DOI:
10.1039/D0QO00631A
(Review Article)
Org. Chem. Front., 2020,
7, 3266-3283
Recent advances in organocatalytic asymmetric oxa-Michael addition triggered cascade reactions
Received
25th May 2020
, Accepted 13th August 2020
First published on 14th August 2020
Abstract
The oxa-Michael cascade reaction, as an important part of the Michael reaction, has also been significantly developed over the recent decade. This is because the problem of the reactivity and selectivity of the addition reactions of oxygen nucleophiles to conjugated systems has been solved by different organocatalysts and reaction conditions. Therefore, using various efficient strategies, many novel and potentially bioactive chiral compounds with excellent yields and stereoselectivities have been synthesized. In this review, we summarize the recent advances in organocatalytic asymmetric oxa-Michael addition triggered cascade reactions for the stereoselective synthesis of heterocyclic compounds.
 Yu Wang | Yu Wang was born in Sichuan, P.R. of China in 1994. He received his B.S. degree from Southwest Petroleum University in 2016. He is currently pursuing postgraduate study at the Beijing Institute of Technology under the supervision of Prof. Da-Ming Du. His current interests are focused on organocatalytic asymmetric development of novel methodologies. |
 Da-Ming Du | Da-Ming Du received his B.Sc. from Zhengzhou University in 1989. He received his M.Sc. and Ph.D. from Nankai University in 1992 and 1995. He took a Lecturer position at Shandong University in 1995. He was promoted to Associate Professor in 1997 at Shandong University, and during this period, he was a Visiting Scholar at The Chinese University of Hong Kong from 1996 to 1997 and then a Postdoctoral Visiting Scholar at Hong Kong University of Science & Technology from 1998 to 1999. He joined the College of Chemistry and Molecular Engineering of Peking University as an Associate Professor in 2001 after working as Postdoctoral Fellow. He moved to Beijing Institute of Technology and was promoted to full Professor in September 2008. He received the Thieme Journal Award 2009. His research interests are focused on the catalytic asymmetric synthesis, the design of new ligands and catalysts, the development of new synthetic methodologies and the synthesis of bioactive heterocycles. |
1 Introduction
The Michael addition reaction is one of the most powerful and valuable tools for the formation of C–X (X = C, O, N, S) bonds, which plays a vital role in organic synthetic chemistry.1–3 It is one of the most typical reactions between a nucleophile called a Michael donor and an activated Michael acceptor, and a huge number of optically functional structures and natural products have been prepared by this important transformation over the years. Meanwhile, the nucleophilicity and reactivity of alcoholic hydroxyl are relatively poor, but the oxa-Michael reaction, as an important part of the Michael reaction for the construction of the C–O bond, has also received considerably more attention from researchers after solving the problems involved in recent years.4,5
Small organic molecules capable of catalyzing reactions have been known for many decades, with the result that asymmetric organocatalysis has become a hot field of research. Moreover, because of the high efficiency, selectivity, low cost, environmental benignness and other characteristics of organocatalysts, more and more research groups are engaged in the development of organocatalytic asymmetric reactions to obtain the corresponding enantiomer-enriched products. In past decades, the rapid development of organocatalysis has greatly changed the profile of asymmetric catalysis.6,7 Many types of chiral organocatalysts, such as chiral prolinol analogues, chiral bifunctional squaramides and thioureas, and other chiral compounds, have been applied to oxa-Michael cascade reactions to synthesize many diverse and versatile oxygen-containing heterocyclic, bicyclic, spiro, and polycyclic structures with multiple stereocenters. In addition, these reaction protocols greatly decrease the number of laboratory operations, save time, cost and work steps,8 and have a high synthetic efficiency, thereby substantially broadening the scope of oxa-Michael reactions.
Therefore, this review aims to illustrate the developments in asymmetric oxa-Michael addition triggered cascade reactions over the last decade. Particular attention will be paid to organocatalytic reaction strategies and their applications in the synthesis of chiral oxygen-containing heterocycles.
2 Organocatalytic enantioselective oxa-Michael initiated cascade reactions
2.1 Reactions catalyzed by chiral prolinol analogues
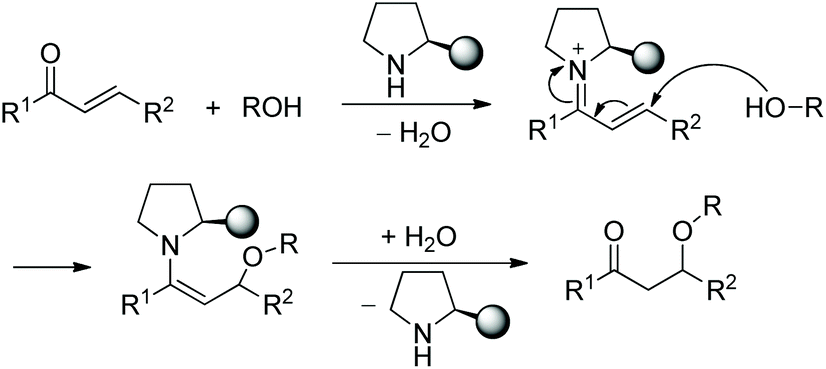
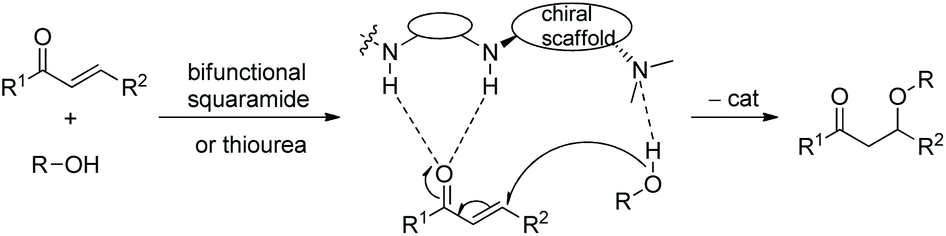
In this sort of catalytic process, the chiral secondary amine will activate the electrophile and generate an iminium–enamine intermediate, which then undergoes conjugate addition and hydrolysis steps to release the product and amine catalyst (Scheme 1). In general, some additives will be used to facilitate this process. It turns out that the proline-derived chiral catalysts have performed with outstanding results in oxa-Michael addition cascade reactions.9
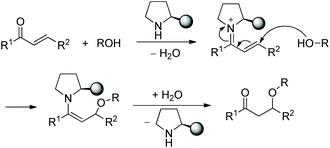 |
| | Scheme 1 Chiral prolinol analogue catalytic general mechanism of the oxa-Michael addition reaction. | |
2.1.1 Oxa-Michael cascade reactions for the synthesis of chiral chromenes.
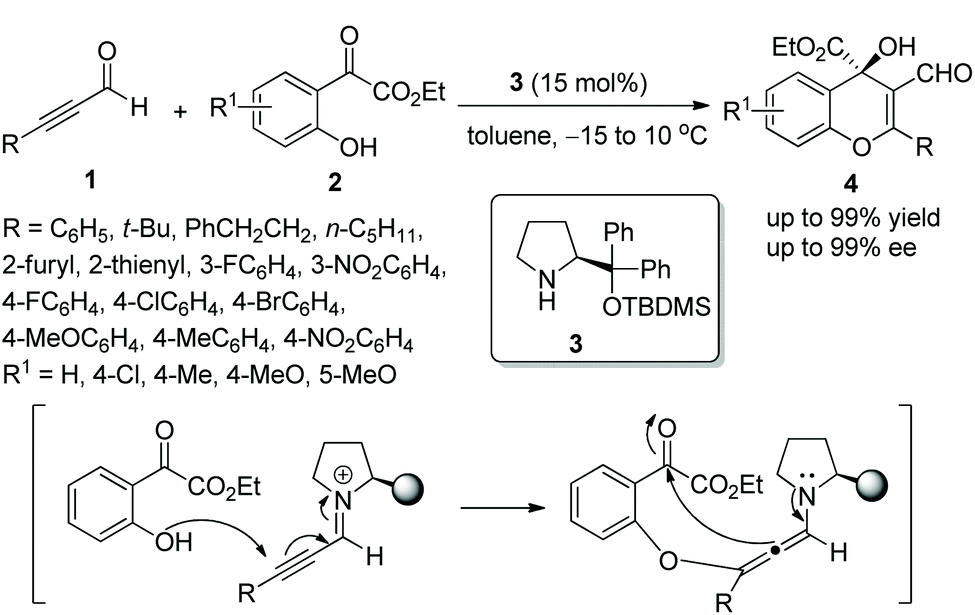
In 2010, Wang's group10 explored a novel mode of generating diverse 4H-chromenes 4 bearing a versatile α-hydroxy carboxylate moiety by using chiral diarylprolinol TBDMS ether 3 as a catalyst. The oxa-Michael/aldol cascade reactions of alkynals 1 with o-hydroxyacetophenone esters 2 tolerated a wide range of substrates and afforded the desired products with excellent yields (up to 99%) and enantioselectivities (up to 99% ee) under mild reaction conditions (Scheme 2). It was noteworthy in the scope of substrates that the less reactive alkyl-substituted alkynals (R = PhCH2CH2 or n-C5H11) also could take part effectively in this cascade reaction and provided corresponding products with satisfactory enantioselectivities (97 and 99% ee, respectively), even though this process needed more reaction time and a higher catalyst loading (25 mol%) than other aryl substituents. Additionally, when the substituent of the aromatic structures was electron-withdrawing Cl, the high enantioselectivity would be ensured by using quinine as a cocatalyst which possibly interacted with the carbonyl moiety of the ketoester. In this cascade process, the iminium-allenamine activation mode is a quite efficient way to generate functionalized chromenes and successfully expand the scope of the organocatalytic oxa-Michael-aldol cascade sequence.
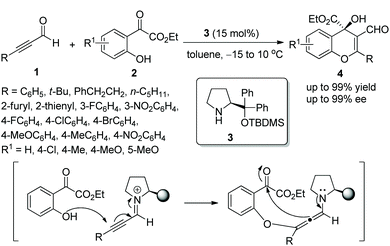 |
| | Scheme 2 Oxa-Michael/aldol cascade reaction for the synthesis of chromenes. | |
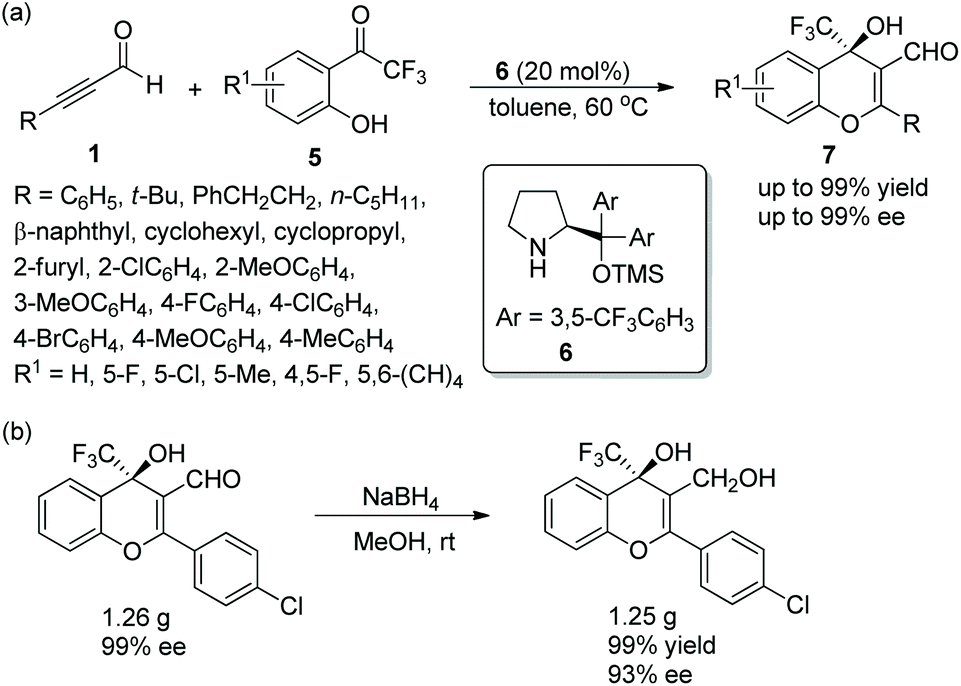
In consideration of the special properties of trifluoromethylated compounds in the pharmaceutical chemistry, it is very important to synthesize biologically active derivatives containing trifluoromethyl substituents. Then, there was the significant work in which Huang and coworker11 first synthesized chiral 4H-chromenes 7 bearing a trifluoromethyl group in 2015. The alkynals 1 and o-hydroxy trifluoroacetophenones 5 underwent an oxa-Michael/aldol cascade process catalyzed by catalyst 6 in toluene at 60 °C, when the desired products 7 were obtained with high yields (up to 99%) and enantioselectivities (up to 99% ee) (Scheme 3a). In the scope of the reaction, both electron-donating and electron-withdrawing group substituted aromatic alkynals were revealed to be efficient reactants and afforded satisfactory results, while furyl-substituted alkynal showed lower yield (40%) and enantioselectivity (84% ee) than other groups. The position of the substituents on the aromatic ring of aromatic alkynals had an effect on the reaction: for example, meta-substituted alkynal afforded better yield and enantioselectivity than ortho-substituted alkynal; and electron-donating substituents on the aromatic ring of 5 provided better reactivity than electron-withdrawing substituents. Furthermore, the reaction product could be further reduced to other valuable building blocks on a gram-scale without sacrificing reaction yield or enantioselectivity (Scheme 3b).
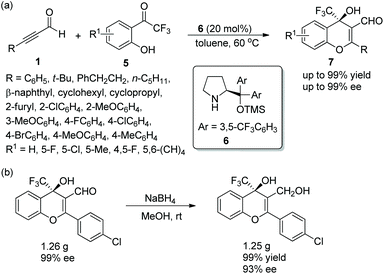 |
| | Scheme 3 Oxa-Michael/aldol cascade reaction for the synthesis of 4H-chromenes bearing a trifluoromethyl and the derivatization of chiral chromene. | |
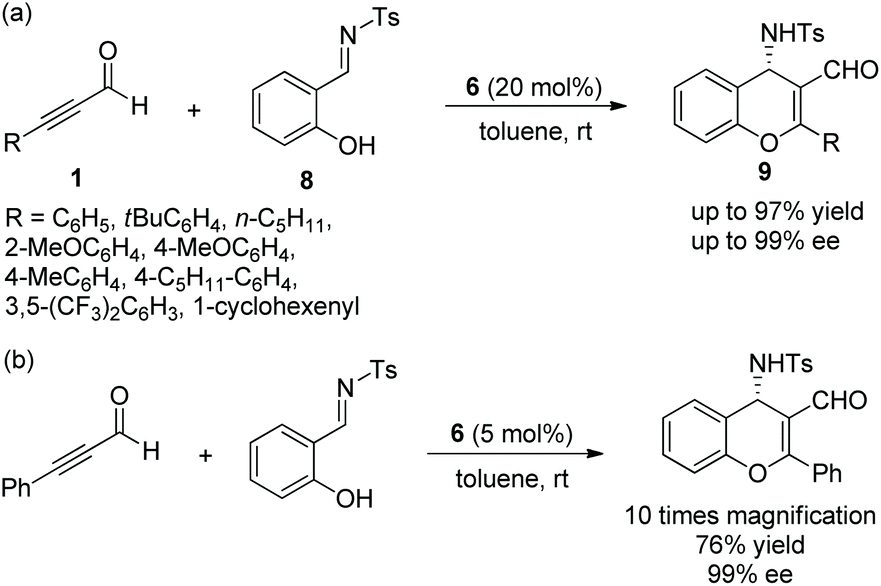
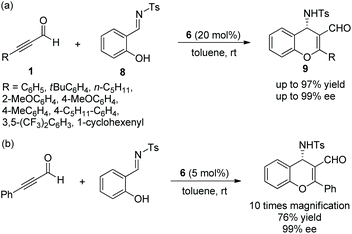
Alemán and co-workers12 also reported a similar method for the highly enantioselective synthesis of 4-amino-4H-chromenes 9 in 2010. Alkynals 1 and salicyl N-tosylimine 8, as the starting materials, could undergo an oxa-Michael/aza-Baylis–Hillman (aza-BH) cascade process catalyzed by (S)-diarylprolinol TMS ether 6 to generate the corresponding products with excellent yields (up to 97%) and enantioselectivities (up to 99% ee) in toluene at room temperature (Scheme 4a). In the scope of substrates, almost all substituents maintained high stereoselectivities; compared with aliphatic substituents, aromatic substituents afforded corresponding products with better yields; a para-substituent on the aromatic ring provided a better result than an ortho-substituent. However, the reactivity of the strong electron-poor aromatic ring 3,5-(CF3)2C6H3 of alkynal was substantially decreased (only 55% yield), even when the catalyst loading had been increased to 40 mol% and the reaction time had been prolonged. Additionally, in the large-scale reaction, this process provided the corresponding product in moderate yield without losing enantiopurity (Scheme 4b).
 |
| | Scheme 4 Oxa-Michael/aza-BH cascade reaction for the synthesis of 4-amino-4H-chromenes and the large-scale preparation reaction. | |
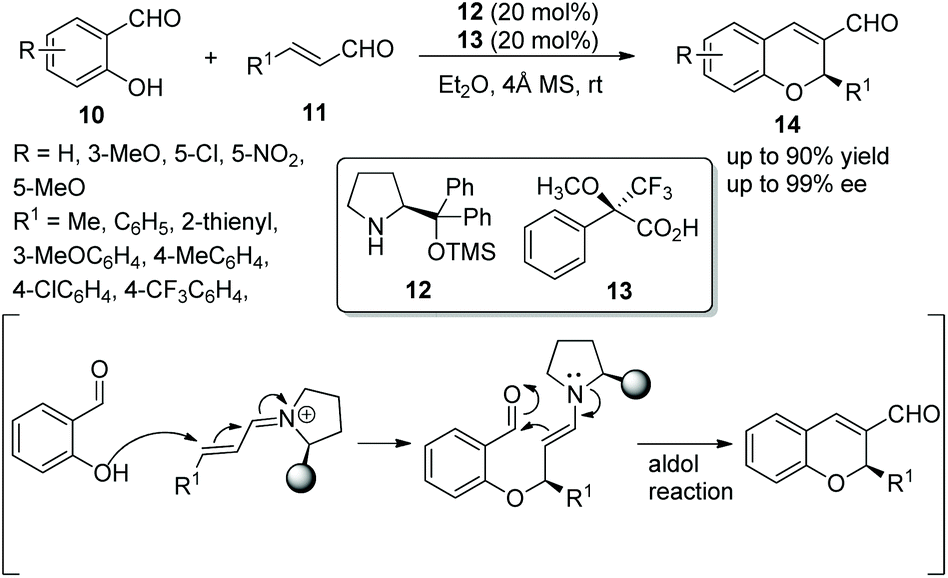
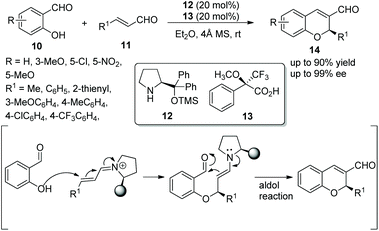
Xu's group13 investigated an oxa-Michael/aldol cascade reaction for the synthesis of chiral 2H-chromenes catalyzed by a chiral amine/chiral acid organocatalytic system in 2009. Salicylic aldehydes 10 and α,β-unsaturated aldehydes 11 in diethyl ether at room temperature, under the catalysis of catalyst 12 and (S)-Mosher's acid 13, afforded the desired products 14 in good yields (up to 90%) with excellent enantioselectivities (up to 99% ee) (Scheme 5). Compared with a single organocatalyst, the chiral amine/chiral acid organocatalytic system efficiently increased the enantioselectivity under the same conditions. In the scope of substrates, compared with aliphatic-substituted unsaturated aldehydes, aromatic-substituted unsaturated aldehydes provided the corresponding products with better yields and enantioselectivities. The electronic property and position of the substituents on the aromatic ring of salicylaldehydes had an effect on the yields and enantioselectivities of the products: for example, compared with 5-methoxy and 5-nitro groups, the electron-donating substituent 3-methoxy provided the corresponding product with the best yield and enantiopurity.
 |
| | Scheme 5 Oxa-Michael/aldol cascade reaction for the synthesis of chromene derivatives. | |
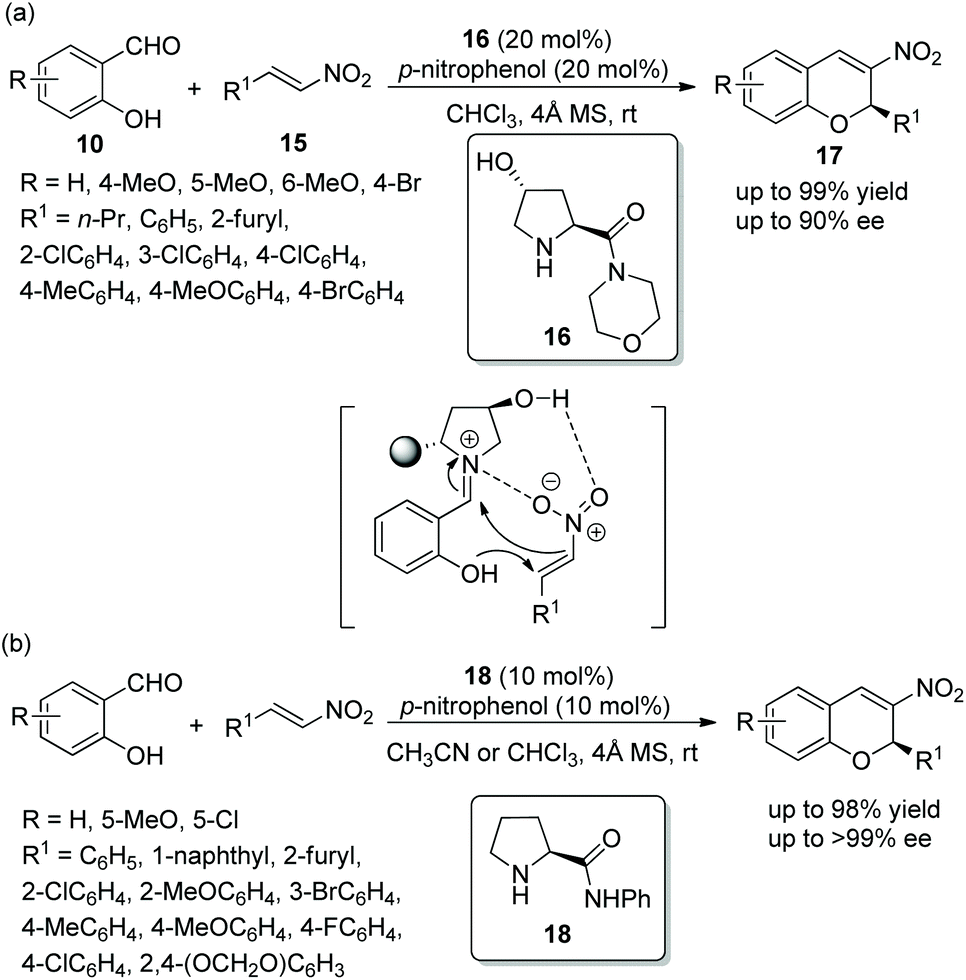
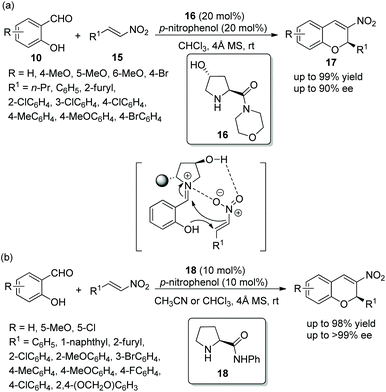
An efficient method for the synthesis of chiral chromenes via an oxa-Michael/Henry cascade reaction was reported by Chen's group14 in 2013. In this cascade procedure, salicylaldehydes 10 and nitroalkenes 15 were employed as starting materials catalyzed by a 4-hydroxy-L-proline derived chiral aminocatalyst 16 and the additive p-nitrophenol, which afforded the desired chromenes 17 in excellent yields (up to 99%) with high enantioselectivities (up to 90% ee) under mild conditions (Scheme 6a). In the scope of substrates, the reactivity of the electron-donating substituents on the aromatic ring was better than that of electron-withdrawing substituents. The position of the substituents on the aromatic ring had a significant influence on the enantioselectivity of the desired products: for example, 4-methoxy-substituted salicylaldehyde only afforded the desired product with 54% ee, but 5-methoxy-substituted salicylaldehyde provided the corresponding product with 89% ee. Additionally, it should be noted that, besides the iminium ion, the formation of hydrogen-bonding also took part in the transition state of the cascade process. A few years later, similar work was performed by Bez's group.15 Another catalyst 18 was employed in this cascade process (Scheme 6b). In general, in the substrate scope of this cascade reaction, regardless of electron-withdrawing or electron-donating substituents on the β-nitrostyrene derivatives, all of them had little effect on the yield or enantioselectivity of the products. However, it should be noted that the solvent still affected the result under optimal conditions. For example, the reaction of salicylaldehyde with p-fluoro-β-nitrostyrene afforded the corresponding product with 86% ee in CHCl3 and 99% ee in acetonitrile.
 |
| | Scheme 6 Oxa-Michael/Henry cascade reaction for the synthesis of chromene derivatives. | |
2.1.2 Oxa-Michael/Michael cascade reaction for the synthesis of chiral chromans.
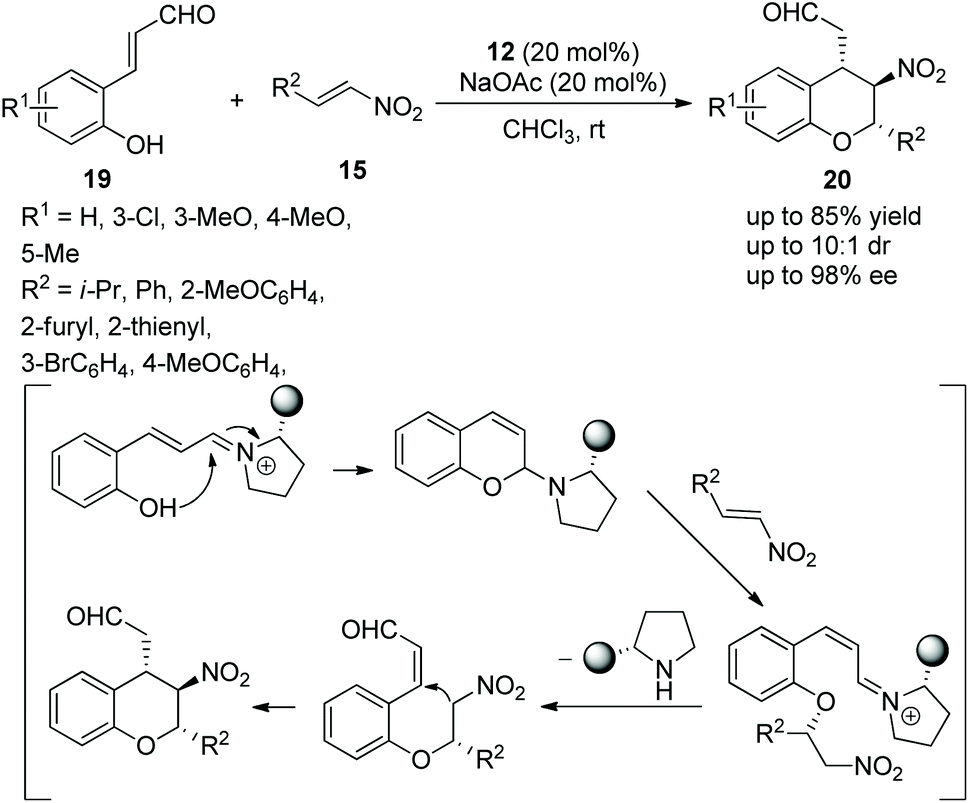
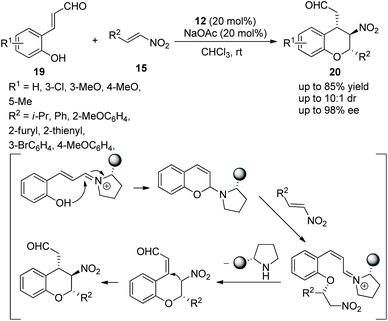
Wang's group16 developed a valuable and efficient strategy for the highly enantioselective synthesis of chiral chromans via an oxa-Michael/Michael cascade reaction in 2009. 2-Hydroxy cinnamaldehydes 19 and nitroolefins 15 catalyzed by chiral (S)-diarylprolinol TMS ether 12 afforded the desired products 20 in good yields (up to 85%) with excellent enantioselectivities (up to 98% ee) in chloroform at room temperature (Scheme 7). In the scope of substrates, all the substituents, regardless of whether they were electron-rich or electron-poor groups, provided the corresponding products with excellent enantiopurity; the reactivity of unsubstituted 2-hydroxy cinnamaldehyde was better than 2-hydroxy cinnamaldehyde bearing substituents; and compared with alkyl-substituted nitroolefins, aryl-substituted nitroolefins gave a better result.
 |
| | Scheme 7 Oxa-Michael/Michael cascade reaction for the synthesis of trisubstituted chromans. | |
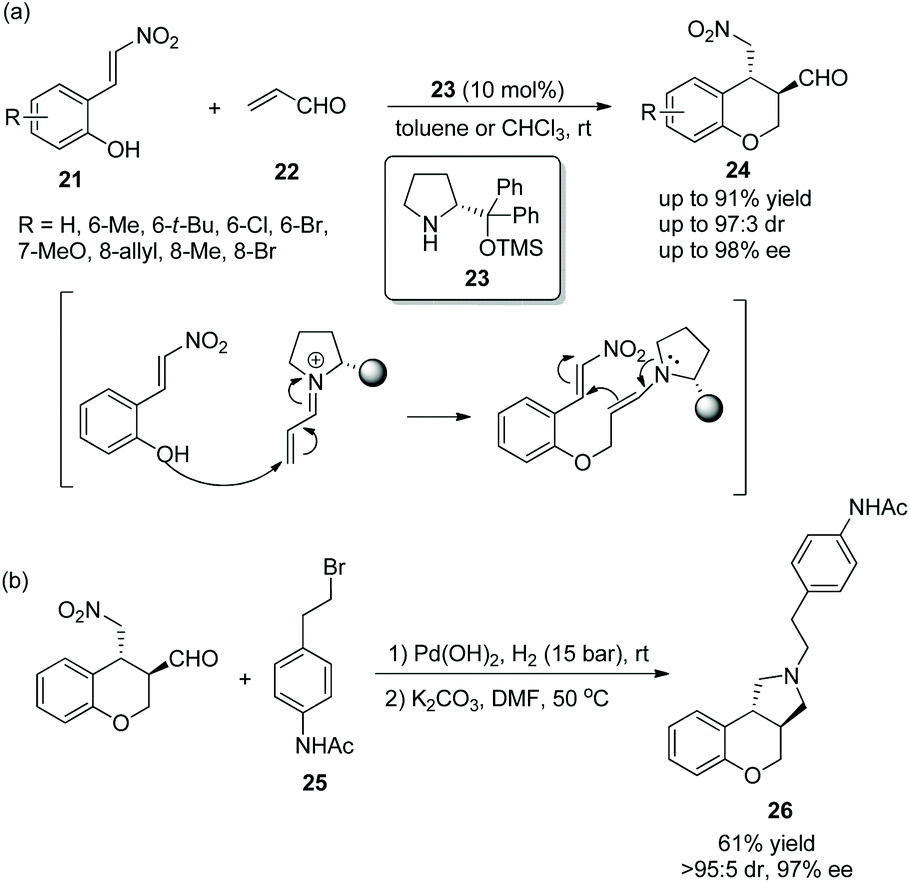
A short and convenient strategy for the synthesis of 3,4-disubstituted chromans via an oxa-Michael/Michael cascade reaction was reported by Enders’ group17 in 2012. Nitrovinylphenols 21 and acrolein 22 were employed to undergo the cascade process catalyzed by (R)-diphenylprolinol TMS ether 23 and provided the desired products 24 in good yields (up to 91%) with great stereoselectivities (up to 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 dr and 98% ee) in toluene or chloroform at room temperature (Scheme 8a). The reason why there were two kinds of solvents is that some substrates 21 bearing substituents, such as 6-Me, 6-Br, 8-Br, or 8-allyl, have poor solubility in toluene and thus the corresponding reactions were conducted in chloroform. Then, in the limited scope of substrates, all of them afforded the corresponding products with excellent stereoselectivities, except the nitrovinylphenol-bearing an electron-rich methoxy-substituent which did not generate the desired product; electron-donating substituents showed better reactivity than electron-withdrawing substituents; and compared with other positions, the reactivity of the methyl substituent at the C-7 position was the best. Furthermore, through a two-step reductive amination/N-alkylation sequence, they successfully transformed the cascade product to trans-N-alkylated benzopyrano[3,4-c]pyrrolidine 26, an antagonist of the dopamine-D3-receptor and potential antipsychotic medicament, without losing much enantiopurity (Scheme 8b).
:3 dr and 98% ee) in toluene or chloroform at room temperature (Scheme 8a). The reason why there were two kinds of solvents is that some substrates 21 bearing substituents, such as 6-Me, 6-Br, 8-Br, or 8-allyl, have poor solubility in toluene and thus the corresponding reactions were conducted in chloroform. Then, in the limited scope of substrates, all of them afforded the corresponding products with excellent stereoselectivities, except the nitrovinylphenol-bearing an electron-rich methoxy-substituent which did not generate the desired product; electron-donating substituents showed better reactivity than electron-withdrawing substituents; and compared with other positions, the reactivity of the methyl substituent at the C-7 position was the best. Furthermore, through a two-step reductive amination/N-alkylation sequence, they successfully transformed the cascade product to trans-N-alkylated benzopyrano[3,4-c]pyrrolidine 26, an antagonist of the dopamine-D3-receptor and potential antipsychotic medicament, without losing much enantiopurity (Scheme 8b).
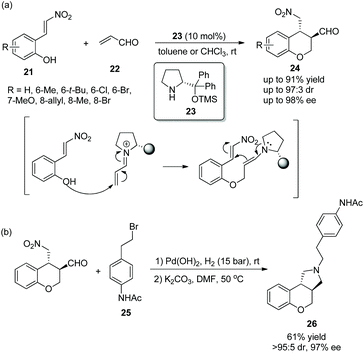 |
| | Scheme 8 Oxa-Michael/Michael cascade reaction for the synthesis of 3,4-trans-disubstituted chromans and the derivatization of chiral chroman. | |
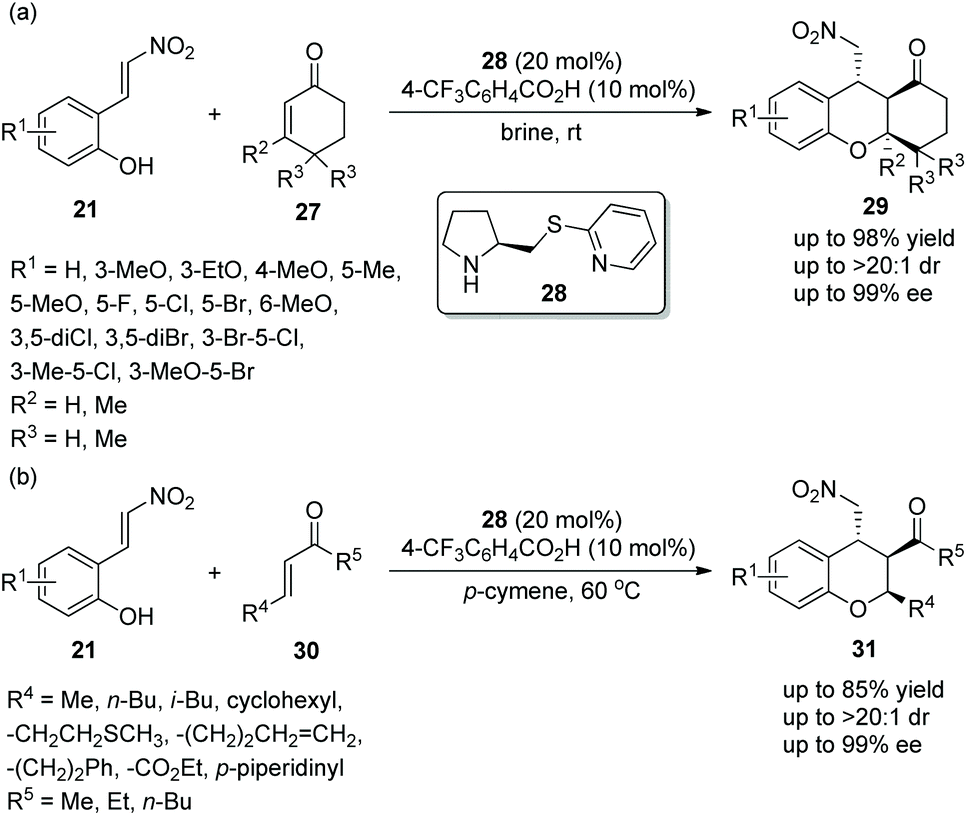
In view of the good result reported by Enders’ group, similar work for the construction of functionalized tetrahydroxanthenones and chromans was finished by Xu's group18 in 2014. Under the catalysis of prolinol thioether catalyst 28 and 4-trifluoromethylbenzoic acid additive, nitrovinylphenols 21 and cyclohexenones 27 gave the products tetrahydroxanthenones 29 with excellent yields (up to 98%) and stereoselectivities (up to >20:1 dr and 99% ee) in brine at room temperature (Scheme 9a). Interestingly, while maintaining the stereoselectivity of this cascade reaction, brine took a shorter reaction time than organic solvents. In the scope of substrates, the electronic property and position of the substituents had little effect on the stereoselectivities of the reaction, and all of them afforded the corresponding products with excellent stereoselectivities. Compared with other positions on the aromatic ring, the reactivity of a methoxy substituent at the C-6 position was lowest, and the reason could be that methoxy is too close to the carbon–carbon double bond to affect the electrophilic site of the reaction. Furthermore, nitrovinylphenols 21 and linear unsaturated ketones 30 were also employed to generate chiral chromans 31via the same catalytic cascade process (Scheme 9b). In general, although the corresponding products were obtained with excellent stereoselectivities, the reactivity of linear unsaturated ketones 30 with nitrovinylphenols 21 in p-cymene was not as good as the reactivity of cyclohexenones 27 with nitrovinylphenols 21 in brine.
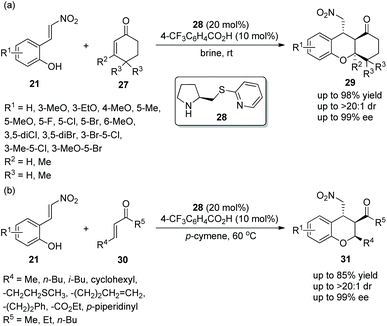 |
| | Scheme 9 Oxa-Michael/Michael cascade reaction for the synthesis of tetrahydroxanthenones and chromans. | |
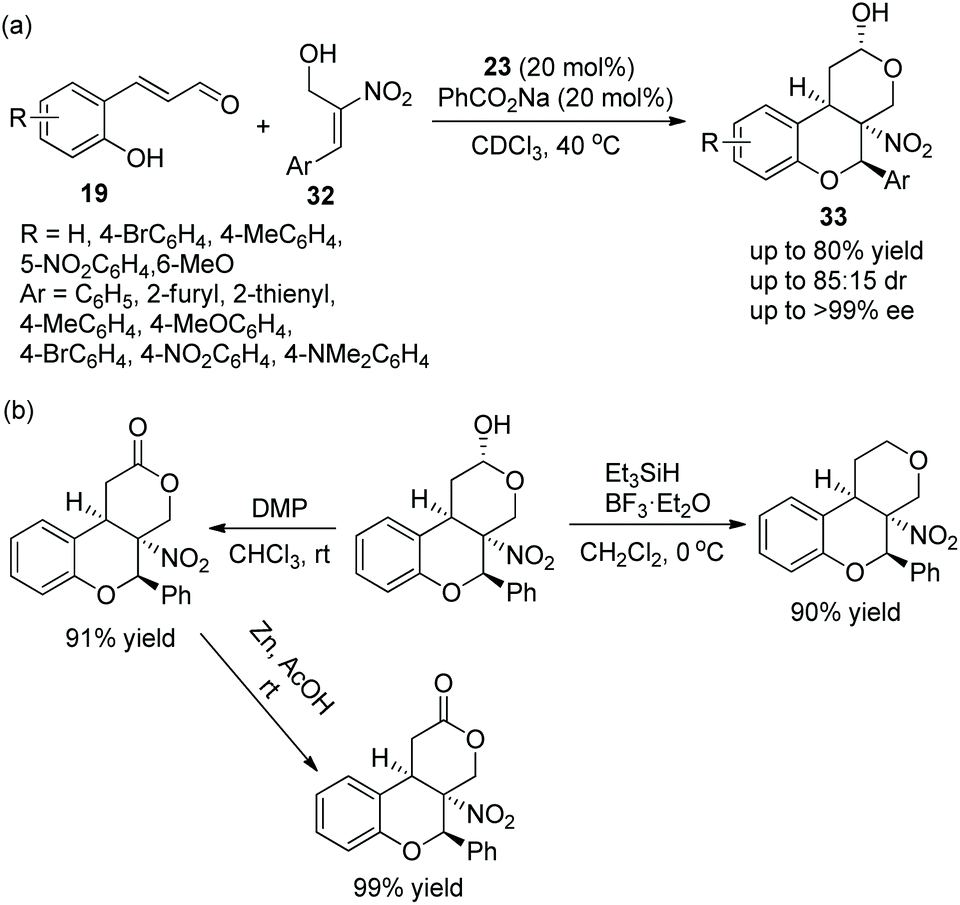
Tricyclic chroman, as an important structure, has attracted the attention of many researchers, especially in the synthesis of enantiomerically enriched chroman derivatives with this structure. A study for synthesizing functionalized tricyclic chromans with four stereocenters via an oxa-Michael/Michael/nucleophilic ring-closing cascade process was reported by Jørgensen's group19 in 2014. They applied o-hydroxycinnamic aldehydes 19 and 2-nitroallylic alcohols 32 to this cascade process and, at the same time, PhCO2Na was added to improve the reactivity and yield. Under the catalysis of chiral diarylprolinol TMS ether 23, the cycloadducts 33 were obtained in good yields (up to 80%) with excellent enantioselectivities (up to >99% ee) in CDCl3 at 40 °C (Scheme 10a). In the substrate scope of this cascade reaction, heteroaryl-substituted 2-nitroallylic alcohols afforded the corresponding products with the best enantioselectivity. The electron-donating groups on the phenyl ring of the o-hydroxycinnamic aldehydes gave better yields and stereoselectivities than the electron-withdrawing groups. Furthermore, they demonstrated that the cascade product could be selectively transformed into other structures, such as the dehydroxylation reaction of chromans with Et3SiH and BF3·Et2O, the oxidation of the hydroxyl group under Dess–Martin conditions and further selective reduction of the nitro group. Each reaction provided the corresponding product with high isolated yield (Scheme 10b).
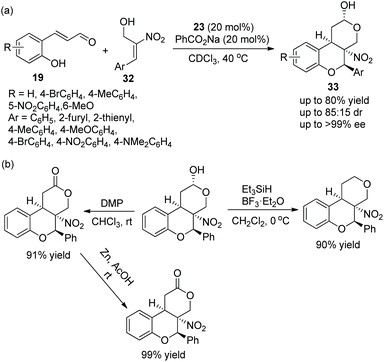 |
| | Scheme 10 Oxa-Michael/Michael/nucleophilic ring-closing cascade reaction for the synthesis of chromans and the derivatizations of chiral chroman. | |
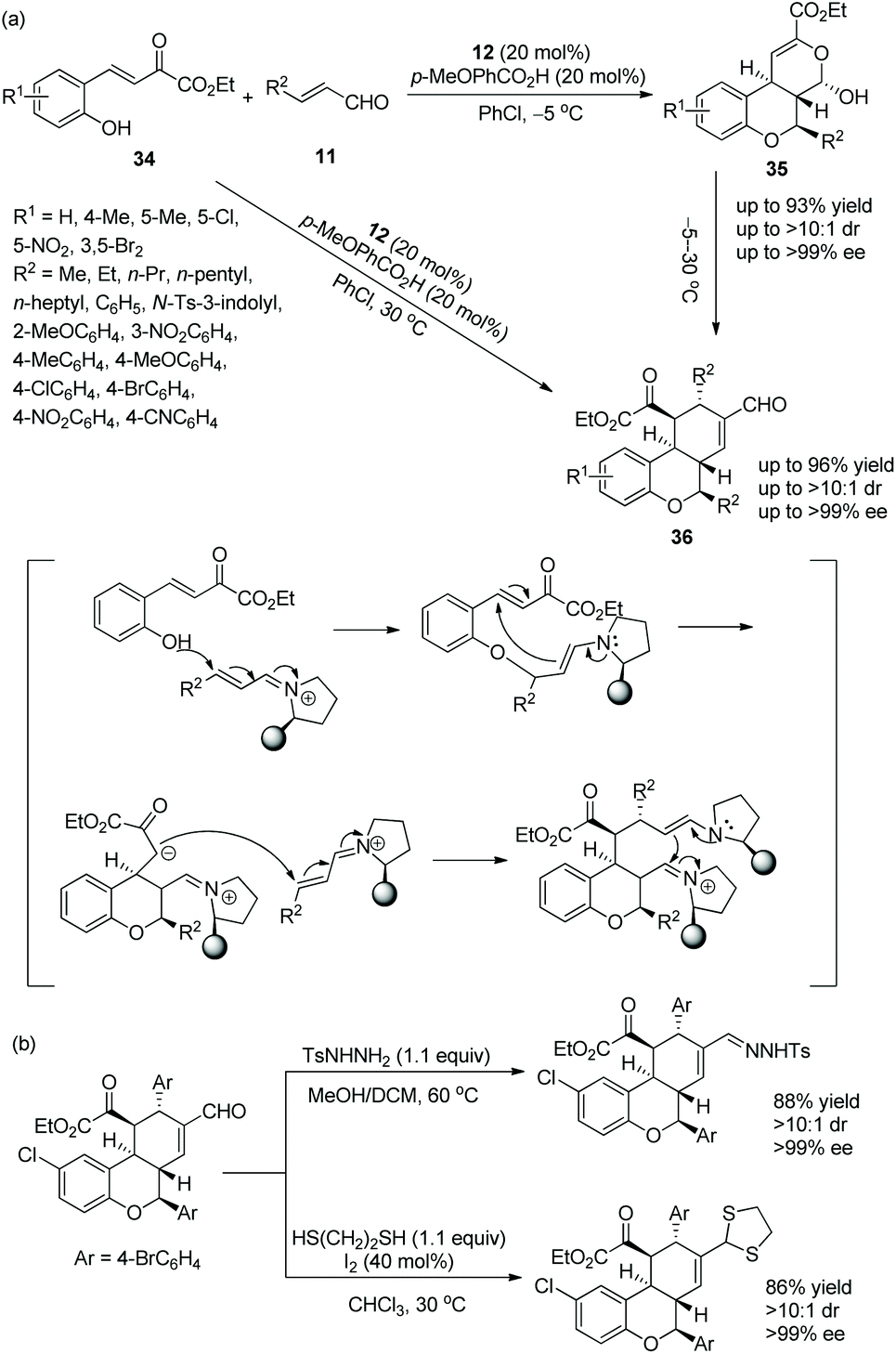
In 2014, Wang's group20 conceived an effective strategy to synthesize tricyclic chroman derivatives under the catalysis of chiral diarylprolinol TMS ether 12 under adjustable reaction conditions. What is noteworthy is that the reaction temperature is a vitally important factor. When the cascade reaction of (E)-2-hydroxyaryl-2-oxa-but-3-enoates 34 with enals 11 catalyzed by catalyst 12 occurred in PhCl at −5 °C, the corresponding products 35 were obtained in high yields with excellent stereoselectivities, but when the reaction temperature was continuously elevated to 30 °C, products 36 would be generated. Obviously, if the cascade reaction catalyzed by catalyst 12 was performed in PhCl at 30 °C, the chroman derivatives 36, with great yields and stereoselectivities, would be synthesized exclusively (Scheme 11a). It is amazing that, irrespective of whether there are electron-withdrawing or electron-donating groups in the substrate scope of the cascade reaction, all of them offered the corresponding products with excellent stereoselectivities (>10:1 dr and >98% ee); electron-withdrawing substituents on the aromatic ring of 34 showed better reactivity than electron-donating substituents; compared with aliphatic-substituted enals, aromatic-substituted enals displayed better reactivity. Furthermore, it was demonstrated that the chiral product could be converted to the corresponding hydrazine by reaction with p-toluenesulfonohydrazide in a mixed solvent of methanol and dichloromethane at 60 °C, but it could also be transformed to the corresponding mercaptal by reaction with 1,2-ethanedithiol in CHCl3 at 30 °C, both without losing enantiomeric purity (Scheme 11b).
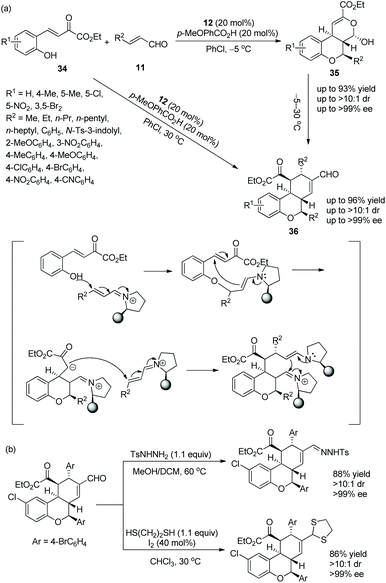 |
| | Scheme 11 Oxa-Michael-IED/HDA-Michael-aldol cascade reaction for the synthesis of chroman derivatives and the derivatizations of chiral chroman. | |
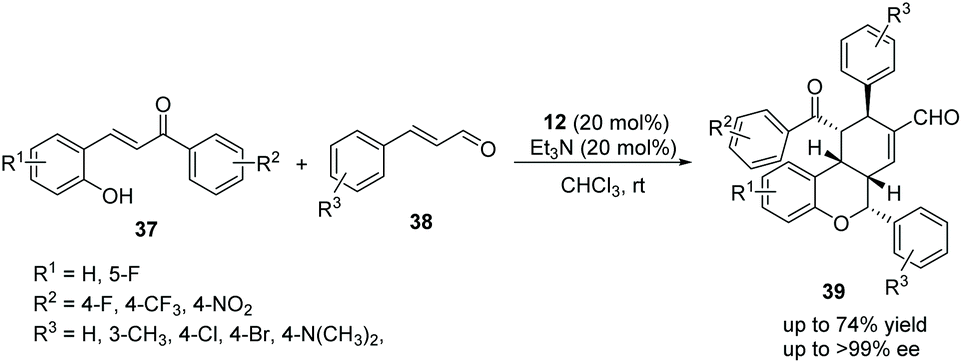
In view of the satisfactory study by Wang's group, Li's group21 also reported some similar work in 2014. o-Hydroxychalcones 37 and cinnamaldehydes 38 were used as the starting materials to synthesize chiral chroman derivatives 39 catalyzed by the same organocatalyst 12. In this procedure, they added Et3N to activate the cascade reaction, and eventually gained a satisfying result (up to 74% yield and >99% ee) (Scheme 12). Nevertheless, the scope of substrates for this cascade reaction is relatively limited owing to the fine-tuning of the electronic properties of the chalcone substrates, and the positions of the substituents have a great influence on the reaction. For instance, the reaction would hardly proceed when R2 was a strong electron-withdrawing nitro group, and no reaction would be observed if the position of the electron-withdrawing group (R1 = F) was changed from the 5-position to the 3-position. From the perspective of the mechanism, the carbon anion of the intermediate does not have sufficient nucleophilic ability to attack the cinnamaldehyde intermediate activated by the organocatalyst under the influence of these factors.
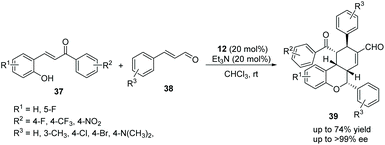 |
| | Scheme 12 Oxa-Michael cascade reaction for the synthesis of chiral tricyclic chromans. | |
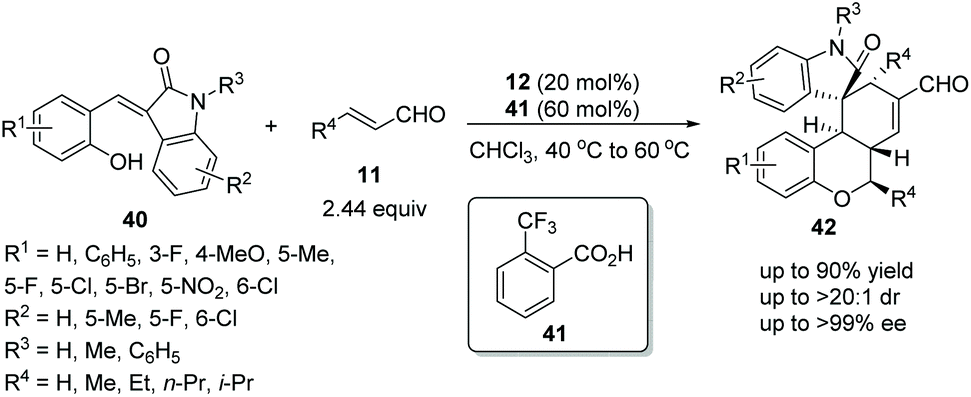
Wang's group22 in 2017 reported a straightforward and efficient protocol for the construction of highly stereoselective polycyclic spiro-fused carbocyclic oxindoles via an oxa-Michael addition triggered cascade reaction. The introduction of a hydroxy group on the alkylideneoxindoles 40, successively performed as nucleophiles and electrophiles under the catalysis of catalyst 12 and additive o-trifluoromethylbenzoic acid 41 in chloroform at 40 °C to 60 °C, and effectively facilitated the cascade reaction with unsaturated aldehydes 11. The desired products 42 were obtained in good yields (up to 90%) with excellent stereoselectivities (up to >20:1 dr and >99% ee) (Scheme 13). It is noteworthy that it was actually a three-component cascade process. After an oxa-Michael/Michael addition reaction had occurred between alkylideneoxindoles 40 and unsaturated aldehydes 11, the intermediate product bearing a new nucleophilic site at the 3-position of oxindole would continue to undergo Michael/aldol condensation with unsaturated aldehydes 11 to generate the desired products 42. Additionally, in the scope of substrates, the worst result (55% yield, 1:1.25 dr and 64% ee) was obtained when there was no substituent on the reactants, but the rest of the other substituents afforded satisfactory stereoselectivities; electron-donating group substituted R1 showed better reactivity than electron-withdrawing group substituted R1, but electron-withdrawing group substituted R2 displayed better reactivity; and the position of the substituents had little effect on the reactivity of the reaction.
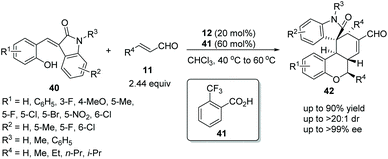 |
| | Scheme 13 Oxa-Michael/Michael cascade reaction for the synthesis of polycyclic spiro-fused carbocyclic oxindoles. | |
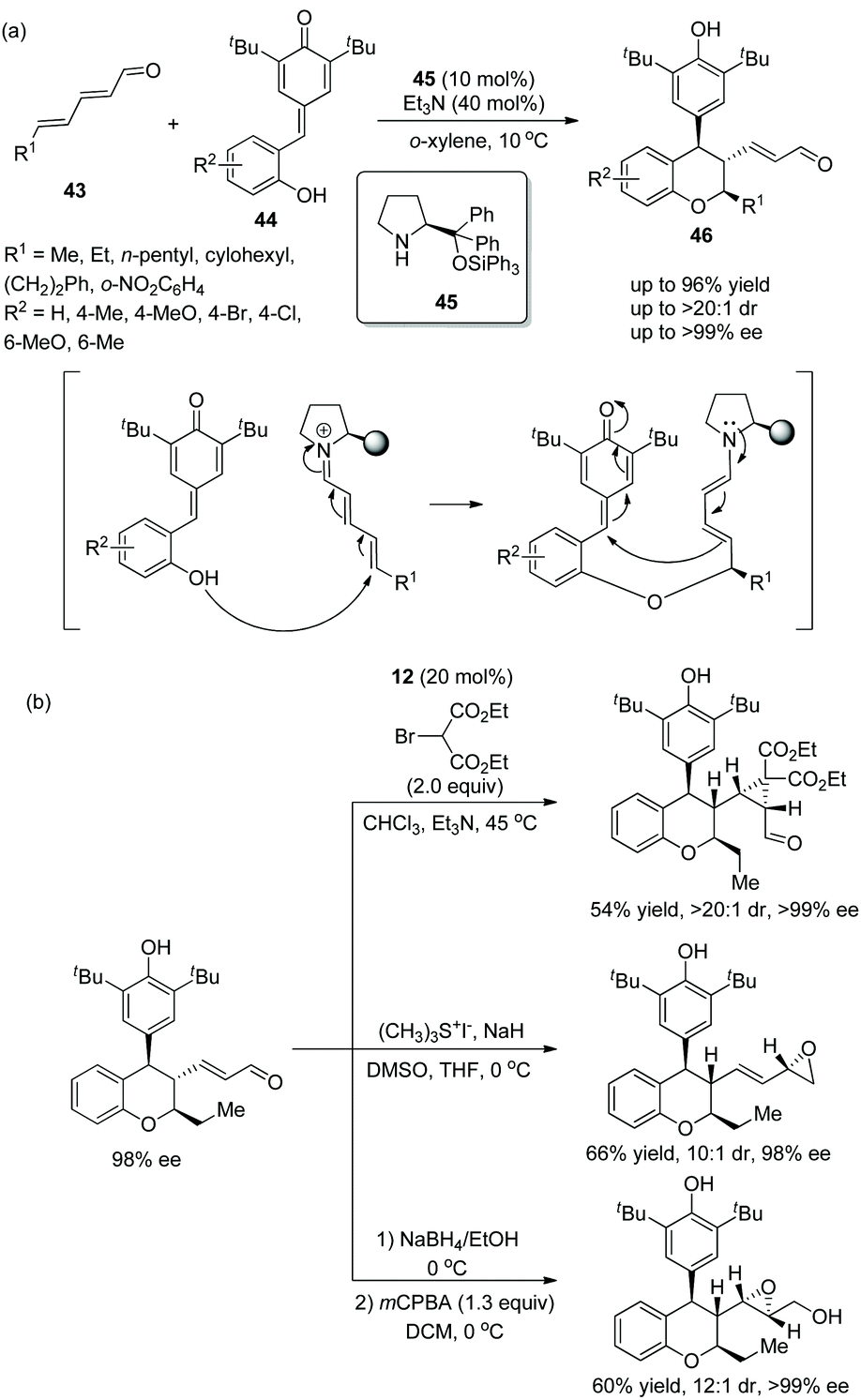
An efficient and promising strategy for the synthesis of chiral chroman derivatives via an oxa-Michael/1,6-addition cascade reaction was reported by Chatterjee's group23 in 2020. Under the catalysis of organocatalyst 45 and triethylamine, α,β,γ,δ-unsaturated enals 43 and ortho-hydroxyphenyl-substituted para-quinone methides (p-QMs) 44 were used as the starting materials and underwent the oxa-Michael/1,6-addition cascade process to generate the desired chromans 46 with excellent yields (up to 96%) and stereoselectivities (up to >20:1 dr and >99% ee) in o-xylene at 10 °C (Scheme 14a). In the scope of substrates, the position and properties of substituents on ortho-hydroxyphenyl-substituted p-QMs had little effect on the enantioselectivity of the corresponding products but affected the reactivity. For instance, 4-Me-substituted 44 and electron-donating substituents afforded the corresponding products with better yields than 6-Me-substituted 44 and electron-withdrawing substituents, respectively. Various unsaturated enals 43 with aliphatic substituents like methyl, n-pentyl, and cyclohexyl groups showed better results than unsaturated enals 43 with aromatic substituents. Additionally, due to the presence of an α,β-unsaturated system of chroman derivatives, some transformations for the construction of more complex molecular scaffolds were performed completely, such as cyclopropanation, the Corey–Chaykovsky reaction and epoxidation, providing the corresponding products without losing enantiopurity (Scheme 14b).
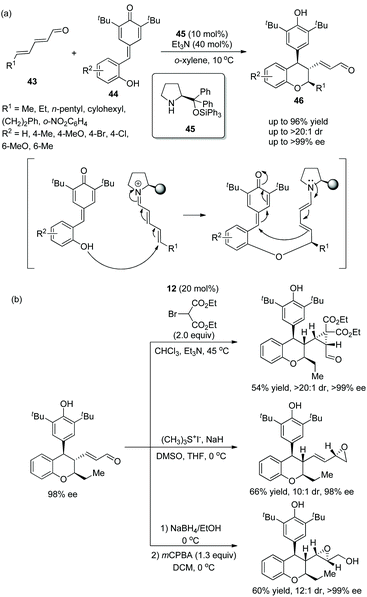 |
| | Scheme 14 Oxa-Michael/1,6-addition cascade reaction for the synthesis of chromans and the derivatization of chiral chroman. | |
2.1.3 Oxa-Michael/Michael cascade reaction of p-hydroxyl cycloenones with enals.
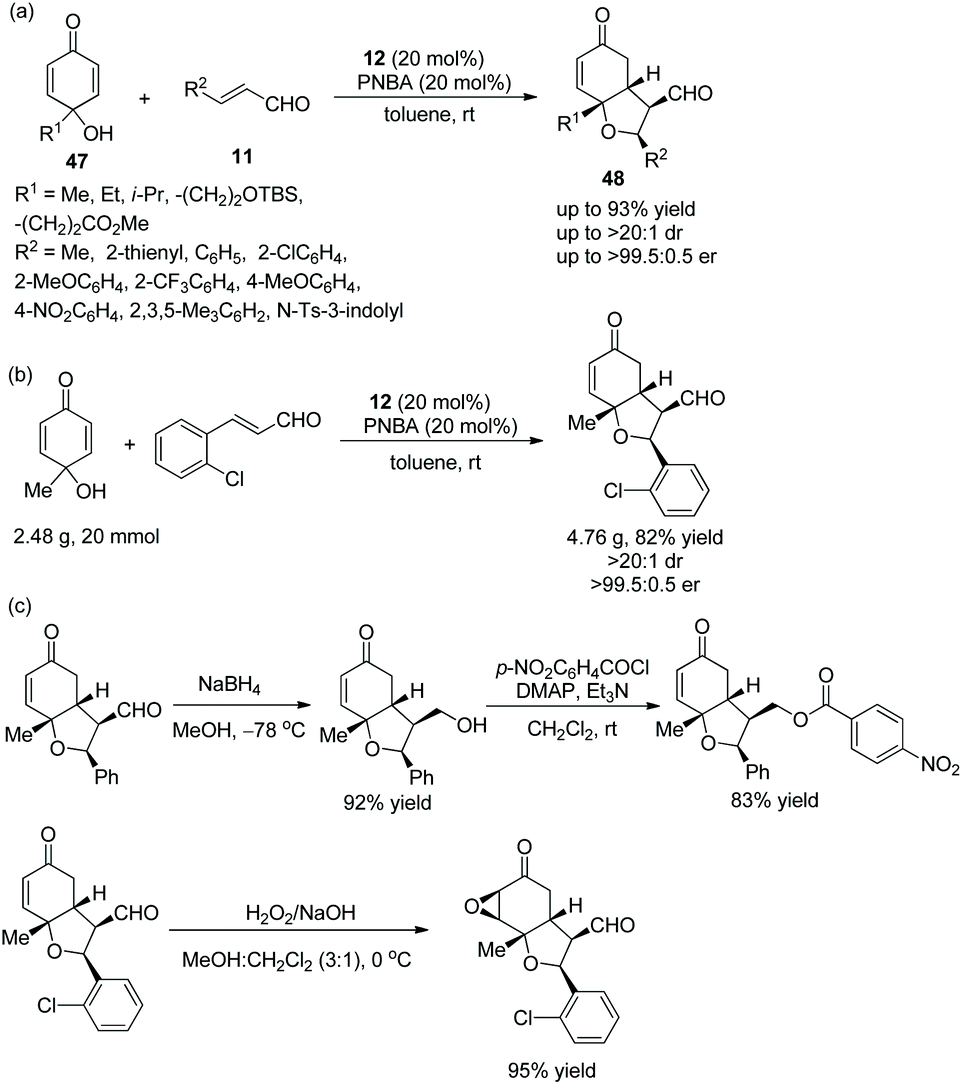
An efficient strategy for the highly stereoselective synthesis of hindered cyclic dialkyl ethers via an oxa-Michael/Michael cascade reaction was reported by Johnson and co-worker24 in 2013. Under the catalysis of catalyst 12 and additive 4-nitrobenzoic acid (PNBA), p-quinols 47 and unsaturated aldehydes 11 afforded the desired products 48 with excellent yields (up to 93%) and stereoselectivities (up to >20:1 dr and >99.5:0.5 er) in toluene at room temperature (Scheme 15a). In the scope of substrates, all of the substituents afforded the corresponding products with great stereoselectivities, although the electron-withdrawing substituents gave a better result than electron-donating substituents, and the methyl-substituted unsaturated aldehyde showed lower reactivity than aryl-substituted unsaturated aldehydes. Moreover, this cascade reaction was performed on a gram-scale without losing enantiopurity (Scheme 15b). In addition, some further transformations were performed, such as chemoselective reduction, the formation of a new ester, and epoxidation (Scheme 15c), which successfully demonstrated the applicability of this methodology.
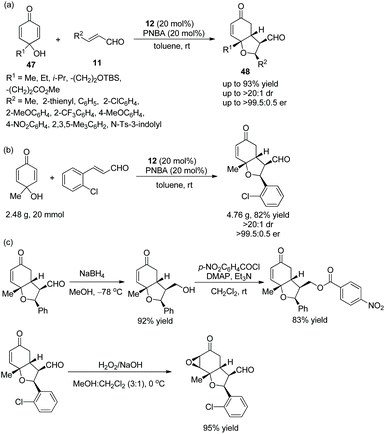 |
| | Scheme 15 Oxa-Michael/Michael cascade reaction for the synthesis of cyclic dialkyl ethers and the derivatization of the chiral adduct. | |
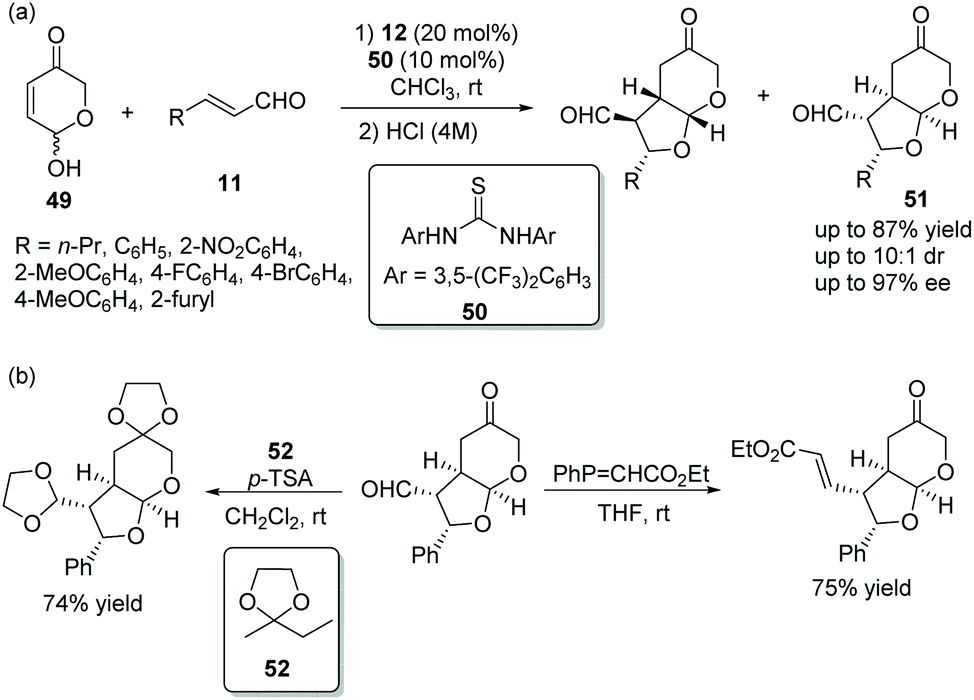
Vicario's group25 in 2017 reported a positive and effective method for the construction of bicyclic furo[2,3-b]pyranes via an oxa-Michael/Michael cascade reaction. Hydroxypyranones 49, active chiral racemic O-pronucleophiles, reacted with unsaturated aldehydes 11 providing the desired products 51 in good yields (up to 87%) with high stereoselectivities (up to 10:1 dr and 97% ee) under the catalysis of catalyst 12 and cocatalyst 50 in chloroform (Scheme 16a). Noticeably, the formation of diastereomers was minimized by stirring the crude reaction mixture with aqueous HCl. In the scope of substrates, compared with aromatic substituents, aliphatic substituents showed higher reactivity and lower stereoselectivity. The electronic property and position of the substituents on the aromatic ring had little effect on the cascade reaction, but electron-withdrawing substituents also showed better reactivity than electron-donating substituents. In addition, some transformations were performed to construct functionalized chiral building blocks, which successfully demonstrated the application of this method (Scheme 16b).
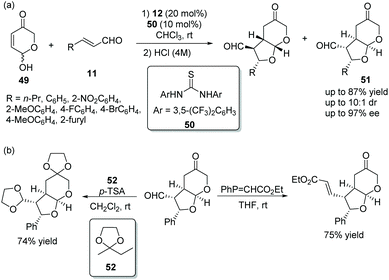 |
| | Scheme 16 Oxa-Michael/Michael cascade reaction for the synthesis of bicyclic furo[2,3-b]pyrans and the derivatization of the chiral adduct. | |
2.1.4 Oxa-Michael cascade reaction for the synthesis of chiral alicyclic compounds.
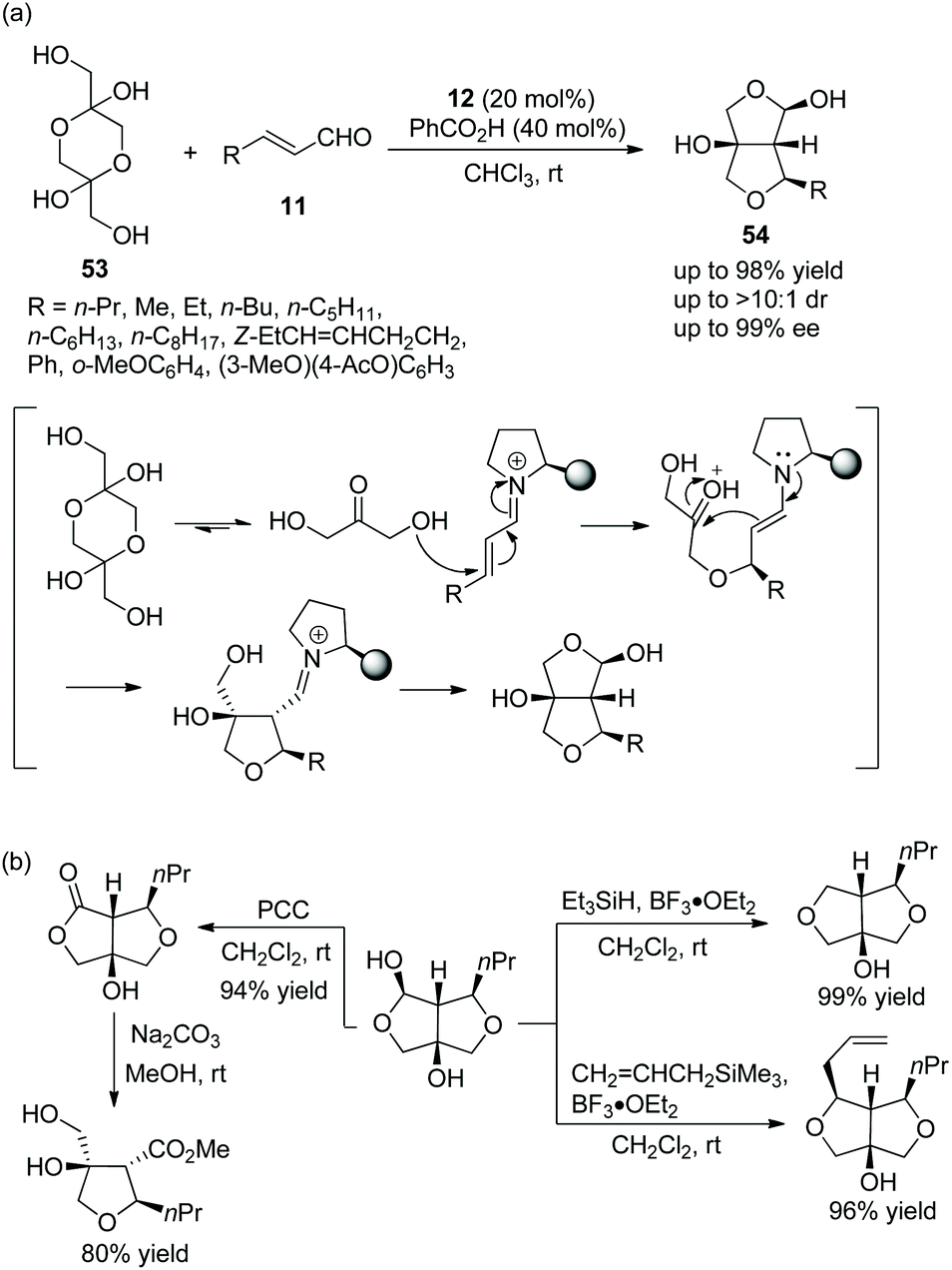
An unprecedented and efficient method for the construction of polysubstituted furofurans via an oxa-Michael cascade reaction was reported by Vicario and coworkers.26 Dihydroxyacetone dimer 53 as an oxygen nucleophile was reacted with α,β-unsaturated aldehydes 11 under the catalysis of catalyst 12 and benzoic acid in chloroform at room temperature. After an initial oxa-Michael addition reaction, a subsequent intramolecular aldol reaction, and lastly a hemiacetalization step, the desired products 54 were generated with excellent yields (up to 98%) and stereoselectivities (up to >10:1 dr and 99% ee) (Scheme 17a). In the scope of substrates, all of the substituents afforded the corresponding products with high stereoselectivities; and the reactivity of aliphatic-substituted unsaturated aldehydes was better than that of aromatic-substituted unsaturated aldehydes. In order to illustrate the applicability of this strategy, some further transformations were performed, such as replacement of functionalized groups, oxidation of hydroxyl, and ring-opening reactions, which afforded the corresponding products with high yields (Scheme 17b).
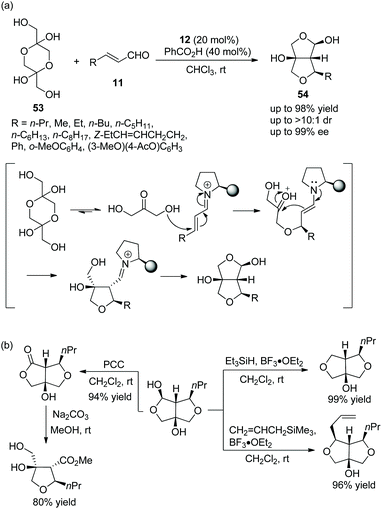 |
| | Scheme 17 Oxa-Michael/aldol/hemiacetalization cascade reaction for the synthesis of furofurans and the derivatization of the chiral adduct. | |
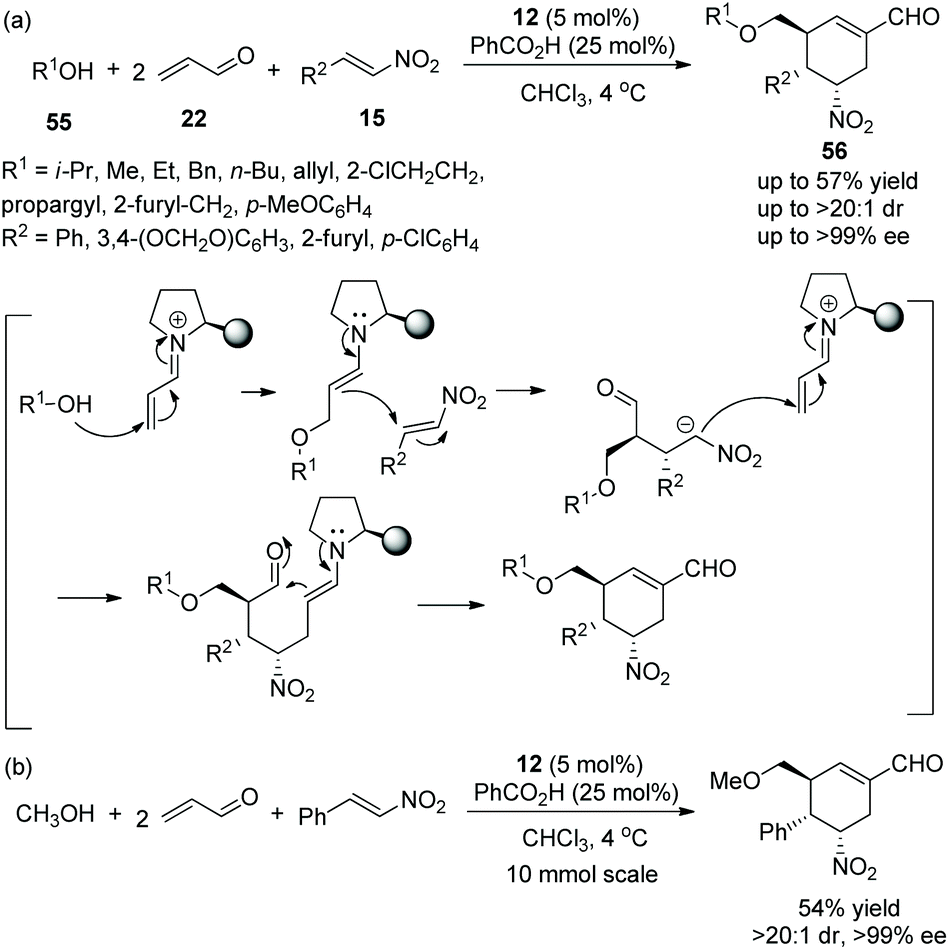
An asymmetric multicomponent cascade reaction catalyzed by organocatalysts for the construction of functionalized structures always receives considerable attention. Gong's group27 in 2009 reported a valuable and efficient protocol for the synthesis of chiral polysubstituted cyclohexenes via a multicomponent cascade reaction. Under the catalysis of catalyst 12 and benzoic acid, simple alcohol 55, a double equivalent of acrolein 22, and nitroalkenes 15 underwent an oxa-Michael/Michael/Michael/aldol condensation cascade process to generate the desired products 56 in moderate yields (up to 57%) with excellent stereoselectivities (up to >20:1 dr and >99% ee) in chloroform at 4 °C (Scheme 18a). In the scope of substrates, regardless of whether the substituents were aliphatic or aromatic, all of them afforded the corresponding products with excellent stereoselectivities; the longer the carbon chain of the alcohol, the lower the yield of the corresponding products. In addition, in a 10-times scale reaction, the desired chiral polysubstituted cyclohexene was obtained without losing any enantiopurity (Scheme 18b).
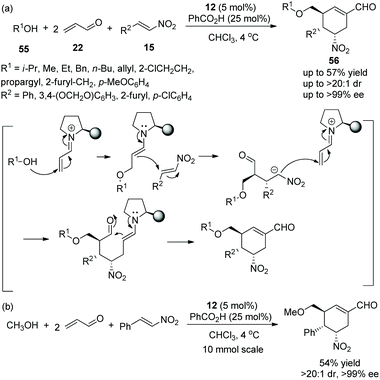 |
| | Scheme 18 Multicomponent cascade reaction for the synthesis of cyclohexene carbaldehydes. | |
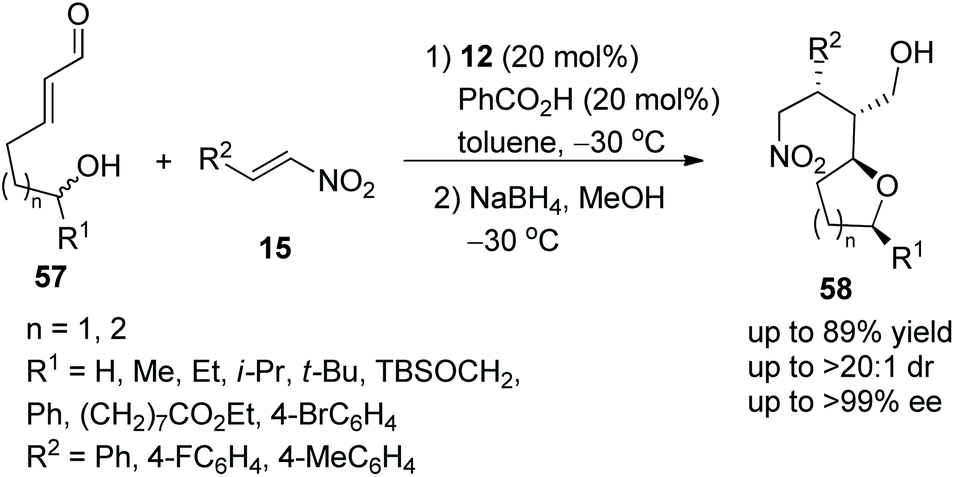
Brenner-Moyer and co-workers28 reported a new strategy for the highly enantioselective construction of functionalized tetrahydropyrans and tetrahydrofurans via an oxa-Michael/Michael cascade reaction in 2012. Under the catalysis of catalyst 12 and benzoic acid, linear unsaturated aldehyde 57 bearing hydroxyl underwent an intramolecular oxa-Michael addition followed by an intermolecular Michael addition with nitroalkenes 15; then the desired products 58 were obtained in good yields (up to 89%) with excellent stereoselectivities (up to >20:1 dr and >99% ee) in toluene at −30 °C (Scheme 19). In the scope of the tetrahydropyran-forming substrates, although the reactivity of substituted 57 was low, the enantiopurity of the corresponding products was well maintained. Then in the scope of tetrahydrofuran-forming substrates, the substituents R1 had little influence on the enantioselectivity of this reaction, substrate 57 with aliphatic-substituted R1 afforded the corresponding products with better yield than substrate 57 with aromatic-substituted R1; and unsubstituted 57 gave a single cascade product in moderate yield (62%) with excellent enantioselectivity (>99% ee). Furthermore, some computational studies were performed to illustrate this catalytic cascade process.
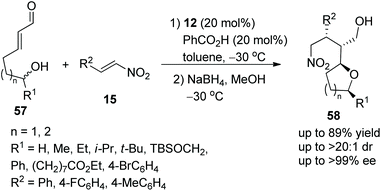 |
| | Scheme 19 Oxa-Michael/Michael cascade reaction for the synthesis of chiral tetrahydropyrans and tetrahydrofurans. | |
2.1.5 Oxa-Michael cascade reaction of γ-hydroxyl enones.
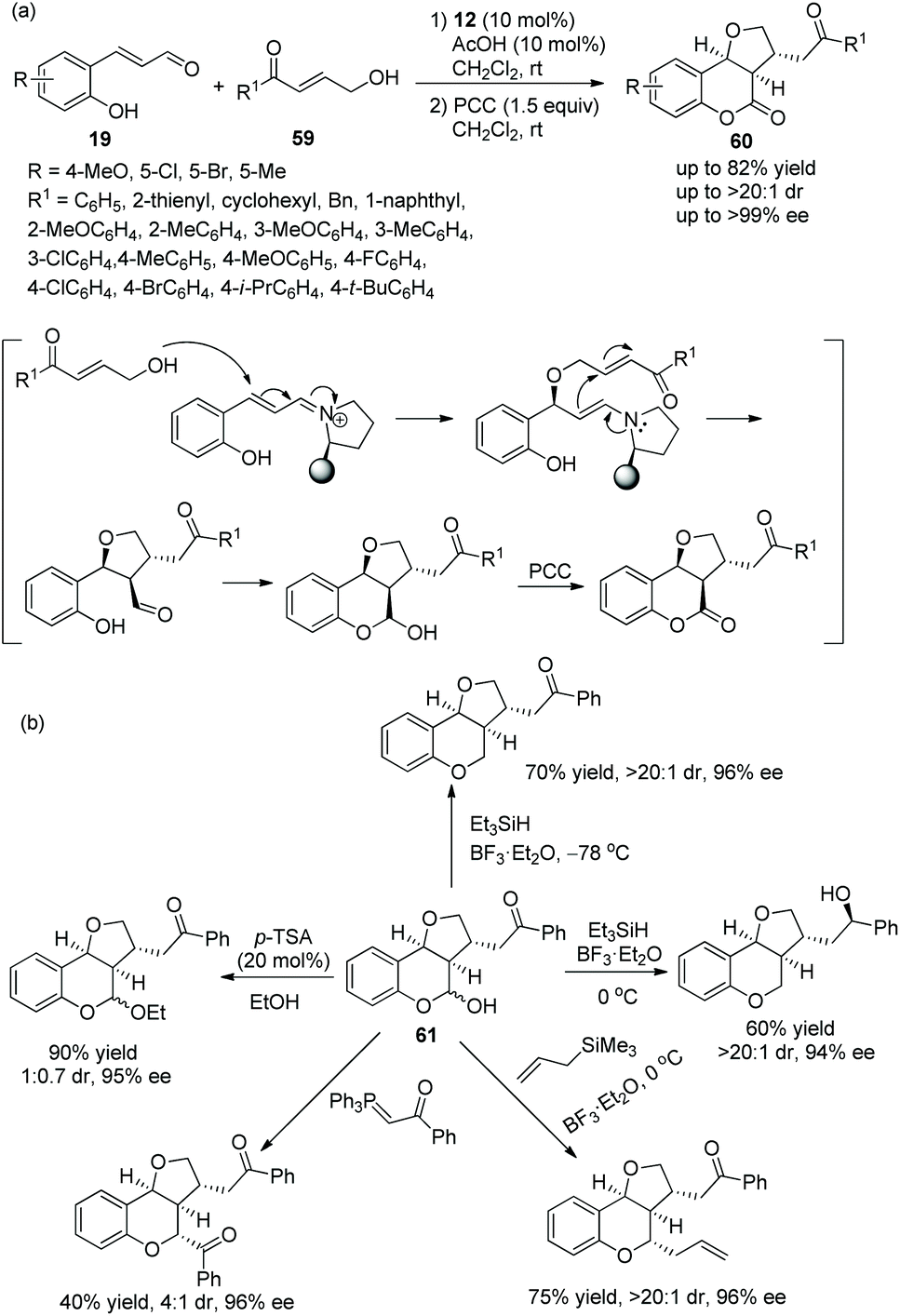
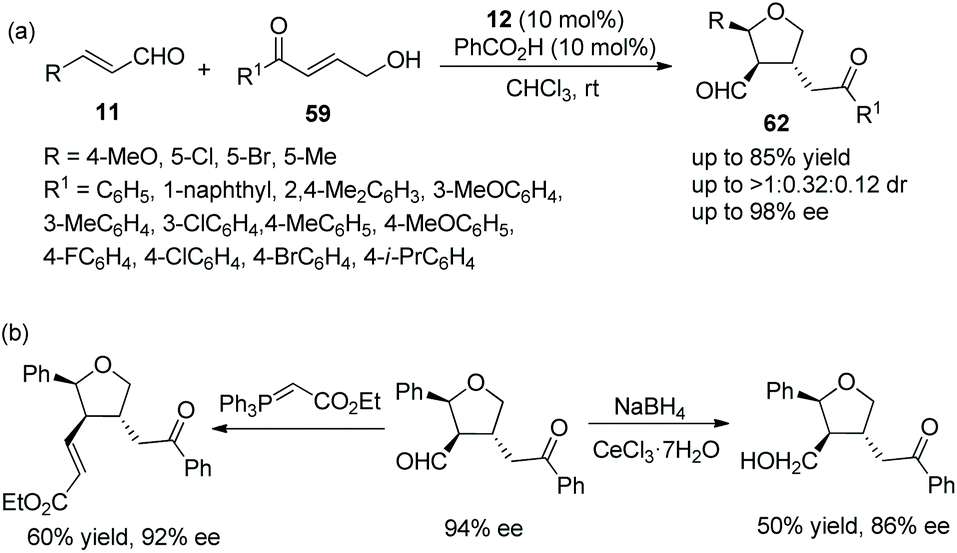
Pan and coworker29 developed a convenient strategy for the asymmetric synthesis of dihydrocoumarin derivatives bearing a tetrahydrofuranylated carbon stereocenter via an oxa-Michael cascade reaction in 2018. In this cascade reaction between o-hydroxy cinnamaldehydes 19 and γ-hydroxy enones 59, the desired products 60 were obtained in good yields (up to 82%) with satisfactory diastereoselectivities (up to >20:1 dr) and enantioselectivities (up to >99% ee) under the combined action of chiral catalyst 12 and acetic acid (Scheme 20a). Nevertheless, in the scope of γ-hydroxy enones, though the thienyl-substituted γ-hydroxy enone could participate in the cascade reaction, the corresponding product was obtained with the lowest yield (62%) and enantioselectivity (46% ee); the electron-donating group on the aromatic substituents of 59 showed better reactivity than the electron-withdrawing group; and ortho-substituents on the aromatic ring of 59 provided the corresponding products with lower yields than other positions. No reaction occurred with γ-hydroxy enones bearing aliphatic substituents. In addition, they converted the unoxidized hemiacetal 61 to some valuable organic structures, such as the formation of acetal, the introduction of an allyl group and the reaction with Wittig reagent. Interestingly, the same reaction of dehydroxylation treated with triethyl silane and BF3·OEt2 at different temperatures would generate different products, and all their enantioselectivities were satisfactory (Scheme 20b). In the same year,30 they again explored the synthesis of tetrahydrofuran derivatives via this cascade reaction using α,β-unsaturated aldehydes 11 and γ-hydroxy enones 59. They used another additive, benzoic acid, to promote the process in CHCl3 solvent, and the corresponding products 62 were obtained with increased yields and enantioselectivities (Scheme 21a). In the scope of enals, when the methyl-substituted enal was employed, the corresponding product was obtained with the lowest enantioselectivity (57% ee). At the same time, in the scope of γ-hydroxy enones, no reaction was observed when the alkyl-substituted γ-hydroxy enone was utilized. Additionally, two further transformations were established subsequently, which illustrated the applicability of this strategy and afforded the corresponding products without losing much enantiomeric purity (Scheme 21b).
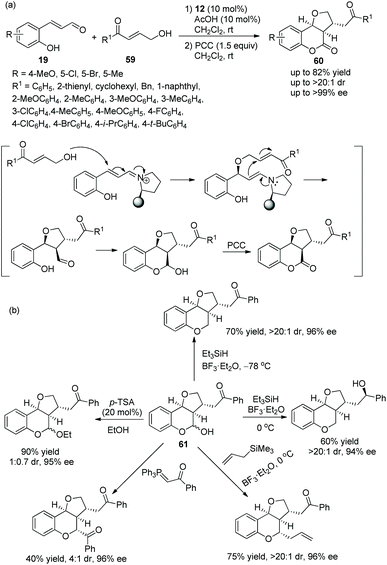 |
| | Scheme 20 Oxa-Michael cascade reaction for the synthesis of chiral dihydrocoumarin derivatives and the derivatization of hemiacetal. | |
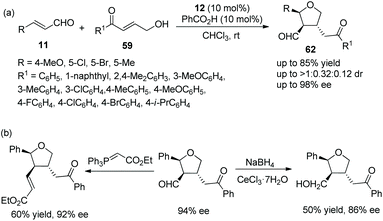 |
| | Scheme 21 Oxa-Michael cascade reaction for the synthesis of chiral tetrahydrofuran derivatives and the derivatization of chiral tetrahydrofuran. | |
2.2 Reactions catalyzed by chiral bifunctional squaramides
As we know, chiral squaramide, as a type of bifunctional chiral catalyst with good catalytic activity, has been of interest to a wide range of organic researchers and has been successfully applied in many asymmetric catalytic cascade reactions, including the oxa-Michael cascade reaction, which has achieved high enantioselectivities.31 The catalytic process of bifunctional squaramides is similar to bifunctional thioureas, which catalyze conjugate addition by forming hydrogen bonds with the reactants (Scheme 22).
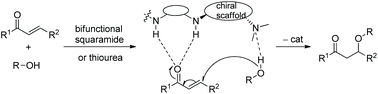 |
| | Scheme 22 Chiral bifunctional squaramide-catalyzed general mechanism of the oxa-Michael reaction. | |
2.2.1 Oxa-Michael cascade reaction for the synthesis of chiral chromans.
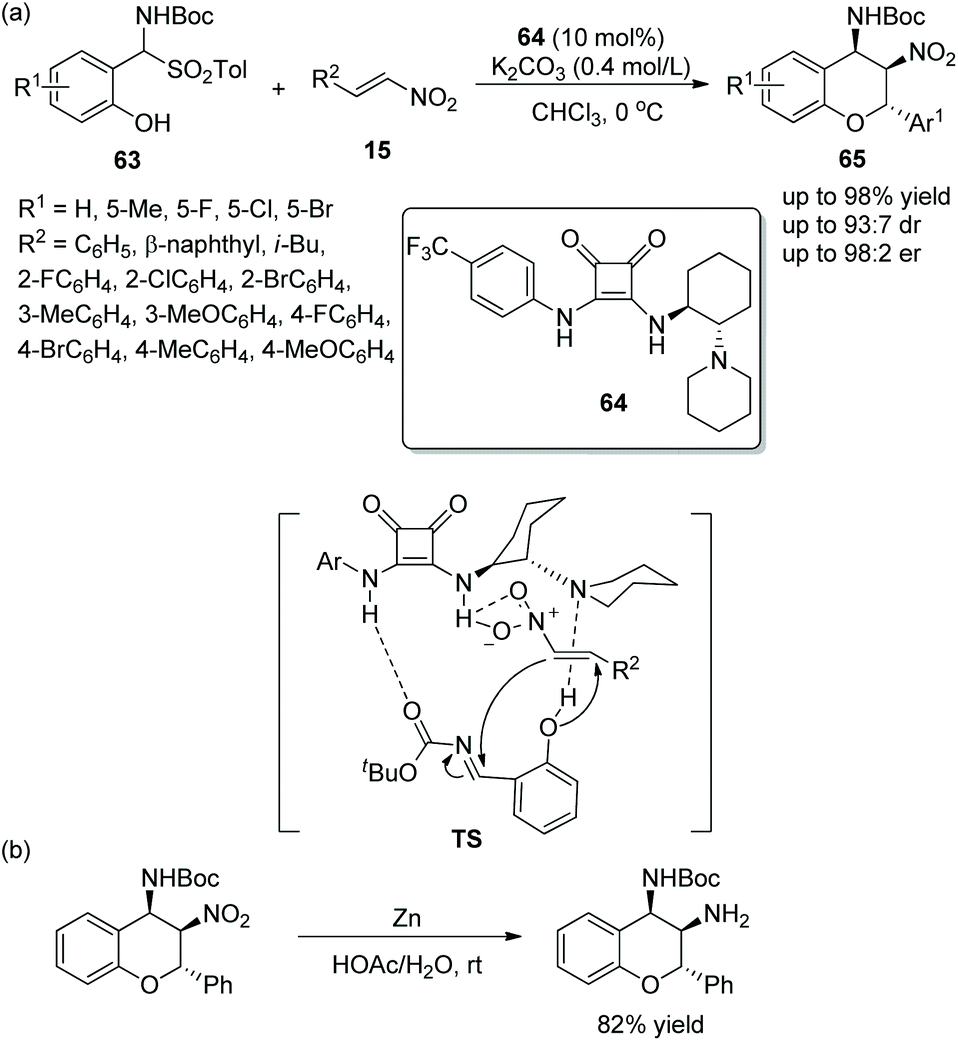
Due to the significance of chromans in pharmaceutical structures and natural products, many novel methods for the construction of these chiral structures via an oxa-Michael cascade reaction catalyzed by various chiral bifunctional squaramides have been developed. Peng's group32 developed an effective approach to synthesize the chroman derivatives with high enantioselectivities in 2014. The o-hydroxyaryl-substituted α-amido sulfones 63 and nitroolefins 15 catalyzed by squaramide 64 bearing chiral cyclohexane underwent an oxa-Michael/aza-Henry cascade process and afforded the products 65 with excellent yields (up to 98%) and stereoselectivities (up to 93:7 dr and 98:2 er) (Scheme 23a). It should be noticed that all the reaction times were relatively long in the scope of this cascade reaction; in particular, isobutyl-substituted nitroolefin generated the lowest yield (46%), but other substituted nitroolefins gave satisfactory results; and the position and electronic property of substituents on the aromatic ring of 63 had little effect on the reaction. Moreover, in order to demonstrate the applicability of this strategy, they tried a reduction reaction of a chiral product using Zn/HOAc at room temperature, which transformed the nitro group to an amino group (Scheme 23b).
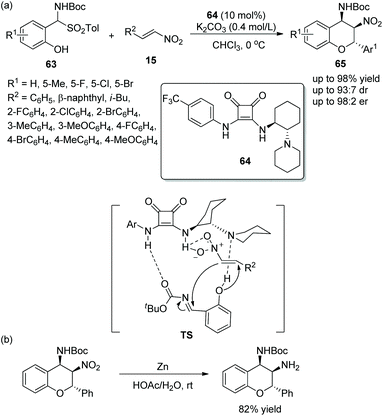 |
| | Scheme 23 Oxa-Michael/aza-Henry cascade reaction for the synthesis of benzopyran derivatives and the derivatization of the chiral product. | |
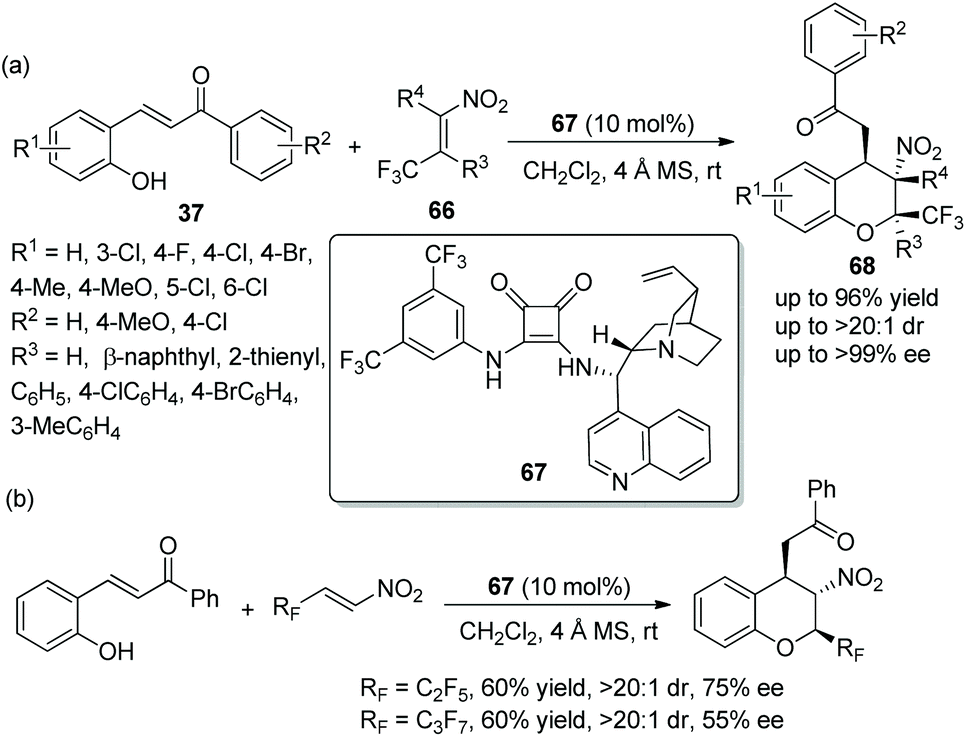
An admirable piece of work was reported by Wang's group33 for the synthesis of 2-CF3 chromans in 2015. In this cascade reaction, β-CF3-nitroalkenes 66 and o-hydroxychalcones 37 were selected as the starting materials, which were catalyzed by a chiral squaramide 67 for the construction of optically active trifluoromethylated chromans 68. The products 68 bearing three contiguous stereocenters were obtained in high yields (up to 96%) with excellent stereoselectivities (up to >20:1 dr and >99% ee) (Scheme 24a). It has to be said that in the scope of substrates, all the substituents, whether electron-withdrawing or electron-donating groups, afforded the corresponding products with excellent enantioselectivities, even though the reaction times were different. The position of the substituents had an effect on the reactivity: for example, if the Cl was moved from the 6-position to the 3-position of o-hydroxychalcone, the relevant reaction time would be prolonged. Compared with unsubstituted R4, methyl-substituted R4 gave the corresponding product with better yield. In addition, the authors further performed this cascade reaction by utilizing perfluoroalkyl substituted β-nitroalkenes (Scheme 24b). The result displayed that pentafluoroethyl- and heptafluoropropyl-substituted nitroalkenes showed lower reactivity and afforded the products with lower yields and enantioselectivities than β-CF3 nitroalkenes, but it is still of great significance for the construction of chiral polyfluorine-substituted chromans.
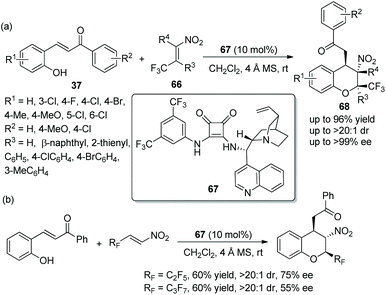 |
| | Scheme 24 Oxa-Michael cascade reaction for the synthesis of chromans bearing trifluoromethyl. | |
Xu's group34 in 2018 developed an efficient strategy for the construction of chiral chroman derivatives via an oxa-Michael cascade reaction catalyzed by chiral bifunctional squaramide 69. The desired products 70 containing two nitro groups were generated in good yields with excellent diastereo- and enantioselectivities by the oxa-Michael/Michael cascade process of 2-(E)-(2-nitrovinyl)phenols 21 and nitroolefins 15 under mild conditions (Scheme 25a). In the scope of this cascade reaction, the position of the substituents on the aromatic ring of 21 had little effect on the reaction, but the electron-donating substituents provided the corresponding products with better yields and enantioselectivities than electron-withdrawing substituents; and aliphatic-substituted nitroolefins generally provided adducts in higher yields than did aromatic substituents. In aromatic-substituted nitroolefins, compared with electron-withdrawing groups, electron-donating groups afforded better enantioselectivities. Additionally, they further examined the synthetic applicability of this cascade reaction by using trans-α-Me-β-nitroolefin, which showed a good result with moderate yield and stereoselectivity (Scheme 25b). Later, Pedrosa's group35 developed a similar strategy for the construction of chiral trisubstituted chroman derivatives via an oxa-Michael triggered cascade reaction catalyzed by 4-vinylphenyl-substituted squaramides. Under the catalysis of 4-vinylphenyl-substituted squaramides 72 or 73, nitrovinylphenols 21 and multi-substituted nitroalkenes 71 afforded desired products 75 with good yields (up to 88%) and stereoselectivities (up to >99:1 dr and >99:1 er) in dichloromethane at room temperature (Scheme 25c). In the scope of substrates, compared with nitroolefins with electron-donating groups, nitroolefins with electron-withdrawing groups showed better reactivity, but afforded the corresponding products with lower stereoselectivities; and if trisubstituted nitroolefins were applied to the reaction, such as R4 being replaced by trifluoromethyl or R5 being replaced by methyl, worse results were obtained. Additionally, another catalyst 74 as the polymeric homolog of 72 was also employed, but no better results were obtained. In general, catalyst 72 showed the best catalytic ability.
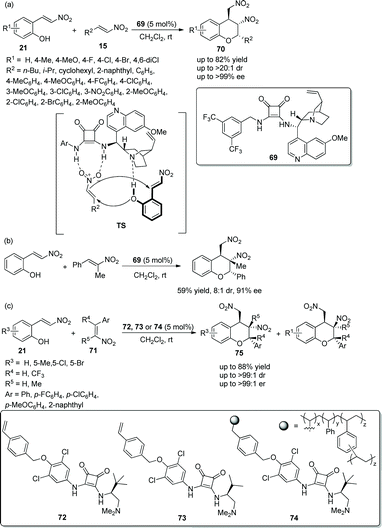 |
| | Scheme 25 Oxa-Michael cascade cyclization reaction for the synthesis of chiral dinitro chromans. | |
2.2.2 Oxa-Michael cascade reaction for the synthesis of chiral spiro cyclic compounds.
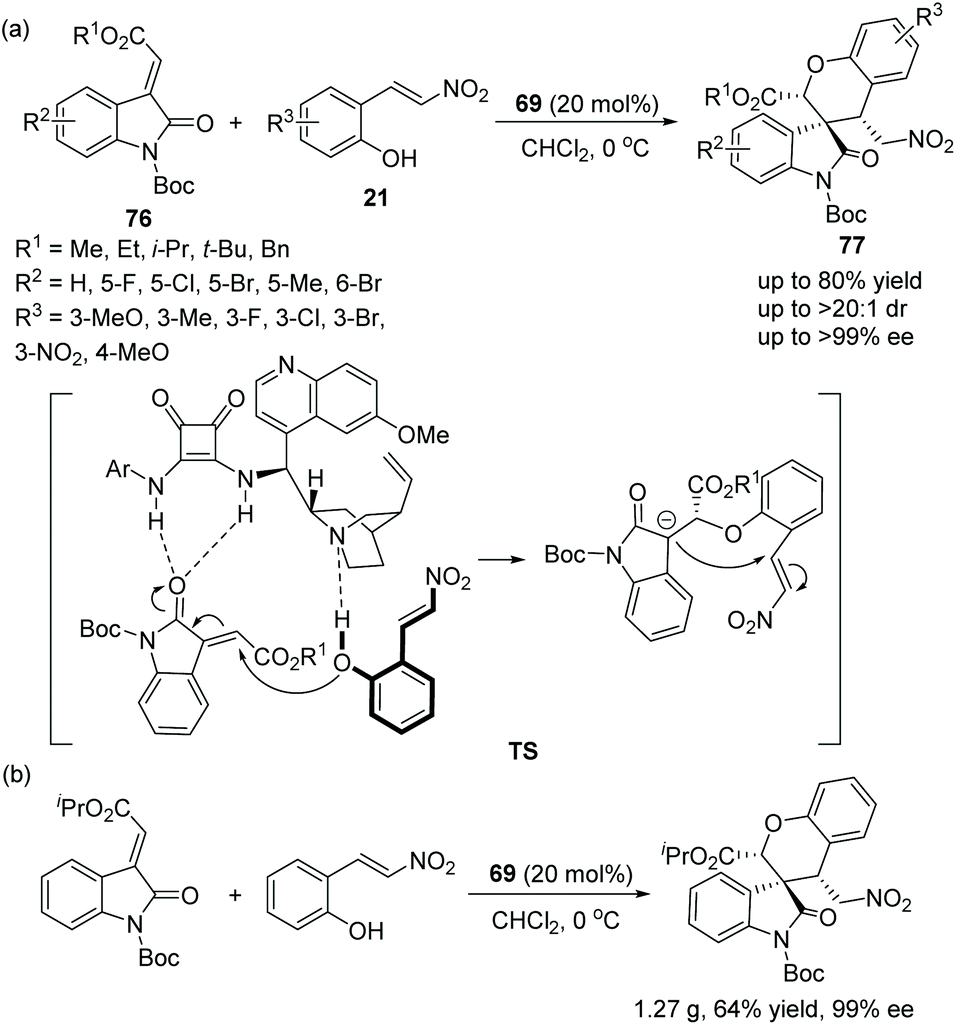
Zhu's group36 described a novel, efficient and valuable methodology for the synthesis of spirooxindoles 77 bearing benzopyran structures via oxa-Michael cascade reactions in 2013. It is worth noting that the cascade reaction of methyleneindolinones 76 with nitrovinylphenols 21 catalyzed by chiral bifunctional squaramide 69 was highly determined by the N-protecting group. Only when the N-protecting group of the oxindoles was a Boc group could the desired products be obtained. Satisfactorily, the reactions afforded good yields and great stereocontrol (>20:1 dr, >99% ee) under mild conditions (Scheme 26a). In an exploration of the substrate scope, the position and electronic property of the substituents on the aromatic ring of 21 and 76 had little effect on the reaction; it is noted that reactant 21, in which R3 was substituted by the electron-withdrawing nitro group, needed the longest reaction time to generate the desired product in the lowest yield (53%), even though the enantioselectivity achieved was 98% ee. Furthermore, they successfully performed this cascade reaction on a gram-scale without losing enantioselectivity and demonstrated the applicability of this strategy (Scheme 26b).
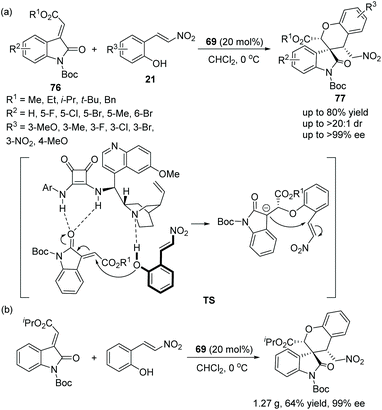 |
| | Scheme 26 Oxa-Michael cascade reaction for the synthesis of benzopyran-spirooxindoles. | |
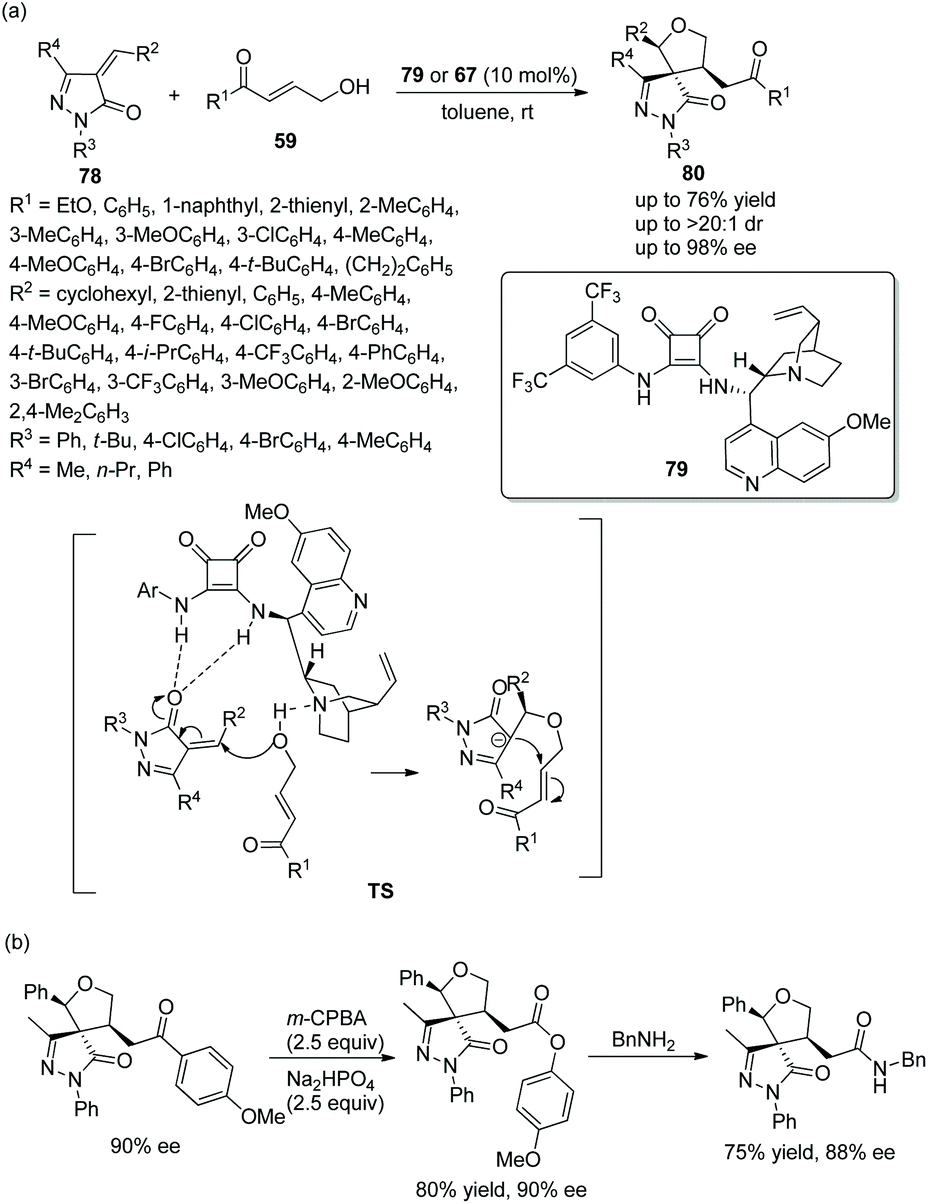
Given the pharmaceutical and medicinal applications of pyrazolone derivatives, an effective method to prepare chiral spiro pyrazolones with three consecutive stereocenters was reported by Pan's group37 in 2018. They employed unsaturated pyrazolones 78 and γ-hydroxyenones 59 to catalytically synthesize chiral spiro tetrahydrofuran-pyrazolones 80. Whether chiral bifunctional squaramide 79 or 67 was employed, the desired products were attained in good yields with excellent stereoselectivities (up to >20:1 dr and 98% ee) (Scheme 27a). The reason why two catalysts were used is that, when examining the generality of unsaturated pyrazolones in this cascade reaction, it was found that cinconidine-derived catalyst 67 showed better enantioselective control than quinine-derived catalyst 79. In the scope of substrates, aryl-substituted γ-hydroxyenones showed better results than alkyl-substituted γ-hydroxyenones; in the scope of pyrazolones with different benzylidene substituents, all the substituents provided the corresponding products with high enantioselectivities, and the reactivity of the electron-donating groups was better than that of the electron-withdrawing groups; in the scope of N-substituents, no desired product was observed when the N-substituent was tert-butyl. In addition, a few further transformations, selective oxidation and a substitution reaction with benzylamine, were performed and afforded the corresponding products without losing much enantiopurity, which proved the synthetic utility of the cascade strategy (Scheme 27b).
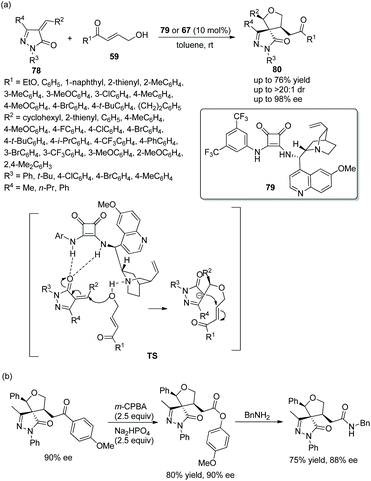 |
| | Scheme 27 Oxa-Michael/Michael cascade reaction for the synthesis of spiro tetrahydrofuran-pyrazolones and the derivatization of the chiral product. | |
2.3 Reactions catalyzed by chiral bifunctional thioureas
Since chiral thioureas have been shown to have an ability to participate in molecular recognition processes by selectively forming hydrogen bonds, they have been employed as comparatively effective organocatalysts in many conversions besides oxa-Michael reactions. The chiral bifunctional thiourea catalytic general mechanism of oxa-Michael is similar to that of chiral squaramides (Scheme 22).
2.3.1 Oxa-Michael/Michael cascade reaction of ethylene β-keto esters with nitroolefins.
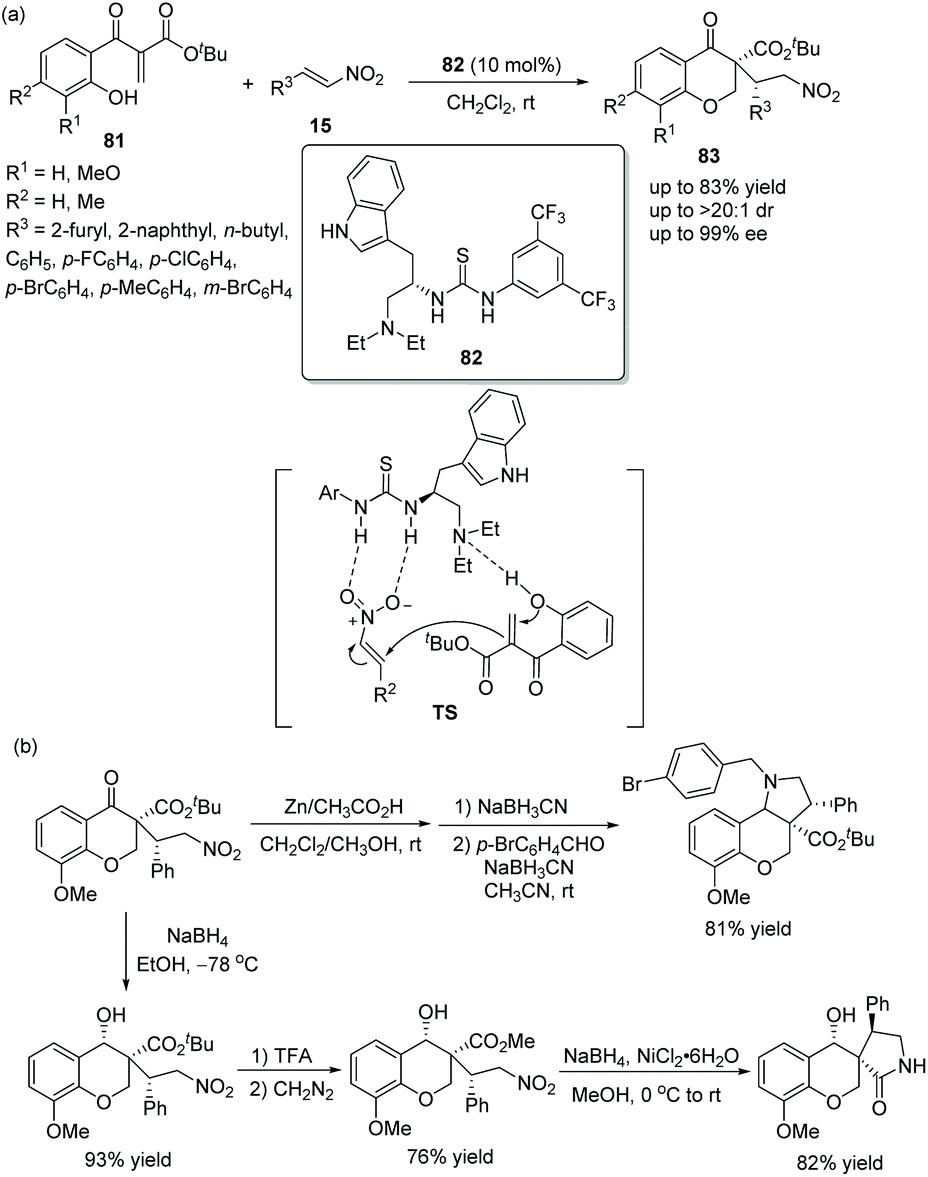
A novel and efficient strategy for the construction of highly functionalized chiral chromanones via an oxa-Michael/Michael cascade reaction was reported by Lu's group38 in 2011. Ethylene β-keto esters 81, as starting materials would first be employed in a cascade reaction; then reaction with nitroolefins 15 in dichloromethane could smoothly generate the desired adducts 83 in good yields (up to 83%) with excellent stereoselectivities (up to >20:1 dr and 99% ee) under the catalysis of a tryptophan-derived tertiary amine-thiourea catalyst 82 (Scheme 28a). In the scope of substrates, n-butyl substituted nitroolefin showed the worst reactivity (41% yield) and enantioselectivity (89% ee), but other substituted nitroolefins provided satisfactory results; and electron-rich substituents on the aryl ring of ethylene β-keto esters 81 promoted the reaction more efficiently. Furthermore, the 3,3-disubstituted 4-chromanone products could be further functionalized, such as reductive amination of the nitro group and the formation of lactam (Scheme 28b), which successfully demonstrated the applicability of this methodology.
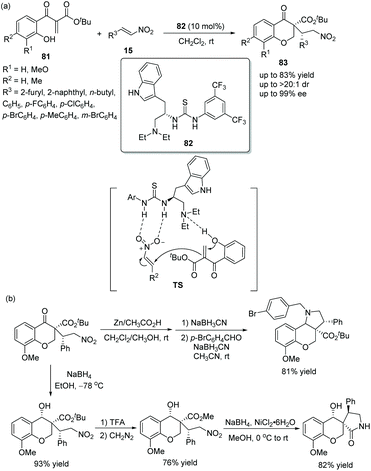 |
| | Scheme 28 Oxa-Michael/Michael cascade reaction for the synthesis of chiral chromanones and the derivatization of the chiral product. | |
2.3.2 Oxa-Michael cascade reactions for the synthesis of chiral chromans.
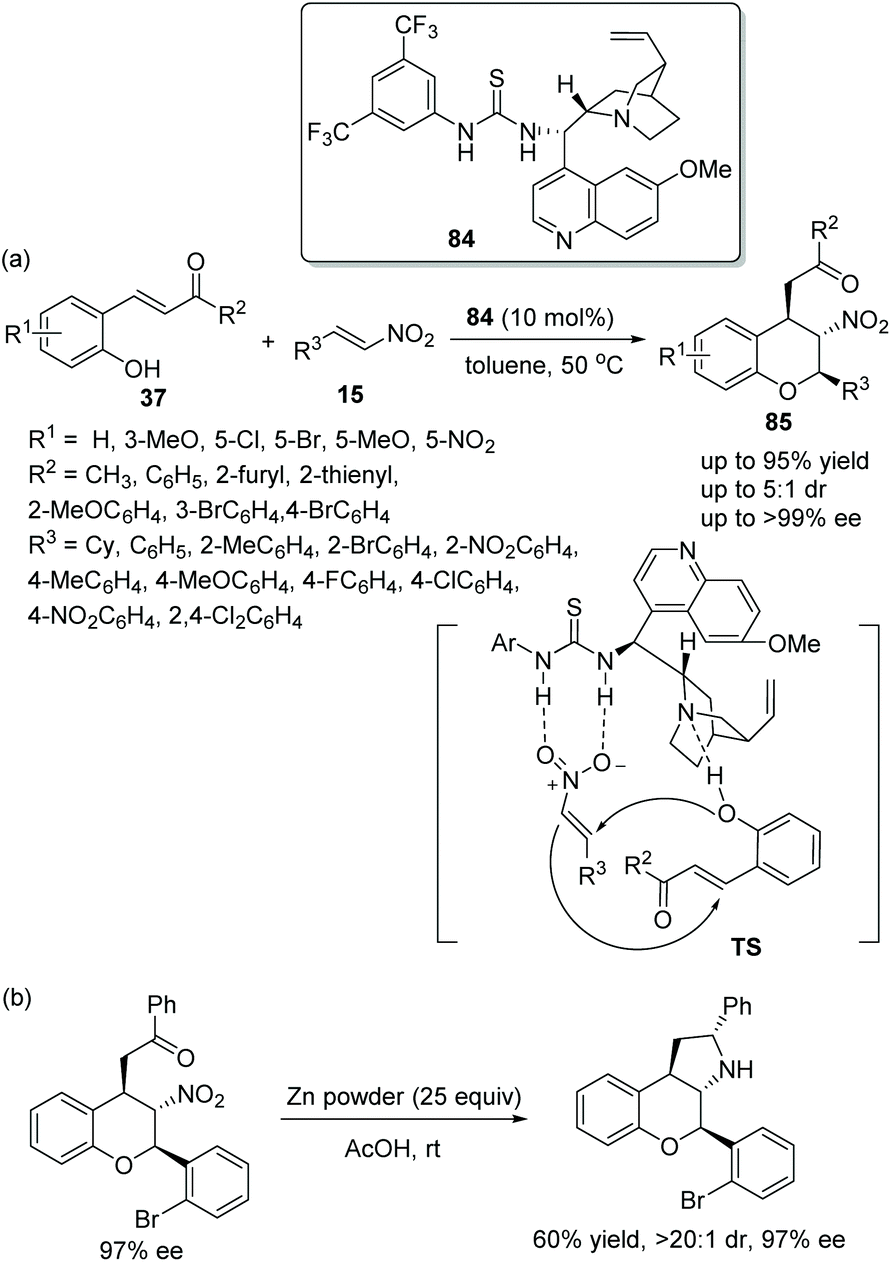
Another effective and simple strategy for the construction of chromans bearing multiple stereocenters was proposed by Singh's group39 in 2015, which expanded the field of synthesis of chiral chromans. The desired products 85 were obtained with excellent yields (up to 95%) and stereoselectivities (up to 5:1 dr and >99% ee) via an oxa-Michael/Michael cascade reaction catalyzed by a chiral bifunctional aminothiourea 84 (Scheme 29a). And in the substrate scope of this cascade reaction, the β-nitrostyrenes 15 bearing electron-donating groups, such as methyl and methoxy, would take a longer reaction time than the β-nitrostyrenes bearing electron-withdrawing groups to generate the corresponding products. Moreover, in the scope of the substrates 37, irrespective of whether substituents on the aromatic ring were electron-donating or electron-withdrawing, all of them provided the corresponding products with high enantioselectivities. However, for substrate 37 with a nitro group on the aromatic ring, no product was obtained, which might be due to the hydrogen bond formed by the nitro group and the bifunctional catalyst hindering the cascade process. Additionally, to display the synthetic utility of this cascade method, a transformation to generate tricyclic chroman was performed, and the corresponding product was obtained without losing enantioselectivity (Scheme 29b).
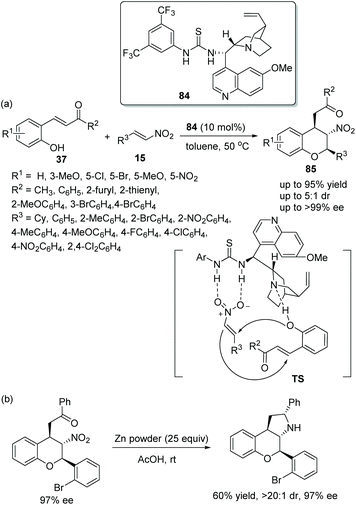 |
| | Scheme 29 Oxa-Michael/Michael cascade reaction for the synthesis of chiral chromans and the derivatization of the chiral product. | |
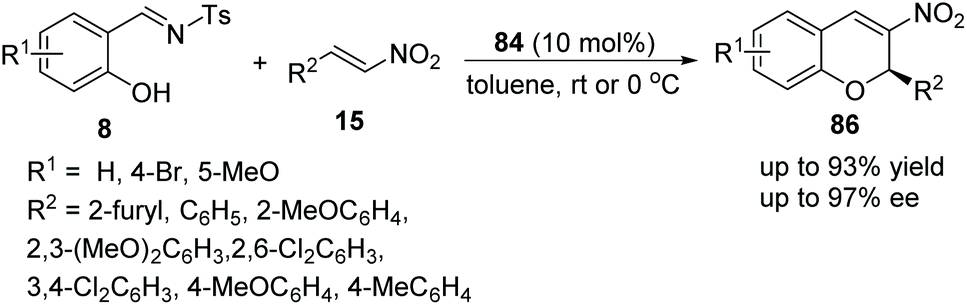
Schreiner's group40 reported a simple and feasible protocol for the construction of 2H-nitrochromenes via an oxa-Michael cascade reaction. Under the catalysis of bifunctional thiourea 84 in toluene, salicyl N-tosylimines 8 and nitrostyrenes 15 underwent an oxa-Michael/aza-Henry/desulfonamidation cascade process and afforded the desired products 86 with high yields and enantioselectivities (Scheme 30). In the scope of substrates, compared with electron-withdrawing substituents, electron-donating-group-substituted salicyl N-tosylimines provided the corresponding products with better enantiopurity; when the reaction temperature was decreased to 0 °C, the enantioselectivity of the corresponding products was promoted but the yield was decreased.
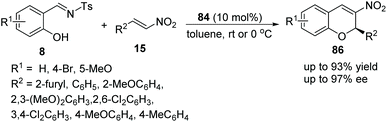 |
| | Scheme 30 Oxa-Michael/aza-Henry/desulfonamidation cascade reaction for the synthesis of chiral 2H-chromenes. | |
2.3.3 Oxa-Michael/Michael cascade reaction for the synthesis of chiral spirocyclic compounds.
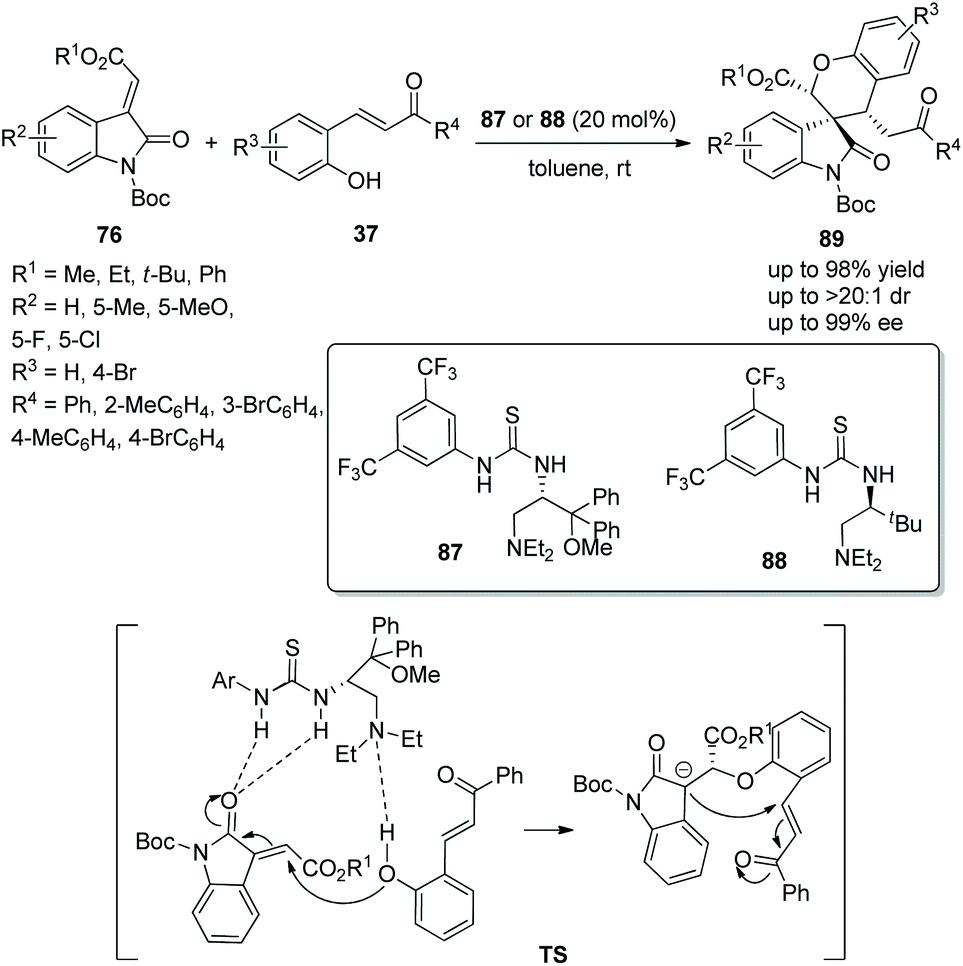
Spirocyclic compounds as a vital structure in natural products and organic synthesis has always attracted the attention of many researchers, which has resulted in various functionalized spiro compounds being synthesized. Zhao's group41 in 2014 reported an available and efficient strategy for the construction of chiral spirooxindole-chromans via an oxa-Michael/Michael cascade reaction. Under the catalysis of amino-acid-derived thioureas 87 or 88, methyleneindolinones 76 reacted with o-hydroxychalcones 37 and provided the corresponding products 89 with excellent yields (up to 98%) and stereoselectivities (up to >20:1 dr and 99% ee) in toluene at room temperature (Scheme 31). In the scope of substrates catalyzed by organocatalyst 87, the electronic property and position of substituents on the aromatic ring of 76 had little effect on the stereoselectivity, but electron-withdrawing substituents showed better reactivity than electron-donating substituents; ester and ketone groups at the carbon–carbon double bond could also be tolerated well. Noticeably, except for the N-protecting group Boc, other N-protecting groups could not participate in the process. When organocatalyst 88 was used, the corresponding products were obtained with higher yields but lower enantioselectivities.
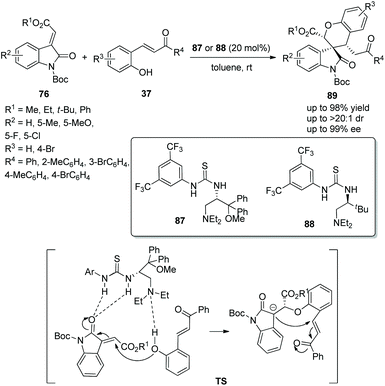 |
| | Scheme 31 Oxa-Michael/Michael cascade reaction for the synthesis of chiral spiro benzohydropyran-oxindoles. | |
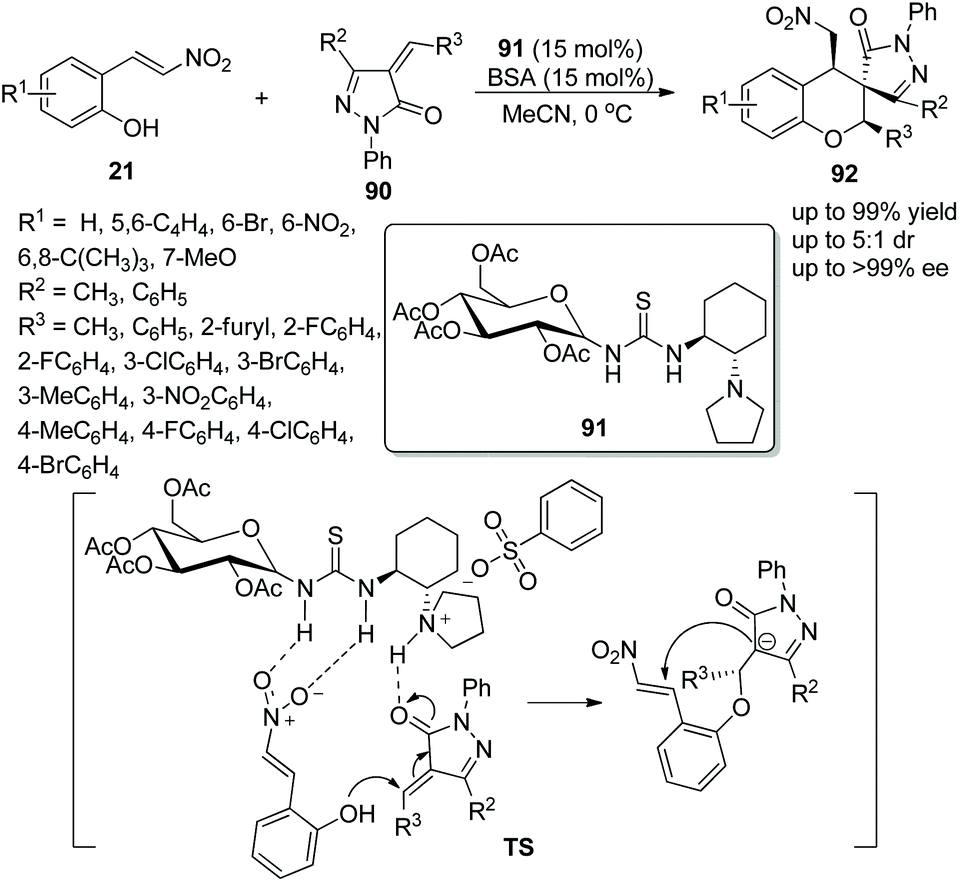
A suitable and effective proposal for the construction of functional spiro chroman-prazolones via a bifunctional aminothiourea 91 catalyzed oxa-Michael/Michael addition cascade reaction was presented by Miao's group42 in 2015. The nitrovinyl phenols 21 and 4-alkenyl pyrazolin-3-ones 90 afforded the target products 92 bearing three contiguous stereocenters with excellent yields (up to 99%) and enantioselectivities (up to >99% ee) under the catalysis of catalyst 91 and benzene sulfonic acid (BSA) additive in MeCN at 0 °C (Scheme 32). In the substrate scope of this cascade reaction, the good reactivity of all the pyrazolones with electron-donating or electron-withdrawing groups at any position of the benzene ring furnished the corresponding products with high yields and enantioselectivities; a methyl substituent at R2 showed better reactivity and stereoselectivity than a phenyl substituent. Moreover, an alkyl substituted at R3 of unsaturated pyrazolone provided the corresponding product with the best enantioselectivity in a comparably short reaction time.
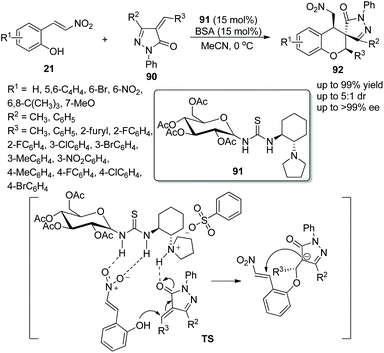 |
| | Scheme 32 Oxa-Michael/Michael cascade reaction for the synthesis of chiral spiro benzohydropyran-pyrazolones. | |
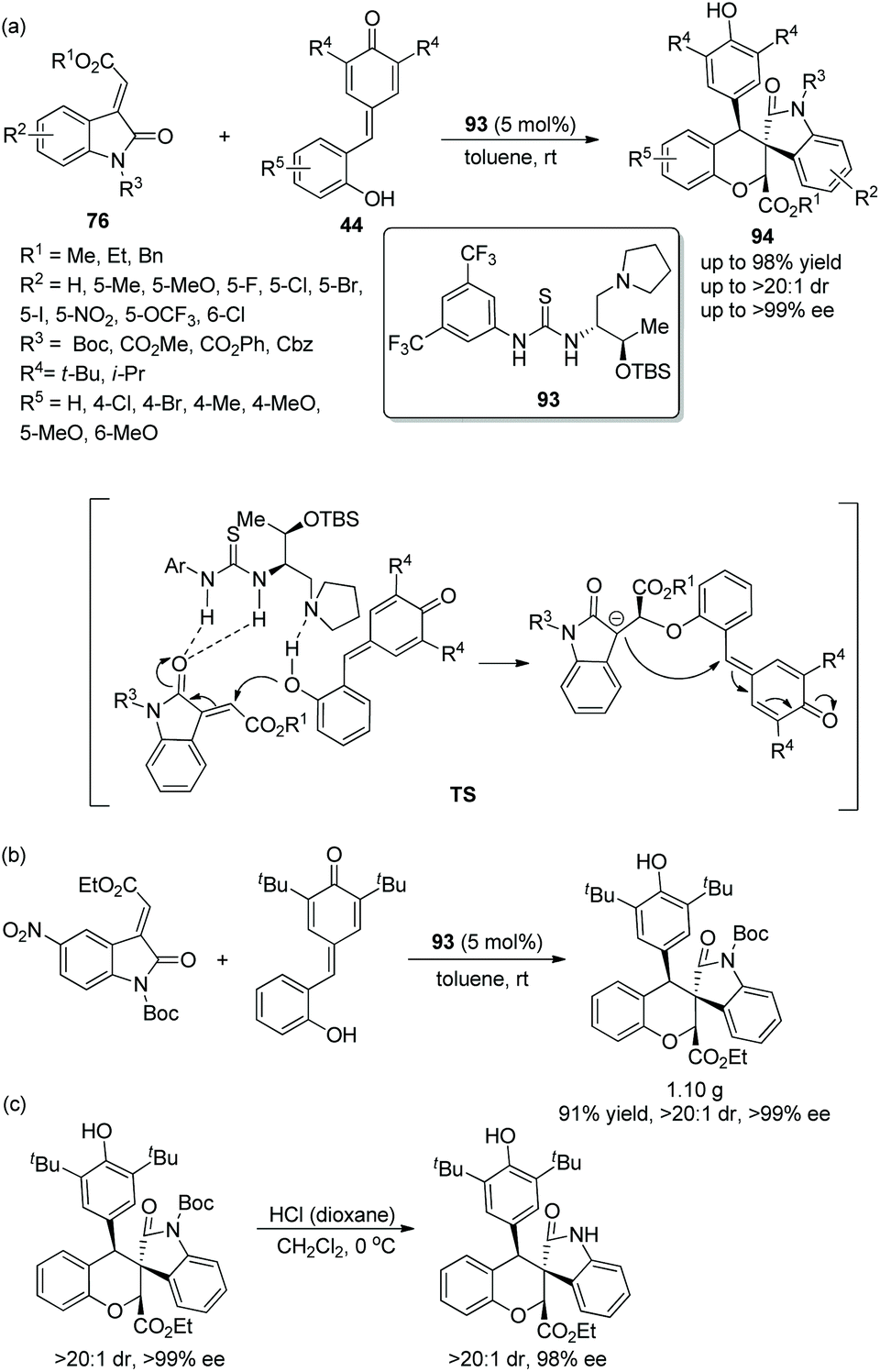
Since there were no precedents for employing o-hydroxyphenyl substituted p-quinone methides 44 as donor–Michael acceptor synthons in domino addition procedures before 2016, a viable and elegant approach to fill the gap was developed by Enders’ group.43 After the methyleneindolinones 76 and o-hydroxyphenyl substituted p-quinone methides were catalyzed by chiral bifunctional aminothiourea 93 and underwent an oxa-Michael addition and intramolecular 1,6-addition, the desired chiral functionalized spirocharomans 94 were generated with excellent yields and stereoselectivities under mild conditions (Scheme 33a). In the substrate scope of this cascade reaction, electron-donating and electron-withdrawing substituents were both tolerated well, and all of them furnished the corresponding products with excellent enantioselectivities; electron-withdrawing substituents on the aromatic ring of 76 or 44 showed better reactivity than electron-donating substituents. Nevertheless, when the R4 substituent was replaced by an isopropyl group, the stereoselectivity of this reaction was not influenced, but the reactivity was decreased, which afforded the product with the lowest yield (43%). The cascade reaction on a gram-scale was accomplished under optimum conditions and maintained the stereoselectivity of the corresponding product (Scheme 33b). In addition, a deprotection transformation to remove the N-Boc group was smoothly finished without losing much enantiopurity (Scheme 33c).
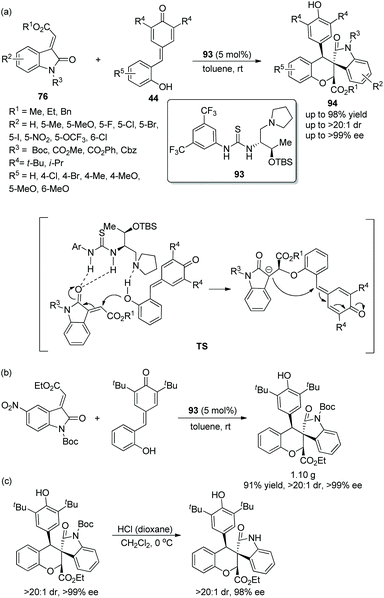 |
| | Scheme 33 Oxa-Michael/1,6-addition cascade reaction for the synthesis of spiro chromans-oxindoles and the derivatization of the chiral product. | |
2.4 Reactions catalyzed by other chiral organocatalysts
There are some other organocatalytic asymmetric oxa-Michael addition triggered cascade reactions, for example chiral cinchona alkaloid derived catalysts, which cannot be strictly classified into the preceding catalyst groups but still produce a satisfactory result. Moreover, from the perspective of the reaction mechanism, they activate an oxa-Michael cascade reaction by forming hydrogen-bond intermediates.
2.4.1 Oxa-Michael cascade cyclization reaction of β-carboxy substituted α,β-unsaturated ketones with hydroxylamine catalyzed by chiral quinidine-based catalyst.
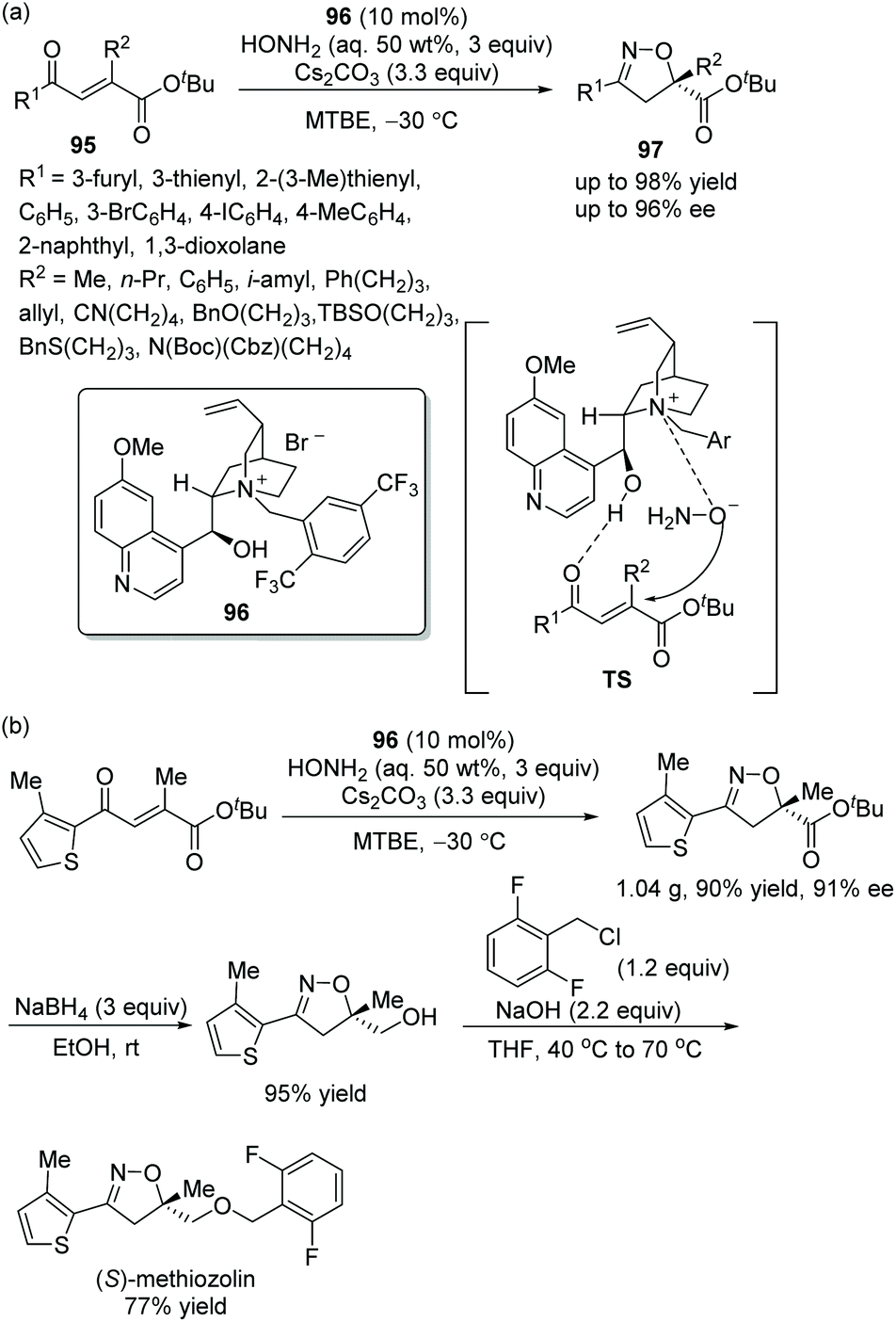
Since isoxazoline derivatives play a significant role in bioactive heterocycles, and the synthesis of some chiral isoxazoline derivatives with potential bioactivity is of great benefit to the pharmaceutical industry and medicine, Cho's group44 displayed an efficient and useful strategy for the construction of chiral carboxy-substituted isoxazolines via an oxa-Michael cascade cyclization reaction in 2018. The β-carboxy substituted α,β-unsaturated ketones 95 and hydroxylamine were phase-transfer-catalyzed by a combination of a chiral quinidine-derived catalyst 96 and cesium carbonate (Cs2CO3), and the corresponding products 97 were generated with high yields and enantioselectivities in methyl tert-butyl ether (MTBE) at −30 °C (Scheme 34a). In the substrate scope of this reaction, all of the substituents provided the corresponding products with satisfactory yields and enantioselectivities, and electron-donating groups were proven to show a better result. Moreover, a gram-scale reaction was carried out and afforded the corresponding product without losing much yield or enantioselectivity. A further transformation for the synthesis of (S)-methiozolin in two steps was performed, which demonstrated well the applicability of this cascade strategy (Scheme 34b).
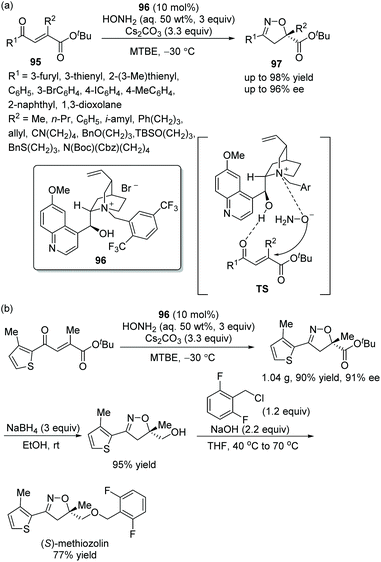 |
| | Scheme 34 Oxa-Michael cascade reaction for the synthesis of chiral isoxazolines and the derivatization of the chiral product. | |
3 Conclusions
In summary, we have introduced recent advances in the field of organocatalytic asymmetric oxa-Michael addition triggered cascade reactions and have illustrated that it is a comparatively valuable and powerful tool for the rapid formation of functionalized oxygen heterocyclic molecules containing stereocenters. A great variety of novel and potentially bioactive compounds with excellent stereoselectivities have been synthesized via various oxa-Michael addition triggered cascade reactions catalyzed by a diverse number of organocatalysts under different conditions, which has indeed expanded the scope of the methodology of the Michael addition cascade reaction. Many further transformations have been performed to display the applicability of the corresponding oxa-Michael addition triggered cascade reactions. However, there is still a lot of work needed to promote and optimize the process, such as more novel and recyclable organocatalysts, more diversified adducts, more convenient reaction processes and so on. It is expected that many more interesting and valuable developments will be established to increase the diversity of oxa-Michael triggered reactions in the near future.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- M. I. Gutierrez-Jimenez, C. Aydillo, C. D. Navo, A. Avenoza, F. Corzana, G. Jimenez-Oses, M. M. Zurbano, J. H. Busto and J. M. Peregrina, Bifunctional Chiral Dehydroalanines for Peptide Coupling and Stereoselective S-Michael Addition, Org. Lett., 2016, 18, 2796 CrossRef CAS PubMed.
- C. D. Navo, N. Mazo, P. Oroz, M. I. Gutierrez-Jimenez, J. Marin, J. Asenjo, A. Avenoza, J. H. Busto, F. Corzana, M. M. Zurbano, G. Jimenez-Oses and J. M. Peregrina, Synthesis of Nbeta-Substituted alpha,beta-Diamino Acids via Stereoselective N-Michael Additions to a Chiral Bicyclic Dehydroalanine, J. Org. Chem., 2020, 85, 3134 CrossRef CAS PubMed.
- N. Umekubo, Y. Suga and Y. Hayashi, Pot and time economies in the total synthesis of Corey lactone, Chem. Sci., 2020, 11, 1205 RSC.
- C. F. Nising and S. Bräse, The oxa-Michael reaction: from recent developments to applications in natural product synthesis, Chem. Soc. Rev., 2008, 37, 1218 RSC.
- C. F. Nising and S. Brase, Recent developments in the field of oxa-Michael reactions, Chem. Soc. Rev., 2012, 41, 988 RSC.
- D. Enders, M. R. Huttl, C. Grondal and G. Raabe, Control of four Stereocentres in a Triple Cascade Organocatalytic Reaction, Nature, 2006, 441, 861 CrossRef CAS PubMed.
- Y. Deng, S. Kumar and H. Wang, Synergistic–cooperative Combination of Enamine Catalysis with Transition Metal Catalysis, Chem. Commun., 2014, 50, 4272 RSC.
- P. Chauhan, S. Mahajan, U. Kaya, D. Hack and D. Enders, Bifunctional Amine-Squaramides: Powerful Hydrogen-Bonding Organocatalysts for Asymmetric Domino/Cascade Reactions, Adv. Synth. Catal., 2015, 357, 253 CrossRef CAS.
- J. L. Vicario, D. Badía and L. Carrillo, Organocatalytic Enantioselective Michael and Hetero-Michael Reactions, Synthesis, 2007, 2065 CrossRef CAS.
- C. Liu, X. Zhang, R. Wang and W. Wang, “One-Pot” Access to 4H-Chromenes with Formation of a Chiral Quaternary Stereogenic Center by a Highly Enantioselective Iminium-allenamine Involved Oxa-Michael–Aldol Cascade, Org. Lett., 2010, 12, 4948 CrossRef CAS PubMed.
- J. Zhang, M. J. Ajitha, L. He, K. Liu, B. Dai and K.-W. Huang, Enantioselective Organocatalyzed Oxa-Michael–Aldol Cascade Reactions: Construction of Chiral 4H-Chromenes with a Trifluoromethylated Tetrasubstituted Carbon Stereocenter, Adv. Synth. Catal., 2015, 357, 967 CrossRef CAS.
- J. Alemán, A. Núñez, L. Marzo, V. Marcos, C. Alvarado and J. L. G. Ruano, Asymmetric Synthesis of 4-Amino-4H-Chromenes by Organocatalytic Oxa-Michael/Aza-Baylis–Hillman Tandem Reactions, Chem. – Eur. J., 2010, 16, 9453 CrossRef PubMed.
- S.-P. Luo, Z.-B. Li, L.-P. Wang, Y. Guo, A.-B. Xia and D.-Q. Xu, Chiral Amine/chiral Acid as an Excellent Organocatalytic System for the Enantioselective Tandem Oxa-Michael-aldol Reaction, Org. Biomol. Chem., 2009, 7, 4539 RSC.
- G. Yin, R. Zhang, L. Li, J. Tian and L. Chen, One-Pot Enantioselective Synthesis of 3-Nitro-2H-chromenes Catalyzed by a Simple 4-Hydroxyprolinamide with 4-Nitrophenol as Cocatalyst, Eur. J. Org. Chem., 2013, 5431 CrossRef CAS.
- R. Mohanta and G. Bez, Augmentation of Enantioselectivity by Spatial Tuning of Aminocatalyst: Synthesis of 2-Alkyl/aryl-3-nitro-2H-chromenes by Tandem Oxa-Michael–Henry Reaction, J. Org. Chem., 2020, 85, 4627 CrossRef CAS PubMed.
- L. Zu, S. Zhang, H. Xie and W. Wang, Catalytic Asymmetric oxa-Michael–Michael Cascade for Facile Construction of Chiral Chromans via an Aminal Intermediate, Org. Lett., 2009, 11, 1627 CrossRef CAS PubMed.
- C. Wang, X. Yang, G. Raabe and D. Enders, A Short Asymmetric Synthesis of the Benzopyrano[3,4-c]pyrrolidine Core via an Organocatalytic Domino Oxa-Michael/Michael Reaction, Adv. Synth. Catal., 2012, 354, 2629 CrossRef CAS.
- A.-B. Xia, C. Wu, T. Wang, Y.-P. Zhang, X.-H. Du, A.-G. Zhong, D.-Q. Xu and Z.-Y. Xu, Enantioselective Cascade Oxa-Michael–Michael Reactions of 2-Hydroxynitrostyrenes with Enones Using a Prolinol Thioether Catalyst, Adv. Synth. Catal., 2014, 356, 1753 CrossRef CAS.
- D. C. Cruz, R. Mose, C. V. Gómez, S. V. Torbensen, M. S. Larsen and K. A. Jørgensen, Organocatalytic Cascade Reactions: Towards the Diversification of Hydroisochromenes and Chromenes through Two Different Activation Modes, Chem. – Eur. J., 2014, 20, 11331 CrossRef PubMed.
- Z. C. Geng, S. Y. Zhang, N. K. Li, N. Li, J. Chen, H. Y. Li and X. W. Wang, Organocatalytic Diversity-oriented Asymmetric Synthesis of Tricyclic Chroman Derivatives, J. Org. Chem., 2014, 79, 10772 CrossRef CAS PubMed.
- L. Liu, Y. Zhu, K. Huang, B. Wang, W. Chang and J. Li, Asymmetric Organocatalytic Quadruple Cascade Reaction of 2-Hydroxychalcone with Cinnamaldehyde for the Construction of Tetrahydro-6H-benzo[c]chromene Containing Five Stereocenters, Eur. J. Org. Chem., 2014, 4342 CrossRef CAS.
- W. Ren, X.-Y. Wang, J.-J. Li, M. Tian, J. Liu, L. Ouyang and J.-H. Wang, Efficient Construction of Biologically Important Functionalized Polycyclic Spiro-fused Carbocyclicoxindoles via an Asymmetric Organocatalytic Quadruple-cascade Reaction, RSC Adv., 2017, 7, 1863 RSC.
- S. Roy, S. Pradhan, K. Kumar and I. Chatterjee, Asymmetric Organocatalytic Double 1,6-Addition: Rapid Access to Chiral Chromans with Molecular Complexity, Org. Chem. Front., 2020, 7, 1388 RSC.
- M. T. Corbett and J. S. Johnson, Enantioselective Synthesis of Hindered Cyclic Dialkyl Ethers via Catalytic Oxa-Michael/Michael Desymmetrization, Chem. Sci., 2013, 4, 2828 RSC.
- A. Orue, U. Uria, D. Roca-López, I. Delso, E. Reyes, L. Carrillo, P. Merino and J. L. Vicario, Racemic Hemiacetals as Oxygen-centered Pronucleophiles Triggering Cascade 1,4-Addition/Michael Reaction through Dynamic Kinetic Resolution under Iminium Catalysis. Development and mechanistic insights, Chem. Sci., 2017, 8, 2904 RSC.
- E. Reyes, G. Talavera, J. L. Vicario, D. Badía and L. Carrillo, Enantioselective Organocatalytic Domino Oxa-Michael/Aldol/Hemiacetalization: Synthesis of Polysubstituted Furofuranes Containing Four Stereocenters, Angew. Chem., Int. Ed., 2009, 48, 5701 CrossRef CAS PubMed.
- F.-L. Zhang, A.-W. Xu, Y.-F. Gong, M.-H. Wei and X.-L. Yang, Asymmetric Organocatalytic Four-Component Quadruple Domino Reaction Initiated by Oxa-Michael Addition of Alcohols to Acrolein, Chem. – Eur. J., 2009, 15, 6815 CrossRef CAS PubMed.
- P. G. McGarraugh, R. C. Johnston, A. Martínez-Muñoz, P. H.-Y. Cheong and S. E. Brenner-Moyer, Organocatalytic Kinetic Resolution Cascade Reactions: New Mechanistic and Stereochemical Manifold in Diphenyl Prolinol Silyl Ether Catalysis, Chem. – Eur. J., 2012, 18, 10742 CrossRef CAS PubMed.
- B. Mondal and S. C. Pan, Organocatalytic Asymmetric Cascade Reaction betweeno-Hydroxycinnamaldehydes andγ/δ-Hydroxyenones: A Route to Tetrahydrofuran/Tetrahydropyran-Fused 3,4-Dihydrocoumarins, Adv. Synth. Catal., 2018, 360, 4348 CrossRef CAS.
- B. Mondal, M. Balha and S. C. Pan, Organocatalytic Asymmetric Synthesis of Highly Substituted Tetrahydrofurans and Tetrahydropyrans via Double Michael Addition Strategy, Asian J. Org. Chem., 2018, 7, 1788 CrossRef CAS.
- B. Ravindra, B. G. Das and P. Ghorai, Organocatalytic, Enantioselective, Intramolecular Oxa-Michael Reaction of Alkoxyboronate: A New Strategy for Enantioenriched 1-Substituted 1,3-Dihydroisobenzofurans, Org. Lett., 2014, 16, 5580 CrossRef CAS PubMed.
- B. Zheng, W. Hou and Y. Peng, Asymmetric oxa-Michael-aza-Henry Cascade Reaction of 2-Hydroxyaryl-Substituted α-Amido Sulfones and Nitroolefins Mediated by Chiral Squaramides, ChemCatChem, 2014, 6, 2527 CrossRef CAS.
- Y. Zhu, X. Li, Q. Chen, J. Su, F. Jia, S. Qiu, M. Ma, Q. Sun, W. Yan, K. Wang and R. Wang, Highly Enantioselective Cascade Reaction Catalyzed by Squaramides: the Synthesis of CF3-Containing Chromanes, Org. Lett., 2015, 17, 3826 CrossRef CAS PubMed.
- C.-K. Tang, K.-X. Feng, A.-B. Xia, C. Li, Y.-Y. Zheng, Z.-Y. Xu and D.-Q. Xu, Asymmetric Synthesis of Polysubstituted Chiral Chromans via an Organocatalytic Oxa-Michael-nitro-Michael Domino Reaction, RSC Adv., 2018, 8, 3095 RSC.
- J. M. Andrés, A. Maestro, M. Valle, I. Valencia and R. Pedrosa, Diastereo- and Enantioselective Syntheses of Trisubstituted Benzopyrans by Cascade Reactions Catalyzed by Monomeric and Polymeric Recoverable Bifunctional Thioureas and Squaramides, ACS Omega, 2018, 3, 16591 CrossRef PubMed.
- H. Mao, A. Lin, Y. Tang, Y. Shi, H. Hu, Y. Cheng and C. Zhu, Organocatalytic oxa/aza-Michael–Michael Cascade Strategy for the Construction of Spiro [Chroman/Tetrahydroquinoline-3,3′-oxindole] Scaffolds, Org. Lett., 2013, 15, 4062 CrossRef CAS PubMed.
- B. Mondal, R. Maity and S. C. Pan, Highly Diastereo- and Enantioselective Synthesis of Spiro-tetrahydrofuran-pyrazolones via Organocatalytic Cascade Reaction between γ-Hydroxyenones and Unsaturated Pyrazolones, J. Org. Chem., 2018, 83, 8645 CrossRef CAS PubMed.
- H. Wang, J. Luo, X. Han and Y. Lu, Enantioselective Synthesis of Chromanones via a Tryptophan- Derived Bifunctional Thiourea-Catalyzed Oxa-Michael–Michael Cascade Reaction, Adv. Synth. Catal., 2011, 353, 2971 CrossRef CAS.
- P. Saha, A. Biswas, N. Molleti and V. K. Singh, Enantioselective Synthesis of Highly Substituted Chromans via the Oxa-Michael–Michael Cascade Reaction with a Bifunctional Organocatalyst, J. Org. Chem., 2015, 80, 11115 CrossRef CAS PubMed.
- Z. Zhang, G. Jakab and P. R. Schreiner, Enantioselective Synthesis of 2-Aryl-3-nitro-2H-chromenes Catalyzed by a Bifunctional Thiourea, Synlett, 2011, 1262 Search PubMed.
- Y. Huang, C. Zheng, Z. Chai and G. Zhao, Synthesis of Spiro[chroman/tetrahydrothiophene-3,3′-oxindole] Scaffolds via Heteroatom-Michael–Michael Reactions: Easily Controlled Enantioselectivity via Bifunctional Catalysts, Adv. Synth. Catal., 2014, 356, 579 CrossRef CAS.
- W. Zheng, J. Zhang, S. Liu, C. Yu and Z. Miao, Asymmetric Synthesis of Spiro[Chroman-3,3′-pyrazol] Scaffolds with An All-Carbon Quaternary Stereocenter via a Oxa-Michael–Michael Cascade Strategy with Bifunctional Amine-Thiourea Organocatalysts, RSC Adv., 2015, 5, 91108 RSC.
- K. Zhao, Y. Zhi, T. Shu, A. Valkonen, K. Rissanen and D. Enders, Organocatalytic Domino Oxa-Michael/1,6-Addition Reactions: Asymmetric Synthesis of Chromans Bearing Oxindole Scaffolds, Angew. Chem., Int. Ed., 2016, 55, 12104 CrossRef CAS PubMed.
- H.-J. Lee, B. Eun, E. Sung, G. T. Hwang, Y. K. Ko and C.-W. Cho, Catalytic Enantioselective Synthesis of Carboxy-substituted 2-isoxazolines by Cascade Oxa-Michael-Cyclization, Org. Biomol. Chem., 2018, 16, 657 RSC.
|
| This journal is © the Partner Organisations 2020 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *
*