
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Modulators of protein–protein interactions as antimicrobial agents
Rashi
Kahan
,
Dennis J.
Worm
,
Guilherme V.
de Castro
,
Simon
Ng
and
Anna
Barnard
 *
*
Department of Chemistry, Molecular Sciences Research Hub, Imperial College London, 82 Wood Lane, London W12 0BZ, UK. E-mail: a.barnard@imperial.ac.uk
First published on 3rd February 2021
Abstract
Protein–Protein interactions (PPIs) are involved in a myriad of cellular processes in all living organisms and the modulation of PPIs is already under investigation for the development of new drugs targeting cancers, autoimmune diseases and viruses. PPIs are also involved in the regulation of vital functions in bacteria and, therefore, targeting bacterial PPIs offers an attractive strategy for the development of antibiotics with novel modes of action. The latter are urgently needed to tackle multidrug-resistant and multidrug-tolerant bacteria. In this review, we describe recent developments in the modulation of PPIs in pathogenic bacteria for antibiotic development, including advanced small molecule and peptide inhibitors acting on bacterial PPIs involved in division, replication and transcription, outer membrane protein biogenesis, with an additional focus on toxin–antitoxin systems as upcoming drug targets.
 Rashi Kahan | Rashi Kahan graduated Chemistry with Biomedicine (MSc) studying at King's College London where she developed great interest in using chemical tools to study and target proteins and their interactions. She carried out her final year research project with Dr Manuel Müller involving the synthesis and chemical evaluation of p53 heterotetramers, investigating the effects of post-translational modifications on their functionality. She started her PhD in 2019 at Imperial College London jointly supervised by Dr Anna Barnard and Dr Louise Walport, focused on novel cyclic peptide inhibitors of toxin proteins involved in bacterial persistence. |
 Dennis J. Worm | Dennis J. Worm obtained a BSc in Biochemistry from the University of Tübingen (Germany) and a MSc in Biochemistry from Ruhr University Bochum (Germany). He then joined the Beck-Sickinger Lab at Leipzig University (Germany), where he was developing novel GPCR-targeting peptide–drug conjugates for advanced cancer therapy and obtained his PhD in Chemistry in 2018. Since 2019, he is a postdoctoral research associate in the Barnard group at Imperial College London focussing on the development of novel peptide inhibitors of protein–protein interactions involved in bacterial persistence. |
 Guilherme V. de Castro | Guilherme V. de Castro obtained his BSc and MSc in Chemistry from the Universidade Federal do ABC and PhD in Life Sciences from the University Dundee. Under the supervision of Prof. Alessio Ciulli, his PhD studies focused on a combination of chemical and biophysical approaches to probe subunits of E3 ligases with fragments, in the search of novel interaction sites for chemical degraders. In 2019 he became a postdoctoral research associate in the group of Dr Anna Barnard at Imperial College London to work on the development of methods to study and modulate protein–protein interactions relevant to antimicrobial research. |
 Simon Ng | Simon Ng is an MRes in Drug Discovery and Development student at Imperial College London, having previously studied Biochemistry at the University of Birmingham. His area of research is toxin–antitoxin interactions in tuberculosis. |
 Anna Barnard | Anna Barnard obtained her PhD from the University of York under the supervision of Prof. David Smith. Following postdoctoral positions at the University of Leeds in the group of Prof. Andrew Wilson and Imperial College London with Prof. Edward Tate, she was awarded an Imperial College Junior Research Fellowship. In 2019, she was awarded a Wellcome Trust Sir Henry Dale Fellowship to start her own laboratory at Imperial which focuses on new methods for targeting protein–protein interactions including key interactions in bacteria. |
1 Introduction
Antibiotic resistance and the recurrence of bacterial infections are two of the most serious threats to future global public health, with the potential to affect anyone in any country in the world. The Centre of Disease Control (CDC) estimates that 2.8 million people acquire an antibiotic-resistant infection in the US every year, already resulting in more than 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 fatalities.1 Without effective antibacterial compounds, infections by multi-drug resistant or tolerant bacteria could result in life-threatening conditions in people with minor injuries. In hospital settings, recalcitrant infections compromise the success of surgery and the therapy of immunosuppressed patients. In addition, the health care costs for patients with resistant infections are significantly higher than for other patients, due to obligatory isolation, longer hospital stays and the use of more expensive drugs.2
000 fatalities.1 Without effective antibacterial compounds, infections by multi-drug resistant or tolerant bacteria could result in life-threatening conditions in people with minor injuries. In hospital settings, recalcitrant infections compromise the success of surgery and the therapy of immunosuppressed patients. In addition, the health care costs for patients with resistant infections are significantly higher than for other patients, due to obligatory isolation, longer hospital stays and the use of more expensive drugs.2
Although antibiotic treatment is naturally linked to the occurrence of resistance in bacteria, misuse in humans and animals has accelerated the generation of antibiotic-resistant bacterial strains around the globe. Furthermore, the discovery of innovative anti-infective agents has been sparse, also attributed to the lack of investment in antimicrobial research by major pharmaceutical companies.3 Most of the antibiotics marketed in the last decades are based on existing drugs, which aim to overcome the resistance acquired by bacteria against their parent compounds.4 However, derivatisation of existing drug scaffolds that are acting on already targeted bacterial cell machineries risks rapid emergence of novel resistance mechanisms, requiring a constant compound optimization.
The major challenge associated with recalcitrant infection is the ability of bacteria to evade antibiotic treatment by three different mechanisms: resistance, tolerance and persistence. While resistance is acquired on the genetic level, allowing the bacterial population to grow in the presence of the antibiotic, tolerance and persistence enable bacteria to transiently survive antibiotic exposure.5 The distinction between the latter two being that tolerance is homogenously displayed by the whole bacterial population, while persistence describes the formation of a subpopulation of bacteria able to survive antibiotic treatment.5
Current antibiotics mostly act on classical targets such as bacterial enzymes, ribosomal RNA, cell wall construction and cell membrane function. It is clear, however, that antibacterial agents with innovative modes of action are urgently needed to tackle mechanisms leading to treatment failure, and to explore unconventional paths in the fight against resistant and recurrent infections. Bacterial protein–protein interactions (PPIs) are highly promising and, thus far, underexplored targets for antibiotic drug discovery.
1.1 Targeting PPIs
PPIs have, until recently, been considered unsuitable drug targets, due to their large, flat interaction surfaces (∼1000–2000 Å2).6 There is however growing research that refutes this notion, and PPIs are now being investigated as targets for novel therapeutics.7 A subset of the amino acid residues involved in the formation of a PPI, known as the hotspots, primarily drive their formation and confer the majority of their binding energy,8 making the challenge of developing targeting molecules less daunting.Various techniques have been utilised to identify lead compounds that can target PPIs. Structurally diverse small molecule compounds have been identified by traditional high throughput screening (HTS), fragment-based lead discovery and also structure-based rational design, as some PPIs have small-molecule sized patches of around 250–900 Å2 formed by clusters of hotspots.9,10 Computational methods such as virtual screening, in silico experiments and homology modelling are frequently used to aid the design of PPI-targeting small molecules.11 Peptides offer the possibility to cover larger surface areas and are often developed into cyclic variants or peptidomimetic compounds to achieve more drug-like properties.12–14 Furthermore, HTS methods using linear and macrocyclic peptide libraries are increasingly used to identify novel PPI inhibitors.15
Targeting PPIs has already yielded new treatments for cancer. Trials for idasanutlin, which targets the MDM2–p53 interaction, and apabetalone, which targets bromodomains, are underway for cancer treatments.16 Venetoclax, which is prescribed to treat chronic lymphocytic leukaemia (CLL), is a BH3-mimetic, resulting in apoptosis of CLL cells due to inhibition of BCL-2 activity.17 Furthermore, stapled peptides targeting MCL-1 protein, a member of the BCL-2 family, are being developed as apoptosis inducers in cancer cells18 and ALRN-6924, a stapled peptide which targets both MDM2 and MDMX, entered clinical trials in 2017 with promising initial results.19
1.2 PPIs in infection
Many crucial cellular functions in viruses, bacteria and other pathogens rely on PPIs. The interactions can be pathogen–pathogen, host–pathogen or host–host, all of which are interesting targets for novel anti-infective agents. Targeting host–pathogen interactions has been successful, with drugs such as antiretroviral drugs enfuvirtide and maraviroc for the treatment of HIV/AIDS.20Many PPIs are linked to bacterial processes absent in eukaryotic cells, making them attractive targets for the discovery of new pathogen-selective leads. The bacterial interactome has the potential to enable the identification of numerous targets for novel therapies.21 As PPIs are linked to crucial processes within bacteria, disrupting these interactions can lead to inhibition of bacterial cell growth and/or result in cell death. Many PPIs have already been identified and targeted in pathogenic bacteria. These are involved in a wide range of vital functions, such as bacterial division (FtsZ and SSBs), transcription (RNA polymerases) and toxin–antitoxin pairs whereby toxins have been implicated in programmed cell death as well as persistence. Both small molecule and peptide inhibitors have been developed and multiple screening techniques have been applied for their identification.
This review focuses on PPIs relating directly to functions in bacteria, i.e. pathogen–pathogen interactions. We present an overview of bacterial PPIs that have been identified as anti-infective targets and the approaches used to identify and progress small molecules and peptides targeting different classes of interaction, highlighting the most promising compounds currently in development.
2 PPIs as antibacterial targets
2.1 Targeting division and replication
Because of their essential role in sustaining cell viability, multiple members of the DNA replication and cell division machinery have been targeted for antimicrobial therapies.6,16,22 Despite efforts described in the literature, inhibitors of components of topoisomerase II represent the only marketed drugs targeting this class of proteins.22 These antibiotics can be divided in two groups: inhibitors of the ATPase site of DNA gyrase, that can effectively block the relaxation of supercoiled DNA (e.g. novobiocin);23,24 and stabilisers of the complex between cleaved DNA strands and topoisomerase II, which are capable of triggering cell death via the accumulation of DNA fragments (e.g. ciprofloxacin).25 Although these compounds are amongst the most widely used antibiotics in the clinic, many limitations of their administration have been identified in recent years. The development of novel aminocoumarins (typical DNA gyrase inhibitors) is often restricted by their poor drug-like physicochemical properties.24 On the other hand, the widespread use fluoroquinolones (well-characterised inducers of fragmented DNA build-up) to treat multiple infections has caused genetically resistant strains to arise. Fluoroquinolone-resistant strains of Campylobacter and Salmonella spp. were highlighted by the WHO as high-priority targets for antimicrobial research.26Due to the multi-protein complexity of the replisome and the divisome, and the poor conservation with eukaryotic homologs, PPIs have been explored as attractive routes for the development of inhibitors of cell division and DNA replication. Notably, three approaches have been investigated in recent years: (i) disruption of the PPI between FtsZ (filamenting temperature-sensitive protein Z) and ZipA (FtsZ interacting protein A), (ii) interference with the PPI network of SSB (single-stranded DNA-binding protein) and (iii) prevention of the recruitment of essential enzymes by the β-subunit of DNA polymerase III (β-sliding clamp) during replication. Reviews describing small-molecule development for each of these approaches can be found elsewhere,6,16,22,27 therefore focus will be given to the key findings for these strategies.
2.1.i FtsZ–ZipA inhibition
Inhibition of the FtsZ–ZipA contact is so far the most advanced strategy targeting cell division PPIs. FtsZ is a tubulin homolog responsible for the formation of a ring-like structure (Z-ring) that acts as a scaffold for the assembly of a number of proteins. ZipA bridges FtsZ and the bacterial membrane, allowing the Z-ring to guide the synthesis and reshaping of the peptidoglycan wall during division. Therefore, inhibition of this PPI would lead to incomplete division, subsequently triggering bacterial cell death.28 The C-terminal α-helix of FtsZ was identified as the key region of interaction with a large hydrophobic surface on ZipA (Fig. 1A).29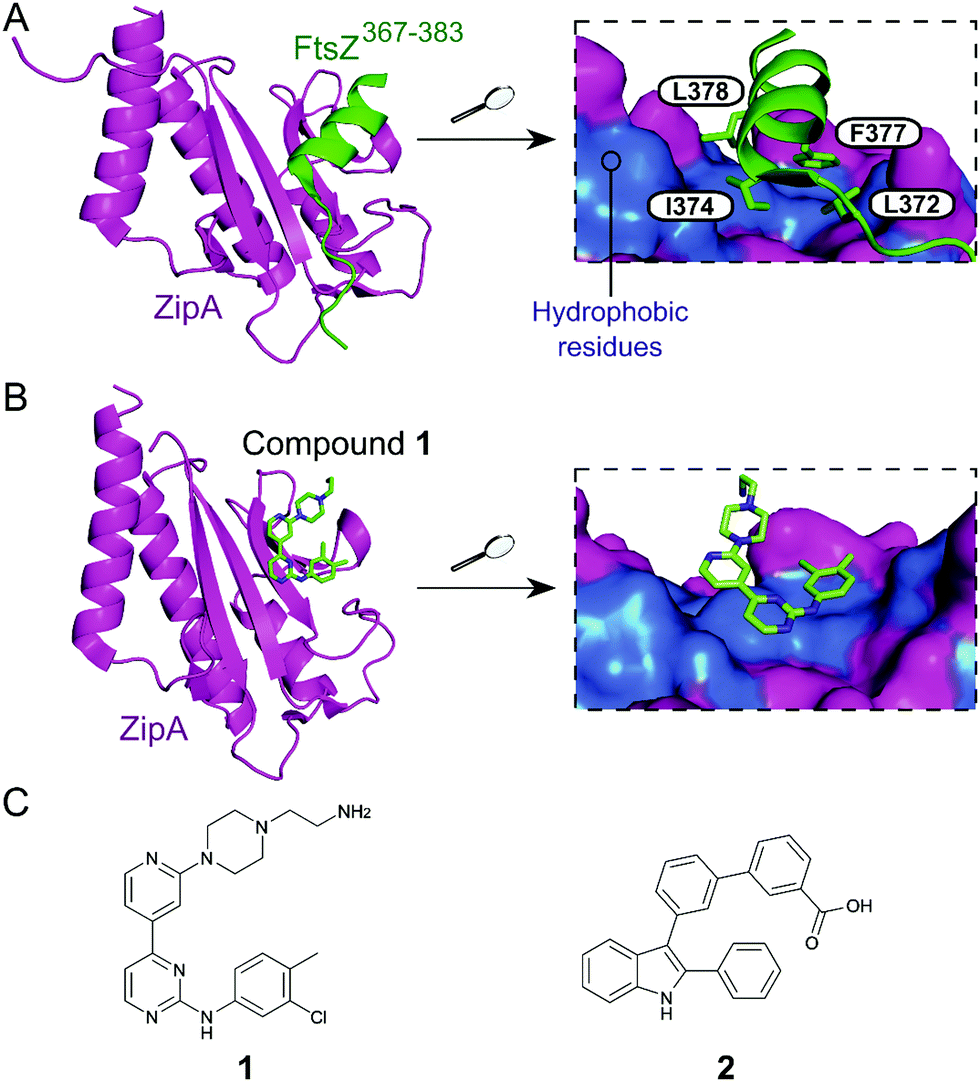 | ||
| Fig. 1 (A) Crystal structure (PDB: 1F47)29 of the C-terminus of E. coli FtsZ (green) bound to E. coli ZipA (magenta). The interaction is driven by a solvent exposed hydrophobic patch (blue surface, right chart) present in ZipA. (B) Crystal structure (PDB: 1Y2F)31 of inhibitor 1 bound to E. coli ZipA. The compound interacts at the same site as FtsZ (right chart). (C) Chemical structures of FtsZ–ZipA PPI inhibitors. | ||
Kenny et al. employed a high-throughput fluorescence polarisation (FP) assay to identify competitors of the interaction between ZipA and a 17-mer C-terminal FtsZ peptide (KZipAd = 7 μM). Compound 1 (Fig. 1B and C) was selected as the most active hit (Ki = 12 μM) and confirmed to bind to ZipA on the expected PPI interface.30 Despite the promising start, high cytotoxicity in Candida albicans indicated that 1 could also harm eukaryotic cells.31 In order to overcome this issue, multiple follow-up studies introducing novel scaffolds via fragment screening and computational methods were performed, establishing structure–activity relationships (SAR).30–34 A fragment-merging approach led to compound 2 (Fig. 1C), which possessed a moderate MIC (10–100 μM) against many bacterial pathogens (e.g. MIC Escherichia coli imp = 20 μM) and promisingly no toxicity to C. albicans (MIC > 300 μM). Two-dimensional NMR experiments confirmed binding to the expected site and an IC50 of 192 μM for inhibition of the PPI was determined by FP.32 The discrepancy between PPI inhibition and activity indicates that further mode-of-action studies are required before progressing the series forward. As MIC values were considerably lower than the IC50 (determined at non-limiting ZipA concentration of 500 nM), 2 and related compounds might act by additional unknown mechanisms beyond ZipA-FtsZ inhibition.
The exonuclease I (ExoI) was identified early on as a promising SSB-interacting protein to target due to its well-characterised role in DNA repair during replication.37 Disruptive mutations of the ExoI–SSB contact were shown to be lethal to E. coli, validating the relevance of targeting this PPI. The co-crystal structure of ExoI in complex with a SSB Ct peptide (Fig. 2A) revealed two independent binding sites (commonly referred as sites A and B). Site B was identified as essential for SSB-mediated ExoI activity, therefore formed the main focus for the development of PPI inhibitors.37 Keck et al. performed an HTS by FP and identified four promising hits able to displace an E. coli SSB mimetic peptide (Fluorescein-WMDFDDDIPF-COOH, KExoId = 136 nM) with an IC50 below 100 μM.37–39 Co-crystal structures of ExoI in complex with two of these compounds (3 and 4 shown in Fig. 2B and C, respectively) confirmed they were binding to the SSB site B. Additionally, these compounds possessed a growth inhibitory effect on a large panel of Gram-positive and Gram-negative bacteria, without demonstrating any toxicity to the eukaryotic Saccharomyces cerevisiae.
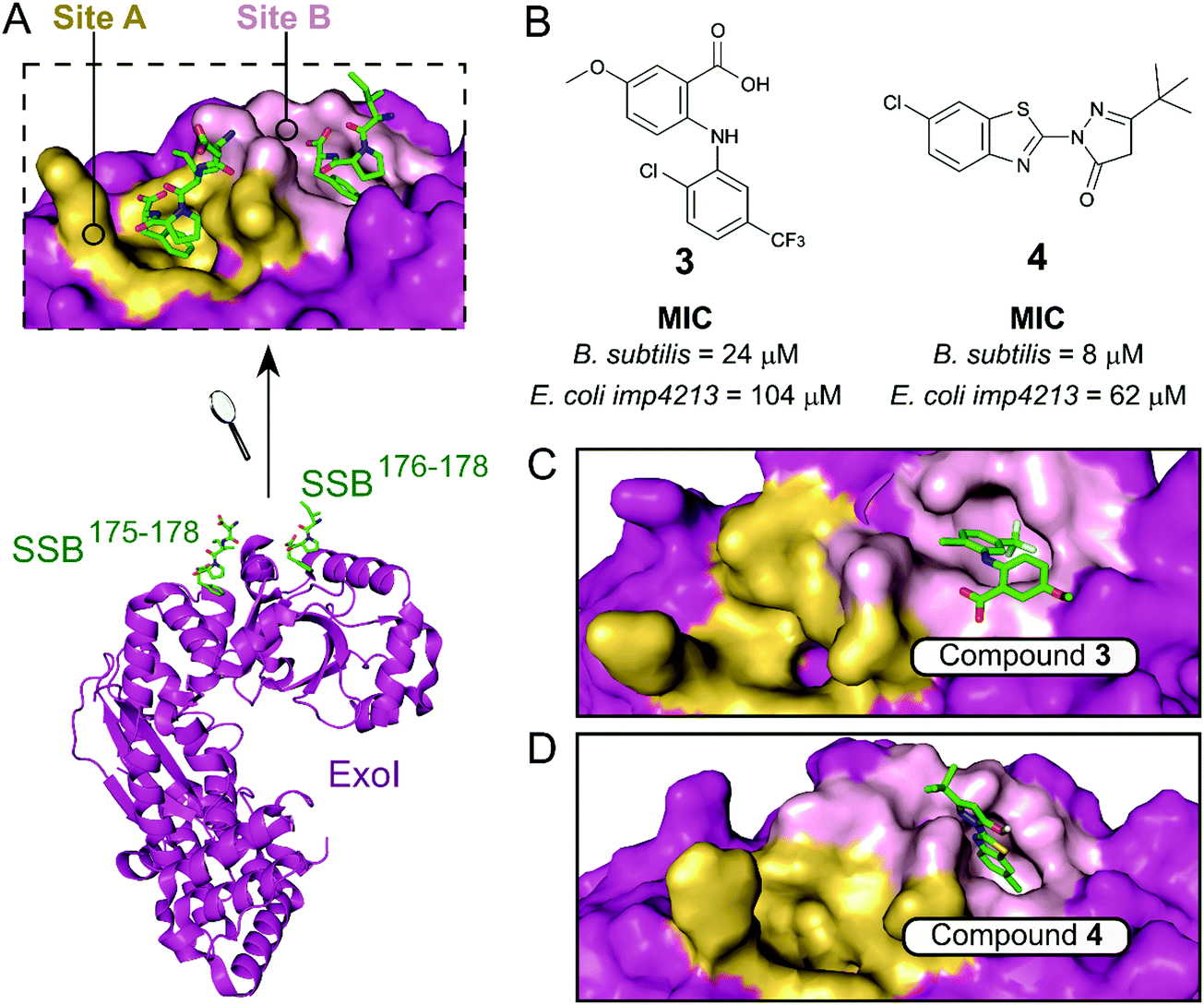 | ||
| Fig. 2 (A) Crystal structure (PDB: 3C94)37 of E. coli ExoI (magenta) bound to a E. coli SSB Ct peptide (green). ExoI can interact simultaneously with two peptide molecules via the binding sites A (yellow surface) and B (pink surface), as shown in the upper chart. (B) Chemical structures and activity of inhibitors 3 and 4. The E. coli ExoI-bound crystal structures of inhibitors 3 (PDB: 3HP9)38 and 4 (PDB: 3HL8)38 are shown in (C) and (D), respectively. | ||
Beyond ExoI, the inhibition of other SSB binding partners has also been investigated: (i) a fragment screening on the DNA primase DnaG with a combination of protein- and ligand-observed NMR experiments;40 (ii) HTS of the helicase PriA via AlphaScreen.41 Interestingly, due to the promiscuous nature of the SSB C-terminal tail, selected hits from these independent studies were found to bind to multiple SSB binding partners, indicating the potential for pan-selective inhibition of the SSB interactome.40,41
The E. coli β-sliding clamp specifically recognises proteins carrying a linear QL[S/D]LF consensus sequence. Crystal structures with peptides mimicking the DNA polymerase II, III and IV (Pol II, Pol III and Pol IV, respectively) revealed the interactions to be highly hydrophobic and divided into two subsites (Fig. 3).44–46 In the search for inhibitors of specific DNA polymerase interactions with the clamp complex, Georgescu et al. performed a HTS using FP,46 based on the displacement of a fluorescent Pol III 20-mer peptide (Kd = 2.7 μM). Compound 5 (Fig. 4A) was selected as a promising hit (Ki = 10 μM) with good activity in replication assays, and a co-crystal structure (Fig. 4B) revealed interactions with subsite 1 of the pocket. However, the presence of the promiscuous rhodamine core in the structure and poor antibacterial activity held back further development of this series.
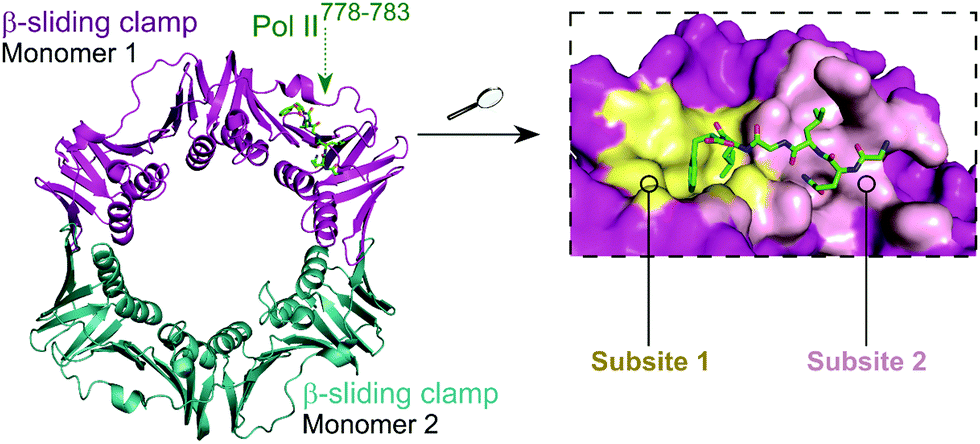 | ||
| Fig. 3 Crystal structure (PDB: 3D1E)46 of the E. coli β-sliding clamp dimer (one monomer is shown in magenta and the second monomer is shown in cyan) bound to the E. coli Pol II C-terminus peptide (green). The binding site is divided into two subsites (1 and 2, shown in yellow and pink, respectively) with the Pol II peptide extending over both regions (right chart). | ||
 | ||
| Fig. 4 (A) Chemical structures of small molecule and peptidic inhibitors of the β-sliding clamp PPI site. The crystal structures of 5, 6, 8 and 10 bound to the E. coli β-sliding clamp (PDB accession codes 3D1G,463QSB,474PNU48 and 3Q4L,50 respectively) are shown in (B), (C), (D) and (E), respectively. Subsites 1 and 2 are shown as yellow and pink surface, respectively, and the remaining regions are shown as magenta surface. | ||
Nevertheless, this work motivated multiple follow-up studies that were able to identify novel scaffolds for inhibition at the same site. Compound library filters for structures with similar properties to the subsite 1 binding motif (consensus sequence residues DLF) identified five novel inhibitors (IC50 ≈ 40–400 μM) that did not possess the rhodamine ring (e.g. biaryl oxime 6, Fig. 4A).47 Fragment-based screening via computational docking and FP also introduced a 1,2,3,4-tetrahydrocarbazole core (7, Fig. 4A) with good in vitro activity (IC50 = 115 μM) and confirmed binding to subsite 1.48 Additionally, fragment 7 demonstrated reasonable MIC (40–80 μM) against a range of Gram-negative and Gram-positive bacteria.48,49 SAR studies ultimately led to compound 8 (Fig. 4A), where the fragment core was extended by a phenylalanine motif to make further interactions in the pocket (Fig. 4D).49 Compound 8 possessed both improved affinity to the β-sliding clamp (Ki = 15 μM) and inhibition of DNA replication (IC50 = 31 μM), although no antimicrobial data was reported.
Besides small-molecule inhibition, potent peptide inhibitors of the β-sliding clamp PPI hub have also been identified (Fig. 4A).47,50,51 Structure-guided optimisation of the pentapeptide consensus sequence 9 using surface plasmon resonance (SPR) (ICSPR50 = 12.4 μM) led to compound 10 (ICSPR50 = 77 nM), a potent binder that covers both subsites of the pocket (Fig. 4B). The acetylation of the terminal glutamine residue establishes a hydrogen bond with the backbone of arginine-365 of the protein. The introduction of the hydrophobic sidechains of cyclohexyl-L-alanyl and 3,4-dichlorophenylalanine provided a further increase in affinity, with the latter presenting the highest contribution in the stability of the peptide–protein complex.
Interestingly, studies on the bactericidal cyclic peptide griselimycin (11a, Fig. 4A) revealed this class of natural products to act by inhibiting the PPI site of the β-sliding clamp.52 Despite its antibacterial activity being known for more than 60 years,53 the poor metabolic stability of griselimycin hindered its development. However, its potent activity against drug-resistant Mycobacterium tuberculosis (Table 1) and disclosed mode of action has revived the interest of many researchers. Proline-8 was identified as the main labile site responsible for the degradation of 11ain vivo. Insertion of a single methyl group at position 4 of the pyrrole ring (11b) significantly increased microsomal stability (Table 1). The addition of a cyclohexyl group at the same position (11c) provided stability and higher lipophilicity, boosting the antibacterial activity.52
2.2 Targeting bacterial transcription
Transcription is an essential cellular process involving the synthesis of RNA from a genomic DNA template, either as mRNA that is translated into proteins or as rRNA that is assembled into the 70S ribosome. Rifamycin and fidaxomicin are the only marketed antibiotics that act on bacterial transcription. The former targets the active site in bacterial RNA polymerase (RNAP) while the latter targets the RNAP clamp and both are highly susceptible to resistance.54,55 Since a number of transcription factors form PPIs that regulate all aspects of transcription in bacteria, these interactions are promising targets for the development of novel antibiotics.2.2.i RNA polymerase (RNAP)
Initiation of transcription of RNA in bacteria requires the association of the RNAP core enzyme (2α, β, β′ and ω subunits) with the transcription factor Sigma (σ) to form the RNAP holoenzyme. The upstream interaction with σ increases the specificity of RNAP for promotor regions, enabling transcription to start at the correct sites.56 Both the RNAP core and σ transcription factors are conserved across Gram-negative and Gram-positive bacterial strains and the main σ factors (termed σ70 in Gram-negative and σA in Gram-positive bacteria) do not have direct eukaryotic homologs,57 making potential inhibitors of this PPI promising broad spectrum antibacterial agents. Although RNAP and σ interact via an extensive network of contacts between regions 1–4 in σ and RNAP subunits β and β′, mutagenesis studies have identified hot-spot residues in the β′ clamp-helix (CH) region of RNAP and the small σ2.2 region that contribute most of the binding enthalpy.58,59 The CH region of RNAP consists of two antiparallel α-helices connected by a loop (Fig. 5A) and forms multiple electrostatic and hydrophobic contacts with σ2.2, therefore, disruption of this binding interface enables PPI inhibition.60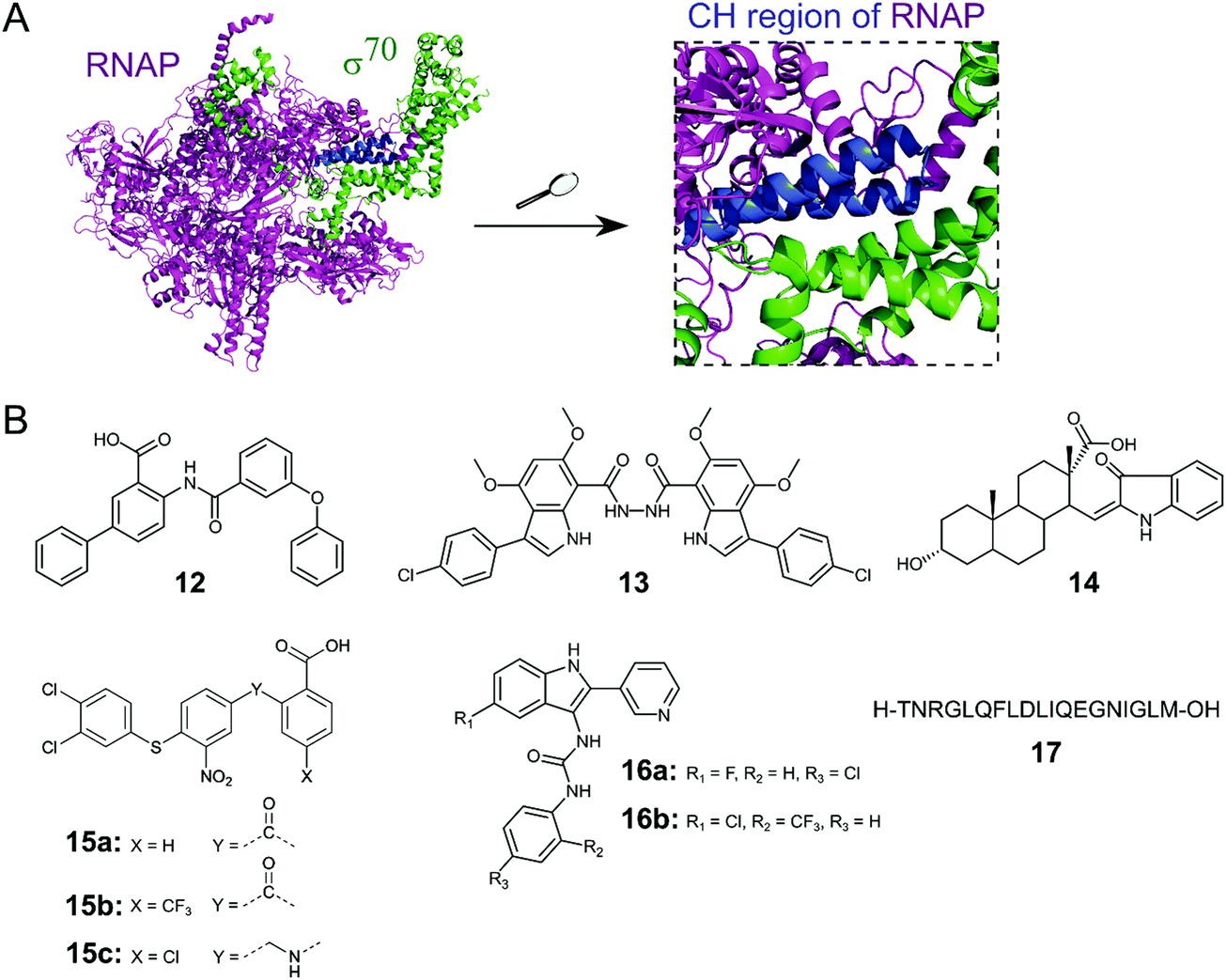 | ||
| Fig. 5 (A) Crystal structure of E. coli RNAP holoenzyme (PDB: 4LJZ)62 with RNAP core enzyme coloured in magenta and σ70 coloured in green. Enlarged picture (right chart) shows the critical interaction of the β′ clamp-helix (CH) region (blue) of RNAP with the N-terminal domain of σ70. (B) Small molecule and peptide inhibitors of RNAP–σ PPI. | ||
Several small molecule inhibitors of the RNAP–σ interaction have been identified. Hinsberger et al. used a pharmacophore model based on the alignment of known RNAP inhibitors for the screening of a 2000-compound in-house library.61 From 64 hits, 11 compounds were confirmed to be transcription inhibitors and three compounds were chosen for derivatisation to improve inhibitory activity. The best compound 12 (Fig. 5B) exhibited an IC50 of 13 μM in an RNAP transcription assay and disruption of the PPI between the RNAP β′ subunit and σ70 as mode of action for 12 was confirmed in an ELISA-based RNAP assembly assay (IC50: 47 μM). Furthermore, 12 displayed good antibacterial activity against Gram-positive Bacillus subtilis and Staphylococcus aureus with MICs of approximately 7 and 15 μM, respectively, but activity against Gram-negative E. coli K12 and Pseudomonas aeruginosa was low.61
The Lewis group has published several studies on novel RNAP–σ inhibitors. Key residues from σA2.2 that are required for interaction with the β′-CH region were used for the generation of a pharmacophore model.60 With this model, a novel in-house library of peptidomimetic compounds was screened and bis-indole 13 (Fig. 5B) was further characterized. Compound 13 was found to bind to the β′-CH region and inhibit RNAP–σA-mediated transcription in vitro with a Ki of 6 nM. Growth inhibition of cultured Gram-negative E. coli and Gram-positive S. aureus USA300 could be observed, but only at very high concentrations of 13 (>1 mM), suggesting that cellular penetration of 13 is likely to be poor.60 In order to obtain smaller β′–CH–σ inhibitors with potentially enhanced cellular permeability, 39 new small molecules based on mono-indole and mono-benzofuran scaffolds were synthesized.63 The new molecules displayed lower potencies in an ELISA-based β′–σA inhibition assay compared to the previously described bis-indoles, but higher antibacterial activities against B. subtilis and/or E. coli in a growth inhibition assay were observed for several compounds.63
In a study by Ma et al., a more comprehensive pharmacophore model for the β′–CH–σ2.2 interaction was established and the model was used to screen the mini-Maybridge library (53000 compounds).64 Out of 27 hits, 14 (Fig. 5B) was found to be the most active compound. In a RNAP transcription assay, 14 exhibited an IC50 of around 50 nM. Inhibition of the RNAP β′–σA PPI by 14 was confirmed by a competitive ELISA and 14 demonstrated specificity for bacterial RNAP. Compound 14 also showed promising antibacterial activity against Gram-positive B. subtilis with a MIC of <50 μM, however, activity against Gram-negative S. aureus strain USA300 was much lower with a MIC of around 94 μM.64 A second hit compound from the described screen was used in a recent study to generate a small series of derivatives.65 Out of the new series, 15a (Fig. 5B) was found to be the most interesting, with a higher β′–CH–σA inhibitory activity than the parent compound and promising antibacterial activity against different Gram-positive cocci (Streptococcus pyogenes, Streptococcus agalactiae and Staphylococcus epidermidis) with MICs of 36 μM.65 Two structural optimization studies of triaryl compound 15a were reported very recently. para-Substitution of the benzoic acid ring of 15a with a trifluoromethyl group (15b) in the first study led to a compound with micromolar β′–CH–σ2.2 inhibitory activity and strong antibacterial activity against S. epidermidis with a MIC of 1 μM.66 In the second study, substitution of the carbonyl linker of 15a with a methylamine linker in combination with para-substitution of the benzoic acid with a chlorine moiety led to 15c with high activity against Streptococcus pneumoniae and S. pyogenes (MICs of 2 μM), comparable to vancomycin.67
Sartini et al. developed and optimized a bioluminescence resonance energy transfer (BRET) assay in yeast as a novel screening platform for the discovery of RNAP β′–σ70 inhibitors.68 Using their assay, 5000 compounds, in silico selected from a larger 34000 compound library, were screened and 7 hits were obtained. Two indol-3-yl-urea derivatives (16a/b) of one hit compound were further investigated and found to preferentially bind to the RNAP β′ subunit and inhibit RNAP transcription in vitro with IC50 values of around 30 μM. The two derivatives 16a/b also exhibited antibacterial activity against Gram-positive B. subtilis, S. aureus and Listeria monocytogenes with MIC values in the range of around 20–100 μM.68
Two studies have described peptides targeting the RNAP–σ interaction. Hüsecken et al. rationally designed 16 peptides covering different regions of the E. coli σ70:core interface and examined them as potential PPI inhibitors.69 Peptide 17 (Fig. 5B), derived from the σ702.2 region, was found to be the most active compound with an IC50 of 5 μM in a RNAP transcription inhibition assay. Structural investigation by molecular dynamics simulation combined with mutagenesis studies revealed that 17 binds to the β′–CH region and also interacts with the β′ lid–rudder-system, with both sites being important for potent PPI inhibition. A recent study attempted to further optimize peptide 17.70 An alanine scan of 17 was performed to identify the core sequence and stapled variants of 17 using copper-catalysed azide–alkyne cycloaddition were synthesized with the aim of improving inhibitory potency. However, despite an increase in α-helicity, stapling of 17 almost completely abolished its inhibitory activity.70
2.2.ii N-utilization substances (Nus) B and E
The Nus transcription factors are conserved across bacterial species and are essential for viability. The proteins NusA, NusB, NusE, NusG and SuhB form the Nus factor complex, which interacts with rRNA and RNAP to promote expression of rRNA, prevents Rho-dependent transcription termination and regulates correct rRNA folding during ribosome assembly.71–74 Studies have shown that the NusB–NusE PPI is the key nucleation point for the formation of the bacterial rRNA transcription complex, rendering this interaction an interesting target for novel antibiotics.75 Structural investigation of the PPI interface (∼1600 Å2) revealed that the α1-helix and the β2-strand of NusE interact with the binding surface of NusB, formed by two helical bundles, via multiple hydrophobic and hydrophilic contacts (Fig. 6A).76–79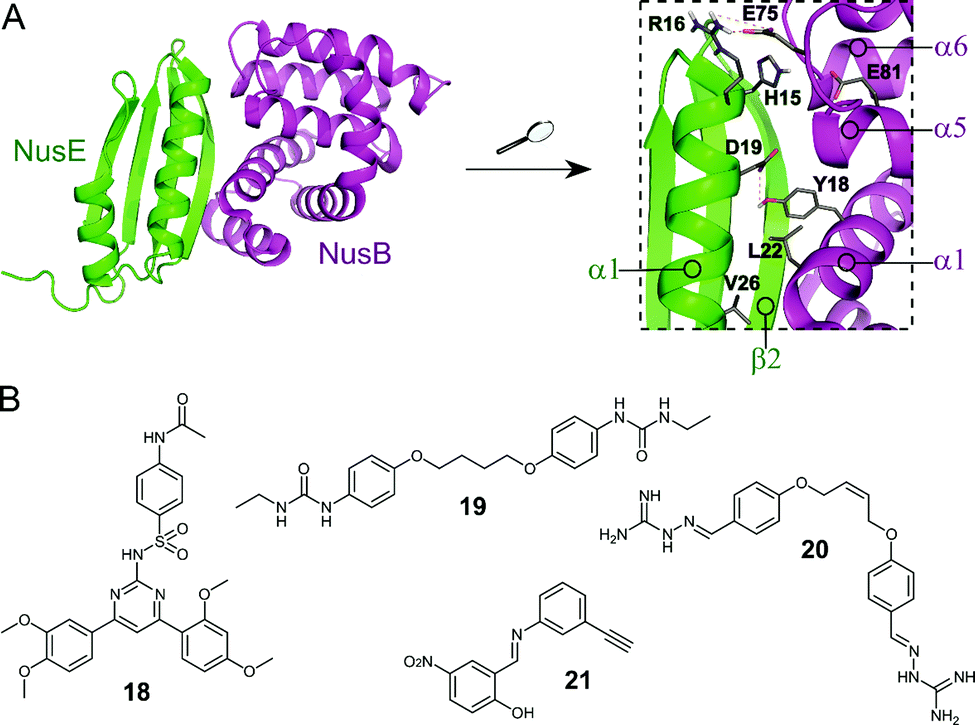 | ||
| Fig. 6 (A) Crystal structure of the E. coli NusB–NusE complex (PDB: 3D3B)77 with NusE coloured in green and NusB in magenta. Enlarged picture (right chart) shows the NusB–NusE PPI interface with residues (shown as sticks) H15, R16, D19 and V26 of the α1-helix of NusE interacting with Y18, L22, E75 and E81 of NusB; key hydrogen bonds are indicated by dashed orange lines. (B) Small molecule inhibitors of the NusB–NusE PPI. | ||
Cossar et al. used a pharmacophore model based on the NusE α1-helix for a virtual screening of the mini-Maybridge 53000 compound library and five synthetically accessible hit compounds were further evaluated.79 Of these five, compounds 18 and 19 (Fig. 6B) showed inhibition of the B. subtilis NusB/NusE PPI in an ELISA assay with IC50 values of 6.1 μM and 19.8 μM, respectively. Pyrimidine 18 also displayed moderate inhibition of the growth of Gram-positive B. subtilis (9%) and Gram-negative E. coli (21%) at a concentration of 200 μM.79 In a follow-up study, the same group synthesized four focused compound libraries based on bis-ether 19 and 34 new derivatives were evaluated as NusB–NusE PPI inhibitors, leading to the identification of cis-butene iminoguanidine derivative 20 (Fig. 6B) as the most promising compound.80 Compound 20 showed good inhibition of the NusB–NusE interaction (55% at 25 μM) and potent antibacterial activity against the Gram-positive pathogens methicillin-resistant S. aureus (MRSA) and S. pneumoniae (MIC ≤ 7 μM), as well as promising activity against the difficult to target, Gram-negative strains P. aeruginosa and Acinetobacter baumannii (MIC ≤ 125 μM). However, 20 also exhibited considerable cytotoxicity in different human cancer cell lines.80
Yang et al. used a novel pharmacophore model to perform an in silico screen of a combination of the mini-Maybridge compound library and the Enamine antibacterial library.81 Seven hit compounds were further evaluated and diarylamine 21 (Fig. 6B) was found to be a potent inhibitor of the NusB–NusE PPI with an IC50 of 35 μM. The antibacterial activity of 21 was tested against a panel of representative pathogen strains (Enterococcus faecalis, Klebsiella pneumonia, A. baumannii, P. aeruginosa, Enterobacter cloacae, E. coli, Proteus vulgaris and S. aureus) and preferential activity of 21 against S. aureus was found with a MIC of 60 μM for healthcare-acquired MRSA ST239. Interestingly, 21 displayed no cytotoxicity in mammalian cell lines.81
Recently, Qiu et al. published two SAR studies based on 21. In the first study, 60 diarylimine and amine derivatives were synthesized by modifying the left or right benzene ring of 21.82 All new compounds were tested for their antimicrobial activity against a panel of WHO priority pathogens and a series of compounds was tested against further Gram-positive bacteria. Several compounds showed promising antibacterial activities against Gram-negative A. Baumannii (lowest MIC 64 μM) and Gram-positive strains S. pneumonia (lowest MIC around 27 μM), S. epidermidis (MIC ∼ 3 μM), Staphylococcus saprophyticus (MIC ∼ 3 μM), S. pyogenes (MIC ∼ 27 μM) and S. agalactiae (MIC ∼ 27 μM), but none of the compounds were active against Gram-negative P. aeruginosa and E. cloacae. Selected compounds were also tested against 14 globally spread healthcare-acquired and community-acquired MRSA strains, with some showing stable antibacterial activity with MICs down to around 7 μM. However, it should be noted that the highly active compounds containing an amine linker displayed mild cytotoxicity in human cancer cell lines.82 In a second study, a further 38 derivatives of 21, now named nusbiarylins, with more diverse modifications of the left benzene ring and the linker were prepared.83 The new compounds were tested against a panel of clinically significant bacterial strains and some promising antibacterial activities against Gram-positive pathogens were observed, with MIC values as low as 3 μM against S. saprophyticus and 13 μM against S. epidermidis. As in the previous study, selected compounds were also tested against 14 MRSA strains and consistent antibacterial activities could be observed with MICs down to 7–15 μM making the general class of nusbiarylins promising lead compounds for the development of transcription-targeting anti-MRSA agents.83 Very recently, 21 and some of its derivatives were also shown to attenuate the release of staphylococcal virulence factors such as the exoproteins α-toxin and panton-valentine leucocidin in vitro, thereby protecting red blood cells from lysis and injury.84
2.3 Targeting outer membrane protein complexes
Gram-negative bacteria have an outer membrane (OM) that acts as a permeability barrier into the periplasm and excludes many current antibiotics.85 Crucial to the barrier function is a network of β-barrel outer membrane proteins (OMPs).86 The OMPs are transported to the OM by three essential pathways: lipopolysaccharide transport (Lpt), β-barrel assembly machine (Bam), and the localization of lipoproteins (Lol).87 Each of these pathways is comprised of protein complexes and their function is necessary for cell viability. There are currently no antibiotics in clinical use targeting these pathways, so disrupting the PPIs of these complexes is a potential strategy for development of new antibacterial drugs.88,89Hagan et al. reported a peptide that inhibits OMP folding by targeting the BamA–BamD interaction in E. coli.101 Initially, they found that BamD binds to unfolded BamA at the C-terminal of its β-barrel.101,104 The authors then used an in vitro assay involving the reconstitution of BamA by the BAM complex in proteoliposomes to identify the effect of this interaction on the folding of BamA. It was found that the introduction of a peptide comprised of residues 765–779 of the C-terminus of BamA (22, Fig. 7A and B) inhibited the folding of BamA.101 Using photo-cross-linking the peptide was confirmed to bind to BamD in vivo.101 They concluded that peptide 22 mimics the interaction of BamA to BamD, effectively inhibiting the BamA–BamD PPI. Peptide 22 was also found to be toxic when expressed in the E. coli periplasm, causing growth defects on plates.101 By increasing the permeability of the OM, the sensitivity of the bacteria to antibiotics that cannot normally cross the membrane is enhanced. These promising results demonstrate that peptide 22 is a good starting point for the development of peptidomimetics targeting BamA–BamD that interfere with OMP assembly.
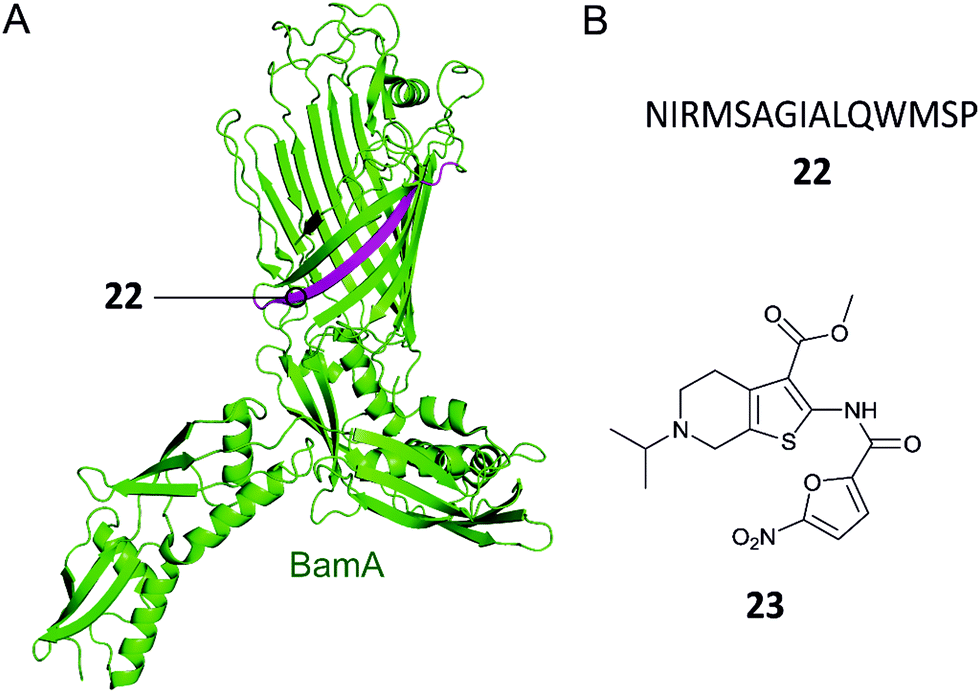 | ||
| Fig. 7 (A) Crystal structure of the E. coli BamA with the region mimicked by peptide 22 highlighted in magenta (PDB: 5D0Q).91 (B) Peptide and small molecule inhibitors of the BamA–BamD complex. | ||
In a recent study, the Si group screened for small molecule compounds disrupting the BamA–BamD PPI in E. coli using a yeast two-hybrid (Y2H) screening system.102 Of the 25000 screened compounds, IMB-H4 (23, Fig. 7B) was identified as the most specific inhibitor of PPI, where a biolayer interferometry assay identified it as a BamA binder. Further experiments demonstrated that treatment of E. coli cells with 23 led to significant damage of the OM. Importantly, 23 was shown to inhibit the growth of E. coli ATCC 25922 strain (MIC 10 μM) and other drug-resistant Gram-negative bacteria strains (MIC range 10–80 μM).
Thanatin (24, Fig. 8A) is a natural product first isolated in 1996 from the hemipteran insect Podisus maculiventris (spined soldier bug).119 Antimicrobial activity against several Gram-negative bacteria was reported with strong MICs (<1.5 μM).11924 has since been identified to target LptA and LptD in E. coli using photolabeling and MS-based proteomic analysis.120 Further studies using a bacterial-two-hybrid screening assay were used to confirm that 24 also inhibits the LptC–LptA interaction in a dose-dependent manner (MIC 0.7–1.4 μM).121In vitro FP studies identified 24 as binding to LptA and LptD with nanomolar affinities and SPR and NMR measurements confirmed that 24 disrupts the LptC–LptA interaction.120,121 The NMR solution structure of LptA–24 complex was solved, where 24 was found to interact with the first N-terminal β-strand in the β-jellyroll of LptA (Fig. 8C).120 The crystal structure of LptA2 shows how the LptA subunits interact head-to-tail through their N- and C-β-strands (Fig. 8B).107 The binding site of 24 overlaps with the LptA–LptA binding interface, therefore blocking the dimerization of the LptA monomers.120 Both the N-terminal β-strands of LptD and the C-terminal β-strands of LptC share similarities with LptA, where residues involved in LptA–24 binding were found to be highly conserved.108
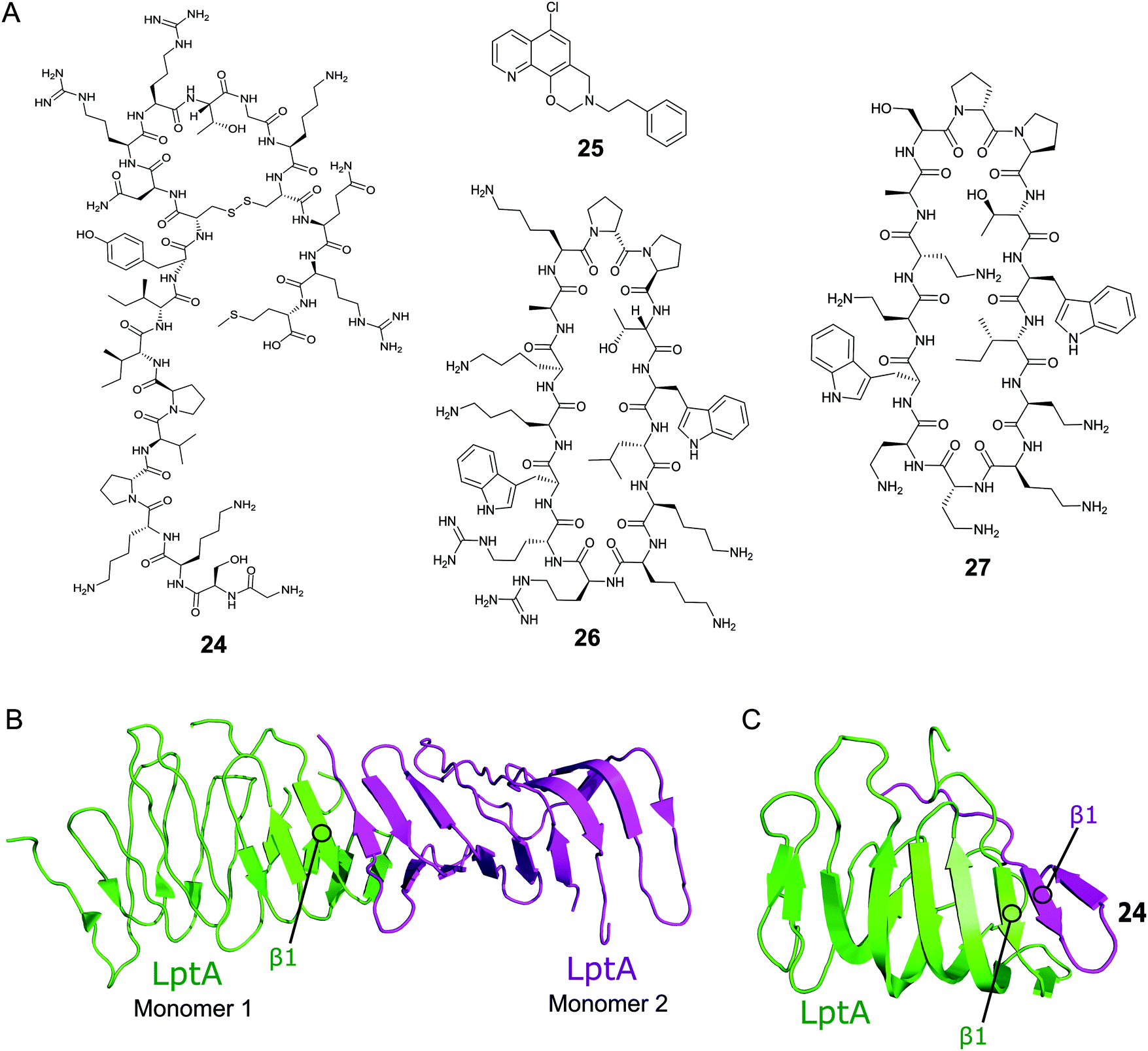 | ||
| Fig. 8 (A) Compounds targeting PPIs of the Lpt complex. (B) Crystal structure of the E. coli homodimer LptA complex (PDB: 2R1A).107 (C) NMR solution structure of the E. coli LptA bound to compound 24, the N-terminal β1 strand of LptA binds to the N-terminal β1 strand of 24 in a similar manner to the LptA–LptA binding interface (PDB: 6GD5).120 | ||
The Si group used a Y2H screening system in E. coli to identify small molecule IMB-881 (25, Fig. 2A) targeting LptA–LptC.122 Using SPR, 25 was found to disrupt the LptA–LptC interaction by specifically binding to LptA.122 Electron microscopy demonstrated that treatment of E. coli cells with 25 led to significant damage of the OM, and antibody detection indicated a higher level of LPS in the periplasm, suggesting 25 inhibits its transport in vivo. 25 was then found to greatly inhibit the growth of E. coli ATCC 25922 strain (MIC of 19 μM), and other clinical multidrug-resistant strains (MIC range of 19–150 μM).
A macrocyclic β-hairpin peptidomimetic, L27-11 (26, Fig. 8A), was initially optimised via a number iterative peptidomimetic library synthesis cycles.123,124 Further optimisation led to murepavadin (27, Fig. 8A), a clinical candidate with an MIC of 5.15 nM against >1000 clinical multi-drug resistant P. aeruginosa strains.124,125 It reached stage three of clinical trials, but trials are currently suspended due to nephrotoxicity.88 Photoaffinity experiments identified LptD as the target for this new family of compounds and mechanistic studies confirmed they inhibit LPS transport.124,126 Photolabeling suggested that the peptidomimetics bind the periplasmic domain of LptD and point mutations indicate that the point of interaction is close to the β-jellyroll domain.127,128 This suggests that the compounds may bind to LptD at the LptD–LptA binding interface and inhibit the PPI.
2.4 Targeting toxin–antitoxin systems
The toxin–antitoxin (TA) systems are present in a large variety in bacteria cells.129 There are many TA families broadly categorised into six types (I–VI) based on the inhibition of toxin function by the antitoxin.130,131 The function of toxins is not fully understood, they were originally identified as conferring plasmid stabilisation and phage resistance.132,133 Since then a number of functions for TA systems have been proposed, such as programmed cell death, biofilm formation,134 regulation of virulence factors in pathogenic bacteria,130 persistence and dormancy.135 The TA systems are ubiquitously present in bacterial genomes providing a class of interactions that can be targeted in multiple bacteria systems.136Type II TA systems are the most abundant in bacteria and the most well characterised.131 They are comprised of a proteic toxin and a proteic antitoxin where the antitoxin directly binds to and inhibits the toxin. The type II toxins can exert toxicity in several ways including inhibition of translation or replication and interruption of cell wall synthesis, although the majority affect translation. In normal growth conditions, the stable toxin is prevented from exerting its lethal effect through tight binding with its less stable antitoxin partner. The antitoxin also regulates expression of the type II TA operon by binding to its operator site.133
Type II TA systems have, in recent years, gained much interest as targets for antibiotic agents as well as to counter persistence.137 A number of reviews have identified TAs as a possible target for new antibiotics with the aim to activate the toxin, essentially allowing bacterial suicide.136,138–142 The toxin can be liberated by targeting the TA PPI via either (a) competitive binding to the antitoxin or (b) dissociation of the existing TA complex. Additionally, identifying molecules that bind to the toxin of a TA system, either mimicking the antitoxin binding at the PPI interface or targeting an allosteric position, is a potential strategy for combatting bacterial persistence.131,143
In this section, recent studies involving the rational design and high throughput screening of peptides and small molecule compounds to perturbate TA PPI interfaces will be summarised. For clarity, the TA systems have been categorised by the targeted cellular function and structural class.
2.4.i.1 VapBC. In a comprehensive systematic genomic analysis of the TA genome in M. tuberculosis, 47 of 88 putative TA systems were identified to belong to the VapBC family.145 The VapC toxins have a characteristic PIN (PilT N-terminus) domain in which four acidic amino acid residues are conserved. The conserved residues are crucial for ribonuclease activity, and the VapC toxins inhibit translation via mRNA cleavage. Each VapB antitoxin inhibits the ribonuclease activity of its cognate toxin. The VapBC family is an attractive target for novel antibacterial therapies since liberating the toxin could lead to cell death due to hindered translation. Structural information for VapBC3, VapBC5, VapBC11, VapBC15, VapBC26 and VapBC30 has been acquired and efforts to rationally design peptides to disrupt the TA complexes have been carried out.146
Lee et al. characterised the structures of the M. tuberculosis VapBC30 in 2015 and VapBC26 in 2017 and identified the key residues involved in binding at the TA interface.147,148 They then rationally designed peptides to mimic the binding interface and interfere with the TA PPI thereby activating the ribonuclease activity of the VapC toxin. The VapBC30 complex comprises three structural components: α1-helix of VapB30 (residues 49–62), α2-helix of VapC30 (residues 17–27), and α4-helix of VapC30 (Fig. 9).147 The authors rationally designed three peptides to mimic each of these components to inhibit TA binding of VapBC30. Peptide 28 mimics the α1-helix of VapB30, designed to compete with VapB30 for interaction with VapC30 (Table 2). Peptides 29 and 30 mimic the α2- and α4-helical regions of VapC30 respectively, designed to compete with VapC30 for interaction with VapB30 (Table 2 and Fig. 9). The ability of the peptides to disrupt the TA complex was determined with a fluorescence reporter assay measuring the ribonuclease activity of VapC30. The inhibitory potency was calculated considering the relative ribonuclease activity, where the toxin alone (10 μM) has a 0% inhibitory value and the TA complex (10 μM) represents 100% inhibition. The peptides were able to disrupt the TA complex in a concentration dependent manner (10–100 μM). At the highest concentration (protein to peptide 1:10) the VapC30 toxin-mimicking peptides 29 and 30 showed a greater inhibitory effect of 53% and 73% compared to the antitoxin-mimicking peptide 28 at 43%.
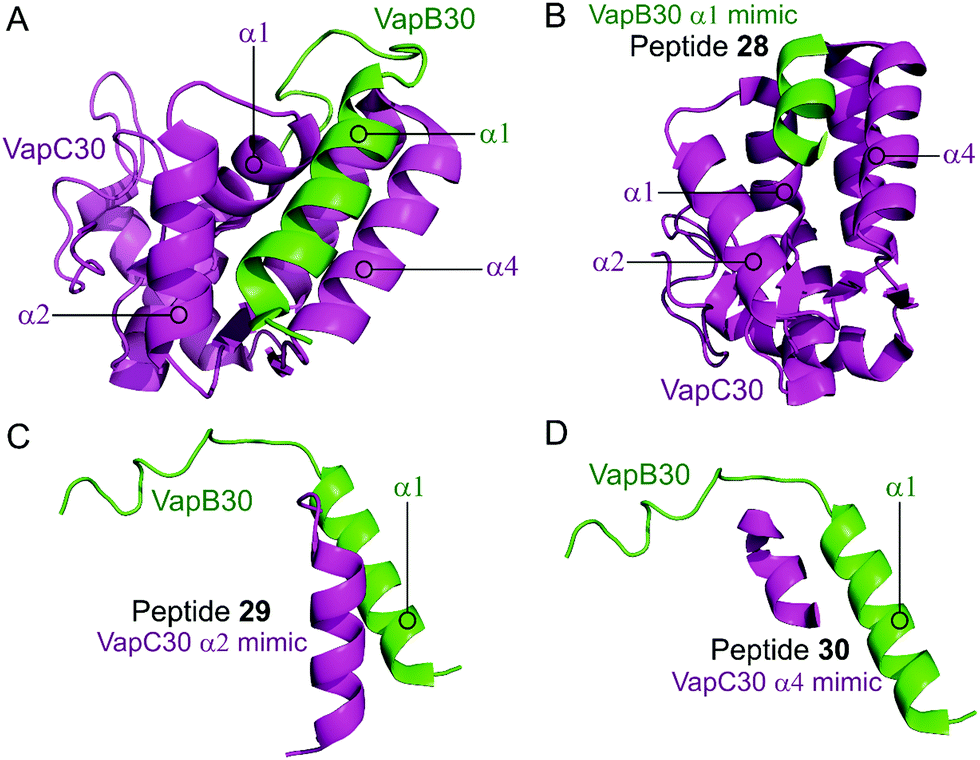 | ||
| Fig. 9 (A) Crystal structure (PDB: 4XGQ)147 of the complex between VapB30 (green) and VapC30 (magenta). A representation of the regions selected for the design of peptides 28 (VapB30 helix α1 mimic), 29 (VapC30 helix α2 mimic) and 30 (VapC30 helix α4 mimic) is shown (B), (C) and (D), respectively. | ||
| TA complex | Mimicked protein | Mimicked region | Peptide (number in reference) | Peptide sequence (residues) | Peptide concentration (μM) | % inhibition of PPI |
|---|---|---|---|---|---|---|
| a NR: not reported. b Toxin and TA complex concentration 10 μM. c Toxin and TA complex concentration 2.5 μM. | ||||||
| VapBC30 147 | VapB30 | α1 | 28 (I) | ELAAIRHR (52–59) | 100 | 43b |
| VapC30 | α2 | 29 (I) | DEPDAERFEAAVEADHI (14–30) | 100 | 53b | |
| VapC30 | α4 | 30 (III) | RPGEPGGRE (48–56) | 100 | 73b | |
| VapBC26 148 | VapB26 | Coil between α2 and α3 | 31 (I) | PPPRGGLYAGSEPIA (44–58) | — | — |
| VapB26 | α3 | 32 (II) | VDELLAGF (61–68) | — | — | |
| VapC26 | α1 | 33 (III) | ALLAYFDAAEP (7–17) | NR | NRa | |
| VapC26 | α3 | 34 (IV) | PYVVAELDYLVATRVG (37–52) | NR | NR | |
| VapC26 | α4 | 35 (V) | DAELAVLRELAG (54–65) | 200 | 80c | |
| VapC26 | Partial α3 and α4 | 36 (VI) | YLVATRVGVDAELAV (45–59) | NR | NR | |
| VapC26 | Whole α3 and α4 | 37 (VII) | PYVVAELDYLVATRVGVDAELAVLRELAG (37–65) | NR | NR | |
In the VapBC26 complex, the active site of VapC26, composed of α1-, α3- and α4-helices, interacts with the α3-helix and the C-terminal region of VapB26.148 The authors designed seven peptides (31–37) to disrupt the TA pair, based on the binding interface (Table 2 and Fig. 10). The inhibitory potency was calculated considering the relative ribonuclease activity, where the toxin alone (2.5 μM) has a 0% inhibitory value and the TA complex (2.5 μM) represents 100% inhibition. Peptides 31 and 32 mimicking the VapB26 antitoxin did not show any effect on enzymatic activity. The remaining five peptides were designed to mimic the VapC26 toxin to compete with VapC26 for interaction with VapB26. Peptides 34 and 35, mimicking the α3- and α4-helices of the VapC26 toxin, respectively, and peptide 37, composed of both the α3- and α4-helices increased the ribonuclease activity in a concentration dependent manner (2.5–1 mM). Peptide 35 demonstrated the best ribonuclease activity and was able to disrupt TA binding of VapBC26 by 80% at 200 μM (protein to peptide 1:80).
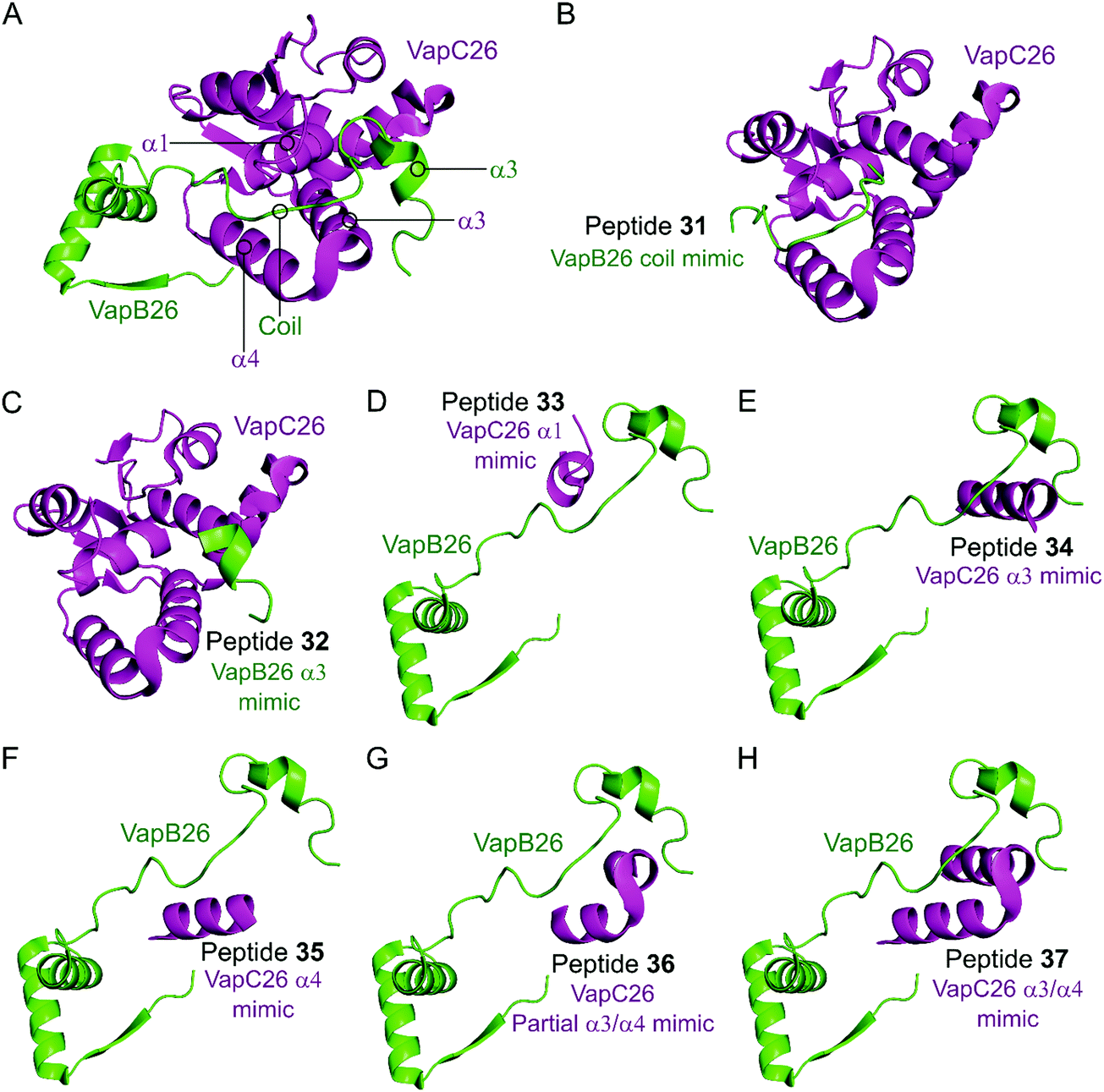 | ||
| Fig. 10 (A) Crystal structure (PDB: 5X3T)148 of the complex between VapB26 (green) and VapC26 (magenta). A representation of the regions selected for the design of peptides 31 (VapB26 coil mimic), 32 (VapB26 helix α3 mimic), 33 (VapC26 α1 mimic), 34 (VapC26 α3 mimic), 35 (VapC26 α4 mimic), 36 (VapC26 partial α3/α4 mimic) and 37 (VapC26 α3/α4 mimic) is shown in (B), (C), (D), (E), (F), (G) and (H), respectively. | ||
Deep et al. carried out a similar experiment, characterising the VapBC11 TA complex of M. tuberculosis, and designed peptide inhibitors of the interface.149 The authors in this case were trying to identify peptides that could inhibit ribonuclease activity. Considering bacterial persistence and tolerance, they postulated that the inactivation of VapC11 could allow a bacterial cell to be more susceptible to clearance. Based on a crystal structure, it was found that the TA interface covers a large area of 1375 Å2. The C-terminal residues of VapB11 wrap around a VapC11 cavity formed by its α1-, α3- and α4-helices. Four overlapping peptides (38–41, 10–14 residues long) mimicking the VapB11 antitoxin were designed (Table 3 and Fig. 11) and the change in ribonuclease activity was measured using in vitro tRNA-LeuCAG cleavage assays. All four peptides inhibited the activity of VapC11 to some degree, the inhibition was dependent on a 100-fold greater amount of peptide compared to protein.
| TA complex | Mimicked protein | Mimicked region | Peptide (number in reference) | Peptide sequence (residues) |
|---|---|---|---|---|
| VapBC11 149 | VapB11 | α3 | 38 (I) | LSREFLLGLE (41–50) |
| VapB11 | α3 and flexible linker | 39 (II) | EGVGWEGDLDD (50–60) | |
| VapB11 | Flexible linker and α4 | 40 (III) | WEGDLDDLRSDRPD (54–67) | |
| VapB11 | α4 | 41 (IV) | LDDLRSDRPD (58–67) |
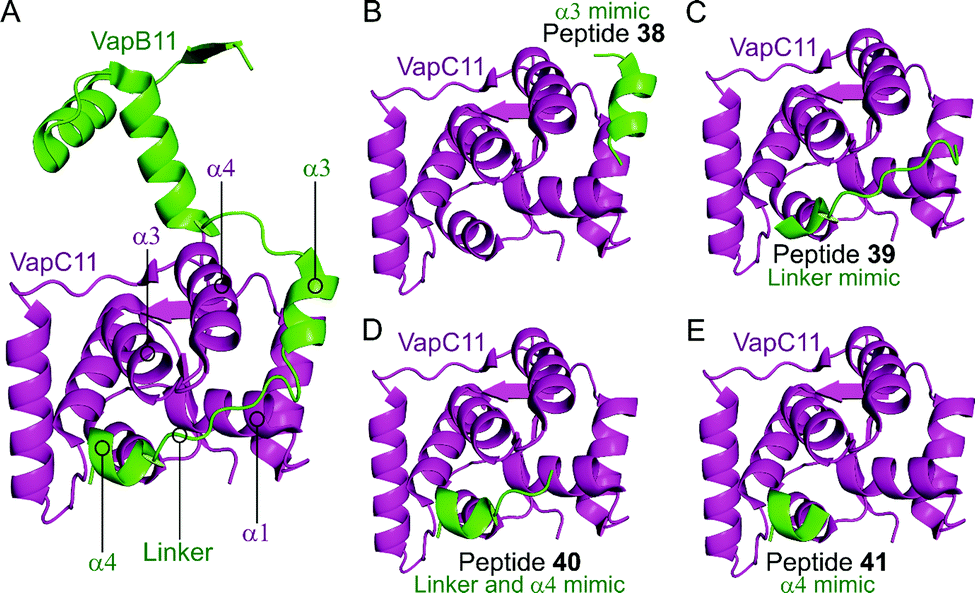 | ||
| Fig. 11 (A) Crystal structure (6A7V)149 of the complex between VapB11 (green) and VapC11 (magenta). A representation of the regions selected for the design of VapB11 peptides 38 (helix α3 mimic), 39 (linker mimic), 40 (linker and helix α4 mimic) and 41 (helix α4 mimic) is shown in (B), (C), (D) and (E), respectively. | ||
In a recent study, Sundar et al. designed peptide inhibitors 42–47 mimicking the published toxin structures of VapBC3, VapBC5, VapBC11, VapBC15, VapBC26 and VapBC30 using the Peptiderive server (Table 4).150 The protein–peptide docking was performed using the Cluspro server to identify the binding affinity of the peptides to the toxins. Peptides with a strong interface score were found for each target. Interestingly, the peptides for VapBC26 and VapBC30 reported here have lower binding energy compared to the ones previously identified, however, this has only been measured in silico and further biochemical characterisation is necessary to confirm binding affinities.147,148,150
2.4.i.2 HicBA. HicBA is a newly identified, relatively uncharacterised type II TA system.151 The Lee group characterised the structure of HicBA from S. pneumonia and designed peptides to target the TA interface in a similar manner to their studies of VapBC in M. tuberculosis.147,148,152 The ribonuclease activity of HicA toxins relies on a RNA binding domain, which contains a conserved histidine residue (H36 in S. pneumoniae).152,153 The N-terminal region of the HicB antitoxin of S. pneumoniae sterically blocks the toxin active site, thereby covering 1183 Å2 of the toxin. The earlier studies targeting VapBC26 and VapBC30 suggested toxin mimicking peptides had the greatest inhibitory effect of the interaction. The authors therefore designed four peptides (48–51) mimicking the α2-helix of HicA (Table 5 and Fig. 12), the inhibitory potency was calculated considering the relative ribonuclease activity, where the toxin alone (4 μM) has a 0% inhibitory value and the TA complex (4 μM) represents 100% inhibition. Peptide 48 demonstrated a concentration dependent inhibition of the interaction and increased ribonuclease activity (2–16 μM), 80% at the highest concentration (protein to peptide 1
:4). Peptide 48 also demonstrated potent antibacterial activity against the Gram-positive pathogens B. subtilis, S. aureus and S. epidermis (MIC 6–12.5 μM), as well as promising activity against the difficult to target, Gram-negative strain P. aeruginosa (MIC 6 μM). Peptide 48 contains 11 residues of the full α2-helix, including seven residues that interact with HicB, and possesses the highest helicity out of the four peptides tested.
| TA complex | Mimicked protein | Mimicked region | Peptide | Peptide sequence (residues) | Peptide concentration (μM) | % inhibition of PPI |
|---|---|---|---|---|---|---|
| a NR: not reported. b Toxin and TA complex concentration 4 μM. | ||||||
| HicBA 152 | HicA | α2 | 48 | ELNKYTERGIRKQAG (53–67) | 16 | 80b |
| HicA | α2 | 49 | GELNKYTERGIRKQAG (52–67) | NR | NRa | |
| HicA | α2 | 50 | ELNKYTERGIRKQAGL (53–68) | NR | NR | |
| HicA | α2 | 51 | GELNKYTERGIRKQAGL (52–68) | NR | NR | |
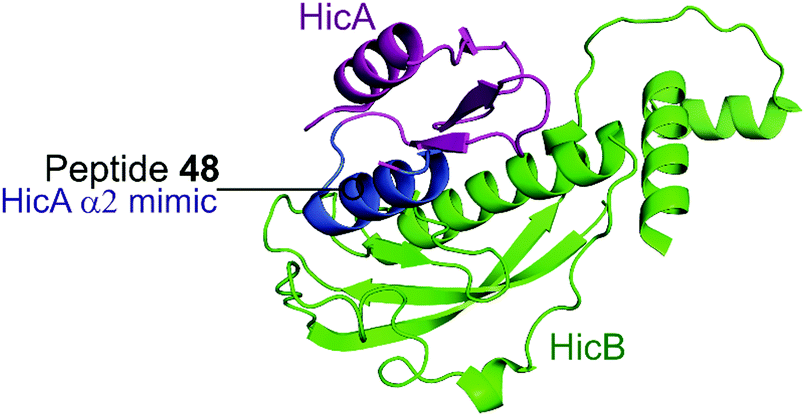 | ||
| Fig. 12 Crystal structure (PDB: 5YRZ)152 of the complex between HicA (magenta) and HicB (green) from S. pneumoniae. The region of HicA mimicked by peptide 48 is highlighted in blue. | ||
2.4.i.3 PemK-like protein family. The PemK-like protein family includes a number of homologs which have been identified in most bacterial systems including the MazF toxin in E. coli, P. aeruginosa, S. aureus, B. subtilis and M. tuberculosis and the MoxT toxin in Bacillus anthracis.154 The toxins share a ribonuclease SH3-like barrel fold domain and an antitoxin with a RHH motif at the N-terminus. Three or four α-helices link the two β-sheets of the toxin monomer. The MazEF toxin–antitoxin pair is very well characterised in pathogenic bacteria.155 The MazF toxin superfamily members are proposed to have a conserved mode of binding to their antitoxin using two binding pockets located on the β1–β2 and β3–β4 linkers. The disordered C-terminus of the antitoxin binds both via a hydrophobic group to site 1 and via an extensive interface with the less conserved site 2.156 Site 2 is the known ribonuclease site, but disruption of either pocket is predicted to affect the ribonuclease activity.
The MazEF pair has been implicated in the programmed cell death (PCD) of E. coli bacteria as a suicide module under stress conditions via activation of the toxin.157 This cell death requires activation by a linear peptide termed the extracellular death factor (EDF), a quorum sensing molecule.158 Using a continuous fluorescence reporter assay the authors found that the EDF amplified the endoribonucleolytic activity of the MazF toxin (0.25 μM) in a concentration dependent manner (1.5–7.5 μM), up to 57% increased activity.159 Additionally, the EDF prevented the inhibitory effect of the antitoxin MazE, and in the presence of MazE (0.025 μM) and increasing concentrations of the EDF (0–15 μM), MazF (0.25 μM) had activity close to 100% (1:60 MazF to EDF). The EDF (sequence: NNWNN) was shown by computational modelling to bind to MazF in a similar way to MazE-MazF binding, with parallel contacts between the EDF with MazF and the binding site of MazE 71–75 (IDWGE) (Fig. 13).159 It has been suggested that MazE binds to one mRNA binding site of MazF and this initial binding interrupts the other mRNA binding site, resulting in MazF inhibition. The EDF, however, only binds to one binding site and the other site is still available to bind and cleave mRNA thereby allowing MazF activity and preventing inhibition by MazE.
 | ||
| Fig. 13 Crystal structure of the E. coli MazEF TA complex (PDB: 1UB4)160 with MazF toxin monomers shown in magenta and cyan, and MazE antitoxin in green. Enlarged picture (right chart) highlights MazE residues 71–75 (IDWGE, in green) that are crucial for the interaction with MazF (magenta and cyan surfaces represent, respectively, monomers 1 and 2). | ||
There are seven MazEF homologs in M. tuberculosis, of which several are proposed to have the same effect on PCD as in E. coli. A linear pentapeptide EDF (sequence: ELWDR) was identified for MazEF4 of M. tuberculosis.161 Interestingly, the charge distribution of the MazEF4 binding site is opposite to that of the MazEF of E. coli. The final positive arginine residue of the EDF reflects this shift and binds to the negative pocket of MazF, otherwise based on NMR interaction studies the EDF binds in a similar manner as in the E. coli homolog. This EDF was also identified as enhancing the ribonuclease activity of MazF4 and disrupting the MazEF4 complex.
There is divergence in the EDF sequences in different species or homologs within a species and other EDFs discovered from B. subtilis and P. aeruginosa were found to enhance activity of MazF in E. coli.162 However, only the EDF isolated from E. coli directly interrupts MazE–MazF binding of E. coli.163 The EDFs of E. coli and P. aeruginosa were found to enhance the activity of MazF-mt3 and MazF-mt6 of M. tuberculosis, although the effect was mainly observed from the EDF of E. coli.164 The specific EDF peptides for each MazEF pair are a possible starting point for a new class of antibiotic peptides to target MazEF mediated cell death. Furthermore, the ability of EDFs from other species to enhance the activity of homologs may be a useful starting point to design a peptide capable of targeting multiple organisms.
In 2007, Agarwal et al. identified a PemK-like toxin, later termed MoxT, in B. anthracis.166 In a series of papers the group rationally designed peptides targeting the MoxX antitoxin/MoxT toxin interface, some mimicking EDFs.165,167,168 The MoxXT PPI was initially modelled based on E. coli MazEF and the C-terminus of the MoxX antitoxin was implicated in toxin binding. In the first study, six peptides (52–57, Table 6) mimicking the C-terminus of either the MoxX or MazE antitoxins were designed to disrupt the toxin–antitoxin interaction at sites 1 or 2 (Fig. 14).167 ELISA was used to identify the effect on the peptides on the TA interaction, the cells were coated with MoxX (200 ng) and preincubated MoxT (200 ng) with peptide (2 μM) was added. The peptides targeting site 2 demonstrated the greatest inhibition (up to 35%) of the TA interaction compared to those targeting site 1. Peptides 54 and 56 (mimicking site 2 of MoxX) inhibited the interaction most effectively but also inhibited the activity of the toxin. In a second study, following homology modelling of the MoxXT complex, a novel peptide (58) was designed to mimic MoxX binding to site 2 (Table 6).168 This peptide (2 μM) was able to inhibit the TA interaction by 42%, however it also demonstrated inhibition of MoxT ribonuclease activity. To preserve the ribonuclease activity, site 1 rather than site 2 must be targeted, however in the above studies the peptides designed to mimic site 1 had little effect on the toxin–antitoxin interaction.
| TA complex | Mimicked protein | Mimicked region | Peptide (number in reference) | Peptide sequence (residues) | Peptide concentration (μM) | % inhibition of PPI |
|---|---|---|---|---|---|---|
| a Toxin and antitoxin 200 ng. b Toxin 0.23 μM and antitoxin 300 ng. | ||||||
| MoxXT 167 | MoxX | Site 1 | 52 (I) | VERLVSGG (88–95) | 2 | 11a |
| MazE | Site 1 | 53 (II) | NLHRNIW (66–73) | 2 | 20a | |
| MoxX | Site 2 | 54 (III) | LLFQHLTE (44–51) | 2 | 35a | |
| MoxX | Site 2 | 55 (IV) | KRYQHESM (52–59) | 2 | 25a | |
| MoxX | Site 2 | 56 (V) | RRGYIEMG (60–67) | 2 | 30a | |
| MazE | Site 2 | 57 (VI) | KAELVNDI (55–62) | 2 | 22a | |
| MoxXT 168 | MoxX | Site 2 | 58 | SKIGAWAS | 2 | 42a |
| MoxXT 165 | E. coli EDF | Site 1 | 59 (I) | NNWDN (1–5) | 1 | 37b |
| E. coli EDF | Site 1 | 60 (II) | NNWNN (1–5) | 1 | 44b | |
| E. coli EDF | Site 1 | 61 (III) | DNWNN (1–5) | 1 | 22b | |
| B. anthracis EDF | Site 1 | 62 (IV) | SIWNK | 1 | 40b | |
| MazE | Site 1 | 63 (V) | HENIDW (68–73) | 1 | 31b | |
| MazE | Site 1 | 64 (VI) | ENIDWGEP (69–76) | 1 | 27b | |
| MoxX | Site 1 | 65 (VII) | RGYIEMG (61–67) | 1 | 46b | |
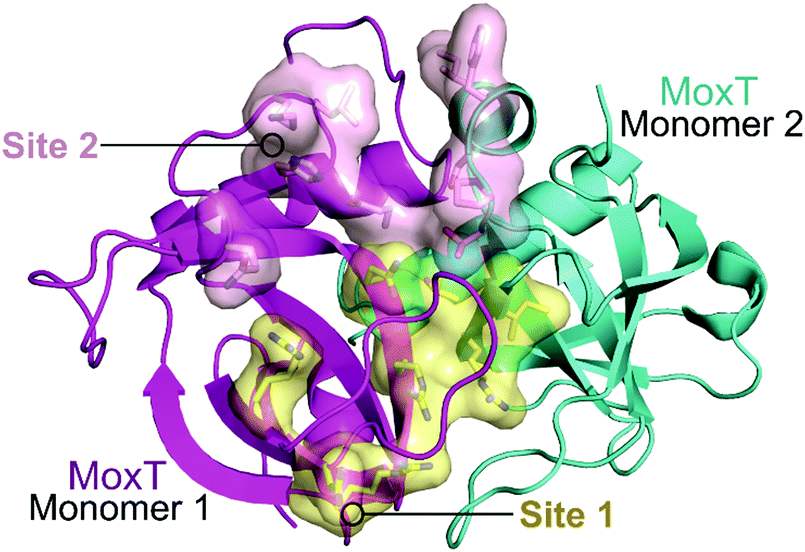 | ||
| Fig. 14 Crystal structure of MoxT dimer (PDB: 4HKE)165 with monomers 1 and 2 shown in magenta and cyan, respectively. Interaction sites 1 and 2 are shown as yellow and pink surfaces, respectively. | ||
A third study involved the rational design of peptides to disrupt the TA interaction of MoxXT and importantly aimed to stimulate the activity of MoxT by targeting site 1.165 Peptides 60–65 were designed to mimic the antitoxins MoxX and MazE of E. coli as well as the EDF of E. coli and predicted EDF of B. anthracis (Table 6). ELISA was used to identify the effect on the peptides on the TA interaction, the cells were coated with MoxX (300 ng) and preincubated MoxT (0.23 μM) mixed with peptide at increasing concentrations (0.2–1 μM) was added. All inhibited the MoxXT interaction, with maximum efficacy reached at 1 μM, peptides 60 and 65 proved to be most successful with inhibition of 44% and 46%, respectively. A continuous fluorometric assay, whereby fluorophores attached to RNA probes emit fluorescence when cleaved, identified an increase in ribonuclease activity in the presence of 40 equivalents of peptide compared to MoxT. Peptides 59, 62 and 64 demonstrated the greatest enhancement in the ribonuclease activity of MoxT. The authors proposed that the helical structures reported in the first studies too closely mimic the antitoxin and bind at site 2 rather than site 1. The non-helical (C-shaped or extended structure) peptides with one aromatic residue that are reported in their later study cause an increase in MoxT activity.
Recently, a number of papers have reported in silico approaches to the design of peptides and peptidomimetics to interfere with the MazEF interface.150,169,170 Sundar et al. designed peptide 66 using the Peptiderive server to mimic the EDF of MazF4 of M. tuberculosis (Table 7).150 Unfortunately the in silico experiments using the Cluspro docking server identified the peptide to have worse binding affinity to MazF than its EDF. Using the same Peptiderive and Cluspro server, Mohammadzadeh et al. designed peptides 67–69 mimicking MazE of the MazEF TA system in L. monocytogenes.169 It is not clear whether these peptides bind with a similar affinity to MazF as MazE, but they interact at the same binding site on the toxin.
Farhadi et al. modelled MazF of A. baumannii and designed peptidomimetics based on the E. coli EDF using the pep:MMs:MIMIC tool.170 The compounds were docked with MazF and the nine highest ranking mimetics had low binding energies of less than −6.8 kcal mol−1, lower than the original peptide (−5.2 kcal mol−1), demonstrating favourable docking. Several mimetics formed two hydrogen bonds with MazF, compared to one formed by the original peptide. Some of the in silico designed peptides and peptidomimetics may be a starting point for novel drugs to target MazF and mediate cell death. Considering the results of the Bhatnagar group suggesting that only targeting site 1 results in enhanced MazF activity, biochemical experiments of peptide–protein affinity and importantly their effect on MazF activity must first be considered.
2.4.i.4 HipAB. The HipAB TA system was the first TA system to be implicated in persistence in E. coli and it has since been identified in a number of pathogenic bacteria.171 The structure of E. coli HipAB was characterised in 2009 by Schumacher et al. (Fig. 15).172 The HipA toxin was found to have a serine kinase-like fold and has kinase activity. It was originally suggested to phosphorylate the elongation factor thermo unstable (EF-Tu).172 It was later found the target was further upstream in the translation process, it phosphorylates, and therefore inactivates, the glutamyl-tRNA-synthetase (GltX).173,174 This leads to inhibition of protein biosynthesis, a process often linked to the initiation of bacterial dormancy and persistence.173,174 In the presence of the HipB antitoxin its activity is neutralised; the HipA N-terminal domain interacts with one HipB subunit whilst the C-terminal domain interacts with the other HipB subunit. Strangely the HipB monomers do not directly interact or occlude the HipA active site, suggesting that HipB simply locks HipA into an open inactive conformation that prevents binding of protein substrates.
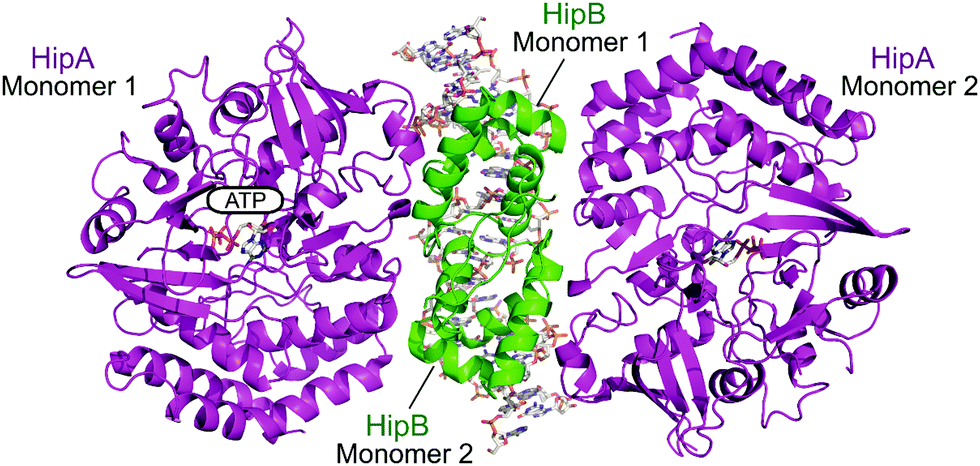 | ||
| Fig. 15 Crystal structure of the E. coli HipAB TA complex bound to DNA (PDB: 3HZI)172 with HipA toxin monomers shown in magenta and HipB antitoxin monomers shown in green. The DNA and HipA-bound ATP are shown as sticks. | ||
Li et al. used in silico structure based computational screening followed by in vitro experiments to identify novel small molecules inhibiting HipA, thereby preventing persistence.175 The Chemdiv kinase and SPECS compounds libraries were used together with docking with Glide to identify tight binders. SPR was used to measure hit HipA binding affinity, and the tightest binders were tested for their effect on persistence. The number of colonies present following addition of hit molecules before and after antibiotic treatment were compared to quantify the persister fraction. 14 molecules reduced E. coli persistence at 250 μM and did not cause cytotoxicity. The EC50 values of the four most potent compounds (70–73, Fig. 16) were found to be less than 126 μM, with compound 72 having the highest anti-persister activity (46 μM, Table 8). The Kd values also demonstrated 72 as the tightest binder (270 nM). These molecules were found to target the active site of the HipA toxins, and bind in a similar manner to ATP, rather than targeting the PPI interface. A similar methodology, perhaps screening for peptides targeting the HipAB interface, would be a strategy to identify other molecules that could target HipA mediated persistence.
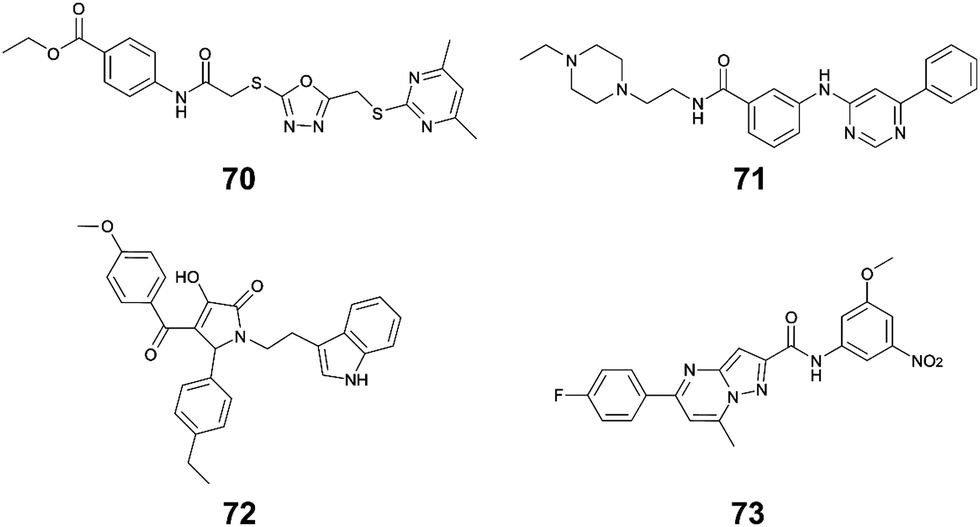 | ||
| Fig. 16 Small molecule inhibitors of HipA toxin. | ||
| TA complex | Species | Compound (number in reference) | K d (μM) | EC50 (μM) |
|---|---|---|---|---|
| HipAB 175 | E. coli | 70 (1) | 54 | 126 |
| E. coli | 71 (2) | 43 | 84 | |
| E. coli | 72 (3) | 0.27 | 46 | |
| E. coli | 73 (4) | 35 | 116 |
2.4.ii.1 ε2ζ2/PezAT. The zeta toxin–epsilon antitoxin complex (ε2ζ2) is a TA system often found in multidrug-resistant bacteria pathogens and is also present in S. pneumoniae or Streptococcus suis as PezAT.139,176 The ζ/PezT toxin is a kinase with an ATP/GTP binding site and a core fold of phophotransferases which phosphorylates and inactivates uridine diphosphate-N-acetylglucosamine (UNAG) amino sugars, preventing their involvement in cell wall peptidoglycan biosynthesis.177 In stable cell conditions the complex is found in a heterotetrameric form, and the ζ toxin is neutralised by the ε antitoxin. In absence of the antitoxin, the toxin induces reversible proliferation of cells in a population due to inhibited peptidoglycan synthesis.178 A prolonged disruption of the complex leads to irreversible proliferation by cell wall autolysis.
The crystal structure of the S. pyogenes ε–ζ system was characterised in 2003.179 The ε2ζ2 binding interface involves the α-helix of ε binding to a groove of the ζ toxin composed of three α-helices. Lioy et al. used a cell-based HTS assay using BRET technology to identify a disruptor of the S. pyogenes ε–ζ PPI.180 They used the crystal structure to carry out molecular dynamic studies to define the main interacting residues of the PPI. They theorised that a compound mimicking the toxin residues that interacts with the antitoxin would free the toxin to trigger growth inhibition. In silico modelling of peptide profiles was used to direct the choice for an extensive collection of peptide libraries that were then used to screen for hits targeting the interaction. They fused the N-terminus of ε and C-terminus of ζ with reporter genes and tested for a decrease in BRET signal relative to controls to identify hit compounds interrupting the ε–ζ interaction. Two of 17-residue peptide sub-libraries resulted in a decrease in BRET signal which was lost upon sub-fractionation of the libraries. Therefore, the authors proposed that the disruption was a result of more than one binder, that when combined can disrupt the PPI. Their experiment validated ε2ζ2 as a potential target, proving it can be disrupted with oligopeptides. It is not clear whether ε or ζ is targeted and if the residues involved in the PPI are the same as those involved in peptide binding. It is possible that the peptides block ATP binding rather than restoring kinase activity of the toxin.
Based on the crystal structure from S. pyogenes, and validation that the TA pair can be disrupted, Fernández-Bachiller et al. rationally designed peptides to target the ε–ζ PPI.181 They initially designed three peptides 74–76 based on the three α-helices of the ζ toxin to target ε (Table 9 and Fig. 17). Due to their high propensity for aggregation, several analogues of 74–76 were designed, possessing shorter sequences and amino acid substitutions in order to improve aqueous solubility. The binding affinity of each peptide to the ε antitoxin was determined by FP. Peptide 77 (Table 9 and Fig. 17), a shorter analogue of 73 where Leu20 was replaced by a His residue, was found to have the tightest binding (Kd = 75 nM) amongst all peptides. This substantial improvement when compared to the affinity of the ε–ζ complex (Kd = 1 μM) making peptide 77 a promising starting point for the design of an antibacterial agent that can trigger ζ-toxin mediated autolysis.
| TA complex | Mimicked protein | Mimicked region | Peptide (number in reference) | Peptide sequence (residues if applicable) | K d (nM) |
|---|---|---|---|---|---|
| ε 2 ζ 2 181 | ζ | α1 | 74 (I) | TDKQFENRLNDNLEELIQ (8–25) | — |
| ζ | α2 | 75 (II) | GSGKTSLRSAIFEETQ (43–58) | — | |
| ζ | α3 | 76 (III) | INSYLGTIERYETMYADD (149–165) | — | |
| ζ | α1 | 77 (Ia) | LNDNHEELIQ | 74.5 |
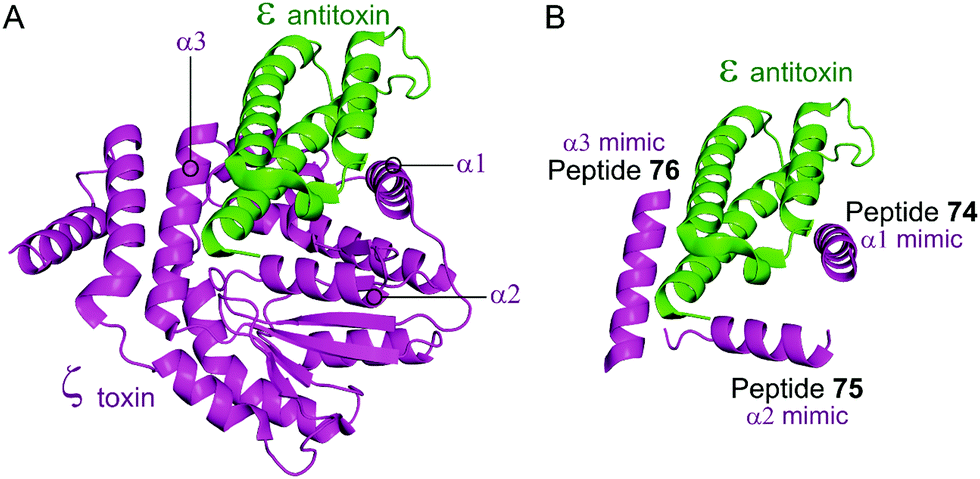 | ||
| Fig. 17 (A) Binding interface between the ε antitoxin (green) and the ζ toxin (magenta) observed in the crystal structure of the tetrameric ε2ζ2 complex (PDB: 1GVN).179 (B) Representation of the regions selected for the design of ζ-derived peptides 74 (helix α1 mimic), 75 (helix α2 mimic) and 76 (helix α3 mimic). | ||
2.4.iii.1 TplE–TplEi. TplE is a phospholipase family protein belonging to the Tle4 ligase family. Its antibacterial lipolytic activity is neutralised by the TlpEi antitoxin.182 The crystal structure of P. aeruginosa TplE–TlpEi has recently been characterised.183 Gao et al. carried out a structural approach to design a peptide mimicking the antitoxin TplEi to free the toxic TplE based on the success of the approaches described above with MoxXT and VapBC systems.147,148,165,184
Residues 82–108 of TplE were identified as the binding crucial region with TplEi composed of α1- and α2-helices linked with a disordered loop which binds to a negatively charged groove in TplEi (Fig. 18).184 The authors termed this the ‘L’ peptide (78) and, using isothermal calorimetry, identified the TplEi–78 interaction to have tighter binding than the wild type complex (Kd = 125 nM, Table 10). E. coli toxicity assays demonstrated that cell growth was inhibited in the presence of 78, with the peptide likely competitively binding to TlpEi, freeing the toxic TplE. Structural analysis identified residue K100 as forming four hydrogen bonds with TplEi and using point mutations of the interacting residues in TplEi identified all to be crucial for the tight interaction. Additionally, 78 with a K100E mutation did not inhibit E. coli cell growth as strongly as wildtype 78.
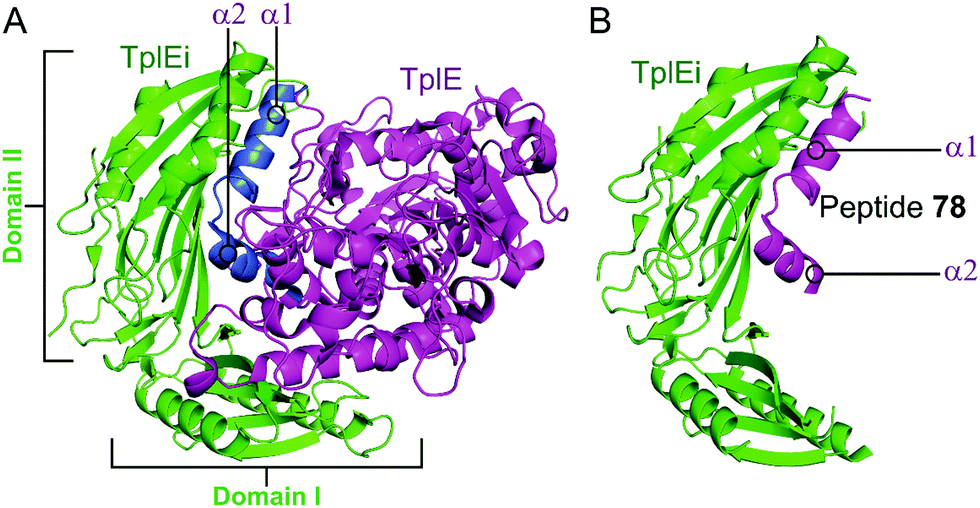 | ||
| Fig. 18 (A) Crystal structure (PDB: 4R1D)183 of P. aeruginosa TplEi (green) in complex with P. aeruginosa TplE (magenta). The antitoxin TplEi can be subdivided in two domains (I and II), with TplE helices α1 and α2 (blue) shown to interact with domain II. (B) Crystal structure (PDB: 5H7Y)184 of P. aeruginosa TplE-derived L-peptide 78 (magenta) bound to P. aeruginosa TplEi (green). | ||
4 Conclusion
From a development and regulatory point of view, antibiotics are drugs that can be easily progressed to the market. Well-established and simple animal models are available and new antibiotics do not require superior performance to existing drugs, a requirement that allows introduction of new compounds that tackle antimicrobial resistance.185 The main reason the antibiotic development pipeline has been dry for many years is a lack of drug discovery campaigns, as targeting pathogenic bacteria poses specific challenges. Drugs with novel modes of action are required to overcome all bacterial mechanisms (tolerance, persistence and resistance) leading to antibiotic treatment failure.Over the last few decades, PPIs have emerged as promising drug targets and intensive development has led to the transition of PPI modulators as next-generation therapeutics for e.g. cancer treatment into the clinic. Naturally, an abundance of PPIs are also present in bacteria and they are therefore increasingly explored as promising antibiotic targets. Bacterial PPIs, exemplified by those presented in this review, are often involved in essential cellular processes, including division and replication (SSB, β-sliding clamp, FtsZ–ZipA), transcription (RNAP, NusB–NusE) and outer membrane protein biosynthesis (BamA–BamD, Lpt). Toxin–antitoxin systems that act on translation, cell wall synthesis or lipolysis are another rich pool of interactions within bacteria that can be utilized for the development of advanced antibiotics. In addition, a number of crucial bacterial PPIs have been well characterised structurally and biophysically, but no inhibitors of these interactions have been reported so far. As PPIs relating directly to functions in bacteria are often conserved in prokaryotes and do not have close human homologues, it may be possible to develop selective drugs without strong side effects in humans and drugs with broad-spectrum activity against high-priority pathogens. Whilst traditional antibiotics that target enzymes are susceptible to resistance formation, by the occurrence of a single mutation in the drug binding pocket of the enzyme that perturbs drug binding but maintains enzyme activity, a mutation of a single hot-spot residue at a PPI interface would not only disrupt drug binding but also disturb the PPI itself and in turn affect bacterial viability. Such a mutation would most likely not persist so readily in the bacterial population, suggesting that PPI-targeting antibiotics might be less susceptible to resistance mechanisms.
Despite all the possibilities that targeting bacterial PPIs offers for the development of effective antibiotics, there are still major obstacles that must be overcome for successful transition of bacterial PPI inhibitors into the clinic. When exploring bacterial PPIs as new targets, it should be verified that their biological function is crucial enough to cause a bacteriostatic or bactericidal effect. This is an ongoing problem as the validation of an observed antibiotic effect as a direct consequence of the inhibition of the PPI in the bacterial cells is still technically challenging and this data is absent in most studies. Additionally, the binding modes of potential PPI inhibitors should be analysed in detail, i.e. if they bind competitively with a protein partner or at an allosteric site, as this might influence the probability of resistance formation. Toxin–antitoxin systems as an emerging class of bacterial PPIs for antibiotic development have their own specific challenges. Most reported approaches that target TA systems aim to liberate the toxin to induce bacterial cell death, either by competitive binding to the antitoxin or designing compounds capable of disrupting the TA PPI without inhibiting the activity of the toxin. However, the release of toxins from certain TA systems has been also suggested to induce persistence in bacteria, and an oppositional strategy has been reported that aims to inhibit the toxin to prevent bacterial persister formation. More detailed biological characterization of TA systems is therefore required to assess the consequences of targeting the toxin or antitoxin and to decide if an antibiotic or anti-persistence development strategy can be pursued.
A crucial factor when targeting bacterial PPIs is also the choice of the structural type of the inhibitor. Many PPIs possess binding pockets formed by clusters of hot-spot residues that can be targeted by small molecules.10 Inhibitory small molecule compounds have been discovered by in silico and high throughput screening, fragment-based lead discovery and rational, structure-based design and are further optimized by intensive SAR development. Small molecule inhibitors have the advantage of high biological stability, potential oral bioavailability and inexpensive synthesis, but the selectivity of small molecule inhibitors for their target proteins needs to be carefully examined. Peptides might be especially useful for bacterial PPIs in which hot-spot residues are distributed over a larger interaction interface due to their potential high affinity and selectivity.186 Guided by structural information, linear peptides are often designed as first step to probe the inhibition of a PPI, but their low biological stability necessitates further development into cyclic analogues or peptidomimetics, which may be able to compete with the bioavailability of small molecules. In addition to rational design, HTS using cyclic peptide libraries is increasingly used to identify more drug-like peptidic inhibitors of target PPIs.
One of the biggest challenges, however, that both small molecule and peptidic inhibitors face as potential antibiotics is uptake into bacteria to reach the PPI target. Penetration into Gram-positive bacteria is often achievable, as their main permeability barrier is the plasma membrane which can be penetrated by hydrophobic molecules. In contrast, uptake into Gram-negative bacteria is a major challenge. The lipopolysaccharide (LPS)-rich, anionic outer membrane prevents entry of hydrophobic compounds, while the inner phospholipid bilayer membrane hinders hydrophilic molecules, creating the need for amphipathic compounds. In addition, both membranes are rich in multidrug-resistance pumps that are highly effective in shuttling out undesired compounds and lateral therapy might be required to block these. As cytosolic targets are difficult to reach in Gram-negative bacteria, periplasmic targets or outer membrane PPI targets, such as Fts protein interactions in cell division, can be more attractive. Small molecule inhibitors have traditionally fared better than peptide-based molecules, however, modern cyclic peptides are increasingly able to rival the cell penetration of small molecules.
Overall, the multitude of PPIs specific to bacteria makes them promising targets for antibiotic development and more detailed understanding of the structural features of bacterial PPIs and their exact roles in bacterial biology is highly desired. In combination with the further development of structurally diverse drug compounds with optimized pharmacokinetic and bacterial cell uptake properties, this is expected to pave the way for antibiotics with novel modes of action, much needed in the fight against multidrug-resistant and multidrug-tolerant pathogens.
Conflicts of interest
There are no conflicts of interest to declare.References
- CDC, Antibiotic Resistance Threats in the United States, 2019, U.S. Department of Health and Human Services, CDC, Atlanta, GA, 2019.
- World Health Organization, Global action plan on antimicrobial resistance, World Health Organization, Geneva, 2015.
- C. Årdal, M. Balasegaram, R. Laxminarayan, D. McAdams, K. Outterson, J. H. Rex and N. Sumpradit, Nat. Rev. Microbiol., 2020, 18, 267–274 CrossRef.
- C. K. Shore and A. Coukell, Nat. Microbiol., 2016, 1, 16083 CrossRef CAS.
- N. Q. Balaban, S. Helaine, K. Lewis, M. Ackermann, B. Aldridge, D. I. Andersson, M. P. Brynildsen, D. Bumann, A. Camilli, J. J. Collins, C. Dehio, S. Fortune, J.-M. Ghigo, W.-D. Hardt, A. Harms, M. Heinemann, D. T. Hung, U. Jenal, B. R. Levin, J. Michiels, G. Storz, M.-W. Tan, T. Tenson, L. V. Melderen and A. Zinkernagel, Nat. Rev. Microbiol., 2019, 17, 441–448 CrossRef CAS.
- P. J. Cossar, P. J. Lewis and A. McCluskey, Med. Res. Rev., 2020, 40, 469–494 CrossRef CAS.
- A. E. Modell, S. L. Blosser and P. S. Arora, Trends Pharmacol. Sci., 2016, 37, 702–713 CrossRef CAS.
- A. A. Bogan and K. S. Thorn, J. Mol. Biol., 1998, 280, 1–9 CrossRef CAS.
- D. E. Scott, A. R. Bayly, C. Abell and J. Skidmore, Nat. Rev. Drug Discovery, 2016, 15, 533–550 CrossRef CAS.
- M. R. Arkin, Y. Tang and J. A. Wells, Chem. Biol., 2014, 21, 1102–1114 CrossRef CAS.
- C. Sheng, G. Dong, Z. Miao, W. Zhang and W. Wang, Chem. Soc. Rev., 2015, 44, 8238–8259 RSC.
- H. Bruzzoni-Giovanelli, V. Alezra, N. Wolff, C.-Z. Dong, P. Tuffery and A. Rebollo, Drug Discovery Today, 2018, 23, 272–285 CrossRef CAS.
- T. A. F. Cardote and A. Ciulli, ChemMedChem, 2016, 11, 787–794 CrossRef CAS.
- O. N. Akram, D. J. DeGraff, J. H. Sheehan, W. D. Tilley, R. J. Matusik, J.-M. Ahn and G. V. Raj, Mol. Cancer Res., 2014, 12, 967–978 CrossRef CAS.
- Z. Qian, P. G. Dougherty and D. Pei, Curr. Opin. Chem. Biol., 2017, 38, 80–86 CrossRef CAS.
- L. Carro, Beilstein J. Org. Chem., 2018, 14, 2881–2896 CrossRef CAS.
- M. A. Anderson, J. Deng, J. F. Seymour, C. Tam, S. Y. Kim, J. Fein, L. Yu, J. R. Brown, D. Westerman, E. G. Si, I. J. Majewski, D. Segal, S. L. Heitner Enschede, D. C. S. Huang, M. S. Davids, A. Letai and A. W. Roberts, Blood, 2016, 127, 3215–3224 CrossRef CAS.
- A. M. Ali, J. Atmaj, N. Van Oosterwijk, M. R. Groves and A. Dömling, Comput. Struct. Biotechnol. J., 2019, 17, 263–281 CrossRef CAS.
- F. Meric-Bernstam, M. N. Saleh, J. R. Infante, S. Goel, G. S. Falchook, G. Shapiro, K. Y. Chung, R. M. Conry, D. S. Hong, J. S.-Z. Wang, U. Steidl, L. D. Walensky, V. Guerlavais, M. Payton, D. A. Annis, M. Aivado and M. R. Patel, J. Clin. Oncol., 2017, 35, 2505 Search PubMed.
- V. G. Kramer, S. M. Schader, M. Oliveira, S. P. Colby-Germinario, D. A. Donahue, D. N. Singhroy, R. Tressler, R. D. Sloan and M. A. Wainberg, Antimicrob. Agents Chemother., 2012, 56, 4154–4160 CrossRef CAS.
- A. Gagarinova, S. Phanse, M. Cygler and M. Babu, Expert Rev. Proteomics, 2017, 14, 779–797 CrossRef CAS.
- E. van Eijk, B. Wittekoek, E. J. Kuijper and W. K. Smits, J. Antimicrob. Chemother., 2017, 72, 1275–1284 CAS.
- A. Sugino, N. P. Higgins, P. O. Brown, C. L. Peebles and N. R. Cozzarelli, Proc. Natl. Acad. Sci. U. S. A., 1978, 75, 4838–4842 CrossRef CAS.
- T. Khan, K. Sankhe, V. Suvarna, A. Sherje, K. Patel and B. Dravyakar, Biomed. Pharmacother., 2018, 103, 923–938 CrossRef CAS.
- T. D. M. Pham, Z. M. Ziora and M. A. T. Blaskovich, Med. Chem. Commun., 2019, 10, 1719–1739 RSC.
- WHO publishes list of bacteria for which new antibiotics are urgently needed, https://www.who.int/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed, accessed 5 June 2020.
- W. Vollmer, Appl. Microbiol. Biotechnol., 2006, 73, 37–47 CrossRef CAS.
- M. C. F. van Teeseling, M. A. de Pedro and F. Cava, Front. Microbiol., 2017, 8, 1264 CrossRef.
- L. Mosyak, Y. Zhang, E. Glasfeld, S. Haney, M. Stahl, J. Seehra and W. S. Somers, EMBO J., 2000, 19, 3179–3191 CrossRef CAS.
- C. H. Kenny, W. Ding, K. Kelleher, S. Benard, E. G. Dushin, A. G. Sutherland, L. Mosyak, R. Kriz and G. Ellestad, Anal. Biochem., 2003, 323, 224–233 CrossRef CAS.
- T. S. Rush, J. A. Grant, L. Mosyak and A. Nicholls, J. Med. Chem., 2005, 48, 1489–1495 CrossRef CAS.
- A. G. Sutherland, J. Alvarez, W. Ding, K. W. Foreman, C. H. Kenny, P. Labthavikul, L. Mosyak, P. J. Petersen, T. S. R. Iii, A. Ruzin, D. H. H. Tsao and K. L. Wheless, Org. Biomol. Chem., 2003, 1, 4138–4140 RSC.
- L. D. Jennings, K. W. Foreman, T. S. Rush, D. H. H. Tsao, L. Mosyak, S. L. Kincaid, M. N. Sukhdeo, A. G. Sutherland, W. Ding, C. H. Kenny, C. L. Sabus, H. Liu, E. G. Dushin, S. L. Moghazeh, P. Labthavikul, P. J. Petersen, M. Tuckman and A. V. Ruzin, Bioorg. Med. Chem., 2004, 12, 5115–5131 CrossRef CAS.
- D. H. H. Tsao, A. G. Sutherland, L. D. Jennings, Y. Li, T. S. Rush, J. C. Alvarez, W. Ding, E. G. Dushin, R. G. Dushin, S. A. Haney, C. H. Kenny, A. Karl Malakian, R. Nilakantan and L. Mosyak, Bioorg. Med. Chem., 2006, 14, 7953–7961 CrossRef CAS.
- A. H. Marceau, Methods Mol. Biol., 2012, 922, 1–21 CAS.
- A. Costes, F. Lecointe, S. McGovern, S. Quevillon-Cheruel and P. Polard, PLoS Genet., 2010, 6, e1001238 CrossRef.
- D. Lu and J. L. Keck, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9169–9174 CrossRef CAS.
- D. Lu, D. A. Bernstein, K. A. Satyshur and J. L. Keck, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 633–638 CrossRef CAS.
- D. Lu, in Single-Stranded DNA Binding Proteins: Methods and Protocols, ed. J. L. Keck, Humana Press, Totowa, NJ, 2012, ch. 10, pp. 155–159 Search PubMed.
- Z. Chilingaryan, S. J. Headey, A. T. Y. Lo, Z.-Q. Xu, G. Otting, N. E. Dixon, M. J. Scanlon and A. J. Oakley, Antibiotics, 2018, 7, 14 CrossRef.
- A. F. Voter, M. P. Killoran, G. E. Ananiev, S. A. Wildman, F. M. Hoffmann and J. L. Keck, SLAS Discovery, 2018, 23, 94–101 CAS.
- N. Y. Yao and M. O’Donnell, Curr. Biol., 2017, 27, R174–R176 CrossRef CAS.
- A. S. Altieri and Z. Kelman, Front. Mol. Biosci., 2018, 5, 87 CrossRef CAS.
- K. A. Bunting, S. M. Roe and L. H. Pearl, EMBO J., 2003, 22, 5883–5892 CrossRef CAS.
- D. Y. Burnouf, V. Olieric, J. Wagner, S. Fujii, J. Reinbolt, R. P. P. Fuchs and P. Dumas, J. Mol. Biol., 2004, 335, 1187–1197 CrossRef CAS.
- R. E. Georgescu, O. Yurieva, S.-S. Kim, J. Kuriyan, X.-P. Kong and M. O’Donnell, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11116–11121 CrossRef CAS.
- G. Wijffels, W. M. Johnson, A. J. Oakley, K. Turner, V. C. Epa, S. J. Briscoe, M. Polley, A. J. Liepa, A. Hofmann, J. Buchardt, C. Christensen, P. Prosselkov, B. P. Dalrymple, P. F. Alewood, P. A. Jennings, N. E. Dixon and D. A. Winkler, J. Med. Chem., 2011, 54, 4831–4838 CrossRef CAS.
- Z. Yin, L. R. Whittell, Y. Wang, S. Jergic, M. Liu, E. J. Harry, N. E. Dixon, J. L. Beck, M. J. Kelso and A. J. Oakley, J. Med. Chem., 2014, 57, 2799–2806 CrossRef CAS.
- Z. Yin, L. R. Whittell, Y. Wang, S. Jergic, C. Ma, P. J. Lewis, N. E. Dixon, J. L. Beck, M. J. Kelso and A. J. Oakley, J. Med. Chem., 2015, 58, 4693–4702 CrossRef CAS.
- P. Wolff, V. Oliéric, J. P. Briand, O. Chaloin, A. Dejaegere, P. Dumas, E. Ennifar, G. Guichard, J. Wagner and D. Y. Burnouf, J. Med. Chem., 2011, 54, 4627–4637 CrossRef CAS.
- C. André, I. Martiel, P. Wolff, M. Landolfo, B. Lorber, C. Silva da Veiga, A. Dejaegere, P. Dumas, G. Guichard, V. Oliéric, J. Wagner and D. Y. Burnouf, ACS Infect. Dis., 2019, 5, 1022–1034 CrossRef.
- A. Kling, P. Lukat, D. V. Almeida, A. Bauer, E. Fontaine, S. Sordello, N. Zaburannyi, J. Herrmann, S. C. Wenzel, C. König, N. C. Ammerman, M. B. Barrio, K. Borchers, F. Bordon-Pallier, M. Brönstrup, G. Courtemanche, M. Gerlitz, M. Geslin, P. Hammann, D. W. Heinz, H. Hoffmann, S. Klieber, M. Kohlmann, M. Kurz, C. Lair, H. Matter, E. Nuermberger, S. Tyagi, L. Fraisse, J. H. Grosset, S. Lagrange and R. Müller, Science, 2015, 348, 1106–1112 CrossRef CAS.
- H. Noufflard-Guy-Loé and S. Berteaux, Rev. Tuberc. Pneumol., 1965, 29, 301–326 Search PubMed.
- C. Ma, X. Yang and P. J. Lewis, Microbiol. Mol. Biol. Rev., 2016, 80, 139–160 CrossRef CAS.
- B. P. Goldstein, J. Antibiot., 2014, 67, 625–630 CrossRef CAS.
- J. Lee and S. Borukhov, Front. Mol. Biosci., 2016, 3, 73 Search PubMed.
- F. Werner and D. Grohmann, Nat. Rev. Microbiol., 2011, 9, 85–98 CrossRef CAS.
- D. G. Vassylyev, S. Sekine, O. Laptenko, J. Lee, M. N. Vassylyeva, S. Borukhov and S. Yokoyama, Nature, 2002, 417, 712–719 CrossRef CAS.
- E. B. Johnston, P. J. Lewis and R. Griffith, Protein Sci., 2009, 18, 2287–2297 CrossRef CAS.
- C. Ma, X. Yang, H. Kandemir, M. Mielczarek, E. B. Johnston, R. Griffith, N. Kumar and P. J. Lewis, ACS Chem. Biol., 2013, 8, 1972–1980 CrossRef CAS.
- S. Hinsberger, K. Hüsecken, M. Groh, M. Negri, J. Haupenthal and R. W. Hartmann, J. Med. Chem., 2013, 56, 8332–8338 CrossRef CAS.
- B. Bae, E. Davis, D. Brown, E. A. Campbell, S. Wigneshweraraj and S. A. Darst, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19772–19777 CrossRef CAS.
- M. Mielczarek, R. V. Thomas, C. Ma, H. Kandemir, X. Yang, M. Bhadbhade, D. S. Black, R. Griffith, P. J. Lewis and N. Kumar, Bioorg. Med. Chem., 2015, 23, 1763–1775 CrossRef CAS.
- C. Ma, X. Yang and P. J. Lewis, ACS Infect. Dis., 2016, 2, 39–46 CrossRef CAS.
- J. Ye, A. J. Chu, L. Lin, X. Yang and C. Ma, Molecules, 2019, 24, 2902 CrossRef CAS.
- J. Ye, A. J. Chu, L. Lin, S. T. Chan, R. Harper, M. Xiao, I. Artsimovitch, Z. Zuo, C. Ma and X. Yang, Eur. J. Med. Chem., 2020, 208, 112671 CrossRef CAS.
- J. Ye, A. J. Chu, R. Harper, S. T. Chan, T. L. Shek, Y. Zhang, M. Ip, M. Sambir, I. Artsimovitch, Z. Zuo, X. Yang and C. Ma, J. Med. Chem., 2020, 63, 7695–7720 CrossRef CAS.
- S. Sartini, E. Levati, M. Maccesi, M. Guerra, G. Spadoni, S. Bach, M. Benincasa, M. Scocchi, S. Ottonello, S. Rivara and B. Montanini, ACS Chem. Biol., 2019, 14, 1727–1736 CrossRef CAS.
- K. Hüsecken, M. Negri, M. Fruth, S. Boettcher, R. W. Hartmann and J. Haupenthal, ACS Chem. Biol., 2013, 8, 758–766 CrossRef.
- A. A. M. Kamal, M. Habib, J. Haupenthal, R. W. Hartmann and M. Empting, Biol. Chem., 2019, 400, 333–342 CAS.
- K. B. Arnvig, S. Zeng, S. Quan, A. Papageorge, N. Zhang, A. C. Villapakkam and C. L. Squires, J. Bacteriol., 2008, 190, 7251–7257 CrossRef CAS.
- R. Sen, J. Chalissery and G. Muteeb, EcoSal Plus, 2018 DOI:10.1128/ecosalplus.4.5.3.1.
- M. Bubunenko, D. L. Court, A. A. Refaii, S. Saxena, A. Korepanov, D. I. Friedman, M. E. Gottesman and J.-H. Alix, Mol. Microbiol., 2013, 87, 382–393 CrossRef CAS.
- N. Singh, M. Bubunenko, C. Smith, D. M. Abbott, A. M. Stringer, R. Shi, D. L. Court and J. T. Wade, mBio, 2016, 7, e00114 CAS.
- S. J. Greive, A. F. Lins and P. H. V. Hippel, J. Biol. Chem., 2005, 280, 36397–36408 CrossRef CAS.
- R. Das, S. Loss, J. Li, D. S. Waugh, S. Tarasov, P. Wingfield, R. A. Byrd and A. S. Altieri, J. Mol. Biol., 2008, 376, 705–720 CrossRef CAS.
- X. Luo, H.-H. Hsiao, M. Bubunenko, G. Weber, D. L. Court, M. E. Gottesman, H. Urlaub and M. C. Wahl, Mol. Cell, 2008, 32, 791–802 CrossRef CAS.
- J. R. Stagno, A. S. Altieri, M. Bubunenko, S. G. Tarasov, J. Li, D. L. Court, R. A. Byrd and X. Ji, Nucleic Acids Res., 2011, 39, 7803–7815 CrossRef CAS.
- P. J. Cossar, C. Ma, C. P. Gordon, J. I. Ambrus, P. J. Lewis and A. McCluskey, Bioorg. Med. Chem. Lett., 2017, 27, 162–167 CrossRef CAS.
- P. J. Cossar, M. K. Abdel-Hamid, C. Ma, J. A. Sakoff, T. N. Trinh, C. P. Gordon, P. J. Lewis and A. McCluskey, ACS Omega, 2017, 2, 3839–3857 CrossRef CAS.
- X. Yang, M. J. Luo, A. C. M. Yeung, P. J. Lewis, P. K. S. Chan, M. Ip and C. Ma, Biochemistry, 2017, 56, 5049–5052 CrossRef CAS.
- Y. Qiu, S. T. Chan, L. Lin, T. L. Shek, T. F. Tsang, N. Barua, Y. Zhang, M. Ip, P. K. Chan, N. Blanchard, G. Hanquet, Z. Zuo, X. Yang and C. Ma, Eur. J. Med. Chem., 2019, 178, 214–231 CrossRef CAS.
- Y. Qiu, S. T. Chan, L. Lin, T. L. Shek, T. F. Tsang, Y. Zhang, M. Ip, P. K. Chan, N. Blanchard, G. Hanquet, Z. Zuo, X. Yang and C. Ma, Bioorg. Chem., 2019, 92, 103203 CrossRef CAS.
- A. J. Chu, Y. Qiu, R. Harper, L. Lin, C. Ma and X. Yang, Int. J. Mol. Sci., 2020, 21, 5772 CrossRef CAS.
- H. Nikaido, Microbiol. Mol. Biol. Rev., 2003, 67, 593 CrossRef CAS.
- H. I. Zgurskaya and V. V. Rybenkov, Ann. N. Y. Acad. Sci., 2020, 1459, 5–18 CrossRef.
- T. J. Silhavy, D. Kahne and S. Walker, Cold Spring Harbor Perspect. Biol., 2010, 2, a000414 Search PubMed.
- K. M. Lehman and M. Grabowicz, Antibiotics, 2019, 8, 163 CrossRef CAS.
- J. A. Robinson, Front. Chem., 2019, 7, 45 CrossRef CAS.
- J. Bakelar, S. K. Buchanan and N. Noinaj, Science, 2016, 351, 180–186 CrossRef CAS.
- Y. Gu, H. Li, H. Dong, Y. Zeng, Z. Zhang, N. G. Paterson, P. J. Stansfeld, Z. Wang, Y. Zhang, W. Wang and C. Dong, Nature, 2016, 531, 64–69 CrossRef CAS.
- L. Han, J. Zheng, Y. Wang, X. Yang, Y. Liu, C. Sun, B. Cao, H. Zhou, D. Ni, J. Lou, Y. Zhao and Y. Huang, Nat. Struct. Mol. Biol., 2016, 23, 192–196 CrossRef CAS.
- N. Noinaj, A. J. Kuszak, J. C. Gumbart, P. Lukacik, H. Chang, N. C. Easley, T. Lithgow and S. K. Buchanan, Nature, 2013, 501, 385–390 CrossRef CAS.
- H. T. Bergal, A. H. Hopkins, S. I. Metzner and M. C. Sousa, Structure, 2016, 24, 243–251 CrossRef CAS.
- T. Wu, J. Malinverni, N. Ruiz, S. Kim, T. J. Silhavy and D. Kahne, Cell, 2005, 121, 235–245 CrossRef CAS.
- D. P. Ricci, C. L. Hagan, D. Kahne and T. J. Silhavy, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 3487 CrossRef CAS.
- K. M. Storek, M. R. Auerbach, H. Shi, N. K. Garcia, D. Sun, N. N. Nickerson, R. Vij, Z. Lin, N. Chiang, K. Schneider, A. T. Wecksler, E. Skippington, G. Nakamura, D. Seshasayee, J. T. Koerber, J. Payandeh, P. A. Smith and S. T. Rutherford, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 3692 CrossRef CAS.
- Y. Imai, K. J. Meyer, A. Iinishi, Q. Favre-Godal, R. Green, S. Manuse, M. Caboni, M. Mori, S. Niles, M. Ghiglieri, C. Honrao, X. Ma, J. J. Guo, A. Makriyannis, L. Linares-Otoya, N. Böhringer, Z. G. Wuisan, H. Kaur, R. Wu, A. Mateus, A. Typas, M. M. Savitski, J. L. Espinoza, A. O’Rourke, K. E. Nelson, S. Hiller, N. Noinaj, T. F. Schäberle, A. D’Onofrio and K. Lewis, Nature, 2019, 576, 459–464 CrossRef CAS.
- E. M. Hart, A. M. Mitchell, A. Konovalova, M. Grabowicz, J. Sheng, X. Han, F. P. Rodriguez-Rivera, A. G. Schwaid, J. C. Malinverni, C. J. Balibar, S. Bodea, Q. Si, H. Wang, M. F. Homsher, R. E. Painter, A. K. Ogawa, H. Sutterlin, T. Roemer, T. A. Black, D. M. Rothman, S. S. Walker and T. J. Silhavy, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 21748 CrossRef CAS.
- M. Urfer, J. Bogdanovic, F. Lo Monte, K. Moehle, K. Zerbe, U. Omasits, C. H. Ahrens, G. Pessi, L. Eberl and J. A. Robinson, J. Biol. Chem., 2016, 291, 1921–1932 CrossRef CAS.
- C. L. Hagan, J. S. Wzorek and D. Kahne, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2011 CrossRef CAS.
- Y. Li, X. Zhu, J. Zhang, Y. Lin, X. You, M. Chen, Y. Wang, N. Zhu and S. Si, Front. Microbiol., 2020, 11, 1252 CrossRef.
- N. Mori, Y. Ishii, K. Tateda, S. Kimura, Y. Kouyama, H. Inoko, S. Mitsunaga, K. Yamaguchi and E. Yoshihara, J. Antimicrob. Chemother., 2012, 67, 2173–2181 CrossRef CAS.
- C. L. Hagan, D. B. Westwood and D. Kahne, Biochemistry, 2013, 52, 6108–6113 CrossRef CAS.
- E. Freinkman, S. Okuda, N. Ruiz and D. Kahne, Biochemistry, 2012, 51, 4800–4806 CrossRef CAS.
- D. J. Sherman, R. Xie, R. J. Taylor, A. H. George, S. Okuda, P. J. Foster, D. J. Needleman and D. Kahne, Science, 2018, 359, 798 CrossRef CAS.
- M. D. L. Suits, P. Sperandeo, G. Dehò, A. Polissi and Z. Jia, J. Mol. Biol., 2008, 380, 476–488 CrossRef CAS.
- A. X. Tran, C. Dong and C. Whitfield, J. Biol. Chem., 2010, 285, 33529–33539 CrossRef CAS.
- I. Botos, N. Majdalani, S. J. Mayclin, J. G. McCarthy, K. Lundquist, D. Wojtowicz, T. J. Barnard, J. C. Gumbart and S. K. Buchanan, Structure, 2016, 24, 965–976 CrossRef CAS.
- S. Qiao, Q. Luo, Y. Zhao, X. C. Zhang and Y. Huang, Nature, 2014, 511, 108–111 CrossRef CAS.
- H. Dong, Q. Xiang, Y. Gu, Z. Wang, N. G. Paterson, P. J. Stansfeld, C. He, Y. Zhang, W. Wang and C. Dong, Nature, 2014, 511, 52–56 CrossRef CAS.
- M. Bollati, R. Villa, L. J. Gourlay, M. Benedet, G. Dehò, A. Polissi, A. Barbiroli, A. M. Martorana, P. Sperandeo, M. Bolognesi and M. Nardini, FEBS J., 2015, 282, 1980–1997 CrossRef CAS.
- N. Ruiz, L. S. Gronenberg, D. Kahne and T. J. Silhavy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5537 CrossRef CAS.
- P. Sperandeo, F. K. Lau, A. Carpentieri, C. De Castro, A. Molinaro, G. Dehò, T. J. Silhavy and A. Polissi, J. Bacteriol., 2008, 190, 4460 CrossRef CAS.
- S. Okuda, E. Freinkman and D. Kahne, Science, 2012, 338, 1214 CrossRef CAS.
- M. P. Bos, B. Tefsen, J. Geurtsen and J. Tommassen, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9417 CrossRef CAS.
- T. Wu, A. C. McCandlish, L. S. Gronenberg, S.-S. Chng, T. J. Silhavy and D. Kahne, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11754 CrossRef CAS.
- C. Laguri, P. Sperandeo, K. Pounot, I. Ayala, A. Silipo, C. M. Bougault, A. Molinaro, A. Polissi and J.-P. Simorre, Sci. Rep., 2017, 7, 9715 CrossRef.
- P. Fehlbaum, P. Bulet, S. Chernysh, J. P. Briand, J. P. Roussel, L. Letellier, C. Hetru and J. A. Hoffmann, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 1221 CrossRef CAS.
- S. U. Vetterli, K. Zerbe, M. Müller, M. Urfer, M. Mondal, S.-Y. Wang, K. Moehle, O. Zerbe, A. Vitale, G. Pessi, L. Eberl, B. Wollscheid and J. A. Robinson, Sci. Adv., 2018, 4, eaau2634 CrossRef CAS.
- E. C. C. M. Moura, T. Baeta, A. Romanelli, C. Laguri, A. M. Martorana, E. Erba, J.-P. Simorre, P. Sperandeo and A. Polissi, Front. Microbiol., 2020, 11, 909 CrossRef.
- X. Zhang, Y. Li, W. Wang, J. Zhang, Y. Lin, B. Hong, X. You, D. Song, Y. Wang, J. Jiang and S. Si, Int. J. Antimicrob. Agents, 2019, 53, 442–448 CrossRef CAS.
- J. A. Robinson, S. C. Shankaramma, P. Jetter, U. Kienzl, R. A. Schwendener, J. W. Vrijbloed and D. Obrecht, Bioorg. Med. Chem., 2005, 13, 2055–2064 CrossRef CAS.
- N. Srinivas, P. Jetter, B. J. Ueberbacher, M. Werneburg, K. Zerbe, J. Steinmann, B. Van der Meijden, F. Bernardini, A. Lederer, R. L. A. Dias, P. E. Misson, H. Henze, J. Zumbrunn, F. O. Gombert, D. Obrecht, P. Hunziker, S. Schauer, U. Ziegler, A. Käch, L. Eberl, K. Riedel, S. J. DeMarco and J. A. Robinson, Science, 2010, 327, 1010 CrossRef CAS.
- H. S. Sader, R. K. Flamm, G. E. Dale, P. R. Rhomberg and M. Castanheira, J. Antimicrob. Chemother., 2018, 73, 2400–2404 CrossRef CAS.
- M. Werneburg, K. Zerbe, M. Juhas, L. Bigler, U. Stalder, A. Kaech, U. Ziegler, D. Obrecht, L. Eberl and J. A. Robinson, ChemBioChem, 2012, 13, 1767–1775 CrossRef CAS.
- S. U. Vetterli, K. Moehle and J. A. Robinson, Bioorg. Med. Chem., 2016, 24, 6332–6339 CrossRef CAS.
- G. Andolina, L.-C. Bencze, K. Zerbe, M. Müller, J. Steinmann, H. Kocherla, M. Mondal, J. Sobek, K. Moehle, G. Malojčić, B. Wollscheid and J. A. Robinson, ACS Chem. Biol., 2018, 13, 666–675 CrossRef CAS.
- S. Ronneau and S. Helaine, J. Mol. Biol., 2019, 431, 3462–3471 CrossRef CAS.
- D. Lobato-Márquez, R. Díaz-Orejas and F. G. Portillo, FEMS Microbiol. Rev., 2016, 40, 592–609 CrossRef.
- K. Y. Lee and B. J. Lee, Toxins, 2016, 8, 305 CrossRef.
- A. Harms, D. E. Brodersen, N. Mitarai and K. Gerdes, Mol. Cell, 2018, 70, 768–784 CrossRef CAS.
- S. M. Kang, D. H. Kim, C. Jin and B. J. Lee, Toxins, 2018, 10, 515 CrossRef CAS.
- Y. Wen, E. Behiels and B. Devreese, Pathog. Dis., 2014, 70, 240–249 CrossRef CAS.
- N. P. Coussens and D. A. Daines, Exp. Biol. Med., 2016, 241, 1332–1342 CrossRef CAS.
- S. J. Unterholzner, B. Poppenberger and W. Rozhon, Mobile Genet. Elem., 2013, 3, e26219 CrossRef.
- S. Shapiro, J. Antibiot., 2013, 66, 371–386 CrossRef CAS.
- J. J. Williams and P. J. Hergenrother, Trends Microbiol., 2012, 20, 291–298 CrossRef CAS.
- H. Mutschler and A. Meinhart, J. Mol. Med., 2011, 89, 1183–1194 CrossRef CAS.
- W. T. Chan, D. Balsa and M. Espinosa, FEMS Microbiol. Rev., 2015, 39, 522–540 CrossRef.
- Y. Tanouchi, A. J. Lee, H. Meredith and L. You, Trends Microbiol., 2013, 21, 265–270 CrossRef CAS.
- F. Hayes and B. Kędzierska, Toxins, 2014, 6, 337–358 CrossRef CAS.
- P. Paul, B. R. Sahu and M. Suar, Mol. Oral Microbiol., 2019, 34, 97–107 CrossRef CAS.
- D. Jurėnas and L. V. Melderen, Front. Genet., 2020, 11, 262 CrossRef.
- H. R. Ramage, L. E. Connolly and J. S. Cox, PLoS Genet., 2009, 5, e1000767 CrossRef.
- A. Khusro, C. Aarti, A. Barbabosa-Pliego and A. Z. M. Salem, Microb. Pathog., 2018, 114, 80–89 CrossRef CAS.
- I.-G. Lee, S. J. Lee, S. Chae, K.-Y. Lee, J.-H. Kim and B.-J. Lee, Nucleic Acids Res., 2015, 43, 7624–7637 CrossRef CAS.
- S.-M. Kang, D.-H. Kim, K.-Y. Lee, S. J. Park, H.-J. Yoon, S. J. Lee, H. Im and B.-J. Lee, Nucleic Acids Res., 2017, 45, 8564–8580 CrossRef CAS.
- A. Deep, P. Tiwari, S. Agarwal, S. Kaundal, S. Kidwai, R. Singh and K. G. Thakur, Nucleic Acids Res., 2018, 46, 11639–11655 CrossRef CAS.
- S. Sundar, M. P. Rajan and S. Piramanayagam, Int. J. Pept. Res. Ther., 2019, 25, 1467–1475 CrossRef CAS.
- K. S. Makarova, N. V. Grishin and E. V. Koonin, Bioinformatics, 2006, 22, 2581–2584 CrossRef CAS.
- D. H. Kim, S. M. Kang, S. J. Park, C. Jin, H. J. Yoon and B. J. Lee, Nucleic Acids Res., 2018, 46, 6371–6386 CrossRef CAS.
- M. G. Jørgensen, D. P. Pandey, M. Jaskolska and K. Gerdes, J. Bacteriol., 2009, 191, 1191–1199 CrossRef.
- N. Chopra, Saumitra, A. Pathak, R. Bhatnagar and S. Bhatnagar, Genome Biol. Evol., 2013, 5, 2268–2284 CrossRef.
- L. Zhu, Y. Zhang, J. S. Teh, J. Zhang, N. Connell, H. Rubin and M. Inouye, J. Biol. Chem., 2006, 281, 18638–18643 CrossRef CAS.
- G. M. Cook, J. R. Robson, R. A. Frampton, J. McKenzie, R. Przybilski, P. C. Fineran and V. L. Arcus, Biochim. Biophys. Acta, Gene Regul. Mech., 2013, 1829, 523–531 CrossRef CAS.
- H. Engelberg-Kulka, B. Sat, M. Reches, S. Amitai and R. Hazan, Trends Microbiol., 2004, 12, 66–71 CrossRef CAS.
- I. Kolodkin-Gal, R. Hazan, A. Gaathon, S. Carmeli and H. Engelberg-Kulka, Science, 2007, 318, 652–655 CrossRef CAS.
- M. Belitsky, H. Avshalom, A. Erental, I. Yelin, S. Kumar, N. London, M. Sperber, O. Schueler-Furman and H. Engelberg-Kulka, Mol. Cell, 2011, 41, 625–635 CrossRef CAS.
- K. Kamada, F. Hanaoka and S. K. Burley, Mol. Cell, 2003, 11, 875–884 CrossRef CAS.
- D.-H. Ahn, K.-Y. Lee, S. J. Lee, S. J. Park, H.-J. Yoon, S.-J. Kim and B.-J. Lee, J. Biol. Chem., 2017, 292, 18832–18847 CrossRef CAS.
- S. Kumar, I. Kolodkin-Gal and H. Engelberg-Kulka, mBio, 2013, 4, 314 CrossRef.
- S. Kumar and H. Engelberg-Kulka, Curr. Opin. Microbiol., 2014, 21, 22–27 CrossRef CAS.
- A. Nigam, S. Kumar and H. Engelberg-Kulka, mBio, 2018, 9, 685 CrossRef.
- S. Verma, S. Kumar, V. P. Gupta, S. Gourinath, S. Bhatnagar and R. Bhatnagar, J. Biomol. Struct. Dyn., 2015, 33, 606–624 CrossRef CAS.
- S. Agarwal, S. Agarwal and R. Bhatnagar, FEBS Lett., 2007, 581, 1727–1734 CrossRef CAS.
- S. Agarwal, N. K. Mishra, S. Bhatnagar and R. Bhatnagar, J. Biol. Chem., 2010, 285, 7254–7270 CrossRef CAS.
- N. Chopra, S. Agarwal, S. Verma, S. Bhatnagar and R. Bhatnagar, J. Comput.-Aided Mol. Des., 2011, 25, 275–291 CrossRef CAS.
- R. Mohammadzadeh, A. Shivaee, E. Ohadi and B. S. Kalani, Int. J. Pept. Res. Ther., 2020, 26, 613–623 CrossRef CAS.
- T. Farhadi, S. M. Hashemian and Z. Farhadi, Int. J. Pept. Res. Ther., 2020, 26, 1061–1071 CrossRef CAS.
- H. S. Moyed and K. P. Bertrand, J. Bacteriol., 1983, 155, 768–775 CrossRef CAS.
- M. A. Schumacher, K. M. Piro, W. Xu, S. Hansen, K. Lewis and R. G. Brennan, Science, 2009, 323, 396–401 CrossRef CAS.
- E. Germain, D. Castro-Roa, N. Zenkin and K. Gerdes, Mol. Cell, 2013, 52, 248–254 CrossRef CAS.
- I. Kaspy, E. Rotem, N. Weiss, I. Ronin, N. Q. Balaban and G. Glaser, Nat. Commun., 2013, 4, 3001 CrossRef.
- T. Li, N. Yin, H. Liu, J. Pei and L. Lai, ACS Med. Chem. Lett., 2016, 7, 449–453 CrossRef CAS.
- H. Mutschler, J. Reinstein and A. Meinhart, J. Biol. Chem., 2010, 285, 21797–21806 CrossRef CAS.
- H. Mutschler, M. Gebhardt, R. L. Shoeman and A. Meinhart, PLoS Biol., 2011, 9, e1001033 CrossRef CAS.
- M. M. Álamo, M. Tabone, V. S. Lioy and J. C. Alonso, Front. Microbiol., 2017, 8, 1130 CrossRef.
- A. Meinhart, J. C. Alonso, N. Sträter and W. Saenger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1661–1666 CrossRef CAS.
- V. S. Lioy, O. Rey, D. Balsa, T. Pellicer and J. C. Alonso, Plasmid, 2010, 63, 31–39 CrossRef CAS.
- M. I. Fernández-Bachiller, I. Brzozowska, N. Odolczyk, U. Zielenkiewicz, P. Zielenkiewicz and J. Rademann, Toxins, 2016, 8, 222 CrossRef.
- F. Jiang, X. Wang, B. Wang, L. Chen, Z. Zhao, N. R. Waterfield, G. Yang and Q. Jin, Cell Rep., 2016, 16, 1502–1509 CrossRef CAS.
- D. Lu, Y. Zheng, N. Liao, L. Wei, B. Xu, X. Liu and J. Liu, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 3233–3243 CrossRef CAS.
- X. Gao, Z. Mu, B. Qin, Y. Sun and S. Cui, Front. Cell. Infect. Microbiol., 2017, 7, 411 CrossRef.
- K. Lewis, Cell, 2020, 181, 29–45 CrossRef CAS.
- L. Nevola and E. Giralt, Chem. Commun., 2015, 51, 3302–3315 RSC.
| This journal is © The Royal Society of Chemistry 2021 |