
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFine-tuning of two-dimensional metal–organic nanostructures via alkali–pyridyl coordination†
Kun
Zhou
a,
Huifang
Liang
b,
Miao
Wang
c,
Shuaipeng
Xing
a,
Honghe
Ding
d,
Yang
Song
a,
Yuxu
Wang
a,
Qian
Xu
d,
Jing-Hui
He
e,
Junfa
Zhu
 d,
Wei
Zhao
*c,
Yu-qiang
Ma
*af and
Ziliang
Shi
*a
d,
Wei
Zhao
*c,
Yu-qiang
Ma
*af and
Ziliang
Shi
*a
aCenter for Soft Condensed Matter Physics & Interdisciplinary Research, School of Physical Science and Technology, Soochow University, 215006 Suzhou, China. E-mail: phzshi@suda.edu.cn
bInstitute of Functional Nano & Soft Materials (FUNSOM), Soochow University, Suzhou 215123, China
cInstitute for Advanced Study, Shenzhen University, 518060 Shenzhen, China. E-mail: weizhao@szu.edu.cn
dNational Synchrotron Radiation Laboratory, University of Science and Technology of China, 230029 Hefei, China
eCollege of Chemistry, Chemical Engineering and Materials Science, Collaborative Innovation Center of Suzhou Nano Science and Technology, Soochow University, 215123 Suzhou, China
fNational Laboratory of Solid State Microstructures and Department of Physics, Nanjing University, 210093 Nanjing, China. E-mail: myqiang@nju.edu.cn
First published on 8th April 2020
Abstract
Herein, we report a fine-tuning of the two-dimensional alkali–pyridyl coordination assemblies facilely realized by surface reaction between tetrapyridyl-porphyrin molecules and alkali halides on Ag(111) under a solventless ultrahigh vacuum condition. High-resolution scanning tunneling topography and X-ray photoelectron spectra reveal the formation of alkali–pyridyl coordination and the induced conformational tuning of the porphyrin macrocycle cores. Furthermore, employing other different alkali halide substitutes, we demonstrate a fine-tuning of the metal–organic nanostructures at the sub-Å scale. Postdeposition of Fe onto the as-formed precursor layer yields a two-dimensional bimetallic framework structure, manifesting a functionalization of the metal–organic interfaces.
1 Introduction
Bottom-up engineering two-dimensional (2D) metal–organic nanostructures on surfaces has attracted intensive attention, due to the resulting nanostructures promising various chemical/physical novelties customizable at the single-molecule level.1–8 A variety of metal species, including alkali, transition and even lanthanide metals, have been employed to direct the metal–organic assembly.7,8 In particular, alkali metals have unique functions when binding with organic ligands.9–18 The low ionization energy of alkali metals introduces a long-range electrostatic interaction, which can be employed to steer ionic self-assembly with organic ligands.19–21 The resulting structures present large-scale ordering and high thermal stability.9,13,14 For instance, alkali-carboxylate bonded Na-terephthalic acid (TPA) networks on the Cu(100) surface exhibited excellent thermal stability up to 165 °C.13 Moreover, the well-defined periodic trend of alkali metals offers opportunities for precisely manipulating the metal–organic interactions, the structures, and the interface work function for the 2D nanomaterials.11,12,22 For instance, the interfacial dipoles were induced by interactions between alkali metals and TCNQ molecules, demonstrating a fine-tuning of the work-function shift of the metal–organic interface.22 In addition, various alkali metals or salts are widely available and inexpensive, attractive for practical applications.However, the organic ligands explored in the 2D alkali organic salt systems (on metal surfaces) to date are limited to a few species that have been often chosen due to intrinsic electronic negativity (i.e., hard bases) or molecular polarity.9–18 Assemblies of a large library of organic ligands have not been well investigated in such systems. This hinders a comprehensive understanding of such nanostructures, especially when considering that their functionalities are highly associated with the conformational, chemical and electronic characteristics of the organic constituents.
Herein, we describe a self-assembled nanostructure driven by coordination between the alkali ion and a borderline/soft base ligand, pyridyl (py), resulting from the codeposition of py-functionalized porphyrin ligands TPyP (5,10,15,20-tetra(4-pyridyl)-porphyrin) with alkali halides on metal surfaces under a solventless ultrahigh vacuum condition. The well-known intramolecular conformational adaptability and the macrocyclic core of porphyrin-based molecules readily accessible for chemical modifications allow for fine tuning the structures and properties of the resulting assembly.23–37 Employing high-resolution scanning tunneling microscopy (STM) and X-ray photoelectron spectra (XPS), we demonstrate that the alkali–py coordination steers the assemblies of the porphyrin-based molecules. By choosing three other different alkali halide substitutes, we further utilize the alkali–py coordination as a facile protocol to precisely tune the topology of metal–organic assemblies at a sub-Å scale. In addition, following the postdeposition of Fe onto the Na–porphyrin precursor layer, we demonstrate the functionalization of the metal–organic interfaces, as manifested by the Na–Fe bimetallic frameworks resulting from the metalation of porphyrins.
2 Results and discussion
2.1 Determination of the assembly structure and Na–py coordination
TPyP forms a monolayer of close-packed (CP) structures on the pristine Ag(111) surface (Fig. 1a). In the CP domains, molecules appear in a twofold symmetric saddle-shape and thus exhibit two orientations. The same oriented molecules are arranged in linear rows, and every two rows are packed alternatively, constituting an extended monolayer. The unit cell measures a = 1.40 ± 0.02 nm, b = 2.75 ± 0.02 nm and γ = 93 ± 2° (Fig. 1b). This CP structure was previously reported by W. Auwärter et al.,29 where the structure was demonstrated to be stabilized by the intermolecular N⋯H hydrogen bonds. | ||
| Fig. 1 (a) STM overview of the TPyP close-packed (CP) monolayer on a pristine Ag(111) surface and (b) high-resolution image of the unit cell (a = 1.40 ± 0.02 nm, b = 2.75 ± 0.02 nm, γ = 93 ± 2°). Inset in (a), the chemical structure of TPyP. (c) Na–TPyP chessboard (CB) structure. Inset, the fast Fourier transformation (FFT) spectra. (d) High-resolution STM topograph of the CB structure. The unit cell (a = 1.40 ± 0.02 nm, b = 1.45 ± 0.02 nm) is illustrated by four TPyP molecular models (C, gray; N, blue; H, white) interlinked with Na (purple). | ||
In constrast, the deposition of NaCl onto the sample led to a drastic change in the assembly pattern. Fig. 1c provides a typical STM overview displaying a chessboard (CB) structure established by a quasi-square tessellation out of TPyP molecules. The fast Fourier transformation (FFT) of the CB monolayer reveals an orthorhombic pattern; see the inset in Fig. 1c. The islands of the CB structure extend in three different orientations, in alignment with, or ±15° separated from the Ag(111) surface vectors <112(−)>. Similar to the CP structure, the alignment of CB domains with the substrate reflects the weak adsorption of the molecules.29
Similar CB structures were also observed when we reversed the deposition sequence of the assembly constituents; see Fig. S1 in the ESI.† In both deposition sequences, postannealing treatments (293–433 K) promoted the growth of CB islands and increased the coverage of CB structures (given the extra presence of TPyP or NaCl) as well. The resulting CB domains as large as ∼300 nm were visible. These results indicate that the CB structure is a thermodynamically favorable phase when both TPyP and NaCl coexist on Ag(111).
A close inspection of the CB structure (Fig. 1d) reveals twofold symmetric molecular units, similar to those in CP structures, which however exhibit much pronounced intramolecular features (vide infra). The molecules assemble in a side-to-side manner, while each molecule is rotated by 90° with respect to its four nearest neighbors, leading to the formation of a chessboard pattern. Considering that the CB structure emerges exclusively in the presence of NaCl, combining with the well-known repulsion between py endgroups and the coordinative nature of the py terminals, we propose that the CB structure is stabilized by Na–py coordination.
Accordingly, we propose a structural model (Fig. 1d) for the CB structure, in which Na is positioned in between four adjacent TPyP molecules. Based on our high-resolution images, the parameters of a unit cell are determined to be: a = 1.40 ± 0.02 nm, b = 1.45 ± 0.02 nm and γ = 90°. This leads to a projected length of 2.2 ± 0.1 Å for the Na–N bond. Considering that the Na ion may lie out of the molecular plane due to its adsorption on the substrate, a longer bond length is feasible and may fall in the typical Na–N length range (2.4–2.8 Å) derived in bulk.20,21 The alternative rotation of the molecular units can be ascribed to the steric repulsive force between the peripheral H atoms of the four py terminals which are linked at a single Na center.37,38
To reveal the nature of the Na–py interaction, we have conducted XPS experiments, and examined Na 2p and N 1s spectra. In the top panel of Fig. 2a, we show Na 2p spectra of NaCl submonolayers on a Ag(111) surface, which indicate two Na species on the surface. This can be interpreted by the growth mechanism of the NaCl on the Ag(111). NaCl grows with the first adlayer composed of two atomic layers, in which half the Na atoms adsorb on the surface in the 1st layer close to the surface, while the other half of Na atoms reside in the 2nd upper layer.39 Therefore, the smaller peak at 31.9 eV can be assigned to the Na in the 1st layer (labeled Na1) due to the damping effect, and the peak at 31.3 eV (labeled Na2) corresponds to the Na in the other layers on top of the 1st layer.
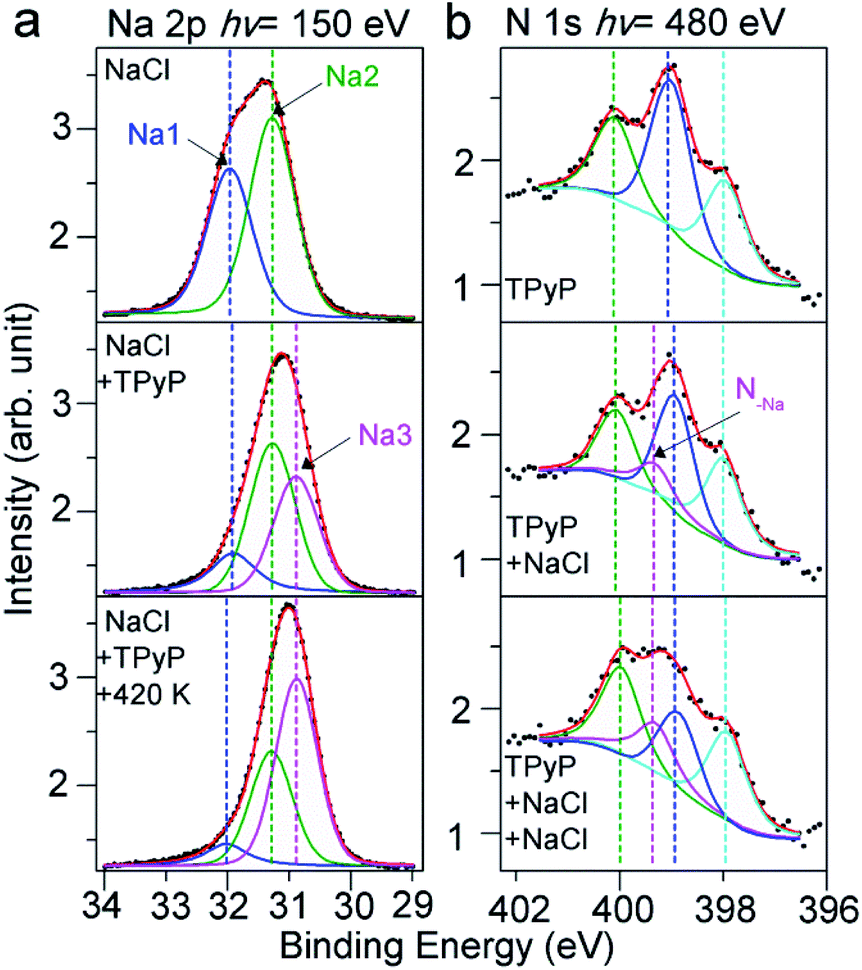 | ||
| Fig. 2 (a) XPS spectra of Na 2p of pure NaCl submonolayers on Ag(111) (top), the spectra after deposition of TPyP (middle) and after the subsequent thermal annealing at 420 K (bottom). (b) XPS spectra of N 1s of pure TPyP submonolayer on Ag(111) (top) and the spectra acquired after stepwise deposition of NaCl (middle and bottom). | ||
After the deposition of TPyP molecules, we observed that in the XPS spectra (Fig. 2a, middle) a new species (labeled Na3) rises at a lower BE (30.9 eV) at the cost of Na1 and Na2 species. The new Na3 at the lower BE indicates a less positively charged state compared to Na1 and Na2 in NaCl, implying a dissociation of the NaCl compound induced by TPyP molecules. The dissociated Na ions coordinate with N in the py ligands, as the model shows in Fig. 1d. The less electronegative affinity of N compared to Cl allows the Na in the Na–N coordination bond to be less positively charged than the Na in Na–Cl ionic bonds, and thus Na3 in Na–N appears at a lower BE. Further annealing (420 K) of this sample resulted in more Na3 species (Fig. 2a, bottom), i.e. more CB assemblies, in agreement with our STM observation.
Our XPS experiments about the N 1s signals have also confirmed the coordination of N with Na. First, we have collected the N 1s spectra for the TPyP submonolayers on Ag(111) in the absence of NaCl. As shown in Fig. 2b (top panel), three peaks at 400.1, 399.0 and 398.0 eV are visible, which are assigned to the pyrrolic, pyridylic, and iminic N atoms, respectively.31,40 After the deposition of NaCl onto the sample, an additional peak at 399.3 eV (labeled N–Na) emerges at the cost of the pyridylic–N (Fig. 2b, middle). Such an observation indicates that the py end groups induce the dissociation of NaCl on Ag(111) and coordinate with Na ions. A deposition of more NaCl further increased the intensity of the N–Na species (Fig. 2b, bottom), in agreement with the evolutions of Na 2p spectra and our STM observations.
The evolution of Cl 2p spectra is shown in Fig. S2 in the ESI.† It is suggested that in the presence of TPyP molecules, Cl ions dissociate from NaCl, adsorb on the Ag(111) surface, and then desorb or diffuse into the substrate during annealing at elevated temperatures later, similar to the phenomena reported previously.13 Our high-resolution XPS data unambiguously demonstrate the dissociation of NaCl on Ag(111) induced by py ligands and the formation of Na–N coordination bonds. Previously, S. Tait et al.14 found the dissociation of NaCl on Cu(100) and the formation of Na–carboxylate ionic bonds. However, what we observed here, i.e., the dissociation of NaCl on metal surfaces under a solventless ultrahigh vacuum condition induced by the coordination bond, has not been reported elsewhere yet. More interestingly, an intramolecular conformational change induced by a Na–N coordination bond has been observed in this work, see the details as given below.
2.2 Intramolecular conformational change due to Na–py coordination
As depicted in a close inspection provided in Fig. 3a, the appearance of TPyP molecules in CB domains distinctly differs from that in CP structures. Under a typical STM tunneling condition (U = −0.9 V and I = 0.1 nA), the molecular unit within CP domains (TPyP∈CP) bears four bright lobes corresponding to its four py end groups (Fig. 3b) that are rotated with a dihedral angle from the molecular macrocycle plane. The macrocycle core appears as a circular ring, reflecting an almost planar shape, in agreement with the STM topographs of native TPyP resolved at the molecular occupied orbitals.29 In contrast, the molecular unit within CB domains (TPyP∈CB) presents two extra bright protrusions (Fig. 3c), each on the two opposite pyrrole rings, respectively, while the other two rings appear in dark depressions. The difference of the macrocycles is clearly manifested by the line profiles (Fig. 3d) along the short axis (defined by the rectangular unit29 represented by the four “bright” py lobes) of the two types of molecular units, respectively.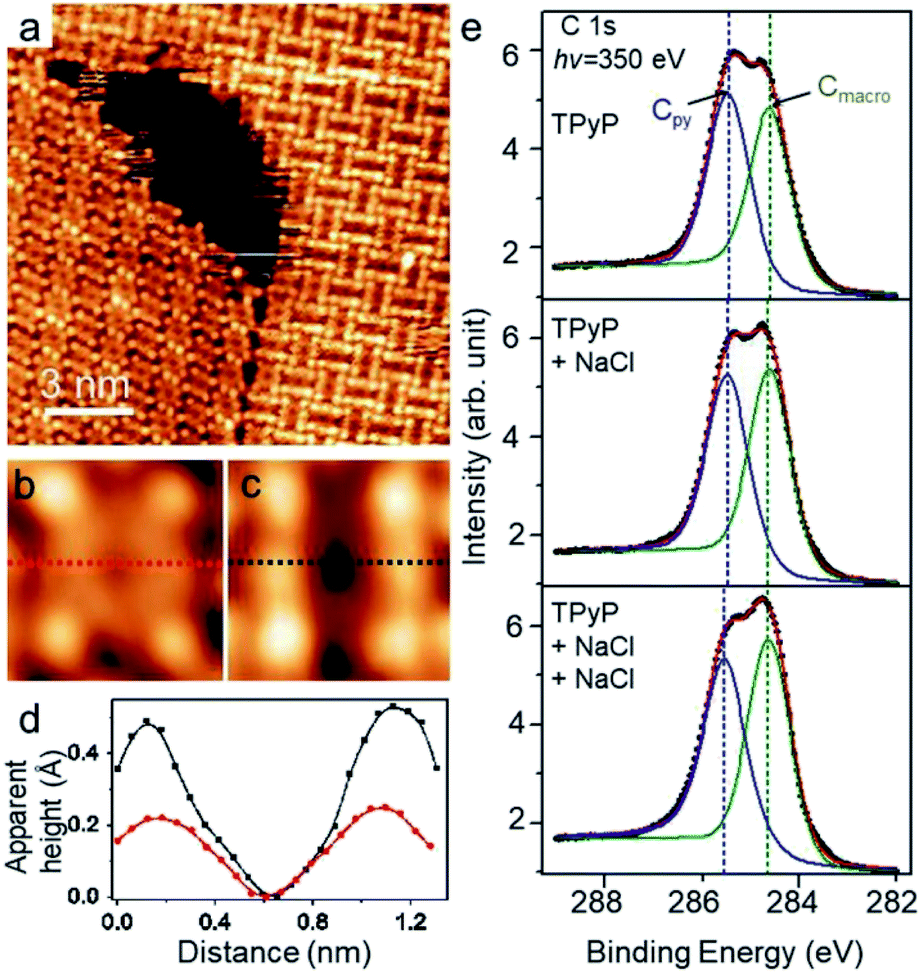 | ||
| Fig. 3 (a) STM topograph (U = −0.9 V and I = 0.1 nA) displaying both CP and CB structures. Molecules show different appearances: (b) TPyP∈CP, (c) TPyP∈CB. (d) The line profiles along the molecular short axes highlighted by the dash lines in (b) and (c) respectively. TPyP∈CP, red; TPyP∈CB, black. (e) XPS spectra of C 1s for the TPyP submonolayer on Ag(111) (top), and for the sample with stepwise deposition of NaCl (middle and bottom). | ||
Because of the physisorption for the TPyP on Ag(111) and of the topograph of both types of TPyP obtained at the same bias voltage (near the molecular HOMO levels),29 we ascribe the change of the molecular appearances to the conformational change of the macrocycles. Thereby, the extra bright protrusions on the macrocycle in the TPyP∈CB are assigned to the two opposite pyrroles whose outer parts tilt upwards from the substrate. Notably, this interpretation agrees with the analysis of the intramolecular conformation of the TPyP molecule: the dihedral rotation of py end groups is inherently coupled with the tilt of pyrroles via an intramolecular steric repulsion between the hydrogen atoms belonging to py terminals and pyrrole rings respectively;31,41 as a result, the tilting-upward pyrrole rings must lie at the short axis of a TPyP molecule, and the other two pyrroles tilt downwards correspondingly.
Such a conformational change induced by alkali–py coordination has also been revealed by our XPS measurements. The C 1s spectra were monitored during the stepwise deposition of NaCl to Ag(111) preadsorbed with TPyP. In the absence of NaCl, the spectra (Fig. 3e, top) show two peaks at 285.4 eV and 284.6 eV, which can be fairly ascribed to the C at py (labeled Cpy) and macrocycle (labeled Cmacro) moieties, respectively. After the deposition of NaCl, the two peaks do not shift, while the intensity of Cmacro increases with respect to that of Cpy, and becomes apparently higher than the latter after the second deposition of NaCl (Fig. 3e, bottom). Since no new peak emerges, such an increment of the intensity of the Cmacro peak suggests a conformational change of the TPyP molecule. According to the previous study by Diller et al.,40 the peaks at 284.6 eV and 285.4 eV can be assigned to the carbon atoms in the macrocycle and the py rings, respectively. This assignment gives the intensity ratio of the two peaks to be approximate 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, as shown in the C 1s spectrum of TPyP/Ag(111) (Fig. 3e, top). Upon the subsequent deposition of NaCl, NaCl is dissociated and connected with the N atoms in py rings steered by metal–organic coordination interaction. Meanwhile, the py endgroups of the TPyP molecules rotate along the axis of the N–Na bond, and the plane of the py ring becomes more perpendicular to Ag(111) surface, as shown in STM images from Fig. 3b and c. Interestingly, the two opposite pyrrole rings in the macrocycle also become brighter as two new protrusions appear in Fig. 3c. It is proposed that two opposite pyrrole rings are tilted as the N atoms bind to the Ag surface and the other carbon atoms point out of surface.42 Therefore in C 1s spectra, the intensity of Cmacro increases because the lifted up carbon atoms in pyrrole rings get closer to X-ray photons. However, the average vertical height of carbon atoms in py rings stays almost the same relative to the Ag surface when the py rings rotate along the axis of the N–Na bond. Thus the intensity of Cpy remains almost the same, but the intensity of Cmacro increases. We have also checked all other possible interpretations but find that only the conformational change explained above agrees with our STM and XPS data well.
:1, as shown in the C 1s spectrum of TPyP/Ag(111) (Fig. 3e, top). Upon the subsequent deposition of NaCl, NaCl is dissociated and connected with the N atoms in py rings steered by metal–organic coordination interaction. Meanwhile, the py endgroups of the TPyP molecules rotate along the axis of the N–Na bond, and the plane of the py ring becomes more perpendicular to Ag(111) surface, as shown in STM images from Fig. 3b and c. Interestingly, the two opposite pyrrole rings in the macrocycle also become brighter as two new protrusions appear in Fig. 3c. It is proposed that two opposite pyrrole rings are tilted as the N atoms bind to the Ag surface and the other carbon atoms point out of surface.42 Therefore in C 1s spectra, the intensity of Cmacro increases because the lifted up carbon atoms in pyrrole rings get closer to X-ray photons. However, the average vertical height of carbon atoms in py rings stays almost the same relative to the Ag surface when the py rings rotate along the axis of the N–Na bond. Thus the intensity of Cpy remains almost the same, but the intensity of Cmacro increases. We have also checked all other possible interpretations but find that only the conformational change explained above agrees with our STM and XPS data well.
2.3 Structural and chemical tuning
In this section, we demonstrate the coordination of py with other alkali metals. Fig. 4a–c show the CB structures (coexisting with CP structures or NaCl islands) resulting from the codeposition of TPyP (or ZnTPyP) with KBr, CsCl and NaCl, respectively. All three assemblies adopt the same coordination mode (see Fig. 4d) as that for the Na–TPyP structures. The unit cell parameters are determined based on the high-resolution STM images, which exhibit a clear tendency that the size of the lattice of CB structures increases with the atomic number of alkali metals (insets in Fig. 4a–c). The derived bond lengths are: d(K–N) = 2.5 Å, d(Cs–N) = 2.9 Å and d(Na–N) = 2.3 Å (in the Na–ZnTPyP structure), respectively. We have summarized these structural parameters (including that for Na–TPyP structures) in Table 1. It reveals that both the sizes of the unit cells and the bond lengths increase as the atomic number of alkali metals increases. This is due to the well-defined periodic trend of alkali metals, i.e., the ionic radius of Na, K and Cs shows an increase from 1.02, 1.38 and 1.67 Å.20,21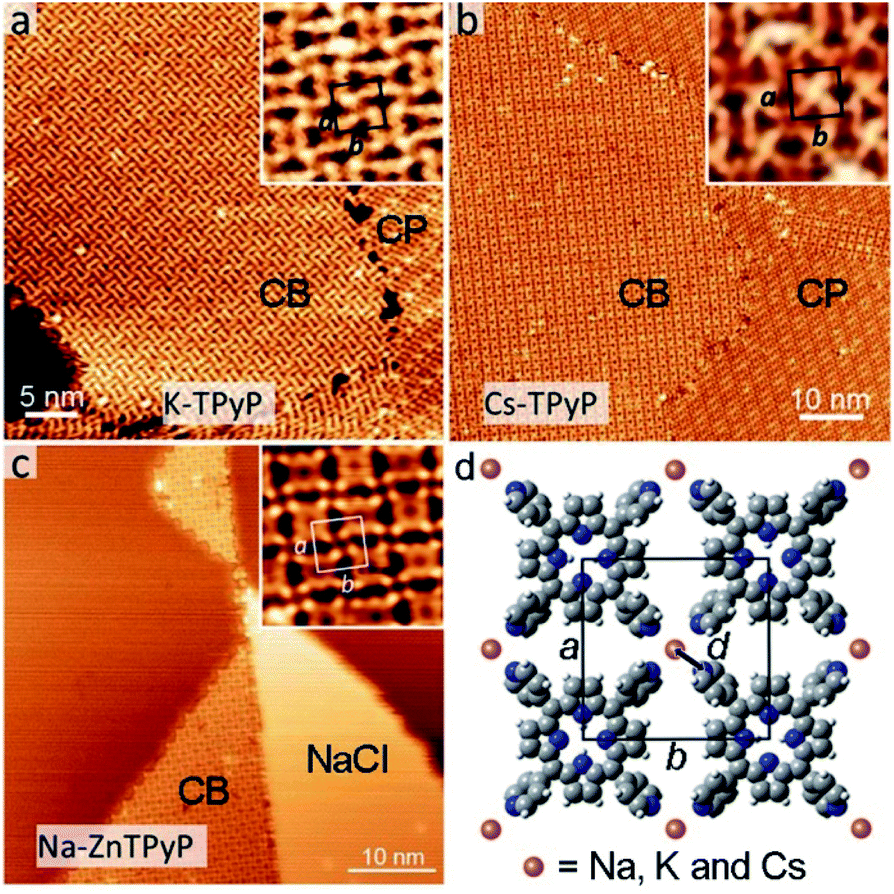 | ||
| Fig. 4 CB structures resulting from the codeposition of TPyP with KBr (a), CsCl (b) and the codeposition of ZnTPyP with NaCl (c). The unit cells are denoted in the insets, respectively. (d) The structural model of CB structures. d denotes the bond length of an alkali–N coordination bond. | ||
| Structure | Na–TPyP | Na–ZnTPyP | K–TPyP | Cs–TPyP |
|---|---|---|---|---|
| a R(alkali) refers to the ionic radius of alkali metals. The data are from ref. 20 and 21. | ||||
| a (nm) | 1.40 ± 0.02 | 1.42 ± 0.02 | 1.44 ± 0.02 | 1.48 ± 0.02 |
| b (nm) | 1.45 ± 0.02 | 1.46 ± 0.02 | 1.49 ± 0.02 | 1.56 ± 0.02 |
| d(alkali–N) (Å) | 2.2 ± 0.1 | 2.3 ± 0.1 | 2.5 ± 0.1 | 2.9 ± 0.1 |
| R(alkali) (Å)a | 1.02 | 1.38 | 1.67 | |
In previously reported 2D ionic assemblies, diverse structural phases coexisted, because the ionic nature of the alkali–ligand bond adds flexibility to the coordination linkages.9 In this work, there exists the CB structure solely, and all structures from the three different alkali metals can be formed with large extensive domains, in spite of their different lattice parameters. The excellent shape persistency presented here can be attributed to the steric constraint exerted by the square shape of the porphyrin-based molecular constituents. Notably, these assemblies reveal no dependence on the type of halogens (Cl or Br), proving that the latter do not interplay with the assembly, presumably because of their fast dissociation or diffusion into the substrate.11,13,14 In addition, the use of ZnTPyP, whose assembly with NaCl is similar to that of TPyP, suggests that the chemical state of the porphyrin macrocycles has little influence on the alkali–py coordination.
Last, we describe the functionalization of alkali–py CB monolayers by modifying the chemical state of the porphyrin macrocycles. To this end, we have deposited the Fe atoms on the as-formed Na–TPyP CB precursor layers. Fig. 5a shows that the molecular cores present two distinct features-dark depression (white/black squares) or bright protrusion (white/black dashed squares). According to ref. 30, we assign the bright protrusion to an Fe ion sitting on the porphyrin macrocycle through substituting with the pyrrolic protons, while the dark depression indicates a native molecule. At room temperature, such Fe metalation occurs in both CB and CP structures without perturbing the assembled structures, implying a close energy cost for the metalation reaction of TPyP∈CP and ∈CB, though the molecules within the two structures adopt different conformations.
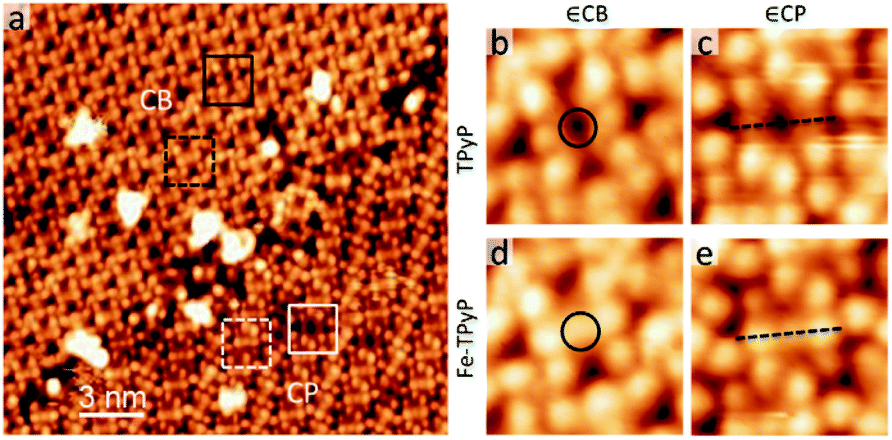 | ||
| Fig. 5 (a) STM topograph (U = −1.0 V, I = 0.07 nA) of the sample with a dosage of Fe. In both CP and CB domains, Fe–TPyPs are marked by white/black dashed squares; TPyPs are marked with white/black squares. (b–e) High-resolution STM images of TPyP and Fe–TPyP in both domains. Circles in (b and d) mark the molecular cores. Dash lines in (c and e) highlight the deformation of macrocycles upon Fe-metalation. | ||
Interestingly, apart from the variation of the contrast at the molecular cores (circles in Fig. 5b and d), the topographic features of the molecule Fe–TPyP∈CB have less noticeable change in comparison with those of TPy∈CB, while the case drastically changes for the molecules in CP (dash lines in Fig. 5c and e). The molecule Fe–TPyP∈CP appears with two extra bright protrusions (i.e. the two pyrrolic rings tilting upwards), reflecting a geometric deformation due to the ligand field effects caused by Fe metalation.43,44 The fact that Fe–TPyP∈CP and ∈CB have similar topographic features corroborate our interpretation on the macrocycle deformation of the Na–TPyP CB structures. Last, in terms of the metallic species, the Na–Fe–TPyP CB structure includes both alkali and transition metals, that afford distinct chemical and charge states, representing a bimetallic nano-lattice.34 The catalytic or spintronic properties of this bimetallic framework are important for further investigations.
3 Conclusions
In conclusion, we have described the self-assembly, the intramolecular deformation, the shape persistency and the functionalization of the 2D alkali–py coordination structures that are facilely realized via the solventless surface reaction of alkali halides with porphyrin-based ligands on Ag(111). Considering the previous reports on similar ionic/coordinative systems mostly based on hard base ligands, the use of borderline/soft py ligands largely increases the understanding of the solventless ionic assembly in vacuum–solid interfaces. The shape persistency realized by porphyrin-based ligands and the structural/chemical modification tailorable at the sub-Å level provide new opportunities for the fine-tuning of electronic, catalytic and spintronic properties of the 2D metal–organic nanomaterials.4 Experimental
4.1 STM
Sample preparation processes were carried out in an ultrahigh vacuum (UHV) system (SPECS GmbH) at a base pressure of ∼3.0 × 10−10 mbar. The single-crystal Ag(111) substrate (MaTeck, 99.999%) was cleaned by cycles of Ar+ ion sputtering at an energy of 900 eV and annealing at 800 K. The molecules (TPyP, TCI, purity>97%; ZnTPyP (zinc 5,10,15,20-tetra(4-pyridyl)-porphyrin), Sigma-Aldrich, purity ∼90%) and the alkali halides (NaCl, Sigma-Aldrich, 99.999%; KBr, Strem Chemicals, 99.999%; CsCl, Sigma-Aldrich, 99.999%) were evaporated separately by organic molecular beam epitaxy (DODECON Nanotechnology GmbH). The sublimation temperatures are 400 °C, 360 °C, 350 °C, 500 °C and 500 °C, respectively. Fe atoms were evaporated from an iron rod (Puratronic, 99.995%) by an electron-beam evaporator.All STM experiments were performed using an Aarhus SPM apparatus controlled by Nanonis electronics. Topographic data were acquired in the constant current mode with the bias voltage applied to the sample. All STM images were taken at room temperature (298 K), and analysed using WSxM.
4.2 XPS
The XPS experiments were performed in situ at the catalysis and surface science endstation at the BL11U beamline in the National Synchrotron Radiation Laboratory (NSRL) (Hefei, China). The Na 2p, N 1s, C 1s and Cl 2p spectra were measured with a photon energy of 150, 480, 350 and 350 eV, respectively. The spectra deconvolution was carried out by using the XPS Peak 41 program with Gaussian Functions after subtraction of a Shirley background.Author contributions
WZ, YQM and ZLS conceived and designed the project. KZ, HFL, SPX, YS, YXW, JHH and ZLS performed STM measurements, analyzed data and prepared the manuscript. MW, HHD, QX and JFZ performed XPS measurements and analyzed the data. All authors discussed the results and contributed in the manuscript preparation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the NSFC (21303113, 21972101, 21972096, and 21773222), the Shenzhen Science and Technology Program (JCYJ20190808150615285), the Natural Science Foundation of Jiangsu Province (BK20130285), the National Key R&D Program of China (2017YFA0403403) and the China Postdoctoral Science Foundation (2013M540460). The work was also funded by Collaborative Innovation Center of Suzhou Nano Science and Technology (CIC-NANO), Soochow University, by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD), and by the 111 Project. Z. S. thanks Dr Nabi Aghdassi for fruitful discussions.Notes and references
- J. V. Barth, G. Costantini and K. Kern, Nature, 2005, 437, 671–679 CrossRef CAS PubMed.
- S. De Feyter and F. C. De Schryver, J. Phys. Chem. B, 2005, 109, 4290–4302 CrossRef CAS PubMed.
- J. V. Barth, Annu. Rev. Phys. Chem., 2007, 58, 375–407 CrossRef CAS PubMed.
- N. Lin, S. Stepanow, M. Ruben and J. V. Barth, Top. Curr. Chem., 2009, 287, 1–44 CAS.
- J. V. Barth, Surf. Sci., 2009, 603, 1533–1541 CrossRef CAS.
- K. Ariga, M. V. Lee, T. Mori, X.-Y. Yu and J. P. Hill, Adv. Colloid Interface Sci., 2010, 154, 20–29 CrossRef CAS PubMed.
- L. Dong, Z. A. Gao and N. Lin, Prog. Surf. Sci., 2016, 91, 101–135 CrossRef CAS.
- Y.-F. Geng, P. Li, J.-Z. Li, X.-M. Zhang, Q.-D. Zeng and C. Wang, Coord. Chem. Rev., 2017, 337, 145–177 CrossRef CAS.
- S. Stepanow, R. Ohmann, F. Leroy, N. Lin, T. Strunskus, C. Wöll and K. Kern, ACS Nano, 2010, 4, 1813–1820 CrossRef CAS PubMed.
- W. Xu, J.-g. Wang, M. Yu, E. Lægsgaard, I. Stensgaard, T. R. Linderoth, B. Hammer, C. Wang and F. Besenbacher, J. Am. Chem. Soc., 2010, 132, 15927–15929 CrossRef CAS PubMed.
- C. Wäckerlin, C. Iacovita, D. Chylarecka, P. Fesser, T. A. Jung and N. Ballav, Chem. Commun., 2011, 47, 9146–9148 RSC.
- N. Abdurakhmanova, A. Floris, T.-C. Tseng, A. Comisso, S. Stepanow, A. De Vita and K. Kern, Nat. Commun., 2012, 3, 940 CrossRef PubMed.
- D. Skomski, S. Abb and S. L. Tait, J. Am. Chem. Soc., 2012, 134, 14165–14171 CrossRef CAS PubMed.
- D. Skomski and S. L. Tait, J. Phys. Chem. C, 2013, 117, 2959–2965 CrossRef CAS.
- T. K. Shimizu, J. Jung, H. Imada and Y. Kim, Angew. Chem., Int. Ed., 2014, 53, 13729–13733 CrossRef CAS PubMed.
- M. Yu, W. Xu, N. Kalashnyk, Y. Benjalal, S. Nagarajan, F. Masini, E. Lægsgaard, M. Hliwa, X. Bouju, A. Gourdon, C. Joachim, F. Besenbacher and T. Linderoth, Nano Res., 2012, 5, 903–916 CrossRef CAS.
- J. Hieulle, D. Peyrot, Z. Jiang and F. Silly, Chem. Commun., 2015, 51, 13162–13165 RSC.
- C. Zhang, L. Wang, L. Xie, H. Kong, Q. Tan, L. Cai, Q. Sun and W. Xu, ChemPhysChem, 2015, 16, 2099–2105 CrossRef CAS PubMed.
- C. F. J. Faul and M. Antonietti, Adv. Mater., 2003, 15, 673–683 CrossRef CAS.
- K. M. Fromm, Coord. Chem. Rev., 2008, 252, 856–885 CrossRef CAS.
- D. Banerjee and J. B. Parise, Cryst. Growth Des., 2011, 11, 4704–4720 CrossRef CAS.
- A. Floris, A. Comisso and A. De Vita, ACS Nano, 2013, 7, 8059–8065 CrossRef CAS PubMed.
- J. Elemans, R. Van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS.
- J. Otsuki, Coord. Chem. Rev., 2010, 254, 2311–2341 CrossRef CAS.
- S. Mohnani and D. Bonifazi, Coord. Chem. Rev., 2010, 254, 2342–2362 CrossRef CAS.
- H. Marbach, Acc. Chem. Res., 2015, 48, 2649–2658 CrossRef CAS PubMed.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- W. Auwärter, A. Weber-Bargioni, A. Riemann, A. Schiffrin, O. Gröning, R. Fasel and J. V. Barth, J. Chem. Phys., 2006, 124, 194708 CrossRef PubMed.
- W. Auwärter, A. Weber-Bargioni, S. Brink, A. Riemann, A. Schiffrin, M. Ruben and J. V. Barth, ChemPhysChem, 2007, 8, 250–254 CrossRef PubMed.
- F. Klappenberger, A. Weber-Bargioni, W. Auwärter, M. Marschall, A. Schiffrin and J. V. Barth, J. Chem. Phys., 2008, 129, 214702 CrossRef CAS PubMed.
- Z. Shi and N. Lin, J. Am. Chem. Soc., 2009, 131, 5376–5377 CrossRef CAS PubMed.
- Z. Shi and N. Lin, ChemPhysChem, 2010, 11, 97–100 CrossRef CAS PubMed.
- Y. Li, J. Xiao, T. E. Shubina, M. Chen, Z. Shi, M. Schmid, H.-P. Steinrück, J. M. Gottfried and N. Lin, J. Am. Chem. Soc., 2012, 134, 6401–6408 CrossRef CAS PubMed.
- J. I. Urgel, D. Écija, W. Auwärter, D. Stassen, D. Bonifazi and J. V. Barth, Angew. Chem., Int. Ed., 2015, 54, 6163–6167 CrossRef CAS PubMed.
- J. I. Urgel, M. Schwarz, M. Garnica, D. Stassen, D. Bonifazi, D. Ecija, J. V. Barth and W. Auwärter, J. Am. Chem. Soc., 2015, 137, 2420–2423 CrossRef CAS PubMed.
- Y. Wang, K. Zhou, Z. Shi and Y.-Q. Ma, Phys. Chem. Chem. Phys., 2016, 18, 14273–14278 RSC.
- X. Chen, S. Lei, C. Lotze, C. Czekelius, B. Paulus and K. J. Franke, J. Chem. Phys., 2017, 146, 092316 CrossRef.
- F. Matthaei, S. Heidorn, K. Boom, C. Bertram, A. Safiei, J. Henzl and K. Morgenstern, J. Phys.: Condens. Matter, 2012, 24, 354006 CrossRef.
- K. Diller, F. Klappenberger, M. Marschall, K. Hermann, A. Nefedov, C. Wöll and J. Barth, J. Chem. Phys., 2012, 136, 014705 CrossRef CAS PubMed.
- F. Buchner, I. Kellner, W. Hieringer, A. Gorling, H.-P. Steinruck and H. Marbach, Phys. Chem. Chem. Phys., 2010, 12, 13082–13090 RSC.
- M. Lepper, J. Kobl, T. Schmitt, M. Gurrath, A. de Siervo, M. A. Schneider, H.-P. Steinrück, B. Meyer, H. Marbach and W. Hieringer, Chem. Commun., 2017, 53, 8207–8210 RSC.
- F. Buchner, K. Flechtner, Y. Bai, E. Zillner, I. Kellner, H.-P. Steinrück, H. Marbach and J. M. Gottfried, J. Phys. Chem. C, 2008, 112, 15458–15465 CrossRef CAS.
- G. Di Santo, C. Castellarin-Cudia, M. Fanetti, B. Taleatu, P. Borghetti, L. Sangaletti, L. Floreano, E. Magnano, F. Bondino and A. Goldoni, J. Phys. Chem. C, 2011, 115, 4155–4162 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional STM images and XPS spectra. See DOI: 10.1039/d0na00091d |
| This journal is © The Royal Society of Chemistry 2020 |