
C–CN bond formation: an overview of diverse strategies
Sandeep
Pimparkar†
 a,
Adithyaraj
Koodan†
a,
Siddhartha
Maiti
b,
Nesreen S.
Ahmed
c,
Mohamed Mokhtar M.
Mostafa
*d and
Debabrata
Maiti
*a
a,
Adithyaraj
Koodan†
a,
Siddhartha
Maiti
b,
Nesreen S.
Ahmed
c,
Mohamed Mokhtar M.
Mostafa
*d and
Debabrata
Maiti
*a
aDepartment of Chemistry, IIT Bombay, Powai, Mumbai 400076, India. E-mail: dmaiti@iitb.ac.in
bVIT Bhopal University, Bhopal-Indore Highway, Kothrikalan, Sehore, Madhya Pradesh-466114, India
cDepartment of Therapeutic Chemistry, National Research Centre, Dokki, Cairo-12622, Egypt
dChemistry Department, Faculty of Science, King Abdulaziz University, P.O. Box-80203, Jeddah-21589, Saudi Arabia. E-mail: mmoustafa@kau.edu.sa
First published on 21st January 2021
Abstract
Nitrile or cyano compounds are an important part of structural motifs in dyes, agrochemicals, medicinal compounds, and electronic materials. Also, aryl nitrile is an important intermediate in the preparation of numerous compounds via transformations such as hydrolysis, hydration, reduction, cycloadditions, and nucleophilic additions. Such methods are beneficial for introducing sensitive functional groups in various positions in the multi-step synthesis of natural products and medicinal compounds. In the past decades, various cyanation methods have been reported in the vast arena of chemistry, which have made several building blocks accessible. Previously reported cyanation reviews, letters, and perspectives are written in parts. Thus, today a comprehensive review that will be able to guide readers through the vast pool of C–CN bond forming reactions via different approaches is obligatory. The present feature article depicts the various areas of cyanation methodologies that are based on the metal catalyst used, directed, non-directed, electrochemical, photochemical, asymmetric, and radical based approaches. This feature article will serve as a comprehensive tool to navigate the C–CN (cyanation) reactions across the vast area in synthetic chemistry.
 Sandeep Pimparkar | Dr. Sandeep Pimparkar was born and brought up in Nasik (Maharashtra) India. After his MSc in organic chemistry, he joined as a junior research fellow at IISER Pune, India with Dr. M. Jeganmohan. Later, in 2015, he joined Prof. Debabrata Maiti's research group for PhD at IIT Bombay (IITB-Monash Research Academy). During his PhD, he mainly studied the design & discovery of templates for distal C–H activation of aliphatic and aromatics by using transition metal catalysis. |
 Adithyaraj Koodan | Mr. Adithyaraj Koodan was born and brought up in Kerala, India. He obtained his Integrated MSc in chemistry from Integrated Science Education and Research Centre, Visva-Bharati, India. He did his final year MSc dissertation project under Prof. Debabarata Maiti from Indian Institute of Technology Bombay, India on transition metal catalyzed remote C–H functionalization reactions. |
 Siddhartha Maiti | Prof. Dr Siddhartha Maiti was born in India. In 2016 for his PhD at the University of Massachusetts Dartmouth (USA), he studied the development of fluorescent sensor for the bioimaging of iron(II) ions under the supervision of Prof. Maolin Guo. He then studied the amidosulfates-mediated peptide formation under potential early-earth conditions at the Indian Institute of Technology, Bombay with Prof. Samir Maji, and Prof. Debabrata Maiti. He is now appointed as Assistant Professor at Vellore Institute of Technology, Bhopal, India in the Bioengineering department. |
 Nesreen S. Ahmed | Prof. Dr Nesreen S. Ahmed completed her Master of Science in 1996 from Ain Shams University, Cairo, Egypt and a PhD in 2000 from the same University. She was an assistant Professor of Organic Chemistry at the Department of Therapeutic Chemistry, Pharmaceutical, and Drug Industries Research Division, National Research Center (NRC), Dokki, Cairo, Egypt. She was delegated as Associate Professor (2006–2018) in the Department of Chemistry at the King Abdulaziz University (KAU), Jeddah, Saudi Arabia. Her scientific interest is in medicinal chemistry, heterocyclic compounds, and green chemistry. She is experienced in the preparation, elucidation, and biological tests of new organic compounds. |
 Mohamed Mokhtar M. Mostafa | Prof. Dr Mohamed Mokhtar got his Master's degree in 1993 from Zagazig University and PhD in 1997 from Cairo University, Cairo, Egypt. He was a researcher from DAAD, in Karlsruhe, Germany in 1998–1999. He was invited as a research fellow to the Surface Chemistry and Catalysis Department, Ulm University, Ulm, Germany in 2003 and at Erlangen-Nuremberg University (FAU), Germany in 2007. His dual roles at the NRC and the KAU are just the latest in a string of leadership roles he has held in the scientific research field for almost 30 years. His focus is in advanced materials and heterogeneous catalysis. |
 Debabrata Maiti | Prof. Dr Debabrata Maiti received his PhD from John Hopkins University (USA) in 2008 under the supervision of Prof. Kenneth D. Karlin. After postdoctoral studies at the Massachusetts Institute of Technology (MIT) with Prof. Stephen L. Buchwald (2008–2010), he joined the Department of Chemistry at IIT Bombay in 2011. His research interests are focused on the development of new and sustainable synthetic and catalytic methods, aliphatic & aromatic distal C–H activation, photocatalysis, electrocatalysis, heterocycle synthesis, and lignin valorisation. |
Introduction
Nitrile or cyano (CN) is one of the versatile synthons in synthetic chemistry because of its ability to transform into other functional groups such as carbonyls and amines.1a–c Also, the introduction of the nitrile group on a bioactive molecule or another functional molecule can alter its properties.1d Cyano group is found as an integral part of natural products, dyes, herbicides, agrochemicals, and pharmaceuticals.1e,f Sandmeyer and Rosenmund–von Braun reactions were the most promising methods for the cyanation of arenes at laboratory and industrial scales but these past couple of decades have seen a remarkable development of newer approaches and newer sources of –CN to prepare nitriles from various coupling partners. In general, aryl cyanides are prepared from activated aryl–X coupling partners (where X can be halides or OTf (trifluoromethanesulfonate)) by transition metal catalysis with various cyanation sources such as CuCN, KCN, NaCN, Zn(CN)2, and organic cyanation sources, namely, TMSCN (trimethylsilylcyanide), acetone cyanohydrin, DMF (dimethylformamide), and NCTS (N-cyano-N-phenyl-p-toluenesulfonamide). Not only activated but also un-activated arenes have been cyanated by transition metal catalysis utilizing suitable directing groups, which could also promote cyanation at the distal positions of arenes. The present review depicts all the important types of cyanation for aromatic, aliphatic, and heterocyclic compounds by classifying them in various categories including transition metal-catalyzed transformation of aryl halides, cyanation of directed & non-directed arene/heteroarenes, asymmetric, electro-catalyzed and photocatalyzed cyanation of arenes (Scheme 1).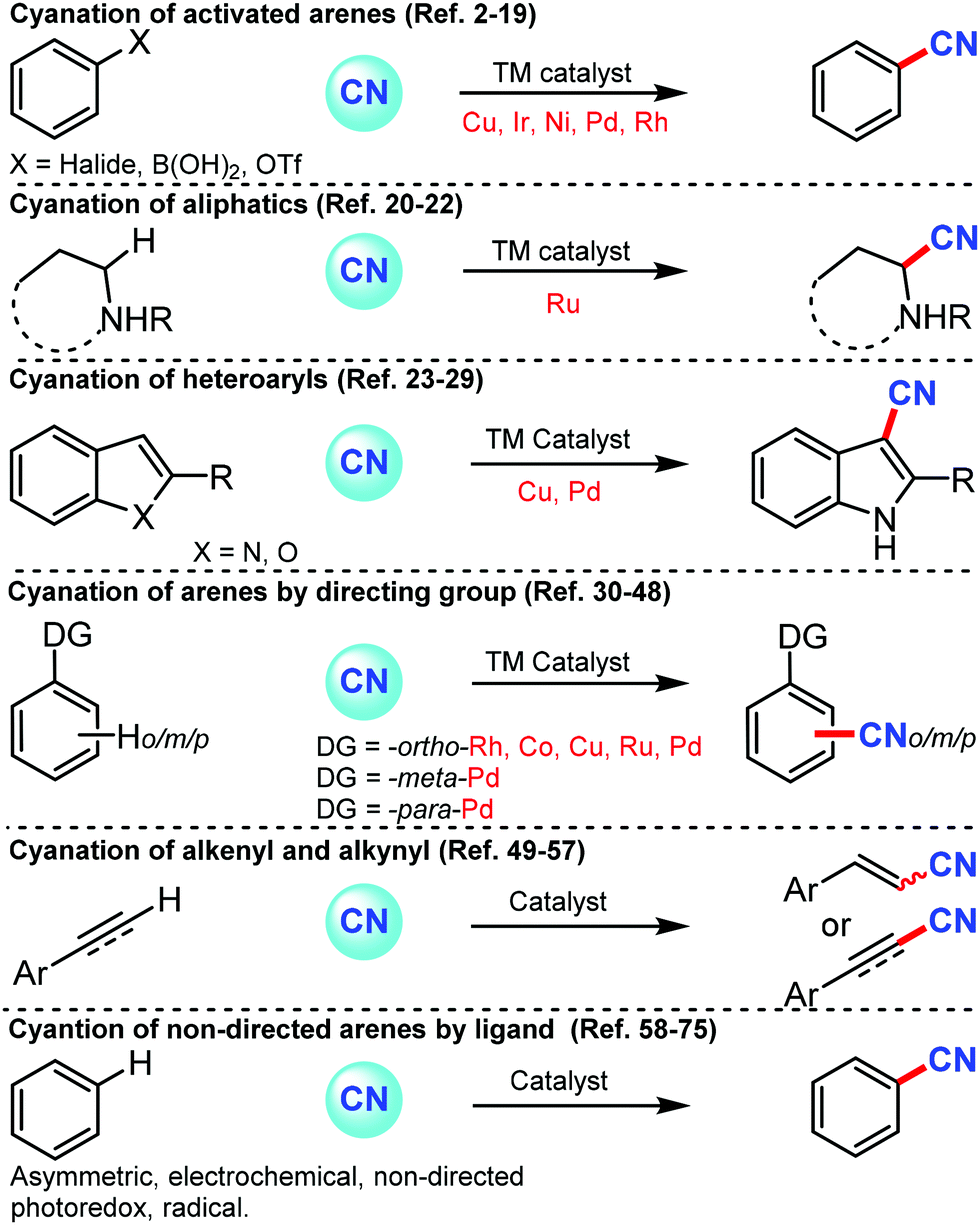 | ||
| Scheme 1 Contextual overview of the present feature article on the different approaches of C–CN bond formation. | ||
1. Transition metal based cyanation of activated arenes
1.1. Copper-catalyzed cyanation of activated arenes
A combination of DMF and NH4HCO3 as a safe cyanide source for the copper-mediated cyanation of aryl halides was developed by Cheng and co-workers in 2011.2 Notably, expensive palladium catalyst or a large excess of ammonia was not required for this transformation (Scheme 2). Aryl iodides with –OMe, benzyloxy, –OAc, and –OH groups could yield the corresponding nitriles. However, the alkyl, alkenyl, and alkynyl iodides did not work for this transformation.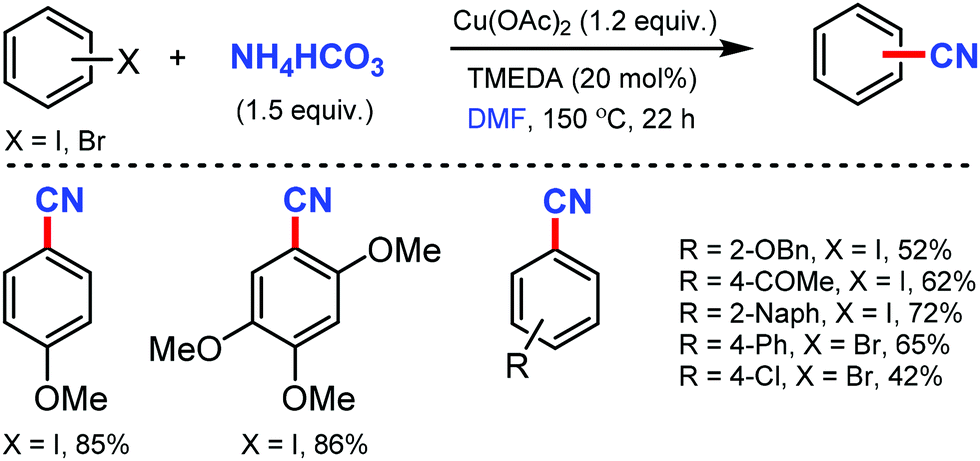 | ||
| Scheme 2 Copper mediated cyanation of aryl halides with the combined cyanide source. | ||
In 2012, Chang and co-workers reported the copper-mediated cyanation of boronic acids, boronate esters, borate salts, and electron-rich arenes under oxidative conditions using ammonium iodide and DMF as the cyanation source.3 The reaction proceeds via a two-step process: initial iodination and subsequent cyanation, where NH4I plays a dual role in supplying iodide and nitrogen for cyanation, thus being the first example of utilizing both cationic and anionic species of ammonium salts in metal-mediated reactions (Scheme 3). Aryl pinacolboronate and phenylborate salts could also afford the corresponding nitriles along with cyanation of electron-rich arenes such as 1,3,5-trimethoxybenzene and 1,2,4-trimethoxybenzene.
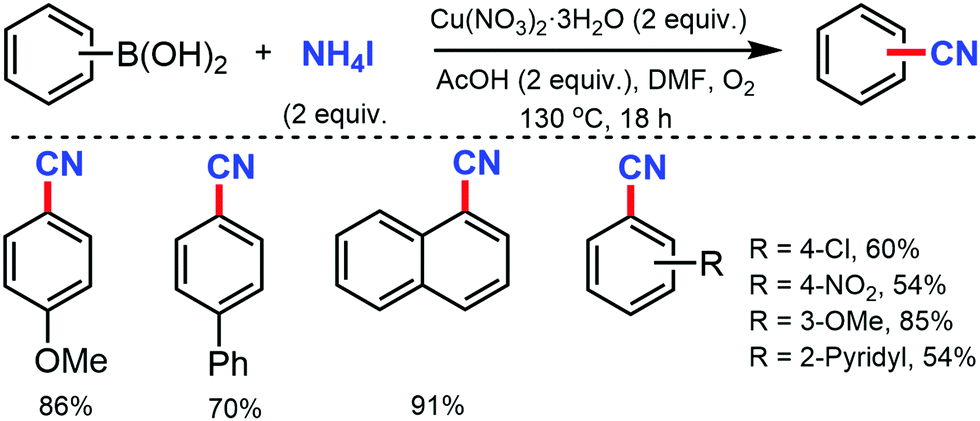 | ||
| Scheme 3 Copper-mediated sequential cyanation of aryl C–B/arene C–H bonds using NH4I and DMF. | ||
A copper-catalyzed strategy for the cyanation of aryl halides using DMF as a single source of cyanide was developed by Wang and co-workers in 2015 by using a stoichiometric amount of Cu(NO3)2·3H2O (Scheme 4).4 Substrates such as methyl, biphenyl, naphthyl, and pyrenyl iodides were well tolerated under this protocol but aryl iodides with carbonyl and amino resulted in a decreased yield. Aryl bromides with fused structures, such as 1-naphthyl, 1-pyrenyl, 2-naphthalenyl, 9-anthracenyl, and 9-phenathlenyl bromides proceed smoothly to produce the corresponding nitriles in moderate to good yields.
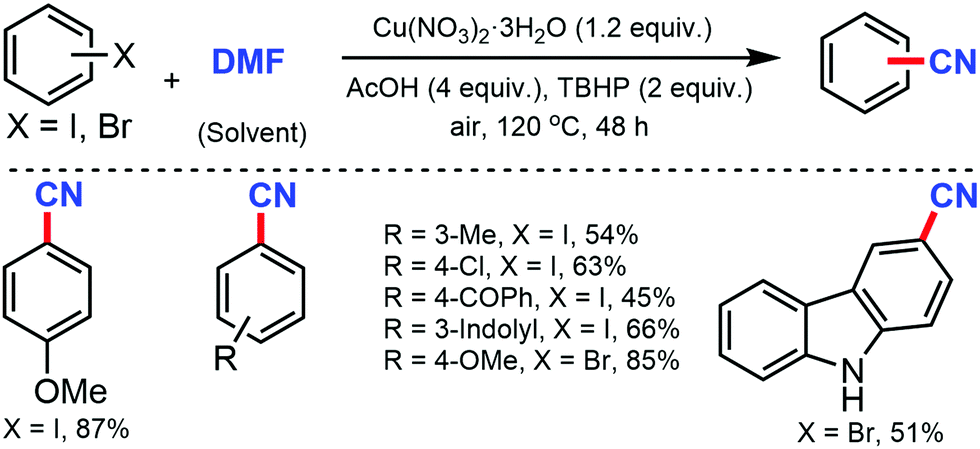 | ||
| Scheme 4 Cu(NO3)2·3H2O mediated cyanation of aryl halides DMF. | ||
Later, an efficient cyanide-free protocol for the cyanation of aryl halides with CO2 and NH3 as sources of cyanation was disclosed by Li and co-workers in 2018 using Cu2O/DABCO as the catalyst.5 Substrates bearing o-substitution were tolerated better than meta- or para-substitution on the aryl ring (Scheme 5). A slightly lower yield was observed in the presence of alkyl, chloro, or alkoxy substituents, indicating that the electronic effect of the substituents varies while hydroxyl, ester, amino, cyano, and amide groups were also favorable for this transformation.
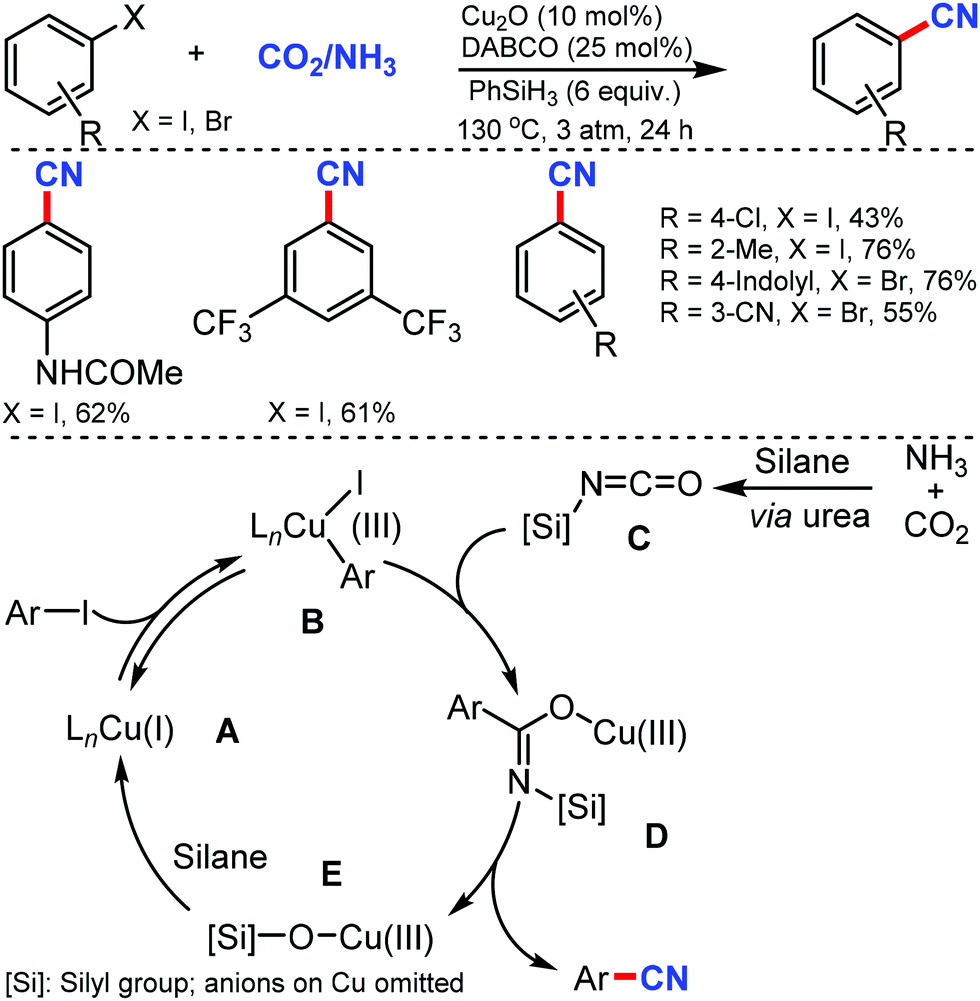 | ||
| Scheme 5 Cyanide-free catalytic cyanation using CO2 and NH3 and the plausible isocyanate mediated cyanation mechanism. | ||
However, in case of activated arenes, mechanistic studies have suggested that the oxidative addition of Cu(I) intermediate A to aryl iodides led to the active Cu(III) species B. Silyl isocyanate C formation was followed by the copper–carbon insertion to generate a transient imidate species D, which gives the cyano product via a plausible 1,3-silyl N-to-O migration, whereby the Cu(III) intermediate E was released and rapidly reduced by silanes to Cu(I) species. The insertion of isocyanate intermediates into Cu(III)–aryl was crucial for the high chemoselectivity.
1.2. Iridium catalyzed cyanation of activated arenes
In 2010, Hartwig and co-workers first reported the tandem cyanation of arenes by iridium/copper catalytic system for di or tri-substituted arenes/heteroarenes (Scheme 6).6 The reaction showed tolerance towards alkyl, alkoxy, halides, alkylcarbonyl, aminocarbonyl, alkoxycarbonyl, or protected phenols as well as 2,6-disubstituted pyridines. Also, arylboronic acids with electron-donating or electron-withdrawing groups produced the corresponding benzonitriles in 67–70% yield. The regioselectivity observed in this reaction resulted from the steric effects that controlled the C–H borylation step.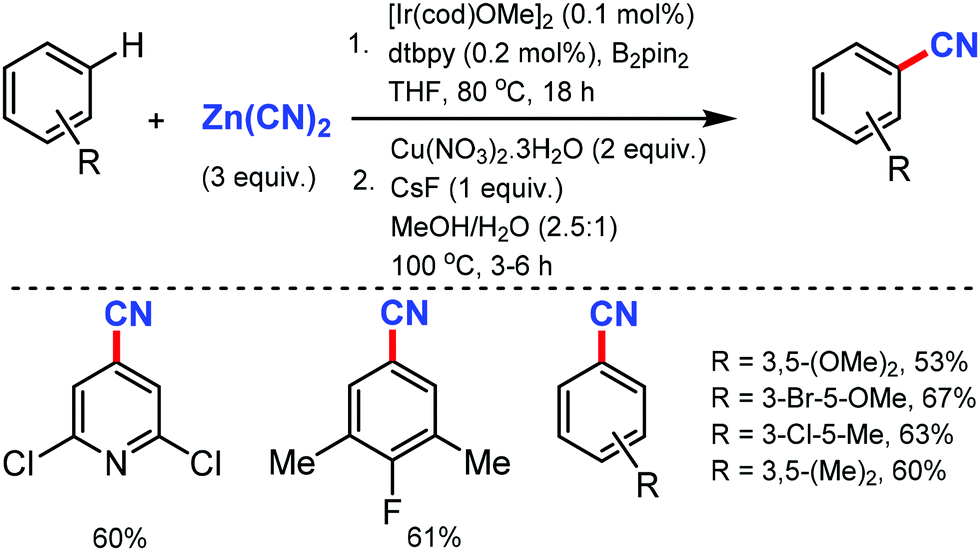 | ||
| Scheme 6 Copper mediated cyanation via Ir-catalyzed borylation. | ||
Recently, an efficient route for the synthesis of α-amino nitrile from a wide range of (hetero)aromatic and aliphatic tertiary amides, and N-alkyl lactams by exploiting the iridium-catalyzed reductive Strecker reaction was reported by Dixon and co-workers in 2017.7 The chemo-selective reduction of the amide and lactam by IrCl(CO)[P(C6H5)3]2 (Vaska's complex) in the presence of tetramethyldisiloxane (TMDS) as a reductant to generate a hemiaminal species on the substitution by cyanide using TMSCN was developed (Scheme 7). This protocol was also suitable for furanyl heterocycles, cinnamamides, aliphatic carboxylic acids, diethyl amines, 1-methylpiperidine, and boc-piperazine.
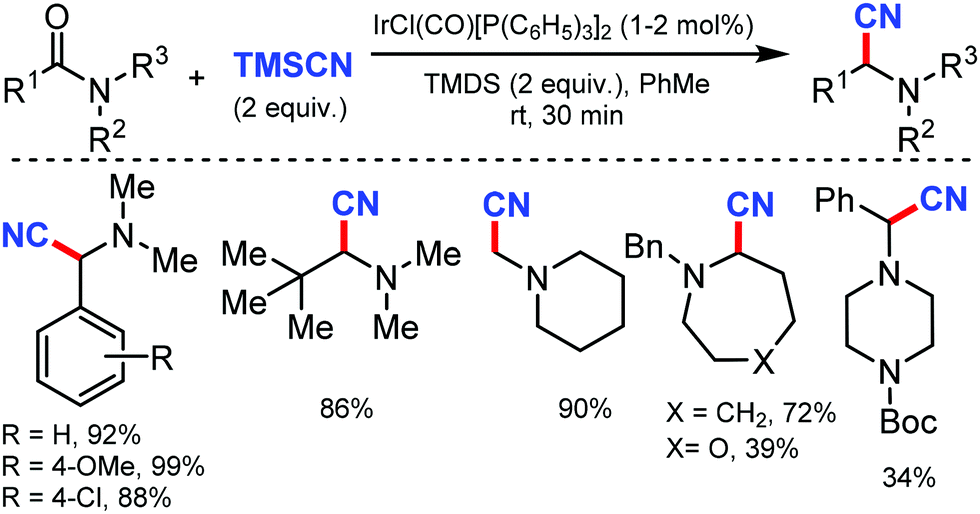 | ||
| Scheme 7 Iridium-catalyzed reductive Strecker reaction for late-stage amide and lactam cyanation. | ||
1.3. Nickel-catalyzed cyanation of activated arenes
The merging of transfer hydro-functionalization and cross-coupling was employed for the synthesis of aryl nitriles from a wide range of aryl chlorides and aryl/vinyl triflates by Morandi and co-workers in 2017 using butyronitrile as the cyano source, which prevents catalyst poisoning.8 Both electron-donating and withdrawing groups tolerated the reaction conditions well (Scheme 8). Naphthyl, 9-phenanthryl chlorides, heterocycles including pyrrolidine, dioxole, carbazole, pyrazole, quinoline, and several medicinally important heterocycles could afford the cyanated products in good yields.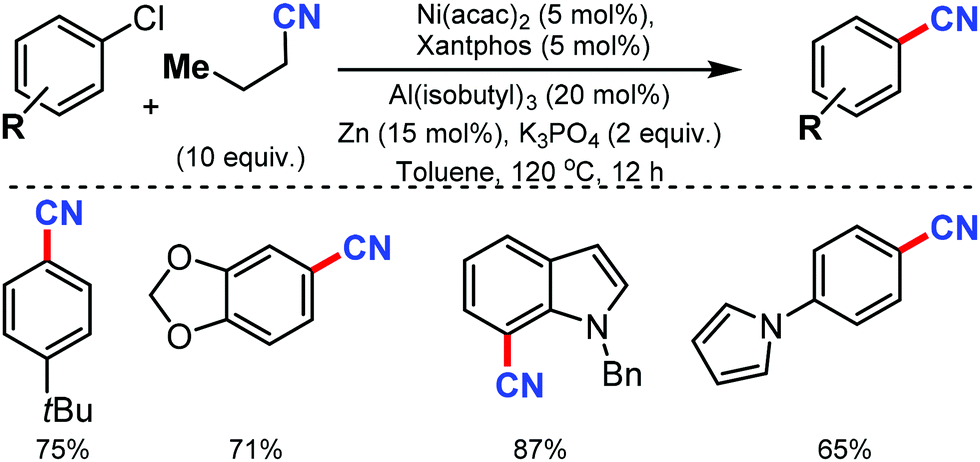 | ||
| Scheme 8 Nickel-catalyzed cyanation of aryl chlorides. | ||
Later, Liu and co-workers developed an inexpensive NiCl2·6H2O/dppf(1,1′-bis(diphenylphosphino)ferrocene)/Zn catalytic system for the cyanation of hetero(aryl) chlorides using less toxic Zn(CN)2 as the cyanide source.9 Employing DMAP as the additive, under mild conditions, various aromatic and heteroaromatic chlorides were well tolerated (Scheme 9). The use of Zn(CN)2 was expected to prevent catalyst de-activation due to its low solubility in most organic solvents. n-Bu, t-Bu, TBSOCH2, 3,5-dimethoxy, and trimethylsilyl-substituted arylchlorides produced the corresponding nitriles in 81–90% yield.
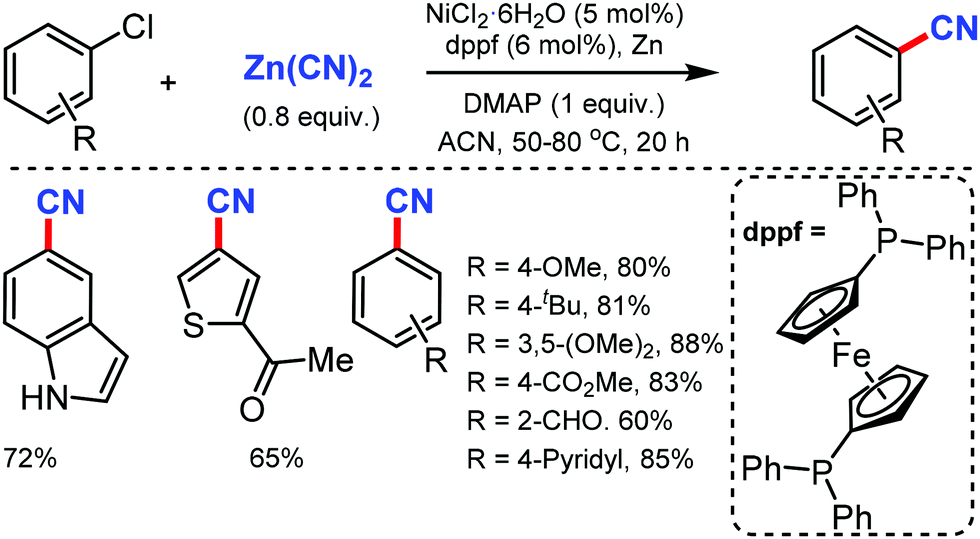 | ||
| Scheme 9 Nickel-catalyzed cyanation of aryl/heteroaryl chlorides with Zn(CN)2. | ||
This cyanation protocol was also applicable to several aryl bromides and iodides. The mechanistic studies suggested that the use of DMAP is crucial for the reaction efficiency and the reaction proceeds via a Ni(0)/Ni(II) catalytic pathway.
In 2020, Liao and co-workers reported the cyanation of aryl halides using nickel catalysis with inexpensive and non-toxic 4-cyanopyridine-N-oxide under mild conditions.10 A broad spectrum of aryl halides bearing different electron-neutral, donating, and withdrawing groups could afford the cyanated products in moderate to good yields (Scheme 10). This Ni catalytic system, with a little modification, was also exploited for the hydrocyanation of alkynes with good regioselectivity. Diaryl alkynes substrates furnished an excellent E/Z selectivity while exclusively Markovnikov vinyl nitriles were obtained in case of terminal alkynes. In the plausible mechanism, the pre-catalyst NiIIX2 may first get reduced by zinc to generate the bipyridine ligand-chelated Ni0A, followed by the oxidative addition of aryl halide generating NiII complex B, which was then reduced by zinc to give aryl NiI complex C. The subsequent oxidative addition of cyano source with assistance from TFAA resulted in the NiIII species D, which then undergoes reductive elimination to yield a cyanation product and the NiI complex E.
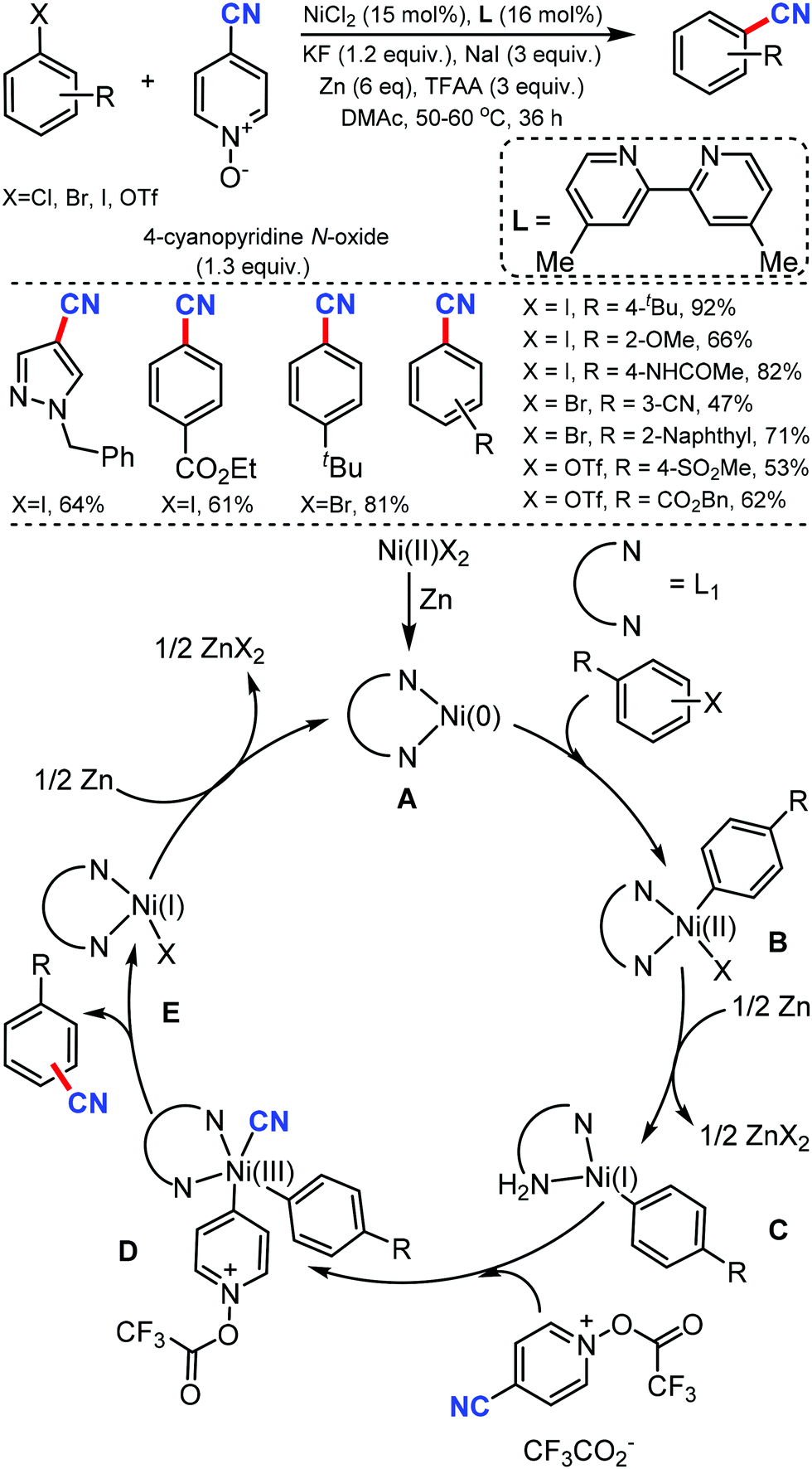 | ||
| Scheme 10 Nickel-catalyzed cyanation of aryl halides via C–CN bond cleavage and the cyano transfer mechanism. | ||
1.4. Palladium-catalyzed cyanation of activated arenes
The development of phenolic derivatives as suitable electrophilic coupling partners for cyanation has made the use of less expensive and stable aryl mesylates or sulfonates as cyanation substrates highly desirable (Scheme 11). In 2010, Kwong and co-workers reported the efficient Pd-catalyzed cyanation of aryl mesylates using an environment friendly solvent such as water or a water/tBuOH solvent mixture under mild reaction conditions.11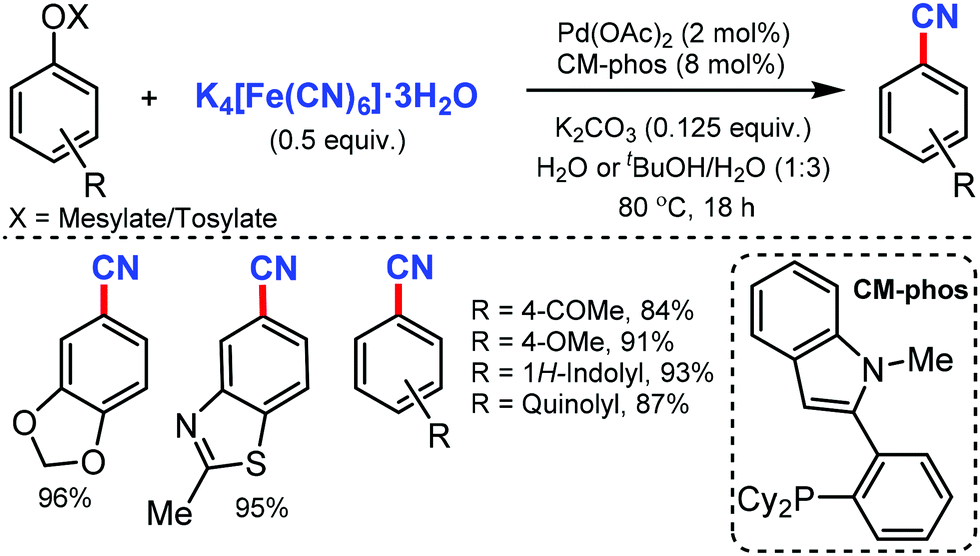 | ||
| Scheme 11 Palladium-catalyzed cyanation of aryl mesylates and tosylates. | ||
This showed ample functional-group tolerance towards substrates bearing nitrile, ester, keto, aldehyde, amine, and heterocyclic groups to give the cyanated products efficiently.
Later, in 2011, the Pd/CM-phos catalyzed cyanation of aryl chloride was developed by Kwong and co-workers using K4[Fe(CN)6]·3H2O as the cyanide source.12 Various aryl chlorides, bearing different functional groups such as carbonyls, nitrile, and amine, were cyanated. Heterocyclic groups such as benzothiazolyl, quinolyl, and N–H indoles were well tolerated under the reaction conditions to afford the cyanated product in good to excellent yields. Also, sterically hindered aryl chlorides proceeded smoothly under this cyanation method. Fortunately, water was found to be essential as a cosolvent for the reaction (Scheme 12).
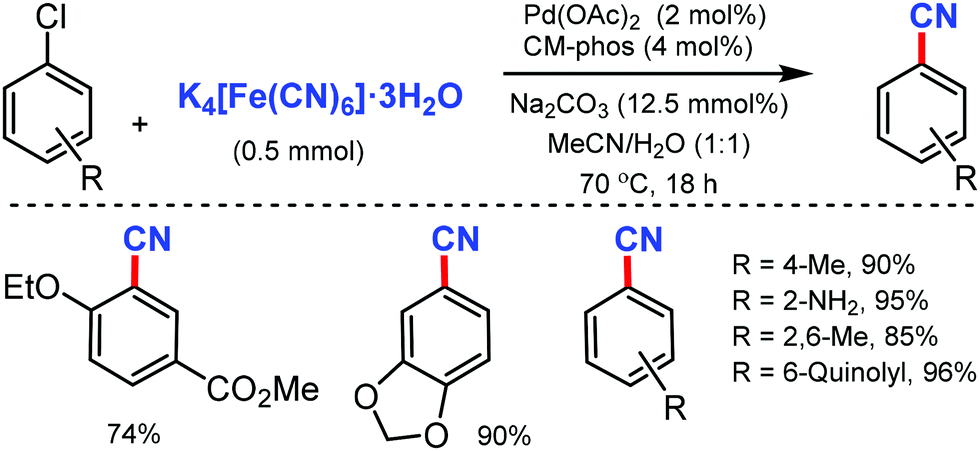 | ||
| Scheme 12 Palladium-catalyzed cyanation of aryl chlorides. | ||
In 2012, Shen and co-workers developed Pd-catalyzed cyanation using inexpensive and user-friendly ethyl cyanoacetate.13 Different aryl halides with various substituents at the ortho-, meta-, and para-positions could smoothly produce the corresponding nitriles. The reactivity of aryl halides decreased as the bond-dissociation energy of the C–X bonds increased (reactivity: I > Br > Cl). However, the presence of two or more strong electron-withdrawing groups was found to be unfavorable. Also, a relatively high loading of the palladium catalyst was required for this transformation (Scheme 13).
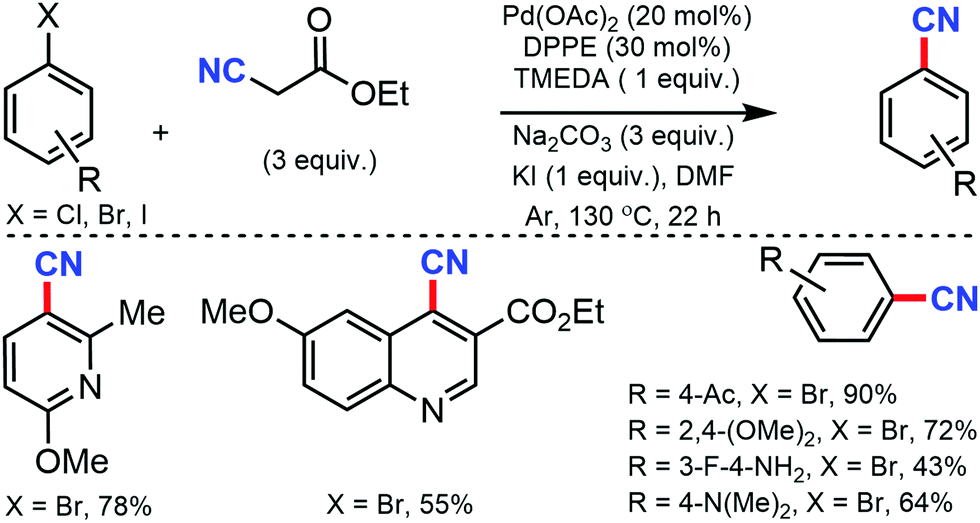 | ||
| Scheme 13 Pd-Catalyzed cyanation with ethyl cyanoacetate. | ||
An efficient Pd(PPh3)4/DBU catalytic system for the cyanation of aryl and heteroaryl bromides using inexpensive and easily handled K4[Fe(CN)6]·3H2O was developed by Liu and co-workers in 2012.14
The use of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) enhanced the release of the cyanide ion as a promoter and reduced the inactivation of Pd as the co-catalyst (Scheme 14). Various bromo-aminopyridines smoothly underwent this reaction protocol, giving corresponding nitriles in good to excellent yields. Substituents such as –CF3, –F, and –Cl were also found to be compatible with this transformation. In addition, different aryl bromides bearing electron-donating groups, such as alkoxy and free amine and electron-withdrawing keto groups at the meta- and para-positions were also favorable for this reaction to proceed.
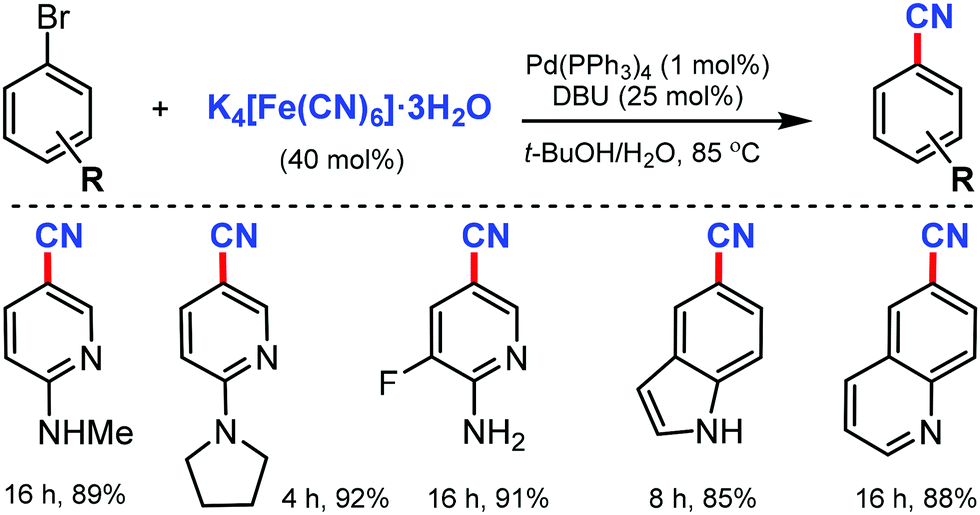 | ||
| Scheme 14 Pd/DBU mediated cyanation of aryl/heteroaryl bromides. | ||
Later, in 2013, Buchwald and co-workers reported a convenient Pd-catalyzed protocol for the cyanation of aryl/heterocyclic halides using non-toxic K4[Fe(CN)6]·3H2O.15 Electron-donating, electron-withdrawing, and di-ortho-substituted aryl chlorides were smoothly cyanated under this approach. Interestingly, substrates bearing free NH/OH groups (primary amides), sulfonamides, anilines, and benzylic alcohols could also afford the cyanated products using less than 1 mol% of Pd (Scheme 15). Furthermore, various heterocycles including indole, thiophenes, thiazole, pyrroles, pyrazoles, and indazoles efficiently transformed their halides into the corresponding nitriles in good to excellent yields (64–99%).
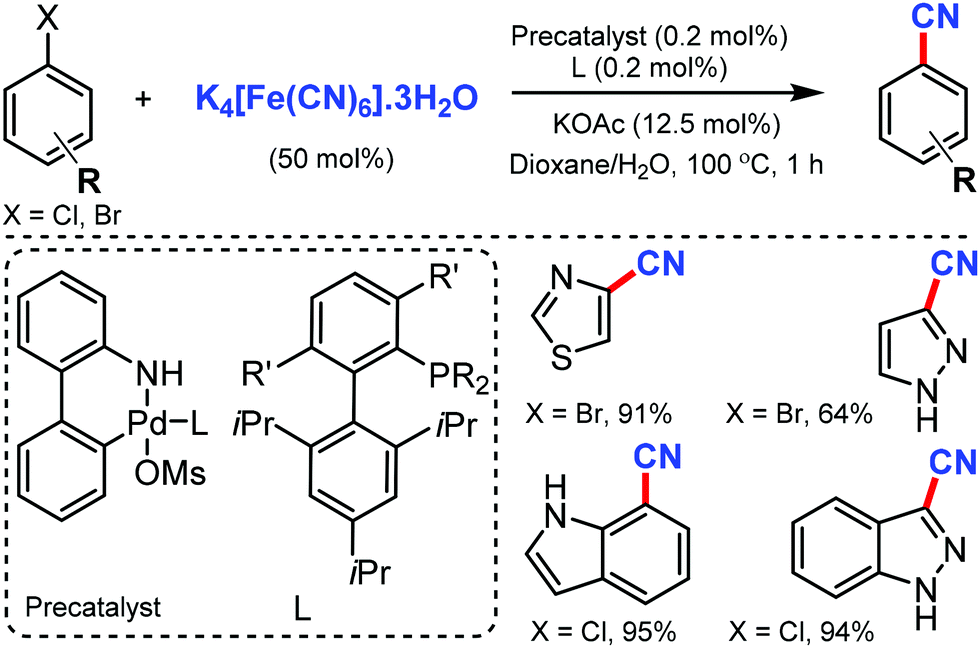 | ||
| Scheme 15 Palladium-catalyzed cyanation of (hetero)aryl chlorides and bromides. | ||
Later, in 2015, a general and efficient room temperature palladium-catalyzed method for the cyanation of (hetero)aryl halides and triflates was developed by Buchwald and co-workers using Zn(CN)2 in aqueous media.16 A wide range of aryl halides/triflates, five/six-membered heterocycles, and natural product derivatives were efficiently cyanated in good to excellent yields (77–99%) (Scheme 16). In addition, an ample scope of heterocycles including indoles, benzothiophene, benzofuran, thiophene, pyrazole, quinoline, and pyridine were readily tolerated under the reaction conditions. Interestingly, the cyanation of natural products derivatives such as estrone, coumarin, and δ-tocopherol triflates could also be carried out efficiently using this method.
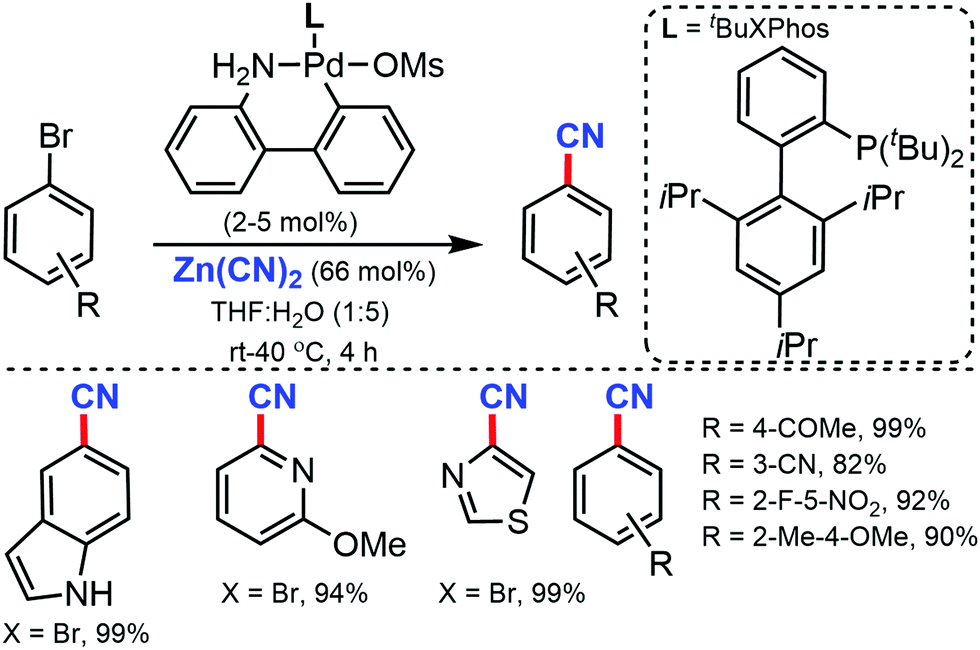 | ||
| Scheme 16 Cyanation of (hetero)aryl halides and triflates. | ||
The scope of the Catellani reaction by Pd-catalyzed norbornene-mediated ortho-C–H substitution of iodoarenes, followed by terminal cross-coupling, was reported for a tandem ortho-C–H amination/ipso-cyanation by Ranu and co-workers in 201617 and Lautens and co-workers18 at the same time using the user-friendly cyanating agent K4Fe(CN)6·3H2O and Zn(CN)2, respectively. Different ortho-substituted aryl iodides bearing both electron-donating (–CH3, –Et, –OCH3, –N(CH3)2, –OCH2Ph) and electron-withdrawing groups (–F, –Cl, –CO2CH3, –OCF3, –CF3) were smoothly cyanated to produce the corresponding 2-aminobenzonitriles (Scheme 17). However, in the case of ortho-unsubstituted iodoarenes, double ortho-C–H amination and ipso-C–I-cyanation was observed. With Zn(CN)2 also, a broad variety of aryl iodides bearing electron-donating and electron-withdrawing groups were found to be compatible with the reaction conditions and yielded the corresponding nitriles in moderate to good yields (Scheme 18).18 Substituted N-benzoyloxyamines such as secondary and cyclic N-benzoyloxyamines were the appropriate nitrogen source for this multicomponent reaction.
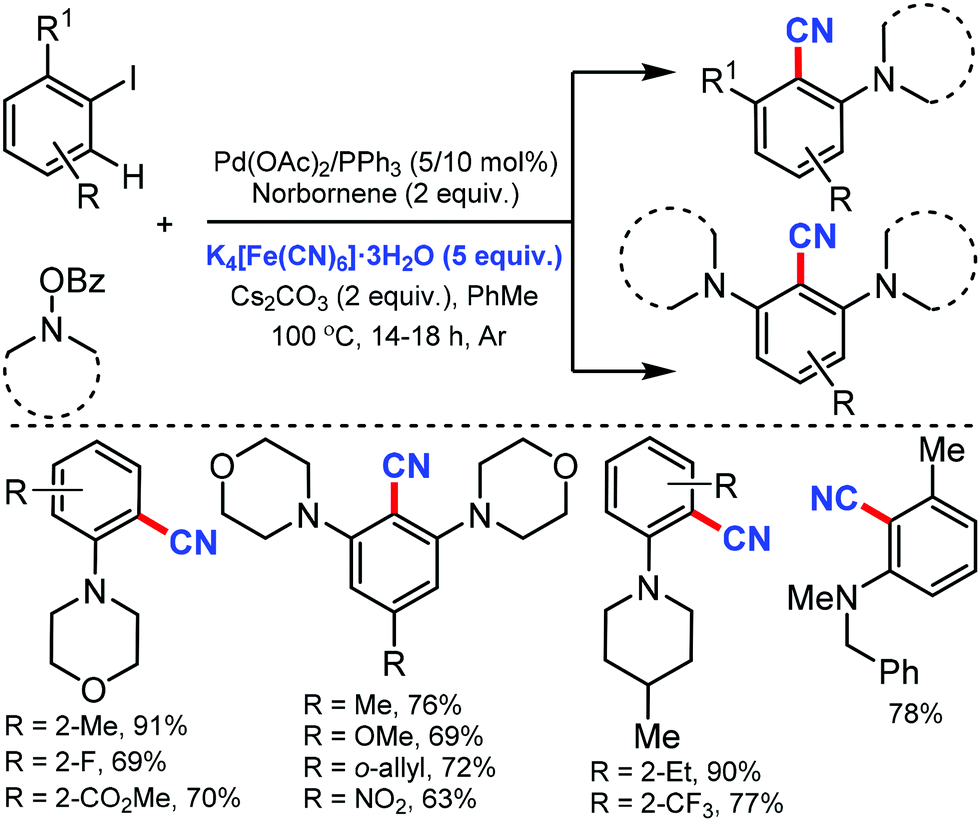 | ||
| Scheme 17 Palladium-catalyzed norbornene mediated tandem ortho-C–H amination/ipso-cyanation of iodoarenes. | ||
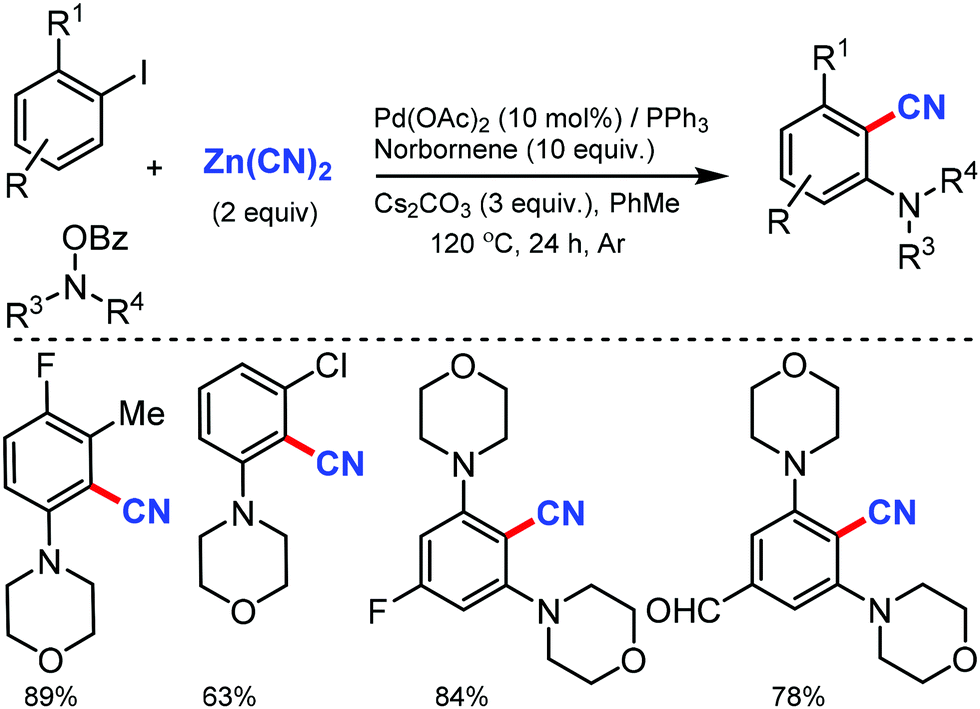 | ||
| Scheme 18 Norbornene-mediated Pd-catalyzed tandem amination/cyanation. | ||
1.5. Rhodium-catalyzed cyanation of activated arenes
In 2011, Beller and co-workers reported the first Rh-catalyzed cyanation of aryl and alkenyl boronic acids using N-cyano-N-phenyl-p-toluenesulfonamide (NCTS) as the cyanating agent and with K2CO3 in 1,4-dioxane solvent.19 Sterically demanding as well as non-hindered boronic acids were efficiently transformed into the corresponding nitriles under mild reaction conditions (Scheme 19). Moreover, electronically different and more challenging functionalized aryl boronic acids were also found to be compatible. The reaction is expected to proceed through the transmetalation of the aryl boronic acid with the active rhodium(I) species, which leads to the formation of the aryl–rhodium species A, which upon coordination with N–CN reagent forms the intermediate B. The transfer of the aryl motif to the nitrile carbon atom generates species C; then, the rearrangement of C results in the formation of the cyanated product.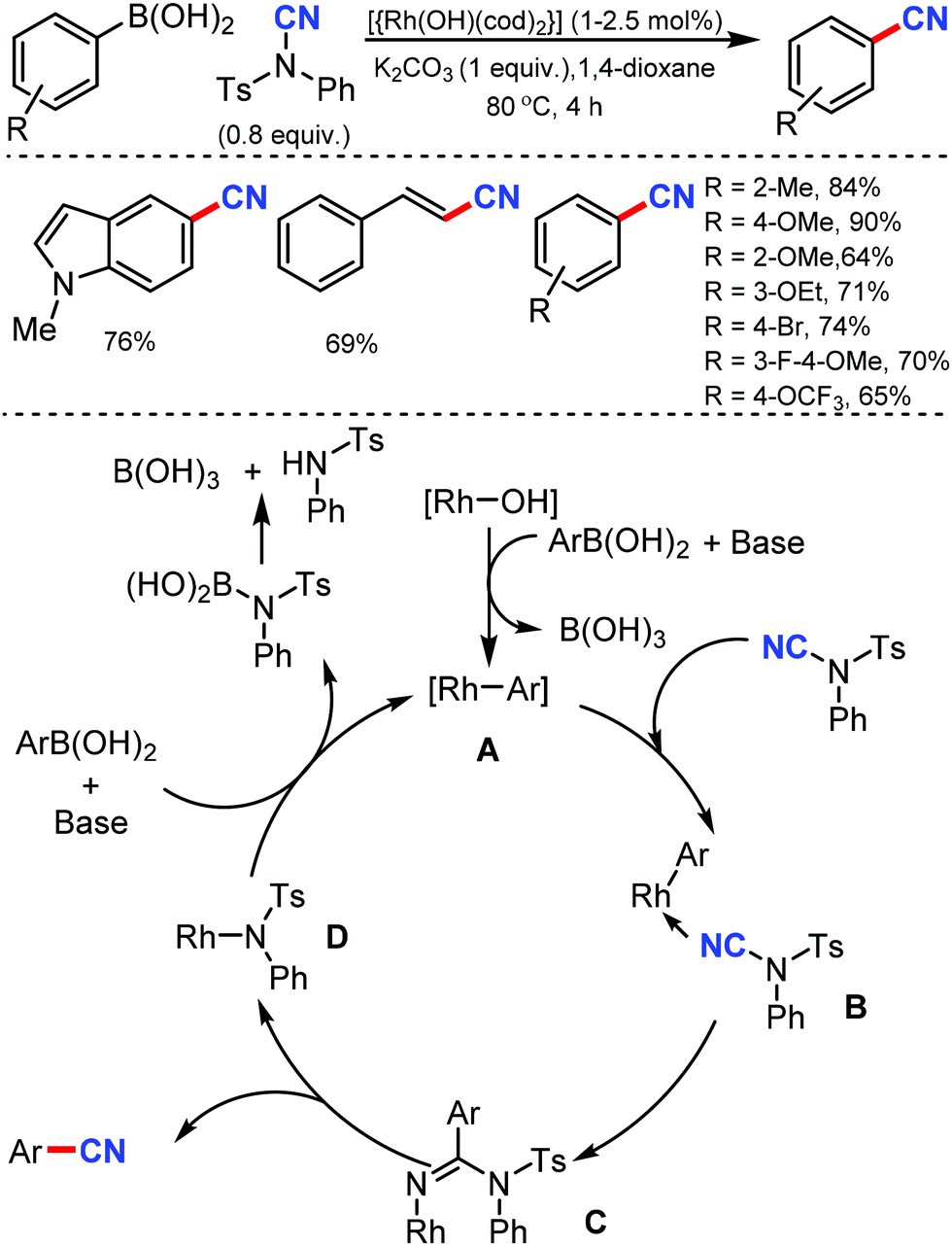 | ||
| Scheme 19 Rh-Catalyzed cyanation of boronic acids. | ||
2. Transition metal-catalyzed aliphatic cyanation
In 2003, Nakae et al. reported the ruthenium-catalyzed oxidative cyanation of tertiary amines with NaCN for regioselective cyanation of the substituted N,N-dimethylanilines with electron-donating or withdrawing groups.20 Even cyclic amines such as tetrahydroisoquinoline were cyanated by this transformation. In this reaction, the oxo–ruthenium species seem to be formed as an active species to generate the iminium ion intermediate and kinetic studies revealed that electron transfer from amine to ruthenium would take place at the initial step (Scheme 20).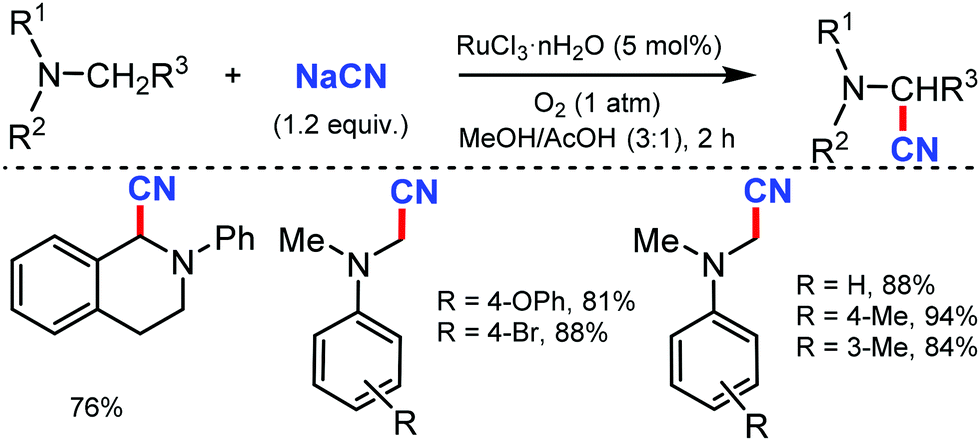 | ||
| Scheme 20 RuCl3·nH2O-Catalyzed cyanation of tertiary amines. | ||
In 2005, Terai et al. reported similar results on ruthenium-catalyzed oxidative cyanation with H2O2 and NaCN or HCN as the cyanation source for tertiary amines.21 This was the first attempt for direct C–H activation and C–C bond formation under H2O2 oxidative conditions. Both electron-donating/withdrawing substituents reacted well under this condition. In the presence of other alkyl groups, N-methyl reacts predominantly along with cyclic amines such as piperidines, pyrrolidines, and tetrahydroisoquinolines to give α-cyanoamines (Scheme 21). While probing the mechanistic analysis, relative rates for the oxidative cyanation of four para-substituted N,N-dimethylanilines with H2O2 in the presence of NaCN was found to be R2 = 0.998 (determined by 1H NMR). The ρ-value of −3.61 indicates the presence of cationic intermediate in the rate determining step. Moreover, intramolecular and intermolecular deuterium isotope effect was found to be 4.1 and 3.7, respectively. These data suggested that low-valent Ru(II) undergoes reaction with H2O2 to give the oxo–ruthenium species [Run+2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O], which produces the iminium ion intermediate by electron and hydrogen transfer, followed by nucleophilic attack by HCN to give the corresponding α-cyanated product and water and the RuII species, which completes the catalytic cycle.
O], which produces the iminium ion intermediate by electron and hydrogen transfer, followed by nucleophilic attack by HCN to give the corresponding α-cyanated product and water and the RuII species, which completes the catalytic cycle.
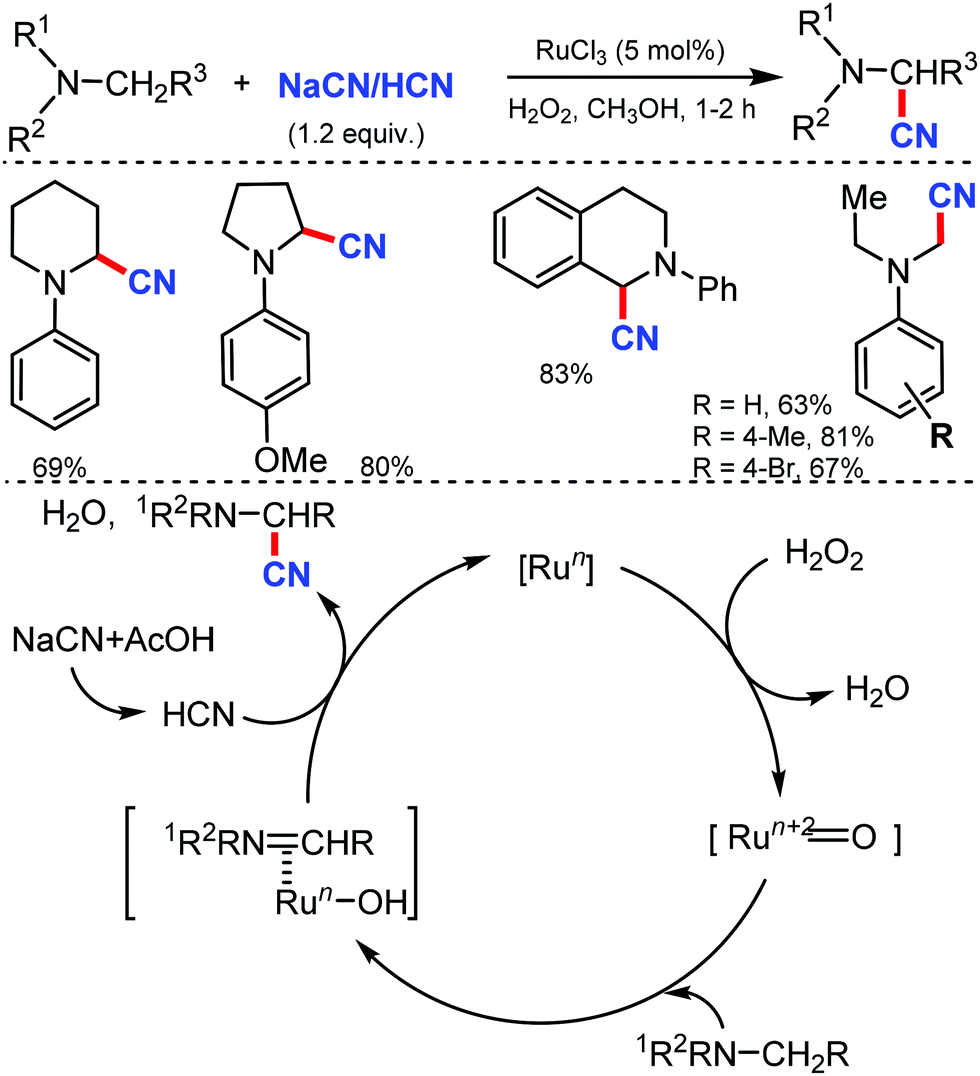 | ||
| Scheme 21 RuCl3-Mediated cyanation of tertiary amines with H2O2. | ||
In 2012, Seidel's group reported the redox-neutral α-cyanation of secondary cyclic amines in the presence of benzoic acid as the catalyst under microwave irradiation.22 The reaction could convert pyrrolidines and benzaldehyde in presence of TMSCN to give α-aminonitrile in 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in 61% yield (Scheme 22). Other cyclic amine such as piperidine and azapane could give α-aminonitriles in moderate yields in the presence of 20 mol% of 2-ethylhexanoic acid (2-EHA) as the catalyst.
:1 in 61% yield (Scheme 22). Other cyclic amine such as piperidine and azapane could give α-aminonitriles in moderate yields in the presence of 20 mol% of 2-ethylhexanoic acid (2-EHA) as the catalyst.
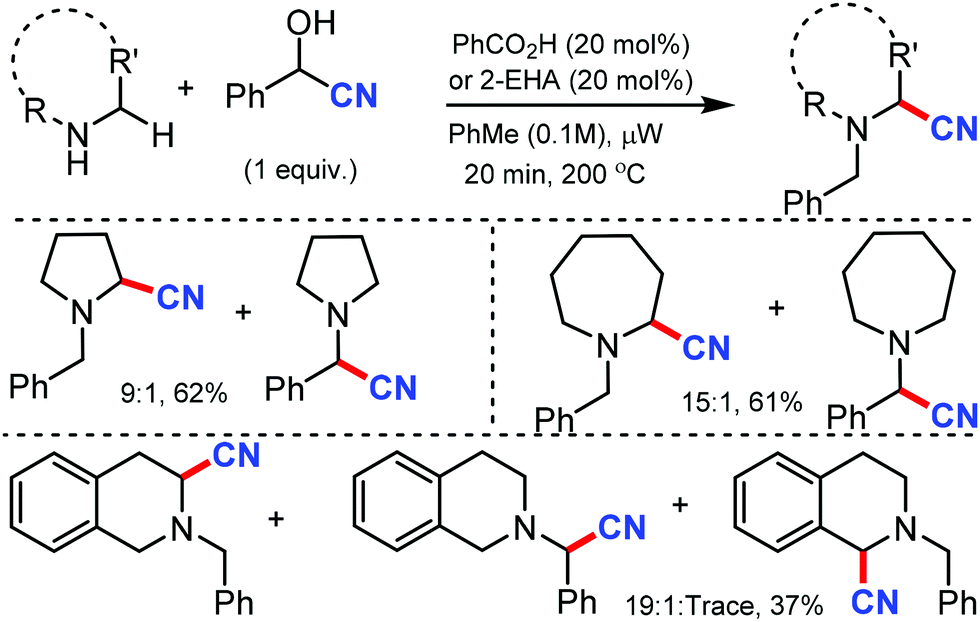 | ||
| Scheme 22 Redox neutral α-cyanation of cyclic amine. | ||
3. Cyanation of heterocycles by various cyanation sources
The copper-catalyzed regioselective cyanation of aromatic heterocycles was developed by Daugulis and co-workers in 2010 using NaCN as the cyanide source.23 Several heterocycles including benzoxazole, benzothiazole, benzimidazole, caffeine, and triazoles were cyanated in reasonable to good yields (Scheme 23). Notably, pyridine derivatives with electron-withdrawing (fluorine) or electron-donating (methoxy) substituents were also tolerated under these reaction conditions.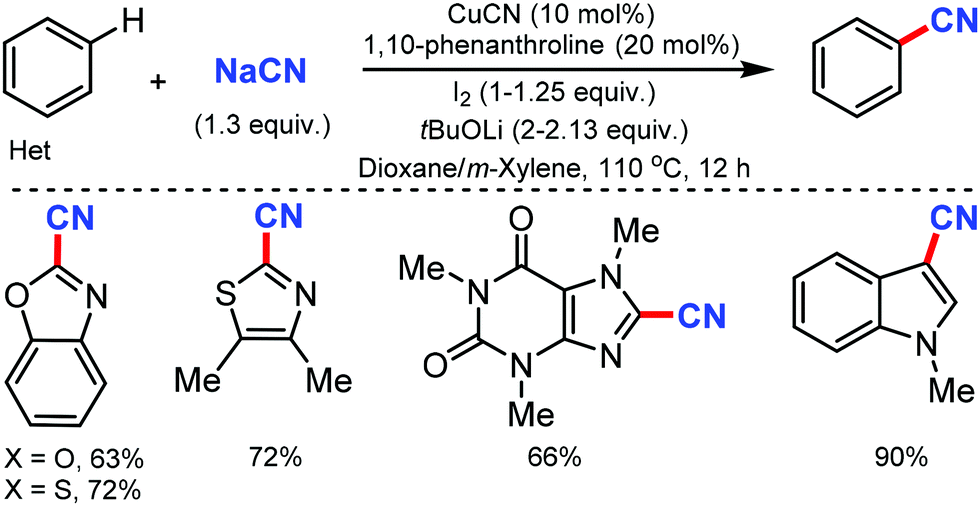 | ||
| Scheme 23 Copper-catalyzed cyanation of heterocycles. | ||
Similarly, in 2010, Wang and co-workers disclosed palladium-catalyzed C–H cyanation of indoles using less toxic and easily available K4[Fe(CN)6]. N-Methylindole with electron-donating substituents underwent the reaction without difficulty (Scheme 24). 2-Substituted N-alkyl- and N-arylindoles afforded cyanation in high yields but N-acetyl, N-phenylsulfonyl, or N-Boc substituents did not. N-Substituted indoles were compatible, albeit with less product yield.24
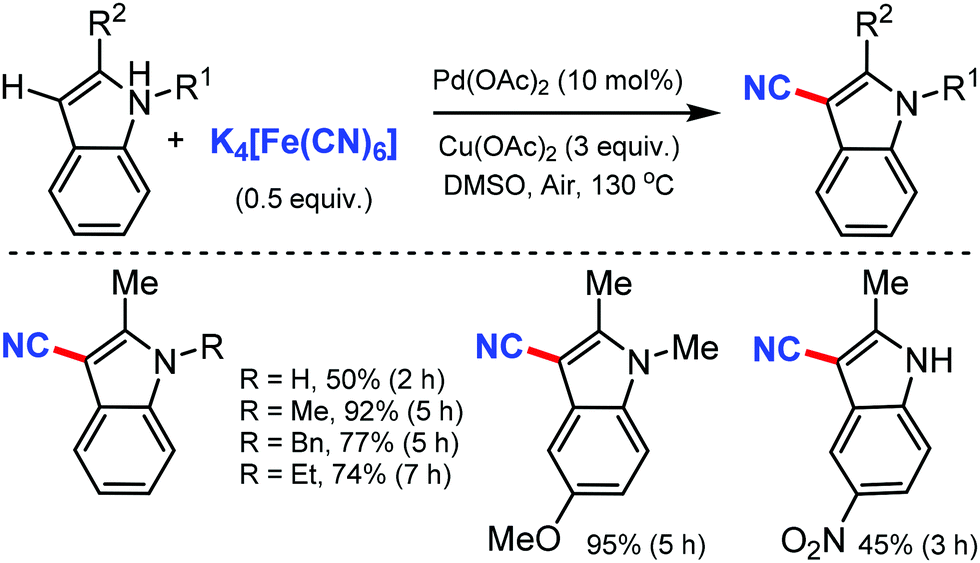 | ||
| Scheme 24 Palladium-catalyzed direct cyanation of indoles. | ||
Jiao and co-workers, in 2011, reported the cyanation of indoles and benzofurans using DMF as the cyano reagent and solvent.25 Electron-donating or electron-withdrawing substituents on 2-aryl indole were tolerated under the reaction conditions. 2 and 3-phenylbenzofuran could also be cyanated in moderate yields. Mechanistic studies indicated that both nitrogen and carbon of the CN group were provided by DMF (Scheme 25).
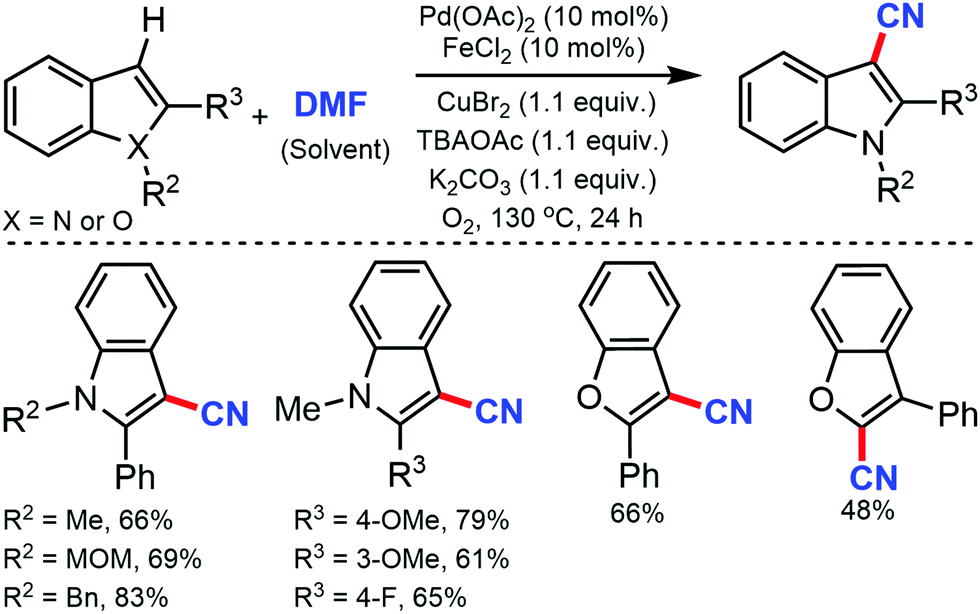 | ||
| Scheme 25 Pd-Catalyzed cyanation of heteroarenes using DMF. | ||
Later, a synthetic route to cyanoindoles/pyrroles using a Lewis acid catalyzed protocol was developed by Wang and co-workers in 2011.26 Benign, bench-stable electrophilic cyanating agent NCTS was employed along with BF3·OEt2 as the catalyst under mild reaction conditions (Scheme 26) to C-3 cyanate with various indole and pyrrole substrates.
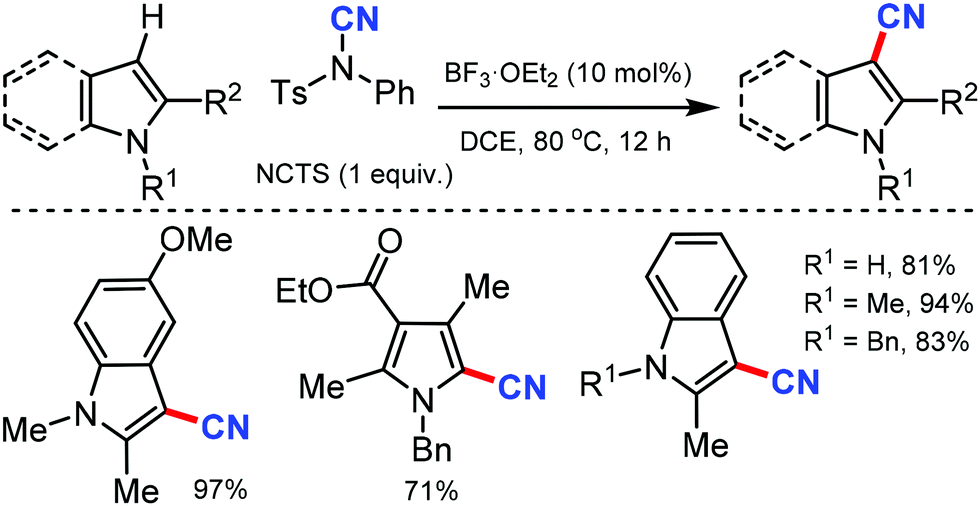 | ||
| Scheme 26 Lewis acid catalyzed cyanation of indoles and pyrroles. | ||
Interestingly, indole with or without substitution at the C-2 position afforded the cyanated indoles in excellent yields by avoiding the formation of the homocoupling by-products observed with similar Pd catalyzed methods.
The combination of NH4I and DMF as the CN source for the copper-catalyzed regioselective cyanation of indoles was reported by Chang and co-workers in 2012.27 Various substituted indole such as N-Ph and N-Bn were cyanated at the C-3 position selectively. The N-carbonyl group substituted indoles were accompanied by decarbonylation, leading to the formation of 3-cyano-1H-indole (Scheme 27). Mechanistic studies indicated that the reaction proceeds through electrophilic iodination, followed by cyanation.
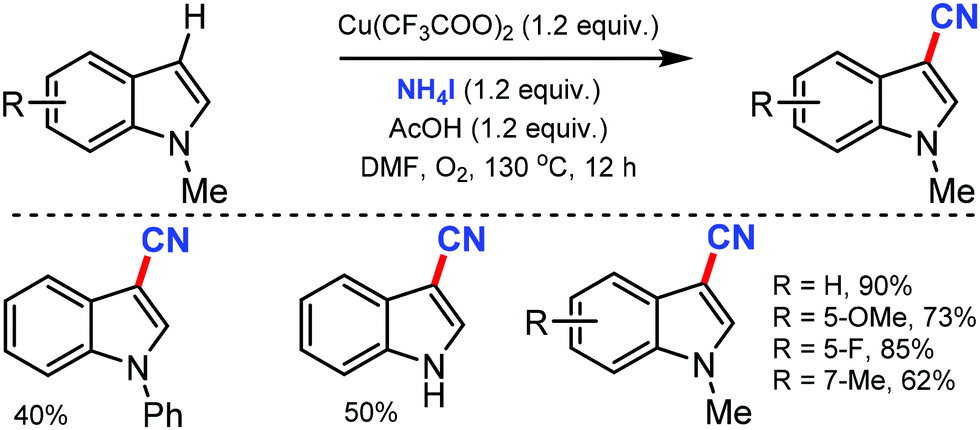 | ||
| Scheme 27 Copper-mediated regioselective cyanation of indoles. | ||
Two different methods to synthesize hetero(aryl)nitriles under Pd catalysis using t-butyl isocyanide as the CN source was developed by Xu and co-workers in 201228 and again by Zhu29 at the same time. Both these C–H cyanation reactions were suitable to cyanate a wide range of indoles, pyrroles, and aromatic rings with high regioselectivity (Scheme 28).
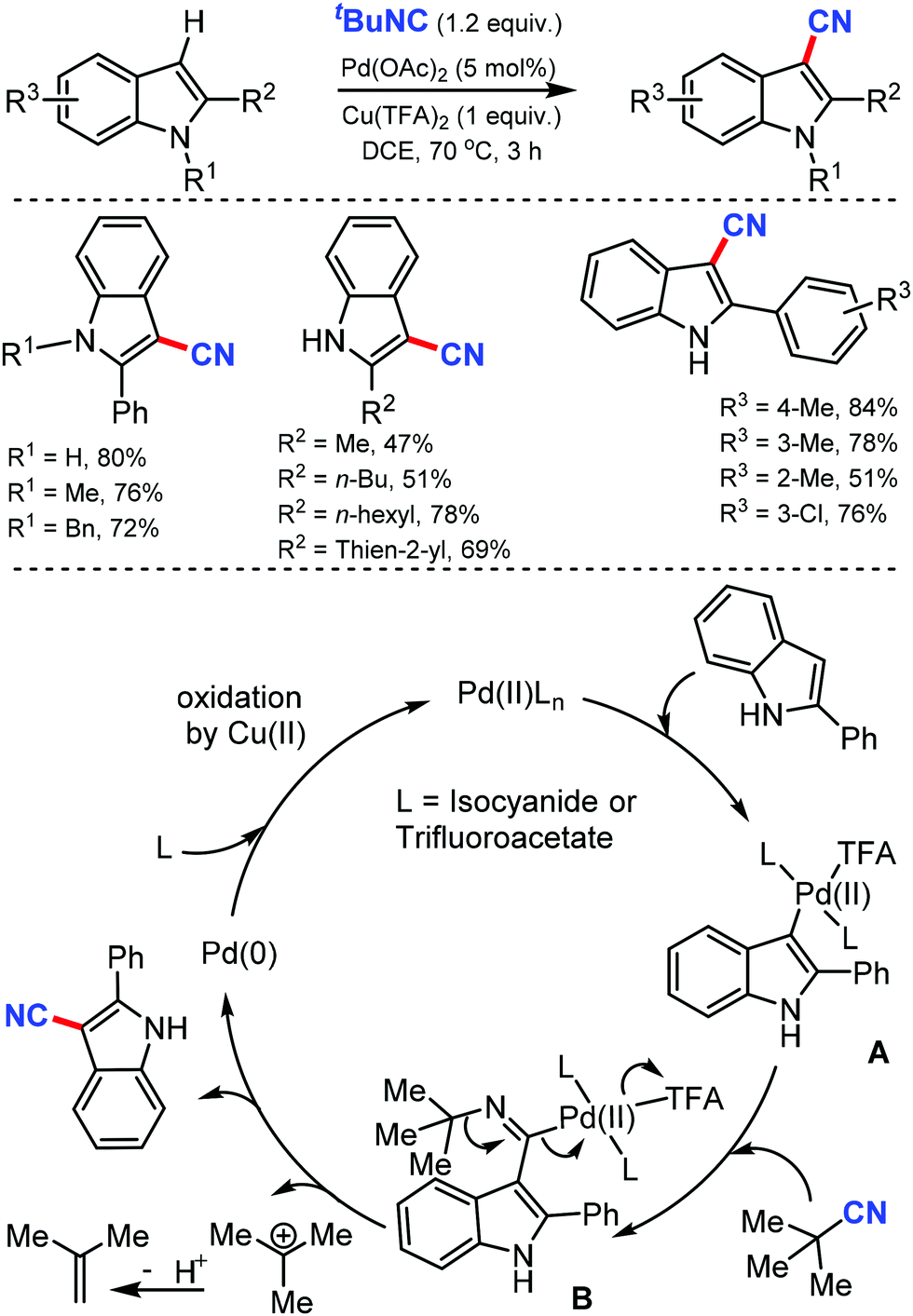 | ||
| Scheme 28 Palladium-catalyzed cyanation by tert-BuNC and the reaction mechanism. | ||
Due to the direct electrophilic palladation at C-3 indole, C-3-palladated intermediate forms, which leads to C-3 cyanated indoles. Furthermore, N-substituted pyrroles, arylpyridines, and arylpyrimidines were also compatible with this palladium catalytic system, producing the corresponding nitriles regioselectively. Zhu's method used a stoichiometric oxidant, the trifluoroacetate counter-ion. Various electron-rich 2-alkyl(aryl)indoles, and electron-poor 2-arylpyridine cyanated as well. Indoles with electron-deficient groups either at the C-5 position or on the 2-phenyl ring demanded an additional requirement of PivOH as the additive. The high electrophilicity of Pd(II) and the stability of the tertiary carbon were beneficial in breaking the C–N bond, which led to the formation of cyanated products (Scheme 28). The mechanistic studies indicated that the electrophilic palladation of Pd(II) on C-3 of the indole forms the σ-indolylpalladium(II) intermediate A. The subsequent migratory insertion of isocyanide generates the key imidoyl palladium(II) intermediate B, in which the loosely-coordinated trifluoroacetate makes the palladium center more electrophilic. The high electrophilicity of Pd(II) and the stability of the tertiary carbocation are crucial for the cleavage of the C–N bond, leading to the cyanation product.
4. Directing group approach for C–H cyanation
4.1. ortho-C–H cyanation
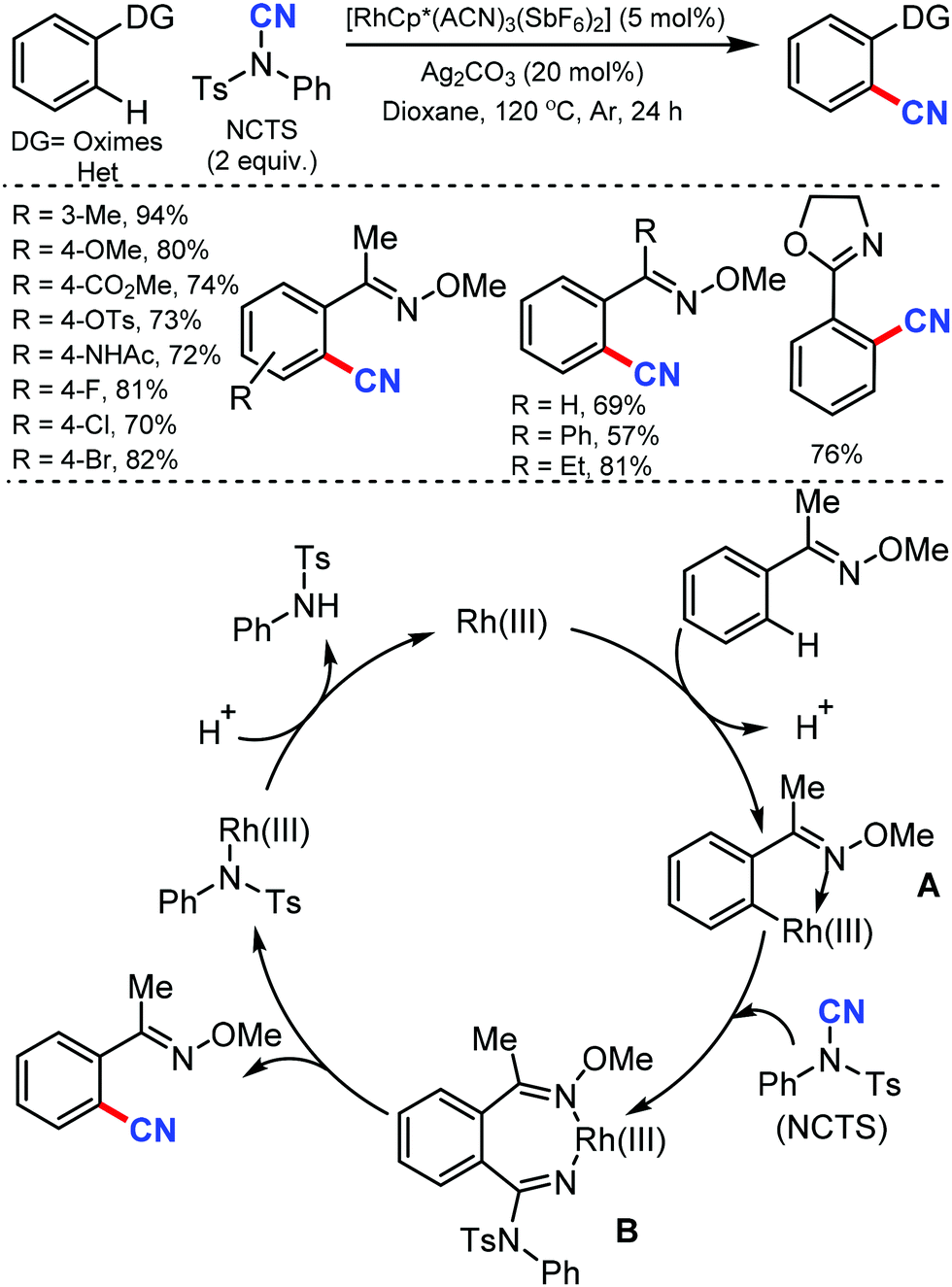 | ||
| Scheme 29 Rh-Catalyzed ortho-cyanation of oximes with a plausible mechanism. | ||
Lastly, the product was formed by the elimination of a tosylaniline-coordinated Rh(III) complex from B.
Similarly, the chelation-assisted rhodium-catalyzed cyanation of C–H bonds using NCTS was reported by Anbarasan and co-workers, which yielded various benzonitriles in good to excellent yield.31 Chelating groups such as pyridine, isoquinoline, benzoquinoline, pyrazine, and pyrimidine were employed in this strategy with low catalyst loading and catalytic additives (Scheme 30). Electron-withdrawing, electron-donating, and sterically-hindered substituted phenyl substrates were also found to be tolerable. Substrates with substitution on the pyridine ring and steric factors played a crucial role in determining the feasibility of the reaction.
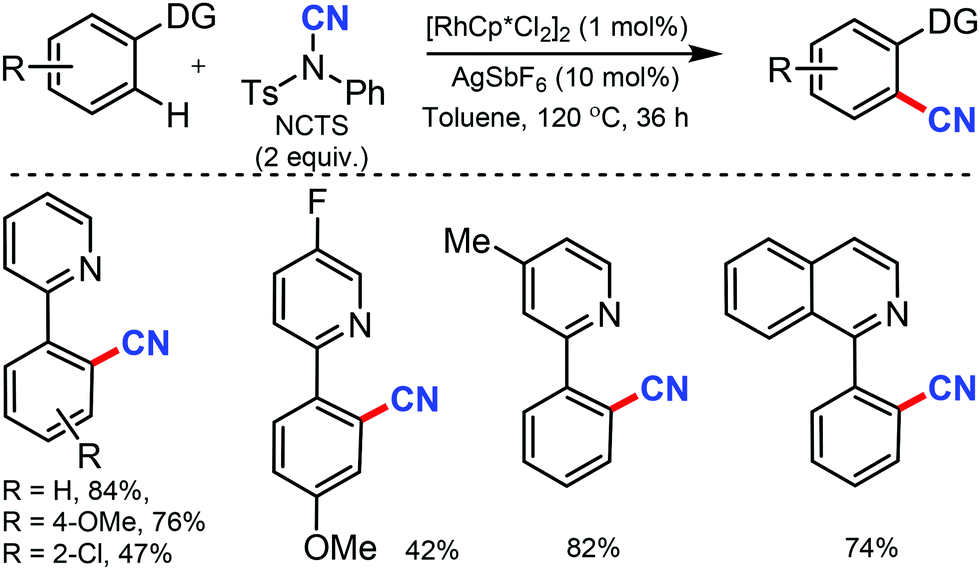 | ||
| Scheme 30 Rh-Catalyzed cyanation for heterocycles. | ||
In 2015, Fu and co-workers reported the synthesis of alkenyl nitriles by Rh(III)-catalyzed cyanation of vinylic C–H bonds employing NCTS.32 The combination of NaOAc and Ag2CO3 as additives considerably accelerated the reaction and arenes with both electron-withdrawing and electron-donating groups were cyanated successfully (Scheme 31). Pharmaceutically prominent fluorinated –CF3, –OCF3, and synthetically important heterocycles such as furan and dibenzofuran derivatives were also cyanated effectively under the optimized conditions.
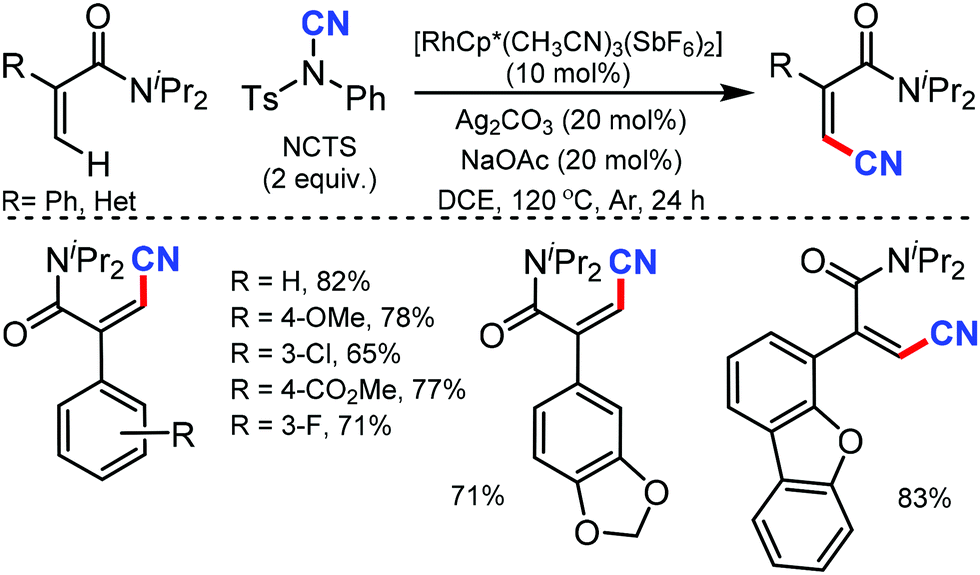 | ||
| Scheme 31 Rh-Mediated C–H cyanation of vinylic amides. | ||
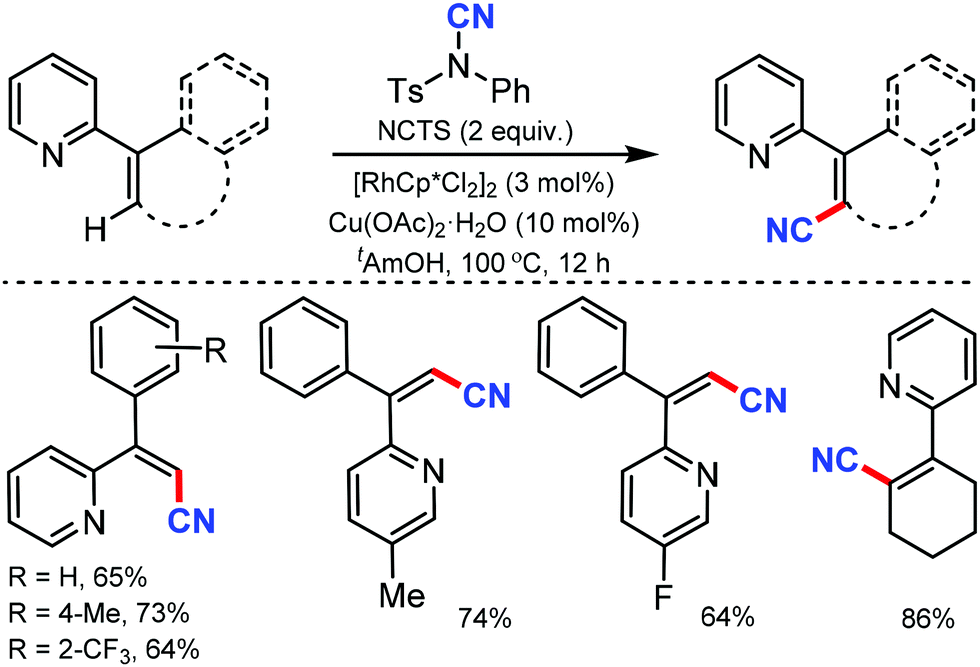 | ||
| Scheme 32 Rh-Catalyzed cyanation of alkenes. | ||
Many of the typical synthetic routes to acrylonitrile derivatives suffer from limited substrate scope and/or poor stereoselectivity. In 2015, Anbarasan and co-workers reported an efficient and selective rhodium-catalyzed cyanation of alkenes by chelation assistance using the readily available NCTS.33 Electron-donating groups (–OMe, –SMe) substituted and sterically-hindered ortho-substituted aryl groups possessing acrylonitriles underwent this reaction smoothly (Scheme 32). Nevertheless, strongly electron-withdrawing and electron-chelating nitro and acetyl-substituted aryl containing alkenes were found to be incompatible for the desired transformation.
Less toxic and highly stable ionic liquids (ILs) have been used as recyclable reaction media in organic synthesis in recent times. Wu and co-workers developed rhodium-catalyzed C–H bond cyanation using 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide ([BMIM]NTf2), an ionic liquid, as the recyclable medium.34
2-Phenylpyridine derivatives and aryl ketoneoxime ether derivatives were successfully cyanated through this protocol near room temperature. Both electron-donating and electron-withdrawing groups were found to be suitable for the cyanation of 2-phenyl pyridine, pyrazole, and triazole scaffolds (Scheme 33). Notably, the kinetic experiments indicate that C–H bond cleavage is the rate-determining step in this transformation.
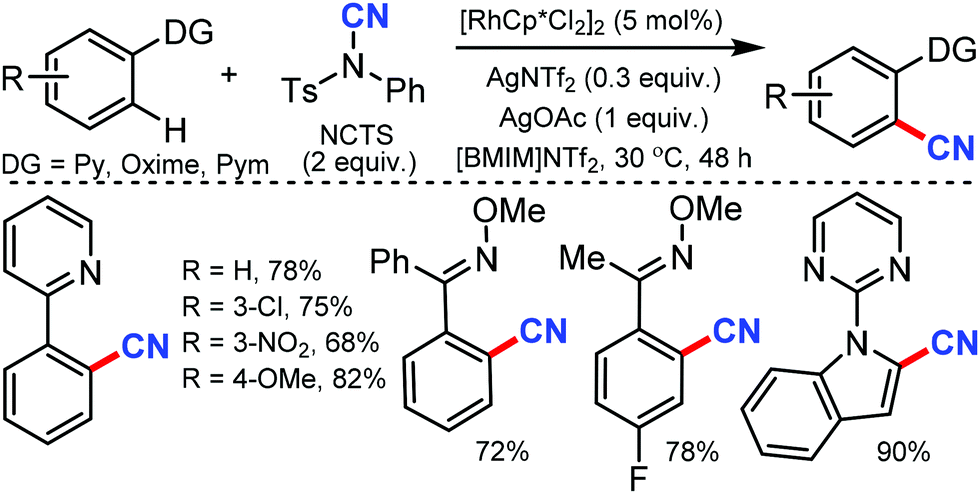 | ||
| Scheme 33 Rh-Catalyzed cyanation of heterocycles. | ||
In 2019, dimethylmalononitrile (DMMN), an easily available and less toxic organic cyanating reagent, was employed for the rhodium-catalyzed aromatic C–H bond cyanation by Bao and co-workers.35 Pyridine, quinoline, pyrimidine, and pyrazole as DG could afford the cyanated products with the use of copper oxide as a promotor (Scheme 34). Moderate to satisfactory yields of products were obtained from the reactions of substrates bearing substituted pyridine, irrespective of their electronic properties. Heterocycles such as quinoline, pyrimidine, and pyrazole could also afford the cyanated products.
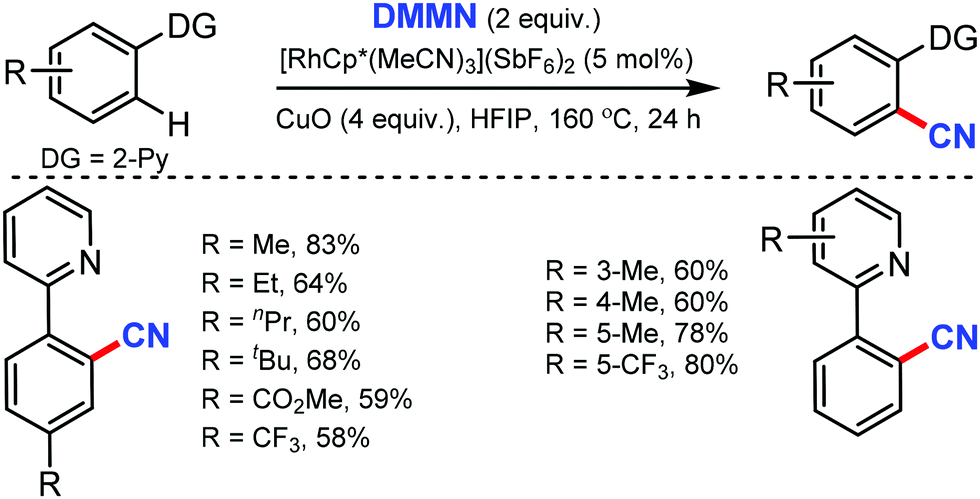 | ||
| Scheme 34 Rh-Catalyzed cyanation by dimethylmalononitrile. | ||
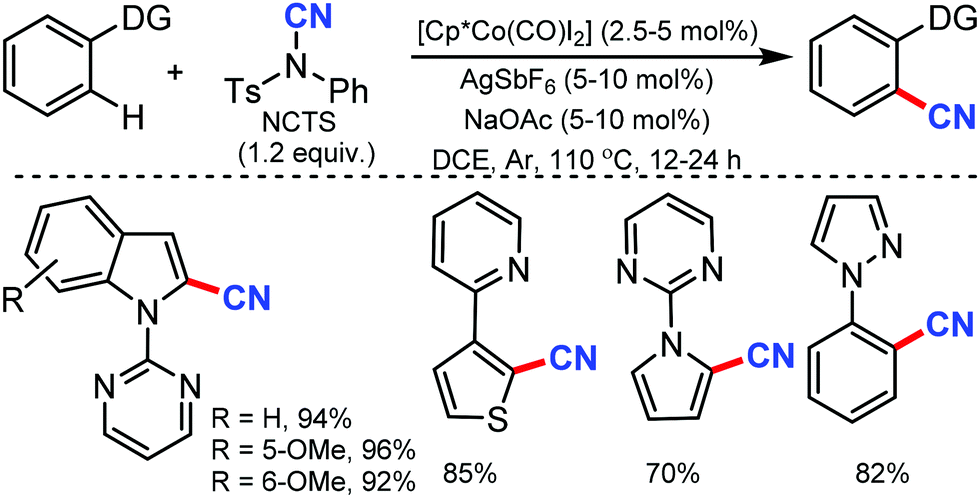 | ||
| Scheme 35 Cobalt-mediated cyanation of indole and heterocycles. | ||
Ackermann and co-workers utilised in situ generated cationic cobalt complex as an efficient catalyst for C–H cyanation using NCTS as the CN source.37 Diverse electrophilic groups such as esters or ketones yielded the desired products with notable chemoselectivity (Scheme 36). Pyridines (py), pyrimidine (pym), pyrazole, pyrrole and thiophene could be employed as the directing groups (DG) using this protocol. Mechanistic probing for kinetic studies suggested that C–H metalation is not the rate-determining step.
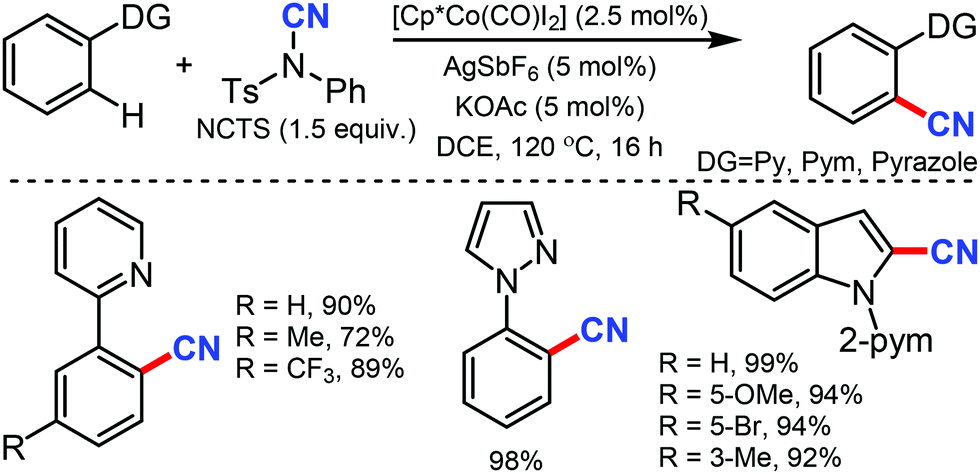 | ||
| Scheme 36 Cobalt-mediated cyanation of 2-phenylpyridines and indoles. | ||
N-Cyanosuccinimide, an easily available and bench-stable electrophilic cyanating reagent, was employed for the cobalt mediated C–H cyanation of arenes and heteroarenes by Chang and co-workers in 2015.38 In general, electron-donating groups, hydroxyl-protecting groups such acetate, silyl, methoxymethyl, p-methoxybenzyl (PMB) halides, heterocycles such as pyrazole, pyrimidine, thiophenyl, furanyl, pyrrolyl, and 6-arylpurines could also be cyanated efficiently (Scheme 37).
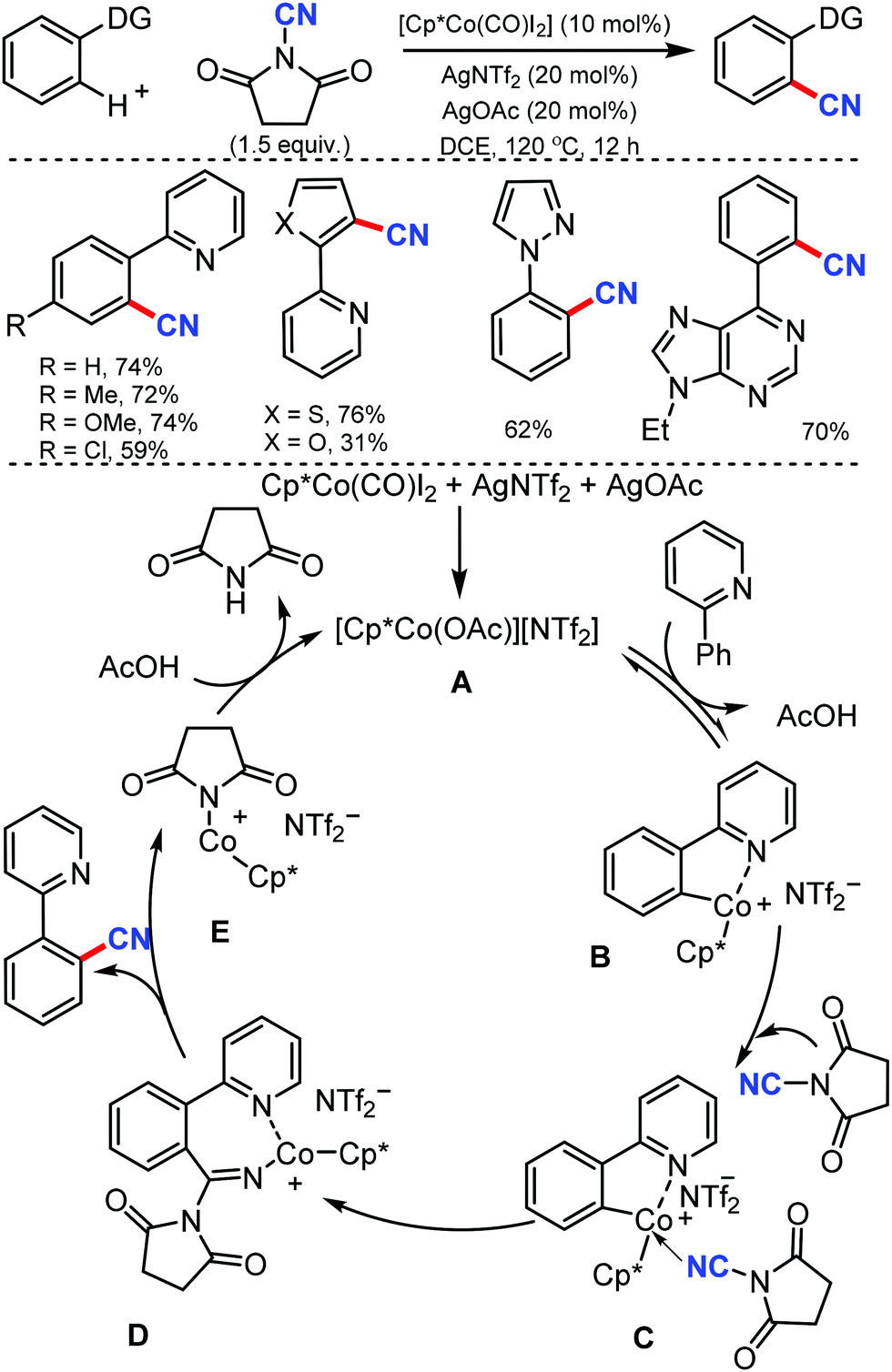 | ||
| Scheme 37 Cobalt-catalyzed cyanation of (hetero)arenes. | ||
Mechanical studies suggest that C–H bond cleavage may not be involved in the rate-limiting step and the reaction may proceed through a key imido intermediate. The cationic Co(III) species A is generated in situ from Cp*Co(CO)I2 and AgNTf2 in the presence of AgOAc reacts reversibly with 2-phenylpyridine to form a cobaltacycle B. The cyanating reagent coordinates to the cobalt center of B and then the migratory insertion of cyano (CN) into the metallacycle would lead to a key imido intermediate D.
The cyanated product is liberated from D. Kinetic experiments suggested that the C–H activation step could be reversible but might not be involved in the rate-limiting step.
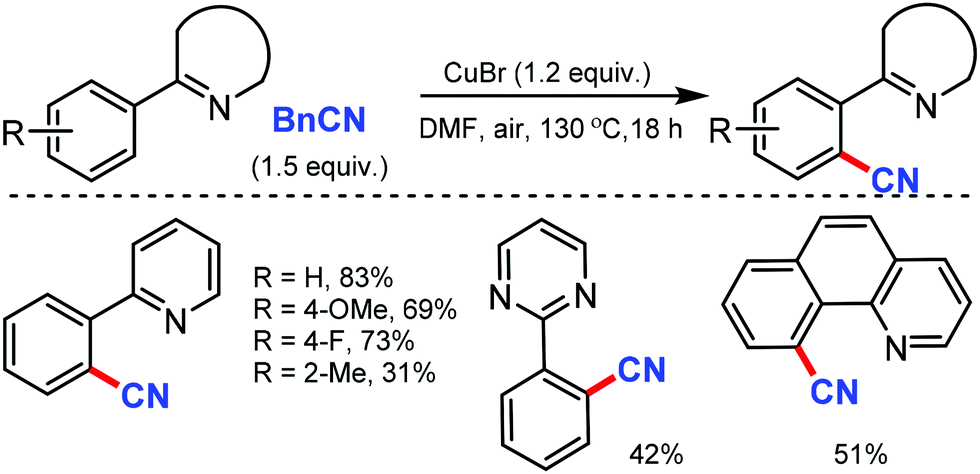 | ||
| Scheme 38 Cu-Catalyzed cyanation by BnCN. | ||
Venugopal and co-workers reported hydroxyapatite [HAP: Ca5(PO4)3(OH)]-supported Cu catalyst as an efficient and reusable heterogeneous catalyst for the cyanation of arenes with the combination of NH4HCO3 and DMF as the CN source.40 The surface basicity and Cu metal surface area of the catalysts were crucial for this cyanation reaction up to five catalytic cycles, which showed consistent activity and selectivity (Scheme 39). Interestingly, pyridine, isoquinolines, quinoline, and benzo[h]quinoline could act as the directing groups, delivering the corresponding aryl nitriles in good yields.
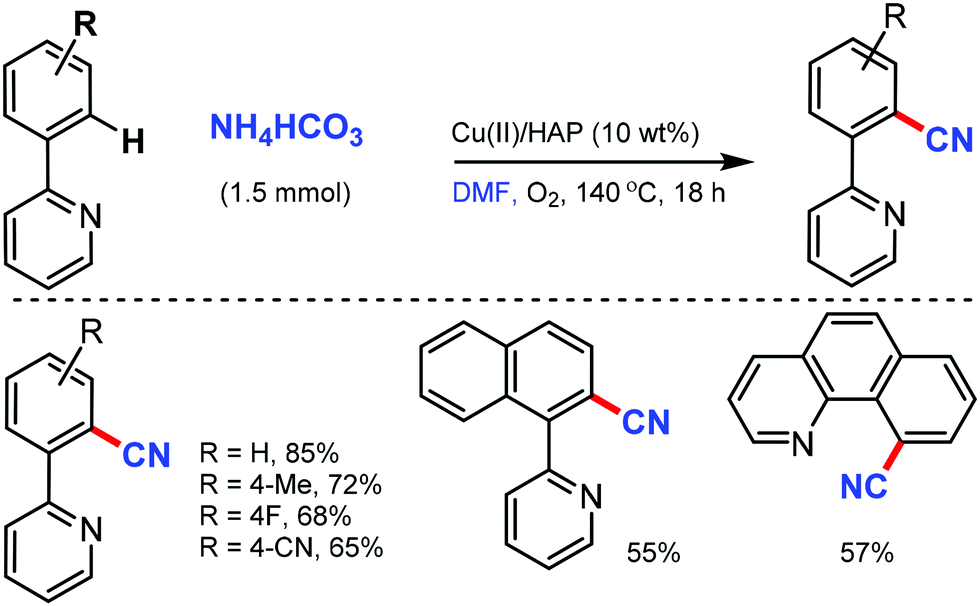 | ||
| Scheme 39 Cu-Mediated cyanation by NH4HCO3 and DMF. | ||
Later, an efficient method for the synthesis of (hetero)aryl nitriles mediated by copper was developed by Jiang and co-workers in 2017 using non-toxic and easily available ethyl (ethoxymethylene)cyanoacetate as the cyanide source with oxygen as the oxidant.41 2-Arylpyridines bearing either electron-donating or electron-withdrawing groups at the para-position of the aryl ring underwent the reaction smoothly, giving mono-cyanated products in moderate to high yields (Scheme 40). The ortho- and meta-substituted substrates could also afford the less hindered ortho-cyanated products regioselectively. For this transformation, kinetic studies suggested that C–H bond cleavage might be involved in the rate limiting step. The initial coordination of the nitrogen atom of the substrate to Cu(OAc)2 and the subsequent irreversible cyclocupration give the Cu(II) species A, which would undergo coordination exchange with the in situ generated cyanide anion to form the intermediate B. After disproportionation with another equivalent of Cu(OAc)2, intermediate B is then oxidized to the Cu(III) species C. Subsequently, the reductive elimination of C yields the cyanated product along with CuOAc.
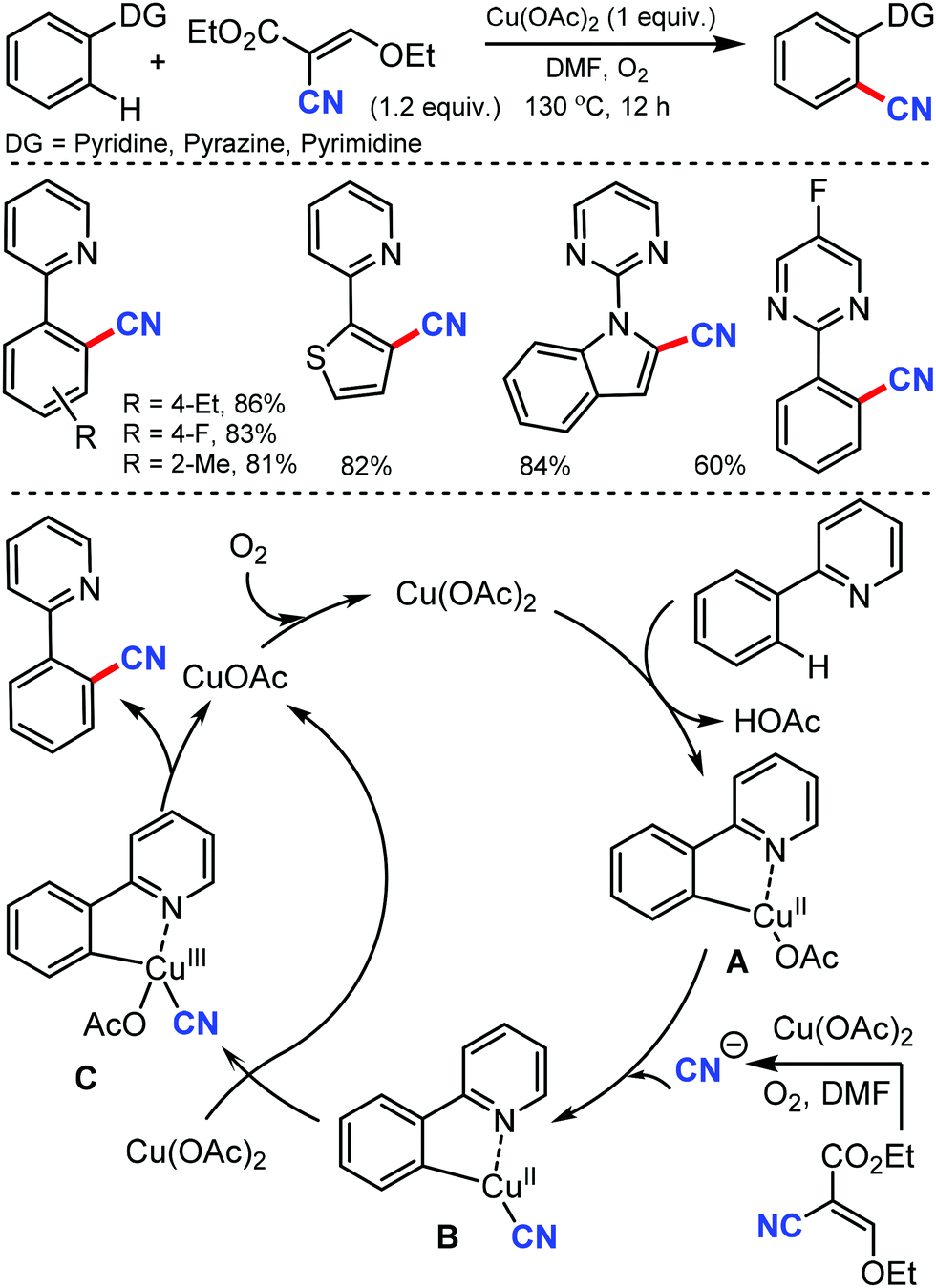 | ||
| Scheme 40 Cu-Catalyzed cyanation of 2-phenylpyridines with the plausible reaction mechanism. | ||
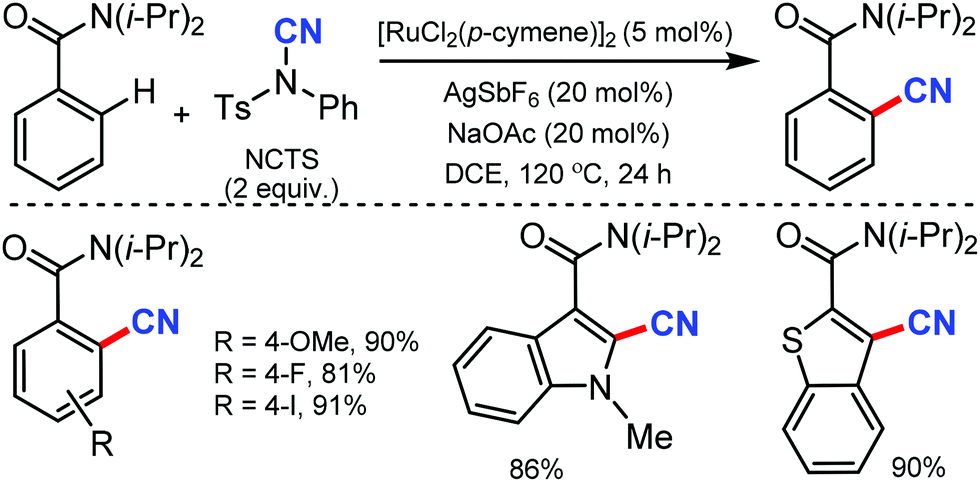 | ||
| Scheme 41 Ruthenium-catalyzed ortho C–H cyanation of amides. | ||
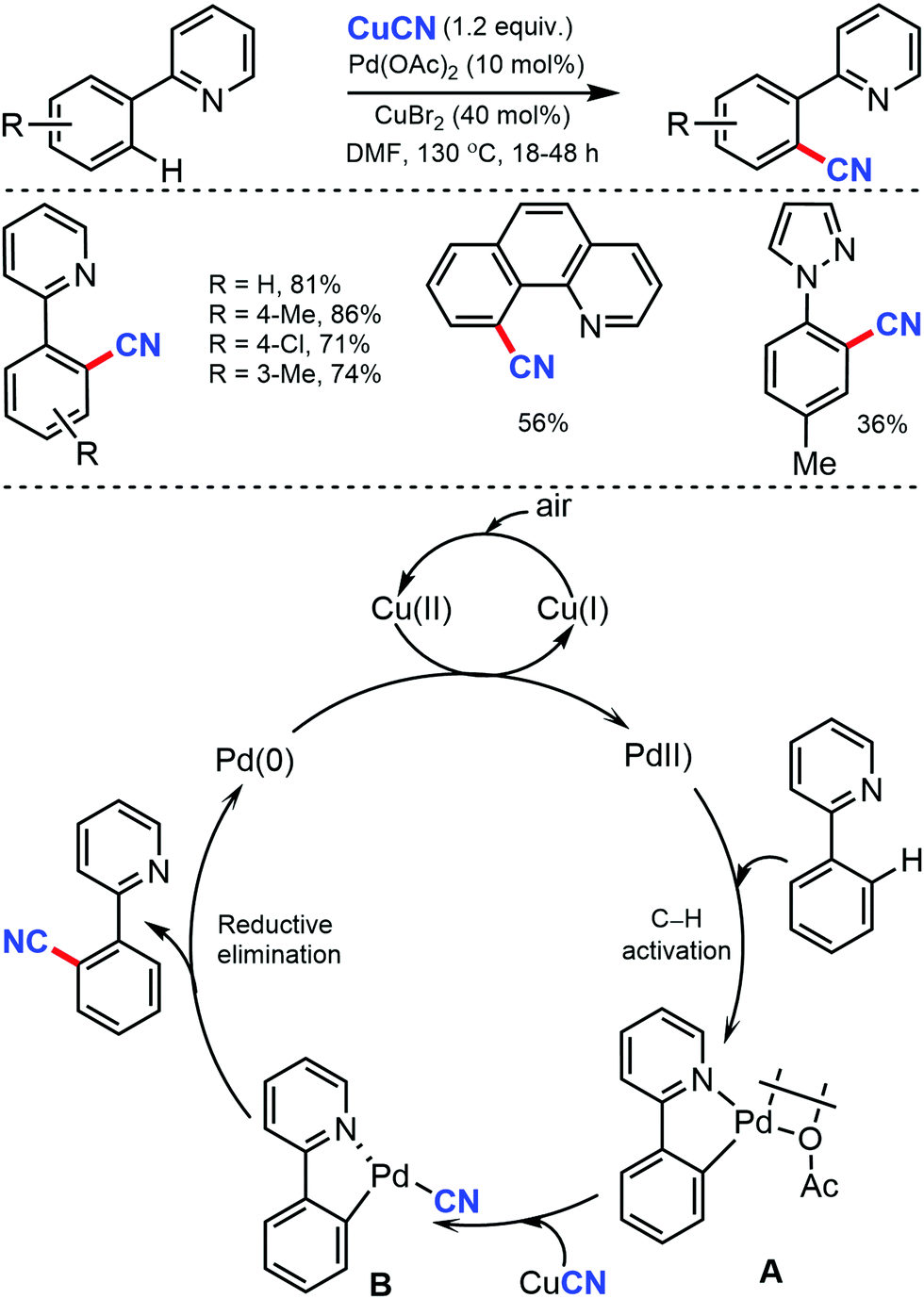 | ||
| Scheme 42 Palladium-catalyzed ortho-cyanation of arenes. | ||
In the proposed catalytic cycle, the cyclopalladated intermediate A is formed through the chelate-directed C–H activation of 2-phenylpyridine. Subsequent ligand exchange of CN− affords the Pd(II) species B, which undergoes the carbon–carbon bond forming reductive elimination to deliver the product and Pd(0), which on oxidation by Cu(II) and/or air, regenerates Pd(II).
Combining DMF and NH3 as the effective CN source for the palladium-catalyzed C–H cyanation was revealed by Chang and co-workers in 2010.44 Intriguingly, the carbon source of “CN” was provided by the dimethylamino moiety rather than the formyl group of DMF. However, the electron-deficient groups resulted in decreased product yields. Notably, substrates such as 1-phenylisoquinoline and benzo[h]quinoline also proceeded smoothly (Scheme 43).
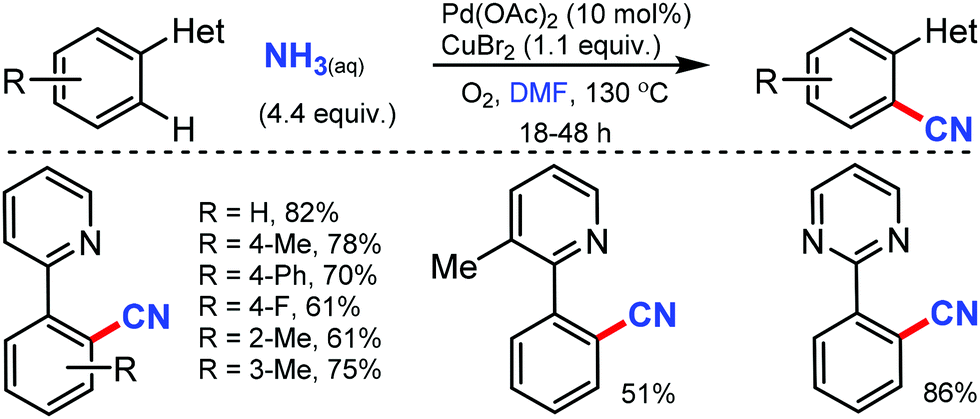 | ||
| Scheme 43 Palladium-catalyzed cyanation of aryl by NH3 and DMF. | ||
4.2. meta-C–H cyanation by the meta-directing template
In 2017, Maiti and co-workers disclosed the palladium-catalyzed meta-C–H cyanation of arenes by the removable pyrimidine-based directing group and CuCN.45 H-bonding interaction with pyrimidine and HFIP is hypothesized to decrease the basicity of the DG and synergistically increases the π-acidity of the palladium center. Electron-rich, electron-poor, and sterically-encumbered para-substituted benzylsilane, benzylsulfonates, benzylphosphonates, phenethylsulfonates, and phenethyl ether derivatives could afford the mono-cyanated products with high yield and selectively (Scheme 44).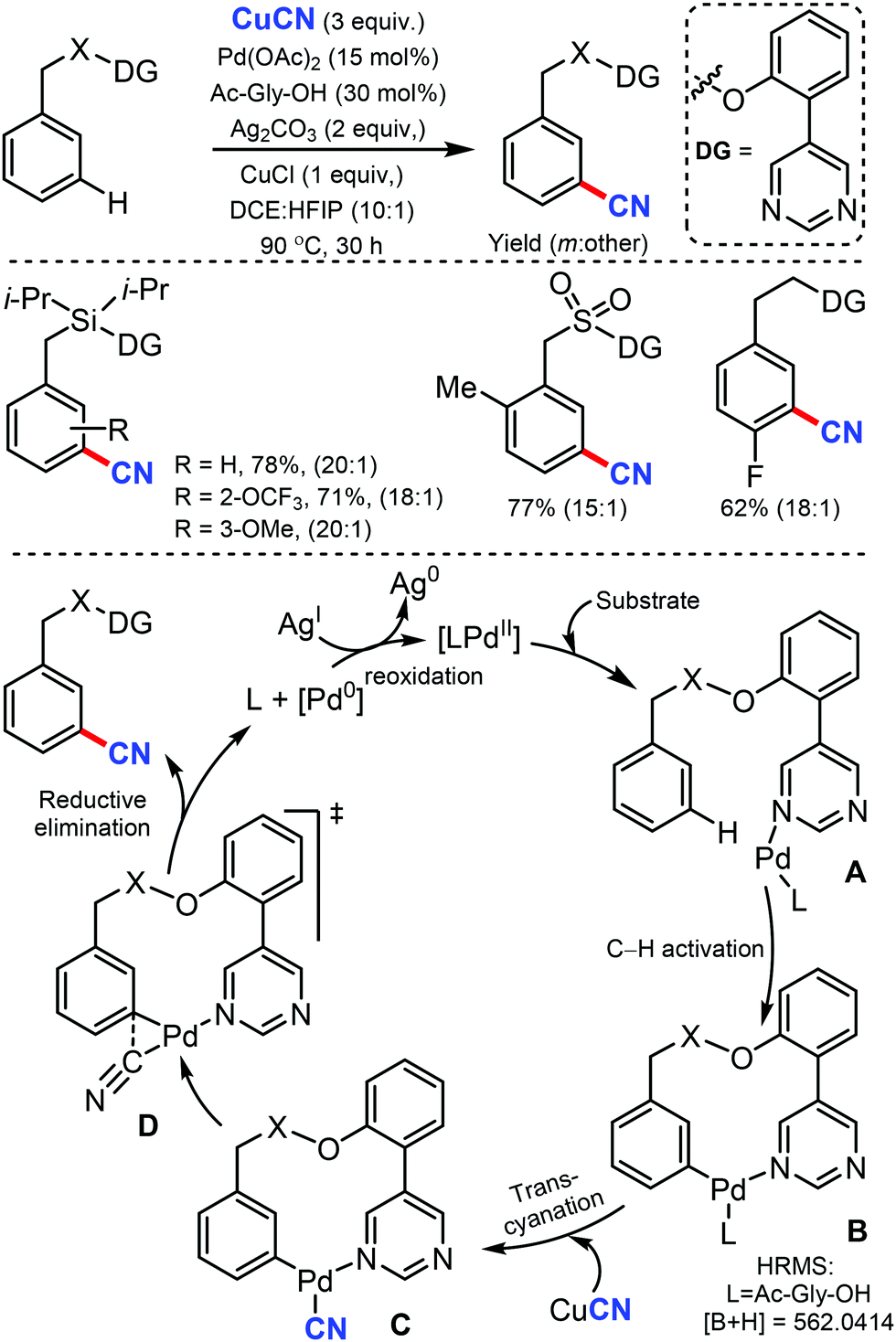 | ||
| Scheme 44 Remote meta C–H cyanation of arenes. | ||
The detailed NMR study by varying the amount of 1,1,1,3,3,3-hexafluoro-propan-2-ol (HFIP) in the presence of meta-substrate in CDCl3 revealed that hydrogen bonding is present in between the pyrimidine-directing group (DG) and HFIP and the kinetic isotope effect (KIE) experiment reported kH/kD = 1.25 and the intermolecular competition experiment PH/PD was found to be 1.15. In the probable catalytic cycle, first, the pyrimidine directing group coordinates with the mono-protected amino acid (MPAA)-ligated palladium catalyst (A) and the close proximity activates the meta-C–H bond (most probably via concerted metallation–deprotonation) to afford the macrocyclic transition state B. The ligand exchange of CN− between copper(I) cyanide and cyclopalladated intermediate forms C. The reductive elimination from Cvia the complex transition state D is presumed to provide the desired cyanation product.
Later, the same group developed the meta-C–H cyanation of arenes containing long alkyl chains using an ether-tethered, conformationally flexible, pyrimidine-based directing group using CuCN.46 Various arene/phenols scaffolds with chains ranging from propyl to octyl were selectively functionalized to afford the meta-cyanated product (Scheme 45). Electron-donating, electron-withdrawing, and sterically encumbered α-methyl propylbenzene were also tolerated. Along with cyanation, alkylation, olefination, and acetoxylation were also facile under the same reaction condition. The control experiments indicated that the pyrimidine-based template has a crucial role in the formation of the macrocyclic transition state for palladium-catalyzed C–H bond activation.
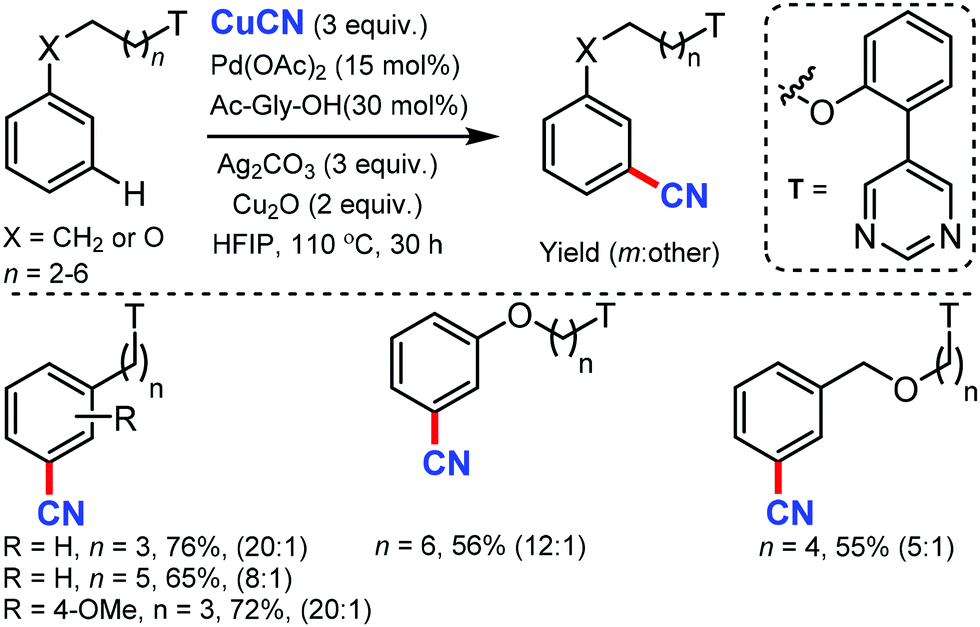 | ||
| Scheme 45 Palladium-catalyzed meta-C–H cyanation of arenes. | ||
Again in 2020, Maiti and co-workers reported the palladium-catalyzed meta-C–H cyanation of amides with the assistance of a pyrimidine-based directing group and CuCN under mild reaction conditions.47 Differently substituted phenylacetamides were selectively cyanated using this strategy. Notably, the cyanation of Ibuprofen could also be afforded with good yield and excellent meta-selectivity. Other than cyanation, diverse remote meta-C–H functionalizations of arenes such as allylation, alkylation, alkynylation, and deuteration were well executed under these reaction conditions (Scheme 46).
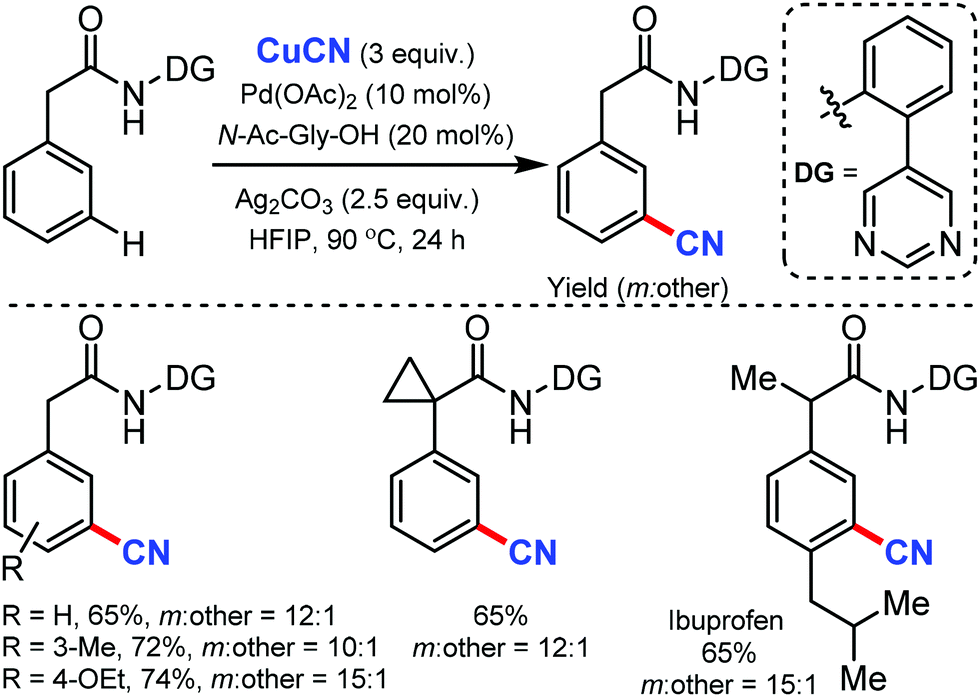 | ||
| Scheme 46 Palladium-catalyzed meta-C–H cyanation of amides. | ||
4.3. para-C–H cyanation by the para-directing template
Recently in 2020, Maiti's group reported a new method for the distal para-selective C–H cyanation of toluenes and phenols by using nitrile-based para-directing template using Pd(OAc)2 & monoprotected amino acid (MPAA) as ligand, Ag2CO3 as base and CuCN as the cyano source (Scheme 47).48 Groups such as CF3, OCF3, OMe, or Me on the same ring gave the para-cyanation product in good yield and with high para-selectivity. The higher para selectivity for this reaction was due to the design of the directing group, which has two methoxy group (Scheme 48), which forms hydrogen bonding with 1,1,1,2,2,2-hexafluoro-propan-2-ol (HFIP). The reaction proceed via C–H activation, which is not the rate determining step of the reaction (probed by experimental and computational studies), followed by CN coordination assisted by Cu and subsequent reductive elimination gives the para-cyanated product.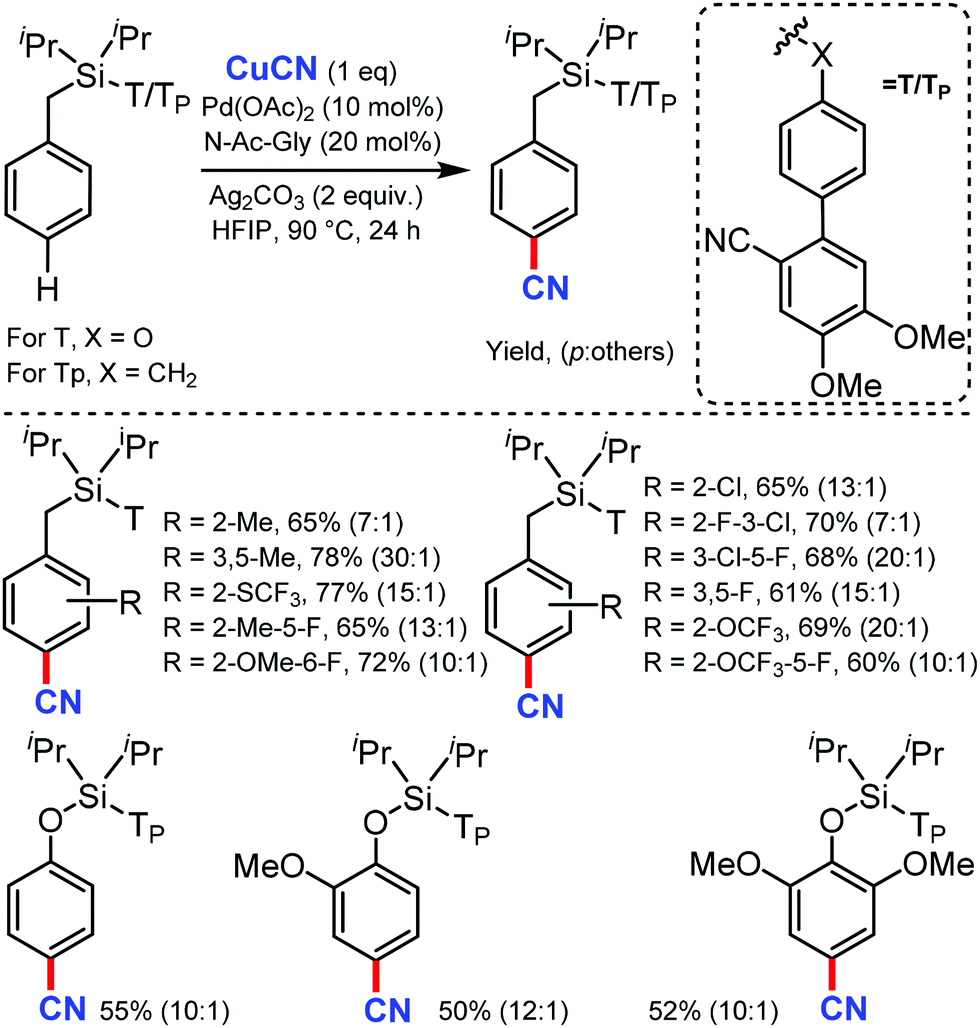 | ||
| Scheme 47 para-Selective cyanation by para-directing templates. | ||
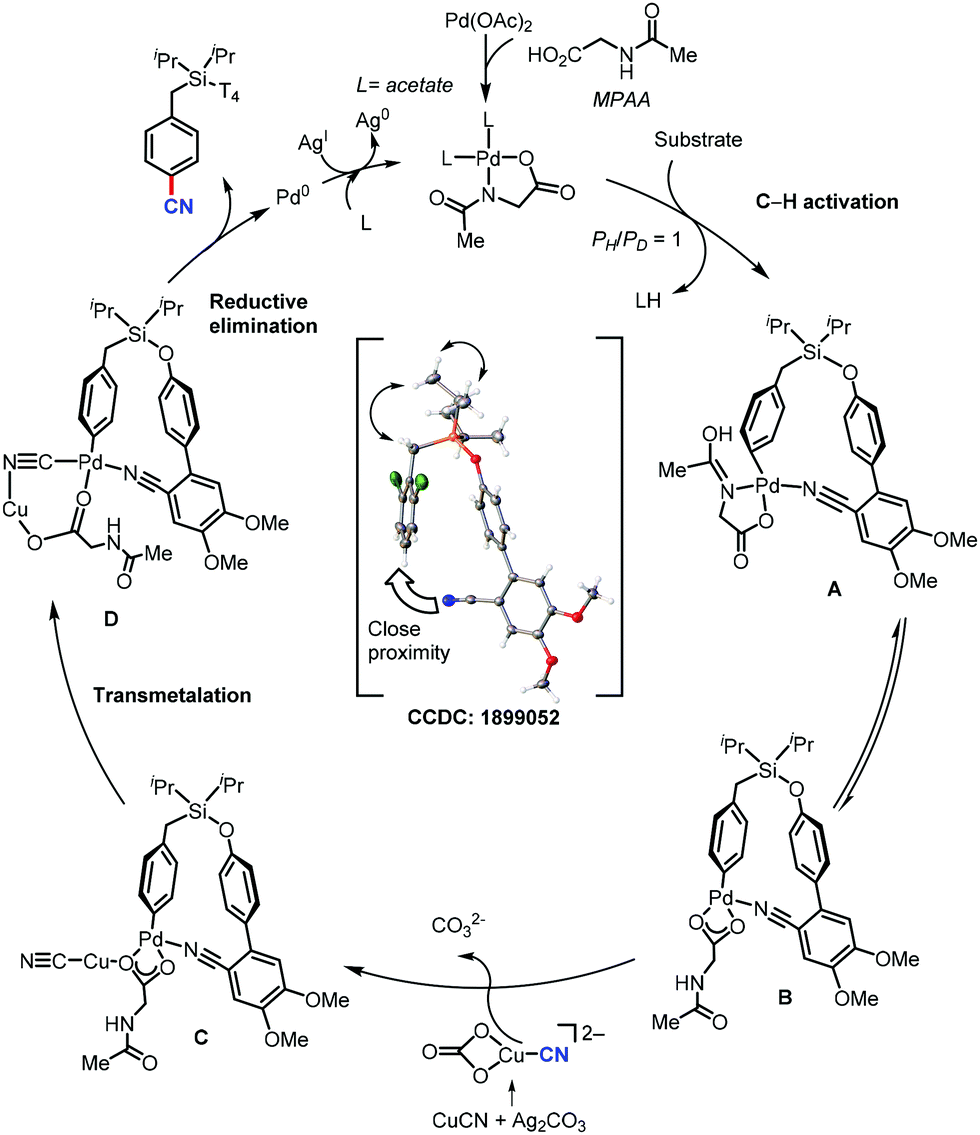 | ||
| Scheme 48 Plausible mechanism for para-C–H cyanation. | ||
This method is superior due to the iterative multi-functionalization and post synthetic modifications.
To understand the mechanism for para-cyanation, a detailed study using density functional theory (DFT) was carried out. Unlike other para-functionalizations reported earlier, mono-N-protected amino acid (MPAA) ligand promotes para-C–H activation via the concerted metalation deprotonation (CMD) pathway. The N-acyl group of MPAA ligand acts as the base to form A. Intermediate A then tautomerizes into a more stable palladacycle B with the carboxylic group bound in the κ2 fashion to the Pd center. Density function theory (DFT) computations for a series of possible CuCN complexes with anions showed that CuCN(CO3)2− is the most stable species. Later, CuCN coordinates to the carboxylic group of palladacycle B to form intermediate C. The resulting palladacycle D smoothly undergoes C–C reductive elimination to form the cyanation product (Scheme 48).
Also, C–H activation was found to be reversible, which was consistent with the experimental observation. The partial solubility of CuCN was very crucial for this reactivity. Due to the favorable interaction with tBuOH (ΔG = −7.3 kcal mol−1), CuCN is soluble in tBuOH with a relatively high –CN concentration. This increases the possibility of catalyst deactivation, leading to a low yield with tBuOH as the solvent. In contrast, the interaction of CuCN with HFIP is thermodynamically neutral (ΔG = 0.2 kcal mol−1), which provided an optimum cyanide concentration for the reactivity rather than catalyst deactivation.
5. Alkenyl and alkynyl C–H cyanation
In 2003, Suginome's group reported Pd2(dba)3 and Ni(COD)2 catalyzed intramolecular cyanoboration of alkynes. In this transformation, terminal and internal alkynes with various substituted groups reacted well to yield the cyanoboration complexes. This complex, when subjected to various synthetic procedures, yielded multi-substituted cyanoalkenyl products (Scheme 49).49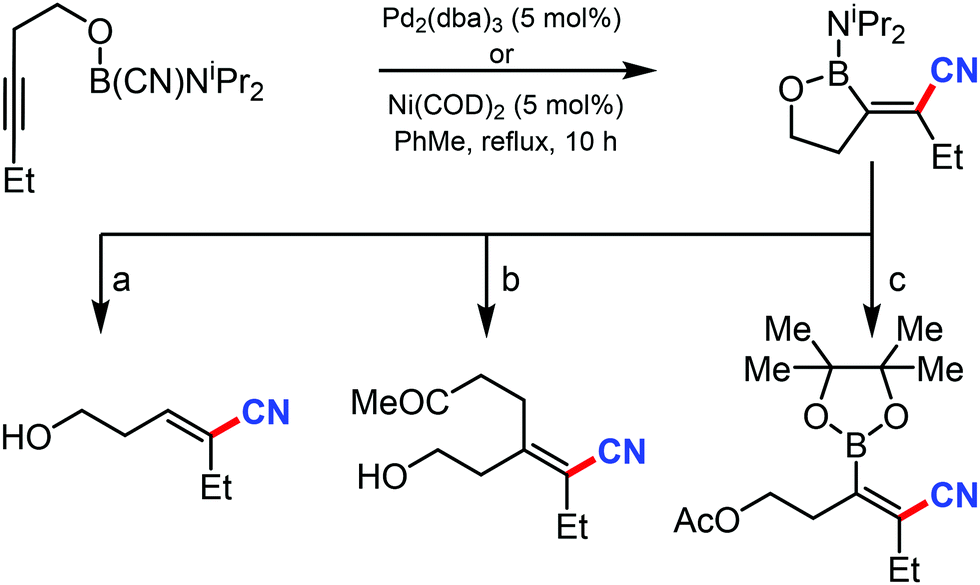 | ||
| Scheme 49 Pd/Ni-catalyzed cyanoboration of alkynes. (a) Rh(acac)(COD)/dppb (3 mol%), H2O, dioxane, 50 °C (b) methyl vinyl ketone (1 equiv.), Rh(acac)(COD)/dppp (3 mol%), MeOH, dioxane, 50 °C (c) pinacol (1.2 equiv.), Ac2O (1.2 equiv.), THF, 40 °C. | ||
The gallium-catalyzed bromocyanation of alkynes with cyanogen bromide was developed by Ohe and coworkers in 2011, which opened up a novel route to synthesize (Z)-β-bromoacrylonitriles regio- and stereoselectively.50 Several arylacetylenes having aromatic rings gave bromocyanated products with high regioselectivity while some internal aliphatic or alicyclic alkynes gave complex mixtures (Scheme 50). NMR studies indicated the probability of formation of a complex between BrCN and GaCl3 during the reaction.
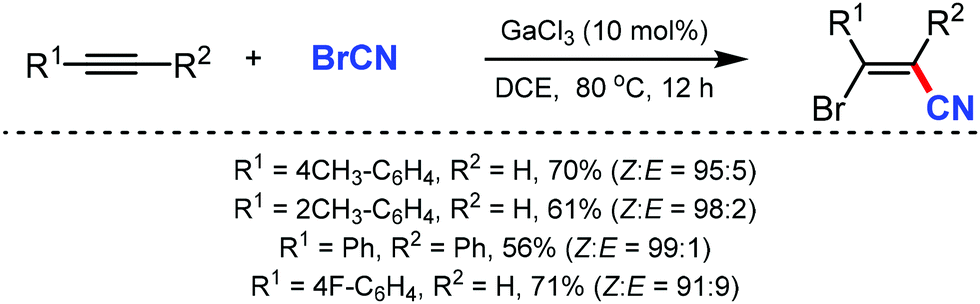 | ||
| Scheme 50 Gallium-catalyzed bromocyanation of alkynes with cyanogen bromide. | ||
An excellent strategy for the intramolecular oxycyanation of alkenes was reported by Nakao and coworkers in 2012.51 Under palladium/BPh3 catalysis, the cleavage of O–CN bonds and the subsequent insertion of double bonds was carried out. Using this method, the simultaneous incorporation of a tetra-substituted carbon and cyano group through O–CN bond activation was accomplished, which gave access to different substituted dihydrobenzofurans with high regioselectivity (Scheme 51).
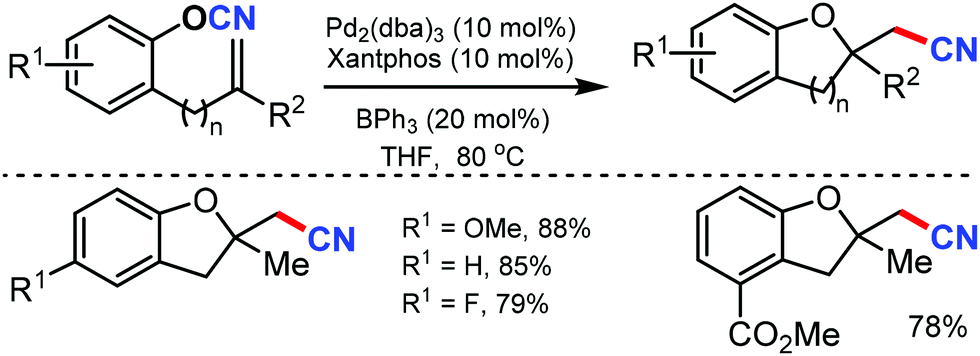 | ||
| Scheme 51 Intramolecular oxycyanation of alkenes by cooperative Pd/BPh3 catalysis. | ||
An easy access to various 1,2-thiobenzonitriles under mild reaction conditions and palladium catalysis was put forward by Werz and coworkers in 2015.52 Palladium mediated activation of carbon–sulfur bonds helps in aryne insertion into aryl thiocyanates to make the new C–SAr and C–CN bond simultaneously. Employing an oxygen atmosphere could reasonably increase the yields and was helpful in minimizing the side reactions (Scheme 52).
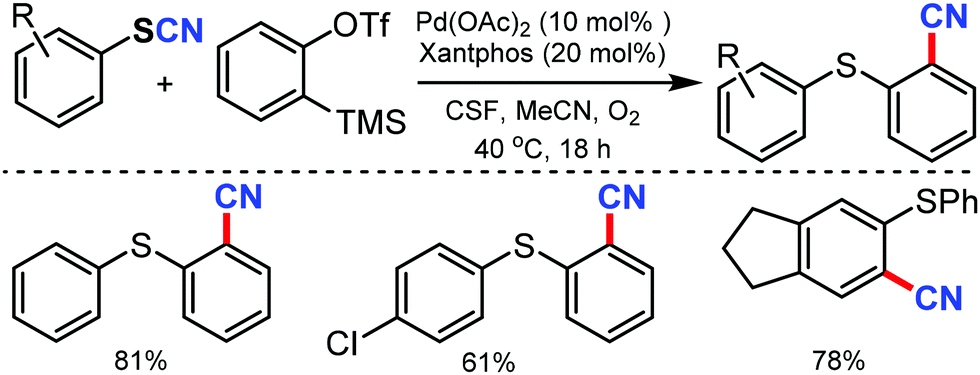 | ||
| Scheme 52 Synthesis of 1,2-thiobenzonitriles through the activation of aryl thiocyanates, followed by aryne insertion. | ||
In 2016, Maiti and co-workers reported an efficient strategy for the synthesis of a wide range of aryl nitriles by incorporating a metal-free cyanation of terminal alkynes by tert-butyl nitrite (tBuONO) under mild reaction conditions53 (Scheme 53). Phenylacetylenes bearing electron-rich groups as well as phenanthrene and pyrene acetylenes could afford the desired product in good to excellent yields. Functional groups such as 4-OMe, 2-Me-4-OMe, 4-pentoxy, 4-Br, esters, amides, and ketones were well tolerated.
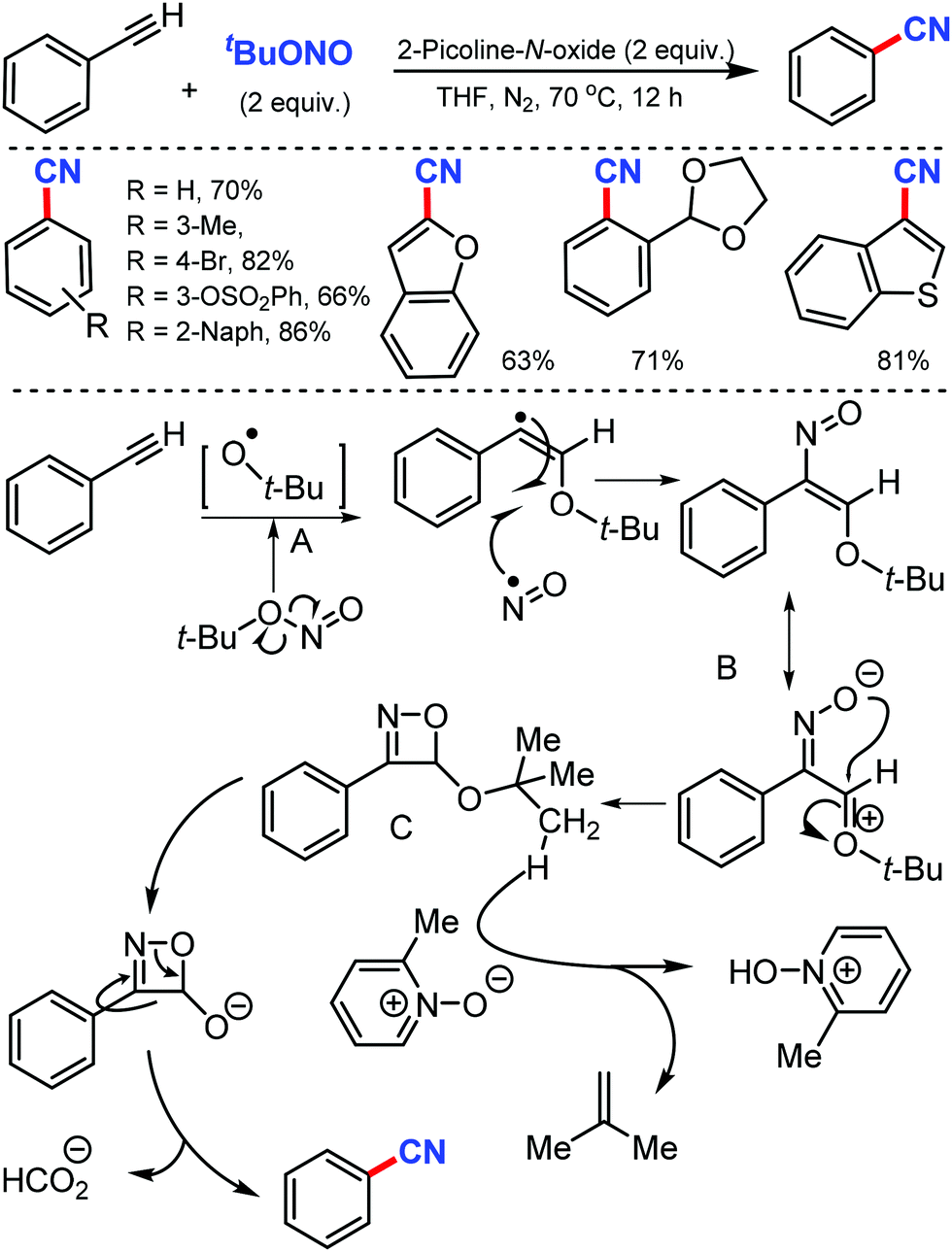 | ||
| Scheme 53 Synthesis of aryl nitriles from phenylalkynes and the radical induced mechanism. | ||
However, the internal alkynes and aliphatic alkynes were not suitable substrates for this cyanation method. Based on the kinetic studies, a plausible mechanism was proposed, in which tert-butyloxy and nitroso radical were formed by the in situ homolysis of tert-butyl nitrite. The alkyne forms a phenyl-substituted vinyl radical A by reacting with the tert-butyloxy radical. The subsequent cyclization of B then provides the strained four-membered intermediate C with the elimination of formic acid, leading to the formation of benzonitrile. Similarly, silver-mediated direct cyanation of terminal alkynes was developed by Bi and co-workers in 2017, employing non-toxic and facile N-isocyanoiminotriphenylphosphorane (NIITP) as the cyanating agent (Scheme 54).54
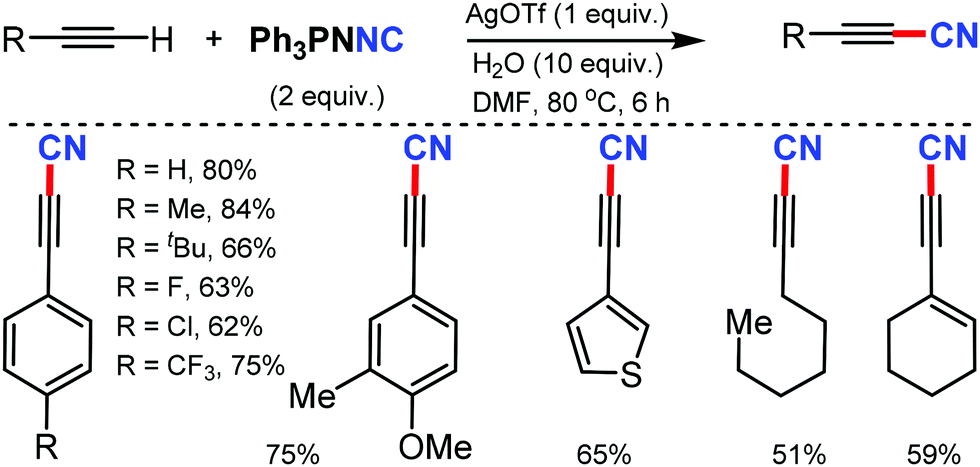 | ||
| Scheme 54 Silver-mediated C–H cyanation of terminal alkynes with NIITP. | ||
A metal-free approach for the direct cyanation of alkenes using TMSCN and [bis(trifluoroacetoxy)iodo]arene was reported by Studer and co-workers in 2018.55 Under mild reaction conditions, various 1,1-disubstituted, 1,2-disubstituted, and trisubstituted alkenes could be cyanated in moderate to good yields with high diastereoselectivity (Scheme 55). Notably, the selectivity was found to reduce as the size of the α-alkyl substituent increased. Mechanistic studies suggest that the electrophilic activation of alkene by the cyano iodine(III) species generated in situ from the [bis(trifluoroacetoxy)iodo]arene reagent is involved in this transformation.
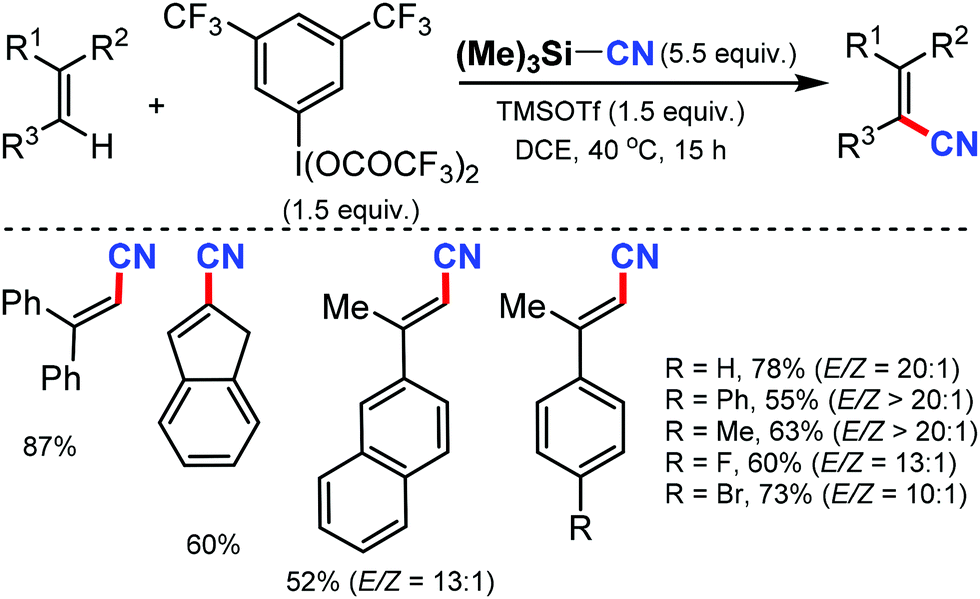 | ||
| Scheme 55 Metal-free direct C–H cyanation of alkenes. | ||
Recently, the realm of cyano-functionalization was further explored by Werz and coworkers by palladium catalysis and was utilized for the cyanosulfenylation56 and cyanoselenylation57 of internal alkynes independently in 2020 (Scheme 56). Both aromatic and aliphatic thiocyanates underwent the syn-1,2-cyanosulfenylation of internal alkynes and could access the tetra-substituted double bonds with sulphur and cyano in adjacent positions. The reaction could tolerate the addition of aromatic and aliphatic thiocyanates in an intra- and intermolecular fashion. Under similar reaction conditions, cyano-selenylation was accomplished, which gave novel access to the intra- and intermolecular synthesis of selenium-substituted acyclic and heterocyclic acrylonitrile derivatives with the assistance of palladium catalysis and with high functional group tolerance. X-ray studies indicate the occurrence of short non-covalent chalcogen–chalcogen (Se⋯O) interaction.
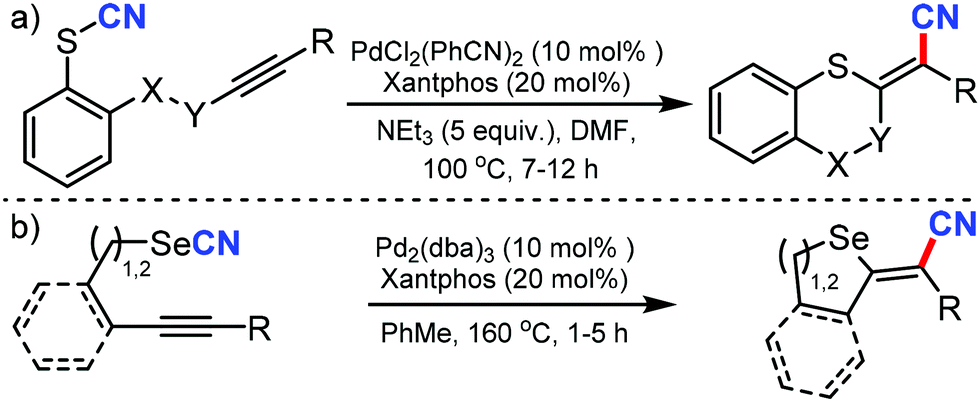 | ||
| Scheme 56 Palladium-catalyzed cyanosulfenylation (a) and cyanoselenylation (b) of internal alkynes. | ||
6. Asymmetric C–H cyanation
An efficient enantioselective cyanation of benzylic C–H bonds assisted by the copper catalyzed radical pathway was illustrated by Liu and co-workers in 2016.58 Various alkyl naphthalenes, alkyl arenes, and heterocycle containing alkyl arenes were tolerated under this protocol (Scheme 57). The mechanistic studies suggested that hydrogen-atom abstraction provides an achiral benzylic radical that undergoes asymmetric C(sp3)–CN bond formation upon reaction with a chiral copper catalyst. The proposed mechanism suggests that the ethylbenzene-derived radical reacts with CuII in an (L)CuII(CN)2 species to afford the benzyl-CuIII species. Subsequent C(sp3)–CN reductive elimination generates the benzylic nitrile product.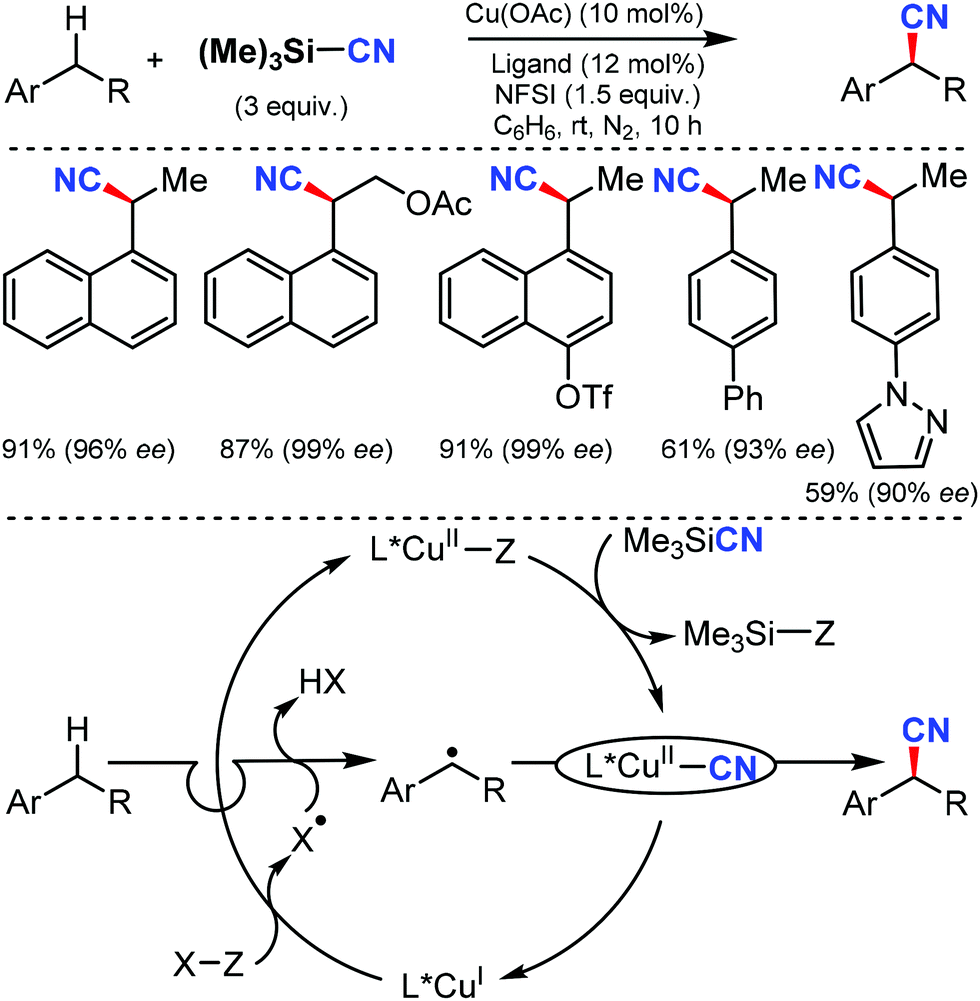 | ||
| Scheme 57 Enantioselective cyanation of benzylic C–H bonds. | ||
In 2016, Zhao and co-workers developed a chiral dipeptide-derived multifunctional organophosphine-based dual-reagent catalytic system for the asymmetric cyanation of ketoimines derived from isatins.59 Importantly, a zwitterion intermediate, which is generated in situ by mixing a chiral multifunctional organophosphine with methyl acrylate, functions as an efficient Lewis-base catalyst in this transformation (Scheme 58).
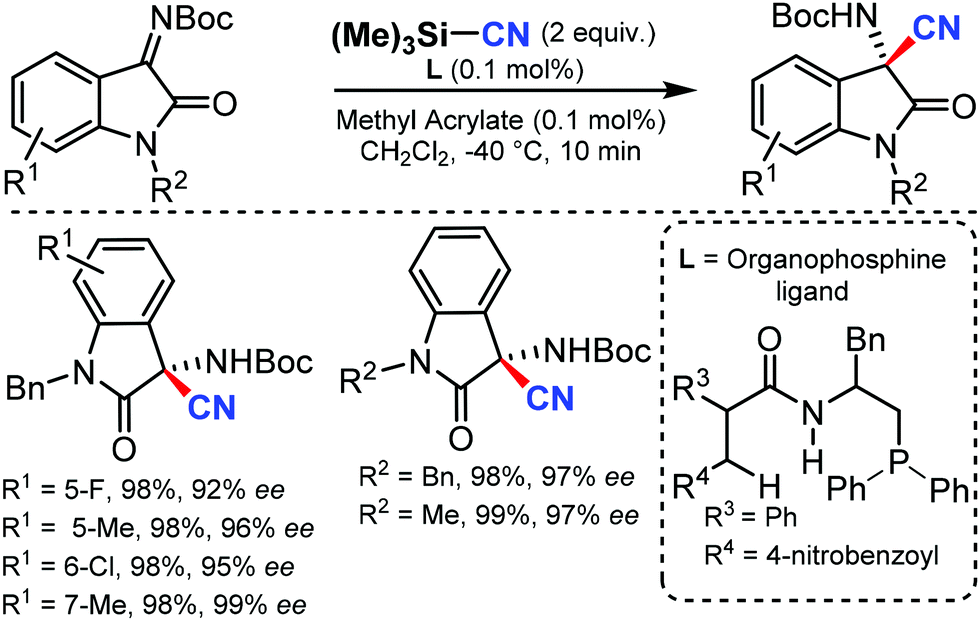 | ||
| Scheme 58 Asymmetric cyanation of ketoimines. | ||
The broad substrate scope of ketoimines derived from isatins and azomethine aldimines was asymmetrically cyanated in excellent yield and enantioselectivity with very low catalyst loading; the kinetic resolution of racemic 3-substituted azomethine imines was also carried out.
7. Electro-catalyzed C–H cyanation
Using 9-azabicyclononane N-oxyl (ABNO) as the catalytic mediator, the electrochemical α-cyanation of secondary piperidines was developed by Stahl and co-workers in 2018.60 The cyanation of the heterocycle adjacent to nitrogen without requiring protection or substitution of the N–H bond was accomplished through this strategy by using TMSCN as the source of cyanide nucleophile. Employing ABNO as a hydride-transfer mediator helps the reaction to proceed at low electrode potentials and is compatible with a wide range of functional groups. The reaction mechanism involved the electrochemical oxidation of ABNO, which produces the corresponding oxoammonium species that promotes the dehydrogenation of the secondary piperidine to the cyclic imine, followed by the addition of cyanide (Scheme 59).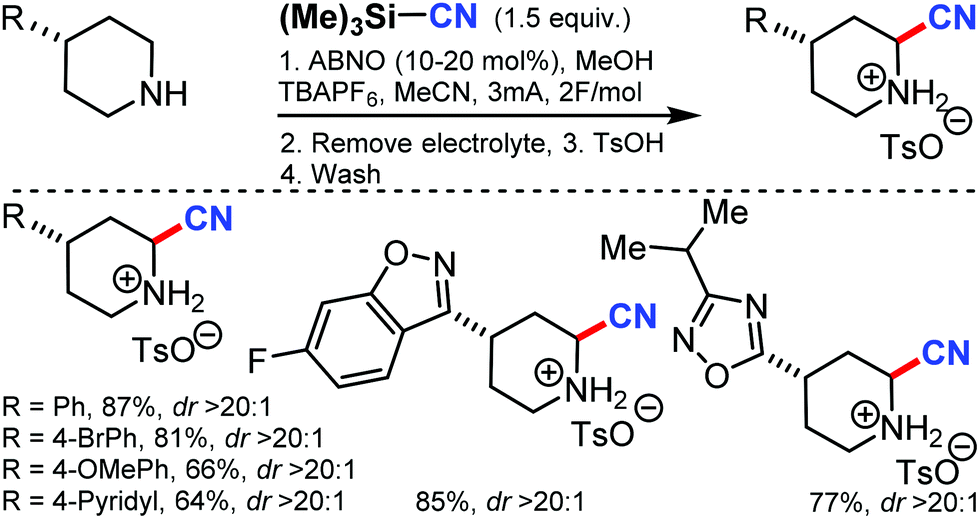 | ||
| Scheme 59 Electrochemical ABNO-mediated α-cyanation of secondary piperidines. | ||
8. Non-directed C–H cyanation of unactivated arenes
Ohe and co-workers reported simple direct cyanation of aromatic and heteroaromatic C–H bonds under gallium catalysis using cyanogen bromide (BrCN) as the CN source.61 Electron-donating groups substituted arenes and polycyclic aromatic compounds such as anthracene and pyrene were tolerated well in moderate to high yields. The scope of this protocol was also applicable to several substituted heterocycles such as pyrrole, furan, thiophene, indole, and benzofuran (Scheme 60).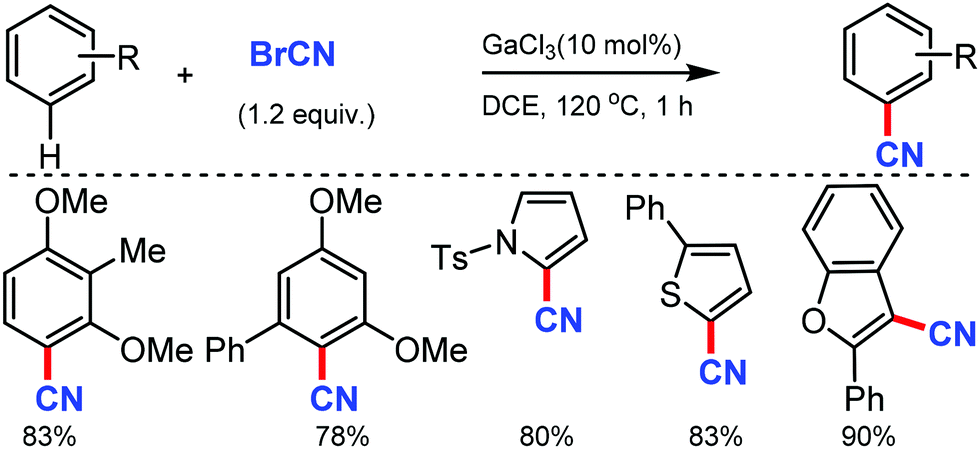 | ||
| Scheme 60 Gallium-catalyzed electrophilic cyanation of arenes. | ||
An efficient approach for the direct conversion of methyl arenes into aromatic nitriles using Pd(OAc)2 and N-hydroxyphthalimide (NHPI) as the catalysts was developed by Wang and co-workers in 2013.62 This ammoxidation method utilizes tert-butyl nitrite as the nitrogen source and oxidant under mild reaction conditions. Interestingly, multi-substituted toluene or oxidative sensitive groups such as Bpin were also compatible with this protocol. The scope of this reaction was also applicable for several polycyclic and heteroaromatic compounds efficiently. Mechanistic studies indicated that the reaction involves the formation of aldoxime as the key intermediate (Scheme 61).
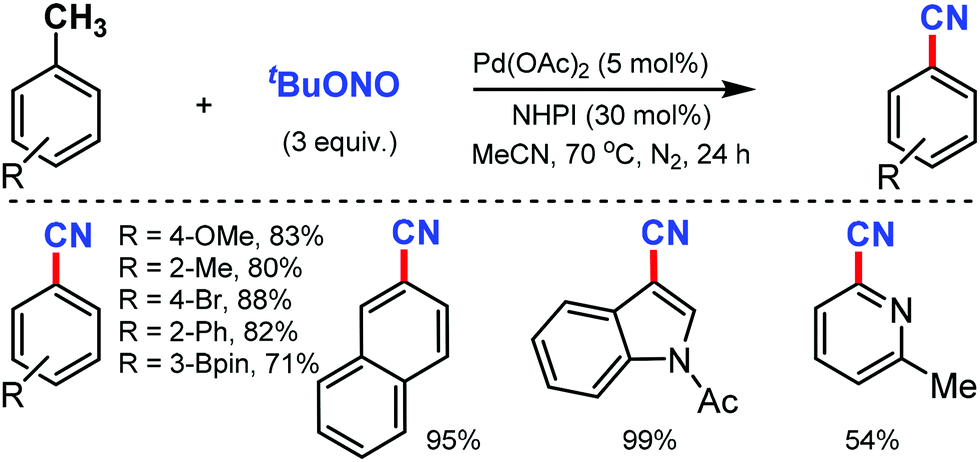 | ||
| Scheme 61 Palladium(II)-catalyzed direct conversion of methyl arenes into aromatic nitriles. | ||
Using 3,5-di(trifluoromethyl)phenyl(cyano)iodonium triflate (DFCT) as the cyanation agent, Wang and co-workers developed the oxidative cyanation of arenes catalyzed by Fe(II).63 Various electron-rich benzene derivatives were smoothly cyanated, with the regioselectivity governed by both electronic and steric effects of the substituents (Scheme 62). In case of heteroaromatic substrates, an additional requirement of 2,6-di-tert-butylpyridine was necessary for the reaction to proceed. The steric effects of the substituents played a crucial role in determining the selectivity of the reaction.
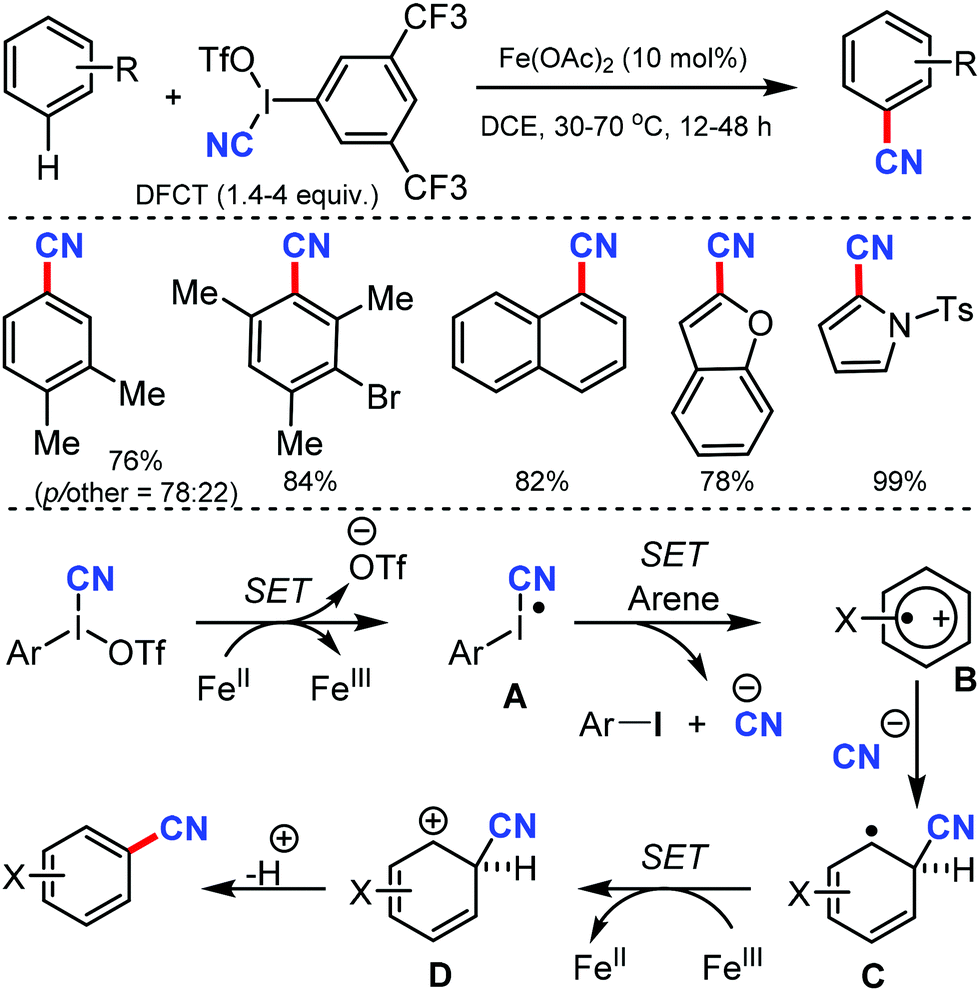 | ||
| Scheme 62 Iron(II)-catalyzed direct cyanation of arenes with DFCT. | ||
The reaction mechanism involves a single-electron transfer pathway (SET), in which a highly reactive radical species A is initially produced. SET from the radical species A to the arene substrate affords intermediate B; nucleophilic addition of cyanide ion to B then generates a radical intermediate C. In the final step, cation intermediate D is formed, the deprotonation of which affords the cyanated product.
Phenol derivatives are generally inexpensive and readily available chemicals in organic synthesis. An environmentally benign protocol for the cyanation of phenol derivatives was disclosed by Yamaguchi and co-workers in 2016 with the help of a nickel-based catalytic system consisting of a unique diphosphine ligand such as dcype (1,2-bis(dicyclohexylphosphino)ethane) or dcypt (3,4-bis-(dicyclohexylphosphino)thiophene) and aminoacetonitrile as the metal-free cyanating agent.64 Various aryl carbamates such as biphenyl, naphthyl, polycyclic, quinoline, carbazole, flavone, estrone, and tyrosine derivatives carbamates could afford the cyanated products in moderate to good yields (Scheme 63). Fortunately, this nickel catalytic system was applicable to tosylates, mesylates, triflates, sulfamates, phosphates as well as enol derivatives and provided the corresponding cyanated products in good yields.
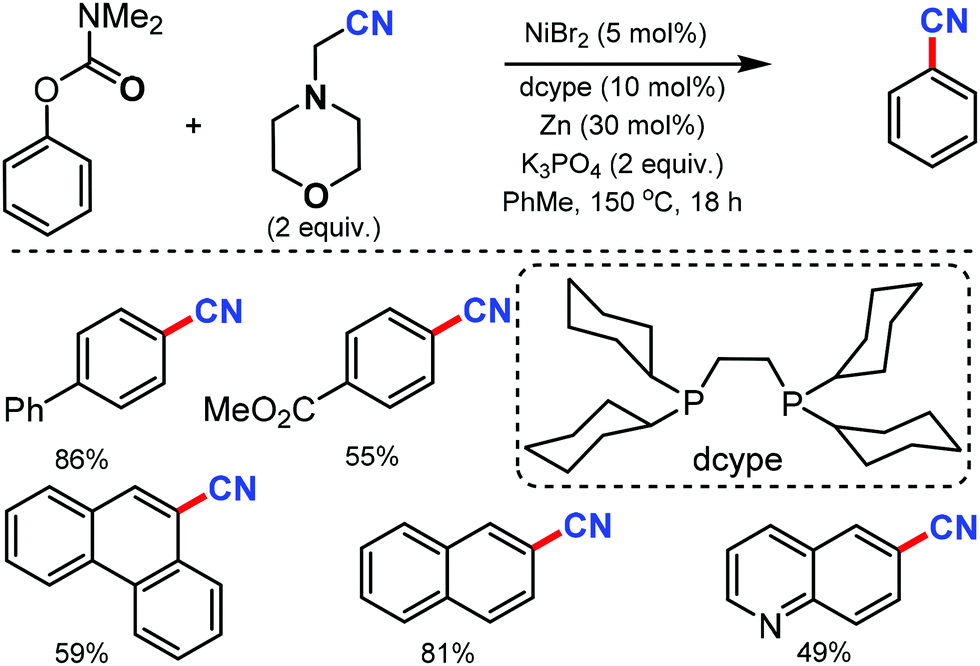 | ||
| Scheme 63 Aryl carbamates’ cyanation with aminoacetonitrile. | ||
Ritter and co-workers in 201965 described an expedient approach for benzonitrile derivatives by the non-directed method with K3Fe(CN)6 as the CN source. With broad substrate scope and functional-group tolerance, this protocol could easily afford the direct cyanation of several marketed small-molecule drugs, common pharmacophores, and organic dyes even though low regioselectivity was observed (Scheme 64).
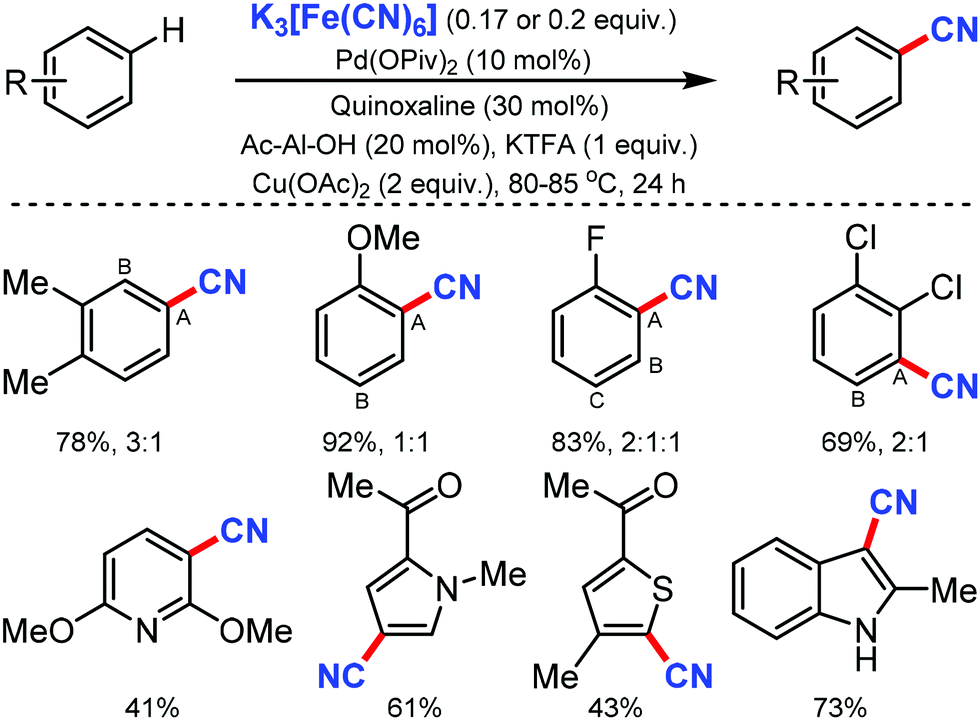 | ||
| Scheme 64 Pd-Catalyzed non-directed C–H cyanation. | ||
Notably, pyridine, thiophene, pyrrole, indoles as well as functional groups including sulfonamides, esters, amides, unprotected hydroxyl groups, and ketones were compatible with the reaction conditions. Both monoprotected amino acids (MPAA) and quinoxaline played a crucial role in accessing the catalyst active sufficiently for C–H metalation.
9. Photocatalysis approaches for C–H cyanation
In 2017, Liu and co-workers reported an enantioselective decarboxylative cyanation by combining photoredox and copper catalysis using TMSCN as the CN source to give the alkyl nitriles.66 A broad range of NHP esters derived from simple carboxylic acids could afford the anticipated products in good yields and excellent enantioselectivities with high functional group tolerance. Mechanistic studies suggested that both benzylic radicals and reactive chiral copper(II) species were generated from the photocatalytic cycle (Scheme 65).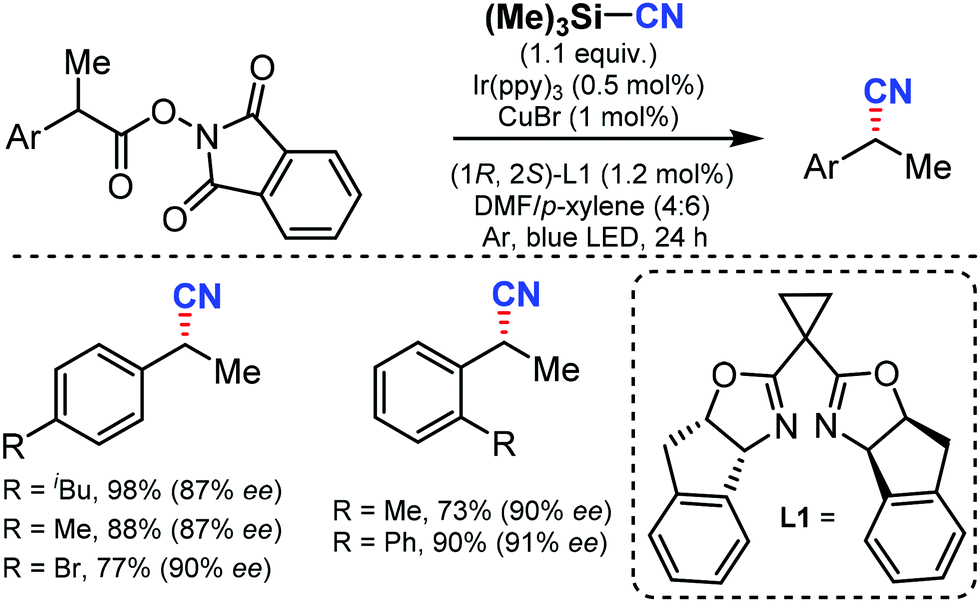 | ||
| Scheme 65 Enantioselective decarboxylative cyanation by cooperative photoredox and copper catalysis. | ||
Later, in 2017, a similar approach for decarboxylative cyanation was developed by Waser and co-workers in 2017.67 A variety of natural and non-natural α-amino, several dipeptides, drug precursors, and α-oxy acids were cyanated smoothly using an iridium photoredox catalyst. Notably, different protecting groups, such as –Cbz, –Boc, and –Fmoc were tolerated for this transformation. It was proposed that single electron transfer (SET) to form the iminium intermediate, followed by cyanide addition, was involved in the reaction mechanism (Scheme 66).
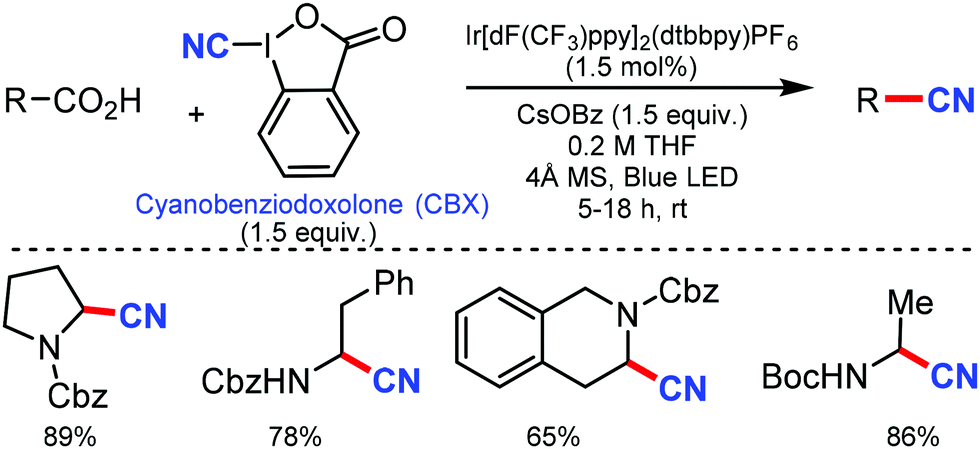 | ||
| Scheme 66 Decarboxylative cyanation of carboxylic acids. | ||
10. Radical mediated cyanation
In 2008, Porta and co-workers reported a method for the synthesis of β-hydroxynitriles by the cross coupling of stabilized radicals and the α-cyanoisopropyl radical.68 In this transformation, Ti(IV) chelates and promotes homolytic C–C cleavage and enhances the captodative effect. This allowed the application of the well-known Ingold–Fischer effect to a wider range of stabilized carbon-centered radicals.Later, a metal-free aerobic oxidative cyanation of tertiary amines to R-aminonitriles using a catalytic amount of azobisisobutyronitrile (AIBN) was disclosed by Yan and co-workers in 2012.69 Several substituted N,N-dimethylanilines, with electron-donating or -withdrawing groups, could afford the corresponding cyanated products in excellent yields (Scheme 67). The reaction proceeds through the formation of highly reactive iminium ion intermediates.
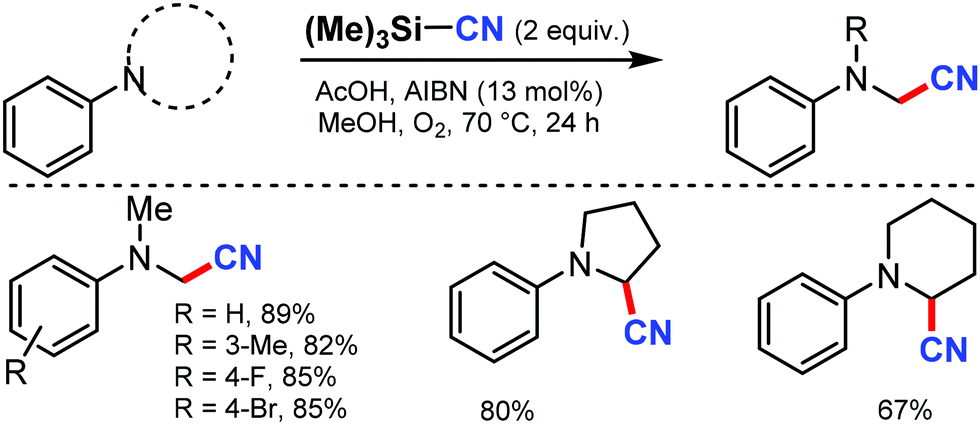 | ||
| Scheme 67 AIBN initiated oxidative cyanation of tertiary amines. | ||
Employing AIBN as a free radical CN source, a direct aryl C–H cyanation method was disclosed by Han and co-workers under Cu catalysis in 2013.70 Various 2-aryl pyridines with electron-donating and -withdrawing groups at the aryl ring gave the corresponding nitriles in moderate to good yields. Several substrates with substitution at the pyridyl ring were also tolerated. In general, electron-rich substrates gave better yield than electron-deficient substrates (Scheme 68). A CN free radical mechanism was involved in this reaction.
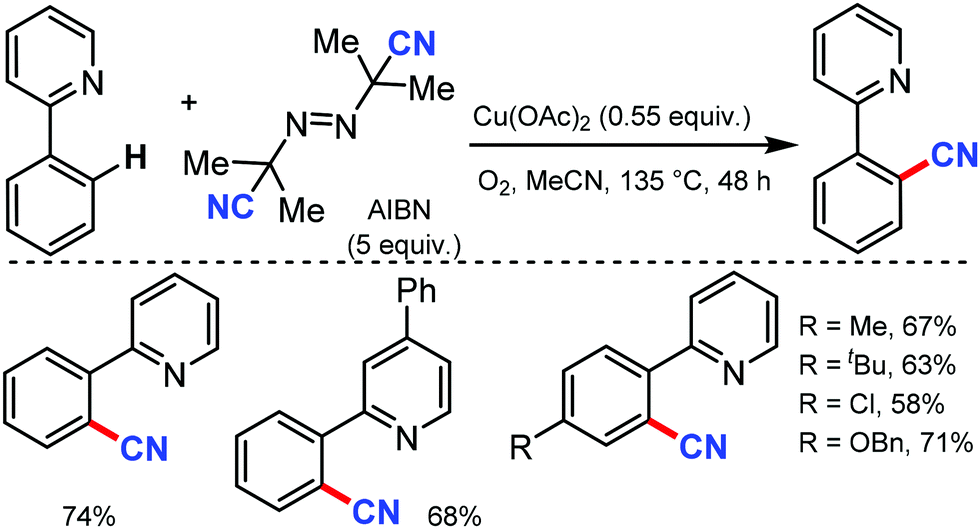 | ||
| Scheme 68 Copper-mediated direct aryl C–H cyanation with AIBN. | ||
The proposed mechanism involves the in situ generation of active catalytic species Cu(I)L by the reduction of Cu(II) with AIBN. The coordination of Cu(I)L with the substrate forms complex A. The subsequent oxidation of B with the assistance of the CN radical and O2 yields the high-valent Cu(III) complex CviaB through electrophilic Cu(III) promoted de-aromatization and re-aromatization. Later, the reductive elimination of C produces the cyanated product (Scheme 69).
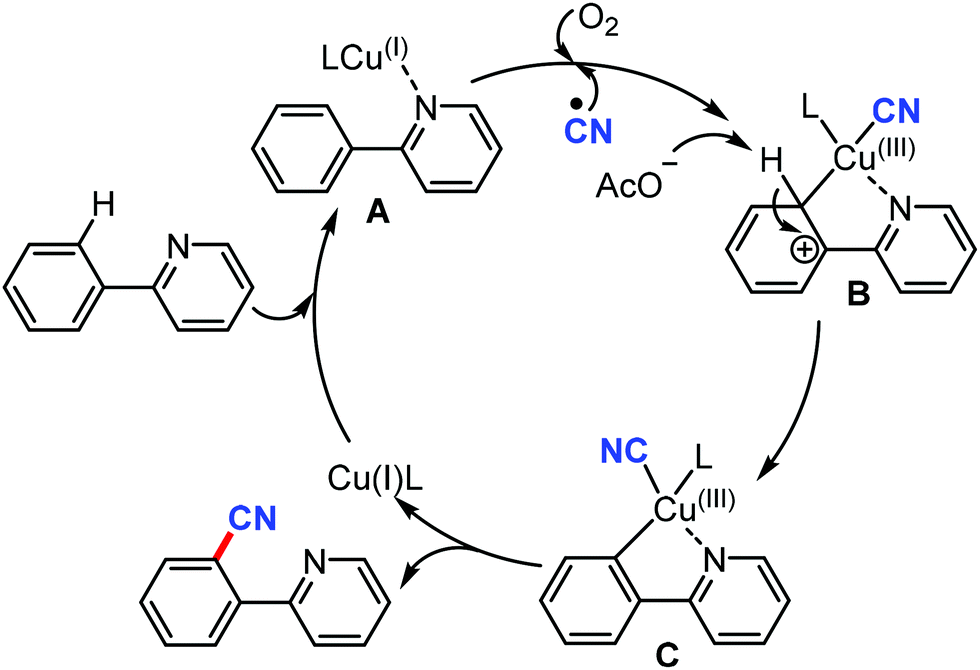 | ||
| Scheme 69 Proposed mechanism for radical mediated cyanation of tertiary amines. | ||
In 2014, Wang and co-workers developed a cost-effective copper catalyzed direct cyanoalkyarylation with AIBN to give the substituted oxyindoles. The thermal decomposition of AIBN released the free 2-cyanoprop-2-yl radical, which mediated the reaction by the single electron transfer (SET) process.71 On a similar note, Tang et al. reported Cu-DTBP (di-tert-butyl peroxide) mediated synthesis of oxyindoles.72 This method could also tolerate different substituted AIBN derivatives. Inter and intramolecular KIE (Kinetic Isotope Effect) of 1 and 1.3 indicated that free radical mechanism is involved. Later, in 2016, Tang and co-workers also reported the aerobic oxidative cyclization of benzamides via meta-selective C–H tert-alkylation using AIBN. This gave rapid access to 7-tert-alkylated isoquinolinediones.73 A radical scavenger such as TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxyl) could suppress the cyclization indicating the radical-mediated mechanism. These methods does not form direct C–CN bonds but involve nitrile/cyano functionalization.
In 2017, Li and co-workers used AIBN with Cu-catalyst for the [2+2+2] annulation of 1,n-enynes for the synthesis of 7,8-dihydrophenanthridine-6,9(5H,6aH)-diones and fluorenes. Enynes with N-Bn, N-Ts, or free NH were well tolerated in this reaction. Unlike previous methods, this reaction also proceeds via radical mediated mechanism. First, it forms the corresponding nitriles, which upon subsequent hydration and annulation, forms diones and fluorenes.74 Recently, Chen's group also described the one-pot construction of benzofuran-2(3H)-one via the radical cascade of para-quinone methides with AIBN/H2O. This method offered an elusive and efficient route to cyano-containing benzofurans as well as 2,3-dialkylating benzofurans. Late-stage functionalizations by non-polar C(aryl)–C(t-butyl) bond cleavage via Cu catalysis provided an interesting synthetic transformation of para-quinone methides.75
11. Conclusion
Undoubtedly, the cyano/nitrile group is a versatile synthetic intermediate owing to its capability to get smoothly transformed into various functional groups such as acids, ketone, amine, aldehyde, and N-heterocycles. Thus, cyanation has become a topic of wide significance in organic/material/medicinal synthesis and has been exploited for the synthesis of various structurally complex molecules. Until a decade ago, the facile introduction of cyano group was achieved by transition metal-catalyzed cyanation of aryl halides. However, since then, approaches developed by various research groups across the globe in recent years showed the diversity of synthetic routes and budding capabilities to discover new routes to make the CN bond. Furthermore, in the near future, the merger of different approaches in one pot for tandem functionalization to synthesize novel motifs, which will serve in agrochemical, pharmaceutical, and fine chemical industries, is highly conceivable. However, more importantly, the design and synthesis of novel organo-cyano reactions in the area of distal C–H activation of aliphatic and aromatic compounds is yet to be achieved.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research activity is supported by SERB (CRG/2018/003951) India. SP thanks IITB-Monash Research Academy for PhD fellowship (IMURA0414).Notes and references
- (a) A. J. Fatiadi, in Preparation and Synthetic Applications of Cyano Compounds, ed. S. Patai and Z. Rappoport, Wiley, New York, 1983, vol. 2, pp. 1057–1303 Search PubMed; (b) J. S. Miller and J. L. Manson, Acc. Chem. Res., 2001, 34, 563 CrossRef CAS; (c) F. F. Fleming and Q. Wang, Chem. Rev., 2003, 103, 2035 CrossRef CAS; (d) F. F. Fleming, L. Yao, P. C. Ravikumar, L. Funk and B. C. Shook, J. Med. Chem., 2010, 53, 7902 CrossRef CAS; (e) P. Anbarasan, T. Schareina and M. Beller, Chem. Soc. Rev., 2011, 40, 5049 RSC; (f) M. Tobisu and N. Chatani, Chem. Soc. Rev., 2008, 37, 300 RSC.
- G. Zhang, X. Ren, J. Chen, M. Hu and J. Cheng, Org. Lett., 2011, 13, 5004 CrossRef CAS.
- J. Kim, J. Choi, K. Shin and S. Chang, J. Am. Chem. Soc., 2012, 134, 2528 CrossRef CAS.
- L. Zhang, P. Lu and Y. Wang, Chem. Commun., 2015, 51, 2840 RSC.
- H. Wang, Y. Dong, C. Zheng, C. A. Sandoval, X. Wang, M. Makha and Y. Li, Chem, 2018, 4, 2883 CAS.
- C. W. Liskey, X. Liao and J. F. Hartwig, J. Am. Chem. Soc., 2010, 132, 11389 CrossRef CAS.
- Á. L. Fuentes de Arriba, E. Lenci, M. Sonawane, O. Formery and D. J. Dixon, Angew. Chem., Int. Ed., 2017, 56, 3655 CrossRef.
- P. Yu and B. Morandi, Angew. Chem., Int. Ed., 2017, 56, 15693 CrossRef CAS.
- X. Zhang, A. Xia, H. Chen and Y. Liu, Org. Lett., 2017, 19, 2118 CrossRef CAS.
- H. Chen, S. Sun, Y. A. Liu and X. Liao, ACS Catal., 2020, 10, 1397 CrossRef CAS.
- P. Y. Yeung, C. M. So, C. P. Lau and F. Y. Kwong, Angew. Chem., Int. Ed., 2010, 49, 8918 CrossRef.
- P. Y. Yeung, C. M. So, C. P. Lau and F. Y. Kwong, Org. Lett., 2011, 13, 648 CrossRef CAS.
- S. Zheng, C. Yu and Z. Shen, Org. Lett., 2012, 14, 3644 CrossRef CAS.
- D. Zhang, H. Sun, L. Zhang, Y. Zhou, C. Li, H. Jiang, K. Chen and H. Liu, Chem. Commun., 2012, 48, 2909 RSC.
- T. D. Senecal, W. Shu and S. L. Buchwald, Angew. Chem., Int. Ed., 2013, 52, 10035 CrossRef CAS.
- D. T. Cohen and S. L. Buchwald, Org. Lett., 2015, 17, 202 CrossRef CAS.
- B. Majhi and B. C. Ranu, Org. Lett., 2016, 18, 4162 CrossRef CAS.
- B. Luo, J.-M. Gao and M. Lautens, Org. Lett., 2016, 18, 4166 CrossRef CAS.
- P. Anbarasan, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2011, 50, 519 CrossRef CAS.
- S.-I. Murahashi, N. Naruyoshi, H. Terai and T. Nakae, J. Am. Chem. Soc., 2003, 125, 15312 CrossRef CAS.
- S.-I. Murahashi, N. Komiya and H. Terai, Angew. Chem., Int. Ed., 2005, 44, 6931 CrossRef.
- L. Ma, W. Chen and D. Seidel, J. Am. Chem. Soc., 2012, 134, 15305 CrossRef CAS.
- H.-Q. Do and O. Daugulis, Org. Lett., 2010, 12, 2517 CrossRef CAS.
- G. Yan, C. Kuang, Y. Zhang and J. Wang, Org. Lett., 2010, 12, 1052 CrossRef CAS.
- S. Ding and N. Jiao, J. Am. Chem. Soc., 2011, 133, 12374 CrossRef CAS.
- Y. Yang, Y. Zhang and J. Wang, Org. Lett., 2011, 13, 5608 CrossRef CAS.
- J. Kim, H. Kim and S. Chang, Org. Lett., 2012, 14, 3924 CrossRef CAS.
- S. Xu, X. Huang, X. Hong and B. Xu, Org. Lett., 2012, 14, 4614 CrossRef CAS.
- J. Peng, J. Zhao, Z. Hu, D. Liang, J. Huang and Q. Zhu, Org. Lett., 2012, 14, 4966 CrossRef CAS.
- T.-J. Gong, B. Xiao, W.-M. Cheng, W. Su, J. Xu, Z.-J. Liu, L. Liu and Y. Fu, J. Am. Chem. Soc., 2013, 135, 10630 CrossRef CAS.
- M. Chaitanya, D. Yadagiri and P. Anbarasan, Org. Lett., 2013, 15, 4960 CrossRef CAS.
- W. Su, T.-J. Gong, B. Xiao and Y. Fu, Chem. Commun., 2015, 51, 11848 RSC.
- M. Chaitanya and P. Anbarasan, Org. Lett., 2015, 17, 3766 CrossRef CAS.
- S. Lv, Y. Li, T. Yao, X. Yu, C. Zhang, L. Hai and Y. Wu, Org. Lett., 2018, 20, 4994 CrossRef CAS.
- H. Li, S. Zhang, X. Yu, X. Feng, Y. Yamamoto and M. Bao, Chem. Commun., 2019, 55, 1209 RSC.
- D.-G. Yu, T. Gensch, F. de Azambuja, S. Vásquez-Céspedes and F. Glorius, J. Am. Chem. Soc., 2014, 136, 17722 CrossRef CAS.
- J. Li and L. Ackermann, Angew. Chem., Int. Ed., 2015, 54, 3435 Search PubMed.
- A. B. Pawar and S. Chang, Org. Lett., 2015, 17, 660 CrossRef CAS.
- J. Jin, Q. Wen, P. Lu and Y. Wang, Chem. Commun., 2012, 48, 9933 RSC.
- B. Venu, B. Vishali, G. Naresh, V. V. Kumar, M. Sudhakar, R. Kishore, J. Beltramini, M. Konarova and A. Venugopal, Catal. Sci. Technol., 2016, 6, 8055 RSC.
- C. Qi, X. Hu and H. Jiang, Chem. Commun., 2017, 53, 7994 RSC.
- W. Liu and L. Ackermann, Chem. Commun., 2014, 50, 1878 RSC.
- X. Jia, D. Yang, S. Zhang and J. Cheng, Org. Lett., 2009, 11, 4716 CrossRef CAS.
- J. Kim and S. Chang, J. Am. Chem. Soc., 2010, 132, 10272 CrossRef CAS.
- S. Bag, R. Jayarajan, U. Dutta, R. Chowdhury, R. Mondal and D. Maiti, Angew. Chem., Int. Ed., 2017, 56, 12538 CrossRef CAS.
- R. Jayarajan, J. Das, S. Bag, R. Chowdhury and D. Maiti, Angew. Chem., Int. Ed., 2018, 57, 7659 CrossRef CAS.
- A. Gholap, S. Bag, S. Pradhan, A. R. Kapdi and D. Maiti, ACS Catal., 2020, 10, 5347 CrossRef CAS.
- S. Pimparkar, T. Bhattacharya, A. Maji, A. Saha, R. Jayarajan, U. Dutta, G. Lu, D. W. Lupton and D. Maiti, Chem. – Eur. J., 2020, 26, 11558 CrossRef CAS.
- A. Yamamoto, M. Murakami and M. Suginome, J. Am. Chem. Soc., 2003, 125, 6358 CrossRef.
- M. Murai, R. Hatano, S. Kitabata and K. Ohe, Chem. Commun., 2011, 47, 2375 RSC.
- D. C. Koester, M. Kobayashi, D. B. Werz and Y. Nakao, J. Am. Chem. Soc., 2012, 134, 6544 CrossRef CAS.
- M. Pawliczek, L. K. B. Garve and D. B. Werz, Org. Lett., 2015, 17, 1716 CrossRef CAS.
- U. Dutta, D. W. Lupton and D. Maiti, Org. Lett., 2016, 18, 860 CrossRef CAS.
- H. Wang, P. Mi, W. Zhao, R. Kumar and X. Bi, Org. Lett., 2017, 19, 5613 CrossRef CAS.
- X. Wang and A. Studer, Angew. Chem., Int. Ed., 2018, 57, 11792 CrossRef CAS.
- M. Bürger, M. N. Loch, P. G. Jones and D. B. Werz, Chem. Sci., 2020, 11, 1912 RSC.
- M. Bürger, S. H. Röttger, M. N. Loch, P. G. Jones and D. B. Werz, Org. Lett., 2020, 22, 5025 CrossRef.
- W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014 CrossRef CAS.
- H.-Y. Wang, C.-W. Zheng, Z. Chai, J.-X. Zhang and G. Zhao, Nat. Commun., 2016, 7, 12720 CrossRef.
- A. J. J. Lennox, S. L. Goes, M. P. Webster, H. F. Koolman, S. W. Djuric and S. S. Stahl, J. Am. Chem. Soc., 2018, 140, 11227 CrossRef CAS.
- K. Okamoto, M. Watanabe, M. Murai, R. Hatanoa and K. Ohe, Chem. Commun., 2012, 48, 3127 RSC.
- Z. Shu, Y. Ye, Y. Deng, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2013, 52, 10573 CrossRef CAS.
- Z. Shu, W. Ji, X. Wang, Y. Zhou, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2014, 53, 2186 CrossRef CAS.
- R. Takise, K. Itami and J. Yamaguchi, Org. Lett., 2016, 18, 4428 CrossRef CAS.
- D. Zhao, P. Xu and T. Ritter, Chem, 2019, 5, 97 CAS.
- D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632 CrossRef CAS.
- F. L. Vaillant, M. D. Wodrich and J. Waser, Chem. Sci., 2017, 8, 1790 RSC.
- R. Spaccini, N. Pastori, A. Clerici, C. Punta and O. Porta, J. Am. Chem. Soc., 2008, 130, 18018 CrossRef CAS.
- L. Liu, Z. Wang, X. Fu and C.-H. Yan, Org. Lett., 2012, 14, 5692 CrossRef CAS.
- H. Xu, P.-T. Liu, Y.-H. Li and F.-S. Han, Org. Lett., 2013, 15, 3354 CrossRef CAS.
- W. Wei, J. Wen, D. Yang, M. Guo, L. Tian, J. You and H. Wang, RSC Adv., 2014, 4, 48535 RSC.
- D. Zhou, Z.-H. Li, J. Li, S.-H. Li, M.-W. Wang, X.-L. Luo, G.-L. Ding, R.-L. Sheng, M.-J. Fu and S. Tang, Eur. J. Org. Chem., 2015, 1606 CrossRef CAS.
- Y.-L. Deng, J. Li, W.-X. Wang, Y.-C. Wang, Z.-Z. Li, L. Yuan, S.-L. Chen, R.-L. Sheng and S. Tang, Chem. Commun., 2016, 52, 4470 RSC.
- B. Liu, C.-Y. Wang, M. Hu, R.-J. Song, F. Chen and J.-H. Li, Chem. Commun., 2017, 53, 1265 RSC.
- J. Yu, H.-X. Sheng, S.-W. Wang, Z.-H. Xu, S. Tang and S.-L. Chen, Chem. Commun., 2019, 55, 4578 RSC.
Footnote |
| † These authors have contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |