
Engineering well-defined rare earth oxide-based nanostructures for catalyzing C1 chemical reactions
Kun
Yuan
and
Ya-Wen
Zhang
 *
*
Beijing National Laboratory for Molecular Sciences, State Key Laboratory of Rare Earth Materials Chemistry and Applications, PKU-HKU Joint Laboratory in Rare Earth Materials and Bioinorganic Chemistry, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, China. E-mail: ywzhang@pku.edu.cn; Fax: +86-10-62756787; Tel: +86-10-62756787
First published on 2nd September 2020
Abstract
C1 chemical reactions have attracted extensive attention in recent decades due to their significant roles in energy transfer and utilization and environmental protection. Among the various catalytic materials, rare earth oxide-based nanocatalysts exhibit superior performances in C1 chemical reactions because of their flexible electronic structures and abundant defect states. In this review, we summarize the nanostructural engineering and applications of rare earth oxide-based nanomaterials with well-defined compositions, crystal phases and shapes for efficiently catalyzing C1 chemical reactions. Initially, we introduce the structural features of rare earth oxides. Subsequently, we present common synthetic approaches and nanostructural engineering strategies toward the preparation of rare earth oxide nanomaterials with well-defined structures. Further, we discuss the structure–function correlation of well-defined rare earth oxide-based nanocatalysts in some important C1 chemical reactions including CO oxidation, water-gas shift reaction, CO2 hydrogenation, methane oxidation and methanol oxidation. Finally, we prospect the challenges and future research trends in this promising field.
 Kun Yuan | Kun Yuan was born in Tianjin, China. He received his BSc Degree in Chemistry from Peking University in 2016. Currently, he is studying for his PhD Degree under the supervision of Prof. Ya-Wen Zhang in the College of Chemistry and Molecular Engineering, Peking University. His current research focuses on the synthesis and investigation of the catalytic properties of ceria-supported noble metal nanocatalysts. |
 Ya-Wen Zhang | Ya-Wen Zhang is a Full Professor and Principle Investigator at the College of Chemistry and Molecular Engineering of Peking University. The research interests of his group include the rational design, controllable synthesis, ordered assembly, catalytic properties and structure–function relationships of rare earth and noble metal nanostructures. He has published more than 150 papers in peer-reviewed scientific journals and several book chapters, and was a Winner of the National Science Fund for Distinguished Young Scholars in 2010. He obtained his BSc Degree, MSc Degree, and PhD Degree from Peking University in 1988, 1994 and 1997, respectively, and did postdoctoral research in the State Key Laboratory of Rare Earth Materials Chemistry and Applications of Peking University during 1998–2000, and was a visiting scholar in the Department of Chemistry of the University of California at Berkeley and Lawrence Berkeley National Laboratory in 2006–2008. |
1. Introduction
As a type of important strategic resource, rare earth elements include 15 lanthanide elements (La–Lu) and two other elements, scandium and yttrium. They usually co-exist in minerals and have similar properties, which make their separation challenging.1 Rare earth elements (except for Sc and Y) have partially unfilled 4f orbitals and abundant electronic energy level structures, thus they have flexible coordination numbers and outstanding catalytic properties for many reactions.2,3 Rare earth materials play a significant role in various applications such as agriculture, military, petrochemical industry, metallurgy, glass, and ceramics.4,5Nanostructured rare earth oxides are the most common rare earth nanomaterials, which are also the most widely studied.6 They are cheap and easily obtained, and they have different oxidation states in various conditions. The earliest report of the synthesis of rare earth oxide nanocrystals with a well-defined architecture dates back to the work of Cao’ s group,7 which involved the preparation of square Gd2O3 nanoplates via a colloidal approach. At almost the same time, Zhang and Yan's group successively developed various methods for the synthesis of well-defined rare earth oxides with zero-dimensional (0D), one-dimensional (1D), two-dimensional (2D) and three-dimensional (3D) nanostructures.8–11 After development for more than 15 years, researchers have developed various synthetic strategies, and successfully prepared many rare earth oxide nanostructures with diverse well-defined morphologies.
Rare earth oxide nanostructures are broadly applied in catalysis, especially heterogeneous catalysis.12–15 On the one hand, rare earth oxide nanostructures by themselves are active for some catalytic reactions such as CO oxidation.12 However, their activity is usually limited due to the weak adsorption of the reagent molecule. On the other hand, rare earth oxide nanostructures can act as supports or promotors to be combined with other metal or metal oxide catalysts.14 The catalytic properties of composite materials are significantly enhanced due to the synergistic effect between the support materials and loading elements. Further, they are used more widely in catalysis than single rare earth oxide nanostructures.
Among the catalytic reactions, C1 chemical reactions are crucial for addressing both energy and environmental problems faced by mankind in this century due to their important roles in producing essential chemicals (e.g. methane and methanol) and controlling pollution (e.g. CO and CO2).16–19 Rare earth oxide-based catalysts are a vital class of catalytic nanomaterials for C1 chemical reactions.20,21 Thus, the development of high-performance rare earth oxide-based catalysts is urgent and significant. Engineering rare earth oxide-based catalysts with well-defined nanostructures is not only beneficial for enhancing their catalytic properties, but also facilitates the study of the relationship between catalytic properties and structure, which can provide guidance for designing catalysts with excellent performances since this is still unclear in many catalytic systems.
Due to the importance of rare earth-based nanomaterials, many groups have reviewed their synthesis and applications in catalysis. For example, Hussein6 published a thermal analysis and applied pyrolysis review about the formation, characterization and catalytic activity of rare earth oxides. In addition, Zhan et al.3 mainly discussed the catalytic applications of rare earth materials in energy production and utilization, and environmental protection. Guo et al.5 commented on the research advances of rare earth-based catalysts in heterogeneous transformation reactions of small molecules. Richard et al.2 focused on the properties and applications of rare earth elements for CO2 hydrogenation to methanol. Besides, Huang et al.1 reviewed the synthetic routes and electrochemical applications of rare earth-based nanomaterials. Similarly, the review by Gao et al.4 also discussed and summarized the recent progress on rare earth-based nanocatalysts, but their key point was the incorporation of rare earth elements with transition metals. Although rare earth oxide-based nanostructures were a significant part introduced in the abovementioned reviews, few of them focused on the shape-control of well-defined rare earth nanostructures and their catalytic applications in C1 chemical reactions. Besides, most of the reported reviews about rare earth oxides mainly focused on the applications of CeO2 nanomaterials,22–30 and rarely discussed other rare earth oxides. Therefore, a review that systematically summarizes the morphological engineering of nanostructured rare earth oxides (not only CeO2) and highlights their structure–function relationships in some important catalytic reactions is still necessary.
Thus, to meet this demand, in the present review, we focus on the shape-controlled synthesis of well-defined rare earth oxide nanostructures and their catalytic structure–activity correlations. Herein, we introduce the structure features of rare earth oxides and summarize the most common synthetic routes for the preparation of well-defined rare earth oxide nanostructures from the perspective of their morphologies. Moreover, we choose some typical C1 chemical reactions to summarize the relationship between the engineered nanostructures of the rare earth oxide-based nanomaterials and their catalytic properties.
2. Structural features of rare earth oxides
Generally, rare earth oxides can be divided into two categories, trivalent oxides and tetravalent oxides. Most of the stable rare earth oxides are trivalent except for CeO2, PrO2 and TbO2. The trivalent RE2O3 usually has three types of structures, which are denoted as A, B, and C (Fig. 1a–c, respectively). The A-type RE2O3 is hexagonal, with the P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 space group. The RE3+ is seven-coordinated with O, in which six O2− form an octahedron and the last O2− is located at one of the facets of the octahedron. The B-type RE2O3 is monoclinic, with the C2/m space group. Similarly, the RE3+ is also seven-coordinated and six O2− form an octahedron, but the last O2− is a little farther from the RE3+. The C-type RE2O3 is cubic, with the Ia space group. The RE3+ is six-coordinated, and the cell structure of C-type RE2O3 is similar to the cubic fluorite structure, in which only two O2− are removed regularly.
m1 space group. The RE3+ is seven-coordinated with O, in which six O2− form an octahedron and the last O2− is located at one of the facets of the octahedron. The B-type RE2O3 is monoclinic, with the C2/m space group. Similarly, the RE3+ is also seven-coordinated and six O2− form an octahedron, but the last O2− is a little farther from the RE3+. The C-type RE2O3 is cubic, with the Ia space group. The RE3+ is six-coordinated, and the cell structure of C-type RE2O3 is similar to the cubic fluorite structure, in which only two O2− are removed regularly.
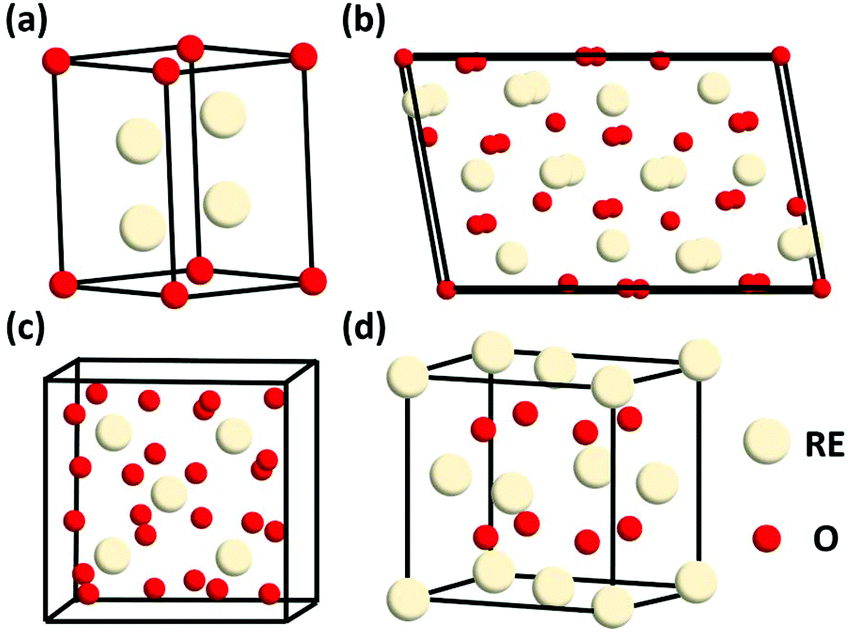 | ||
| Fig. 1 Schematic illustration of the crystal structure of rare earth oxides: (a) A-type RE2O3, (b) B-type RE2O3, (c) C-type RE2O3, and (d) fluorite REO2. | ||
REO2 has a cubic fluorite structure, with the Fmm space group, in which the RE4+ is cubic close packing and coordinated with eight O2−, and the O2− is located in a tetrahedral void and coordinated with four RE4+ (Fig. 1d). The RE in REO2 also has a stable valence of +3, and it can transform between +3 and +4 through the generation and elimination of an oxygen vacancy.
As can be seen in the phase diagram in Fig. 2, RE2O3 (RE = La–Pr) has an A-type structure, RE2O3 (RE = Y, Dy–Lu) has a C-type structure, and the others are C-type at low temperature and become B-type at high temperature.
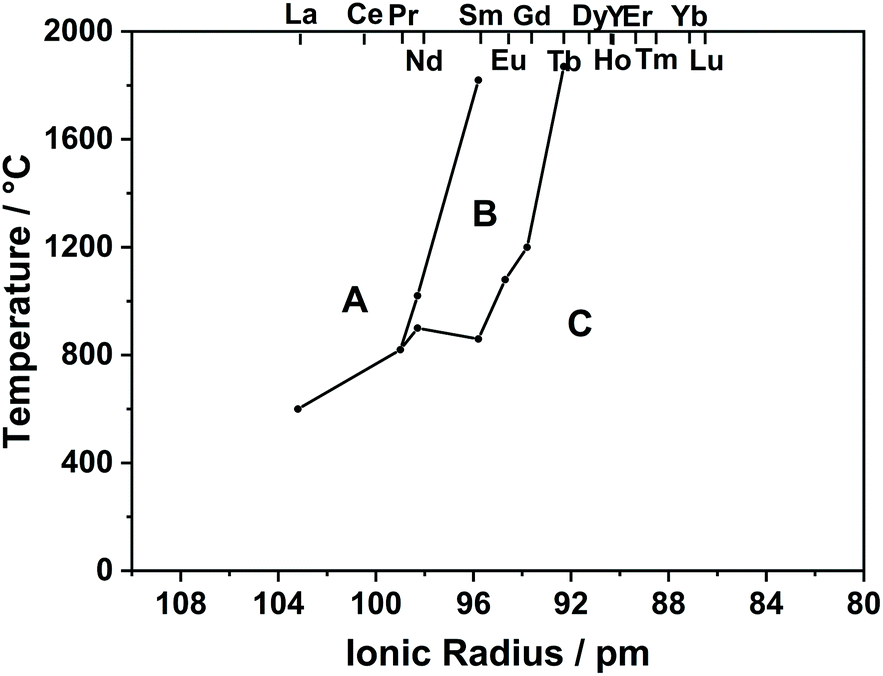 | ||
| Fig. 2 Phase diagram of RE2O3. | ||
3. Synthesis of well-defined rare earth oxide nanostructures
Thus far, researchers have developed numerous methods for the synthesis of rare earth oxide nanomaterials with well-defined structures, including 0D nanocubes and nanooctahedra,8,31–33 1D nanorods and nanowires,34–37 2D nanosheets and nanobelts,38–41 and 3D dendritic and flower-like structures.11,42–44 Optimized strategies such as selective adsorption on specific facets, pH adjustment, templating and self-assembly are widely used in engineering well-defined rare earth oxide nanostructures45,46 (Fig. 3). Rare earth oxide nanomaterials with different shapes usually have diverse physical and chemical properties due to their distinguishable surface structures and chemical states. Hence, controlling the shapes of rare earth oxide nanostructures will support the realization of different functions and applications. In this section, we mainly summarize the synthesis of well-defined rare earth nanostructures considering their morphologies, with the introduction of some typical synthetic examples. Furthermore, it should be noted that most of the rare earth oxides in the cases we discuss below are cubic structure according to their phase diagram. Actually, cubic fluorite CeO2 is the most studied nanomaterial based on the current research status of rare earth oxides.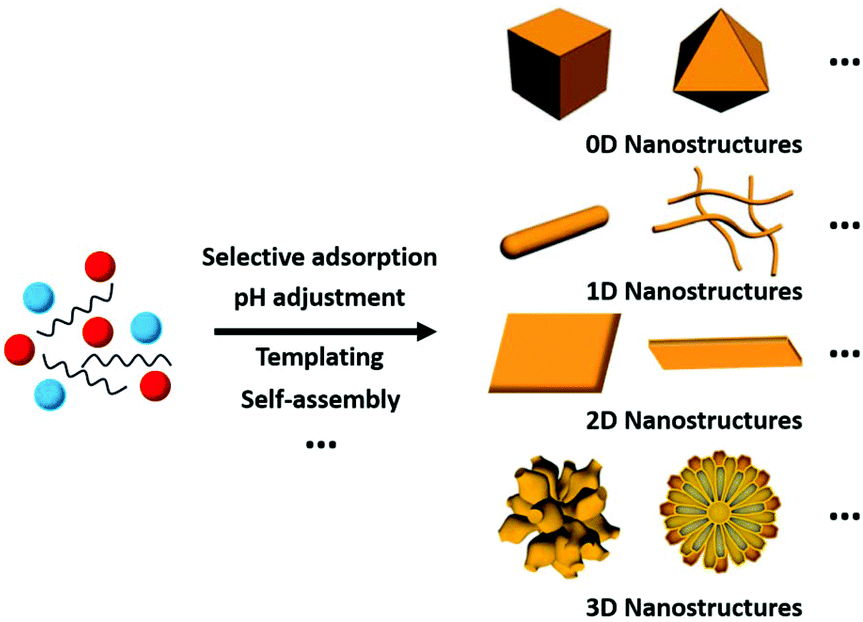 | ||
| Fig. 3 Schematic illustration of the synthesis of rare earth oxide nanostructures with different morphologies. | ||
3.1 0D rare earth oxide nanostructures
0D nanomaterials are defined as nanocrystals with a nanoscale size in three dimensions. They usually contain nanocubes, nanooctahedra, nanotetrahedra, nanopolyhedra, etc. In general, nanostructures with different morphologies will have diverse exposed crystal planes. For instance, nanocubes have exposed {100} facets, whereas nanooctahedra and nanotetrahedra have exposed {111} facets. The key factor for the preparation of rare earth oxide nanocrystals with specific shapes is controlling the relative crystal growth rate of specific facets during either the anisotropic growth mode or isotropic growth mode.8,33Taking the synthesis of rare earth oxide nanocubes as an example, Zhang and Yan's group8 prepared CeO2 nanocubes through pH adjustment using a hydrothermal method. The precipitation reaction occurred between Ce(NO3)3 and NaOH, and the secret of gaining CeO2 nanocubes enclosed with {100} facets was applying a high pH and temperature to achieve a fast dissolution–recrystallization process for the transformation of Ce(OH)3 into CeO2 particles. In contrast, a slow dissolution–recrystallization process with a low pH and temperature resulted in the formation of CeO2 nanopolyhedra with {111} and {100} planes. Besides, the same goal was completed by Dang et al.47 using a liquid–liquid interface with the assistance of oleic acid (OLA). As shown in Fig. 4a, {111}-dominated CeO2 nanooctahedra or truncated nanooctahedra were obtained in the water phase initially owing to the lower surface energy of the {111} planes, then the OLA selectively adsorbed on the {100} planes, resulting in the transfer of the CeO2 nanoparticles from the water phase to the toluene phase. Because of the coating of OLA on the {100} planes, the growth rate of the CeO2 [100] direction was limited, and finally the CeO2 nanoparticles turned into nanocubes (Fig. 4b). This was a typical case that using selective adsorption on a specific facet controlled the morphologies of the rare earth oxide nanostructures. Specially, Miao et al.48 prepared CeO2 nanocubes through the doping of F. They concluded that the generation of CeO2 nanocubes was a result of the etching effect by HF in the crystal growth process.
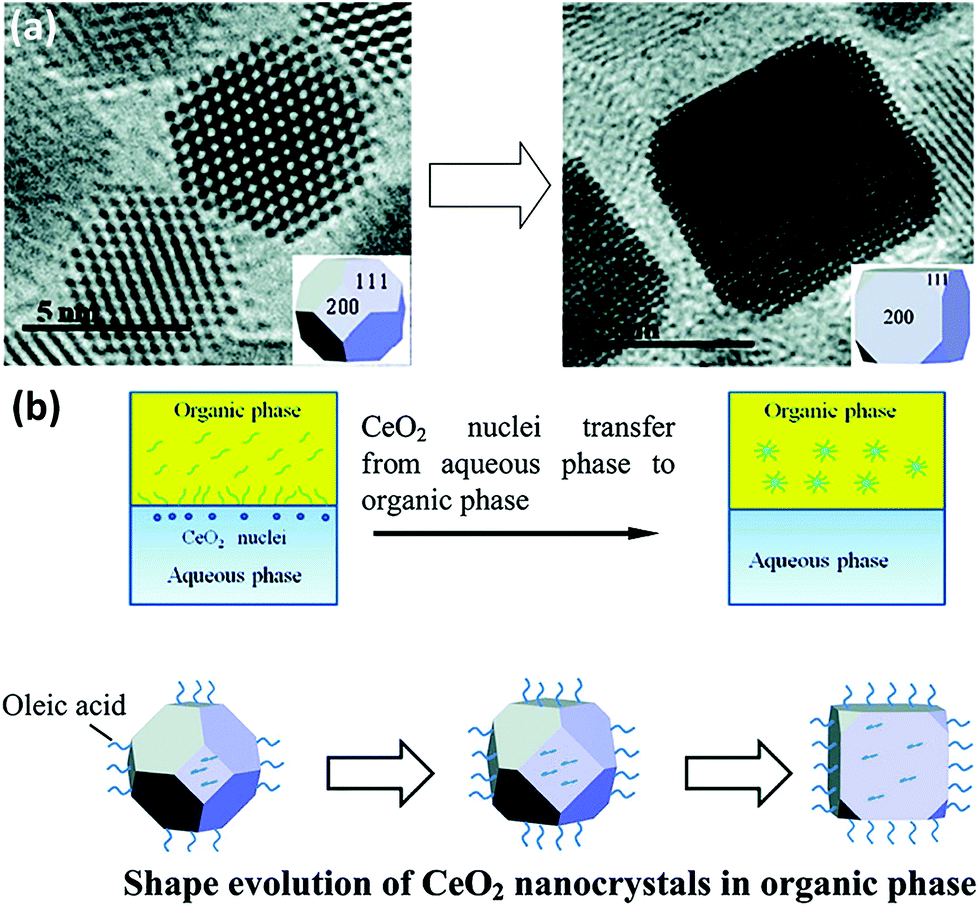 | ||
| Fig. 4 (a) Transmission electron microscopy (TEM) images of the evolution from CeO2 truncated nanooctahedra to nanocube. (b) Schematic illustration of shape evolution of CeO2 nanocrystals in the organic phase. Adapted with permission from ref. 47 Copyright 2010, American Chemical Society. | ||
For the preparation of rare earth oxide nanooctahedra, they are easier to be obtained than nanocubes as a consequence of the lower surface energy of the {111} facets than that of the {100} facets. They can be facilely produced by tuning the precipitation step in the hydrothermal method.32,33 For example, Feng et al.33 synthesized CeO2 nanooctahedra with a size of around 100 nm using the weak base Na3PO4 in the hydrothermal process. The slow growth rate guaranteed the generation of the more stable morphology of nanooctahedra rather than nanocubes. The authors also demonstrated that replacing Na3PO4 with the strong base NaOH would result in the formation of CeO2 nanospheres and nanocubes. Similarly, CeO2 nanooctahedra were successfully synthesized by Ren et al.49 through the hydrothermal route using Ce(NO3)3 and a relatively small amount of NaOH. Also, they also showed that a high concentration of NaOH could yield nanocubes. Specially, single crystalline CeO2 nanooctahedra were obtained through galvanostatic electrodeposition in a conventional three-electrode cell.50 It was also the first time that CeO2 nanooctahedra were prepared via the electrochemical route.
In the case of other 0D rare earth oxide nanostructures, nanopolyhedra with no specific shape (called nanoparticles) are common nanomaterials. Their crystal facets usually consist of {100} and {111} planes, and they are easy to obtain because engineering of their morphology is not required. Furthermore, they can be used as reference samples in the study of the plane effect derived from diverse morphologies and are widely used in applications such as catalysis because of their easy accessibility. For their synthesis, Yang et al.51 used the sol–gel method combined with a solvothermal process to obtain CeO2 nanoparticles with a high surface area. They carried out the solvothermal treatment after they obtained the gel powder, followed by calcination in air to obtain the final product. Additionally, Shiri et al.52 synthesized Sm2O3 nanoparticles using a nitrate bath of Sm(NO3)3via pulse electrochemical deposition assisted by ultrasound. Different from the regular precipitation method, precipitation in reverse microemulsions based on different surfactants was applied to prepare CeO2 nanoparticles by Shlapa et al.53 A schematic of the synthetic route is shown in Fig. 5. Compared with the traditional precipitation method, the precipitation reaction in reverse microemulsions is limited by the surfactant molecules, and the size can be easily controlled by tuning the length of the hydrophilic part of the surfactant. The as-prepared CeO2 nanoparticles had a small and uniform size in the range of 6–10 nm and showed good catalase-like activity.
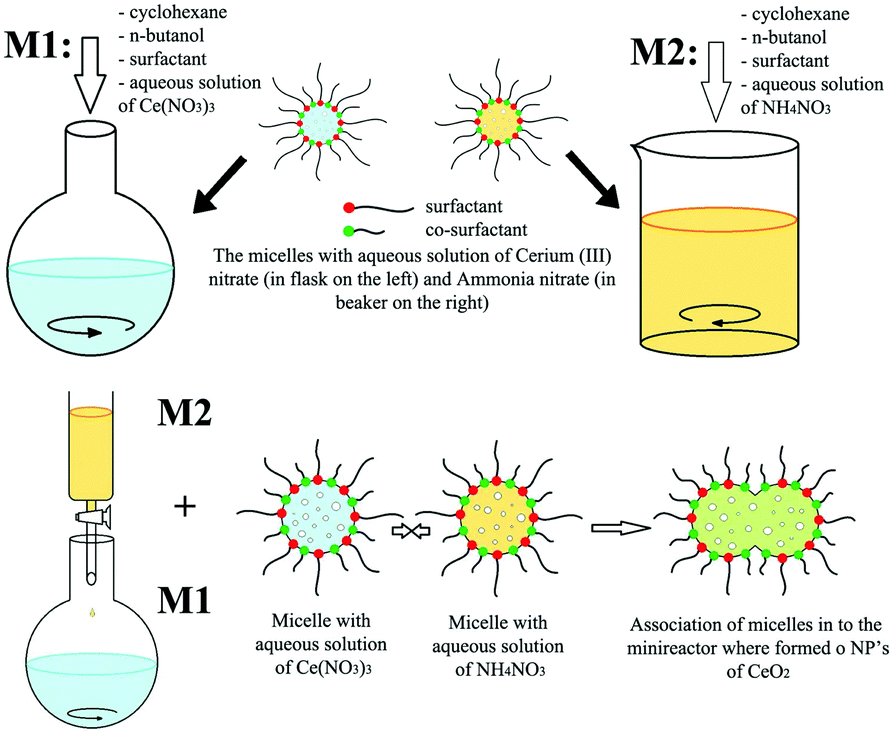 | ||
| Fig. 5 Schematic illustration of the synthesis of CeO2 nanoparticles via precipitation in reverse microemulsions. Adapted with permission from ref. 53. Copyright 2019, Springer Nature. | ||
3.2 1D rare earth oxide nanostructures
Compared with 0D nanostructures, one non-nanoscale dimension is allowed to exist in 1D nanostructures, while the other two dimensions must be on the nanoscale. 1D rare earth oxide nanostructures including nanorods, nanowires and nanotubes are important nanomaterials for catalysis because of their unique exposed crystal planes, abundant defect sites, and good stability, and they can also provide good models for theoretical simulation.34,35 In the synthetic strategies for engineering 1D rare earth oxide nanostructures, pH adjustment in the hydrothermal procedure is the most common means because the acid–base balance can influence many key factors in shape-control such as the crystal growth rate and stability of intermediates.34–37Furthermore, it should be noted that the hydrothermal method for the preparation of rare earth oxides can be divided into two types, alkali and acid methods. The alkali hydrothermal route is very common, for example, CeO2 nanorods with a length in the range of 100–300 nm and width of 13–20 nm were firstly prepared by Zhou et al.34 using Ce(NO3)3 and NaOH. The predominantly exposed facets were the unusually active {001} and {110} planes rather than the {111} plane. The authors provided inspiration for designing and controlling the synthesis of nanocrystals with different shapes. Our group35 synthesized CeO2 nanowires via the hydrothermal method at 180 °C using CeCl3 and NaOH without the addition of any capping agent. In the synthetic process, NaCl was specially used to obtain uniform wire-like nanostructures dominantly enclosed by {110} facets. We also prepared Ln-doped (Ln = La–Lu) CeO2 nanowires using the same method, and the distribution of dopant was very homogeneous. The key was the co-precipitation of Ce3+ and Ln3+ to form Ce(OH)3:Ln(OH)3, which transformed into CeO2:Ln through calcination. The dopant content was about 10%, and the aspect ratio of most of the CeO2:Ln nanowires were smaller than that of the pure CeO2 nanowires due to the difference in the ionic radius between Ce and the dopant ion. As another example, Sohn36 successfully synthesized Yb2O3 nanowires and nanorods via the hydrothermal method at 210 °C using ammonia water as the precipitant, and YbCl3 and Yb(NO3)3 as the precursor, respectively (Fig. 6). The abovementioned cases indicate that besides the vital role of pH adjustment, the formation of 1D rare earth oxide nanostructures also greatly relies on the transformation of the intermediates. It may be difficult to obtain rare earth oxide nanowires or nanorods directly, but it can be easy to obtain their corresponding hydroxides and transform them into oxides. Acid hydrothermal treatment is less used in the synthesis of 1D rare earth oxide nanostructures. As an example, Pr2O3 nanowires were prepared by Sobahi37 using the hydrothermal method by tuning the pH value of the precursor solution of Pr(NO3)3 to 1.3 and adding glycine as the shaping reagent.
 | ||
| Fig. 6 SEM images of as-synthesized Yb2O3 (a) nanowires and (b) nanorods. Adapted with permission from ref. 36. Copyright 2018, Elsevier. | ||
Rare earth oxide nanotubes are another type of important 1D nanomaterials on account of their high surface area and rich defects. Due to the hollow structure of the nanotubes, templating is a very suitable strategy for their preparation. As an example, Eu2O3 nanotube arrays (Fig. 7a) could be obtained using the sol–gel method assisted by porous anode alumina templates.54 According to the synthetic mechanism shown in Fig. 7b, the alumina templates were impregnated in the mixture of Eu(NO3)3 solution and urea, and the solution filled the pores of the templates. After heating the solution, the pH value increased owing to the hydrolysis of urea, resulting in the formation of Eu(OH)3 in both the template pores and solution. The sol particles of Eu(OH)3 were negatively charged, while the templates were positively charged, and therefore the sol particles gathered at the walls of the templates and gradually extended to the center. Finally, the templates were removed from the sol solution and sintered in a furnace, generating Eu2O3 nanotubes. As another example, Tb2O3 nanotubes were synthesized by Tang et al.55 through the hydrothermal method using terbium chloride as the precursor and sodium dodecyl benzenesulfonate as the template reagent. Tb(OH)3 nanotubes were initially obtained via the hydrothermal process after adjusting the solution pH to 12 with NaOH. Finally, they calcined the Tb(OH)3 nanotubes at 800 °C under a reducing atmosphere and obtained Tb2O3 nanotubes. Interestingly, Tb2O3 nanotubes could also be synthesized through a facile precipitation method using Tb(NO3)3 and ammonia.56 The precipitation process involved aging at room temperature for two days, which was necessary for the formation of the tube-like structure under a slow growth rate.
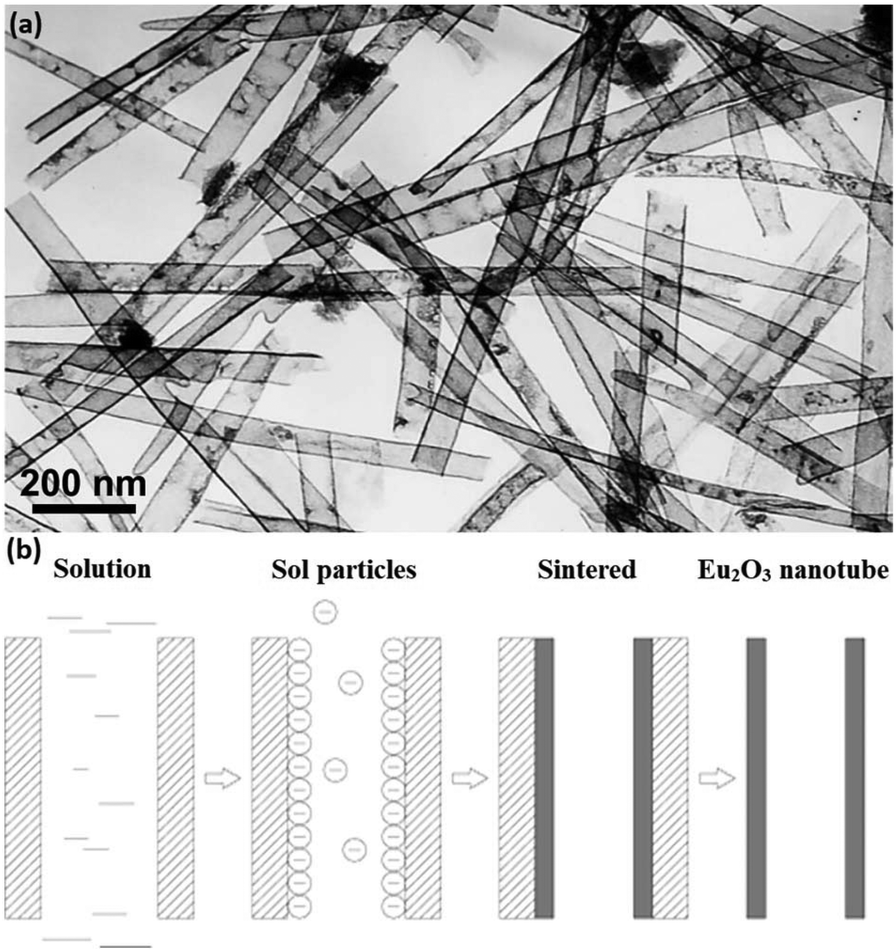 | ||
| Fig. 7 (a) TEM image of Eu2O3 nanotubes. (b) Schematic illustration of the formation mechanism of Eu2O3 nanotubes. Adapted with permission from ref. 54. Copyright 2004, American Chemical Society. | ||
3.3 2D rare earth oxide nanostructures
Similar to the definition of 0D and 1D nanostructures, 2D nanostructures allow the existence of two non-nanoscale dimensions, and thus they usually contain nanosheets, nanoplates, nanobelts, etc. 2D rare earth oxide nanocrystals have many advantages for catalytic applications such as huge surface area, a variety of defects, and high atomic utilization owing to their thin structure.57–59 However, it is challenging to prepare 2D rare earth oxide nanomaterials due to the complex requirement of anisotropic growth. In general, a relatively slow growth rate of a specific crystal plane is preferred to realize anisotropic morphologies.Nanoplates are well-known nanomaterials among the 2D rare earth oxide nanostructures, which have been successfully synthesized via several methods.57,59 The first reported synthetic work on 2D rare earth oxide nanostructures was the Gd2O3 square nanoplates prepared by Cao7 using a solution-phase decomposition method. Soon afterwards, Zhang and Yan's group9,10 prepared a series of cubic rare earth (RE = La–Lu, Y) oxide nanoplates or nanodisks via a non-hydrolytic route using various rare earth complexes as precursors with the assistance of oleylamine (OM), oleic acid (OA) and 1-octadecene (ODE). Our group carried out control experiments to determine the specific effects of different experimental parameters. As shown in Fig. 8a, the whole shape evolution can be divided into two parts. Firstly, the rare earth oleates are formed through ion exchange with OA in solution. Secondly, decomposition of the rare earth oleates occurs via the catalysis of OM. Due to the selective adsorption of OA on the {100} planes, the RE2O3 crystals can realize anisotropic growth to generate 2D structures. Also, they would result in diverse morphologies because of the difference in growth rate, which is affected by the rare earth precursors. As another illustration, La2O3 nanoplates were obtained by Wu et al.60via a combination of precipitation and calcination using La(NO3)3 and formamide. In the precipitation process, La(OH)3 nanoparticles were formed through nucleation and aggregated (Fig. 8b). Then they grew into 1D La(OH)3 nanorods because hexagonal La(OH)3 has an anisotropic crystal structure. During the calcination step, the –OH groups were removed and the nanocrystals underwent reconstruction to achieve the lowest surface energy. Consequently, the La(OH)3 nanorods transformed into hexagonal La2O3 nanoplates under the driving force of calcination. The high-resolution TEM (HRTEM) results showed the {002} exposed facet, suggesting that preferential growth occurred along the [002] direction.
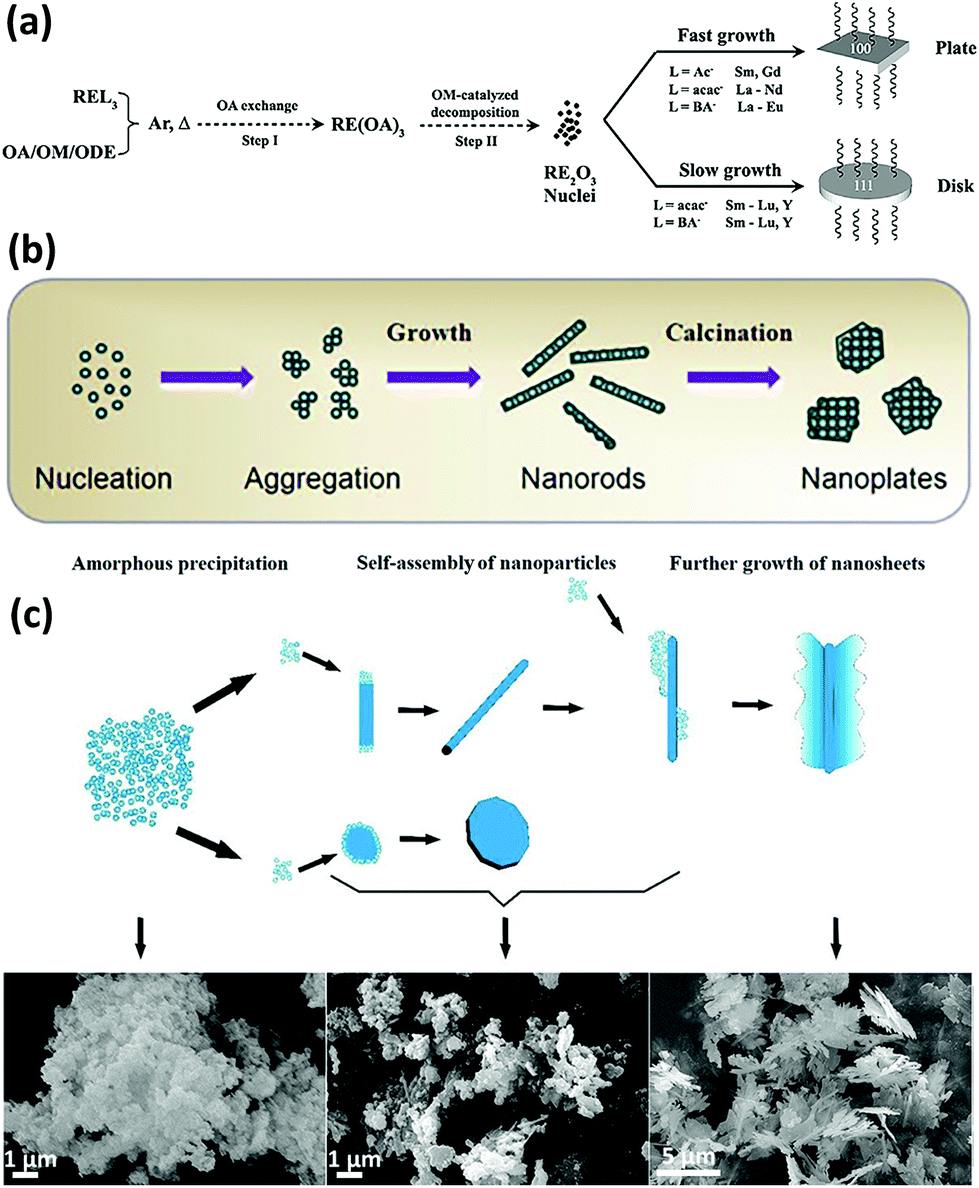 | ||
| Fig. 8 (a) Schematic of the formation mechanism of RE2O3 nanoplates and nanodisks. Adapted with permission from ref. 10. Copyright 2007, American Chemical Society. (b) Schematic illustration of the formation mechanism of La2O3 nanoplates. Adapted with permission from ref. 60. Copyright 2013, Elsevier. (c) Schematic formation evolution of leak-like CeO2 nanosheets and corresponding scanning electron microscopy (SEM) images of the different stages. Adapted with permission from ref. 61. Copyright 2013, Springer Nature. | ||
In the case of engineering of rare earth oxide nanosheets, Hu et al.61 synthesized leaf-like CeO2 nanosheets mainly enclosed with {111} and {200} facets via the hydrothermal method using Ce(NO3)3 as the precursor, NH4HCO3 as the precipitant, and ethylenediamine as the complexant. As displayed in Fig. 8c, the product evolution could be divided into three stages. In the first stage, Ce3+ reacted with NH4HCO3 and generated a large amount of Ce(OH)CO3 nanoparticles, which aggregated to form fluffy particles through self-assembly. Afterwards, nanorods and nanosheets emerged due to the dissolution–recrystallization and self-assembly of the fluffy particles. In the last stage, the nanorods continued to grow until the formation of leaf-like nanosheets. In the whole process, ethylenediamine was coordinated with Ce3+ and controlled the release of Ce3+, thus further controlling the crystal growth direction to form nanosheets. Similarly, Dai et al.62 obtained CeO2 nanosheets using Ce(NO3)3 and NH4HCO3, but they did not add ethylenediamine or any other reagents. As an alternative, they selected a low temperature of 30 °C to carry out the precipitation reaction in order to obtain an adequately slow growth rate for the formation of sheet-like nanostructures. As a supplement, the electrochemical method is also good for synthesizing 2D rare earth oxides. For example, Huang et al.38 obtained Eu-doped CeO2 nanosheets through electrodeposition on a Ti substrate and subsequent calcination in N2. However, the formation mechanism was unclear and needs more exploration.
Rare earth oxide nanobelts is another type of 2D nanomaterials that are promising candidates for the fundamental study of physical and chemical properties in many fields.63,64 However, they are difficult to synthesize due to the preference to form 1D nanostructures or other nanocrystals with lower anisotropy. Some groups successfully achieved their synthesis through the solvothermal method, which is suitable for crystal shape control.63,64 For example, Eu-doped single-crystal Y2O3 nanobelts with an average thickness of ca. 10 nm and width of 40–100 nm were obtained by Li et al.63 using a simple solvothermal method without the addition of any templates. The key factors for controlling the belt-like shape and size were the initial pH of the solution and reaction time of the solvothermal process. As another example, Rao et al.64 obtained mesoporous CeO2 nanobelts via the hydrothermal method, and subsequent calcination without any template or surfactant. The authors researched the effect of the experimental conditions in the hydrothermal process on morphologies of the CeO2 precursors, and they found that the temperature, cationic type of alkali and ratio of alkali/Ce contributed to the final shape of the products (Fig. 9a–e). Fig. 9f further illustrates the possible formation mechanism of the mesoporous CeO2 nanobelts. Briefly, colloidal Ce(OH)3 nuclei were initially formed, followed by dissolution–recrystallization in a mixture of OH− and HCHO. Formates and carbonates were generated in the solution via the disproportionation reactions between OH− and HCHO, and they could coexist due to the high concentration and small radius of Na+, which was vital for the anisotropic growth to form belt-like nanostructures. During the calcination of the nanobelt precursors, they transformed into polycrystalline CeO2 nanobelts and a mass of pores was generated via the decomposition of formates and carbonates, leaving sodium oxide on the surface. After washing with water, the final mesoporous CeO2 nanobelts with a thickness of 10–30 nm and a width of 50–250 nm (Fig. 9g) were obtained. Since rare earth oxide nanobelts are similar to 1D nanostructures to a certain degree, the electrospinning method is also suitable for the preparation of rare earth oxide nanobelts. As an example, Eu2O3 nanobelts with a uniform size were obtained via the electrospinning process.65
 | ||
Fig. 9 (a) Schematic illustration of the synthesis of CeO2 precursors under various conditions. TEM images of CeO2 precursors prepared under various conditions: (b) 140 °C, NaOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ce = 3.3; (c) 140 °C, NaOH:Ce = 10.8; (d) 120 °C, KOH:Ce = 16.3; and (e) 180 °C, NaOH:Ce = 16.3. (f) Schematic illustration of the mechanism for the formation of mesoporous CeO2 nanobelts. (g) TEM image of mesoporous CeO2 nanobelts. Adapted with permission from ref. 64. Copyright 2013, Elsevier. :Ce = 3.3; (c) 140 °C, NaOH:Ce = 10.8; (d) 120 °C, KOH:Ce = 16.3; and (e) 180 °C, NaOH:Ce = 16.3. (f) Schematic illustration of the mechanism for the formation of mesoporous CeO2 nanobelts. (g) TEM image of mesoporous CeO2 nanobelts. Adapted with permission from ref. 64. Copyright 2013, Elsevier. | ||
3.4 3D rare earth oxide nanostructures
3D nanostructures can be regarded as composites of 0D, 1D, and 2D nanomaterials. 3D rare earth oxide nanostructures play a significant role in catalytic applications mainly because of three advantages.11,66–68 First, they usually have good robustness in catalytic processes due to their anti-sintering ability, which is attributed to their large size. Secondly, they possess a large surface area since they consist of small units such as nanoparticles and nanosheets. Thirdly, they are rich in defects and high-index facets due to the oriented attachment of the tiny building blocks. However, unlike 0D, 1D and 2D nanostructures, the 3D rare earth oxide nanostructures hardly have regular morphologies. They have well-known structures such as dendrites and flowers, but most of them are original nanomaterials.66–69As an instructive work, CeO2 nanoflowers obtained via the rapid thermolysis of (NH4)2Ce(NO3)6 in a mixture of OA/OM were reported by Zhang and Yan's group.11 The ceria clusters capped with OA and OM were generated as a result of the hydrolysis of (NH4)2Ce(NO3)6 at a low temperature (<220 °C). When the temperature increased to above 220 °C, the capping agents on the surface of the ceria particles disappeared quickly through the redox reaction. Consequently, the small particles spontaneously aggregated via 3D oriented attachment and generated CeO2 nanoflowers. Self-assembly is often chosen for the construction of 3D rare earth oxide nanostructures, whose driving force is the hydrophobic effect of the surfactants adsorbed on the nanocrystals. For instance, ultrathin nanodisks of Sm2O3 and hierarchical flower-like Gd2O3 and Dy2O3 (Fig. 10a–c) with abundant high-index {400} facets were prepared by Xiao et al.66 through a benzyl alcohol-based nonaqueous sol–gel route using rare earth acetylacetonates as precursors. The X-ray diffraction (XRD) results showed that all of them were in the cubic phase (Fig. 10d). Also, Fig. 10e shows the possible formation mechanism of these rare earth oxide nanostructures, where nanodisks of the three oxides were obtained in the first stage of the sol–gel process, then Sm2O3 stacks were formed through mesocrystal fusion, and flower-like spheres of Gd2O3 and Dy2O3 were generated via self-assembly. The different behaviors in the final stage were possibly due to their diverse inherent properties such as crystal growth behavior.
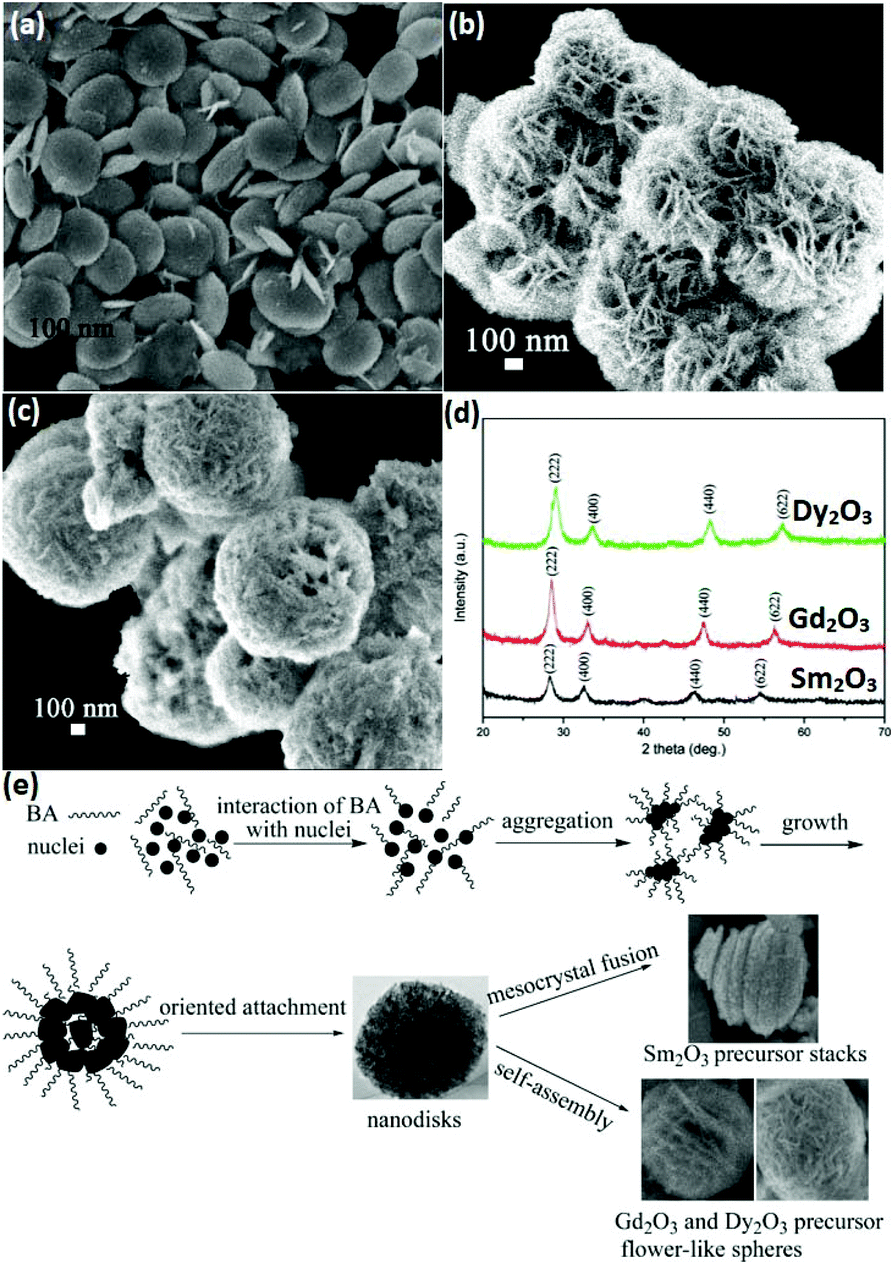 | ||
| Fig. 10 SEM images of (a) Sm2O3 nanodisks; (b) Gd2O3; and (c) Dy2O3 flower-like spheres. (d) XRD patterns and (e) schematic illustration of the possible formation process of Sm2O3 nanodisks or Gd2O3 and Dy2O3 flower-like spheres. Adapted with permission from ref. 66. Copyright 2009, American Chemical Society. | ||
With respect to rare earth oxide dendrites, Zhang et al.67 developed a combination method of hydrothermal treatment and thermal decomposition to prepare CeO2 dendrites with a three-fold shape. In the hydrothermal procedure, the authors chose Ce(NO3)3 and urea as reactants to react in a water–triethanolamine (TEA) mixed solvent. It is established that TEA can steadily coordinate with Ce3+, which will notably lower the concentration of free Ce3+ in solution. Thus, the rate of Ce(OH)CO3 formation would be extremely slow, which is beneficial for the crystallization and separation of the nucleation and growth stage. Consequently, the nuclei aggregated with each other and formed three-fold branched units, then these building units connected together through self-assembly and generated the final dendrites. An identical function as TEA could be realized using ammonia water or hydrazine hydrate in the studies by Zhang et al.43,68 They obtained three-fold and four-fold CeO2 dendrites by intentionally using ammonia water and hydrazine hydrate, respectively. Both reagents had an important effect on engineering the shape of the CeO2 dendrites through decreasing the Ce3+ concentration and lowering the crystal growth rate, which are crucial for obtaining branched architectures.
Besides 3D dendritic and flower-like rare earth oxides, Qu et al.69 obtained La2O3 hollow spheres with multi shells via a one-pot hydrothermal method combined with calcination. They used a mixture of D-glucose and lanthanum nitrate as the precursor solution, and the special evolution mechanism could be ascribed to the template effect of the carbon in the structure. With a continuous increase in the calcination temperature, the carbon contained in the spheres was oxidized to CO2, resulting in the formation of a multi-shell structure. Besides, Dong et al.70 reported the preparation of coral-shaped Dy2O3 through an environmentally friendly solvothermal method combined with calcination. They used dysprosium acetylacetonate and carbamide as reagents and methanol as the solvent. The formation of the coral-like Dy2O3 was based on the spontaneous accumulation of small particles with a size of about 12 nm. Moreover, 3D porous CeO2 with various shapes was obtained by Jiang et al.44via a simple precipitation method using glycine as a soft bio-template. The preparation procedure, which involved stirring at room temperature, drying at 100 °C, and calcination at 500 °C, was very facile, as shown in Fig. 11a. By modulating the reagent ratio of Ce3+, urea and glycine, shuttle-like, bowknot-like, prism-like, and dumbbell-like CeO2 were obtained (Fig. 11b–g). Furthermore, the authors investigated the formation mechanism of the CeO2 products, as shown in Fig. 11h. As a bio-template, the glycine provided place for the reaction of Ce3+ and urea to produce nanospheres since Ce3+ was adsorbed on the surface of glycine. Then the final product was formed through the self-assembly of the nanospheres. The morphologies could be tuned by the reagent ratio because of its influence on the dispersion and hydrolysis rate of Ce3+.
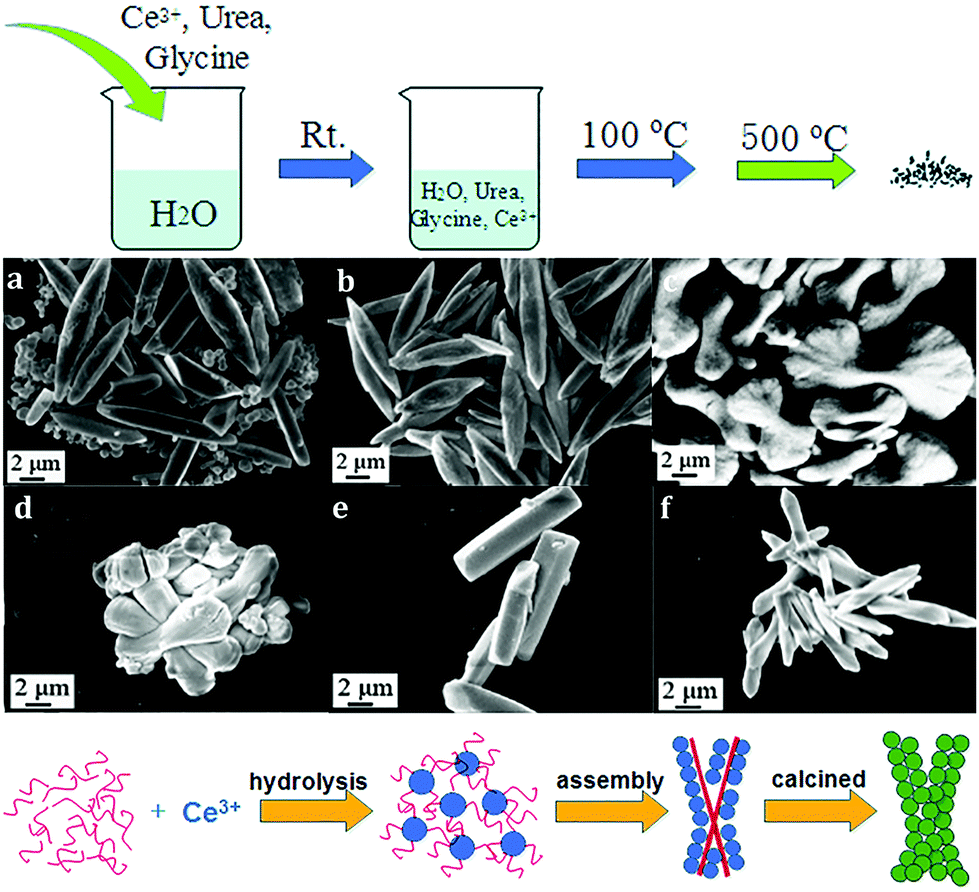 | ||
| Fig. 11 (a) Procedure for the synthesis of 3D porous CeO2. SEM images of CeO2 prepared with different ratios of reagents (Ce3+:urea:glycine) of (b) 3:0:1; (c) 3:1:1; (d) 3:2:1; (e) 3:40:1; (f) 3:4:2; and (g) 6:2:1. (h) Schematic illustration of the formation of the 3D porous CeO2 with a bowknot shape. Adapted with permission from ref. 44. Copyright 2018, Royal Society of Chemistry. | ||
4. Catalytic applications of rare earth oxide nanomaterials in C1 chemical reactions
Rare earth oxide nanomaterials are very promising for catalytic applications due to their abundant electronic structures and flexible oxidation states and coordination numbers.3–5 Also, researchers have already studied and utilized rare earth oxide-based nanocatalysts in many important reactions such as C1 chemical reactions.20,21 C1 chemical reactions are vital because they are closely related with the generation, storage and conversion of energy and environmental protection.16–19 Therefore, the development of novel well-defined rare earth oxide nanomaterials with high catalytic performances in C1 chemical reactions has important implications for alleviating the increasing energy and environment crisis. In this section, we employ several typical C1 chemical reactions including the CO oxidation reaction, water gas-shift reaction (WGSR), CO2 hydrogenation reaction, methane oxidation reaction and methanol oxidation reaction (MOR) to summarize the nanostructural engineering of rare earth oxide nanomaterials in catalytic applications and reveal the corresponding structure–performance relationship, assisted by some representative works.4.1 CO oxidation reaction
The CO oxidation reaction (CO + O2 → CO2) is often used as a probe reaction to study the structure–activity relationship of catalysts owing to its simple reaction formula and mechanism. Besides, it also has applied value in practical applications such as automobile tail gas purification, CO sensors, and indoor air purification.71,72 For the mechanism of the CO oxidation reaction, the Mars-van Krevelen mechanism is accepted by most researchers, that is, a CO molecule reacts with the lattice oxygen of the oxide and in situ generates an oxygen vacancy, and then the oxygen molecule fills the vacancy and finishes the cycle. Consequently, oxygen vacancies play a vital role in the catalytic CO oxidation reaction, which are abundant in many rare earth oxide nanomaterials.Obviously, rare earth oxide nanomaterials can independently catalyze the CO oxidation reaction without modulation with other non-rare earth metals, and their performance is closely correlated with their morphologies.11,34,73 As an example, the CeO2 nanorods synthesized by Zhou et al.34 exhibited a T50 (temperature of 50% CO conversion) of 186 °C towards CO oxidation, which is much lower than that of CeO2 nanoparticles. The enhanced activity was ascribed to the well-defined reactive {001} and {110} planes. Our group73 implemented the dimension-manipulated synthesis of CeO2 nanostructures and obtained 0D uniform crystals, 2D polycrystalline assembly, and 3D mesoporous framework. The 2D polycrystalline assembly sample showed the highest activity (T50 = 310 °C) for CO oxidation, while the 3D mesoporous framework had the lowest activity (T50 = 390 °C), and all three nanocatalysts were much more active than bulk CeO2. In addition, Kang et al.74 prepared hexagonal Pr(OH)3 and cubic Pr6O11 nanorods and tested their activity for CO oxidation. The Pr(OH)3 nanorods had a T10 (temperature of 10% CO conversion) of 525 °C in the first run and then transformed into Pr6O11 nanorods after the first run. Subsequently, in the second run, the T10 was 465 °C due to the transformation from Pr(OH)3 to Pr6O11. Although their activity was lower than that of commercial catalysts, they can be applied in catalysis and sensing. As another example, Zhang et al.75 synthesized 3D ordered porous Pr6O11 and Tb4O7 using a hard template method. The Pr6O11 and Tb4O7 nanostructures showed a T50 of 305 °C and 360 °C, respectively, which were much more active than commercial Pr6O11 (550 °C). The enhanced activity may have originated from their large surface area, abundant oxygen vacancies, and higher low-temperature reducibility.
Doping is a common strategy for enhancing the catalytic activity of rare earth oxide nanomaterials. For instance, the CeO2 nanowires prepared by our group35 could catalyze the CO oxidation reaction with a T50 of 350 °C, which may be ascribed to the facile formation of oxygen vacancies in the CeO2 {110} facet. We successfully doped lanthanide elements in the CeO2 nanowires (Fig. 12a) to enhance the activity of CeO2, and the most active sample of Nd-doped CeO2 nanowires showed an order of magnitude higher activity (TOF at 200 °C = 5.4 × 10−4 s−1) than that of the pure CeO2 nanowires (TOF at 200 °C = 4.1 × 10−5 s−1, Fig. 12b). We also found that the big difference in the activity of the doped catalysts was derived from two aspects. On the one hand, the oxygen vacancy formation energy of the light rare earth-doped samples was much lower than that of the heavy rare earth-doped samples (Fig. 12c), indicating that the former had more oxygen vacancies, which are beneficial for the catalytic activity. On the other hand, the concentration of the active intermediates of unidentate carbonates was also crucial for governing the activity, and Fig. 12d exhibits that CeO2:Nd had the largest amount of these active species. These two factors together resulted in the volcano-like relationship between the catalytic activity and the ionic radius of the dopant elements. Similarly, Li et al.76 also demonstrated that doping with La remarkably enhanced the catalytic activity of CeO2 for the CO oxidation reaction with a T50 of 245 °C because the formation of non-stoichiometric Ce1−xLaxO2−δ increased the formation of oxygen vacancies, and the optimal value of x is 0.5. Besides doping to boost the concentration of oxygen vacancies in rare earth oxides, reduction treatment is also a suitable method for obtaining higher activity for the CO oxidation reaction. As an example, Gao et al.12 enhanced the activity of as-prepared CeO2 nanorods via chemical redox etching with ascorbic acid.
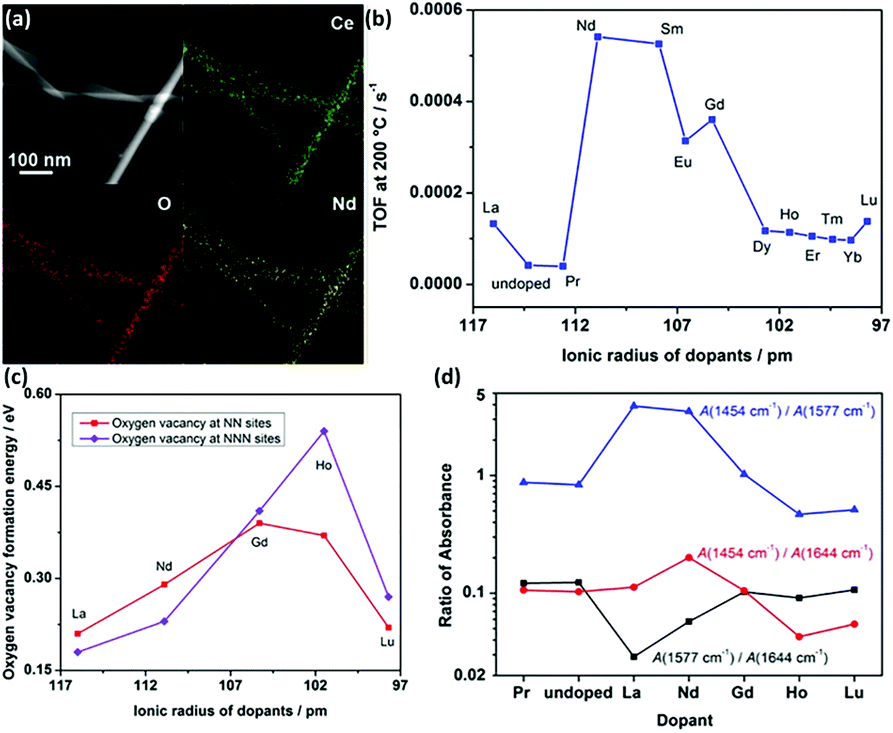 | ||
| Fig. 12 (a) EDS mapping images of CeO2:Nd. (b) Catalytic activity for the oxidation reaction of lanthanide-doped CeO2 nanowires versus the radius of the dopants. (c) Oxygen vacancy formation energy of CeO2:Ln. (d) Variation of IR intensity at 1454 (unidentate carbonates), 1577 (bidentate carbonates), and 1644 cm−1 (bicarbonates) with different dopants. Adapted with permission from ref. 35. Copyright 2013, American Chemical Society. | ||
According to the above examples, it can be concluded that single rare earth oxide nanocatalysts have limited activity for the CO oxidation reaction, and thus the introduction of other metals and engineering the local metal–support structure is a valid and common strategy for boosting their activity, in which rare earth oxide nanomaterials can act as a support.77–79 For example, our group77 systematically investigated the effect of the local coordination structure of Pt/CeO2 nanocatalysts on their activity for CO oxidation. We prepared Pt/CeO2 catalysts with different Pt–O coordination numbers by tuning the reduction temperature of the samples. A decrease in the activity of the catalysts was observed with an increase in the Pt–O coordination number, and the most active sample had a TOF at 50 °C of 2.0 × 10−2 s−1. The Pt atoms were over-stabilized at the catalyst surface, resulting in the inactivation of some of the Pt atoms when the Pt–O coordination number kept increasing, and thus the activity of the catalyst decreased. Specifically, it was the local structural effect that determined the catalytic performance. As another example, Huang et al.78 prepared Pr6O11 nanorods with a diameter of 20–40 nm and length of several microns using a hydrothermal method. They found that the pure Pr6O11 nanorods were not active for CO oxidation until 220 °C. However, when they loaded gold nanoparticles with a size of 8–12 nm on the surface of the Pr6O11 nanorods, the composites could achieve 100% CO conversion at 140 °C, and were also much more active than the Au/Pr6O11 bulk. Similarly, Zhang et al.79 synthesized and studied nanostructured Ag/Pr6O11 for CO oxidation. The authors prepared Pr6O11 nanorods and nanoparticles using different routes, and utilized them as supports to couple with Ag species via the conventional impregnation method (Fig. 13a). The XPS results indicated that the Ag/Pr6O11 nanorods possessed more oxygen vacancies than that of the Pr6O11 nanoparticles (Fig. 13b). Besides, the H2-TPR profiles showed a stronger synergistic effect between Ag and Pr6O11 in the Ag/Pr6O11 nanorods (Fig. 13c), suggesting higher activity for this catalyst. As expected, the Ag/Pr6O11 nanorods showed a better performance than the Ag/Pr6O11 nanoparticles for CO oxidation, where the T50 of the Ag/Pr6O11 nanorods was around 125 °C (Fig. 13d and e).
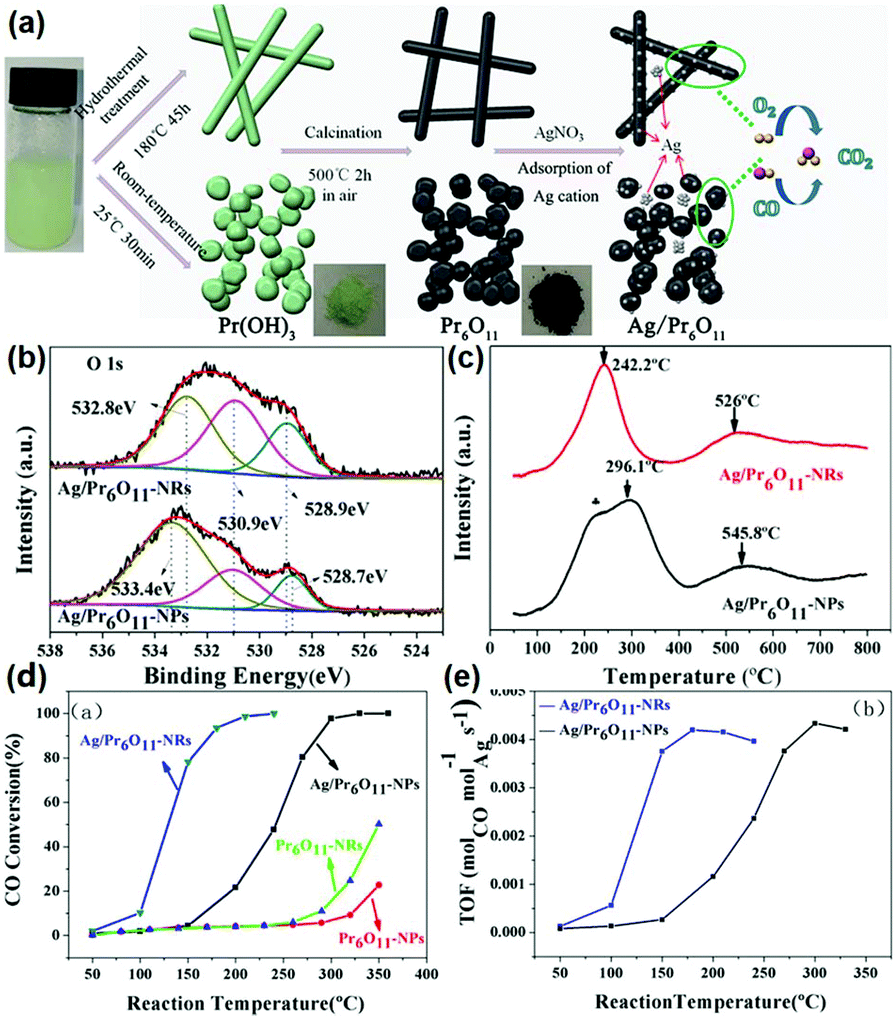 | ||
| Fig. 13 (a) Schematic illustration of the synthetic routes for Pr6O11 nanostructures. (b) O 1s XPS spectra of Ag/Pr6O11 catalysts. (c) H2-TPR profiles of Ag/Pr6O11 catalysts. (d) CO conversion of Pr6O11 and Ag/Pr6O11 samples for CO oxidation. (e) TOF of Pr6O11 and Ag/Pr6O11 samples for CO oxidation. Adapted with permission from ref. 79. Copyright 2017, American Chemical Society. | ||
The well-known shape effect of rare earth oxide nanomaterials also has a vital influence on the catalytic activity for CO oxidation when they act as a support.80,81 Taking the work of Lykaki et al.80 as an example, they prepared CeO2 nanorods (NR), nanopolyhedra (NP), and nanocubes (NC) via the hydrothermal method, and explored the shape effect of Cu/CeO2 on their catalytic properties. The results of the catalytic test showed that the activity (T50) of both the pure CeO2 and Cu/CeO2 followed the same order of NR (320/72 °C) > NP (350/83 °C) > NC (385/92 °C). The authors used multiple characterizations to investigate the structure–function correlation, and found that the activity had a direct correlation with the amount of Cu+ and oxygen defects. They proposed the reaction mechanism based on the Mars-van Krevelen mechanism, which highlighted the significant roles of oxygen vacancies and Cu+ in this benchmark reaction. Furthermore, it is known that the shape effect is equal to the plane effect. Also, NR have mainly exposed {110} and {100} facets, while NP are enclosed with {111} and {100} facets, and NC have exposed {100} facets. Therefore, the CeO2 {110} planes were beneficial for CO oxidation due to their easy generation of oxygen defects.
In general, the nanostructural engineering of rare earth oxide-based catalysts for the CO oxidation reaction is mainly focused on the tuning of oxygen vacancies, the effect of which mainly embodied in stabilizing the metal loading on the surface of the rare earth oxides and providing sites for the adsorption and activation of O2. Also, the strategies for modulating oxygen vacancies usually involve reduction treatment, doping, controlling the size and morphology, etc. Besides, other factors such as the surface area and metallic oxidation state of the rare earth oxide-based catalysts also have a significant effect on the catalytic performance. Thus, researchers should consider all these factors in the design of catalysts with excellent properties for CO oxidation.
4.2 WGSR
The WGSR (CO + H2O → CO2 + H2) is mainly used for converting CO to CO2 and H2 in syngas, which has important application value in hydrogen production, fuel cells and ammonia synthesis.82 Most of the catalysts for the WGSR are supported nanomaterials since single materials hardly exhibit activity. Rare earth oxide nanomaterials are widely used as a support for the WGSR due to their good stability and abundant oxygen vacancies, which are significant for stabilizing the metal loading and activating H2O molecules.83,84 Also, a good way for improving the catalytic performance is through tuning the morphologies of rare earth oxides since the shape effect is also important for the WGSR.Among the various catalytic systems, noble metals such as Au and Pt supported by well-defined rare earth oxide nanostructures have been extensively applied for the WGSR because of their high activity and good stability.83–86 For example, Si et al.87 investigated the catalytic performance of Au/CeO2 with nanorod, nanocube and nanopolyhedra structures. They found that the WGSR performance strongly depended on the exposed facets, and the sample of Au/CeO2 nanorods showed the highest activity among the three samples. This could be explained by the lowest anion vacancy formation energy on the {110} planes, which meant more oxygen vacancies boosted the catalytic activity. As another example, Fu et al.15 prepared two types of Au/CeO2 catalysts, including Au nanoclusters (<2 nm) and Au nanoparticles (3–4 nm), for the WGSR (Fig. 14a and b, respectively). Specially, the Au nanoclusters were in situ generated in the reaction process from Au single atoms. The Au nanoclusters showed higher activity (rate at 200 °C = 5.0 × 102 mol gAu−1 s−1) compared with that of the Au particles (rate at 200 °C = 1.0 × 102 mol gAu−1 s−1) because of their more abundant interfacial sites (Fig. 14c). The results of in situ Raman spectroscopy demonstrated that the oxygen vacancies were involved in the catalytic reaction, which were the most abundant in the Au_cluster/CeOx sample (Fig. 14d). The authors also performed in situ infrared spectroscopy (IR) to investigate the reaction mechanism, and they found that the bridged surface –OH groups were essential for the conversion of the CO adsorbed on the Auδ+ species to CO2, which was verified by the isotopic labelling experiment, where one of the O atoms in CO2 came from the bridged –OH group (Fig. 14e). Fig. 14f shows the whole cycle of the catalytic process, where H2O was activated at the oxygen vacancy and generated a bridged –OH group in the first stage, then the bridged –OH group reacted with the CO adsorbed on the Auδ+ species to produce CO2 in the second stage, and finally the CO2 desorbed and left a new oxygen vacancy. The moderate Auδ+ species, bridged –OH group and oxygen vacancies were essential in the WGSR.
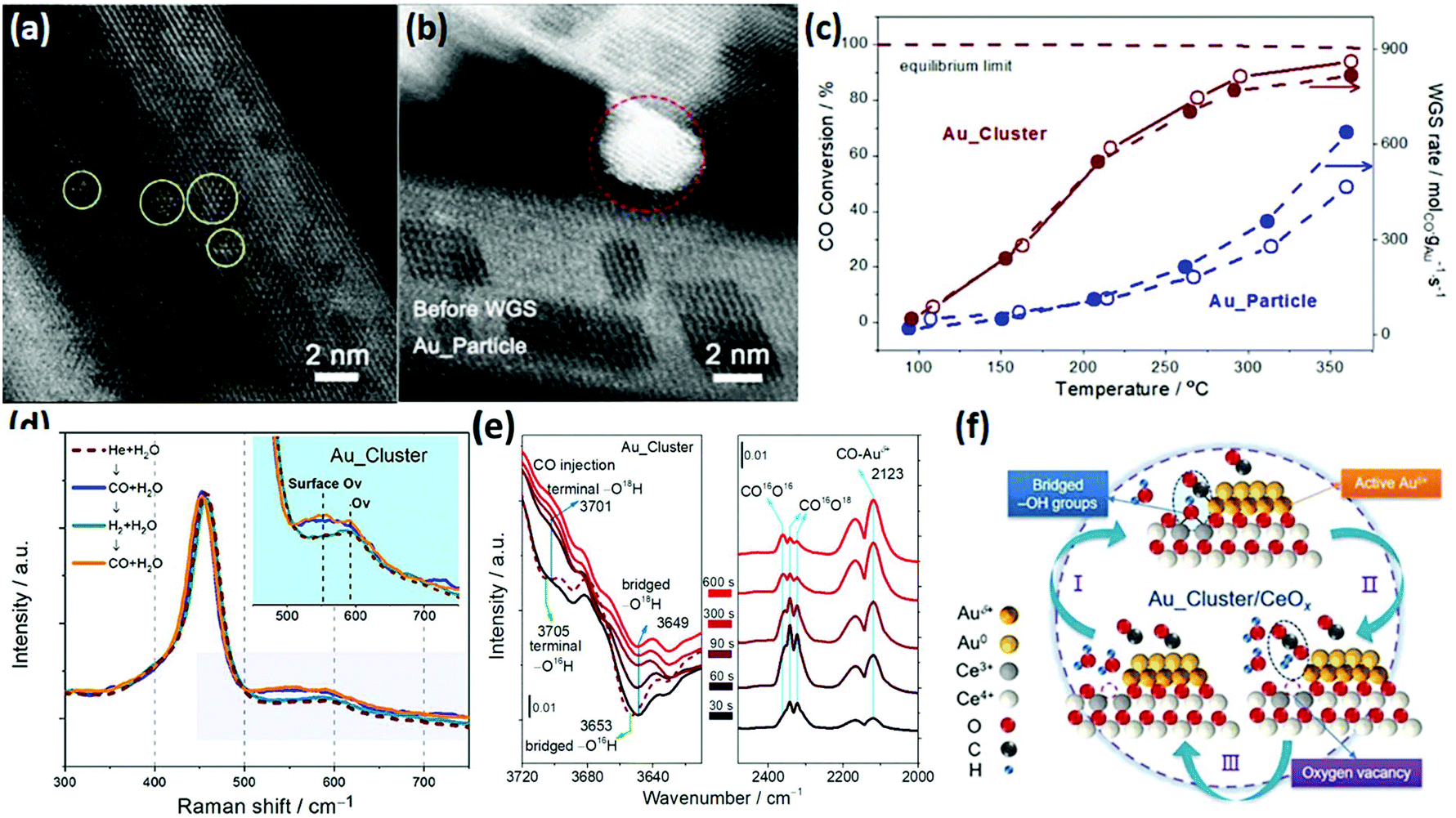 | ||
| Fig. 14 High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images of (a) Au_cluster/CeOx and (b) Au_particle/CeOx catalyst. (c) Catalytic activity for the WGSR by the Au_cluster/CeOx and Au_particle/CeOx catalysts. (d) In situ Raman spectra of Au_cluster/CeOx under different atmospheres at 150 °C. (e) In situ time-sequencing IR spectra of Au_cluster/CeOx under CO injection with isotope prelabeled using H2O.18 (f) Schematic illustration of the reaction mechanism of Au_cluster/CeOx catalyst for the WGSR. Adapted with permission from ref. 15. Copyright 2019, American Chemical Society. | ||
The use of non-Ce rare earth oxides for supporting noble metals has been less applied in the WGSR compared with CeO2. For instance, novel Au/La2O3 and Au/La2O2SO4 catalysts for the low-temperature WGSR were synthesized by Lessard et al.13 through an anion adsorption method. They found that both Au/La2O3 and Au/La2O2SO4 had a good WGSR activity, and Au/La2O2SO4 showed higher activity compared with that of Au/La2O3 which may be because of its higher content of Au3+, as confirmed by the X-ray photoelectron spectroscopy (XPS) results. Besides, the authors carried out chemical leaching of the catalysts using NaCN, and the leached samples exhibited no difference in activity, indicating that the support of La2O3 and La2O2SO4 could well stabilize the Au species. Besides, Pt supported by a composite oxide of Ce0.6Y0.4O2 for the WGSR was obtained by Lee et al.86via a sol–gel method. As can be seen in Fig. 15a, the activity of Pt/Ce0.6Y0.4O2 (TOF at 250 °C = 5.5 × 10−1 s−1) was much higher than that of both Pt/CeO2 (TOF at 250 °C = 1.6 × 10−1 s−1) and Pt/Y2O3 (TOF at 250 °C = 1.3 × 10−1 s−1), and also higher than that of many reported Pt-based WGSR catalysts. The H2-TPR results (Fig. 15b) showed that the introduction of Y to CeO2 obviously enhanced the reducibility and the oxygen mobility of the support. Furthermore, the CO-TPR profiles indicated that there were much more OH species in the Pt/Ce0.6Y0.4O2 sample than the other two catalysts (Fig. 15c and d), which are beneficial for the WGSR. The authors also performed diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) to explore the surface reactions on these catalysts (Fig. 15e). The results clearly showed that the CO adsorption on Pt//Ce0.6Y0.4O2 was weaker than that on Pt/CeO2 and Pt/Y2O3, indicating its more efficient activation of CO. In general, the enhanced reducibility of the support and reduced CO adsorption contributed to the higher activity.
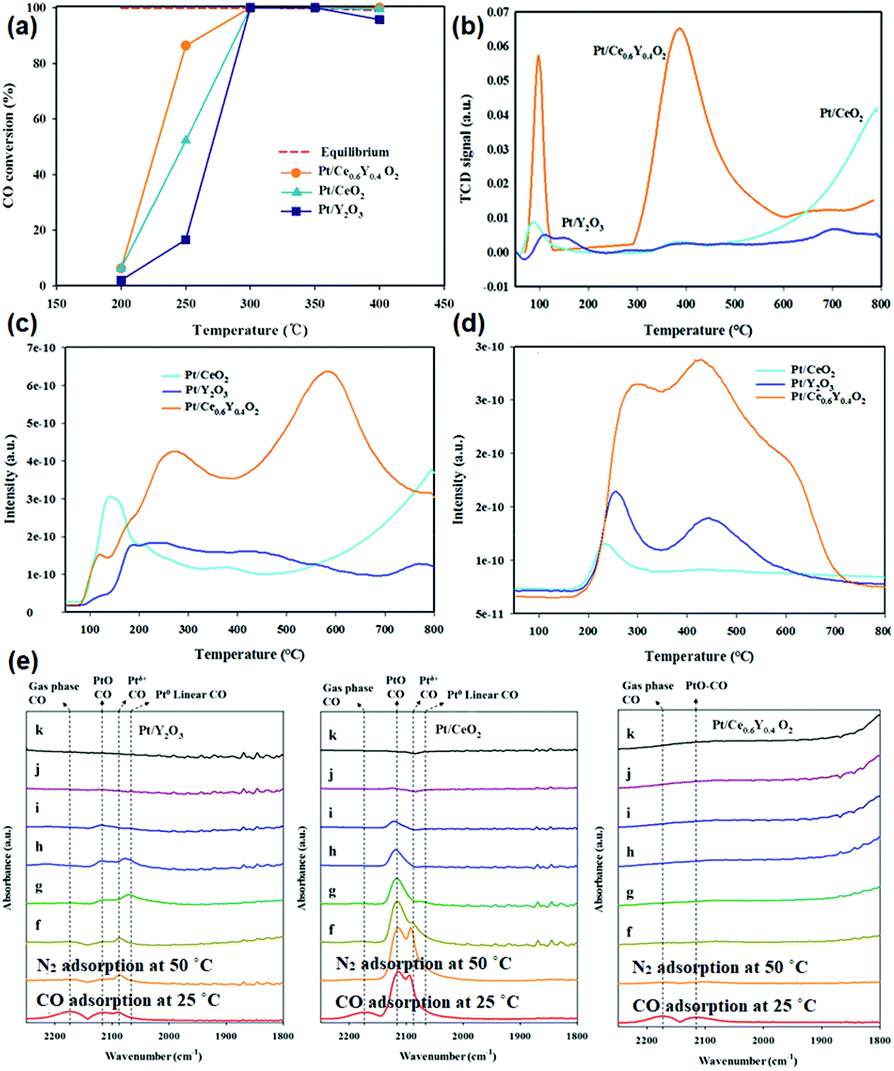 | ||
| Fig. 15 (a) CO conversion of Pt/Y2O3, Pt/CeO2 and Pt/Ce0.6Y0.4O2 in the WGSR. (b) H2-TPR profiles of Pt/Y2O3, Pt/CeO2 and Pt/Ce0.6Y0.4O2. CO-TPR profiles of Pt/Y2O3, Pt/CeO2 and Pt/Ce0.6Y0.4O2: (c) CO2 profile and (d) H2 profile. (e) DRIFTS spectra of Pt/Y2O3, Pt/CeO2 and Pt/Ce0.6Y0.4O2: (f) N2 adsorption at 100 °C; (g) N2 adsorption at 150 °C; (h) N2 adsorption at 200 °C; (i) N2 adsorption at 250 °C; (j) N2 adsorption at 300 °C; and (k) N2 adsorption at 350 °C. Adapted with permission from ref. 86. Copyright 2018, American Chemical Society. | ||
Although supported noble catalysts have superior catalytic properties for the WGSR, their high price limits their large-scale application. Accordingly, nowadays, many researchers aim to use a base metal in the nanostructural engineering of catalysts to obtain high performances for the WGSR.88–90 For instance, Ren et al.49 studied the plane effect on the activity of Cu/CeO2 catalysts for the WGSR. The results showed that the Cu catalysts supported on CeO2 nanooctahedra with {111} facets were more active than that of supported on nanorods and nanocubes, which could be attributed to the best dispersion of Cu, strongest Cu–CeO2 interactions and the largest amount of Cu species with a moderate valence. She et al.88 studied the doping effect of RE2O3 (RE = Y, La, Sm, Nd) on the WGSR activity of CuO/CeO2 catalysts. The evaluation of their catalytic performance showed that the introduction of La2O3 and Nd2O3 was beneficial for enhancing the activity and stability of CuO/CeO2, but the doping of Y2O3 and Sm2O3 exhibited negative effects. The Raman spectra showed that the CuO/CeO2–La2O3 and CuO/CeO2–Nd2O3 samples had more oxygen vacancies than CuO/CeO2, while that of the Y and Sm-doped samples were less. Besides, the authors measured the Cu dispersion using N2O and found out that the order of Cu dispersion was consistent with the order of activity. The H2-TPR results showed three peaks for non-crystalline copper oxide, moderate copper oxide (crystalline) and surface ceria. Interestingly, the order of the peak area for moderate copper oxide was also consistent with that of activity. In summary, the difference in activity mainly originated from the concentration of oxygen vacancies, Cu dispersion, and moderate Cu valence. Another Cu/CeO2 catalyst for the WGSR was synthesized by Chen et al.89 using a co-precipitation method. In order to improve the activity, the authors attempted to calcine the catalyst at 300 °C in air, vacuum and H2. They found that the catalyst annealed in H2 had the highest performance due to the highest amount of oxygen vacancies and strongest synergistic interaction between CuO and CeO2.
Based on the research thus far, it can be concluded that rare earth oxide nanomaterials are rarely used alone for the WGSR on account of their weak adsorption of CO. They are always used as a support and additive to improve the catalytic performance through the construction of well-defined structures and controlling the electronic and geometric structure of the catalysts. The effects of rare earth oxide nanomaterials are mainly reflected in dispersing metal catalysts, providing active sites such as oxygen vacancies and enhancing the metal support interaction and synergistic effect.
4.3 CO2 hydrogenation reaction
The CO2 hydrogenation reaction is not a specific reaction since it has several possible products, such as CO, methane and methanol. It is a significant reaction because it not only can weaken the greenhouse effect, but also ease the energy crisis.91 However, the high chemical stability of the CO2 molecule and the complexity of its products are a big challenge for catalysts for this reaction. The abundant electronic structures and flexible oxidation states of rare earth oxide nanomaterials are beneficial for the CO2 hydrogenation reaction, which is ascribed to their facile activation of CO2 and selectivity control.92 Nevertheless, pure rare earth oxides are less used for catalyzing this reaction due to their poor activity. Here, we summarize the nanostructural engineering of rare earth oxide nanomaterials utilized in the CO2 hydrogenation reaction according to the different products.The CO2 hydrogenation reaction is also known as the reverse water gas shift (RWGS) reaction (CO2 + H2 → CO + H2O) when the product is CO and H2O. The morphologies of rare earth oxides have a significant influence on their catalytic performance.93,94 As an example, CeO2 nanocatalysts with different morphologies synthesized by Dai et al.93 using various methods showed extremely different activities for the RWGS reaction. CeO2 with a 3D porous structure was much more active than the loosely aggregated CeO2 nanoparticles and bulk CeO2 due to its largest surface area and oxygen vacancy concentration. As another example, Lin et al.94 prepared Cu/CeO2 nanorods and Cu/CeO2 nanospheres and investigated their catalytic performance for the CO2 hydrogenation reaction. They found that both catalysts mainly produced CO through the RWGS reaction and the Cu/CeO2 nanorods exhibited higher activity (rate at 250 °C = 1.8 μmol gcat−1 s−1) (Fig. 16a). The results of multiple in situ characterizations showed that the active intermediates of formates and bidentate carbonates were preferentially generated on the CeO2 {110} facet and CO2 could also be activated and dissociated more effectively on the CeO2 {110} facets. Consequently, the Cu/CeO2 nanorods with more abundant {110} terminations were more active, indicating that researchers can design high-performance CeO2-based catalysts by engineering their structure. Additionally, Liu et al.95 synthesized LaNiO3 perovskite catalysts and tested them for the RWGS reaction in a dielectric barrier discharge plasma reactor. After reduction, the Ni species were extracted from the perovskite structure, and the Ni/La2O3 catalysts were thus formed. The highest CO yield among these catalysts was 39.1% at around 100 °C. The activity was closely correlated with the Ni particle size, Ni dispersion and metal–support interactions.
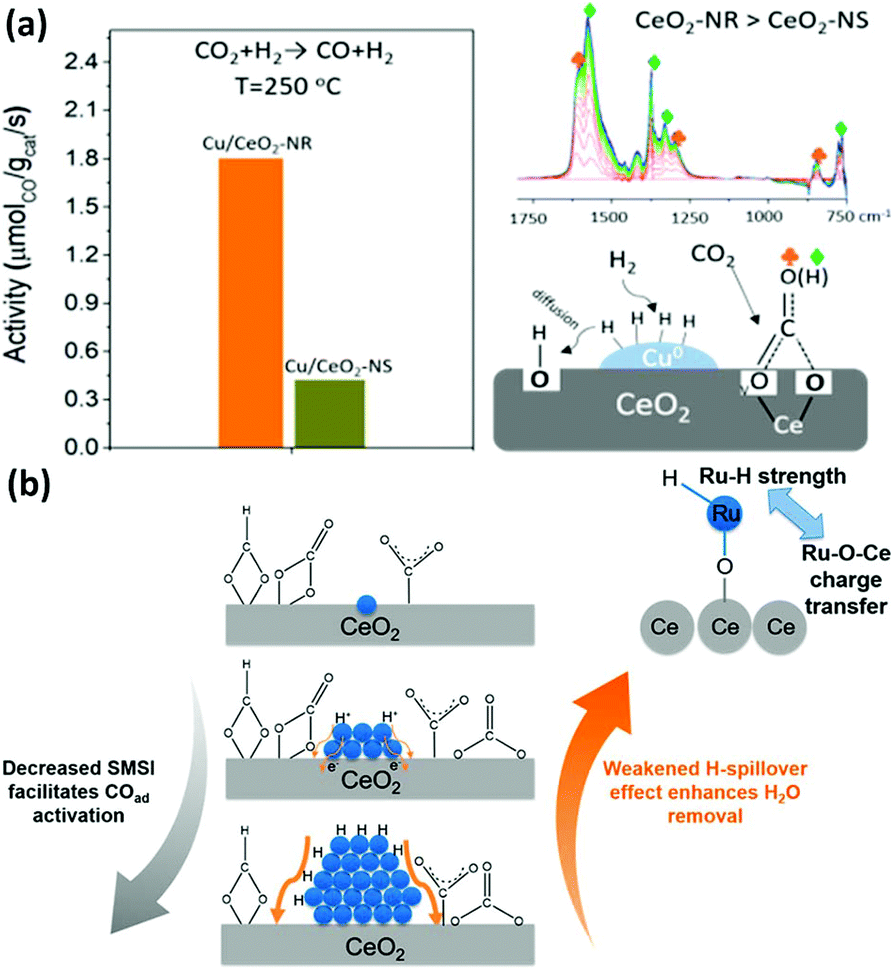 | ||
| Fig. 16 (a) Activity and schematic illustration of the mechanism of the RWGS reaction for Cu/CeO2 nanorods and Cu/CeO2 nanospheres. Adapted with permission from ref. 94. Copyright 2018, American Chemical Society. (b) Schematic illustration of the compete effect between SMSI and H-spillover on Ru/CeO2 catalysts for the CO2 methanation reaction. Adapted with permission from ref. 96. Copyright 2018, American Chemical Society. | ||
The CO2 methanation reaction (CO2 + H2 → CH4 + H2O) is one of the CO2 hydrogenation reactions and its product is methane. Although this reaction is thermodynamically preferred, the 8-electron reduction process is a big hindrance for obtaining high activity and selectivity. Thus, engineering the morphologies of rare earth oxides to obtain well-defined structures is beneficial to solve this problem. For example, our group96 synthesized three types of catalysts containing Ru single atoms, nanoclusters and nanoparticles supported by CeO2 nanowires for the CO2 methanation reaction. We found that the sample of Ru nanoclusters showed the highest activity (TOF at 190 °C = 7.4 × 10−3 s−1), which was also much higher than that of other reported Ru/CeO2 catalysts. Unexpectedly, the order of activity was not consistent with that of the concentration of oxygen vacancies. According to our investigation, the strong metal support interaction (SMSI) decreased with an increase in the Ru size and benefited the activation of adsorbed CO species, while the H-spillover effect weakened with a decrease in the Ru size and facilitated the removal of H2O. Accordingly, the competition between the SMSI and H-spillover effect together determined the activity of the Ru/CeO2 catalysts (Fig. 16b). In the case of other rare earth oxides used in CO2 methanation, Ilsemann et al.97 obtained novel Sm2O3–Ni xerogel catalysts through the sol–gel method for the CO2 methanation reaction. The optimal loading of Ni was 39 wt%, which showed the highest activity. Also, the catalysts had a good score in the stability test, and their activity could easily recover by a reduction treatment. Díez-Ramírez et al.98 studied the effect of different supports (CeO2, ZrO2, Gd2O3, and ZnO) on the activity of Co catalysts for the CO2 methanation reaction. The methane yield of Co/CeO2 was much higher than that of the other catalysts due to the enhanced reducibility related with Co–CeO2 interactions.
The selective hydrogenation of CO2 to methanol has attracted increasing attention in recent years because methanol is an important basic chemical in many chemical industries.99–102 Chen et al.103 synthesized rod-like La2O2CO3 through a facile hydrothermal method (Fig. 17a) and used it as a support for Cu catalysts (denoted as Cu/La2O2CO3-R) in the CO2 hydrogenation to methanol. They also prepared Cu supported by amorphous La2O2CO3 (denoted as Cu/La2O2CO3-A), Cu supported by rod-like CeO2 (denoted as Cu/CeO2) and a co-precipitation catalyst of CuLaxOy (denoted as CuLaxOy-CP) for comparison. The Cu/La2O2CO3-R catalyst had a much better catalytic performance (TOF at 240 °C = 7.0 × 10−3 s−1, selectivity = 92.5%) compared with the control samples and commercial CuZnAl catalyst (TOF at 240 °C = 4.0 × 10−3 s−1, selectivity = 68%) (Fig. 17b). The CO2 temperature-programmed desorption (TPD) results (Fig. 17c) showed that Cu/La2O2CO3-R had a stronger and larger amount of CO2 adsorption, indicating that it could activate CO2 more efficiently. Besides, the order of the Cuδ+ ratio was consistent with that of the activity, as can be seen in Fig. 17d, which suggested that Cuδ+ species were the active sites. The authors also performed in situ DRIFTS to reveal the reaction mechanism, and they found that the HCOO and CH3O species were the main active intermediates (Fig. 17e), which were stable on the catalyst surface and not easy transformed into CO. In brief, it was the synergistic basic sites of the Cuδ+ species tuned by the structure of the metal–support interface that contributed to the high activity. Some groups used rare earth oxide as promotors to adjust the catalyst structure for CO2 hydrogenation to methanol and obtained positive results.104,105 For instance, Ban et al.104 verified that the using of La and Ce as promoters boosted the methanol production of CuZnZr catalyst, especially Ce. However, the introduction of Pr and Nd as additives decreased the activity. The opposite effect of the different rare earth promotors on activity was mainly due to the enhanced synergistic effect and H2 adsorption caused by the addition of La and Ce, which were not found in the catalysts with Pr and Nd as additives. Similarly, Kourtelesis et al.105 found that La2O3 could promote 30% of the methanol yield for the CuO/ZnO/Al2O3 catalyst. La2O3 could be facilely introduced by a co-precipitation method, and the enhanced methanol yield could be attributed to enhanced adsorption of CO2 and active intermediates.
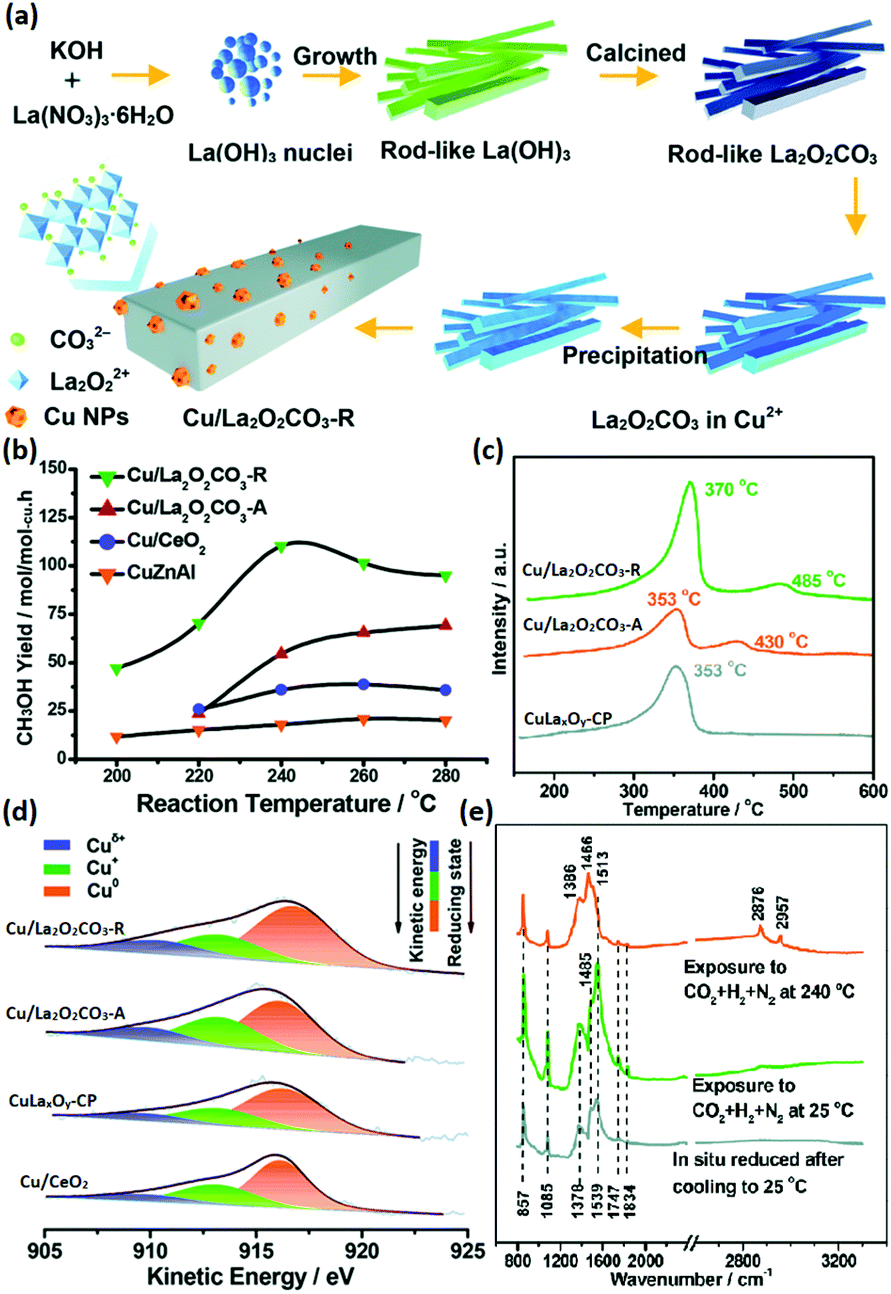 | ||
| Fig. 17 (a) Schematic illustration of the synthetic route for Cu/La2O2CO3-R. (b) CH3OH yield of the catalysts for CO2 hydrogenation to methanol. (c) CO2-TPD profiles of Cu/La2O2CO3-R, Cu/La2O2CO3-A, and CuLaxOy-CP. (d) Cu LMM Auger spectra of Cu/La2O2CO3-R, Cu/La2O2CO3-A, CuLaxOy-CP, and Cu/CeO2. (e) In situ DRIFTS spectra of Cu/La2O2CO3-R under different conditions. Adapted with permission from ref. 103. Copyright 2018, Royal Society of Chemistry. | ||
Considering the complexity of the product of the CO2 hydrogenation reaction, the choice and design of catalysts must be based on selectivity and avoiding side reactions. Thus, reasonably engineering catalysts with well-defined structures is vital. Based on the reported works, using rare earth oxide nanomaterials in the CO2 hydrogenation reaction as support or promotor can usually enhance the adsorption of CO2 and metal support interactions, which will result in improved activity and selectivity.
4.4 Methane oxidation reaction
Methane is an important resource due to its applications such as fuel and chemical synthesis. The methane oxidation reaction can be divided into two types, total oxidation and partial oxidation. The total oxidation of methane can be used for energy and heat production or solving the global warming caused by methane.106,107 The partial oxidation of methane can produce the syngas for further utilization.108–110 Rare earth oxide nanomaterials are widely applied in both reactions111–118 as supports or promotors.In the total oxidation of methane, CeO2 is often used as a catalyst support on account of its high concentration of oxygen vacancies.111,112 Also, the well-defined structures of CeO2-based catalysts are conducive for catalytic activity. For instance, Co3O4 nanoparticles supported by CeO2 nanorods (Fig. 18a and b) for methane combustion were synthesized by Dou et al.111 using a deposition precipitation method. The catalyst of Co3O4/CeO2 (T50 = 475 °C) was found to be much more active than pure Co3O4 and CeO2 due to the synergistic effect between Co3O4 and CeO2, and also the activation energy, which was also much lower in the case of Co3O4/CeO2 (Fig. 18c and d). Pd-based nanomaterials are some of the most effective catalysts for the total oxidation of methane, and using rare earth oxides as a support can be further beneficial due to their unique redox properties. As an example, Ozawa et al.113 tested the influence of the addition of La2O3 and Nd2O3 to PdO/Al2O3 for the combustion of methane. They found that adding La2O3 or Nd2O3 alone had an effect of stabilizing Al2O3 and enhancing the activity. However, when the authors introduced both La2O3 and Nd2O3 to PdO/Al2O3, they observed reduced activity and an extended lifetime. Besides, this group also investigated the stabilizing effect of La2O3, Nd2O3, and ZrO2 on PtPdO/Al2O3 for methane combustion.114 Similarly, the introduction of both La2O3 and Nd2O3 prevented the deactivation of the catalyst, but the addition of ZrO2 caused deactivation. Also, the addition of all three oxides resulted in the longest lifetime among the catalysts. As another example, Danielis et al.115 prepared Pd-embedded CeO2 catalysts via a dry ball-milling method. The Pd–CeO2 catalysts showed a core–shell structure, in which the shell consisted of Pd and CeO2. The as-prepared catalyst could activate methane at a much lower temperature (T10 = 291 °C) compared with that with the conventional Pd/CeO2 catalyst (T10 = 346 °C) synthesized by impregnation (Fig. 18e). The superior performance was ascribed to the creation of highly active and stable sites due to the robust contact between Pd and CeO2.
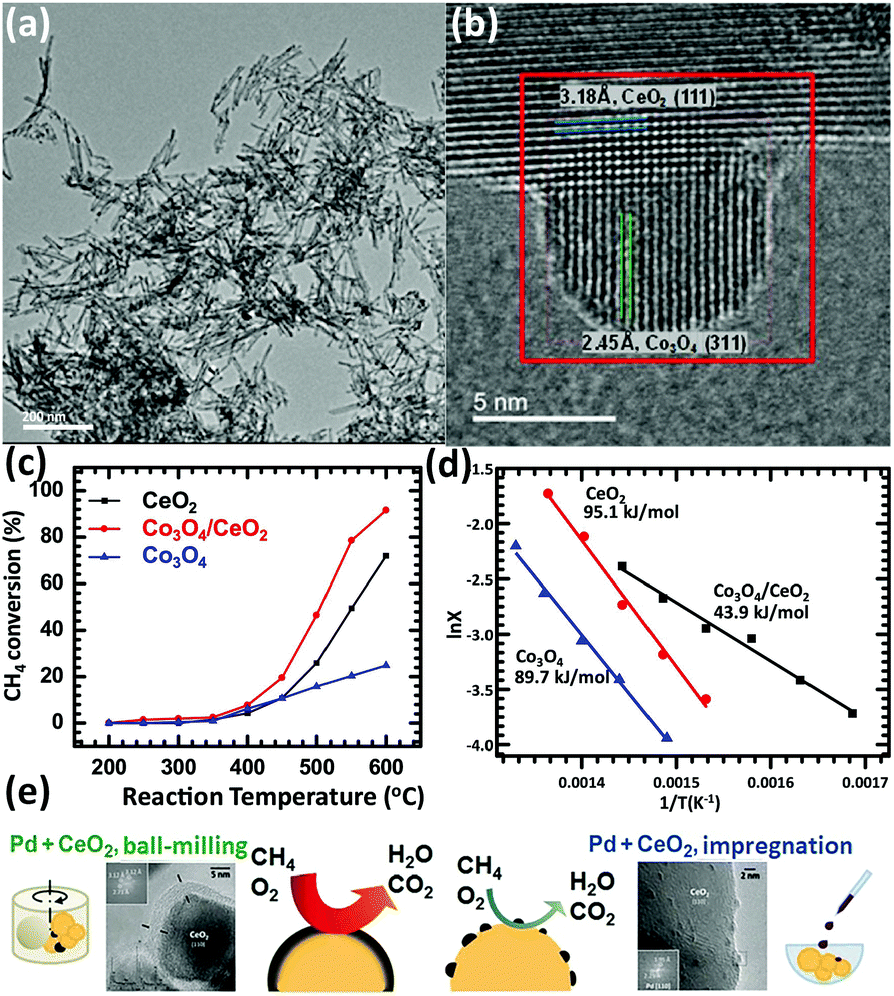 | ||
| Fig. 18 (a) TEM image of CeO2 nanorods. (b) HRTEM image of Co3O4/CeO2 nanocomposite. (c) Catalytic activity of CeO2, Co3O4 and Co3O4/CeO2 for methane combustion. (d) Arrhenius plots and corresponding activation energy of CeO2, Co3O4 and Co3O4/CeO2. Adapted with permission from ref. 111. Copyright 2018, Elsevier. (e) Schematic illustration of the preparation method, catalyst morphology and catalytic activity for the methane combustion reaction using Pd/CeO2 catalysts. Adapted with permission from ref. 115. Copyright 2018, Wiley-VCH. | ||
In the partial oxidation of methane reaction, 1D rare earth oxide nanostructures are often used. For instance, Zhu et al.119 prepared a core–shell Ni/nanorod-CeO2@SiO2 catalyst using a microemulsion method. The as-prepared catalyst showed better activity and stability than that of Ni/nanorod-CeO2 and Ni@SiO2, benefitting from the greater amount of oxygen vacancies and stronger anti-sintering of Ni particles derived from the enhanced metal–support interactions. Even after testing at 750 °C for 140 h, the catalyst maintained a CH4 conversion of 86% and CO selectivity of 94%. Besides, Ru nanoparticles supported over Ce0.5Zr0.5O2 nanorods obtained by Das et al.120 were proven to be more active than Ru/CeO2 and Ru/ZrO2. Specifically, the stronger metal–support interactions and higher oxygen storage capacity of the CeO2–ZrO2 solid solution contributed to the increased catalytic performance. Innovatively, Singha et al.118 reported 3D Pt-CeO2 nanoporous spheres with a bimodal pore structure for the partial oxidation of methane. The unique bimodal pore structure was confirmed by an N2-adsorption study, which resulted in a high surface area. The as-prepared Pt–CeO2 catalysts were highly active and selective for the production of syngas (TOF at 400 °C = 1.3 × 103 s−1, CO selectivity = 50.0%). Briefly, the unique bimodal pore structure resulted in a high surface area and metal–support interactions as well as abundant oxygen vacancies, contributing to the superior catalytic performance for the partial oxidation of methane to produce syngas.
In the case of other rare earth oxides used in the partial oxidation of methane besides CeO2, Ferreira et al.116 tested the catalytic performance of LnNi (Ln = Pr, Gd, Lu) binary oxides. They found that NiO–Gd2O3 and NiO–Lu2O3 exhibited much higher activity than that of NiO–Pr2O3, which was even comparable with that of the commercial 5% Pt/Al2O3 catalyst. The authors believed that there was an unusual synergistic effect between the two oxides, resulting in high activity and selectivity for H2 and CO. Similarly, Liu et al.117 developed another Ni-based catalyst, Ni/Y2O3, for methane oxidation conversion to syngas. They controlled the structural effect by tuning the calcination temperature of the catalysts (Fig. 19a–c), and showed that Ni/Y2O3 calcined at 500 °C had the highest activity and high anti-carbon deposition ability (Fig. 19d–f). All the Ni/Y2O3 catalysts showed an extremely low signal in the NH3-TPD profiles (Fig. 19a) because Y2O3 was a basic oxide. Also, the amount of basic sites and reducible oxygen decreased with an increase in the calcination temperature (Fig. 19b and c). The reason for the highest activity of Ni/Y2O3-500 was its appropriate amount of reducible oxygen and moderate interaction between Ni and Y2O3, and both factors were controlled by the calcination temperature. Importantly, Choudhary et al.121 studied the catalytic properties of Pt- and Pd-based alkaline and rare earth oxide catalysts for the partial oxidation of methane. Among the Pt (Pd)/MgO (CaO, La2O3, Pr6O11, Nd2O3, Sm2O3, Gd2O3, Dy2O3, and Er2O3) catalysts, Pt/Gd2O3 and Pd/Sm2O3 showed the best performance with a CO productivity of 9.5 and 7.9 mol g−1 h−1, respectively. Besides, both catalysts had high CO selectivity but low H2 selectivity due to the RWGS reaction. These results also indicated that the rare earth oxides not only acted as supports, but also played an essential role in deciding the activity and selectivity.
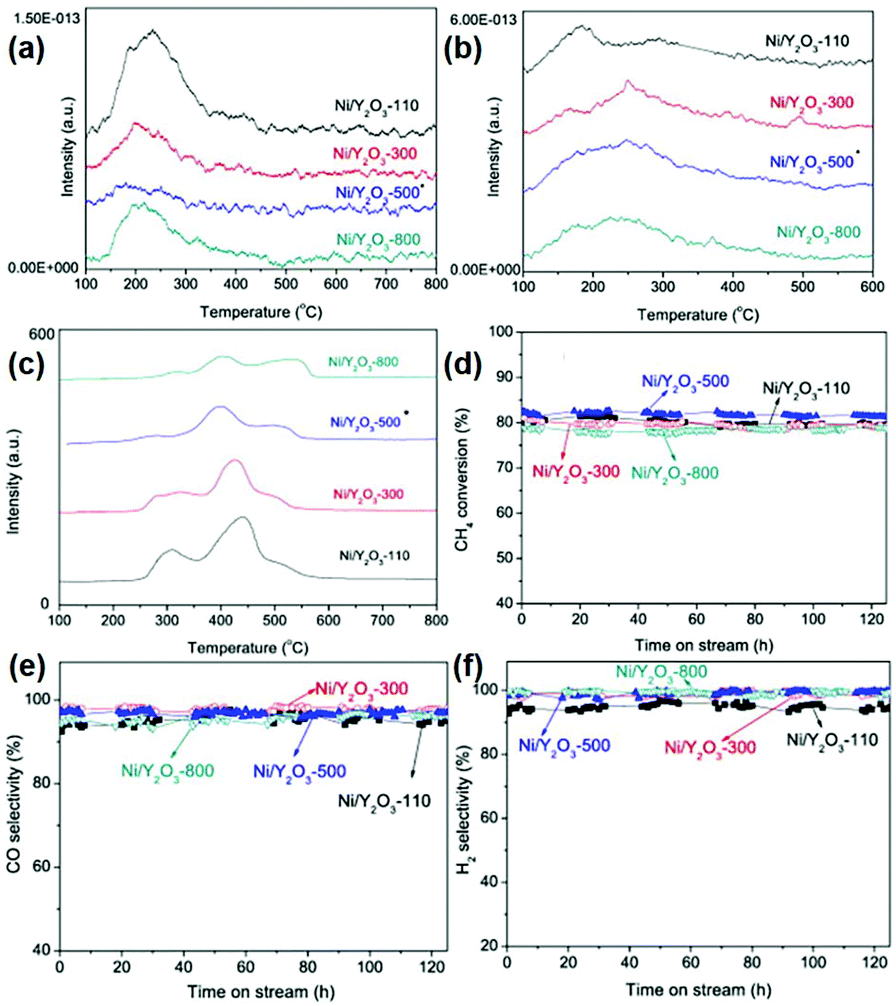 | ||
| Fig. 19 (a) NH3-TPD profiles of the Ni/Y2O3 samples. (b) CO2-TPD profiles of the Ni/Y2O3 samples. (c) TPR profiles of the Ni/Y2O3 samples. Catalytic performance of the Ni/Y2O3 samples in the partial oxidation of methane: (d) CH4 conversion; (e) CO selectivity; and (f) H2 selectivity. Adapted with permission from ref. 117. Copyright 2014, Elsevier. | ||
In many cases, the total and partial oxidation of methane occurs simultaneously; therefore, it is a challenge to control the product selectivity. Different rare earth oxide nanomaterials may have different preferences towards the two reactions. Thus, researchers can realize diverse goals by choosing suitable rare earth oxides with designed well-defined structures as a support or promotor.
4.5 MOR
The MOR discussed below refers to the anode reaction of direct methanol fuel cells (DMFCs), which is very important for alleviating the energy crisis since it can directly transform chemical energy to electrical energy.122–125 Besides, methanol has a high energy density and is easier to store and transport compared with hydrogen. The catalytic application of rare earth oxide nanomaterials in the MOR is not as extensive as in other C1 chemical reactions such as the CO oxidation and CO2 hydrogenation reactions because of their weak conductivity. Although it is difficult for rare earth oxide nanomaterials to be used as a single catalyst in MOR, there are many cases that combine rare earth oxide nanomaterials with other well-conducting catalysts for the MOR.126–133 This not only can enhance the electronic metal support interactions, but also tune the chemical adsorption of the reactants and intermediates. As an example, Li et al.126 studied the introduction of Y2O3 on the MOR activity of the Pd/C catalyst. A 1.5 times higher specific activity (SA = 145 mA cm−2) was observed with the assistance of Y2O3 due to the relatively well-dispersed Pd nanoparticles, enhanced electrochemically active surface area and the synergistic interaction between Pd and Y2O3.In the case that the role of rare earth oxides is a promotor in the MOR, a well-defined structure can also have a positive influence on the catalytic performance. As a good example, Feng et al.33 synthesized three types of well-defined CeO2 nanostructures containing nanooctahedra, nanospheres and nanocubes as promotors for Pt catalysts. Consequently, the Pt catalyst decorated with CeO2 nanospheres exhibited the best performance in both activity and stability. The explanation for the enhanced activity could be divided into two parts. One was that the loosened structure of nanospheres was beneficial for the dispersion of Pt particles, thus generating a strong physical interaction between Pt and CeO2. The other was the oxygen vacancies, which were the most abundant in the nanospheres. The rich oxygen vacancies led to the transfer of surplus electrons from CeO2 to Pt, and thereby increased the intrinsic activity of Pt. Besides, Wang et al.129 investigated the effect of the introduction of La2O3 in the Pd/C catalyst for the MOR. As can be seen in Fig. 20a, the Pd nanoparticles (ca. 2.6 nm) were uniform and well dispersed. Compared with the Pd/C catalyst, Pd–La2O3/C exhibited a lower onset potential and higher current density (Fig. 20b). The XPS results indicated that the introduction of La2O3 resulted in the formation of more metallic Pd species (Fig. 20c and d), resulting in enhanced activity. Furthermore, the authors loaded Pd–La2O3 on chitosan-functionalized carbon nanotubes and obtained a better performance than that with Pd–La2O3/C. In addition, a 3D core–shell nanocatalyst of Au@CeO2@Pt/C for the MOR was obtained by Dao et al.130 through a facile hydrothermal route. The as-prepared catalyst showed much higher activity (MA = 1360 mA mgPt−1) and durability than that of commercial Pt/C (MA = 670 mA mgPt−1) and CeO2@Pt/C (MA = 920 mA mgPt−1) owing to the electronic and synergistic effects among Au, CeO2 and Pt, which could easily remove the poisoner of CO intermediates (Fig. 20e).
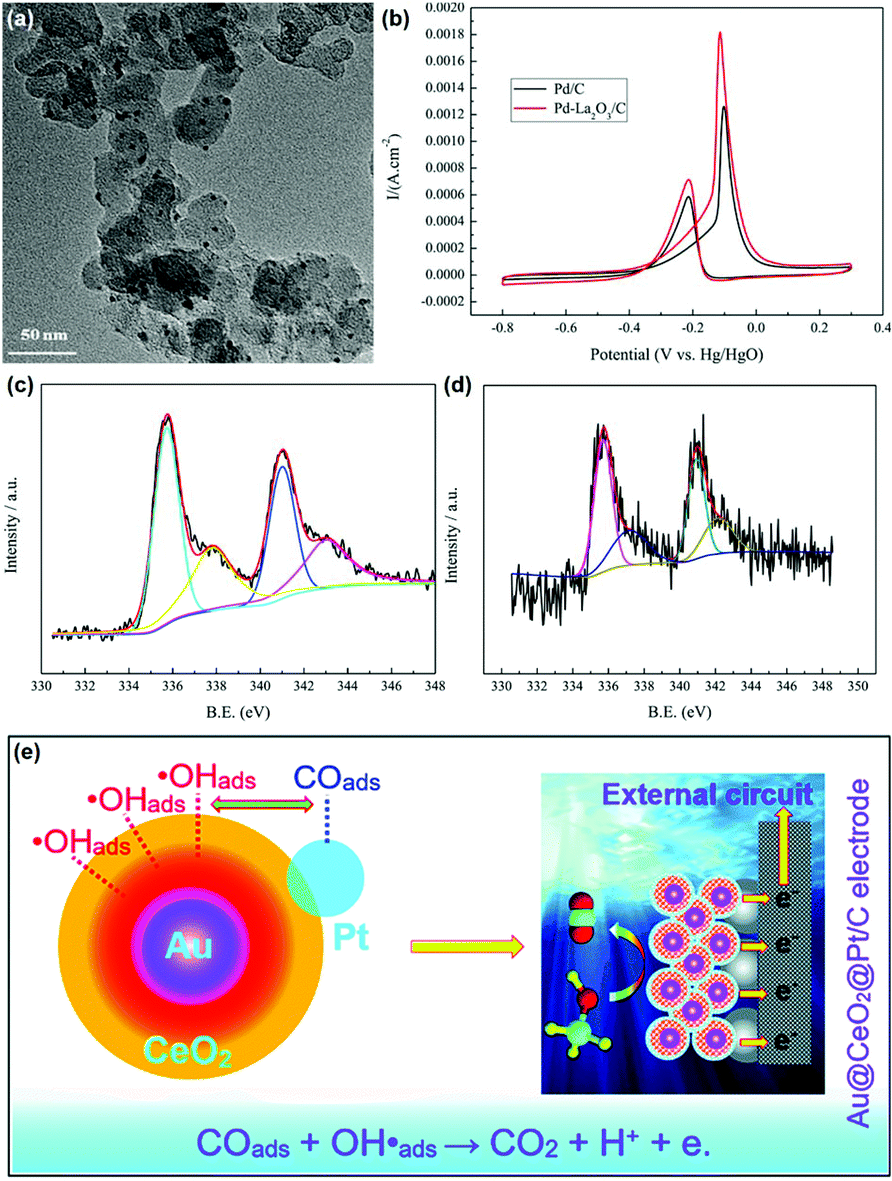 | ||
| Fig. 20 (a) TEM image of Pd–La2O3/C. (b) Cyclic voltammograms of Pd/C and Pd–La2O3/C in 1 M KOH + 1 M CH3OH. XPS spectra of Pd 3d for (c) Pd/C and (d) Pd–La2O3/C. Adapted with permission from ref. 129 Copyright 2019, Elsevier. (e) Schematic illustration of the possible mechanism of Au@CeO2@Pt/C for the MOR. Adapted with permission from ref. 130. Copyright 2019, Royal Society of Chemistry. | ||
When rare earth oxides are used as a support in the MOR, efforts must be made to improve their conductivity. For example, CeO2 nanorod-supported Pt catalysts were prepared by Tao et al.131 and used for the MOR. They used plasma etching of the CeO2 nanorods to modify the surface structure of the CeO2 support (denoted as CeO2-P), and used the untreated CeO2 as a comparison sample (Fig. 21a). The authors found that the abundant oxygen vacancies in Pt/CeO2-P played a significant role in the enhanced activity (MA = 714 mA mgPt−1) and stability compared with the sample without plasma etching (MA = 164 mA mgPt−1) (Fig. 21b and c), which influenced the interactions between Pt and CeO2 and enriched the electronic density of Pt, resulting in the enhanced conductivity of CeO2 and MOR activity. As another example, Tang et al.133 combined the rare earth oxides of Pr and Ce with carbon black to support Pt species for the MOR. They found that the introduction of Pr2O3 and CeO2 significantly improved the catalytic performance (Fig. 21d and e), and the ratio of Pr/Ce had a crucial effect on the activity. According to their observations, Pt/Pr1Ce1Oz-C exhibited the highest activity (SA = 118.3 mA cm−2) and best stability among the samples. The abundant surface oxygen-containing species as well as the high oxygen mobility derived from Pr2O3 and CeO2 contributed to the enhanced CO-tolerance and catalytic performance.
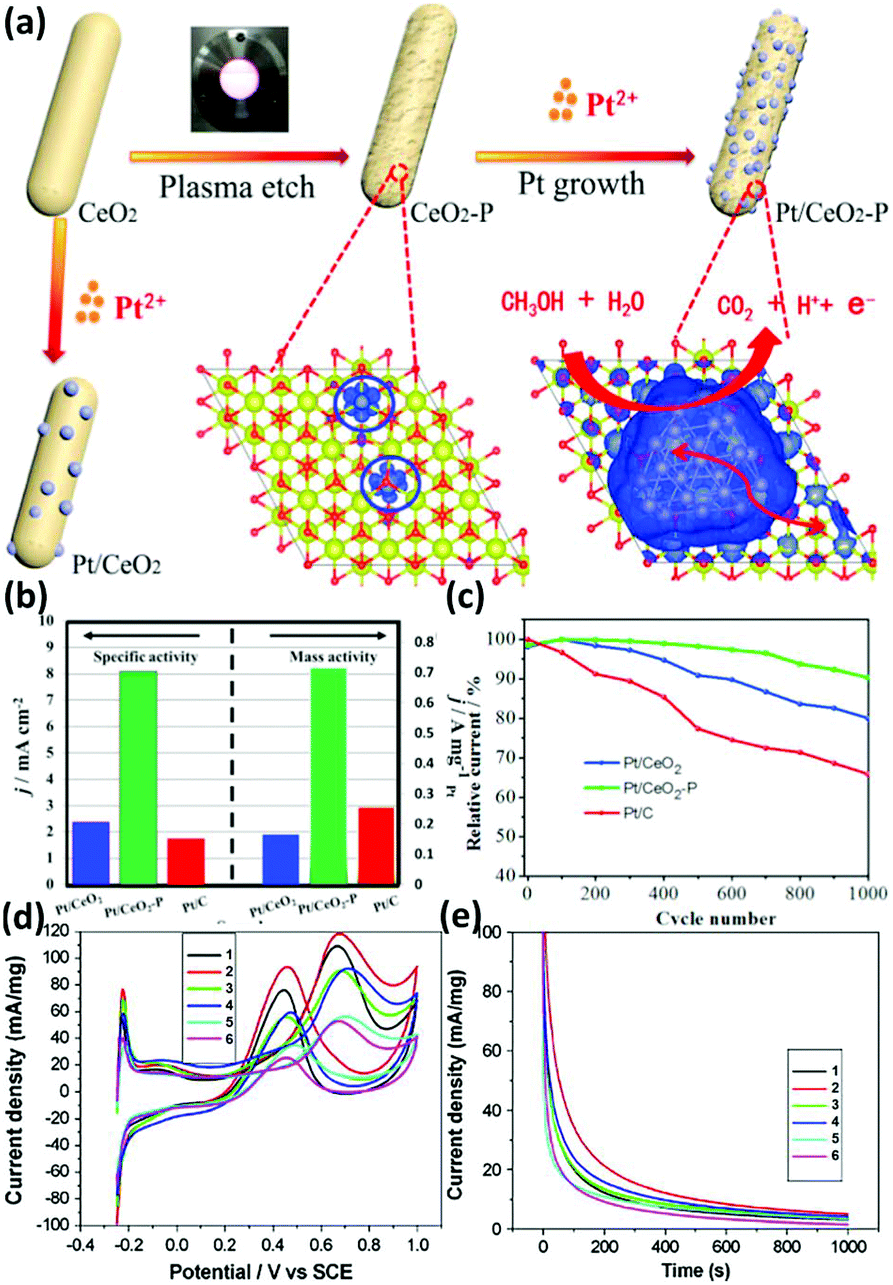 | ||
| Fig. 21 (a) Schematic illustration of the mechanism of the Pt/CeO2 catalyst for the MOR. (b) Mass activity and specific activity for the MOR. (c) Stability test for the MOR. Adapted with permission from ref. 131. Copyright 2018, Elsevier. (d) Cyclic voltammograms and (e) chronoamperometry curves at 0.65 V (vs. SCE) for Pt/C and Pt/RE-C catalysts: Sample 1, Pt/Pr3Ce1Oz-C; Sample 2, Pt/Pr1Ce1Oz-C; Sample 3, Pt/Pr1Ce3Oz-C; Sample 4, Pt3/PrOx-C; Sample 5, Pt3/CeOx-C; and Sample 6, Pt/C. Adapted with permission from ref. 133. Copyright 2008, Elsevier. | ||
In summary, although rare earth oxide nanomaterials have poor conductivity, which limits their activity for the MOR, they are widely used as a promotor or support for the rational engineering of their structures in the MOR since they can enrich the electronic density and improve the CO tolerance through electronic and synergistic effects.
5. Summary and perspective
In this review, we introduced the structural features of the rare earth oxides and summarized the engineering of rare earth oxide nanomaterials with well-defined structures mainly considering their morphology. Then we chose some representative C1 chemical reactions including CO oxidation, water gas-shift reaction, CO2 hydrogenation reaction, methane oxidation reaction and methanol oxidation reaction to summarize the structure–function correlation of rare earth oxide-based nanomaterials applied in catalytic applications. We also presented the reported works in Table 1, including their preparation methods and catalytic properties. In summary, the role of well-defined rare earth oxide-based catalysts in C1 chemical reactions mainly involves three aspects: (1) providing stable sites for the loading and dispersion of other metal catalysts, (2) providing strong metal support interaction and enhancing the electron density of the metal loading, and (3) providing sites for the adsorption and activation of small molecules such as H2O, O2 and CO2. There is no doubt that rare earth oxide-based catalysts play a significant role in C1 chemical reactions, but there are also many problems that need to be solved.| Catalyst | Synthetic route | Application | Conditions | Performance | Key factors in catalysis | Ref. |
|---|---|---|---|---|---|---|
| Ln-Doped CeO2 nanowires (Ln = La–Lu) | Hydrothermal method | CO oxidation reaction | 1% CO–20% O2–He | TOF at 200 °C = 5.4 × 10−4 s−1 | Doping effect | 35 |
| Total speed: 40 mL min−1 | ||||||
| Pr6O11 | Hard template method | CO oxidation reaction | 1% CO–20% O2–N2 | T 50 = 305 °C | Surface area | 75 |
| SV = 10000 mL g−1 h−1 |
Oxygen vacancy | |||||
| Tb4O7 | Hard template method | CO oxidation reaction | 1% CO–20% O2–N2 | T 50 = 360 °C | Surface area | 75 |
| SV = 10000 mL g−1 h−1 |
Oxygen vacancy | |||||
| PtOx nanoclusters/CeO2 | Hydrothermal method | CO oxidation reaction | 13.3% CO–33.3% O2–53.3% He | TOF at 50 °C = 1.3 × 10−2 s−1 | Suitable Pt–O coordination number | 77 |
| Total speed: 15 mL min−1 | ||||||
| Au/Pr6O11 nanorods | Hydrothermal method | CO oxidation reaction | 1% CO–Air | T 100 = 140 °C | Synergistic effect | 78 |
| Total speed: 33.6 mL min−1 | ||||||
| Ag/Pr6O11 nanorods | Hydrothermal method | CO oxidation reaction | 1% CO–20% O2–Ar | T 100 = 210 °C | Surface area | 79 |
| Total speed: 50 mL min−1 | Oxygen vacancy | |||||
| Au/CeO2 nanorods | Hydrothermal method | WGSR | 2% CO–10% H2O–He | T 50 ∼ 190 °C | Plane effect | 87 |
| Total speed: 70 mL min−1 | ||||||
| Au nanoclusters/CeO2 | Hydrothermal method | WGSR | 2% CO–12% H2O–N2 | Rate at 200 °C ∼ 5.0 × 102 mol gAu−1 s−1 | Bridged –OH and Auδ+ | 15 |
| Total speed: 30 mL min−1 | ||||||
| Pt/Ce0.6Y0.4O2 | Sol–gel method | WGSR | 5% CO–15% H2O–N2 | TOF at 250 °C = 0.55 s−1 | Reducibility of the support | 86 |
| WHSV = 30000 mL gcat−1 h−1 |
||||||
| Au/La2O3 | Hydrolysis method | WGSR | 10% CO–3% H2O–He | T 50 ∼ 375 °C | Oxidation state of Au | 13 |
| Au/La2O2SO4 | Surfactant-assisted technique | WGSR | 10% CO–3% H2O–He | T 50 ∼ 350 °C | Oxidation state of Au | 13 |
| CuO/CeO2–La2O3 | Co-precipitation method | WGSR | 5% CO–30% H2–6% CO2–50% H2O–N2 | T 90 ∼ 225 °C | Oxidation state of Cu and oxygen vacancy | 88 |
| Total speed: 207.4 mL min−1 | ||||||
| Cu/CeO2 nanorods | Hydrothermal method | RWGS | 10% CO2–50% H2–He | Rate at 250 °C = 1.8 μmol gcat−1 s−1 | CeO2 (110) facets | 94 |
| Total speed: 20 mL min−1 | ||||||
| Ni/La2O3 | Sol–gel method | RWGS | 50% CO2–50% H2 | ∼100 °C: CO2 conversion = 51.0%; CO yield = 39.1% | Ni dispersion and SMSI | 95 |
| Total speed: 20 mL min−1 | ||||||
| Ru nanoclusters/CeO2 | Hydrothermal method | CO2 methanation | 5% CO2–20% H2–He | TOF at 190 °C = 7.4 × 10−3 s−1 | SMSI and H-spillover | 96 |
| Total speed: 40 mL min−1 | Selectivity ∼100% | |||||
| Ni–Sm2O3 | Sol–gel method | CO2 methanation | 10% CO2–40% H2–Ar | T 100 ∼ 100 °C | Smaller size and more perimeter sites | 97 |
| Total speed: 50 mL min−1 | Selectivity ∼100% | |||||
| Co/Gd2O3 | Impregnation method | CO2 methanation | 10% CO2–90% H2 | TOF at 300 °C = 1.3 min−1 | Reducibility | 98 |
| Total speed: 75 mL min−1 | Selectivity ∼80% | |||||
| Cu/La2O2CO3 nanorods | Deposition–precipitation method | CO2 hydrogenation to methanol | 24% CO2–72% H2–N2 3.0 MPa | TOF at 240 °C = 7.0 × 10−3 s−1 | Cuδ+ and enhanced CO2 adsorption | 103 |
| Selectivity = 92.5% | ||||||
| La2O3 doped CuO/ZnO/ZrO2 | Co-precipitation method | CO2 hydrogenation to methanol | 25% CO2–75% H2 3.0 MPa | CH3OH productivity at 230 °C = 2.7 mol kgcat−1 h−1 | Oxygen vacancy | 104 |
| Selectivity = 49.8% | ||||||
| La2O3–CuO/ZnO/Al2O3 | Co-precipitation method | CO2 hydrogenation to methanol | 10% CO2–90% H2 | 210 °C: CH3OH yield = 0.9%; selectivity ∼40% | Enhanced adsorption of CO2 | 105 |
| Total speed: 50 mL min−1 | ||||||
| La·Nd–PdO/Al2O3 | Impregnation method | Methane combustion reaction | 1% CH4/Ar | TOF at 850 °C = 4.8 mol g−1 s−1 | Stabilizing Pd species | 113 |
| La·Nd–PdO/ZrO2·Al2O3 | Impregnation method | Methane combustion reaction | 1% CH4/Ar | Rate at 800 °C = 6.9 × 102 μmol g−1 s−1 | Stabilizing Pd species | 114 |
| Pd–CeO2 | Ball milling method | Methane combustion reaction | 0.5% CH4–2% O2–He | T 50 ∼ 350 °C | Structural effect | 115 |
| GHSV = 200000 h−1 |
||||||
| NiO–Gd2O3 | Controlled oxidation of the intermetallic compounds | Partial oxidation of methane | 28% CH4–14% O2–N2 | T 50 ∼ 625 °C | Synergistic effect | 116 |
| GHSV = 8500 mL g−1 h−1 | ||||||
| Ni/Y2O3 | Precipitation method | Partial oxidation of methane | 66.7% CH4–33.3% O2 | CH4 conversion at 700 °C ∼ 82%; CO and H2 selectivity at 700 °C ∼ 90% | Basic sites and reducible oxygen | 117 |
| Total speed: 165 mL min−1 | ||||||
| Pt/Gd2O3 | Impregnation method | Partial oxidation of methane | 64% CH4–36% O2 | CO productivity at 700 °C = 9.5 mol g−1 h−1 | Basic sites | 121 |
| SV = 5 × 105 mL g−1 h−1 | ||||||
| Pd/Sm2O3 | Impregnation method | Partial oxidation of methane | 64% CH4–36% O2 | CO productivity at 700 °C = 7.9 mol g−1 h−1 | Basic sites | 121 |
| SV = 5 × 105 mL g−1 h−1 | ||||||
| Pd–Y2O3/C | Microwave-assisted reduction | MOR | 1 M CH3OH + 0.5 M KOH | ECSA = 70 m2 g−1 | Enhanced ECSA and synergistic effect | 126 |
| SA: 145 mA cm−2 | ||||||
| Pd–La2O3/C | Reduction reaction | MOR | 1 M CH3OH + 1 M KOH | SA: 1.8 mA cm−2 | Metallic Pd | 129 |
| Au@CeO2@Pt | Hydrothermal method | MOR | 1 M CH3OH + 0.25 M H2SO4 | ECSA = 80 m2 g−1 | Electronic, bifunctional and synergistic effects | 130 |
| MA: 1360 mA mgPt−1 | ||||||
| Pt/CeO2 | Hydrothermal method plasma etch | MOR | 1 M CH3OH + 0.5 M H2SO4 | MA: 714 mA mgPt−1 | Oxygen vacancy | 131 |
| Pt/Pr1Ce1Oz-C | Precipitation method | MOR | 0.5 M CH3OH + 0.5 M H2SO4 | SA: 118.3 mA cm−2 | Surface oxygen-containing species and oxygen mobility | 133 |
Firstly, optimized synthesis strategies still need to be developed for the large-scale and low-cost production of rare earth oxide materials with high performances. Based on the current developed synthetic routes, on the one hand, methods such as thermal decomposition and precipitation are low-cost and efficient, but they are not suitable for designing and obtaining nanostructures with a specific morphology, which may result in desirable properties. On the other hand, routes such as the hydrothermal and sol–gel methods are suitable for the preparation of functional nanomaterials, but they cannot guarantee their large production and low cost. Besides, the role of rare earth oxides themselves in the catalytic reaction needs a deeper and clearer understanding. Many reported works have demonstrated that rare earth oxides exhibit catalytic activity for C1 chemical reactions. However, when the rare earth oxides are used as a support to form composite materials with other metal or metal oxides, it is not clear if they only work as support or they also provide active sites. In addition, the defects in rare-earth oxides also play a crucial role in C1 chemical reactions, such as stabilizing the active metal loading, providing sites for the adsorption of reactant molecules such as H2O and CO2, and promoting the electron transfer between the support and active metals, especially oxygen defects. However, other defects such as dislocation and grain boundaries in rare earth oxides are rarely discussed in C1 chemical reactions, and thus more research is needed to fill this blank. Furthermore, most of the characterizations of rare earth oxide-based catalysts are restricted to the ex situ level. Nevertheless, the surface structure of the catalyst always changes in the reaction process, and there are many intermediate species generated that disappear and cannot be observed by ex situ techniques. Consequently, in situ/operando characterizations combined with theoretical simulation need to be performed to uncover the real reaction mechanism behind the catalytic reaction. Finally, to date, most of the research on rare earth oxide-based catalysts for C1 chemical reactions has focused on CeO2, but the catalytic properties of other rare earth oxides need to be explored to obtain more novel catalysts with high performances. Also, the powerful function of computational simulation in explaining and predicting the catalytic properties of rare earth oxides should be noted. There is also a problem that most computational works on rare earth oxides are based on CeO2 nanomaterials. However, using theoretical computation to predict properties may result in the development of more non-ceria rare earth oxides nanostructures with high catalytic performances. We believe that all of these problems will be addressed in the future, and rare earth oxide-based nanomaterials will shine brightly at the stage of catalytic applications in C1 chemical reactions.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (no. 21832001 and 21771009), and the National Key Research and Development Program of China (no. 2016YFB0701100).Notes and references
- H. Huang and J. J. Zhu, The electrochemical applications of rare earth-based nanomaterials, Analyst, 2019, 144, 6789–6811 Search PubMed.
- A. R. Richard and M. Fan, Rare earth elements: properties and applications to methanol synthesis catalysis via hydrogenation of carbon oxides, J. Rare Earths, 2018, 36, 1127–1135 Search PubMed.
- W. Zhan, Y. Guo, X. Gong, Y. Guo, Y. Wang and G. Lu, Current status and perspectives of rare earth catalytic materials and catalysis, Chin. J. Catal., 2014, 35, 1238–1250 Search PubMed.
- W. Gao, D. Wen, J. C. Ho and Y. Qu, Incorporation of rare earth elements with transition metal-based materials for electrocatalysis: a review for recent progress, Mater. Today Chem., 2019, 12, 266–281 Search PubMed.
- Y. Guo and Y.-W. Zhang, Research advances on rare earth-based heterogeneous catalysts in transformation reactions of small molecules, J. Rare Earths, 2017, 035, 55–68 Search PubMed.
- G. A. M. Hussein, Rare earth metal oxides: formation, characterization and catalytic activity thermoanalytical and applied pyrolysis review, J. Anal. Appl. Pyrolysis, 1996, 37, 111–149 Search PubMed.
- Y. C. Cao, Synthesis of square gadolinium-oxide nanoplates, J. Am. Chem. Soc., 2004, 126, 7456–7457 Search PubMed.
- H.-X. Mai, L.-D. Sun, Y.-W. Zhang, R. Si, W. Feng, H.-P. Zhang, H.-C. Liu and C.-H. Yan, Shape-selective synthesis and oxygen storage behavior of ceria nanopolyhedra, nanorods, and nanocubes, J. Phys. Chem. B, 2005, 109, 24380–24385 Search PubMed.
- R. Si, Y.-W. Zhang, L.-P. You and C.-H. Yan, Rare-earth oxide nanopolyhedra, nanoplates, and nanodisks, Angew. Chem., Int. Ed., 2005, 44, 3256–3260 Search PubMed.
- R. Si, Y.-W. Zhang, H.-P. Zhou, L.-D. Sun and C.-H. Yan, Controlled-synthesis, self-assembly behavior, and surface-dependent optical properties of high-quality rare-earth oxide nanocrystals, Chem. Mater., 2007, 19, 18–27 Search PubMed.
- H.-P. Zhou, Y.-W. Zhang, H.-X. Mai, X. Sun, Q. Liu, W.-G. Song and C.-H. Yan, Spontaneous organization of uniform CeO2 nanoflowers by 3D oriented attachment in hot surfactant solutions monitored with an in situ electrical conductance technique, Chem. – Eur. J., 2008, 14, 3380–3390 Search PubMed.
- W. Gao, Z. Zhang, J. Li, Y. Ma and Y. Qu, Surface engineering on CeO2 nanorods by chemical redox etching and their enhanced catalytic activity for CO oxidation, Nanoscale, 2015, 7, 11686–11691 Search PubMed.
- J. D. Lessard, I. Valsamakis and M. Flytzani-Stephanopoulos, Novel Au/La2O3 and Au/La2O2SO4 catalysts for the water-gas shift reaction prepared via an anion adsorption method, Chem. Commun., 2012, 48, 4857–4859 Search PubMed.
- X. Yang, X. Cheng, J. Ma, Y. Zou, W. Luo and Y. Deng, Large-pore mesoporous CeO2-ZrO2 solid solutions with in-pore confined Pt nanoparticles for enhanced CO oxidation, Small, 2019, 15, e1903058 Search PubMed.
- X. P. Fu, L. W. Guo, W. W. Wang, C. Ma, C. J. Jia, K. Wu, R. Si, L. D. Sun and C. H. Yan, Direct identification of active surface species for the water-gas shift reaction on a gold-ceria catalyst, J. Am. Chem. Soc., 2019, 141, 4613–4623 Search PubMed.
- C. Mesters, A Selection of Recent Advances in C1 chemistry, Annu. Rev. Chem. Biomol. Eng., 2016, 7, 223–238 Search PubMed.
- P. Li, F. Yu, N. Altaf, M. Zhu, J. Li, B. Dai and Q. Wang, Two-dimensional layered double hydroxides for reactions of methanation and methane reforming in C1 chemistry, Materials, 2018, 11, 221 Search PubMed.
- G. Chen, G. I. N. Waterhouse, R. Shi, J. Zhao, Z. Li, L. Z. Wu, C. H. Tung and T. Zhang, From solar energy to fuels: recent advances in light-driven C1 chemistry, Angew. Chem., Int. Ed., 2019, 58, 17528–17551 Search PubMed.
- J. Bao and N. Tsubaki, Design and synthesis of powerful capsule catalysts aimed at applications in C1 chemistry and biomass conversion, Chem. Rec., 2018, 18, 4–19 Search PubMed.
- W. Keim, Catalysis in C1 Chemistry, Reidel Publishing Company, 1983 Search PubMed.
- W.-G. Cui, G.-Y. Zhang, T.-L. Hu and X.-H. Bu, Metal-organic framework-based heterogeneous catalysts for the conversion of C1 chemistry: CO, CO2 and CH4, Coord. Chem. Rev., 2019, 387, 79–120 Search PubMed.
- C. Sun, H. Li and L. Chen, Nanostructured ceria-based materials: synthesis, properties, and applications, Energy Environ. Sci., 2012, 5, 8475–8505 Search PubMed.
- D. Zhang, X. Du, L. Shi and R. Gao, Shape-controlled synthesis and catalytic application of ceria nanomaterials, Dalton Trans., 2012, 41, 14455–14475 Search PubMed.
- N. Ta, J. Liu and W. Shen, Tuning the shape of ceria nanomaterials for catalytic applications, Chin. J. Catal., 2013, 34, 838–850 Search PubMed.
- Z.-A. Qiao, Z. Wu and S. Dai, Shape-controlled ceria-based nanostructures for catalysis applications, ChemSusChem, 2013, 6, 1821–1833 Search PubMed.
- W. Huang and Y. Gao, Morphology-dependent surface chemistry and catalysis of CeO2 nanocrystals, Catal. Sci. Technol., 2014, 4, 3772–3784 Search PubMed.
- K. Wu, L.-D. Sun and C.-H. Yan, Recent progress in well-controlled synthesis of ceria-based nanocatalysts towards enhanced catalytic performance, Adv. Energy Mater., 2016, 6, 1600501 Search PubMed.
- A. Trovarelli and J. Llorca, Ceria catalysts at nanoscale: how do crystal shapes shape catalysis?, ACS Catal., 2017, 7, 4716–4735 Search PubMed.
- Z. Wang and R. Yu, Hollow micro/nanostructured ceria-based materials: synthetic strategies and versatile applications, Adv. Mater., 2019, 31, 1800592 Search PubMed.
- S. Chen, F. Xiong and W. Huang, Surface chemistry and catalysis of oxide model catalysts from single crystals to nanocrystals, Surf. Sci. Rep., 2019, 74, 100471 Search PubMed.
- X. Hao, A. Yoko, C. Chen, K. Inoue, M. Saito, G. Seong, S. Takami, T. Adschiri and Y. Ikuhara, Atomic-scale valence state distribution inside ultrafine CeO2 nanocubes and its size dependence, Small, 2018, 14, e1802915 Search PubMed.
- H. Tan, J. Wang, S. Yu and K. Zhou, Support Morphology-Dependent Catalytic Activity of Pd/CeO2 for Formaldehyde Oxidation, Environ. Sci. Technol., 2015, 49, 8675–8682 Search PubMed.
- Y.-Y. Feng, H.-S. Hu, G.-H. Song, S. Si, R.-J. Liu, D.-N. Peng and D.-S. Kong, Promotion effects of CeO2 with different morphologies to Pt catalyst toward methanol electrooxidation reaction, J. Alloys Compd., 2019, 798, 706–713 Search PubMed.
- K. Zhou, X. Wang, X. Sun, Q. Peng and Y. Li, Enhanced catalytic activity of ceria nanorods from well-defined reactive crystal planes, J. Catal., 2005, 229, 206–212 Search PubMed.
- J. Ke, J. W. Xiao, W. Zhu, H. Liu, R. Si, Y. W. Zhang and C. H. Yan, Dopant-induced modification of active site structure and surface bonding mode for high-performance nanocatalysts: CO oxidation on capping-free (110)-oriented CeO2:Ln (Ln = La-Lu) nanowires, J. Am. Chem. Soc., 2013, 135, 15191–15200 Search PubMed.
- Y. Sohn, Yb2O3 nanowires, nanorods and nano-square plates, Ceram. Int., 2018, 44, 3341–3347 Search PubMed.
- T. Sobahi, Photocatalytic degradation of herbicides under visible light using Ni-Pr2O3 nanocomposites, J. Alloys Compd., 2017, 695, 1279–1284 Search PubMed.
- Y. Huang, B. Long, M. Tang, Z. Rui, M.-S. Balogun, Y. Tong and H. Ji, Bifunctional catalytic material: an ultrastable and high-performance surface defect CeO2 nanosheets for formaldehyde thermal oxidation and photocatalytic oxidation, Appl. Catal., B, 2016, 181, 779–787 Search PubMed.
- Y. Zhang, F. Hou and Y. Tan, CeO2 nanoplates with a hexagonal structure and their catalytic applications in highly selective hydrogenation of substituted nitroaromatics, Chem. Commun., 2012, 48, 2391–2393 Search PubMed.
- Z.-L. Wang, G.-R. Li, Y.-N. Ou, Z.-P. Feng, D.-L. Qu and Y.-X. Tong, Electrochemical deposition of Eu3+-doped CeO2 nanobelts with enhanced optical properties, J. Phys. Chem. C, 2011, 115, 351–356 Search PubMed.
- Q. Dai, S. Bai, H. Li, W. Liu, X. Wang and G. Lu, Catalytic total oxidation of 1,2-dichloroethane over highly dispersed vanadia supported on CeO2 nanobelts, Appl. Catal., B, 2015, 168–169, 141–155 Search PubMed.
- M. Mousavi-Kamazani, R. Rahmatolahzadeh and F. Beshkar, Facile solvothermal synthesis of CeO2-CuO nanocomposite photocatalyst using novel precursors with enhanced photocatalytic performance in dye degradation, J. Inorg. Organomet. Polym. Mater., 2017, 27, 1342–1350 Search PubMed.
- D. Zhang, W. Wu, X. Cao, S. Li, X. Zhang, G. Han, A. Ying, X. Xu and Z. Tong, Fabrication of three-dimensional dendrite-like CeO2 crystallites via simple template-free solution route, J. Phys. Chem. Solids, 2009, 70, 1348–1352 Search PubMed.
- X. Jiang, X. Huang, W. Zeng, J. Huang, Y. Zheng, D. Sun and Q. Li, Facile morphology control of 3D porous CeO2 for CO oxidation, RSC Adv., 2018, 8, 21658–21663 Search PubMed.
- J. Gu, Y.-W. Zhang and F. Tao, Shape control of bimetallic nanocatalysts through well-designed colloidal chemistry approaches, Chem. Soc. Rev., 2012, 41, 8050–8065 Search PubMed.
- J.-W. Yu, W. Zhu and Y.-W. Zhang, Solution synthesis protocols for shaping mixed valent oxide crystalline particles as robust catalytic materials, Inorg. Chem. Front., 2016, 3, 9–25 Search PubMed.
- F. Dang, K. Kato, H. Imai, S. Wada, H. Haneda and M. Kuwabara, Characteristics of CeO2 nanocubes and related polyhedra prepared by using a liquid-liquid interface, Cryst. Growth Des., 2010, 10, 4537–4541 Search PubMed.
- H. Miao, G.-F. Huang, J.-H. Liu, B.-X. Zhou, A. Pan, W.-Q. Huang and G.-F. Huang, Origin of enhanced photocatalytic activity of F-doped CeO2 nanocubes, Appl. Surf. Sci., 2016, 370, 427–432 Search PubMed.
- Z. Ren, F. Peng, J. Li, X. Liang and B. Chen, Morphology-dependent properties of Cu/CeO2 catalysts for the water-gas shift reaction, Catalysts, 2017, 7, 48 Search PubMed.
- X. H. Lu, X. Huang, S. L. Xie, D. Z. Zheng, Z. Q. Liu, C. L. Liang and Y. X. Tong, Facile electrochemical synthesis of single crystalline CeO2 octahedrons and their optical properties, Langmuir, 2010, 26, 7569–7573 Search PubMed.
- J. Yang, L. Lukashuk, H. Li, K. Fottinger, G. Rupprechter and U. Schubert, High surface area ceria for CO oxidation prepared from cerium t-butoxide by combined sol-gel and solvothermal processing, Catal. Lett., 2014, 144, 403–412 Search PubMed.
- H. M. Shiri, A. Ehsani and M. Jalali Khales, Electrochemical synthesis of Sm2O3 nanoparticles: application in conductive polymer composite films for supercapacitors, J. Colloid Interface Sci., 2017, 505, 940–946 Search PubMed.
- Y. Shlapa, V. Sarnatskaya, I. Timashkov, L. Yushko, I. Antal, B. Gerashchenko, I. Nychyporenko, A. Belous, V. Nikolaev and M. Timko, Synthesis of CeO2 nanoparticles by precipitation in reversal microemulsions and their physical-chemical and biological properties, Appl. Phys. A, 2019, 125, 412 Search PubMed.
- G. Wu, L. Zhang, B. Cheng, T. Xie and X. Yuan, Synthesis of Eu2O3 nanotube arrays through a facile sol-gel templete approach, J. Am. Chem. Soc., 2004, 126, 5976–5977 Search PubMed.
- Q. Tang, J. Shen, W. Zhou, W. Zhang, W. Yu and Y. Qian, Preparation, characterization and optical properties of terbium oxide nanotubes, J. Mater. Chem., 2003, 13, 3103–3106 Search PubMed.
- N. Sabari Arul, J. Vidya, V. Ramya and D. Mangalaraj, Synthesis, characterization and electrochemical sensing of Tb2O3 nanotubes, J. Electron. Mater., 2016, 46, 1072–1078 Search PubMed.
- B. Xu, Z. Liu, W. Qiu, Q. Liu, X. Sun, G. Cui, Y. Wu and X. Xiong, La2O3 nanoplate: an efficient electrocatalyst for artificial N2 fixation to NH3 with excellent selectivity at ambient condition, Electrochim. Acta, 2019, 298, 106–111 Search PubMed.
- C. Wang and Y. Wang, Effects of different surfactants on humidity sensing properties of CeO2 nanobelts thin film prepared by hydrothermal method, Int. J. Appl. Ceram. Technol., 2015, 12, E142–E148 Search PubMed.
- H.-L. Lin, C.-Y. Wu and R.-K. Chiang, Facile synthesis of CeO2 nanoplates and nanorods by [100] oriented growth, J. Colloid Interface Sci., 2010, 341, 12–17 Search PubMed.
- Y. Wu, Y. Chen and J. Zhou, La(OH)3 nanorods and La2O3 nanoplates: facile synthesis and photoluminescence properties, Mater. Lett., 2013, 95, 5–8 Search PubMed.
- W. Hu, F. He, X. Chen and S. Liu, Hydrothermal synthesis of leaf-like CeO2 nanosheets and its MnOx/CeO2 composites for catalytic combustion of chlorobenzene, J. Nanopart. Res., 2018, 21, 6 Search PubMed.
- Q. Dai, Z. Zhang, J. Yan, J. Wu, G. Johnson, W. Sun, X. Wang, S. Zhang and W. Zhan, Phosphate-functionalized CeO2 nanosheets for efficient catalytic oxidation of dichloromethane, Environ. Sci. Technol., 2018, 52, 13430–13437 Search PubMed.
- X. Li, Q. Li, Z. Xia, L. Wang, W. Yan, J. Wang and R. I. Boughton, Growth and characterization of single-crystal Y2O3:Eu nanobelts prepared with a simple technique, Cryst. Growth Des., 2006, 6, 2193–2196 Search PubMed.
- R. Rao, M. Yang, Q. Ling, Q. Zhang, H. Liu, A. Zhang and W. Chen, Mesoporous CeO2 nanobelts synthesized by a facile hydrothermal route via controlling cationic type and concentration of alkali, Microporous Mesoporous Mater., 2013, 169, 81–87 Search PubMed.
- J.-M. Li, X.-L. Zeng, Y.-H. Dong and Z.-A. Xu, White-light emission and weak antiferromagnetism from cubic rare-earth oxide Eu2O3 electrospun nanostructures, CrystEngComm, 2013, 15, 2372–2377 Search PubMed.
- H. Xiao, P. Li, F. Jia and L. Zhang, General nonaqueous sol-gel synthesis of nanostructured Sm2O3, Gd2O3, Dy2O3, and Gd2O3:Eu3+ phosphor, J. Phys. Chem. C, 2009, 113, 21034–21041 Search PubMed.
- D.-E. Zhang, X.-J. Zhang, X.-M. Ni, J.-M. Song and H.-G. Zheng, Fabrication of novel threefold shape CeO2 dendrites: optical and electrochemical properties, Chem. Phys. Lett., 2006, 430, 326–329 Search PubMed.
- D. Zhang, F. Li, S. Li, X. Zhang, G. Han, A. Ying and Z. Tong, Synthesis of four-fold shape CeO2 dendrites by a reduction route, Mater. Chem. Phys., 2013, 142, 496–501 Search PubMed.
- S. Qu, Y. Yu, K. Lin, P. Liu, C. Zheng, L. Wang, T. Xu, Z. Wang and H. Wu, Easy hydrothermal synthesis of multi-shelled La2O3 hollow spheres for lithium-ion batteries, J. Mater. Sci.: Mater. Electron., 2017, 29, 1232–1237 Search PubMed.
- X. Dong, X. Cheng, X. Zhang, L. Sui, Y. Xu, S. Gao, H. Zhao and L. Huo, A novel coral-shaped Dy2O3 gas sensor for high sensitivity NH3 detection at room temperature, Sens. Actuators, B, 2018, 255, 1308–1315 Search PubMed.
- Y. Zhou, Z. Wang and C. Liu, Perspective on CO oxidation over Pd-based catalysts, Catal. Sci. Technol., 2015, 5, 69–81 Search PubMed.
- Y. Cui, L. Xu, M. Chen, C. Lv, X. Lian, C.-E. Wu, B. Yang, Z. Miao, F. Wang and X. Hu, CO oxidation over metal oxide (La2O3, Fe2O3, PrO2, Sm2O3, and MnO2) doped CuO-based catalysts supported on mesoporous Ce0.8Zr0.2O2 with intensified low-temperature activity, Catalysts, 2019, 9, 724 Search PubMed.
- H.-P. Zhou, Y.-W. Zhang, R. Si, L.-S. Zhang, W.-G. Song and C.-H. Yan, Dimension-manipulated ceria nanostructures (0D uniform nanocrystals, 2D polycrystalline assembly, and 3D mesoporous framework) from cerium octylate precursor in solution phases and their CO oxidation activities, J. Phys. Chem. C, 2008, 112, 20366–20374 Search PubMed.
- J.-G. Kang, B.-K. Min and Y. Sohn, Physicochemical properties of praseodymium hydroxide and oxide nanorods, J. Alloys Compd., 2015, 619, 165–171 Search PubMed.
- Y. Zhang, J. Deng, H. Zhang, Y. Liu and H. Dai, Three-dimensionally ordered macroporous Pr6O11 and Tb4O7 with mesoporous walls: Preparation, characterization, and catalytic activity for CO oxidation, Catal. Today, 2015, 245, 28–36 Search PubMed.
- X. Li, C. Ni, X. Lu, S. Zuo, W. Liu and C. Yao, In situ fabrication of Ce1−xLaxO2−δ/palygorskite nanocomposites for efficient catalytic oxidation of CO: effect of La doping, Catal. Sci. Technol., 2016, 6, 545–554 Search PubMed.
- J. Ke, W. Zhu, Y. Jiang, R. Si, Y.-J. Wang, S.-C. Li, C. Jin, H. Liu, W.-G. Song, C.-H. Yan and Y.-W. Zhang, Strong local coordination structure effects on subnanometer PtOx clusters over CeO2 nanowires probed by low-temperature CO oxidation, ACS Catal., 2015, 5, 5164–5173 Search PubMed.
- P. X. Huang, F. Wu, B. L. Zhu, G. R. Li, Y. L. Wang, X. P. Gao, H. Y. Zhu, T. Y. Yan, W. P. Huang, S. M. Zhang and D. Y. Song, Praseodymium hydroxide and oxide nanorods and Au/Pr6O11 nanorod catalysts for CO oxidation, J. Phys. Chem. B, 2006, 110, 1614–1620 Search PubMed.
- X. Zhang, S. Cheng, W. Zhang, C. Zhang, N. E. Drewett, X. Wang, D. Wang, S. J. Yoo, J.-G. Kim and W. Zheng, Mechanistic insight into nanoarchitected Ag/Pr6O11 catalysts for efficient CO oxidation, Ind. Eng. Chem. Res., 2017, 56, 11042–11048 Search PubMed.
- M. Lykaki, E. Pachatouridou, S. A. C. Carabineiro, E. Iliopoulou, C. Andriopoulou, N. Kallithrakas-Kontos, S. Boghosian and M. Konsolakis, Ceria nanoparticles shape effects on the structural defects and surface chemistry: Implications in CO oxidation by Cu/CeO2 catalysts, Appl. Catal., B, 2018, 230, 18–28 Search PubMed.
- W.-W. Wang, W.-Z. Yu, P.-P. Du, H. Xu, Z. Jin, R. Si, C. Ma, S. Shi, C.-J. Jia and C.-H. Yan, Crystal plane effect of ceria on supported copper oxide cluster catalyst for CO oxidation: importance of metal-support interaction, ACS Catal., 2017, 7, 1313–1329 Search PubMed.
- W.-H. Chen and C.-Y. Chen, Water gas shift reaction for hydrogen production and carbon dioxide capture: A review, Appl. Energy, 2020, 258, 114078 Search PubMed.
- C. Schilling and C. Hess, Elucidating the role of support oxygen in the water-gas shift reaction over ceria-supported gold catalysts using operando spectroscopy, ACS Catal., 2018, 9, 1159–1171 Search PubMed.
- J. J. Plata, F. Romero-Sarria, J. Amaya Suarez, A. M. Marquez, O. H. Laguna, J. A. Odriozola and J. Fdez Sanz, Improving the activity of gold nanoparticles for the water-gas shift reaction using TiO2-Y2O3: an example of catalyst design, Phys. Chem. Chem. Phys., 2018, 20, 22076–22083 Search PubMed.
- L. G. Pinaeva, E. M. Sadovskaya, Y. A. Ivanova, T. G. Kuznetsova, I. P. Prosvirin, V. A. Sadykov, Y. Schuurman, A. C. van Veen and C. Mirodatos, Water gas shift and partial oxidation of CH4 over CeO2−ZrO2(–La2O3) and Pt/CeO2−ZrO2(–La2O3): performance under transient conditions, Chem. Eng. J., 2014, 257, 281–291 Search PubMed.
- S. M. Lee, G. J. Kim, S. H. Lee, I. H. Hwang, S. C. Hong and S. S. Kim, Catalytic performance of Ce0.6Y0.4O2-supported platinum catalyst for low-temperature water-gas shift reaction, ACS Omega, 2018, 3, 3156–3163 Search PubMed.
- R. Si and M. Flytzani-Stephanopoulos, Shape and crystal-plane effects of nanoscale ceria on the activity of Au-CeO2 Catalysts for the water–gas shift reaction, Angew. Chem., Int. Ed., 2008, 47, 2884–2887 Search PubMed.
- Y. She, Q. Zheng, L. Li, Y. Zhan, C. Chen, Y. Zheng and X. Lin, Rare earth oxide modified CuO/CeO2 catalysts for the water-gas shift reaction, Int. J. Hydrogen Energy, 2009, 34, 8929–8936 Search PubMed.
- C. Chen, Y. Zhan, J. Zhou, D. Li, Y. Zhang, X. Lin, L. Jiang and Q. Zheng, Cu/CeO2 catalyst for water-gas shift reaction: effect of CeO2 pretreatment, ChemPhysChem, 2018, 19, 1448–1455 Search PubMed.
- S. Sangsong, T. Ratana, S. Tungkamani, T. Sornchamni and M. Phongaksorn, Effect of CeO2 loading of the Ce-Al mixed oxide on ultrahigh temperature water-gas shift performance over Ce-Al mixed oxide supported Ni catalysts, Fuel, 2019, 252, 488–495 Search PubMed.
- N. Podrojková, V. Sans, A. Oriňak and R. Oriňaková, Recent developments in the modelling of heterogeneous catalysts for CO2 conversion to chemicals, ChemCatChem, 2020, 12, 1802–1825 Search PubMed.
- M. Boaro, S. Colussi and A. Trovarelli, Ceria-based materials in hydrogenation and reforming reactions for CO2 valorization, Front. Chem., 2019, 7, 28 Search PubMed.
- B. Dai, S. Cao, H. Xie, G. Zhou and S. Chen, Reduction of CO2 to CO via reverse water-gas shift reaction over CeO2 catalyst, Korean J. Chem. Eng., 2018, 35, 421–427 Search PubMed.
- L. Lin, S. Yao, Z. Liu, F. Zhang, N. Li, D. Vovchok, A. Martínez-Arias, R. Castañeda, J. Lin, S. D. Senanayake, D. Su, D. Ma and J. A. Rodriguez, In situ characterization of Cu/CeO2 nanocatalysts for CO2 hydrogenation: morphological effects of nanostructured ceria on the catalytic activity, J. Phys. Chem. C, 2018, 122, 12934–12943 Search PubMed.
- L. Liu, Z. Zhang, S. Das, S. Xi and S. Kawi, LaNiO3 as a precursor of Ni/La2O3 for reverse water-gas shift in DBD plasma: effect of calcination temperature, Energy Convers. Manage., 2020, 206, 112475 Search PubMed.
- Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan and Y.-W. Zhang, Low-temperature CO2 methanation over CeO2-supported Ru single atoms, nanoclusters, and nanoparticles competitively tuned by strong metal–support interactions and H-spillover effect, ACS Catal., 2018, 8, 6203–6215 Search PubMed.
- J. Ilsemann, A. Sonström, T. M. Gesing, R. Anwander and M. Bäumer, Highly active Sm2O3−Ni xerogel catalysts for CO2 methanation, ChemCatChem, 2019, 11, 1732–1741 Search PubMed.
- J. Díez-Ramírez, P. Sánchez, V. Kyriakou, S. Zafeiratos, G. E. Marnellos, M. Konsolakis and F. Dorado, Effect of support nature on the cobalt-catalyzed CO2 hydrogenation, J. CO2 Util., 2017, 21, 562–571 Search PubMed.
- Z. Shi, Q. Tan and D. Wu, Enhanced CO2 hydrogenation to methanol over TiO2 nanotubes-supported CuO-ZnO-CeO2 catalyst, Appl. Catal., A, 2019, 581, 58–66 Search PubMed.
- B. Ouyang, W. Tan and B. Liu, Morphology effect of nanostructure ceria on the Cu/CeO2 catalysts for synthesis of methanol from CO2 hydrogenation, Catal. Commun., 2017, 95, 36–39 Search PubMed.
- Q. Tan, Z. Shi and D. Wu, CO2 hydrogenation to methanol over a highly active Cu–Ni/CeO2−nanotube catalyst, Ind. Eng. Chem. Res., 2018, 57, 10148–10158 Search PubMed.
- Q. Tan, Z. Shi and D. Wu, CO2 hydrogenation over differently morphological CeO2−supported Cu–Ni catalysts, Int. J. Energy Res., 2019, 43, 5392–5404 Search PubMed.
- K. Chen, X. Duan, H. Fang, X. Liang and Y. Yuan, Selective hydrogenation of CO2 to methanol catalyzed by Cu supported on rod-like La2O2CO3, Catal. Sci. Technol., 2018, 8, 1062–1069 Search PubMed.
- H. Ban, C. Li, K. Asami and K. Fujimoto, Influence of rare-earth elements (La, Ce, Nd and Pr) on the performance of Cu/Zn/Zr catalyst for CH3OH synthesis from CO2, Catal. Commun., 2014, 54, 50–54 Search PubMed.
- M. Kourtelesis, K. Kousi and D. I. Kondarides, CO2 hydrogenation to methanol over La2O3-promoted CuO/ZnO/Al2O3 catalysts: a kinetic and mechanistic study, Catalysts, 2020, 10, 183 Search PubMed.
- A. Toso, S. Colussi, S. Padigapaty, C. de Leitenburg and A. Trovarelli, High stability and activity of solution combustion synthesized Pd-based catalysts for methane combustion in presence of water, Appl. Catal., B, 2018, 230, 237–245 Search PubMed.
- P. Lott, M. Eck, D. E. Doronkin, R. Popescu, M. Casapu, J.-D. Grunwaldt and O. Deutschmann, regeneration of sulfur poisoned Pd–Pt/CeO2–ZrO2–Y2O3–La2O3 and Pd–Pt/Al2O3 methane oxidation catalysts, Top. Catal., 2018, 62, 164–171 Search PubMed.
- C. Wen, Y. Liu, Y. Guo, Y. Wang and G. Lu, Strategy to eliminate catalyst hot-spots in the partial oxidation of methane: enhancing its activity for direct hydrogen production by reducing the reactivity of lattice oxygen, Chem. Commun., 2010, 46, 880–882 Search PubMed.
- T. H. Nguyen, A. Łamacz, A. Krztoń, B. Liszka and G. Djéga-Mariadassou, Partial oxidation of methane over Ni0/La2O3 bifunctional catalyst III. Steady state activity of methane total oxidation, dry reforming, steam reforming and partial oxidation. Sequences of elementary steps, Appl. Catal., B, 2016, 182, 385–391 Search PubMed.
- Y. J. O. Asencios, F. C. F. Marcos, J. M. Assaf and E. M. Assaf, Oxidative-reforming of methane and partial oxidation of methane reactions over NiO/PrO2/ZrO2 catalysts: effect of nickel content, Braz. J. Chem. Eng., 2016, 33, 627–636 Search PubMed.
- J. Dou, Y. Tang, L. Nie, C. M. Andolina, X. Zhang, S. House, Y. Li, J. Yang and F. Tao, Complete oxidation of methane on Co3O4/CeO2 nanocomposite: a synergic effect, Catal. Today, 2018, 311, 48–55 Search PubMed.
- A. F. Zedan and A. S. AlJaber, Combustion synthesis of non-precious CuO-CeO2 nanocrystalline catalysts with enhanced catalytic activity for methane oxidation, Materials, 2019, 12, 878 Search PubMed.
- Y. Ozawa, Y. Tochihara, M. Nagai and S. Omi, Effect of addition of Nd2O3 and La2O3 to PdO/Al2O3 in catalytic combustion of methane, Catal. Commun., 2003, 4, 87–90 Search PubMed.
- Y. Ozawa, Y. Tochihara, A. Watanabe, M. Nagai and S. Omi, Stabilizing effect of Nd2O3, La2O3 and ZrO2 on Pt·PdO/Al2O3 during catalytic combustion of methane, Appl. Catal., A, 2004, 258, 261–267 Search PubMed.
- M. Danielis, S. Colussi, C. de Leitenburg, L. Soler, J. Llorca and A. Trovarelli, Outstanding methane oxidation performance of palladium-embedded ceria catalysts prepared by a one-step dry ball-milling method, Angew. Chem., Int. Ed., 2018, 57, 10212–10216 Search PubMed.
- A. C. Ferreira, A. M. Ferraria, A. M. B. do Rego, A. P. Gonçalves, M. R. Correia, T. A. Gasche and J. B. Branco, Partial oxidation of methane over bimetallic nickel–lanthanide oxides, J. Alloys Compd., 2010, 489, 316–323 Search PubMed.
- H. Liu, H. Wu and D. He, Methane conversion to syngas over Ni/Y2O3 catalysts—effects of calcination temperatures of Y2O3 on physicochemical properties and catalytic performance, Fuel Process. Technol., 2014, 119, 81–86 Search PubMed.
- R. K. Singha, A. Shukla, A. Yadav, T. Sasaki, A. Sandupatla, G. Deo and R. Bal, Pt–CeO2 nanoporous spheres – an excellent catalyst for partial oxidation of methane: effect of the bimodal pore structure, Catal. Sci. Technol., 2017, 7, 4720–4735 Search PubMed.
- S. Zhu, X. Lian, T. Fan, Z. Chen, Y. Dong, W. Weng, X. Yi and W. Fang, Thermally stable core–shell Ni/nanorod-CeO2@SiO2 catalyst for partial oxidation of methane at high temperatures, Nanoscale, 2018, 10, 14031–14038 Search PubMed.
- S. Das, R. Gupta, A. Kumar, M. Shah, M. Sengupta, S. Bhandari and A. Bordoloi, Facile synthesis of ruthenium decorated Zr0.5Ce0.5O2 nanorods for catalytic partial oxidation of methane, ACS Appl. Nano Mater., 2018, 1, 2953–2961 Search PubMed.
- V. R. Choudhary, B. Prabhakar, A. M. Rajput and A. S. Mamman, Oxidative conversion of methane to CO and H2 over Pt or Pd containing alkaline and rare earth oxide catalysts, Fuel, 1998, 77, 1477–1481 Search PubMed.
- A. Moura, J. Fajín, M. Mandado and M. Cordeiro, Ruthenium–platinum catalysts and direct methanol fuel cells (DMFC): a review of theoretical and experimental breakthroughs, Catalysts, 2017, 7, 106 Search PubMed.
- E. Antolini, Photo-assisted methanol oxidation on Pt-TiO2 catalysts for direct methanol fuel cells: A short review, Appl. Catal., B, 2018, 237, 491–503 Search PubMed.
- J. S. Spendelow, P. K. Babu and A. Wieckowski, Electrocatalytic oxidation of carbon monoxide and methanol on platinum surfaces decorated with ruthenium, Curr. Opin. Solid State Mater. Sci., 2005, 9, 37–48 Search PubMed.
- A. Ali and P. K. Shen, Recent advances in graphene-based platinum and palladium electrocatalysts for the methanol oxidation reaction, J. Mater. Chem. A, 2019, 7, 22189–22217 Search PubMed.
- J. Li, S. Wang, B. Zhang, W. Wang and L. Feng, Highly efficient methanol electrooxidation catalyzed by co-action of PdY2O3 in alkaline solution for fuel cells, Int. J. Hydrogen Energy, 2017, 42, 12236–12245 Search PubMed.
- Q. Tan, C. Shu, J. Abbott, Q. Zhao, L. Liu, T. Qu, Y. Chen, H. Zhu, Y. Liu and G. Wu, Highly dispersed Pd-CeO2 nanoparticles supported on N-doped core–shell structured mesoporous carbon for methanol oxidation in alkaline media, ACS Catal., 2019, 9, 6362–6371 Search PubMed.
- H. Wang, Y. Xue, B. Zhu, J. Yang, L. Wang, X. Tan, Z. Wang and Y. Chu, CeO2 nanowires stretch-embedded in reduced graphite oxide nanocomposite support for Pt nanoparticles as potential electrocatalyst for methanol oxidation reaction, Int. J. Hydrogen Energy, 2017, 42, 20549–20559 Search PubMed.
- L. Wang, Y. Wang, A. Li, Y. Yang, J. Wang, H. Zhao, X. Du and T. Qi, Electrocatalysis of carbon black- or chitosan-functionalized activated carbon nanotubes-supported Pd with a small amount of La2O3 towards methanol oxidation in alkaline media, Int. J. Hydrogen Energy, 2014, 39, 14730–14738 Search PubMed.
- D. V. Dao, T. D. Le, G. Adilbish, I.-H. Lee and Y.-T. Yu, Pt-loaded Au@CeO2 core–shell nanocatalysts for improving methanol oxidation reaction activity, J. Mater. Chem. A, 2019, 7, 26996–27006 Search PubMed.
- L. Tao, Y. Shi, Y.-C. Huang, R. Chen, Y. Zhang, J. Huo, Y. Zou, G. Yu, J. Luo, C.-L. Dong and S. Wang, Interface engineering of Pt and CeO2 nanorods with unique interaction for methanol oxidation, Nano Energy, 2018, 53, 604–612 Search PubMed.
- Q. Zhou, Z. Pan, D. Wu, G. Hu, S. Wu, C. Chen, L. Lin and Y. Lin, Pt-CeO2/TiN NTs derived from metal organic frameworks as high-performance electrocatalyst for methanol electrooxidation, Int. J. Hydrogen Energy, 2019, 44, 10646–10652 Search PubMed.
- Z. Tang and G. Lu, Synthesis and characterization of high performance Pt-(PrxCeyOz)/C catalysts for methanol electrooxidation, Appl. Catal., B, 2008, 79, 1–7 Search PubMed.
| This journal is © the Partner Organisations 2020 |