
An MOF-derived C@NiO@Ni electrocatalyst for N2 conversion to NH3 in alkaline electrolytes†
Shijian
Luo
,
Xiaoman
Li
 *,
Wanguo
Gao
,
Haiqiang
Zhang
and
Min
Luo
*
*,
Wanguo
Gao
,
Haiqiang
Zhang
and
Min
Luo
*
State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, School of Chemistry and Chemical Engineering, Ningxia University, Yinchuan, Ningxia 750021, China. E-mail: martinluomin@163.com; lixm2017@nxu.edu.cn
First published on 18th September 2019
Abstract
Today, industrial ammonia synthesis mainly depends on the Haber–Bosch process, which causes a lot of energy consumption and huge CO2 emissions. The electrochemical N2 reduction reaction (NRR) is considered a more sustainable and environmentally benign alternative to produce ammonia, but it requires an efficient catalyst to overcome the difficulty of N2 activation. In this work, we reported that MOF-derived C@NiO@Ni microtubes behaved as a high-efficiency electrocatalyst in 0.1 M KOH electrolyte. This electrocatalyst achieved a high NH3 yield of 43.15 μg h−1 mgcat.−1 and faradaic efficiency of 10.9% at −0.7 V vs. a reversible hydrogen electrode. The experimental results indicated that the excellent NRR performance originated from the oxygen vacancies in NiO. Moreover, the abundant NiO/Ni interfaces were conducive to proton adsorption and further enhanced the NRR performance.
Introduction
As a significant chemical in the world's industry, NH3 has been widely used for the production of fertilizers, medicines, plastics and dyes.1 It is also considered as a portable energy carrier with high energy density that does not cause pollution.2 Today, the industrial production of NH3 largely depends on the Haber–Bosch process under harsh conditions, which causes a lot of energy consumption and huge CO2 emissions.3 Therefore, it is vital to explore eco-friendly and sustainable methods for NH3 synthesis under milder conditions.The electrochemical N2 reduction reaction (NRR), which can be powered by the electric energy that comes from renewable solar and wind sources, has been regarded as a prospective means for N2 fixation.4 Compared with the industrial Haber–Bosch reaction, the electrocatalysis method is energy-efficient and eco-friendly, but it requires an efficient catalyst to overcome the difficulty of N2 activation.5 Depending on the catalytic system, NRR electrocatalysts can be divided into three categories, namely, heterogeneous, homogeneous and biological catalysts. Although homogeneous catalysts exhibit good activity for NRR, the difficulty in recovering and recycling the catalysts limits their further research.6 Most biological catalysts can only work in plants or microorganisms. Although there are reports showing that enzymes can be coupled to light absorbers to produce NH3, the performance of these catalysts is still unsatisfactory and requires substantial improvements.7,8 Despite the system complexity of heterogeneous catalysts, they are currently the most widely researched catalysts for electrochemical NRR due to their high NH3 production and excellent cycling properties.9 Noble metal-based heterogeneous catalysts (Au, Pt, Pd and Ru) exhibit good electrocatalytic activities for NRR, but it is difficult to use them on a large scale because of their high price and low abundance.10 Recently, cheap and abundant transition-metal oxides (TMOs) have been reported as remarkable NRR electrocatalysts in acidic and neutral media, including Cr2O3,11 Mn3O4,12 Fe3O4,13 and Co3O4,14 but they are unstable and show a low NRR performance in alkaline media because they can hardly adsorb protons.15 We considered that alkaline media can suppress the hydrogen evolution reaction (HER) to improve the faradaic efficiency (FE) of NRR. Therefore, it is significant to identify stable TMO-based catalysts in alkaline media and develop new and effective strategies to achieve a higher NH3 yield and FE. Recently, our research revealed that a metal–organic framework (MOF)-derived TMO-based material (Co3O4@NC) has excellent catalytic activities and stability for NRR due to its abundant defects and special structure.14 On this basis, we considered MOF-derived TMO-based materials that could be designed as dual-function catalysts to simultaneously adsorb nitrogen and protons in alkaline electrolytes, thus increasing the FE of NRR. These dual-function catalysts have potential applications for electrocatalytic NRR in alkaline electrolytes.
Herein, we reported an MOF-derived hollow C@NiO@Ni microtube as an efficient electrocatalyst for NRR in alkaline media. The experimental results demonstrated that a low-cost Ni-based MOF (Ni2+ and 1,3,5-benzenetricarboxylic, Ni–BTC)16 could be transformed to the C@NiO@Ni catalyst with the co-existence of Ni and NiO by controlling the annealing process. When tested in 0.1 M KOH, C@NiO@Ni achieved a high NH3 yield of 43.15 μg h−1 mgcat.−1 and FE of 10.9% at −0.7 V vs. a reversible hydrogen electrode (RHE). These values were higher than those of most reported NRR electrocatalysts under ambient conditions. Notably, this electrocatalyst also showed excellent structural stability and long cycle life.
Results and discussion
C@NiO@Ni was prepared from Ni-MOF (Ni–BTC) by a two-step annealing process based on thermogravimetric curves (TG) (Fig. S1†). First, Ni–BTC was synthesized by the solvothermal method, followed by annealing at 600 °C in an N2 atmosphere for 3 h to obtain C@Ni microtubes. Then, C@Ni was annealed at 250 °C in an air atmosphere for 10 or 15 h to prepare C@NiO@Ni or C@NiO. The X-ray diffraction (XRD) method was carried out to investigate the composition of the as-prepared samples. As shown in Fig. 1a, the XRD patterns of C@Ni and C@NiO match well with those of the Ni phase (JCPDS no. 87-0712) and the cubic NiO phase (JCPDS no. 44-1159), respectively. In addition, the XRD pattern of C@NiO@Ni presented the characteristic peaks of both Ni and NiO, indicating the co-existence of the two phases. Raman spectroscopy was performed to further ascertain the NiO and carbon components in the catalyst. As shown in Fig. 1b, the two peaks for the three samples at 1355 cm−1 and 1590 cm−1 are attributed to the D and G bands of carbon, respectively.17 The Raman peaks at 380, 530, 720 and 1100 cm−1 for C@NiO@Ni and C@NiO could be assigned to the NiO phase. The broad and intense peak at 1100 cm−1 was consistent with the 2LO 2 phonon scattering mode of the Ni–O vibration.16 The chemical states for the C@NiO@Ni microtube were characterized by X-ray photoelectron spectroscopy (XPS). As shown in the Ni 2p3/2 XPS spectra (Fig. 1c), the two peaks at 853.8 and 856.1 eV correspond to the typical feature of Ni2+ 2p3/2, which are in good agreement with the Ni2+ peaks of the NiO phase; the peak at 861 eV is attributed to the satellite peak of Ni2+. The binding energy peak at 852.6 eV could be assigned to Ni0, further confirming the existence of both Ni and NiO on the surface of C@NiO@Ni.16 Moreover, in order to investigate the oxidation degree of Ni inside the catalyst, Ar ionic sputtering was conducted to etch the catalyst for 10 nm and analyse the Ni 2p3/2 pattern. As shown in Fig. S2,† the peaks for Ni0 increase significantly, confirming that the Ni0![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ni2+ ratio increases from the surface to the inside of the catalyst. The O 1s spectra of the C@NiO@Ni surface are exhibited in Fig. 1d; the characteristic binding energy values of 529.8, 531.3, 532.1 and 533.7 eV are assigned to the lattice oxygen, oxygen vacancies, adsorbed oxygen and C–O–C bond, respectively.14,18,19 Fig. S3† presents the C 1s peaks of the C@NiO@Ni surface at 284.7 and 286.2 eV, which can be assigned to the C
:Ni2+ ratio increases from the surface to the inside of the catalyst. The O 1s spectra of the C@NiO@Ni surface are exhibited in Fig. 1d; the characteristic binding energy values of 529.8, 531.3, 532.1 and 533.7 eV are assigned to the lattice oxygen, oxygen vacancies, adsorbed oxygen and C–O–C bond, respectively.14,18,19 Fig. S3† presents the C 1s peaks of the C@NiO@Ni surface at 284.7 and 286.2 eV, which can be assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O bonds, respectively.19 As a contrast, the XPS results of C@NiO are also provided in Fig. S4.† The Ni 2p3/2 spectra indicate that there is no peak of Ni0 for the C@NiO sample, proving that Ni0 can be completely oxidized to Ni2+. Notably, the O 1s pattern of C@NiO also exhibited a broad peak of oxygen vacancies, but its area was smaller than that of C@NiO@Ni. These results demonstrated that Ni would be gradually oxidized to NiO by O2 in air and could reduce the oxygen vacancy concentration in the catalyst with an annealing time from 10 to 15 h.
C and C–O bonds, respectively.19 As a contrast, the XPS results of C@NiO are also provided in Fig. S4.† The Ni 2p3/2 spectra indicate that there is no peak of Ni0 for the C@NiO sample, proving that Ni0 can be completely oxidized to Ni2+. Notably, the O 1s pattern of C@NiO also exhibited a broad peak of oxygen vacancies, but its area was smaller than that of C@NiO@Ni. These results demonstrated that Ni would be gradually oxidized to NiO by O2 in air and could reduce the oxygen vacancy concentration in the catalyst with an annealing time from 10 to 15 h.
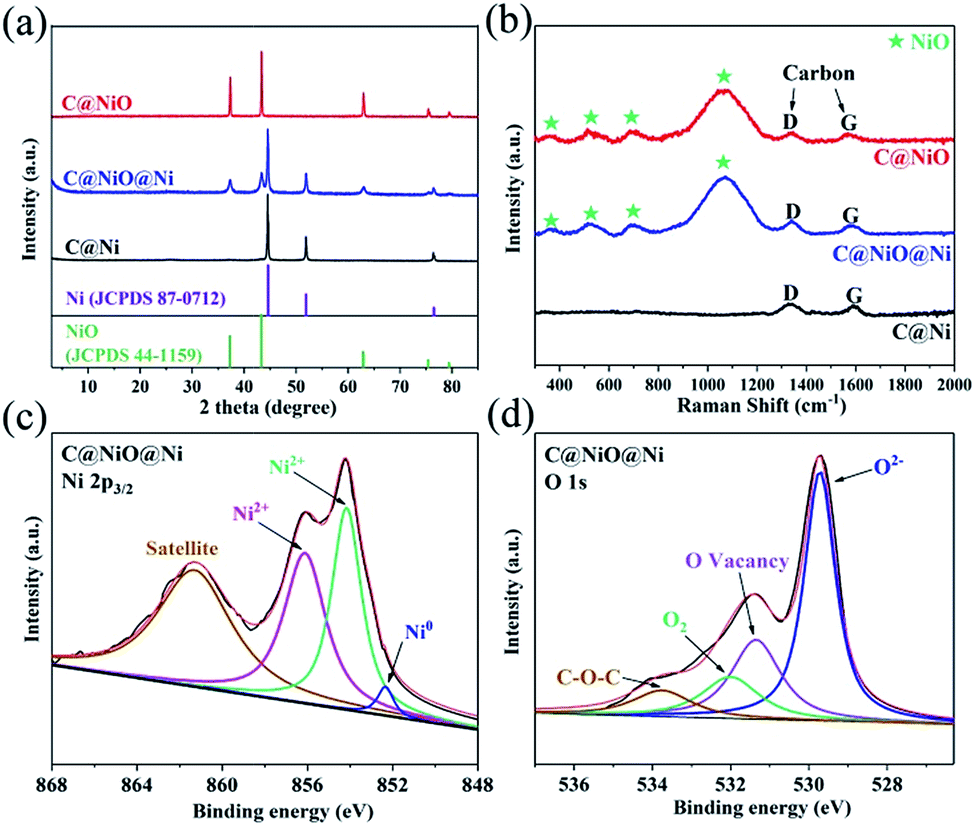 | ||
| Fig. 1 (a) XRD patterns and (b) Raman patterns of C@Ni, C@NiO@Ni and C@NiO. (c) XPS spectra of Ni 2p3/2 of C@NiO@Ni. (d) XPS spectra of O 1s of C@NiO@Ni. | ||
The morphology of the catalysts was explored by scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The Ni–BTC precursor presented a rod-like structure with a diameter of about 2 μm (Fig. S5†). Fig. 2a and b show the SEM and TEM images of C@NiO@Ni; it is clear that C@NiO@Ni basically maintains the size and structure of Ni–BTC. Compared to the Ni–BTC precursor, C@NiO@Ni had a different inner structure and it exhibited hollow microtube morphology. The carbon organic matter inside Ni–BTC continuously decomposed to form a carbon shell on the surface of the C@NiO@Ni nanoparticles; therefore, the hollow structure of the microtube was formed. The inset in Fig. 2b of the TEM image with a high magnification indicates that these microtubes are composed of many small nanoparticles with a size between 20 and 30 nm, and each nanoparticle is composed of Ni and NiO phases partly coated by carbon. Fig. 2c shows the high-resolution TEM (HRTEM) image of C@NiO@Ni; the well-defined lattice fringes with distances of 0.20 and 0.24 nm are attributed to the (111) lattice face of Ni and NiO, respectively.20,21 The lattice fringes with a distance of 0.35 nm could be assigned to the (002) plane of graphite carbon.22 Notably, there were many interfaces between Ni and NiO in the sample, which have been reported to be able to adsorb protons significantly.16,21 Moreover, the SEM, TEM and HR-TEM images of C@Ni and C@NiO are provided in Fig. S6 (C@Ni) and Fig. S7† (C@NiO); it is obvious that C@Ni and C@NiO also have microtube morphologies, but the HR-TEM images indicate that there is no NiO/Ni interface in C@Ni and C@NiO. The energy-dispersive X-ray (EDX) spectra of the three electrocatalysts are provided in Fig. S8† and the elemental mapping images of C@NiO@Ni (Fig. 2d) exhibit the uniform dispersion of the Ni, O and C elements in the sample.
 | ||
| Fig. 2 (a) SEM image, (b) TEM images and (c) HR-TEM image of the C@NiO@Ni catalyst. (d) The EDX element mapping images of Ni, O and C for C@NiO@Ni. | ||
The electrochemical measurements were recorded in a 0.1 M KOH electrolyte solution with a three-electrode system consisting of a Pt sheet as the counter electrode, a saturated calomel electrode (SCE) as the reference electrode and electrocatalysts deposited on a 1 cm2 carbon paper (CP) as the working electrodes. All potentials were converted to the RHE scale through calibration. Fig. 3a exhibits the linear sweep voltammetry (LSV) curves of the three electrocatalysts under an Ar or N2 atmosphere. When the potential moved below −0.27 V, it was obvious that the current density of C@NiO@Ni under N2 increased clearly compared with that under Ar, demonstrating that N2 fixation occurred on C@NiO@Ni with an overpotential of −0.27 V. For comparison, the overpotential for NRR of C@Ni or C@NiO was measured as −0.38 V or −0.34 V, respectively, which was more negative than that of C@NiO@Ni. In addition, the net current density for NRR (jN2–jAr) of C@NiO@Ni was much higher than that of C@Ni or C@NiO at the same overpotential. These results revealed that C@NiO@Ni was more positive for NRR than the other two electrocatalysts. Furthermore, NRR was performed using potentiostatic electrolysis under N2 for 1 h and by measuring the concentration of synthetic NH4+ by the Nessler's reagent method (Fig. S9†).17,23,24 The measured NH3 yield and FE of C@NiO@Ni at different potentials are displayed in Fig. 3b. Clearly, it attained the highest NH3 yield of 43.15 μg h−1 mgcat.−1 with FE of 10.9% at −0.7 V. These values were higher than those of most reported electrocatalysts under ambient conditions; the detailed performance comparisons of various electrocatalysts are shown in Table S1.† When the potential moved below −0.7 V, the NRR performance decreased, which resulted from the competitive adsorption of hydrogen.25 The UV-Vis absorption curves of the electrolyte after the tests of C@NiO@Ni at different potentials are shown in Fig. S10.† The time-dependent current density curves of C@NiO@Ni at different potentials remained almost stable for 3 h (Fig. S11†). For confirmation, ion chromatography was also used to measure the concentration of ammonia and the result was 41.70 μg h−1 mg−1, which was similar to the value calculated from the Nessler's reagent method. Fig. 3c exhibits the NRR performance comparisons of C@NiO@Ni, C@NiO, C@Ni, carbon (etching NiO and Ni on C@NiO@Ni by HNO3, Fig. S12†) and commercial NiO. At −0.7 V, the NH3 yield and FE of C@NiO could attain 26.87 μg h−1 mg−1 and 6.82%, respectively, which were better than those of C@Ni (7.1 μg h−1 mg−1 and 2.49%) and carbon (6.7 μg h−1 mg−1 and 2.31%) but still lower than those of C@NiO@Ni. Commercial NiO only exhibited an NH3 yield of 2.09 μg h−1 mg−1 and FE of 0.98%. It was predicted that the NRR performance mainly originated from the high concentration of the oxygen vacancies in NiO and this was why C@NiO@Ni and C@NiO had better NRR performances than C@Ni, carbon and commercial NiO. According to the experimental results and recently reported results,16,20,21 the abundant NiO/Ni interfacial sites are conducive to proton adsorption in alkaline electrolytes, which can improve the ammonia synthesis rate and is the main reason why C@NiO@Ni shows a higher NRR rate than C@NiO. In addition, Ni0 had almost no N2 fixation activities. Therefore, the NRR rate of C@Ni was attributed to the weak catalytic activities of carbon because C@Ni and carbon had similar NRR performances. The NH3 yield and FE of C@NiO@Ni in acidic and neutral media are also shown in Fig. S13;† the NRR performances in 0.05 M H2SO4 and 0.1 M Na2SO4 were both lower than that in 0.1 M KOH due to the recently reported enhancement effect of alkali metal potassium ions.26 The K+ ion in the electrolyte can bind with nitrogen and enrich the stern layer interaction with the nitrogen molecules, resulting in a higher nitrogen concentration at the catalyst surface.27 To prove this, NRR was also conducted in 0.05 M K2SO4, 0.1 M KCl and 0.1 M KNO3 electrolytes (Fig. S13†). The NH3 yields were similar for 0.1 M KOH, 0.05 M K2SO4, 0.1 M KCl and 0.1 M KNO3, but FE for 0.1 M KOH was much higher than that for the other neutral K+ electrolytes, which further confirmed that an alkaline electrolyte can suppress HER and enhance the efficiency of NRR.
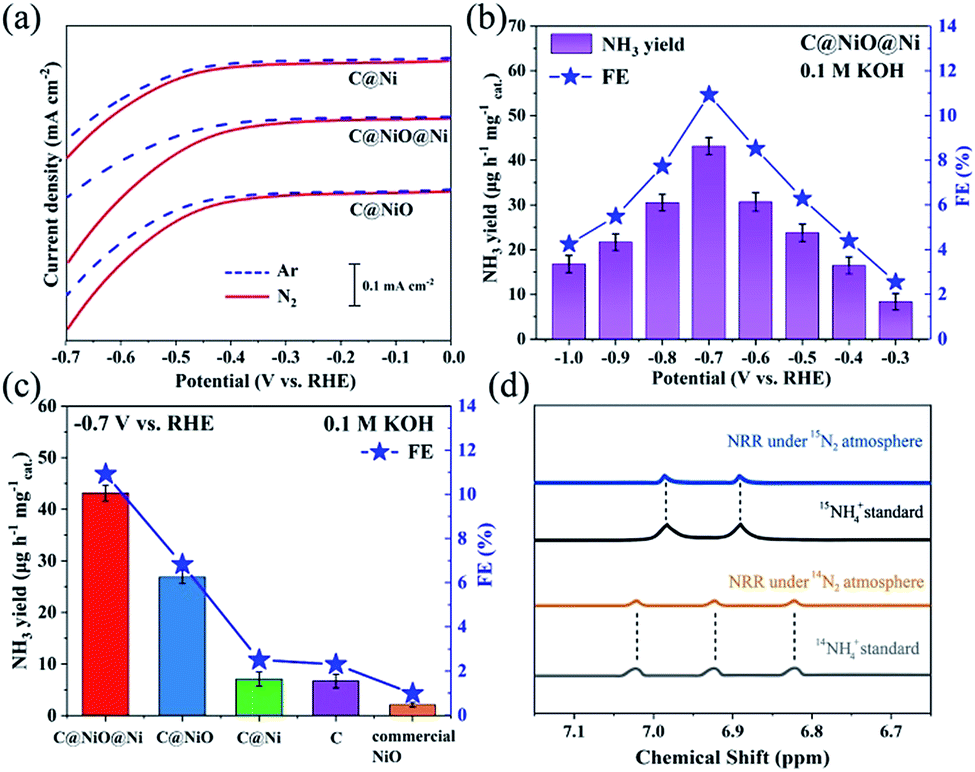 | ||
| Fig. 3 (a) LSV curves of C@Ni, C@NiO@Ni and C@NiO in Ar- or N2-saturated 0.1 M KOH. (b) NH3 yields and FEs of C@NiO@Ni for NRR at various potentials. (c) The NRR performance of different catalysts at −0.7 V. (d) 15N2 and 14N2 isotope labelling experiments. | ||
To attest that the produced ammonia was indeed synthesized from the electrochemical NRR of C@NiO@Ni, control experiments were carried out with alternate 1 hour cycles between Ar and N2 atmospheres at −0.7 V (Fig. S14†). The results indicated that almost no ammonia was generated under the Ar atmosphere. Moreover, the 14N2 and 15N2 isotopic labelling experiments were conducted. As shown in Fig. 3d, the corresponding 1H nuclear magnetic resonance spectrum only exhibits 14NH4+ or 15NH4+ signals after using 14N2 or 15N2 as the feeding gas. The NRR performance under the 15N2 atmosphere was very close to that under the 14N2 atmosphere (Fig. S15†). All these experimental results confirmed that NH3 was synthesized via the NRR process on the electrocatalysts. In addition, the possible by-product N2H4 was measured using the method of Watt and Chrisp (Fig. S16†).28 As presented in Fig. S17,† no N2H4 is produced after NRR on C@NiO@Ni at −0.7 V, demonstrating the excellent selectivity of the C@NiO@Ni catalyst for NH3. Fig. S18† confirms that the Ni-MOF precursor and bare carbon paper have low and negligible NRR activities, respectively.
As an electrocatalyst, stability is an important criterion to evaluate the NRR performance. As observed in Fig. 4a, the NH3 yield and FE of C@NiO@Ni have almost no change during the cycling experiments for 10 times at −0.7 V. Moreover, the current density curve of C@NiO@Ni was stable for 24 h NRR (Fig. 4b) and the NRR performance suggested a negligible change before and after 24, 48 and 72 h NRR (Fig. 4c). We further carried out a time-dependent experiment to assess its stability. As shown in Fig. 4d, the NH3 yield increases almost linearly with the reaction time. Meanwhile, the XRD pattern and SEM image of C@NiO@Ni after 24 h NRR also remained unchanged (Fig. S19†). All these results suggested the excellent durability of C@NiO@Ni in the alkaline electrolytes.
 | ||
| Fig. 4 (a) Durability test for 10 times at −0.7 V. (b) 24 h time-dependent current density curve for C@NiO@Ni. (c) NRR performance for C@NiO@Ni before and after the 24, 48 and 72 h cycling test. (d) NRR performance of C@NiO@Ni versus the reaction time. (e) NH3 yields and FEs against the reaction temperatures. (f) NH3 production and FE against the area of the carbon paper. | ||
Additionally, although there are an increasing number of reports about NRR electrocatalysts, their performance in possible practical applications is rarely discussed. Based on this, we made some explorations. Fig. 4e exhibits the NRR performance of C@NiO@Ni at various temperatures. It was evident that the NRR performance was enhanced with the applied temperature; the NH3 yields at 80 °C were about 5.36 and 2.57 times those at 0 °C and 20 °C, respectively. This was because the mass transfer rate was faster at a higher temperature than that at a lower temperature; these experimental results have also been reported in other recent studies.14,19 Furthermore, the apparent activation energy of C@NiO@Ni for NRR was calculated as 16.15 kJ mol−1 by the Arrhenius plot (Fig. S20a†). This value was much lower than that of the Co3Mo3N catalyst (57 kJ mol−1)29 used in the Haber–Bosch reaction but a little higher than that of some recently reported catalysts based on electrochemical NRR (7.4, 13.5 and 5.9 kJ mol−1 for N-doped carbon,19 Co3O4@NC,14 and Au/TiO2,30 respectively). We considered whether NRR could be performed directly in air to significantly reduce the cost of ammonia production. Fig. S20b† shows the NRR performance of C@NiO@Ni in air; the NH3 yield and FE were 33.01 μg h−1 mg−1 and 7.62%, respectively, which were still higher than those of most reported electrocatalysts under an N2 atmosphere, indicating that ammonia could be efficiently and directly generated in air on C@NiO@Ni. In addition, NRR tests were applied at a larger scale using the C@NiO@Ni catalyst. Fig. 4f suggests that the NH3 yield increases almost linearly with the increase in the electrode area, demonstrating that C@NiO@Ni can be applied on a larger scale. These experiments prove that C@NiO@Ni can be one of the potential candidates in the practical applications for NRR.
A possible reason for the excellent NRR performance on C@NiO@Ni was also discussed. Recent reports emphasize that oxygen vacancies can act as efficient active sites for NRR;31–35 therefore, electron paramagnetic resonance (EPR) was used to investigate the oxygen vacancy concentration of the catalysts. As shown in Fig. 5a, all samples demonstrate an EPR signal at g = 2.004, which can correspond to the electrons captured on the oxygen vacancies.36 The intensity of the signal demonstrated that C@NiO@Ni had a higher oxygen vacancy concentration than C@NiO, which was consistent with the XPS results. Moreover, the oxygen vacancy concentration in C@NiO@Ni was 1.23 times that in C@NiO, but the NH3 yield of C@NiO@Ni was 1.62 times that of C@NiO. Based on these results, we believe that the main factor for the performance difference between C@NiO@Ni and C@NiO is the effect of the NiO/Ni interfaces. Although there were many oxygen vacancies that could act as nitrogen activation sites in C@NiO, the catalyst did not have the ability to capture protons to synthesize ammonia in alkaline electrolytes. However, the abundant NiO/Ni interfacial sites in the C@NiO@Ni catalyst were conducive to proton adsorption. This structure played a vital role in overcoming the difficulty of capturing protons and enhancing the NRR performance in alkaline electrolytes. Thus, C@NiO@Ni and C@NiO showed very different NRR rates. In order to further explore the nitrogen adsorption capacity of the electrocatalysts, a nitrogen temperature-programmed desorption (N2-TPD) experiment was performed. As shown in Fig. 5a, C@Ni, C@NiO@Ni and C@NiO exhibit adsorption peaks between 50 and 380 °C. According to our analysis, these peaks can be assigned to the chemical adsorption of N2 on carbon in the catalysts and the peak area is proportional to the carbon content in the catalysts. The adsorption peaks between 380 and 800 °C were attributed to chemical adsorption on the oxygen vacancies in NiO. By comparison, C@NiO@Ni showed the largest N2 adsorption peak area, indicating that it had the strongest nitrogen adsorption capacity. Moreover, the adsorption peak area between 380 and 800 °C was positively correlated with the oxygen vacancy concentration, which further proved that oxygen vacancies were the main active sites for the NRR process. Additionally, electrochemical impedance spectroscopy (EIS) was performed to assess the conductivity of the catalyst (Fig. S21†). Compared to the C@Ni catalyst, C@NiO@Ni exhibited a steeper straight line in the low frequency region and a smaller semicircle in the high frequency region, which demonstrated that oxygen vacancies could enhance the electron transfer; thus, C@NiO@Ni had lower impedance.35 C@NiO@Ni also exhibited better conductivity than C@NiO, which proved that Ni0 could improve the electronic structure of the catalyst. Furthermore, C@Ni showed lower impedance than commercial Ni0, testifying that the carbon coating could further enhance the conductivity. In brief, conductive C@NiO@Ni not only had a high oxygen vacancy concentration for N2 adsorption and activation, but also possessed abundant NiO/Ni interfaces for proton adsorption. This dual-function electrocatalyst can significantly improve the efficiency of NRR.
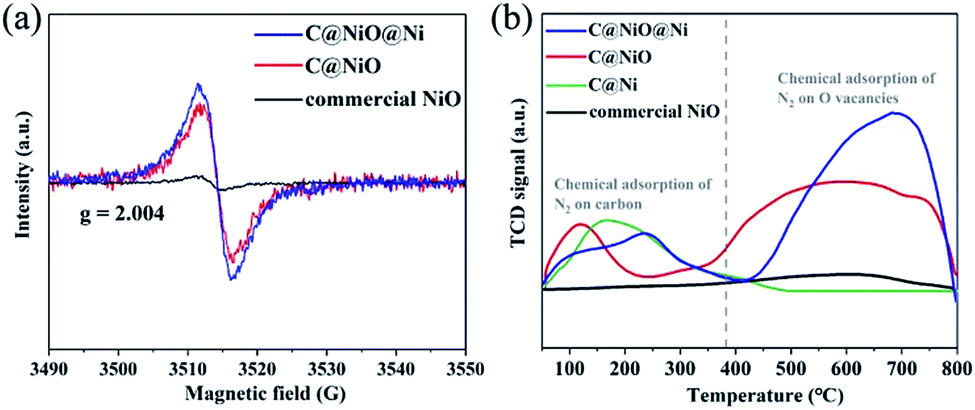 | ||
| Fig. 5 (a) EPR spectra of C@NiO@Ni, C@NiO and commercial NiO. (b) The N2-TPD profiles of C@NiO@Ni, C@NiO, C@Ni and commercial NiO. | ||
Conclusions
In summary, an MOF (Ni–BTC)-derived C@NiO@Ni microtube was proposed as a high-efficiency catalyst for NRR. It achieved an NH3 yield of 43.15 μg h−1 mgcat.−1 with FE of 10.9% at −0.7 V in 0.1 M KOH. The results also suggested excellent structural stability and long cycle life. The experimental data indicated an outstanding NRR performance that was mostly attributed to the high oxygen vacancy concentration in C@NiO@Ni. Moreover, the abundant NiO/Ni interfaces in the catalyst were beneficial for proton adsorption and for overcoming the difficulty of capturing protons in alkaline electrolytes. This work not only develops an effective strategy to achieve a higher NH3 yield and FE, but also opens up new avenues for the rational design of novel dual-function electrocatalysts for NRR applications with alkaline electrolytes.Experimental
Materials
NiCl2·6H2O, 1,3,5-benzenetricarboxylic acid, and commercial NiO were purchased from Aladdin Ltd. (Shanghai, China). N,N-Dimethylformamide and ethanol were purchased from Sinopharm Chemical Reagent Co., Ltd. Nafion (5 wt%) was purchased from Sigma-Aldrich Chemical Reagent Co., Ltd. Deionized water was purified through a Millipore system.Synthesis of precursor Ni-MOF (Ni–BTC) microcrystals
NiCl2·6H2O (1.43 g) was dissolved in a binary mixture of 20 mL of N,N-dimethylformamide and 20 mL of deionized water. 1,3,5-Benzenetricarboxylic acid (0.42 g) was dissolved in a binary mixture of 20 mL of N,N-dimethylformamide and 20 mL of deionized water. The above two solutions were then mixed under vigorous stirring for 10 min. After all the reagents were dissolved completely, the solution was transferred to a 50 mL Teflon-lined autoclave, and the autoclave was sealed and heated at 180 °C for 24 h. The precipitate was washed alternately with deionized water and ethanol by centrifugation, and the solid product was collected and dried in an oven at 60 °C for 12 h.Synthesis of C@Ni, C@NiO@Ni and C@NiO microtubes
Ni-MOF microcrystals were first heated in a furnace under N2 from room temperature to 600 °C at a heating rate of 2 °C min−1 and maintained for 3 h to obtain the C@Ni microtube. Then, C@Ni was heated in air from room temperature to 250 °C at a heating rate of 2 °C min−1 and maintained for 10 h or 15 h. The product after 10 h was marked as C@NiO@Ni and the product after 15 h was marked as C@NiO.Preparation of working electrode
The carbon paper was sonicated for 1 h in an ethanol solution and dried at ambient conditions. Then, 20 mg catalyst and 100 μL of the Nafion solution (5 wt%) were dispersed in a mixed solution containing 750 μL isopropanol and 2 mL H2O by 1 h sonication to form a homogeneous ink. Then, 50 μL catalyst ink was loaded on a 1 × 1 cm2 carbon paper and dried at 60 °C for 12 h.Characterizations
X-ray diffraction (XRD) data were obtained on AXS D8 ADVANCE A25 with Cu Kα radiation (40 kV, 40 mA) at a wavelength of 0.154 nm (Germany). SEM and energy dispersive spectroscopy (EDS) were performed using the ZEISS EVO18 scanning electron microscope at an accelerating voltage of 40 kV (Germany). TEM images were obtained on an FEI Talos 200S transmission electron microscope operated at 200 kV. The sizes of the prepared Ni-MOF, C@Ni, C@NiO, and C@NiO@Ni were measured by the Nano Measurer 1.2 software. XPS was performed on a Thermo ESCALAB 250Xi X-ray photoelectron spectrometer using Mg as the excitation source. Raman spectra were collected on a Thermo Fisher Raman spectrometer under backscattering geometry (λ = 532 nm). Electron paramagnetic resonance (EPR) spectra were obtained on a Bruker A300-10/12 system. Ion chromatography (ICP) was carried out on a Thermo Fisher ICP-500 system with AS23/AG23 and CS12/CG12A chromatographic columns. Temperature programmed desorption (TPD) was carried out on an AutoChem II 2920 system. Nuclear magnetic resonance was tested on a Bruker AVANCE III 600 MHz system. The absorbance data were measured on a Persee TU-19 UV-Vis spectrophotometer.Electrochemical measurements
The N2 reduction experiments were carried out in a two-compartment cell, which was separated by a Nafion 211 membrane. The electrochemical experiments were performed with a CHI 660E electrochemical analyzer (CH Instruments, Inc., Shanghai, China) using three-electrode configuration including a prepared working electrode, a platinum sheet electrode, and a saturated calomel electrode (SCE), which served as the working electrode, counter electrode, and reference electrode, respectively. The potentials reported in this work were converted to the RHE scale via calibration with the following equation: E (vs. RHE) = E (vs. SCE) + 0.2412 + 0.059 × pH. For the N2 reduction experiments, the 0.1 M KOH electrolyte was purged with N2 for 30 min before the measurement. A potentiostatic test was conducted in an N2-saturated 0.1 M KOH solution (70 mL) for 1 hour.Determination of NH4+
NH3/NH4+ concentration analysis was conducted using the Nessler's reagent method.23 First, 50 mL of the solution was placed in a 50 mL flask. Then, 1 mL of the potassium sodium tartrate solution was added to the flask. After blending, 1 mL of the Nessler's reagent was added to the same flask and mixed. Then, the mixture was left to stand for 10 min for full colour processing. Finally, the concentration of NH3/NH4+ was tested using a UV-Vis spectrophotometer at a 420 nm wavelength. The fitting curve (y = 0.1685x + 0.008, R2 = 0.998) showed a good linear relation of the absorbance value with the NH4+ concentration.Determination of N2H4
N2H4 in the electrolyte was estimated using the method by Watt and Chrisp.28 A mixture of C9H11NO (5.99 g), HCl (concentrated, 30 mL) and ethanol (300 mL) was used as a colour reagent. In detail, 5 mL electrolyte was removed from the electrochemical reaction vessel, added into 5 mL of the above prepared colour reagent solution, and stirred for 15 min at room temperature. The absorbance of the resulting solution was measured at 455 nm. The concentration absorbance curves were calibrated using a standard N2H4·H2O solution with a series of concentrations. The fitting curve (y = 1.6331x + 0.0187, R2 = 0.999) showed a good linear relation of the absorbance value with the N2H4 concentration.Calculations of NH3 formation rate and faradaic efficiency (FE)
The rate of formation of NH3 was calculated using the following equation: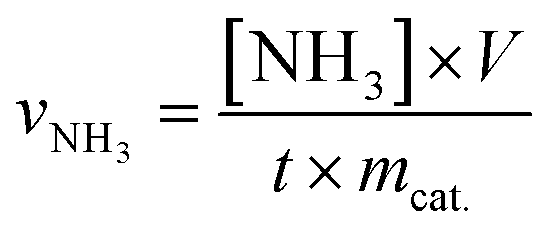
FE was calculated according to the following equation:
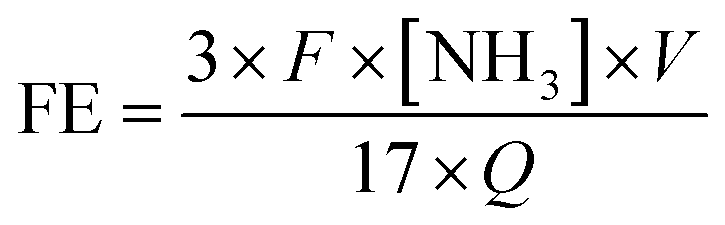
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was sponsored by the National Natural Science Foundation of China (Grant No. 21561026, 21802078, and 21965027) and the National First-rate Discipline Construction Project of Ningxia: Chemical Engineering and Technology (Grant No. NXY-LXK2017A04).Notes and references
- T. Oshikiri, K. Ueno and H. Misawa, Green Chem., 2019, 21, 4443–4448 RSC
.
- B. Cui, J. Zhang, S. Liu, X. Liu, W. Xiang, L. Liu, H. Xin, M. J. Lefler and S. Licht, Green Chem., 2017, 19, 298–304 RSC
- C. Mao, J. Wang, Y. Zou, H. Li, G. Zhan, J. Li, J. Zhao and L. Zhang, Green Chem., 2019, 21, 2852–2867 RSC
- M. A. Shipman and M. D. Symes, Catal. Today, 2017, 286, 57–68 CrossRef CAS
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Catal. Today, 2017, 286, 2–13 CrossRef CAS
- J. Y. Becker and S. Avraham, J. Electroanal. Chem., 1990, 280, 119–127 CrossRef CAS
- J. Schimpl, H. M. Petrilli and P. E. Blöchl, J. Am. Chem. Soc., 2003, 125, 15772–15778 CrossRef CAS PubMed
- K. A. Brown, D. F. Harris, M. B. Wilker, A. Rasmussen, N. Khadka, H. Hamby, S. Keable, G. Dukovic, J. W. Peters, L. C. Seefeldt and P. W. King, Science, 2016, 352, 448–450 CrossRef CAS PubMed
- H. Liu, L. Wei, F. Liu, Z. Pei, J. Shi, Z.-j. Wang, D. He and Y. Chen, ACS Catal., 2019, 9, 5245–5267 CrossRef CAS
- J. Nash, X. Yang, J. Anibal, J. Wang, Y. Yan and B. Xu, J. Electrochem. Soc., 2017, 164, 1712–1716 CrossRef
- Y. Zhang, W. Qiu, Y. Ma, Y. Luo, Z. Tian, G. Cui, F. Xie, L. Chen, T. Li and X. Sun, ACS Catal., 2018, 8, 8540–8544 CrossRef CAS
- X. Wu, L. Xia, Y. Wang, W. Lu, Q. Liu, X. Shi and X. Sun, Small, 2018, 14, e1803111 CrossRef PubMed
- Q. Liu, X. Zhang, B. Zhang, Y. Luo, G. Cui, F. Xie and X. Sun, Nanoscale, 2018, 10, 14386–14389 RSC
- S. Luo, X. Li, B. Zhang, Z. Luo and M. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 26891–26897 CrossRef CAS PubMed
- S. Mukherjee, D. A. Cullen, S. Karakalos, K. Liu, H. Zhang, S. Zhao, H. Xu, K. L. More, G. Wang and G. Wu, Nano Energy, 2018, 48, 217–226 CrossRef CAS
- Y. Yang, X. Sun, G. Han, X. Liu, X. Zhang, Y. Sun, M. Zhang, Z. Cao and Y. Sun, Angew. Chem., Int. Ed., 2019, 58, 10644–10649 CrossRef CAS PubMed
- Y. Liu, Y. Su, X. Quan, X. Fan, S. Chen, H. Yu, H. Zhao, Y. Zhang and J. Zhao, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS
- C. Lv, C. Yan, G. Chen, Y. Ding, J. Sun, Y. Zhou and G. Yu, Angew. Chem., Int. Ed., 2018, 57, 6073–6076 CrossRef CAS PubMed
- K. Chu, Y.-p. Liu, J. Wang and H. Zhang, ACS Appl. Energy Mater., 2019, 2, 2288–2295 CrossRef CAS
- F. Song, W. Li, J. Yang, G. Han, P. Liao and Y. Sun, Nat. Commun., 2018, 9, 4531 CrossRef PubMed
- J. Wang, S. Mao, Z. Liu, Z. Wei, H. Wang, Y. Chen and Y. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 7139–7147 CrossRef CAS PubMed
- Y. Yang, Z. Lun, G. Xia, F. Zheng, M. He and Q. Chen, Energy Environ. Sci., 2015, 8, 3563–3571 RSC
- R. H. Leonard, Clin. Chem., 1963, 9, 417–422 CAS
- X. Li, X. Sun, L. Zhang, S. Sun and W. Wang, J. Mater. Chem. A, 2018, 6, 3005–3011 RSC
- T. Oshikiri, K. Ueno and H. Misawa, Angew. Chem., Int. Ed., 2016, 55, 3942–3946 CrossRef CAS PubMed
- Y. Song, D. Johnson, R. Peng, D. K. Hensley, P. V. Bonnesen, L. Liang, J. Huang, F. Yang, F. Zhang, R. Qiao, A. P. Baddorf, T. J. Tschaplinski, N. L. Engle, M. C. Hatzell, Z. Wu, D. A. Cullen, H. M. Meyer, B. G. Sumpter and A. J. Rondinone, Sci. Adv., 2018, 4, e1700336 CrossRef PubMed
- D. Wu, H. Wang, H. Huang, R. Zhang, L. Ji, H. Chen, Y. Luo, J. You, D. Tang, Z. Zhang and X. Sun, Chem. Commun., 2019, 55, 7546–7549 RSC
- G. W. Watt and J. D. Chrisp, Anal. Chem., 1952, 24, 2006–2008 CrossRef CAS
- R. Kojima and K.-i. Aika, Appl. Catal., A, 2001, 218, 121–128 CrossRef CAS
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed
- H. Xie, H. Wang, Q. Geng, Z. Xing, W. Wang, J. Chen, L. Ji, L. Chang, Z. Wang and J. Mao, Inorg. Chem., 2019, 58, 5423–5427 CrossRef CAS PubMed
- L. Zhang, X.-Y. Xie, H. Wang, L. Ji, Y. Zhang, H. Chen, T. Li, Y. Luo, G. Cui and X. Sun, Chem. Commun., 2019, 55, 4627–4630 RSC
- C. Zhang, S. Liu, T. Chen, Z. Li and J. Hao, Chem. Commun., 2019, 55, 7370–7373 RSC
- J. Yu, C. Li, B. Li, X. Zhu, R. Zhang, L. Ji, D. Tang, A. M. Asiri, X. Sun, Q. Li, S. Liu and Y. Luo, Chem. Commun., 2019, 55, 6401–6404 RSC
- W. Kong, R. Zhang, X. Zhang, L. Ji, G. Yu, T. Wang, Y. Luo, X. Shi, Y. Xu and X. Sun, Nanoscale, 2019 10.1039/c9nr03678d
- I. Nakamura, N. Negishi, S. Kutsuna, T. Ihara, S. Sugihara and K. Takeuchi, J. Mol. Catal. A: Chem., 2000, 161, 205–212 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9se00691e |
| This journal is © The Royal Society of Chemistry 2020 |