
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of a synthesis method for a difluoroglycine derivative based on a path generated by quantum chemical calculations†
Tsuyoshi
Mita
 *ab,
Yu
Harabuchi
abcd and
Satoshi
Maeda
*abce
*ab,
Yu
Harabuchi
abcd and
Satoshi
Maeda
*abce
aInstitute for Chemical Reaction Design and Discovery (WPI-ICReDD), Hokkaido University, Kita 21 Nishi 10, Kita-ku, Sapporo, Hokkaido 001-0021, Japan. E-mail: tmita@icredd.hokudai.ac.jp; smaeda@eis.hokudai.ac.jp
bJST, ERATO Maeda Artificial Intelligence for Chemical Reaction Design and Discovery Project, Kita 10 Nishi 8, Kita-ku, Sapporo, Hokkaido 060-0810, Japan
cDepartment of Chemistry, Faculty of Science, Hokkaido University, Kita 10 Nishi 8, Kita-ku, Sapporo, Hokkaido 060-0810, Japan
dJST, PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
eResearch and Services Division of Materials Data and Integrated System (MaDIS), National Institute for Materials Science (NIMS), Tsukuba, Ibaraki 305-0044, Japan
First published on 22nd May 2020
Abstract
The systematic exploration of synthetic pathways to afford a desired product through quantum chemical calculations remains a considerable challenge. In 2013, Maeda et al. introduced ‘quantum chemistry aided retrosynthetic analysis’ (QCaRA), which uses quantum chemical calculations to search systematically for the decomposition paths of a target product and proposes a synthesis method. However, until now, no new reactions suggested by QCaRA have been reported to lead to experimental discoveries. Using a difluoroglycine derivative as a target, this study investigated the ability of QCaRA to suggest various synthetic paths to the target without relying on previous data or the knowledge and experience of chemists. Furthermore, experimental verification of the most promising path chosen by an organic chemist among the predicted paths led to the discovery of a synthesis method for a difluoroglycine derivative. We emphasize that the purpose of this study is not to propose a fully automated workflow. Therefore, the extent of the hands-on expertise of chemists required during the verification process was also evaluated. These insights are expected to advance the applicability of QCaRA to the discovery of viable experimental synthetic routes.
Introduction
Recent developments in computational algorithms and computer technology have allowed quantum chemical calculations to become an essential tool in the field of organic synthesis.1–5 One of the most important applications of quantum chemical calculations is for the geometry optimization of transition states.6 In many recent studies, reaction mechanisms have been elucidated by calculating the transition state through quantum chemical calculations.1–5 Increasingly, these calculations are being used not only to confirm experimental results but also to design reactions with improved reactivity and selectivity. In general, quantum chemical calculations require an initial guess for the target molecular structure. That is, they can only be used when a reaction mechanism is assumable. Therefore, quantum chemical calculations have never been used to predict a synthesis method for which a similar method was not already available.Concurrently, extensive efforts have been made to predict synthesis methods based on enormous amounts of experimental data.7–15 Some of these efforts are based on a concept similar to retrosynthetic analysis, which is traditionally used in organic synthesis.16 In the conventional retrosynthetic analysis, a synthesis method is proposed by dividing the target molecule into fragments and then determining the synthetic equivalents of each fragment. However, only expert organic chemists, who can often make appropriate choices based on their knowledge and experience, can identify suitable approaches from the vast collection of fragmentation patterns and synthesis methods. Therefore, there has been recent focus on developing the ability of machines to make such choices by learning from huge quantities of experimental data. Moreover, some successful tools of this nature have already been recognized in the field of organic chemistry.
Despite these efforts, it remains unclear whether it is feasible to propose a synthesis method from scratch using quantum chemical calculations. To realize such predictions, we applied ‘quantum chemistry aided retrosynthetic analysis’17 (QCaRA), which was introduced in 2013, as a tool to provide hypothetical paths to a real organic synthesis for the first time. In this paper, the acronym QCaRA has been used for convenience. QCaRA applies quantum chemical calculations to search systematically for the decomposition paths of a target molecule and suggest a synthesis method corresponding to the reverse reaction of the obtained path. This concept is similar to retrosynthetic analysis, as it predicts a reaction path by decomposing the target molecule into fragments and tracing the decomposition pathways backward to generate input molecules. However, QCaRA differs from general retrosynthetic analysis in three ways: (1) the transition state of the decomposition process can be obtained, (2) the effect of the presence of compounds other than the target compound on the decomposition path and its transition state can be predicted, and (3) reactant candidates that are not found in previous data and would typically be considered implausible can be included. Of these, (1) and (2) are both advantageous and disadvantageous. Because the transition state of the decomposition process is also that of the formation process, its energy and structure can guide the design of a synthetic path. Furthermore, executing calculations with a virtual catalyst added to the system enables the selective exploration of paths in which the catalyst effectively lowers the energy barrier. However, because of the vast amount of catalyst options, the result depends on the selection of a suitable catalyst, which is a serious drawback.
Previously, the prediction capability of QCaRA has been considered theoretical, as no new reactions have been discovered experimentally based on the synthetic paths proposed by QCaRA. Maeda et al. introduced QCaRA based on the results of an extensive exploration of the decomposition paths of glycine. Prior to this analysis, they had reported that glycine could be obtained from acetolactone and ammonia or ammonium ylide and CO2 as reactant pairs.18,19 Separately, Mita et al. developed a synthesis method involving the carboxylation of ammonium ylide with CO2.20 Although the findings of Maeda et al. and Mita et al. were separate outcomes in different fields, this case can be considered to demonstrate the possibility of experimentally verifying a prediction made through QCaRA.
Herein, we report the first successful discovery of a synthesis method based on a theoretical path suggested by QCaRA. In this study, to demonstrate the power of QCaRA, we chose a target molecule for which no facile synthesis methods exist, i.e. α,α-difluoroglycine (NH2–CF2–CO2H). The replacement of a hydrogen atom with a fluorine atom in bioactive molecules such as α-amino acids is a promising strategy for enhancing the biological activity and bioavailability via increased lipophilicity and improved stability against enzymatic degradation. However, to our knowledge, no synthesis method of difluoroglycine itself has been reported. Although the basic difluoroglycine core (“N”–CF2–CO2H with “N” = NR2, N3, NO2, etc.) has been constructed by organic synthesis, it can only be performed in inefficient ways using already functionalized starting materials like Br–CF2–CO2R.21 If simple, small, easily accessible molecules could be assembled to create the target structure, such a method would be effective due to its novelty and good performance. It should be emphasized that the theoretical paths, including the one that led the discovery of the synthesis method, were generated solely from quantum chemical calculations without the input of any previous experimental data or human knowledge. Then, the most promising path was chosen by an organic chemist for further computational and experimental verification. The verification process demanded human knowledge and experience. Thus, this paper discusses the contribution of quantum chemical calculations to the prediction of possible synthetic methods and the use of human knowledge for further verification.
Results and discussion
Prediction of synthetic paths using QCaRA
Over the last several years, the development of various automated reaction path search methods has shown that many reaction paths can be predicted.22–24 If previously unknown chemical transformations were to be predicted and such novel chemical transformations achieved experimentally, a series of new chemical reactions could be discovered. This study used QCaRA, proposed by Maeda et al. in 2013, to carry out systematic searches for the decomposition paths of α,α-difluoroglycine as a target molecule and suggest a synthetic path that used the resulting decomposition products or their equivalents as reactants.17QCaRA requires an automated reaction path search method as its reaction path exploration engine, and, in principle, any automated reaction path search method can be used for this purpose. The technical difficulty of QCaRA is that the search target is a reaction path that does not actually proceed. Conventional automated reaction path searches can be performed by assuming thermodynamics or kinetics. In other words, it is not necessary to search for a path leading to the thermodynamically unstable structure or for a path with a high energy barrier. In QCaRA, by contrast, the decomposition product that is energetically less stable than the target molecule is often more favourable in the forward synthetic path (exothermic). Thermodynamically very unstable decomposition products should also be searched for because their equivalents could be candidate reactants for novel reactions. Therefore, in this study, an artificial force induced reaction (AFIR) method,25,26 which has been proven to provide an exhaustive search, including for unstable decomposition products,27 was adopted as an automated reaction path search method for QCaRA.
The AFIR method is one of the automated reaction path search methods implemented in the global reaction route mapping (GRRM) program.17 The AFIR method induces structural changes by applying a virtual artificial force between molecules or fragments within a molecule. The systematic repetition of this process makes it possible to calculate a path for transforming a given reactant into an unknown product. It also allows the prediction of an unknown reaction by analysing the network of obtained reaction paths. The further details of this method are beyond the scope of this paper.25,26 In this study, the AFIR method was only applied to structures with the same bonding pattern as the input molecule and the decomposition path was comprehensively searched.28 After prospective decomposition products were selected from the obtained candidates, the automated reaction path search was also performed using their equivalents as starting materials, where the option to select a path kinetically was used.29 Finally, the predicted synthetic path was experimentally verified. The calculations and experiments are detailed in the ESI.†
Prediction of synthesis methods for difluoroglycine
This study focused on the synthesis of α,α-difluoroglycine, for which an efficient synthesis method has not yet been established. However, obtaining such a synthesis protocol is highly desirable due to the expected usefulness. Therefore, the difluoroglycine derivative was used as an input when executing the automated reaction path search. A number of reactant candidates were obtained through a constrained search, in which the AFIR method was only applied to the equilibrium structures with the same bonding pattern as the input structure, i.e. conformers of difluoroglycine.Of the obtained reactant candidates, the 30 with the lowest energies are shown in Fig. 1. These reactant candidates correspond to decomposed species obtained by the systematic exploration of the dissociation pathways of difluoroglycine. The most stable reactant pair is R1, which consists of difluoromethylamine and CO2. The set with the second lowest energy (R2) consists of fluoroimine, CO2, and hydrogen fluoride. Although most species observed in these 30 reactant candidate sets are stable molecules in which all atoms fulfil the octet rule, there are also some short-lived intermediates like carbene. The energy levels of the reactant candidates are depicted in Fig. 2 together with the reaction path network on which these species were predicted. Notably, the 4 sets with the lowest energies, i.e. R1–R4, cannot be reactant candidates because the reactions from them to the target molecule are endothermic. Among the other 26 reaction candidates, the path from R26 (CF2, NH3, and CO2), highlighted in Fig. 1 and 2, was selected considering the energy barrier for the formation of difluoroglycine and the availability of the reactants. As seen in the reaction profile for this path (Fig. 3), the energy barrier along the path from R26 is reasonably small, and the three simple components, i.e. CF2, NH3, and CO2, convergently react to give difluoroglycine in one step.
 | ||
| Fig. 1 Reactant candidates predicted by the automated reaction path search. | ||
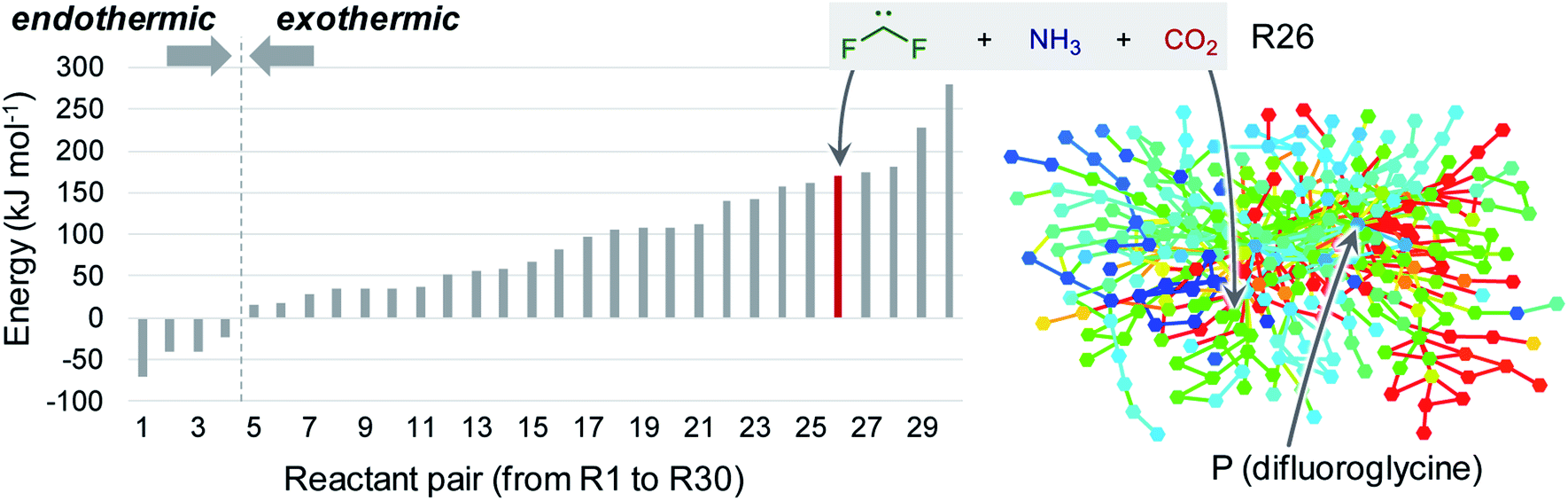 | ||
| Fig. 2 Histogram of the energy levels of the 30 reactant candidates (R1–R30, listed in Fig. 1) (left) and the reaction path network obtained through the automated reaction path search (right). In the reaction path network, each node and edge is colour-coded according to the energy of the corresponding equilibrium structure and transition state (blue: −70.4 kJ mol−1, aqua: 54.5 kJ mol−1, green: 179.5 kJ mol−1, yellow: 304.5 kJ mol−1, and red: 429.5 kJ mol−1). | ||
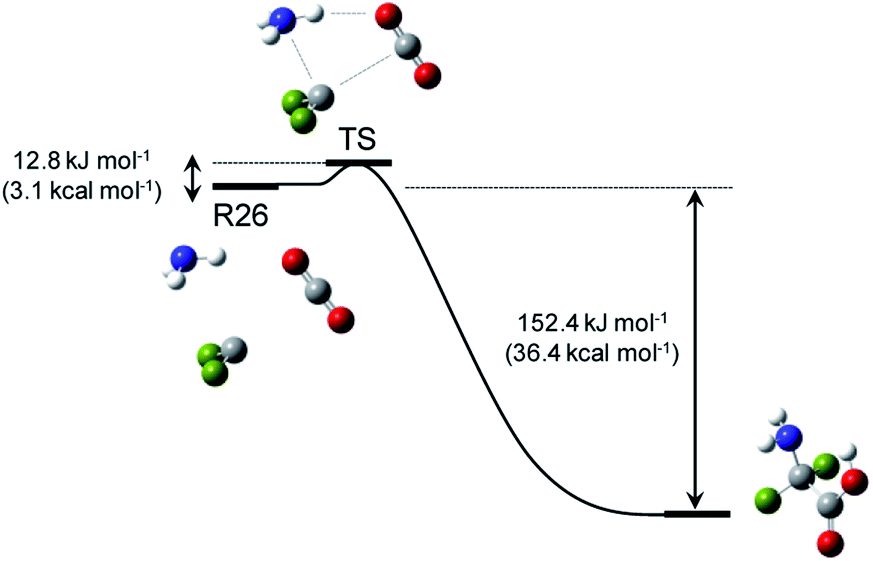 | ||
| Fig. 3 Reaction path for R26, which was selected for experimental demonstration. The H, C, N, O, and F atoms are coloured white, grey, blue, red, and green, respectively. | ||
Among the components of R26, NH3 and CO2 are both commercially available. In particular, CO2 is an abundant, inexpensive, and non-toxic chemical utilized in various organic transformations as a C1 unit.30 Furthermore, difluorocarbene can be generated in situ in several ways.31 Among them, a method using CF3− as a precursor, which is generated from Me3SiCF3 (Ruppert–Prakash reagent) and Ph3SiF2·NBu4 (TBAT), was initially selected owing to the reasonable accessibility of both reagents.32 To evaluate the validity of this synthesis method, the automated reaction path search was executed using CF3− + NH3 + CO2 as reactants, with the constrained search option applying the AFIR method only to equilibrium structures that the system can reach at a reaction temperature of 300 K in a reaction time of 1 h. The resulting reaction path networks are illustrated in Fig. 4a and b, which represent identical networks colour-coded by (a) the energy of the structure to which each node corresponds and (b) the calculated yield of the corresponding structure. The calculated yield was obtained by simulating the propagation of the population from the initial structure using the rate constant matrix contraction method under a reaction temperature of 300 K and a reaction time of 1 h. Fig. 4a shows that the target product, difluoroglycine, is obtained on the network, while Fig. 4b indicates that the calculated yield of difluoroglycine is almost zero because the equilibrium between CF3− and CF2 + F− favours the former. As a result, CF3CO2−, in which CF3− is directly bound to CO2, was obtained as the main product (99.8%).
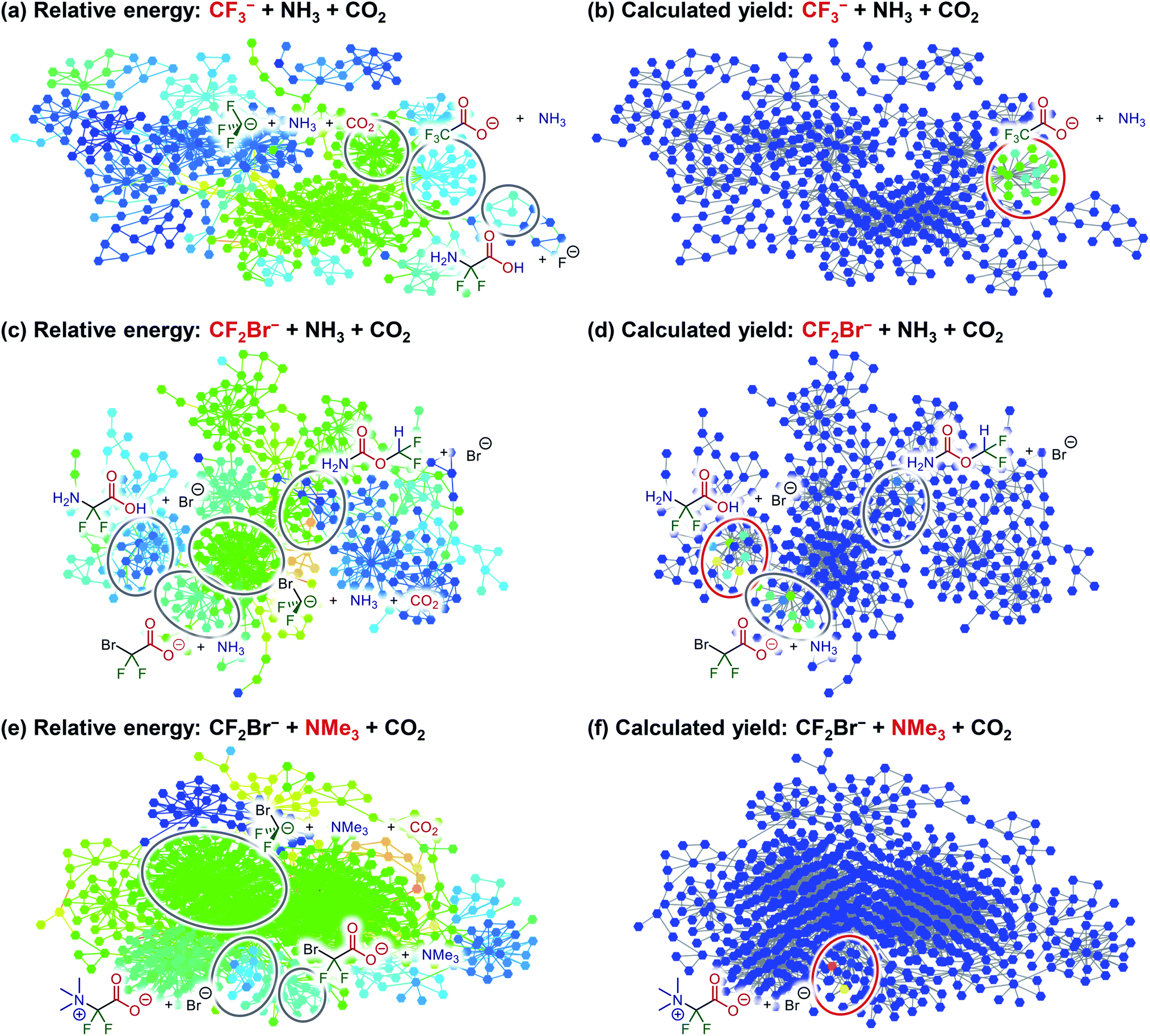 | ||
| Fig. 4 Reaction path networks corresponding to (a and b) CF3− + NH3 + CO2, (c and d) CF2Br− + NH3 + CO2, and (e and f) CF2Br− + NMe3 + CO2. Each node and edge in the reaction path networks of (a), (c), and (e) is colour-coded by the energy (relative to the most stable structure) of the corresponding equilibrium structure and transition state (blue: 0.0 kJ mol−1, aqua: 125.0 kJ mol−1, green: 250.0 kJ mol−1, yellow: 375.0 kJ mol−1, and red: 500.0 kJ mol−1). Each node in the reaction path networks of (b), (d), and (f) is colour-coded by the calculated yield of the corresponding equilibrium structure at a reaction temperature of 300 K and a reaction time of 1 h (blue: 0.0%, aqua: 0.8%, green: 4.0%, yellow: 20.0%, and red: 100.0%). | ||
As the addition of CF3− to CO2 was faster than α-elimination from CF3−, to enhance α-elimination, Me3SiCF2Br was then chosen as the difluorocarbene precursor.33 The improved leaving ability of bromide accelerates α-elimination from the corresponding CF2Br−, which can be generated in the presence of an appropriate silane activator.33 To examine the validity of this method, the reaction path networks were obtained with CF2Br− + NH3 + CO2 as reactants (Fig. 4c and d). The usage of CF2Br− was revealed to shift the equilibrium between CF2Br− and CF2 + Br− towards the latter; thus, difluoroglycine was obtained. However, as shown in Fig. 4d, the generation of CF2BrCO2− (29.3%) competes with the process for forming the target product (69.6%). Furthermore, it was suggested that NH2–CO2–CHF2 was obtained as a minor by-product (0.8%).
To further improve the yield of difluoroglycine, the addition of the amine to difluorocarbene should be accelerated. It is also necessary to reduce proton transfer from the amine to CF2 in order to suppress the formation of NH2–CO2–CHF2. Therefore, to meet both of these requirements simultaneously, a tertiary amine was selected. To test the validity of this hypothesis, the reaction path networks with CF2Br− + NMe3 + CO2 as reactants were obtained, as illustrated in Fig. 4e and f. These results verify that the desired difluoroglycine derivative was selectively obtained when NMe3 was used as a reactant. In addition, the calculated yield of 99.98% could be further increased to 99.99% by lowering the reaction temperature to 250 K and reached almost 100% when the temperature was further lowered to 200 K.
Experimental verification of the predicted synthetic method
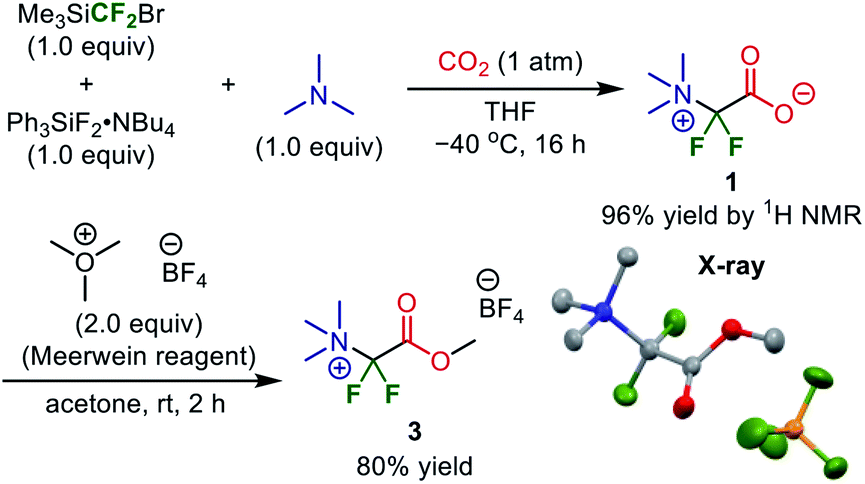
As a calculated yield of 100% was achieved with CF2Br− + NMe3 + CO2 as reactants, we chose to verify the validity of this synthetic path for the difluoroglycine derivative experimentally. We first examined several silane activators for the generation of the difluorocarbene in situ from Me3SiCF2Br in THF under an atmosphere of 1 atm CO2 at room temperature (∼300 K) (Table 1). When CsF was employed as the silane activator, target difluoroglycine derivative 1 was obtained in 10% yield together with compound 2, which was derived from protonation of the ammonium ylide intermediate, in 27% yield. Although F·NBu4 (TBAF) was not a suitable activator, the use of TBAT dramatically improved the yield of 1 to 81% and gave 2 in only 7% yield. Subsequently, the reaction was carried out at a lower temperature (−40 °C = 233 K) for 16 h and the generated precipitates were isolated by filtration. It was found that the obtained solids, as a mixture with Br·NBu4, contained 1 and 2 in 96% and 2% yields, respectively. This result, with 1 obtained in high yield, is consistent with our prediction based on quantum chemical calculations. In addition, we conducted the reaction of Me3SiCF3 (1 equiv.), TBAT (1 equiv.), and NMe3 (1 equiv.) under a CO2 atmosphere (1 atm) in THF at room temperature for 16 h. This reaction gave trifluoroacetate (CF3CO2·NBu4) in 96% yield, which is also consistent with the theoretical prediction that CF3− is consumed quickly by reacting with CO2 rather than providing the difluorocarbene.
Although the reaction occurred in THF, product 1 was obtained as a precipitate that was insoluble in THF. Thus, the precipitate should be dissolved in another solvent for further purification to remove Br·NBu4. However, the dissolution of salt 1 in solvents such as H2O, MeOH, CH3CN, and DMF promoted to varying extents a decarboxylation–protonation process that generated 2. For example, when the obtained product mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 = 98:2) was dissolved in various solvents at room temperature for 1 h, the ratio of 1:2 changed to 87:13 (H2O), 89:11 (MeOH), 62:38 (CH3CN), and 50:50 (DMF). This result indicates that it might be difficult to achieve the purification of 1 at this stage.
:2 = 98:2) was dissolved in various solvents at room temperature for 1 h, the ratio of 1:2 changed to 87:13 (H2O), 89:11 (MeOH), 62:38 (CH3CN), and 50:50 (DMF). This result indicates that it might be difficult to achieve the purification of 1 at this stage.
Therefore, esterification was investigated as a method of isolating and purifying the difluoroglycine derivative without decarboxylation. First, MeI was employed as a methylating reagent. However, established conditions using MeI in DMF or an excess amount of MeI without any solvent (neat conditions) did not afford the corresponding methyl carboxylate. Instead, the undesired decarboxylation–protonation process proceeded to some extent. Thus, esterification using highly electrophilic Me3O·BF4 (Meerwein reagent)34 was investigated. However, as the esterification process also requires redissolution in a highly polar solvent in which salt 1 is soluble, there was some concern that the yield might be decreased. First, CH2Cl2, which is a typical solvent for esterification using the Meerwein reagent, was used (Table 2). Although target methyl ester 3 was obtained in 18% yield, protonated compound 4 was obtained as a by-product in 73% yield. In contrast, when esterification was performed under solvent-free ball-milling conditions, the yield of 3 was increased to 48%. Furthermore, although AcOEt was not a suitable solvent, the use of acetone as a solvent improved the yield of 3 to 81%.
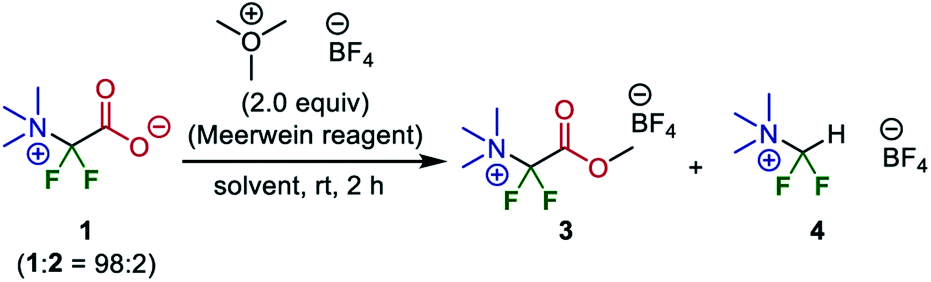
Subsequently, we conducted preparative-scale synthesis using 1 mmol of each reactant under the optimized reaction conditions (Fig. 5). After 1 was obtained as a mixture with Br·NBu4, the obtained solids were treated with Me3O·BF4 in acetone. The resulting product (methyl ester 3) was washed with MeOH to remove Br·NBu4, unreacted Me3O·BF4, and protonated compound 4, affording 3 in 80% yield (205 mg). The structure was confirmed by X-ray crystallography after recrystallization from MeOH (CCDC 1971834). This method is an elegant demonstration of the synthesis of a difluoroglycine derivative from three simple compounds through multi-component assembly under silica-gel-column-chromatography-free conditions. In most reported examples,31 an excess amount of difluorocarbene precursor (>2 equiv.) is necessary, probably due to undesirable carbene dimerization. In contrast, our new protocol achieves a high yield with just 1 equiv. of Me3SiCF2Br, which emphasizes the practicality of this synthesis. Other substrate candidates predicted by the quantum chemical calculations as well as the application of this compound are now being investigated and the results will be reported in due course.35
 | ||
| Fig. 5 Preparative-scale synthesis of a difluoroglycine derivative without silica-gel column chromatography. | ||
We would like to highlight that this synthesis of a difluoroglycine derivative is the first experimental demonstration of a reaction suggested by QCaRA. Moreover, the process by which QCaRA provided predictions, i.e. the process for obtaining reactant candidates (Fig. 1) after deciding the target, did not rely on the experience or intuition of chemists nor was any previous experimental data used in this process. Although the developer version of the GRRM program was used in this study, the same calculation can be performed using the GRRM17 program.28,36 It is noted that a chemist's knowledge was indispensable in selecting difluoroglycine as the target and choosing R26 from the many candidates predicted by QCaRA. Automating these two processes remains a future challenge.
The prediction strongly motivated us to conduct the subsequent experimentation. However, the experience and intuition of chemists also played an important role in validating the predicted synthetic path. First, because the proposed reactant candidates included a molecule (difluorocarbene) that was not commercially available, it was generated in situ using a previously reported method. The calculations to determine what products were actually produced and in what proportion suggested that the yield of the target compound could be improved by changing the type of amine. The use of a tertiary amine based on this prediction was also decided by chemists according to their experience.
The process that demanded the greatest input from chemists was the isolation of products. After determining the optimal reagents and reaction conditions, the product was precipitated as a mixture with Br·NBu4; however, the target difluoroglycine derivative was not sufficiently stable during subsequent isolation processes. Because the calculated yield was approximately 100%, the decision was taken that isolation should be performed after methyl esterification. After examining several solvents for this process, acetone was found to be the most suitable. In fact, the trial and error required for this step was the most time-consuming process in this study.
The selection of both THF as the reaction solvent and the calculation method were also responsible for the success of this demonstration. THF was chosen as the reaction solvent because it is commonly used in CO2 fixation and carbene insertion chemistry. However, if a different solvent had been used, competition with decarboxylation might have prevented the isolation of the target product. For the computational techniques, it is possible that the choice of an unsuitable DFT functional or basis function could lead to incorrect predictions. It is undeniable that the experience and intuition of chemists, or even luck, contributed to appropriate choices being made.
Experimental
General experimental procedure
In an oven-dried round-bottom flask was placed TBAT (539.9 mg, 1.0 mmol, 1 equiv.). The flask was evacuated and backfilled with CO2 (3 times) followed by the addition of THF (10 mL). The mixture was stirred at room temperature until TBAT was completely dissolved. After adding trimethylamine (2 M in THF, 500 μL, 1.0 mmol, 1 equiv.) at 0 °C, the solution was cooled to −40 °C, and then Me3SiCF2Br (156 μL, 1.0 mmol, 1 equiv.) was added dropwise. After the resulting slurry was stirred at −40 °C for 16 h, the precipitate was isolated by filtration, washed with hexane, and dried under vacuum. The approximate yield of carboxylate 1 was 96%, as determined by 1H NMR spectroscopy using pyridine as an internal standard. The solids were then treated with the Meerwein reagent (295.8 mg, 2.0 mmol, 2 equiv.) in acetone (1.5 mL) for 2 h. After the solvent was removed under vacuum, the resulting solids were washed with a small amount of MeOH to afford methyl carboxylate 3 after filtration (1st crop: 176.6 mg, 2nd and 3rd crops: 28.2 mg, 0.803 mmol, 80% yield).White solids; IR (ATR): 3068, 2977, 1787, 1483, 1343 cm−1; 1H NMR (400 MHz, DMSO-d6) δ: 4.05 (s, 3H), 3.40 (s, 9H) ppm; 13C NMR (100 MHz, DMSO-d6) δ: 155.7 (JCF = 31.6 Hz), 112.5 (JCF = 282.7 Hz), 56.5, 49.4 ppm; 19F NMR (376 MHz, DMSO-d6) δ: −105.4 (CF2), −151.4 (BF4) ppm (internal reference: CF3CO2H in DMSO-d6 = −78.5 ppm); HRMS (ESI) m/z calcd for C6H12F2NO2+ [M − BF4−]+: 168.0831, found: 168.0833; calcd for BF4− [M − C6H12F2NO2+]: 87.0035, found: 87.0030.
Computational analysis
All the calculations were performed using the developer version of the GRRM program combined with the Gaussian 16 program.36,37 For all the calculations, the ωB97X-D functional and 6-31+G* basis functions were used and the Grid = FineGrid option was adopted. The Gibbs free energy values at 300 K and 1 atm were estimated by assuming ideal-gas, rigid-rotor, and harmonic vibrational models, where all the harmonic frequencies smaller than 50 cm−1 were set as 50 cm−1 as suggested in the literature.38 The QCaRA calculation was performed under gas-phase conditions, whereas the other calculations were performed in THF, as modelled by the CPCM method, because THF is often chosen as an experimental solvent for reactions involving CO2. Kinetic simulations were performed using the reaction path networks shown in Fig. 4, where the initial populations given to the reactants were propagated using the rate constant matrix contraction method at a reaction temperature of 300 K and a reaction time of 1 h.29 Further details on the reaction path searches and kinetic simulations are available in the ESI.†Conclusions
In this study, we applied QCaRA to a difluoroglycine derivative for which no efficient synthesis method has been established. Out of the 30 synthetic paths proposed for difluoroglycine, those involving a difluorocarbene, an amine, and CO2 were selected for further investigation. The effects of the difluorocarbene generation method (CF3−vs. CF2Br−) and the selected amine (NH3vs. NMe3) on the yield were then verified by calculation to determine a set of reactants that gave a calculated yield of almost 100%. Subsequently, experiments were conducted to verify the predicted synthetic path.It should be emphasized that QCaRA was not used to propose a fully automated workflow. QCaRA is used to explore possible paths leading to a given target structure. In the process of verifying such possibilities, a trial-and-error approach and the hands-on expertise of chemists are still required. After the synthetic target was chosen, the prediction of potential synthetic paths by QCaRA did not require the experience and intuition of chemists or previous experimental data. However, the experience of chemists and trial-and-error experiments were necessary during the verification process. In this example, this hands-on expertise of chemists was needed to select a suitable path from among those predicted by QCaRA, to propose a difluorocarbene production method, to select an appropriate amine, and to isolate the product. In particular, experimental trial and error was key for the purification of the product.
This study demonstrated the effectiveness of QCaRA for predicting new synthesis methods. However, if the procedures used in this study were applied to a more complex molecule, the computational cost would be huge. In our previous applications of the AFIR method, the number of atoms contained in target systems was less than 30 when a small reactive centre was not assumable in the system. In QCaRA, it is usually difficult to assume a reactive centre because a limited search assuming a reactive centre may exclude important paths involving bond rearrangements occurring outside the reactive centre. It follows that the application of QCaRA to systems involving more than 30 atoms is not straightforward. For a catalytic reaction or a reaction involving a leaving group, QCaRA would need to be performed on the system to which the catalyst or the leaving group would be added, which would further limit the applicability of QCaRA. Moreover, to evaluate different catalysts systematically, QCaRA would need to be repeated while considering various metal/ligand combinations. In future, we hope to apply QCaRA to more complex systems by improving the calculation procedures, making a database of the results, and so forth.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Dr Mingoo Jin is greatly acknowledged for assistance with the X-ray crystallographic analysis. This work was financially supported by a Grant-in-Aid for Scientific Research (C) (No. 18K05096), JST-CREST (No. JPMJCR14L5), JST-PRESTO (No. JPMJPR16N8), JST-ERATO (No. JPMJER1903), and JSPS-WPI. T. M. thanks the Astellas Foundation for Research on Metabolic Disorders for financial support. We thank Ms. Takako Homma for editing a draft of this manuscript.Notes and references
- K. N. Houk and P. H.-Y. Cheong, Nature, 2008, 455, 309–313 CrossRef CAS PubMed.
- W. Thiel, Angew. Chem., Int. Ed., 2014, 53, 8605–8613 CrossRef CAS PubMed.
- W. M. C. Sameera, S. Maeda and K. Morokuma, Acc. Chem. Res., 2016, 49, 763–773 CrossRef CAS PubMed.
- K. N. Houk and F. Liu, Acc. Chem. Res., 2017, 50, 539–543 CrossRef CAS PubMed.
- S. Ahn, M. Hong, M. Sundararagan, D. H. Ess and M.-H. Baik, Chem. Rev., 2019, 119, 6509–6560 CrossRef CAS PubMed.
- H. B. Schlegel, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 790–809 CAS.
- E. J. Corey, A. K. Long and S. D. Rubenstein, Science, 1985, 228, 408–418 CrossRef CAS PubMed.
- H. Satoh and K. Funatsu, J. Chem. Inf. Comput. Sci., 1995, 35, 34–44 CrossRef CAS.
- A. I. Lin, T. I. Madzhidov, O. Klimchuk, R. I. Nugmanov, I. S. Antipin and A. Varnek, J. Chem. Inf. Model., 2016, 56, 2140–2148 CrossRef CAS PubMed.
- S. Szymkuć, E. P. Gajewska, T. Klucznik, K. Molga, P. Dittwald, M. Startek, M. Bajczyk and B. A. Grzybowski, Angew. Chem., Int. Ed., 2016, 55, 5904–5937 CrossRef PubMed.
- J. N. Wei, D. Duvenaud and A. Aspuru-Guzik, ACS Cent. Sci., 2016, 2, 725–732 CrossRef CAS PubMed.
- C. W. Coley, W. H. Green and K. F. Jensen, Acc. Chem. Res., 2018, 51, 1281–1289 CrossRef CAS PubMed.
- P. Schwaller, T. Gaudin, D. Lányi, C. Bekas and T. Laino, Chem. Sci., 2018, 9, 6091–6098 RSC.
- M. H. S. Segler, M. Preuss and M. P. Waller, Nature, 2018, 555, 604–610 CrossRef CAS PubMed.
- T. Klucznik, B. Mikulak-Klucznik, M. P. McCormack, H. Lima, S. Szymkć, M. Bhowmick, K. Molga, Y. Zhou, L. Rickershauser, E. P. Gajewska, A. Toutchkine, P. Dittwald, M. P. Startek, G. J. Kirkovits, R. Roszak, A. Adamski, B. Sieredzińska, M. Mrksich, S. L. J. Trice and B. A. Grzybowski, Chem, 2018, 4, 522–532 CAS.
- E. J. Coley, Angew. Chem., Int. Ed. Engl., 1991, 30, 455–465 CrossRef.
- S. Maeda, K. Ohno and K. Morokuma, Phys. Chem. Chem. Phys., 2013, 15, 3683–3701 RSC.
- S. Maeda and K. Ohno, Chem. Lett., 2004, 33, 1372–1373 CrossRef CAS.
- S. Maeda and K. Ohno, Chem. Phys. Lett., 2004, 398, 240–244 CrossRef CAS.
- T. Mita, M. Sugawara and Y. Sato, J. Org. Chem., 2016, 81, 5236–5243 CrossRef CAS PubMed.
- M. Mamone, R. S. B. Gonçalves, F. Blanchard, G. Bernadat, S. Ongeri, T. Milcent and B. Crousse, Chem. Commun., 2017, 53, 5024–5027 RSC.
- A. L. Dewyer, A. J. Argüelles and P. M. Zimmerman, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1354 Search PubMed.
- G. N. Simm, A. C. Vaucher and M. Reiher, J. Phys. Chem. A, 2019, 123, 385–399 CrossRef CAS PubMed.
- Y. Sumiya and S. Maeda, Paths of chemical reactions and their networks: from geometry optimization to automated search and systematic analysis, in Chemical Modelling: Volume 15 (Specialist Periodical Reports), ed. M. Springborg and J.-O. Joswig, Royal Society of Chemistry, 2020, pp. 28–69 Search PubMed.
- S. Maeda and K. Morokuma, J. Chem. Theory Comput., 2011, 7, 2335–2345 CrossRef CAS PubMed.
- S. Maeda, Y. Harabuchi, M. Takagi, T. Taketsugu and K. Morokuma, Chem. Rec., 2016, 16, 2232–2248 CrossRef CAS PubMed.
- S. Maeda and Y. Harabuchi, J. Chem. Theory Comput., 2019, 15, 2111–2115 CrossRef CAS PubMed.
- S. Maeda, Y. Harabuchi, M. Takagi, K. Saita, K. Suzuki, T. Ichino, Y. Sumiya, K. Sugiyama and Y. Ono, J. Comput. Chem., 2018, 39, 233–251 CrossRef CAS PubMed.
- Y. Sumiya and S. Maeda, Chem. Lett., 2019, 48, 47–50 CrossRef CAS.
- A review on the synthesis of α-amino acids from CO2: T. Mita and Y. Sato, Chem.–Asian J., 2019, 14, 2038–2047 CrossRef CAS PubMed.
- (a) C. Ni and J. Hu, Synthesis, 2014, 46, 842–863 CrossRef; (b) A. D. Dilman and V. V. Levin, Acc. Chem. Res., 2018, 51, 1272–1280 CrossRef CAS PubMed. An ammonium ylide generated from NR3 and difluorocarbene was trapped with proton electrophile: (c) E. Nawrot and A. Joñczyk, J. Org. Chem., 2007, 72, 10258–10260 CrossRef CAS PubMed.
- F. Wang, T. Luo, J. Hu, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. K. Surya Prakash and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 7153–7157 CrossRef CAS PubMed.
- L. Li, F. Wang, C. Ni and J. Hu, Angew. Chem., Int. Ed., 2013, 52, 12390–12394 CrossRef CAS PubMed.
- D. J. Raber, P. Gariano Jr, A. O. Brod, A. Gariano, W. C. Guida, A. R. Guida and M. D. Herbst, J. Org. Chem., 1979, 44, 1149–1154 CrossRef CAS.
- A three-component coupling reaction between pyridine, difluorocarbene, and CO2 nicely proceeded to afford C5H5N+–CF2–CO2− in 88% yield under the same conditions. Using the leaving ability of the pyridinium cation, we are now intensively studying new coupling reactions with various nucleophiles.
- S. Maeda, Y. Harabuchi, Y. Sumiya, M. Takagi, K. Suzuki, M. Hatanaka, Y. Osada, T. Taketsugu, K. Morokuma and K. Ohno, GRRM17, http://iqce.jp/GRRM/index_e.shtml, accessed on March 11, 2020 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. F. Ribeiro, A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2011, 115, 14556–14562 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, characterization data, and computational details. See DOI: 10.1039/d0sc02089c |
| This journal is © The Royal Society of Chemistry 2020 |