
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFinding and characterizing a catalytic antibody light chain, H34, capable of degrading the PD-1 molecule†
Emi
Hifumi
 *a,
Hiroaki
Taguchi
b,
Tamami
Nonaka
a,
Takunori
Harada
c and
Taizo
Uda
d
*a,
Hiroaki
Taguchi
b,
Tamami
Nonaka
a,
Takunori
Harada
c and
Taizo
Uda
d
aOita University, Research Promotion Institute, 700 Dannoharu, Oita-shi, Oita 870-1192, Japan. E-mail: e-hifumi@oita-u.ac.jp
bSuzuka University of Medical Science, Faculty of Pharmaceutical Sciences, 3500-3 Minamitamagaki-cho, Suzuka 510-0293, Japan
cOita University, Faculty of Science & Technology, Division of Applied Chemistry, 700 Dannoharu, Oita-shi, Oita 870-1192, Japan
dNanotechnology Laboratory, Institute of Systems, Information Technologies and Nanotechnologies (ISIT), 4-1 Kyudai-shinmachi, Fukuoka 879-5593, Japan
First published on 10th December 2020
Abstract
Programmed cell death 1 (PD-1) is an immune checkpoint molecule regulating T-cell function. Preventing PD-1 binding to its ligand PD-L1 has emerged as an important tool in immunotherapy. Here, we describe a unique human catalytic antibody light chain, H34, which mediates enzymatic degradation of human PD-1 peptides and recombinant human PD-1 protein and thus functions to prevent the binding of PD-1 with PD-L1. H34 degraded one half of the PD-1 molecules within about 6 h under the experimental conditions. Investigating the acquisition of the catalytic function by H34, which belongs to subgroup I and lacks a Pro95 residue in CDR-3, revealed the importance of this sequence, as a Pro95-reconstituted mutant (H34-Pro95(+)) exhibited very little catalytic activity to cleave PD-1. Interestingly, EDTA inhibited the catalytic activity of H34, which could work as a metallo-protease. Zn2+ or Co2+ ions may work as a cofactor. It is meaningfull that H34 was obtained from the human antibody gene taken from a healthy volunteer, suggesting that we potentially have such unique molecules in our body.
Introduction
The immune checkpoint molecule programmed cell death 1 (PD-1) is attracting much attention globally following the report by Honjo et al. that it can control T-cell function.1 Monoclonal antibodies against PD-1 (anti-PD-1 mAb) act as anti-cancer drugs by inhibiting the binding of PD-L1 with PD-1.2–4 However, clinical responses to the drug are only seen in 20–30% of cancer patients, but all may suffer toxic side effects. Deficiency of the PD-1 gene greatly enhances T-cell immune-function,5,6 further suggesting that the control of the interaction between PD-1 and PD-L1 would be important for improving the treatment efficacy and reducing the toxic side effects of anti-PD-1 mAb. It would be desirable to develop novel ways of blocking interactions between PD-1 and PD-L1, for example by destroying the PD-1 molecule itself using a new type of protein degrader.7 Therefore, we have started to develop a molecule capable of degrading PD-1 and inhibiting its binding to PD-L1.Research on catalytic antibodies has revealed that many naturally catalytic antibodies hydrolyze their target peptides,8–11 nucleotides,12–14 and also some physiologically active molecules,15–18 in addition to some viral and bacterial antigens.19–28 Many studies have been conducted since 1989, when the first naturally-occurring catalytic antibody was found to cleave vasoactive intestinal peptide.8 The mechanism by which such catalytic antibodies cleave their targets is mostly thought to be similar to serine proteases, according to studies on site-directed mutagenesis,29,30 X-ray crystallography,31–33 and other approaches. However, no catalytic antibodies active against PD-1 have been reported thus far. In order to generate such desirable catalytic antibodies, over the last 10 years, we have been working on protein banks now containing hundreds of highly purified human antibody light chains. By employing high-throughput screening methods, we have now identified a unique catalytic antibody capable of directly degrading the PD-1 molecule. Here, we describe this antibody, H34, in detail and elucidate the importance of the Pro95 residue for its functional properties.
Experimental
Reagents
Chemical reagents such as Tris, glycine, CuCl2·2H2O, FeCl3·6H2O, Co(NO3)2·6H2O, ZnSO4·7H2O, KCl, Na2HPO4·12H2O, NaCl, KH2PO4, EDTA·2Na and IPTG were purchased from Wako Pure Chemical Industries Ltd, Osaka, Japan (Guaranteed Reagent). CaCl2·2H2O was purchased from Nacalai Tesque (Kyoto, Japan). The synthetic substrate peptidyl-pNA, Arg-pNA, was purchased from Peptides Institute Inc., Osaka, Japan. Tryptone and yeast extract were purchased from Becton-Dickinson and Company, NJ, USA. A commercially available recombinant PD-1 molecule was used (ENZO Life Sciences Inc., product number ENZ-PRT190; PD-1 (aa 25–167) containing a 5′-His-tag, V5 epitope tag spacer, and FLAG-tag; a doublet at 40 kDa and 50 kDa: Farmingdale, NY). In our SDS-PAGE analysis, the doublet was observed at smaller positions (∼36 kDa and ∼42 kDa) than those stated in the description manual. This might be caused by the reducing conditions we employed.Synthesis of the FRET substrates
The syntheses of all FRET peptides were carried out on a solid support using the Fmoc/tBu strategy on the Rink amide resin as previously reported.34,35 Briefly, removal of the Fmoc group was carried out with 20% piperidine in dimethylformamide, whereas chain elongation was achieved with standard HBTU/HOBt chemistry using 3 equivalents of protected amino acids or 7-MCA.34 After completing the synthesis, the protected peptide resin was treated with a TFA/phenol/H2O/thioanisole/1,2-ethanedithiol (82.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:5:2.5, v/v/v/v) mixture. The crude material obtained was purified by HPLC. The structures of the FRET peptides were confirmed by MS.
:5:5:2.5, v/v/v/v) mixture. The crude material obtained was purified by HPLC. The structures of the FRET peptides were confirmed by MS.
Sequencing and molecular modelling
The H34 clone was sequenced with an ABI 3730xl analyser (Applied Biosystems, CA, USA) by using ABI BigDye™ Terminator v3.1 cycle sequencing kits. GENETIX Ver. 8 (GENETIX, Tokyo, Japan) software was used for sequence analysis and deduction of amino acid sequences.Computational analysis of the antibody structures was performed using the deduced antibody light chain amino acid sequences using Discovery Studio (Accelrys Software, San Diego, CA, USA). For the homology modelling, the template structures were made by a BLAST search, following the minimization of the total energy of the molecule by using the CHARMM algorithm. The resulting Protein Data Bank (PDB) data were used for modification of the CDR (complementarity-determining region) structures defined by the Kabat numbering system.
Culture, recovery and purification
The transformant was grown at 18 °C in 1 liter of Luria–Bertani medium containing 100 mg mL−1 ampicillin to an A600 nm of 0.6 and then incubated with 0.01 mM IPTG for 20 h at 18 °C. Cells were harvested by centrifugation (3500g, 4 °C, 10 min) and then re-suspended in a 100 mL solution of 250 mM NaCl and 25 mM Tris–HCl, pH 8.0. The cells were lysed by ultra-sonication three times for 2 min each (×3) in an ice bath, followed by centrifugation (21500g, 4 °C, 20 min). The expressed human light chain was recovered as the supernatant.
The supernatant was first subjected to Ni-NTA column chromatography (Takara, Otsu, Japan) equilibrated with 25 mM Tris–HCl, pH 8.0, containing 250 mM NaCl. Elution was performed by increasing the concentration of imidazole from 0 and/or 30 to 300 mM.
After the Ni-NTA column chromatography was completed, an aliquot of a solution of 100 mM CuCl2 (1.25 eq. for the light chain) was added into the eluent, based on the calculation that an absorbance of A600 nm of 1.0 in UV/vis was regarded as ∼1 mg mL−1 (40 mM light chain). Then, the solution including the light chain and copper ions was dialyzed against a 50 mM Na acetate buffer, pH 5.5, for 22.4 h. After removing some aggregates by centrifugation (21500g, 4 °C, 20 min), the solution was subjected to cation-exchange chromatography using a column of SP-5PW (TOSOH, Japan) with a gradient of NaCl (from 100 to 500 mM) in a 50 mM Na acetate buffer, pH 5.5, on purification apparatus (AKTA system, GE Healthcare-Japan, Tokyo). Then, the eluent was dialyzed against 20 mM Tris–HCl/150 mM NaCl buffer (pH 8.5) for 16.9 h, followed by concentrating the solution using an Amicon Ultra 10000 (Millipore, USA). EDTA was put into the solution to be 50 mM and allowed to react for 1 h under the condition of 4 °C. Then, the solution was dialyzed against 3 L of PBS (pH 7.4) twice. Finally, it was filtered using a 0.2 μm membrane filter (Merck-Millipore) and stored at 4 °C or frozen.
Protein concentrations were determined by the Lowry method using the DC protein assay kit (Bio-Rad).
For each gene of the human antibody light chain cloned, the above procedure (expression and purification) was performed for these ten years. As a result, the number of purified light chains reached hundreds, which are stored as a protein bank in our laboratory (see Fig. S6, ESI†).
Cleavage assays
The reaction mixtures were fractionated by RP-HPLC (Cosmosil type: 5C18-AR-2 (4.6 × 250 mm); MilliQ water in 0.05% TFA:acetonitrile in 0.05% TFA from 90:10 to 30:70 in 60 min (1.0 mL min−1)). The peaks fractionated were analyzed by ESI-MS.
Kinetics
The concentration of the H34 light chain was fixed at 5 μM and that of the FRET-PD1 substrate was varied from 2.5 to 30 μM at 37 °C in the TGT buffer (pH 7.7). The concentration change of the FRET-PD1 substrate within 10% conversion after mixing the H34 light chain and the substrate was regarded as the initial rate of the reaction.(1) Preparation of samples for ELISA. Prior to the ELISA experiment, the following samples rPD-1, H34 and their mixture (55 μL each) were prepared using TGT buffer (pH 7.7). They were incubated at 37 °C for 48 h.
Sample A: the final concentration of rPD-1 was prepared as 0.25 μM in TGT buffer using an RNase free PCR tube (0.2 mL tube, Molecular BioProducts Ltd, San Diego, CA).
Sample B: the final concentrations of PD-1 and H34 were 0.25 μM and 0.5 μM, respectively, in TGT buffer using an RNase free PCR tube.
Sample C: the final concentration of H34 was prepared as 0.5 μM in TGT buffer using an RNase free PCR tube.
The concentration is the final concentration.
(2) ELISA. Fifty microliters of recombinant rPD-L1 (human, recombinant, ENZ-PRT223-0100, ENZO Life Sciences, Inc., Farmingdale, NY) in PBS solution (2 μg mL−1) was fixed on an immunoplate (Nunc, Denmark) at 4 °C overnight. Blocking was performed using 10 μg mL−1 human serum albumin (Sigma-Aldrich, Saint Louis, MO) for 30 min at room temperature. Fifty microliters of each of the above samples left at 37 °C for 48 h incubation was immuno-reacted with PD-L1, followed by a reaction with an HRP-conjugated anti-V5 tag epitope antibody of 1/2000 dilution (Rabbit; Abcam, ab1325, Cambridge, UK). After the substrate reaction using 0.01% H2O2 and 1.3 mg mL−1 of o-phenylenediamine/2HCl (WAKO, biochemical grade, Osaka) dissolved in 0.1 M citric acid/0.2 M phosphate buffer/pH 5.0, the absorption band at 492 nm (reference: 620 nm) was measured using an immunoplate reader (Scanlt 3.1 for Multiskan FC, Thermo Fisher Scientific, MA, USA).
CD spectra
The sample (1.54 for H34 or 1.55 mg mL−1 for H34-Pro95(+)) was dissolved in PBS (pH = 7.4) and its CD spectra were recorded by using a computer-controlled Jasco J-1500 CD spectropolarimeter with a cylindrical cell having an optical path of 0.1 cm for far UV/CD, a resolution of 0.2 nm and accumulation of 16. For near UV/CD, conditions such as an optical path of 1.0 cm, resolution of 0.2 nm and accumulation of 25 were used.Results
(1) FRET-PD1, FRET-Aβ and FRET-Tau substrates
For the high-throughput screening of the human PD-1 molecule and the examination of cross-reactivity, four types of FRET-peptide substrates were synthesized. The structure of a human FRET-PD-1 peptide (octadecapeptide 123GAISLAPKAQIKESLRAE140; this sequence was chosen as it is the epitope of nivolumab (anti PD-1 mAb)) is presented in Fig. 1a (and Fig. S1a, ESI,† showing the aa sequence of human PD-1). Adducts of MCA are included at the N-terminal and DNP at the C terminal. The structures of the three other FRET-peptides (mouse PD-1 (123–140), amyloid beta (Aβ) (26–33) and Tau peptide (391–408)) used in this study are given in Fig. S1b–d (ESI†). These FRET substrates were obtained using the synthesis protocol described in ref. 34.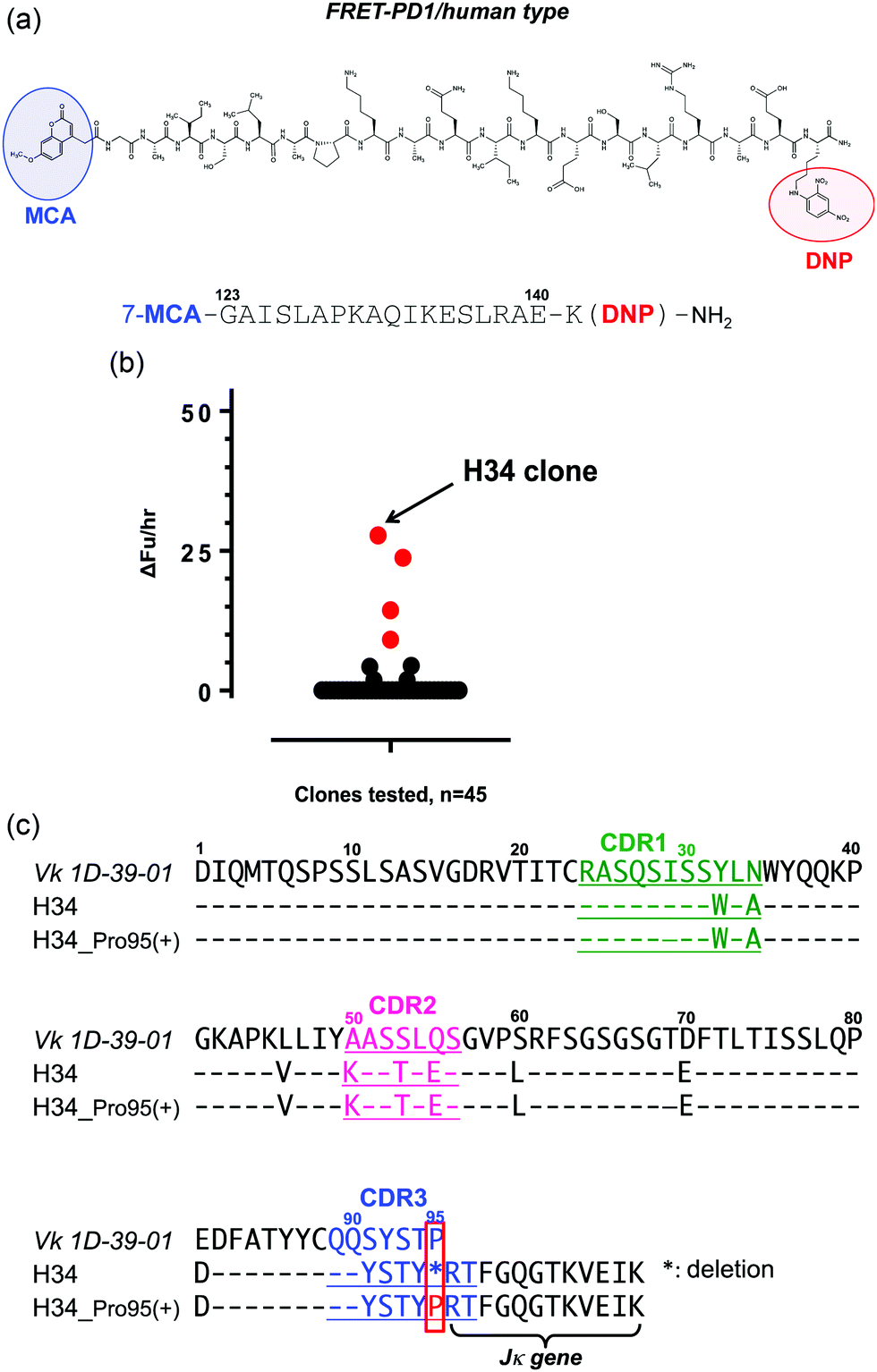 | ||
| Fig. 1 (a) Structure of FRET-PD-1: AA position 123–140 was selected to make the FRET-PD-1 peptide substrate in which MCA (4-methyl-coumaryl-7-amide; fluorescent reagent) was added at the N-terminal and DNP (2,4-dinitrophenyl; quenching reagent) at the C-terminal. Lysine was inserted in order to bind the DNP. (b) Results of screening tests: of 45 clones investigated, the H34 light chain had the highest activity for cleaving the FRET-PD-1 substrate. Light chain: 5 μM, FRET-PD-1: 25 μM, buffer: PBS (pH 7.4), reaction temperature: 37 °C. (c) Amino acid sequences: the amino acid (aa) sequences of the Vk region (1–95) of germline Vk 1D-39-01 (major clone belonging to subgroup I), and variable regions (1–107) of H34 and H34-Pro95(+) are presented. Pro95 is highly conserved in subgroup I. It is characteristic that Pro95 was lacking in H34. | ||
(2) Screening of catalytic light chains from a protein bank
We have constructed a protein bank now containing several hundred human antibody light chains, purified and stored at 4 °C or frozen. In the present study, 45 light chains from this protein bank were randomly screened on the FRET-PD-1 substrate, with the results shown in Fig. 1b. Several antibody light chains exhibited catalytic activity resulting in cleavage of the FRET-PD-1 substrate.Of these, H34 exhibited the strongest FRET-PD-1 catalytic degradation activity.
In Fig. 1c, the aa sequence of a germline of the Vk region (aa 1–95:1D-39-01, a major germline gene of subgroup I) is presented together with its variable region of H34 (aa 1–107; Vk + Jk). Intriguingly, this reveals that H34 is characterized by deletion of the Pro95 residue in CDR3.
(3) H34 cleavage of different substrates
For detailed investigation of the features of the H34 light chain, it was carefully purified again according to the protocol described in Experimental. The results of SDS-PAGE analysis are presented in Fig. 2a, in which a 44 kDa band corresponding to the dimer can be observed under non-reducing conditions, where the monomer band at 23–24 kDa was hardly observed. This fact indicates its presence in solution as the dimer. Only a 29 kDa band corresponding to the monomer was detected under reducing conditions (the monomer band at 23–24 kDa under non-reduced conditions always shifts to 28–30 kDa under reduced conditions, because of the conformational change on cutting the S–S bond using β-ME). No other bands were detected with CBB staining.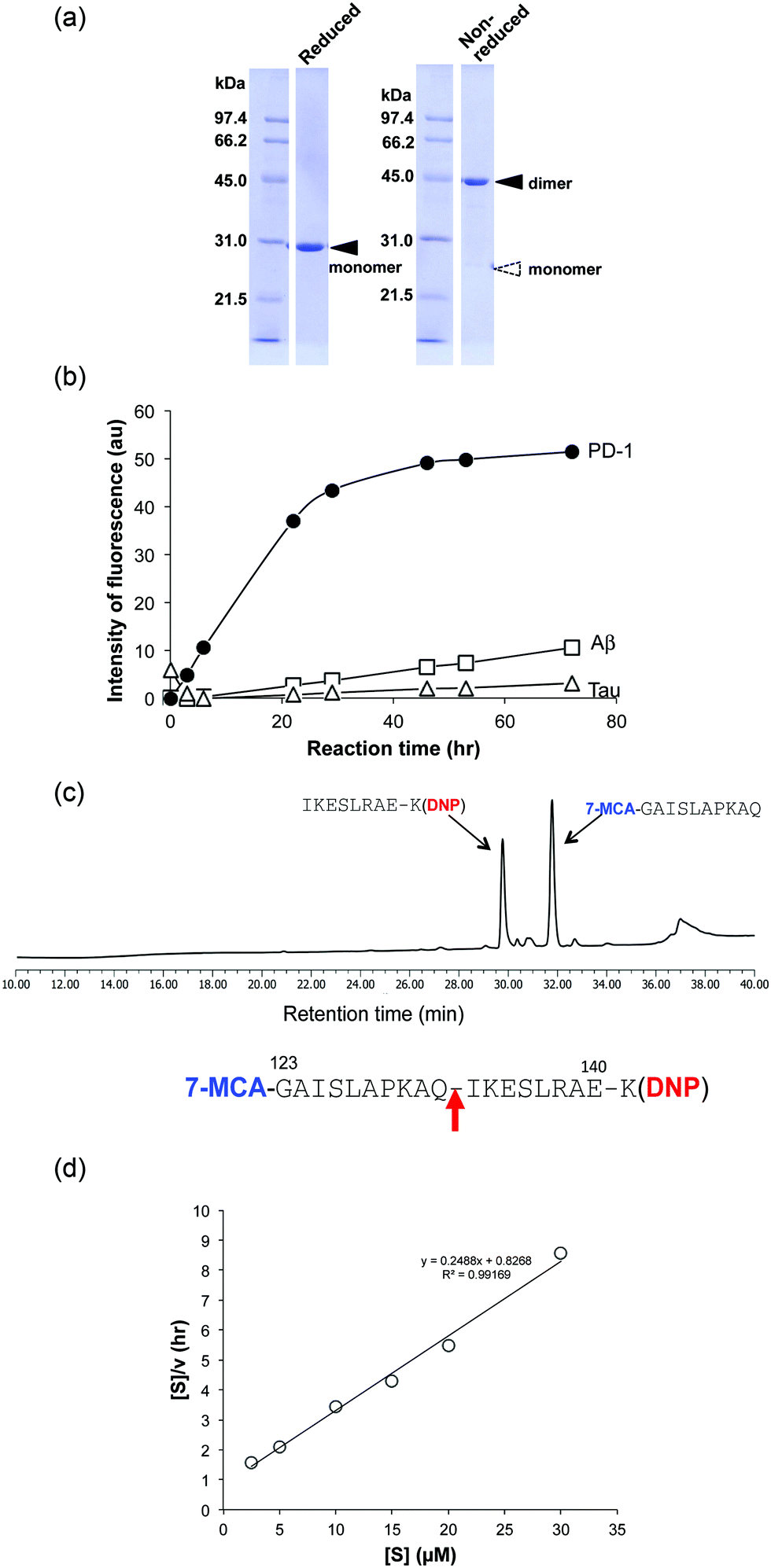 | ||
| Fig. 2 (a) Results of SDS-PAGE analysis: a band of the dimer at ∼44 kDa is observed under non-reduced conditions. Under reduced conditions, only a single ∼29 kDa band corresponding to the monomer is detected. Bands other than the monomer of the light chains were essentially absent under either reducing or non-reducing conditions. (b) Cleavage reaction for FRET substrates: the cleavage reaction was performed with 5 μM H34 using 25 μM of FRET-PD1 (PD-1; aa 123–140), FRET-Aβ (Aβ; aa 26–33) or FRET-Tau (Tau; aa 391–408). H34 exhibited strong catalytic activity for FRET-PD1, but low activity on FRET-Aβ or FRET-Tau. All reactions were carried out in duplicate. (c) Bond cleavage for FRET-PD1: the H34 reaction products were analyzed by HPLC and MS. The fragments were identified as 7-MCA-GAISLAPKAQ-OH (31.7 min) and NH2-IKESLRAEK(DNP)-NH2 (29.6 min), indicating cleavage of the peptide bond between Gln132 and Ile133. (d) Kinetic analysis: [S]: concentration of the FRET-PD1 substrate. [V]: initial rate of the cleavage reaction. The Hanes–Woolf plot demonstrates that cleavage by the H34 light chain fits Michaelis–Menten kinetics, indicating that the reaction is enzymatic. kcat and Km were 1.3 × 10−2 min−1 and 3.32 × 10−6 M, respectively. | ||
The cross-reactivity of the H34 light chain was investigated using other FRET substrates such as FRET-Aβ and FRET-Tau as well as specific FRET-PD1. The reaction profiles using 5 μM of H34 light chain and 25 μM of the FRET substrates are shown in Fig. 2b. Interestingly, H34 rapidly cleaved the FRET-PD-1 substrate (PD1: aa 123rd–141st (7-MCA-GAISLAPKAQIKESLRAE-K(DNP)-NH2)), but had little or no effect on the FRET-Aβ (Aβ: aa 26th–33rd (7-MCA-SNKGAIIGK(DNP)rrr-NH2)) or FRET-Tau (Tau: aa 391st–408th (7-MCA-EIVYKpSPVVSGDTpSPRHLK(DNP)-NH2:pS, phosphorylated serine)) substrates until 24 h later. Although H34 belongs to subgroup I and has no histidine residue in the variable region (Fig. 1c; aa 1–117), it exhibited high FRET-PD-1 substrate degradation activity. In contrast, H34 did not cleave a mouse PD-1 peptide at all (see Fig. S1e, ESI†), suggesting a high degree of specificity for human PD-1.
(4) Identification of the scissile bond in the FRET-PD1 substrate
Because the H34 light chain mediated the catalytic activity, the reaction products were analyzed by means of HPLC and MS, with the results shown in Fig. 2c. Two peaks were present with 29.6 min and 31.7 min retention times, relative to the native FRET-PD-1 substrate at around 37 min. The two peaks at 29.6 min and 31.7 min were analyzed by MS as shown in Fig. S2a and b (ESI†), respectively. The divalent mass of the former was detected at 619.81 m/2z, which was identified as an NH2-I-K-E-S-L-R-A-E-K(DNP)-NH2 fragment (Fig. S2a, ESI†), and for the latter at 586.29 m/2z, identified as a 7-MCA-G-A-I-S-L-A-P-K-A-Q-OH fragment (Fig. S2b, ESI†). These results suggest that the peptide bond between Gln132 and Ile133 was cleaved by H34.(5) Kinetics
Kinetic data were obtained by varying the concentration of the FRET-PD-1 substrate (5–60 mM), while keeping H34 constant at 5 μM. Hanes–Woolf plots (Fig. 2d) revealed that the H34 reactions fitted the Michaelis–Menten equation. Thus, degradation of the FRET-PD-1 substrate by H34 should be enzymatic. The Michaelis constant (Km) was determined to be 3.32 × 10−6 M. The catalytic reaction constant (kcat) was 1.3 × 10−2 min−1 and the catalytic efficiency (kcat/Km) was 1.96 × 103 M−1 min−1. Paul et al.10 reported that an anti-VIP antibody light chain cleaved the substrate VIP with Km = 0.2 × 10−6 M and kcat = 1.1 × 10−2 min−1. Hifumi et al. showed that 41S-2 light chains cleaved the antigenic HIV-gp41 peptide with Km = 0.22 × 10−6 M and kcat = 6 × 10−1 min−1.19 They also reported that the HpU-9 light chain hydrolyzed an antigenic peptide of urease with Km = 1.6 × 10−5 M and kcat = 1.1 × 10−1 min−1.35 The kinetic values for H34 are therefore similar to those of other catalytic antibodies reported so far.(6) Cleavage of the recombinant PD-1 molecule
A recombinant PD-1 molecule (rPD-1, 1 μM), the aa sequence of which is presented in Fig. S3 (ESI†), was mixed with H34 light chains (0.5 μM) in TGT buffer containing 0.02% sodium azide, and incubated at 37 °C. Samples taken after 0, 24, 48 and 72 h were analyzed by SDS-PAGE with silver staining (Fig. 3a and b). In this experiment, human serum albumin (HSA) was used as the negative control. Fig. 3a shows the time course of the reaction of rPD-1 with H34 light chains. Fig. 3b presents several control experiments, (HSA + H34), H34, PD-1 and HSA. In Fig. 3a, two major rPD1 bands were observed at around 38 kDa and 43 kDa. H34 was present as a single band at ∼30 kDa. After 24 h of incubation, bands at ∼28 kDa and ∼17 kDa could be clearly observed and became more prominent up to 72 h of incubation. In contrast, the band at ∼38 kDa gradually faded with time up to 72 h. In the control experiments (Fig. 3b), no fragmented bands were detected in the mixture of HSA and H34 up to 72 h of incubation.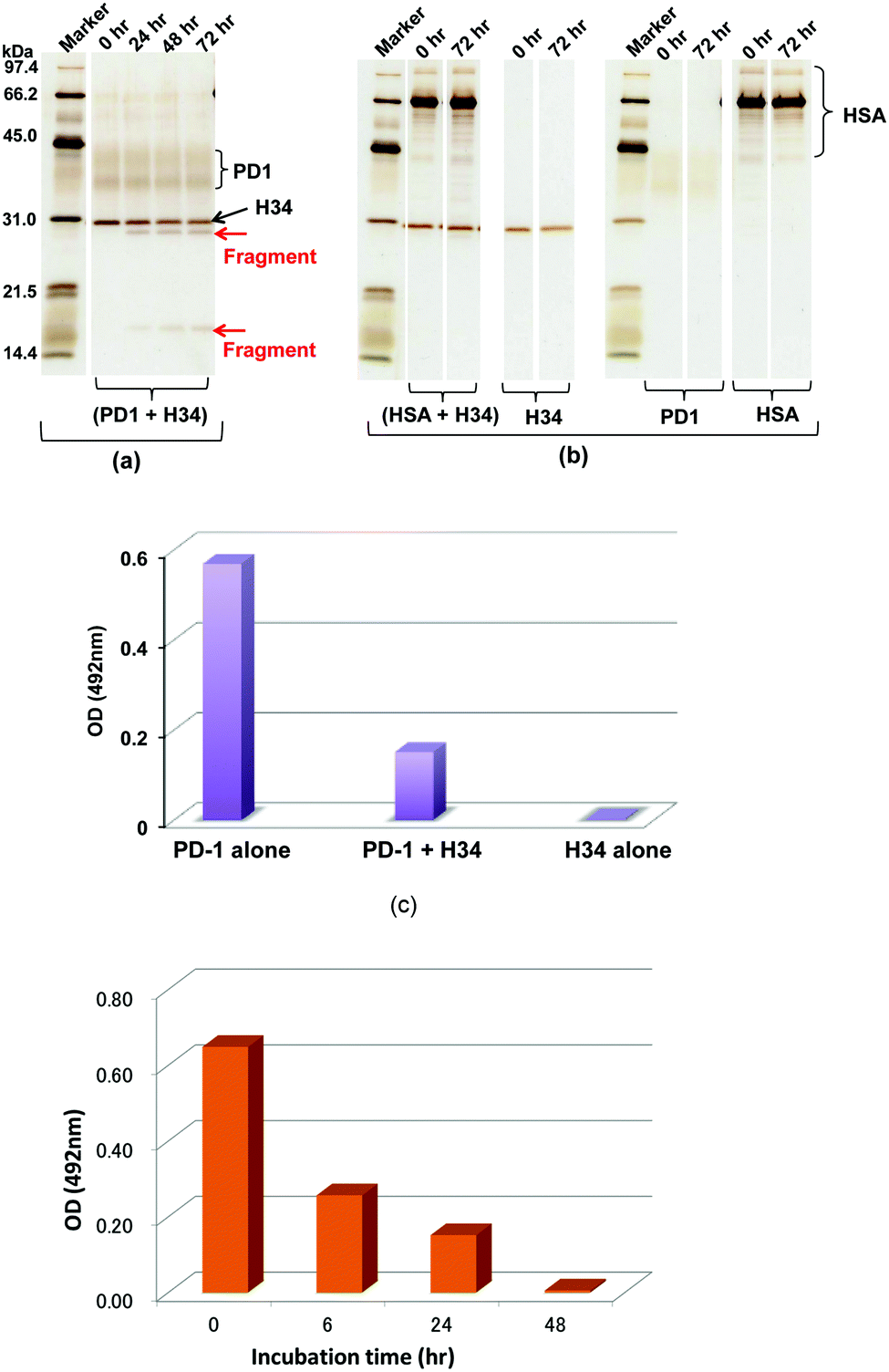 | ||
| Fig. 3 (a and b) SDS-PAGE analysis with silver staining under reduced conditions: (a) cleavage of recombinant PD-1 molecules: recombinant PD1 molecules (rPD1; 1 μM) were incubated with H34 (0.5 μM) at 37 °C for 72 h. The rPD1 bands are around 38 kDa and 43 kDa, whereas H34 is a single band at 30 kDa. After 24 h, bands at 28 kDa and around 17 kDa are visible and become gradually more prominent from 24 h to 72 h. In contrast, the 38–42 kDa PD-1 bands gradually fade with time. (b) Irrelevant protein HSA: HSA is an irrelevant protein used as a control. Only the band at ∼66 kDa and the rPD-1 band were observed. No fragments were detected from H34, PD-1, HSA and a mixture of H34 and HSA up to 72 h. Thus, H34 cleaved the rPD-1 molecule in a time-dependent manner. (c) ELISA-1: first, rPD-1 alone, H34 alone or a mixture of both was incubated in TGT buffer (pH 7.7) at 37 °C for 48 h. Next, 50 μL of each sample was assayed by ELISA on rPD-L1-coated wells at room temperature. The reaction was detected using the HRP-conjugated anti-V5 tag epitope antibody. For rPD-1 alone, the absorbance at 492 nm was 0.569, and 0.003 for H34 alone, suggesting no reaction of H34 with rPD-L1. In contrast, the absorbance for the mixture of rPD-1 and H34 was 0.151. This is lower than the value of rPD-1 alone by a factor of four. It is clear that H34 diminished the binding interaction between PD-1 and PD-L1 by degrading the PD-1 molecule. (d) ELISA-2: a mixture of rPD-1 (0.18 μM) and H34 (0.5 μM) was incubated up to 48 h. A sample at 0, 6, 24, and 48 h was taken and we measured the binding of rPD-1/PD-L1 by ELISA. | ||
The bands corresponding to H34 (30 kDa), PD-1 (∼36 kDa and ∼42 kDa) and HSA (mainly 66 kDa) did not show any changes during incubation. These findings indicate that H34 cleaves the rPD-1 molecule in a time-dependent manner. Considering the molecular size, the band at 17 kDa is attributed to a fragmented peptide from the N-terminus of rPD1 cleaved at Q132–I133. The band at 28 kDa may be a dimer of the above fragmented peptide (see the aa sequences of PD-1, rPD-1 and fragmented PD-1 in Fig. S3a–d, ESI†).
(7) ELISAs
The binding behaviour of rPD-1 with rPD-L1 was examined using an ELISA system. rPD-1 was incubated with the H34 catalytic antibody at 37 °C for 48 h, and rPD-1 or H34 incubated alone under the same conditions served as controls. Thereafter, each was transferred to the wells of ELISA plates coated with rPD-L1 and the binding was visualized by the HRP-conjugated anti V5-epitope tag antibody. The results are presented in Fig. 3c. When rPD-1 was incubated alone, the absorbance at 492 nm was 0.569. For H34 alone, the absorbance was 0.003, suggesting that the cross-reactivity of the antibody with rPD-L1 was negligible. In contrast, rPD-1 incubated with H34 for 48 h yielded an absorbance of 0.151, only a quarter of the value with rPD-1 alone. Thus, it is concluded that the binding of rPD-1 with rPD-L1 was inhibited or blocked by the degradation of rPD-1.In addition, another experiment, where the incubation was carried out for 0, 6, 24 and 48 h for the mixture rPD-1 (0.18 μM) and H34 (0.5 μM), was performed. Then, each sample was submitted to ELISA. About one half of the rPD-1 molecules were degraded at 6 h of incubation. At an incubation time of 48 h, rPD-1 had almost disappeared, suggesting that the event did not occurr by a competitive reaction but degradation (Fig. 3d).
(8) Chemical features of H34
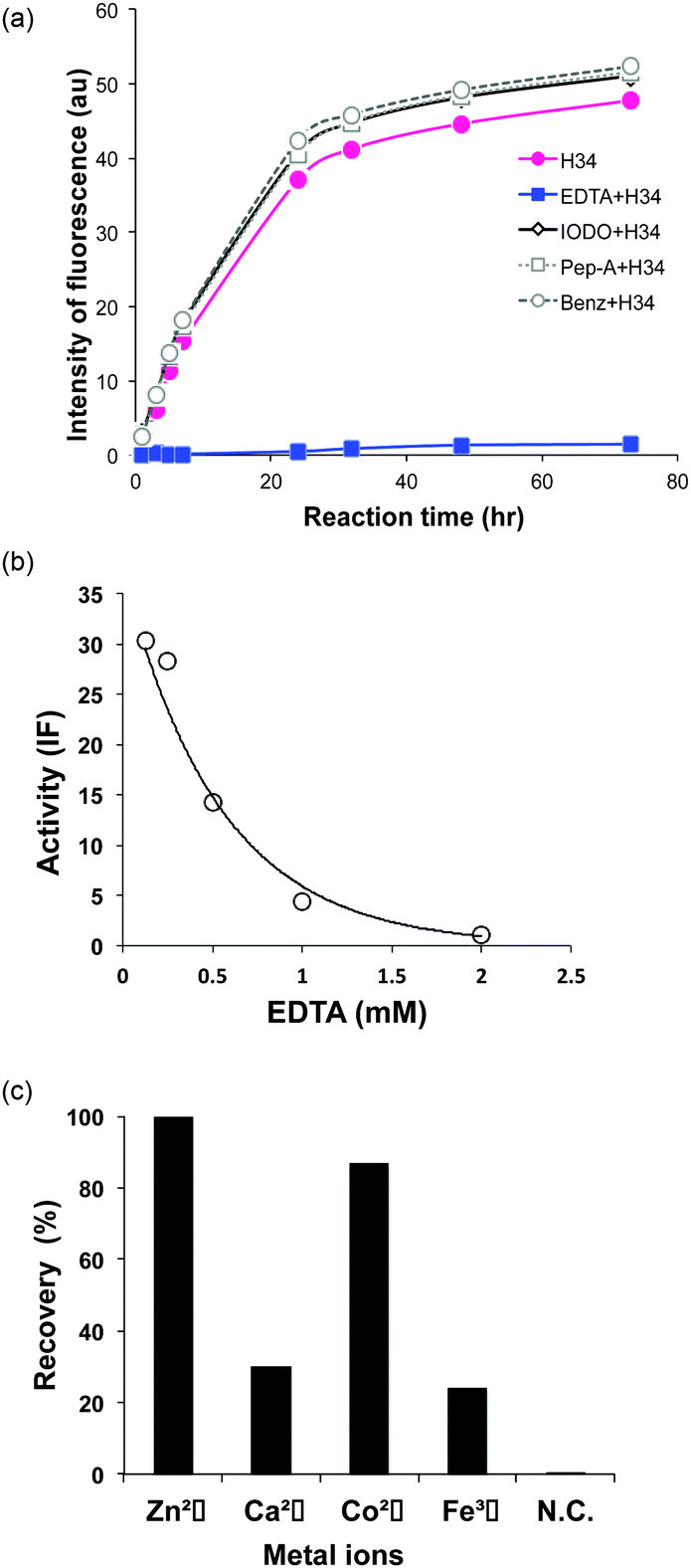 | ||
| Fig. 4 (a) Inhibition of FRET-PD1 cleavage: four inhibitors were tested: EDTA (2 mM; metalloproteinase inhibitor), iodoacetamide (0.1 mM; cysteine proteinase inhibitor), pepstatin A (0.02 mM; aspartic/glutamic protease inhibitor) and benzamidine (1 mM; serine protease inhibitor). Only EDTA suppressed the cleavage reaction. (b) Dependence on the concentration of EDTA: the cleavage reaction was suppressed in a dose-dependent manner. At a concentration >500 mM, the inhibitory effect was clearly observed, and no cleavage occurred at 2 mM. The intensity of fluorescence (IF) was measured as the catalytic activity at 21.1 h of incubation. (c) Effect of metal ions: in the presence of EDTA (2 mM), 3 mM metal ions such as Zn(II), Ca(II), Co(II) and Fe(III) were added to the reaction solution. Zn(II) ions completely recovered the catalytic activity, followed in decreasing order by Co(II), Ca(II) and Fe(III). | ||
(9) Pro95-insertion
It was recently reported that the amino acid residue Pro95, present in the CDR3 of the antibody light chain, plays an important role in the catalytic function.37 Therefore, in this study, a mutant was made with an inserted Pro95 (H34-Pro95(+)) and the effect on the catalytic activity was investigated. H34 (without Pro95) began cleaving the FRET-PD-1 substrate very soon after initial mixing and this gradually increased up to 78 h. On the other hand, the mutant with an inserted Pro95 only weakly cleaved FRET-PD-1 even after 78 h. These profiles up to 78 h for both tested light chains are presented in Fig. 5. Thus, the deletion of the Pro95 residue is important for the observed peptidase activity.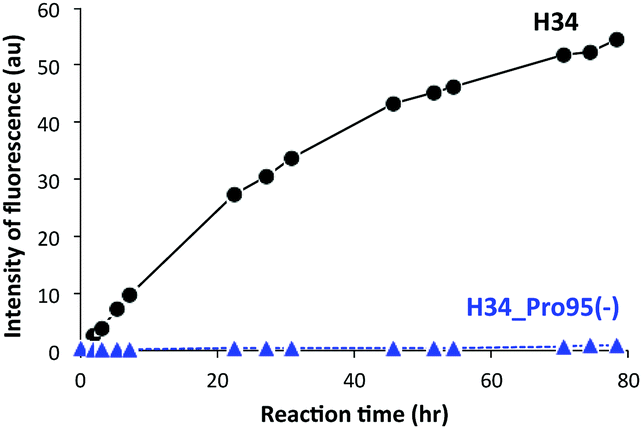 | ||
| Fig. 5 Catalytic activity of H34 with inserted Pro95 (H34-Pro95(+)): reaction profiles for FRET-PD-1 substrate cleavage were investigated for up to 78 h of reaction time using H34 and H34-Pro95(+). The catalytic activity of the H34-Pro95(+) mutant was substantially reduced relative to H34 lacking Pro95. | ||
(10) CD spectra
CD spectrometry is useful to analyze the change of chemical circumstances in the catalytic antibody field, as shown by Gololobov et al.41 In this study, CD spectrometry was undertaken using a concentration of 1.54–1.55 mg mL−1 for both H34 and H34-Pro95(+) samples. Fig. 6a and b show the far UV/CD spectra (for the secondary structure) and the near UV/CD spectra (for the tertiary structure), respectively. The spectrum changes from 200–205 nm and from 225–245 nm in Fig. 6a suggest increasing α-helical content in H34-Pro95(+). In Fig. 6b, the bands with fine structure arising from vibrational transitions from 260 to 270 nm (assigned to Phe) became different.42 This means that the chemical circumstances of Phe should be changed between the two light chains. It is hypothesized that these small conformational changes contribute to making H34 more catalytic.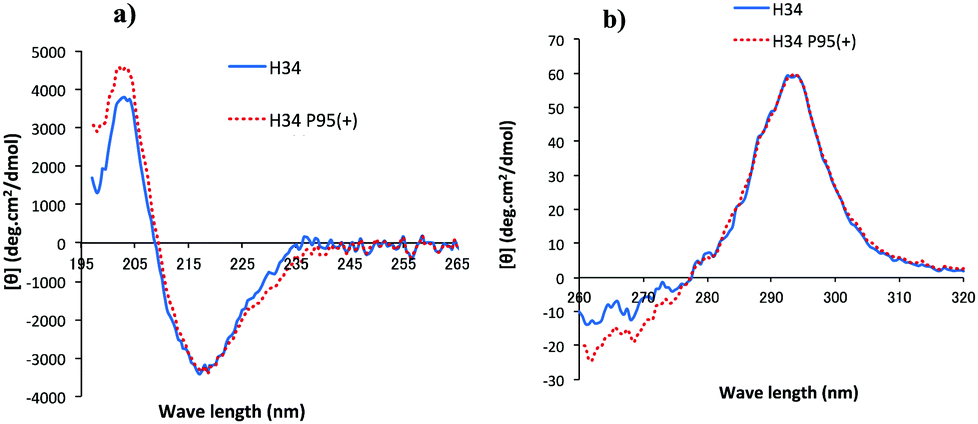 | ||
| Fig. 6 (a and b) CD spectra: (a) Far UV/CD spectra (secondary structure). (b) Near UV/CD spectra (tertiary structure). H34; blue solid line, H34-Pro95(+); red dotted line. From the secondary structure (a), it seems that H34-Pro95(+) has a higher a-helical content than that of H34. In the tertiary structure (b), the two peaks of Trp (295 nm) are almost the same between the two light chains. Differences are observed in the range from 260 to 270 nm, which is assigned to Phe. Thus, the chemical circumstances of Phe should be changed with or without the Pro95 residue. | ||
(11) Molecular modelling
We have succeeded in the X-ray diffraction analysis for only the #4 light chain,38 although many light chains have been submitted to the experiment. It is not difficult to determine the crystal structure of Fab or scFv of the antibody. However, note that the crystallization of only the light chains is very difficult, maybe due to the structural diversity.38 Therefore, in this study, we employed molecular modelling as a tool to interpret the present results.A second possibility, namely that the constant region of the antibody light chain possesses a zinc finger-like motif, should also be considered. This motif is Cys-X3-His (the H34 aa sequence (193–197) is Cys-Glu-Val-Thr-His; see the full sequence of H34 in Fig. S5, ESI†). Interestingly, a complete motif (Cys-X3-His-X15-Cys-X3-His) of a zinc finger is present at aa 193–217.39,40 The sequence is CEVTHQGLSSPVTKSFNRGECLEHH, in which the His residues at positions 216 and 217 are part of a His tag. This motif can take up a divalent metal ion such as Zn2+ or Ni2+.
Discussion
The aims of this study were both to find a new catalytic antibody light chain which can enzymatically and specifically decompose the PD-1 molecule, and characterization of the chemical properties of the catalytic antibody. To approach these goals, we explored the catalytic antibody light chains from our protein bank. We have assembled a protein bank of human antibody kappa light chains mainly belonging to subgroups I, II, and III (according to Kabat's numbering system). Once several hundred had been collected, they were screened for the presence of catalytic antibody light chains capable of degrading the PD-1 molecule. For high-throughput screening, we synthesized a FRET-PD-1 substrate, consisting of the 18-mer GAISLAPKAQIKESLRAE (aa position 123–140 of the PD-1 molecule) with MCA at the N-terminal and Lys (DNP) at the C-terminal. In the present study, we randomly screened 45 of the human kappa light chains from our protein bank for their ability to cleave this FRET-PD-1 peptide. We focused on the most reactive of these, clone H34, which had minimal activity on other substrates such as FRET-Aβ (26–33) and FRET-Tau (391–408), as well as the HSA protein (Fig. 2b). Thus, the H34 catalytic antibody light chain cleaved the human r-PD-1 molecule with high specificity. Note that this is the first finding of a catalytic antibody degrading the PD-1 molecule. And it should be also stressed that H34 existed in human genes donated by a volunteer.Next, we investigated the physicochemical properties of H34 and determined that the kcat and Km values were 1.3 × 10−2 min−1 and 3.32 × 10−6 M, respectively, for FRET-PD1 cleavage. These values were similar to those obtained for several peptide antigens by Paul et al.10 and Hifumi et al.,19,34 where the kcat value was reported to range from 10−1 to 10−2 min−1 and Km from 10−5 to 10−6 M. One of the most important findings in the present study is that the H34 catalytic antibody light chain blocks the binding of PD-1 with its ligand PD-L1 by virtue of its ability to degrade the PD-1 molecule. The mechanism of action of monoclonal antibodies against PD-1 (for example, nivolumab) is thought to be via blocking the binding of PD-1 to PD-L1 through stoichiometric hindrance. In contrast, the mechanism of action of H34 interference with the binding is thought to be quite different, resulting from degradation of the PD-1 molecule itself, a new type of blockade mediator.7
The presence of a metal ion is necessary for H34 to mediate its catalytic activity and cleave PD-1, with Zn(II) or Co(II) being particularly effective. Several metal-dependent catalytic antibodies have been reported by Paul's group43 and Nevinsky's group.44 The catalytic antibody IgV 2E6, described by Paul's group, requires the same metal ions (Zn(II) or Co(II)) as H34. They concluded that the metal ion induced a conformational transition of the catalytic antibody and exerted a favorable effect on a catalytic step after the initial nucleophilic attack on the amyloid-β antigen. Thus, the metal ion acts as a co-factor. In the case of H34, the metal ion may be working as the cofactor, but not the active site. Furthermore, many catalytic antibody light chains function optimally in dimeric form but not as monomers; H34 is also considered to function as a dimer (Fig. 2a). Smider et al. published a paper regarding the relationship between the structure and the catalytic activity of human antibody light chains; in that study, monomers also mediated lower catalytic activity for cleaving the PFR-MCA synthetic substrate than did dimers or tetramers.18 In our case, the metal ion may contribute to dimer formation, which facilitates catalytic function.
The results of H34 CD spectral analysis were also interesting in this respect. Namely, differences between H34 and the H34-Pro95(+) mutant were observed in the ranges from 200–205 nm and from 225–245 nm, suggesting that the conformational change of H34-Pro95(+) slightly occurred so as to increase the α-helical content. The near UV/CD revealed that the chemical circumstances of Phe should be changed. This phenomenon was in good agreement with the result of molecular modelling.
Molecular modelling was performed in order to explore the catalytic site. Asp1 in H34 lacking Pro95 has a tendency to approach near to the CDR3 loop, which is similar to what is observed in other catalytic antibodies such as S38 and T99-Pro95(−).37 This conformational change seems to contribute to the catalytic function.
We have already pointed out the importance of the deletion of the Pro95 residue for acquisition or enhancement of the catalytic activity of antibody light chains belonging to subgroup II.37 However, H34 belongs to subgroup I, which is the major group (∼50% of human light chains). Thus, it is significant that H34 nonetheless exhibited catalytic activity. Note that production of large numbers of catalytic antibodies from subgroup I and II light chains is relatively straightforward by means of deleting Pro95 from many of the monoclonal antibodies produced worldwide since 1974.45
From a biological point of view, it is very interesting that the H34 light chain was included in our protein bank, which has been made from a human antibody gene. In addition, H34 could degrade human PD-1 more specifically than mouse PD-1.
Conclusions
We have identified a catalytic antibody light chain, designated H34, which is capable of degrading the human PD-1 molecule with high specificity. This catalytic antibody may usher in a new form of checkpoint blockade by inhibiting the binding of PD-1 to PD-L1 by a mechanism different from the anti PD-1 antibodies. Finally, we conclude that also subgroup I antibody light chains lacking Pro95 can mediate catalytic activity. These types of catalytic antibody should have a great deal of potential as a new biocatalyst or protein degrader to expand drug development, as they can easily be created from the many currently available well-characterized monoclonal antibodies.Abbreviations
| aa | Amino acid |
| Aβ | Amyloid-beta |
| au | Arbitrary unit |
| β-ME | Beta-mercaptoethanol |
| CBB | Coomassie brilliant blue |
| CD | Circular dichroism |
| DNP | 2,4-Dinitrophenyl |
| eq. | Equivalent |
| FRET | Förster resonance energy transfer |
| HRP | Horseradish peroxidase |
| HSA | Human serum albumin |
| IPTG | Isopropyl-β-D-thiogalactopyranoside |
| mAb | Monoclonal antibody |
| 7-MCA | (7-Methoxycoumarin-4-yl)acetyl |
| MS | Mass spectroscopy |
| PBS | Phosphate buffered saline |
| rPD-1 | Recombinant PD-1 |
| rPD-L1 | Recombinant PD-L1 |
| TGT | 50 mM Tris–100 mM glycine–Tween-20 buffer (pH 7.7) |
| TFA | Trifluoroacetic acid |
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors thank Ms M. Watanabe and Ms Y. Akiyoshi for their assistance with this study. All data needed to evaluate the conclusions in the paper are present in the paper and/or the ESI.† Additional data related to this paper may be requested from the authors. This work was financially supported by JSPS Grant-in Aid for challenging Exploratory Research (Grant no. JP20K21255) and for Scientific Research(A) (Grant no. JP16H02282).Notes and references
- Y. Ishida, Y. Agata, K. Shibahara and T. Honjo, EMBO J., 1992, 11, 3887–3895 CrossRef CAS.
- D. Y. Lin, Y. Tanaka, M. Iwasaki, A. G. Gittis, H.-P. Su, B. Mikami, T. Okazaki, T. Honjo, N. Minato and D. N. Garboczi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 3011–3016 CrossRef CAS.
- L. Zhang, T. F. Gajewski and J. Kline, Blood, 2009, 114, 1545–1552 CrossRef CAS.
- O. Hamid, C. Robert, A. Daud, F. S. Hodi, W.-J. Hwu, R. Kefford, J. D. Wolchok, P. Hersey, R. W. Joseph, J. S. Weber, R. Dronca, T. C. Gangadhar, A. Patnaik, H. Zarour, A. M. Joshua, K. Gergich, J. Elassaiss-Schaap, A. Algazi, C. Mateus, P. Boasberg, P. C. Tumeh, B. Chmielowski, S. W. Ebbinghaus, X. N. Li, S. P. Kang and A. N. Ribas, N. Engl. J. Med., 2013, 369, 134–144 CrossRef CAS.
- Y. Iwai, M. Ishida, Y. Tanaka, T. Okazaki, T. Honjo and N. Minato, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12293–12297 CrossRef CAS.
- C. Blank, I. Brown, A. C. Peterson, M. Spiotto, Y. Iwai, Y. Honjo and T. F. Gajewski, Cancer Res., 2004, 64, 1140–1145 CrossRef CAS.
- A. Mullard, Nat. Rev. Drug Discovery, 2019, 18, 237–239 Search PubMed.
- S. Paul, D. J. Volle, C. M. Beach, D. R. Johnson, M. J. Powell and R. J. Massey, Science, 1989, 244, 1158–1162 CrossRef CAS.
- Q. S. Gao, M. Sun, S. Tyutyukova, D. Webster, A. Rees, A. Tramontano, R. J. Massey and S. Paul, J. Biol. Chem., 1994, 269, 32389–32393 CAS.
- S. Mei, B. Mody, S. H. Eklund and S. Paul, J. Biol. Chem., 1991, 266, 15571–15574 CAS.
- E. Hifumi, H. Kondo, Y. Mitsuda and T. Uda, Biotechnol. Bioeng., 2003, 84, 485–493 CrossRef CAS.
- A. M. Shuster, G. V. Gololobov, O. A. Kvashuk, A. E. Bogomolova, I. V. Smirnov and S. G. Gabibov, Science, 1992, 256, 665–667 CrossRef CAS.
- T. A. Parkhomenko, V. N. Buneva, O. B. Tyshkevich, I. I. Generalov, B. M. Doronin and G. A. Nevinsky, Biochimie, 2010, 92, 545–554 CrossRef CAS.
- M. A. Krasnorutskii, V. N. Buneva and G. A. Nevinsky, Biochemistry, 2011, 76, 1065–1072 CAS.
- S. Lacroix-Desmazes, A. Moreau, Sooryanarayana, C. Bonnemain, N. Stieltjes, A. Pashov, Y. Sultan, J. Hoebeke, M. D. Kazatchkine and S. V. Kaveri, Nat. Med., 1999, 5, 1044–1047 CrossRef CAS.
- Y. Mitsuda, E. Hifumi, K. Tsuruhata, H. Fujinami, N. Yamamoto and T. Uda, Biotechnol. Bioeng., 2004, 86, 217–225 CrossRef CAS.
- E. Hifumi, K. Higashi and T. Uda, FEBS J., 2010, 277, 3823–3832 CrossRef CAS.
- V. Sharma, W. Heriot, K. Trisler and V. Smider, J. Biol. Chem., 2009, 284, 33079–33087 CrossRef CAS.
- E. Hifumi, Y. Mitsuda, K. Ohara and T. Uda, J. Immunol. Methods, 2002, 269, 283–298 CrossRef CAS.
- S. Paul, S. Karle, S. Planque, H. Taguchi, Y. Nishiyama, B. Handy, M. Salas, A. Edmundson and A. Hanson, J. Biol. Chem., 2004, 279, 39611–39619 CrossRef CAS.
- N. A. Ponomarenko, I. I. Vorobiev, E. S. Alexandrova, A. V. Reshetnyak, G. B. Telegin, S. V. Khaidukov, B. Avalle, A. Karavanov, H. C. Morse, 3rd, D. Thomas, A. Friboulet and A. G. Gabibov, Biochemistry, 2006, 45, 324–330 CrossRef CAS.
- E. Hifumi, F. Morihara, K. Hatiuchi, T. Okuda, A. Nishizono and T. Uda, J. Biol. Chem., 2008, 283, 899–907 CrossRef CAS.
- O. M. Durova, I. I. Vorobiev, I. V. Smirnov, A. V. Reshetnyak, G. B. Telegin, O. G. Shamborant, N. A. Orlova, D. D. Genkin, A. Bacon, N. A. Ponomarenko, A. Friboulet and A. G. Gabibov, Mol. Immunol., 2009, 47, 87–95 CrossRef CAS.
- E. Hifumi, S. Takao, N. Fujimoto and T. Uda, J. Am. Chem. Soc., 2011, 133, 15015–15024 CrossRef CAS.
- E. Hifumi, E. Honjo, N. Fujimoto, M. Arakawa, A. Nishizono and T. Uda, FASEB J., 2012, 26, 1607–1615 CrossRef CAS.
- E. Hifumi, N. Fujimoto, M. Arakawa, E. Saito, S. Matsumoto, N. Kobayashi and T. Uda, J. Biol. Chem., 2013, 288, 19558–19568 CrossRef CAS.
- E. Hifumi, M. Arakawa, S. Matsumoto, T. Yamamoto, Y. Katayama and T. Uda, FASEB J., 2015, 29, 2347–2358 CrossRef CAS.
- A. Bowen, M. P. Wear, R. Cordero, S. Oscarson and A. Casadevall, J. Biol. Chem., 2017, 292, 417–434 CrossRef CAS.
- G. Gololobov, M. Sun and S. Paul, Mol. Immunol., 1999, 36, 1215–1222 CrossRef CAS.
- N. Okochi, M. Kato-Murai, T. Kadonosono and M. Ueda, Appl. Microbiol. Biotechnol., 2007, 77, 597–603 CrossRef CAS.
- R. A. Ramsland, S. S. Terzyan, G. Cloud, C. R. Bourne, W. Farrugia, G. Tribbick, H. M. Geysen, C. R. Moomaw, C. A. Slaughter and A. B. Edundson, Biochem. J., 2006, 396, 473–481 CrossRef.
- I. Smirnov, E. Eugénie Carletti, I. Kurkova, F. Nachon, Y. Nicolet, V. A. Mitkevich, H. Débat, B. Avalle, A. A. J. Belogurov, N. Kuznetsov, A. Reshetnyak, P. Masson, A. G. Tonevitsky, N. Ponomarenko, A. A. Makarov, A. Friboulet, A. Tramontano and A. Gabibov, Proc. Natl. Acad. Sci., U. S. A., 2011, 108, 15954–15959 CrossRef CAS.
- N. Ponomarenko, S. D. Chatziefthimiou, I. Kurkova, Y. Mokrushina, A. Stepanova, I. Smirnov, M. Avakyan, T. Bobik, A. Mamedov, V. Mitkevich, A. Belogurov Jr, O. S. Fedorova, M. Dubina, A. Golovin, V. Lamzin, A. Friboulet, A. A. Makarov, M. Wilmanns and A. Gabibov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 708–719 CrossRef CAS.
- E. Hifumi, H. Taguchi, E. Toorisaka and T. Uda, FASEB BioAdv., 2018, 1, 105–114 Search PubMed.
- E. Hifumi, K. Hatiuchi, T. Okuda, A. Nishizono, Y. Okamura and T. Uda, FEBS J., 2005, 272, 4497–4505 CrossRef CAS.
- E. L. Brown, Y. Nishiyama, J. W. Dunkle, S. Aggarwal, K. Watanabe, K. Csencsits-Smith, M. G. Bowden, S. L. Kaplan and S. Paul, J. Biol. Chem., 2012, 287, 9940–9951 CrossRef CAS.
- E. Hifumi, H. Taguchi, H. Tsuda, T. Minagawa, T. Nonaka and T. Uda, Sci. Adv., 2020, 6(13), eaay6 Search PubMed.
- E. Hifumi, H. Taguchi, R. Kato, M. Arakawa, Y. Katayama and T. Uda, Structural diversity problem of antibodies and catalytic antibody light chains and the solving method, in Antibody Engineering, ed. T. Boldicke, InTech Publishers, 2018, ch. 10, pp. 231–257 Search PubMed.
- R. T. Radulescu, Med. Hypotheses, 1995, 44, 139–145 CAS.
- R. T. Radulescu, Immunol. Today, 1998, 19, 288 CrossRef CAS.
- G. V. Gololobov, E. A. Chernova, D. V. Schourov, I. V. Smirnov, I. A. Kudelina and A. G. Gabibov, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 254–257 CrossRef CAS.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 1751, 119–139 CrossRef CAS.
- Y. Nishiyama, H. Taguchi, M. Hara, S. A. Planque, Y. Mitsuda and S. Paul, J. Biotechnol., 2014, 180, 17–22 CrossRef CAS.
- E. S. Odintsova, N. P. Zaksas, V. N. Buneva and G. A. Nevinsky, J. Mol. Recognit., 2010, 24, 45–59 CrossRef.
- G. Kohler and C. Milstein, Nature, 1975, 256, 495–497 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0cb00155d |
| This journal is © The Royal Society of Chemistry 2021 |