
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An alginate-confined peroxygenase-CLEA for styrene epoxidation†
Friederike E. H.
Nintzel‡
 a,
Yinqi
Wu‡
a,
Matteo
Planchestainer
b,
Martin
Held
b,
Miguel
Alcalde
c and
Frank
Hollmann
*a
a,
Yinqi
Wu‡
a,
Matteo
Planchestainer
b,
Martin
Held
b,
Miguel
Alcalde
c and
Frank
Hollmann
*a
aDepartment of Biotechnology, Delft University of Technology, van der Maasweg 9, 2629 HZ, Delft, The Netherlands. E-mail: f.hollmann@tudelft.nl
bDepartment of Biosystems Science and Engineering, ETH Zürich, Mattenstrasse 26, Basel 4058, Switzerland
cDepartment of Biocatalysis, Institute of Catalysis and Petrochemistry (CSIC), Madrid, Spain
First published on 7th May 2021
Abstract
Oxyfunctionalisation reactions in neat substrate still pose a challenge for biocatalysis. Here, we report an alginate-confined peroxygenase-CLEA to catalyse the enantioselective epoxidation of cis-β-methylstyrene in a solvent-free reaction system achieving turnover numbers of 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 for the biocatalyst and epoxide concentrations of 48 mM.
000 for the biocatalyst and epoxide concentrations of 48 mM.
Biocatalytic oxyfunctionalisation reactions are enjoying an increasing interest in organic chemistry.1 Especially the often very high regio- and enantioselectivity of enzymatic oxyfunctionalisation reactions such as hydroxylations or epoxidations offers synthetic chemists straightforward access to chiral building blocks, which with traditional chemical means are difficult to prepare.
Next to the well-known P450 monooxygenases,2 in recent years also peroxygenases3 have been in the centre of attention. P450 monooxygenases reductively activate molecular oxygen to form the catalytically active oxyferryl-heme species. Peroxygenases form this species directly from partially reduced oxygen species (peroxides) and thereby circumvent the complex molecular architectures of P450 monooxygenases (Scheme 1).
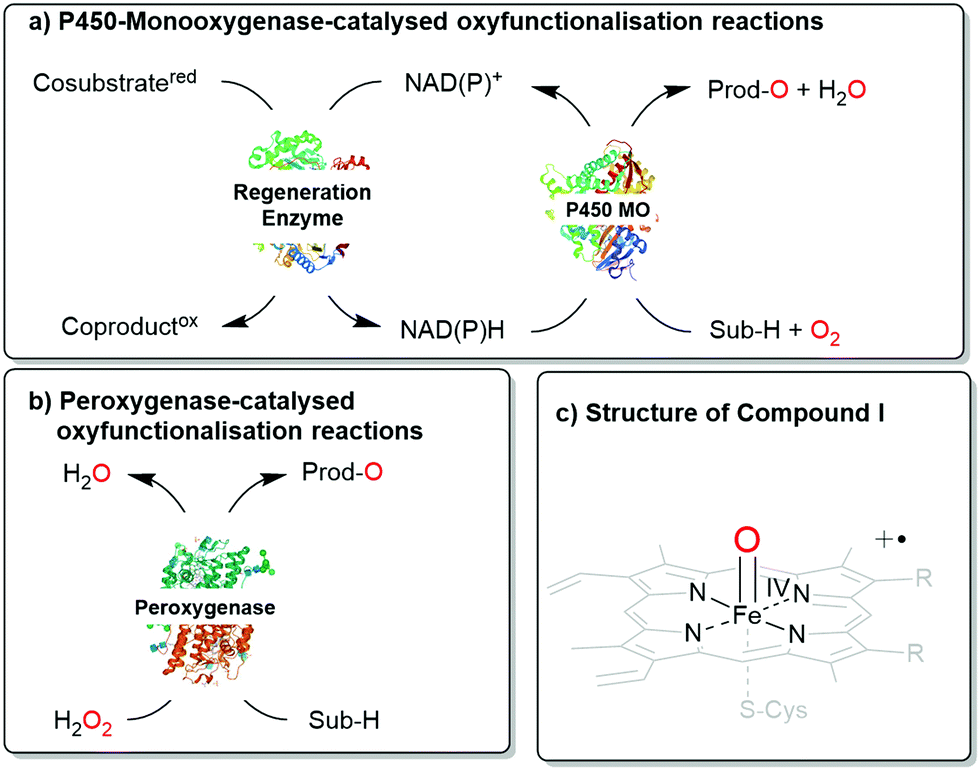 | ||
| Scheme 1 Comparison of (a) P450 monooxygenase-catalysed and (b) peroxygenase-catalysed oxyfunctionalisation reactions. (c) Both enzyme classes utilise Compound I as the oxygenating agent. | ||
The synthetic application of both enzyme classes, however, still suffers from the poor water solubility of the majority of starting materials, resulting in rather dilute reaction mixtures with an enormous water footprint. Therefore, increasing the starting material (and product) concentration is of utmost importance to increase the economic viability and environmental friendliness of such biocatalytic reactions.4
One common approach to increase the substrate loading is to use the so-called two liquid phase approach5 in which an aqueous, biocatalyst-containing layer is contacted with a hydrophobic, organic layer serving as substrate reservoir and product sink. To alleviate possible phase transfer rate limitations of this system, intensive mechanical stirring is needed, which however, also may impair the stability of the biocatalyst.6
The latter issue can be addressed by immobilising the biocatalyst to a heterogeneous carrier material and thereby physically protecting the enzyme. While a limited number of studies report immobilisation of peroxygenases, this technique is not fully explored yet for this enzyme class.7–12 In previous works, we could demonstrate that immobilised peroxygenases in principle can even be applied in neat (i.e. almost water-free) reaction media.9 A drawback of this approach, however, was the very poor specific activity of the immobilised enzyme, possibly due to a combination of activity losses of the enzyme during immobilisation and further activity losses originating from dehydratation of the enzyme surface.
Encapsulating enzymes in alginate matrices may represent an elegant compromise. The so-confined enzymes are mechanically stabilised while still situated in a micro-aqueous environment.8,11,13
We therefore set out to immobilise a peroxygenase in an alginate matrix and evaluate its catalytic activity under non aqueous reaction conditions. As model enzyme we chose the recombinantly expressed, evolved peroxygenase from Agrocybe aegerita (rAaeUPO)14–16 as catalyst for the epoxidation of styrene and cis-β-methylstyrene (Scheme 2). The biocatalyst was obtained from the supernatant of the fermentation broth of recombinant Pichia pastoris and used without further purification.
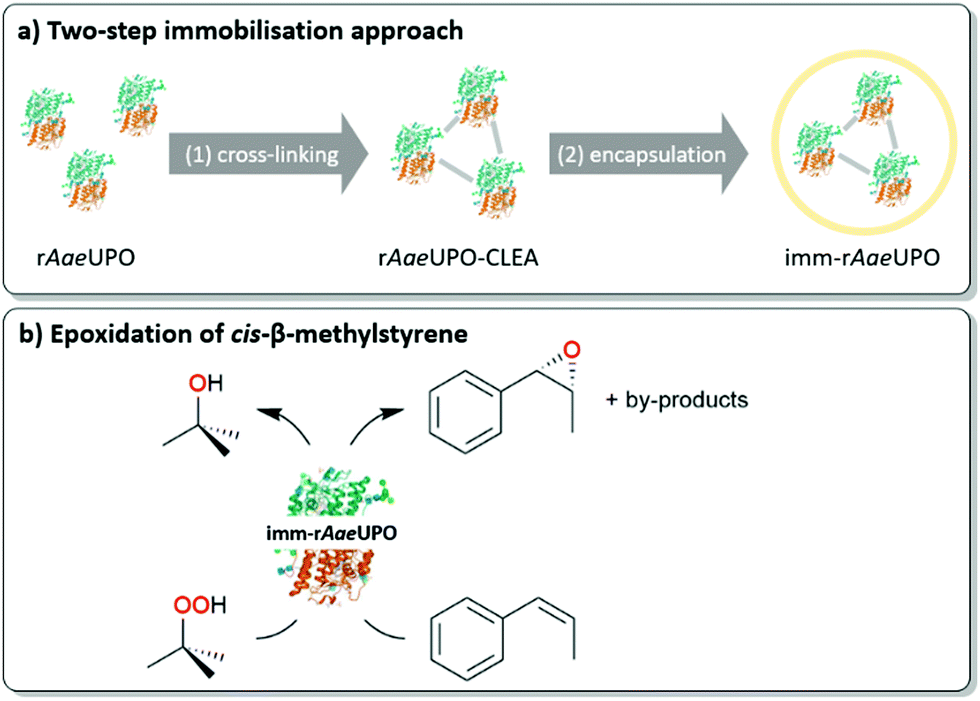 | ||
| Scheme 2 (a) Overview of the utilised immobilisation approach consisting of CLEA formation and alginate confinement. (b) Epoxidation of cis-β-methylstyrene by immobilised rAaeUPO with tert-butyl hydroperoxide (tBuOOH) as oxidant. | ||
Confining rAaeUPO in Ca2+-hardened alginates proved to be feasible. To our surprise, however, the resulting immobilisate showed low, and somewhat irreproducible catalytic activity (Fig. 1a), which most likely was due to leaching of the enzyme from the beads during the immobilisation procedure and storage. To improve the retention of the biocatalyst in the alginate beads, we decided to increase its molecular mass by covalent cross-linking (CLEA formation).17–19
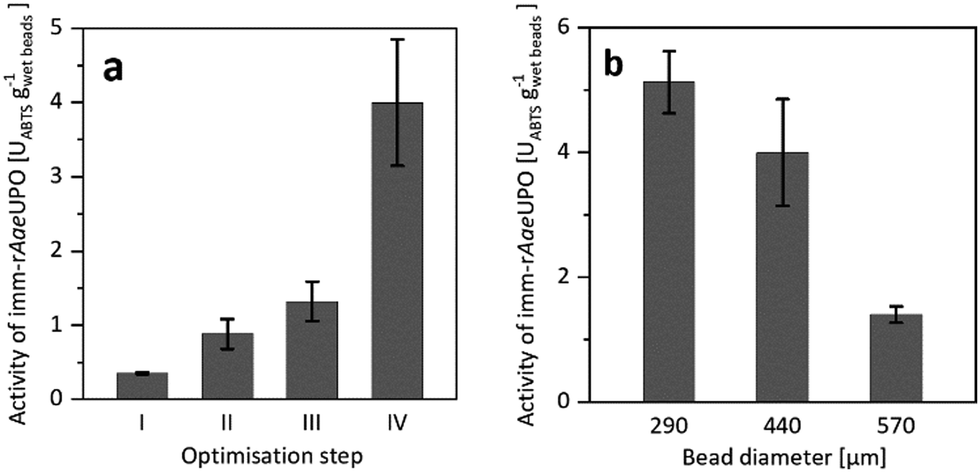 | ||
| Fig. 1 (a) Activity of imm-rAaeUPO during optimisation of the immobilisation procedure, determined by ABTS-activity assay in aqueous environment. Reference (I) is the encapsulation of free rAaeUPO. The catalytic activity of imm-rAaeUPO was improved by CLEA formation (II), by application of chitosan as coagulant (III), and by maximising the enzyme load (IV). (b) Activity of imm-rAaeUPO immobilisates with different diameters, determined by ABTS-activity assay in aqueous environment. Data represents the average of duplicates. Further information on the immobilisation optimisation can be found in the ESI.† | ||
Indeed, CLEA formation more than doubled the catalytic activity of the immobilised peroxygenase (Fig. 1a). Further improvements were achieved by using chitosan as coagulant20–23 and by increasing the enzyme load (Fig. 1a). Additional optimisation steps are reported in the ESI.† It is important to note that the size of the beads had a significant influence on the activity of the immobilised peroxygenase. The larger the beads, the lower the catalytic activity under otherwise identical conditions (Fig. 1b).
Overall, approximately 19% of the enzyme was immobilised (as determined via quantification of the amount of active heme sites using CO-differential spectra) and 11.4% of the peroxidase activity, as judged by the ABTS oxidation activity, was found back in the immobilisates. It is worth mentioning here that rAaeUPO immobilisation also increased its storage stability. While the free enzyme completely lost its catalytic activity after 12 days storage at room temperature, the immobilised version exhibited at least 80% of its initial activity even after two weeks (Fig. S10, ESI†).
Having the immobilised rAaeUPO preparation at hand, we decided to compare its catalytic performance in the epoxidation of cis-β-methylstyrene with the free enzyme in a two liquid phase approach (Fig. 2) using tBuOOH as oxidant.
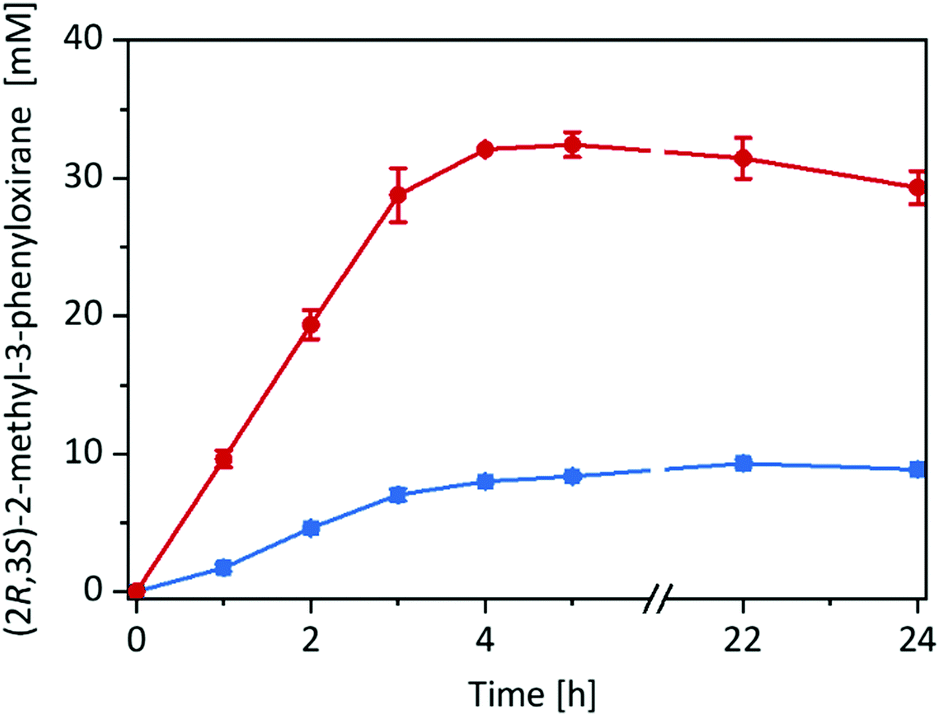 | ||
| Fig. 2 Product formation over time for the epoxidation of cis-β-methylstyrene in different reaction systems: imm-rAaeUPO in neat substrate (180 mg immobilisate in 570 μL cis-β-methylstyrene) (red circles), and free rAaeUPO in a two-liquid phase system (180 μL TRIS-HCl buffer (20 mM, pH 7): 570 μL cis-β-methylstyrene) (blue squares). General reaction conditions: [rAaeUPO] = 0.5 μM, tBuOOH feeding rate = 10 mM h−1, room temperature, shaking at 99 rpm with 60° angle in an overhead rotator. Data represents the average of duplicates. | ||
Very much to our surprise the immobilised enzyme outperformed the free enzyme under otherwise identical conditions (such as volumetric ratio of aqueous or alginate volume to the organic phase and enzyme concentrations). With the immobilised enzyme the product accumulation rate was approx. 10 mM h−1 (corresponding to the tBuOOH feed rate) while with the free enzyme it was only 3 mM h−1. A plausible explanation for this may be the higher surface area of the reactions using alginate-immobilised rAaeUPO, largely eliminating the diffusion rate limitation of tBuOOH and/or cis-β-methylstyrene into the aqueous reaction phase (Fig. S15, ESI†).
Overall, in this experiment, approx. 30 mM of enantiomerically pure (2R,3S)-2-methyl-3-phenyloxirane has been synthesised within 4 h corresponding to a turnover number (TN = molProduct × molrAaeUPO−1) for the enzyme of 60000 and an average (over 4 h) turnover frequency of 4.1 s−1. It should be mentioned here, that under these reaction conditions several side products such as benzaldehyde and phenylacetone were observed (vide infra).
Despite the promising results, the reactions stopped after approximately 5 h. We suspected the irreversible, oxidative inactivation of the heme-containing biocatalyst by the hydroperoxide to account for the low robustness of the reaction.
Therefore, we performed a series of experiments varying the tBuOOH addition rate (Fig. 3). While the initial product formation rate decreased with decreasing tBuOOH feeding rates, the long-term robustness of the reaction increased: at a tBuOOH feeding rate of 20 mM h−1, product formation rates of 9 mM h−1 were observed but the product accumulation ceased after 4 h. Applying a tBuOOH feeding rate of 1 mM h−1 approximately the same product formation rate was observed, albeit for at least 72 h. Also the formation of the undesired side products decreased considerably with lower tBuOOH feeding rates (Fig. S19, ESI†). Both observations are most likely related to each other. At high tBuOOH feeding rates, the peroxide availability exceeds the enzyme's epoxidation capacity resulting in oxidative inactivation of the heme prosthetic group and release of iron ions. The latter catalyse Fenton-like transformations resulting in non-selective oxidation of the cis-β-methylstyrene starting material and formation of the undesired side-products.
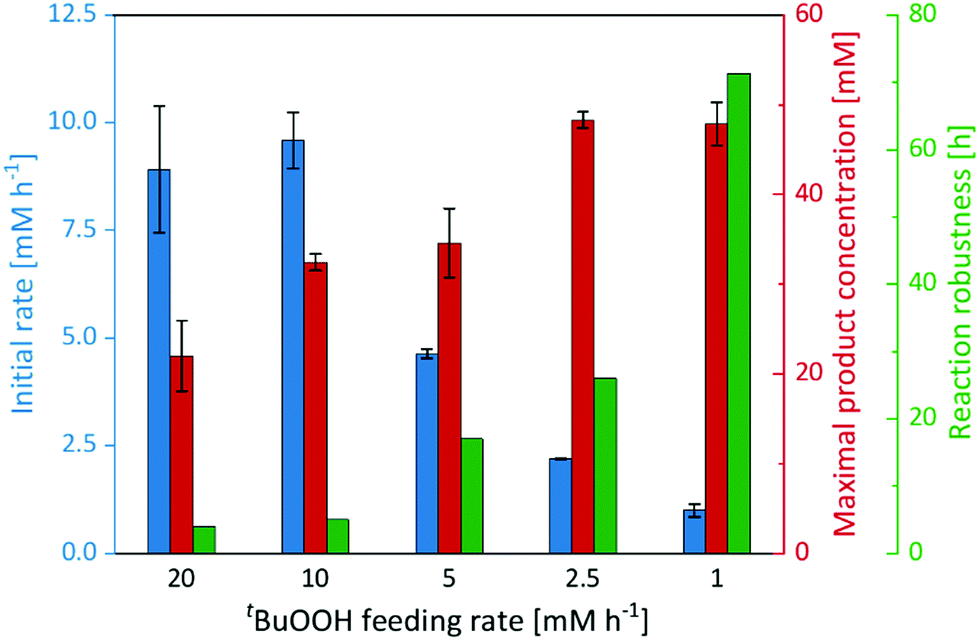 | ||
| Fig. 3 Initial rate, maximal product concentration and reaction robustness of the epoxidation of cis-β-methylstyrene catalysed by imm-rAaeUPO at different tBuOOH feeding rates. Reaction set-up: 570 μL cis-β-methylstyrene, supplemented with 180 mg immobilisate and continuously fed with the indicated tBuOOH rate. General reaction conditions: [rAaeUPO] = 0.5 μM, room temperature, shaking at 99 rpm with 60° angle in an overhead rotator. Data represents the average of duplicates. | ||
In any case, lowering the tBuOOH feeding rate not only increased the robustness of the reaction but also reduced the side-product formation. Under these conditions, 48 mM (6.4 g L−1) of enantiomerically pure (2R,3S)-2-methyl-3-phenyloxirane have been synthesised within 72 h. This corresponds to excellent TN of 96000 for the biocatalyst. This catalytic performance also favourably compares to other enzyme systems such as flavin-dependent monooxygenases.24–27 Admittedly, the current substrate scope of the proposed immobilised rAaeUPO preparation is rather limited, preliminary experiments on styrene epoxidation indicate a similar catalytic potential for this substrate (Fig. S21, ESI†). Future experiments will broaden the synthetic scope of the proposed rAaeUPO preparation.
To obtain a first overview over the environmental impact of the reaction system established in this study, we used Sheldon's E-factor28,29 to estimate the wastes generated in the formation of (2R,3S)-2-methyl-3-phenyloxirane. As shown in Table 1, a total of 153.3 kg of waste was generated per kg of the desired product. 70% of the E-factor contribution stems from non-reacted starting material, which in a putative preparative-scale reaction can be recovered via distillation. The second-largest contributor (24%) is the enzyme preparation. The latter is mostly comprised of the alginate beads and buffer (making 99% of the total mass of the immobilised enzyme).
| a It is assumed that all added tBuOOH converted into tBuOH. The indicated tBuOH mass includes decane which was used for dilution of tBuOOH. b Concentrations and masses of side products were estimated based on the GC calibration line and response factor of (2R,3S)-2-methyl-3-phenyloxirane. c Mass of cis-β-methylstyrene after reaction stop is estimated based on formation of epoxide and side products. d rAaeUPO includes imm-rAaeUPO beads. | ||
|---|---|---|
| Products | (2R,3S)-2-methyl-3-phenyloxirane | 4.8 |
| t BuOHa | 41.0 | |
| Various side productsb | 0.9 | |
| Reactants | cis-β-Methylstyrenec | 515.9 |
| t BuOOHa | 0.0 | |
| Catalyst | rAaeUPOd | 180.0 |
| E-factor | SUM (waste) | 737.9 |
| SUM (epoxide product) | 4.8 | |
| E-factor | 153.3 | |
We are convinced that further optimisation of the immobilisation protocol will improve the rAaeUPO loading in the alginate beads. For example using alginate-in-oil emulsions for the bead preparation will certainly improve the rAaeUPO loading.30 Also the E-factor contribution of the oxidant and its by-product tBuOH, respectively, can be reduced significantly when co-immobilising the formate oxidase from Aspegillus oryzae (AoFOX) to use methanol as sacrificial electron donor for the in situ generation of H2O2.31–34 Implementing this system will eliminate the tBuOH contribution to the E-factor (8.5 kg kg−1) and reduce it to approx. 0.11 kgCO2 kgProduct−1.
Overall, in this contribution we have established alginate-confined peroxygenase-CLEAs as practical enzyme preparations for the selective epoxidation of styrene derivates such as cis-β-methylstyrene to synthesise enantiomerically pure epoxides. In terms of catalyst efficiency (more than 90000 catalytic cycles observed), the current system outperforms comparable reaction systems using chemical catalysts,35 P450 monooxygenases36 or other established enzymatic systems,26,27,37 also compared to our previous efforts using immobilised rAaeUPO.9 We are convinced that further optimisation will bring this approach to maturity and will establish an economically and environmentally feasible reaction system.
Financial support by the China Scholarship Council, the from the German Academic Exchange Service and the German Academic Scholarship Foundation as well as the European Research Council (ERC Consolidator Grant No. 648026) is gratefully acknowledged.
F. E. H. N., Y. W. and M. P. have performed the experiments and analysed the results. The study was conceptualised by M. H., M. A. and F. H. All authors contributed to the manuscript writing.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Dong, E. Fernández-Fueyo, F. Hollmann, C. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes and W. Zhang, Angew. Chem., Int. Ed., 2018, 57, 9238–9261 CrossRef CAS PubMed.
- V. B. Urlacher and M. Girhard, Trends Biotechnol., 2019, 37, 882–897 CrossRef CAS PubMed.
- M. Hobisch, D. Holtmann, P. G. de Santos, M. Alcalde, F. Hollmann and S. Kara, Biotechnol. Adv., 2021, 107615, DOI:10.1016/j.biotechadv.2020.107615.
- Y. Ni, D. Holtmann and F. Hollmann, ChemCatChem, 2014, 6, 930–943 CrossRef CAS.
- M. van Schie, J.-D. Spöring, M. Bocola, P. Dominguez de Maria and D. Rother, Green Chem., 2021 10.1039/D1GC00561H.
- E. Churakova, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2013, 5, 565–568 CrossRef CAS.
- A. Yayci, T. Dirks, F. Kogelheide, M. Alcalde, F. Hollmann, P. Awakowicz and J. E. Bandow, J. Phys. D: Appl. Phys., 2021, 54, 035204 CrossRef CAS.
- M. Hobisch, M. M. C. H. van Schie, J. Kim, K. Røjkjær Andersen, M. Alcalde, R. Kourist, C. B. Park, F. Hollmann and S. Kara, ChemCatChem, 2020, 12, 4009–4013 CrossRef CAS.
- M. C. R. Rauch, F. Tieves, C. E. Paul, I. W. Arends, M. Alcalde and F. Hollmann, ChemCatChem, 2019, 11, 4519–4523 CrossRef CAS PubMed.
- E. Fernández-Fueyo, Y. Ni, A. Gomez Baraibar, M. Alcalde, L. M. van Langen and F. Hollmann, J. Mol. Catal. B: Enzym., 2016, 134, 347–352 CrossRef.
- M. Poraj-Kobielska, S. Peter, S. Leonhardt, R. Ullrich, K. Scheibner and M. Hofrichter, Biochem. Eng. J., 2015, 98, 144–150 CrossRef CAS.
- P. Molina-Espeja, P. Santos-Moriano, E. García-Ruiz, A. Ballesteros, F. J. Plou and M. Alcalde, Int. J. Mol. Sci., 2019, 20, 1627 CrossRef CAS PubMed.
- L. Fernandez-Arrojo, B. Rodriguez-Colinas, P. Gutierrez-Alonso, M. Fernandez-Lobato, M. Alcalde, A. O. Ballesteros and F. J. Plou, Process Biochem., 2013, 48, 677–682 CrossRef CAS.
- R. Ullrich, J. Nüske, K. Scheibner, J. Spantzel and M. Hofrichter, Appl. Environ. Microbiol., 2004, 70, 4575–4581 CrossRef CAS PubMed.
- P. Molina-Espeja, S. Ma, D. M. Mate, R. Ludwig and M. Alcalde, Enzyme Microb. Technol., 2015, 73–74, 29–33 CrossRef CAS PubMed.
- P. Molina-Espeja, E. Garcia-Ruiz, D. Gonzalez-Perez, R. Ullrich, M. Hofrichter and M. Alcalde, Appl. Environ. Microbiol., 2014, 80, 3496–3507 CrossRef PubMed.
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC.
- C. Mateo, J. M. Palomo, L. M. van Langen, F. van Rantwijk and R. A. Sheldon, Biotechnol. Bioeng., 2004, 86, 273–276 CrossRef CAS PubMed.
- L. Q. Cao, F. van Rantwijk and R. A. Sheldon, Org. Lett., 2000, 2, 1361–1364 CrossRef CAS PubMed.
- M.-Q. Xu, S.-S. Wang, L.-N. Li, J. Gao and Y.-W. Zhang, Catalysts, 2018, 8, 460 CrossRef.
- S. Velasco-Lozano, F. López-Gallego, J. C. Mateos-Díaz and E. Favela-Torres, Biocatalysis, 2016, 166–177, DOI:10.1515/boca-2015-0012.
- S. Talekar, A. Joshi, G. Joshi, P. Kamat, R. Haripurkar and S. Kambale, RSC Adv., 2013, 3, 12485–12511 RSC.
- D. Grajales-Hernández, M. Armendáriz-Ruiz, S. Velasco-Lozano, F. López-Gallego and J. C. Mateos-Díaz, Appl. Microbiol. Biotechnol., 2020, 104, 10033–10045 CrossRef PubMed.
- C. E. Paul, D. Tischler, A. Riedel, T. Heine, N. Itoh and F. Hollmann, ACS Catal., 2015, 5, 2961–2965 CrossRef CAS.
- H. Toda, R. Imae, T. Komio and N. Itoh, Appl. Microbiol. Biotechnol., 2012, 96, 407–418 CrossRef CAS PubMed.
- K. Hofstetter, J. Lutz, I. Lang, B. Witholt and A. Schmid, Angew. Chem., Int. Ed., 2004, 43, 2163–2166 CrossRef CAS PubMed.
- A. Schmid, K. Hofstetter, H.-J. Feiten, F. Hollmann and B. Witholt, Adv. Synth. Catal., 2001, 343, 732–737 CrossRef CAS.
- R. A. Sheldon, Green Chem., 2017, 19, 18–43 RSC.
- R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- L. Mazutis, R. Vasiliauskas and D. A. Weitz, Macromol. Biosci., 2015, 15, 1641–1646 CrossRef CAS PubMed.
- S. J. P. Willot, M. D. Hoang, C. E. Paul, M. Alcalde, I. Arends, A. S. Bommarius, B. Bommarius and F. Hollmann, ChemCatChem, 2020, 12, 2713–2716 CrossRef CAS.
- F. Tieves, S. J.-P. Willot, M. M. C. H. van Schie, M. C. R. Rauch, S. H. H. Younes, W. Zhang, P. G. de Santos, J. M. Robbins, B. Bommarius, M. Alcalde, A. Bommarius and F. Hollmann, Angew. Chem., Int. Ed., 2019, 58, 7873–7877 CrossRef CAS PubMed.
- J. M. Robbins, A. S. Bommarius and G. Gadda, Arch. Biochem. Biophys., 2018, 643, 24–31 CrossRef CAS PubMed.
- J. M. Robbins, M. G. Souffrant, D. Hamelberg, G. Gadda and A. S. Bommarius, Biochemistry, 2017, 56, 3800–3807 CrossRef CAS PubMed.
- D. Banerjee, R. V. Jagadeesh, K. Junge, M.-M. Pohl, J. Radnik, A. Brückner and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 4359–4363 CrossRef CAS PubMed.
- M. R. Sarkar, J. H. Z. Lee and S. G. Bell, ChemBioChem, 2017, 18, 2119–2128 CrossRef CAS PubMed.
- H. Toda, R. Imae and N. Itoh, Tetrahedron: Asymmetry, 2012, 22–23, 1542–1549 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed information about synthetic and analytical procedures as well as additional experimental results. See DOI: 10.1039/d1cc01868j |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |