Int. Ed., 2007, 46, 8440–8444 CrossRef CAS.
- B. Heller, B. Sundermann, H. Buschmann, H. Drexler, J. You, U. Holzgrabe, E. Heller and G. Oehme, J. Org. Chem., 2002, 67, 4414–4422 CrossRef CAS PubMed.
- H. Zhang, X. Su and K. Dong, Org. Biomol. Chem., 2020, 18, 391–399 RSC.
- X. Fang, P. Yu and B. Morandi, Science, 2016, 351, 832–836 CrossRef CAS PubMed.
- B. N. Bhawal and B. Morandi, Angew. Chem., Int. Ed., 2019, 58, 10074–10103 CrossRef CAS PubMed.
- A. Bhunia, K. Bergander and A. Studer, J. Am. Chem. Soc., 2018, 140, 16353–16359 CrossRef CAS PubMed.
- P. Orecchia, W. Yuan and M. Oestreich, Angew. Chem., Int. Ed., 2019, 58, 3579–3583 CrossRef CAS.
- D. Kahne and D. B. Collum, Tetrahedron Lett., 1981, 22, 5011–5014 CrossRef CAS.
- H. Shen, J. Li, Q. Liu, J. Pan, R. Huang and Y. Xiong, J. Org. Chem., 2015, 80, 7212–7218 CrossRef CAS PubMed.
- X. Ren, C. Shen, G. Wang, Z. Shi, X. Tian and K. Dong, Org. Lett., 2021, 23, 2527–2532 CrossRef PubMed.
- K. Kiyokawa, T. Nagata and S. Minakata, Angew. Chem., Int. Ed., 2016, 55, 10458–10462 CrossRef CAS PubMed.
- T. Nagata, A. Tamaki, K. Kiyokawa, R. Tsutsumi, M. Yamanaka and S. Minakata, Chem. – Eur. J., 2018, 24, 17027–17032 CrossRef CAS PubMed.
- N. Miwa, C. Tanaka, S. Ishida, G. Hirata, J. Song, T. Torigoe, Y. Kuninobu and T. Nishikata, J. Am. Chem. Soc., 2020, 142, 1692–1697 CrossRef CAS PubMed.
- T. Nagata, H. Matsubara, K. Kiyokawa and S. Minakata, Org. Lett., 2017, 19, 4672–4675 CrossRef CAS PubMed.
- B. Ma, X. Lin, L. Lin, X. Feng and X. Liu, J. Org. Chem., 2017, 82, 701–708 CrossRef CAS.
- N. W. J. Ang, C. S. Buettner, S. Docherty, A. Bismuto, J. R. Carney, J. H. Docherty, M. J. Cowley and S. P. Thomas, Synthesis, 2018, 803–808 CAS.
- J. H. Docherty, K. Nicholson, A. P. Dominey and S. P. Thomas, ACS Catal., 2020, 10, 4686–4691 CrossRef CAS.
- E. Nieto-Sepulveda, A. D. Bage, L. A. Evans, T. A. Hunt, A. G. Leach, S. P. Thomas and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2019, 141, 18600–18611 CrossRef CAS PubMed.
- K. Nicholson, J. Dunne, P. Dabell, A. B. Garcia, A. D. Bage, J. H. Docherty, T. A. Hunt, T. Langer and S. P. Thomas, ACS Catal., 2021, 11, 2034–2040 CrossRef CAS.
- K. Nicholson, T. Langer and S. P. Thomas, Org. Lett., 2021, 23, 2498–2504 CrossRef PubMed.
- D. R. Willcox, G. S. Nichol and S. P. Thomas, ACS Catal., 2021, 11, 3190–3197 CrossRef CAS.
- Y. Matsumoto and T. Hayashi, Synlett, 1991, 349–350 CrossRef CAS.
- D. A. Evans and G. C. Fu, J. Org. Chem., 1990, 55, 5678–5680 CrossRef CAS.
- H. C. Brown and S. Krishnamurthy, Tetrahedron, 1979, 35, 567–607 CrossRef CAS.
- F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 11, 4009–4091 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of materials and methods, characterization of materials and reaction results. See DOI: 10.1039/d1cc03649a |
| This journal is © The Royal Society of Chemistry 2021 |