
Evidence for a kinetically controlled burying mechanism for growth of high viscosity secondary organic aerosol†
Allison C.
Vander Wall
 ,
Véronique
Perraud
,
Lisa M.
Wingen
and
Barbara J.
Finlayson-Pitts
*
,
Véronique
Perraud
,
Lisa M.
Wingen
and
Barbara J.
Finlayson-Pitts
*
Department of Chemistry, University of California, Irvine, CA 92697-2025, USA. E-mail: bjfinlay@uci.edu; Fax: (949) 824-2420; Tel: (949) 824-7670
First published on 31st October 2019
Abstract
Secondary organic aerosol (SOA) particles are ubiquitous in air and understanding the mechanism by which they grow is critical for predicting their effects on visibility and climate. The uptake of three organic nitrates into semi-solid SOA particles formed by α-pinene ozonolysis either with or without an OH scavenger was investigated. Four types of experiments are presented here. In Series A, uptake of the selected organic nitrates (2-ethylhexyl nitrate (2EHN); β-hydroxypropyl nitrate (HPN); β-hydroxyhexyl nitrate (HHN)) into impacted SOA particles was interrogated by attenuated total reflectance (ATR)-FTIR. In this case, equilibrium was reached and partition coefficients (KSOA = [–ONO2]SOA/[–ONO2]air) were measured to be K2EHN = (3.2–11) × 104, KHPN = (4.4–5.4) × 105, and KHHN = (4.9–9.0) × 106. In Series B, SOA particles were exposed on-the-fly to gas phase organic nitrates for comparison to Series A, and uptake of organic nitrates was quantified by HR-ToF-AMS analysis, which yielded similar results. In Series C (AMS) and D (ATR-FTIR), each organic nitrate was incorporated into the SOA as the particles formed and grew. The incorporation of the RONO2 was much larger in Series C and D (during growth), exceeding equilibrium values determined in Series A and B (after growth). This suggests that enhanced uptake of organic nitrates during SOA formation and growth is due to a kinetically controlled “burying” mechanism, rather than equilibrium partitioning. This has important implications for understanding SOA formation and growth under conditions where the particles are semi-solid, which is central to accurately predicting properties for such SOA.
Environmental significanceSecondary organic aerosol (SOA) particles are known to have deleterious effects on human health and to impact climate and visibility. Understanding the processes by which gases are incorporated into these particles to grow them to diameters large enough to have these impacts is therefore critical for predicting and addressing their effects. In this work, we examine how gas phase organic nitrates interact with SOA particles either during the particle growth process in a flow reactor or after particle growth has occurred. It is shown that co-condensation of organics that grow the particles enhances particulate organic nitrate content relative to that expected from equilibrium partitioning, and is best described by a kinetically controlled ‘burying’ mechanism. |
Introduction
Secondary organic aerosol (SOA) is a major contributor to airborne particles, which are known to impact human health,1–7 visibility8–11 and climate.7–9,12,13 Yet the molecular processes that lead to SOA particle formation from the oxidation of volatile organic compounds and the subsequent growth of these particles is not well understood.13–17 Specifically, there is not yet a comprehensive understanding of the mechanism by which gases are taken up into the particles to grow them.18Many models have been developed to explain the mechanisms by which particles grow and the various physical and chemical processes involved.19–34 In some cases, particles may have relatively high viscosity and a semi-solid or glassy physical state, and hence the growth process is subject to diffusion limitations.25,26,35–47 This has important implications for particle growth as well as for chemical reactions and interactions occurring both between the gas and condensed phases, and within the condensed phase.25,26
Organic nitrates are known to be formed by NO3 radical oxidation of volatile organic compounds (VOCs) and OH radical oxidation of VOCs in the presence of NOx.48–54 Additionally, organic nitrates (including alkyl nitrates and multifunctional hydroxy nitrates) have been measured in air.55–66 Recently, initial uptake and bulk partition coefficients for three organic nitrates into several thin film substrates of selected organic compounds and into particles from the ozonolysis of α-pinene were measured.67 The organic nitrates included the alkyl nitrate (2-ethylhexyl nitrate, 2EHN) and two isomeric β-hydroxy nitrates (β-hydroxyhexyl nitrate, HHN, and β-hydroxypropyl nitrate, HPN), shown in Fig. 1. It was found that the trend in uptake coefficients did not uniformly track the trend in partition coefficients, suggesting that interactions controlling initial uptake (e.g., surface of the film) were different from those determining the equilibrium partitioning into the substrates (bulk properties).
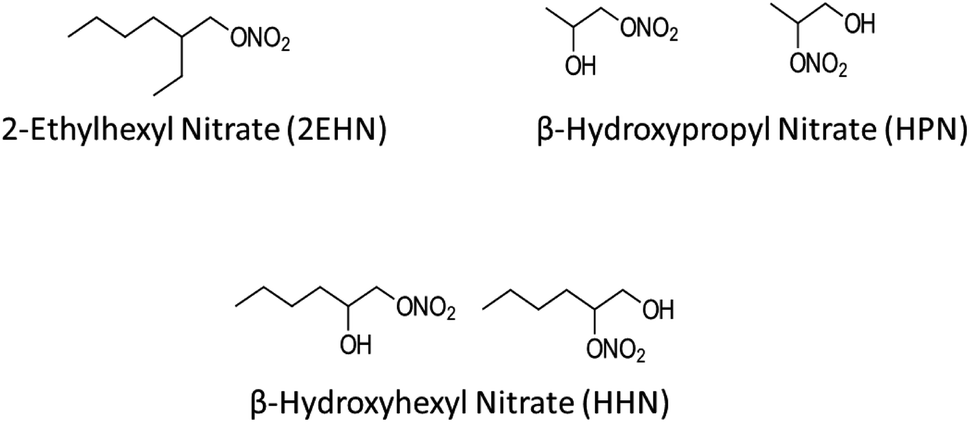 | ||
| Fig. 1 Structures of the gas phase organic nitrates used in this study. The synthesis of the hydroxynitrates resulted in the presence of the two isomers. | ||
The goal of this study was to elucidate the mechanism of incorporation of those three organic nitrates into high viscosity semi-solid SOA particles formed in the α-pinene ozonolysis. This was accomplished by measuring the organic nitrate content of SOA in particles during their formation and growth, and by comparison, the uptake of the organic nitrates into “fully grown” SOA. Zelenyuk et al.68 previously demonstrated that when polycyclic aromatic hydrocarbons (PAHs) were incorporated during SOA formation, they remain trapped inside the particles due to the highly viscous semi-solid nature of the SOA and remained shielded from oxidation and evaporation; however, when the PAHs interacted with the SOA already formed, they remained on the surface and were subject to greater evaporation rates. In the current study, we show that a higher partition coefficient is obtained when the organic nitrate tracer is present during formation and growth than when equilibrium partitioning into fully grown SOA is measured. This suggests that a kinetically controlled “burying” mechanism is responsible for uptake into highly viscous, semi-solid particles.
Experimental
Fig. 2 shows a schematic of the four experimental configurations used in these studies. In Series A, nitrate-free SOA particles formed in the α-pinene ozonolysis were generated in a large volume, slow flow stainless steel aerosol flow reactor69 and impacted on an attenuated total reflectance (ATR) crystal to generate a thin film of SOA particles, over which gas phase organic nitrate was flowed and uptake of the organic nitrate was measured until equilibrium was reached. This method was limited to higher concentrations of organic nitrate (approaching saturation vapor pressure) in order to detect –ONO2 peaks by ATR-FTIR. A benefit of this method over in situ production of organic nitrates is that it avoids oxidation of the organic nitrate compounds from gas phase OH that is generated during ozonolysis in the reactor.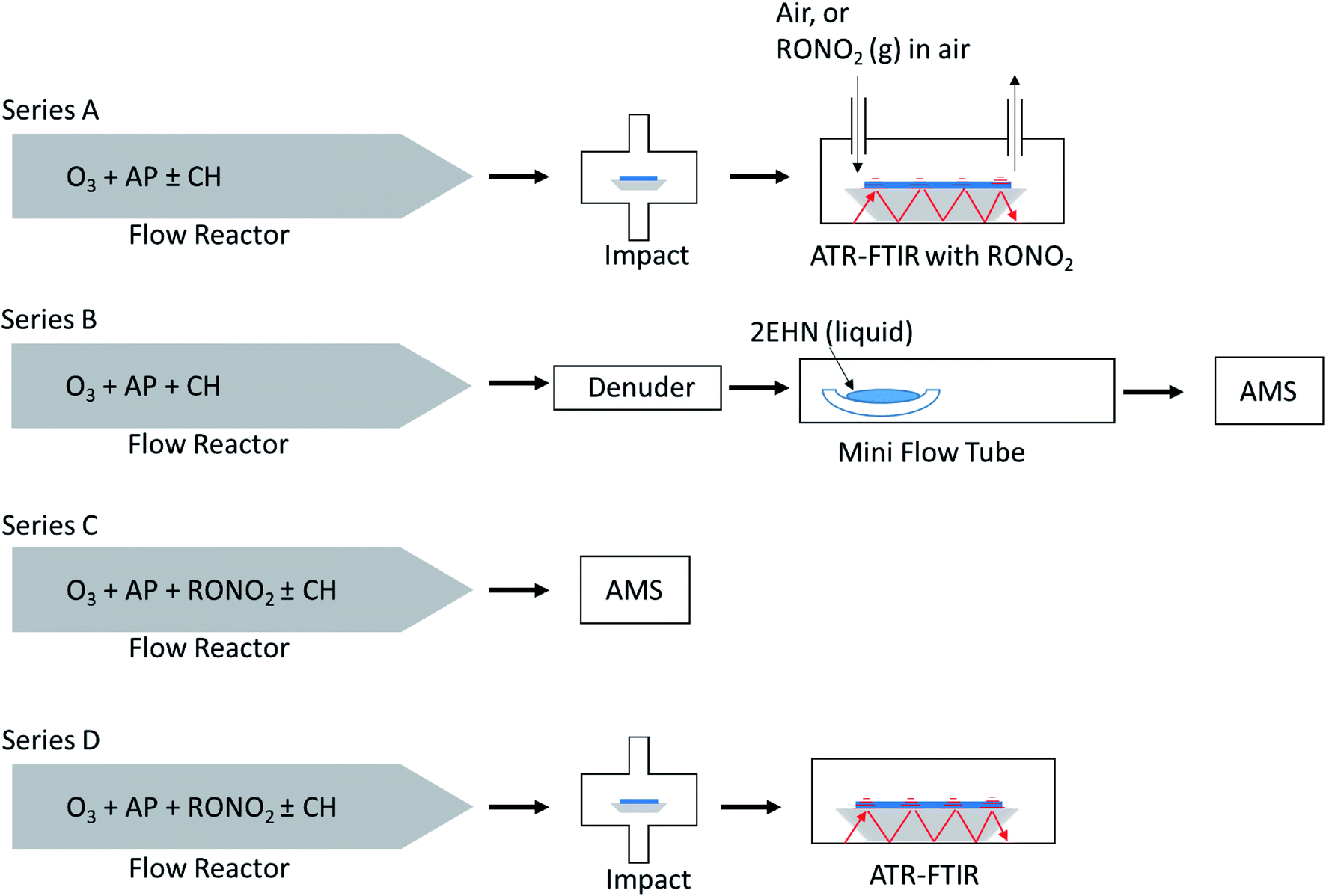 | ||
| Fig. 2 Schematic of the four experiment types. In Series A, SOA particles are formed in the absence of organic nitrates (RONO2) in the stainless steel flow reactor and impacted on an ATR crystal to generate a thin film of particles, over which RONO2 at near saturation vapor pressures (4.7 × 1015 (2EHN), 3.9 × 1015 (HPN) and 1.7 × 1014 (HHN) molecules per cm3) were flowed and the uptake of RONO2 was measured by ATR-FTIR. In Series B, SOA particles were generated in the stainless steel flow reactor and then passed through a charcoal denuder before subsequently being flowed into a 1 m long glass flow tube and exposed to 2EHN through either a reservoir with a pure liquid or a trap flowing air over the pure liquid, and analyzed by HR-ToF-AMS. In Series C, RONO2 at lower levels (1.4 × 1014 (2EHN), 1.2 × 1014 (HPN) and 5.0 × 1012 (HHN) molecules per cm3) were incorporated into SOA particles as they form and grow in the stainless steel flow reactor. These particles were quantified by HR-ToF-AMS. In Series D, the same particles as in Series C were impacted on an ATR crystal and partition coefficients were determined from the quantification of RONO2 by ATR-FTIR. For Series A, the gas-phase organic nitrate concentration was measured using GC-MS from the trap. For Series B, the gas-phase 2EHN concentration was measured directly from the mini flow tube. For Series C/D, the gas-phase organic nitrate concentration is calculated from the measured concentration exiting the trap and accounting for dilution (a factor of 34). | ||
In Series B, SOA particles formed in the α-pinene ozonolysis also from the large flow reactor were first passed through a monolith carbon denuder (NovaCarb™; MAST Carbon, Ltd.) to remove gas phase species and then flowed into a smaller glass flow tube where they were exposed to gaseous 2EHN. This limited series was carried out to ensure that the observations were in agreement with Series A results on impacted particles.
In Series C and D, the organic nitrates were introduced into the stainless steel flow reactor and were incorporated into SOA particles from α-pinene ozonolysis as they formed and grew in the reactor. Smaller concentrations of the organic nitrates than those in Series A were used here due to the much larger volumes of air and hence higher dilution factors that are associated with the large flow reactor. In Series C, the organic nitrate in the particles was quantified on-the-fly by high resolution time-of-flight aerosol mass spectrometry (HR-ToF-AMS). In Series D the same particles as in Series C were simultaneously impacted on an ATR crystal and quantification of each organic nitrate (RONO2) was carried out by FTIR.
Aerosol generation and particle size distributions
SOA from the ozonolysis of α-pinene (AP) was generated in the stainless steel flow reactor with a total flow rate of 34 L min−1, and all reactants were introduced in the initial mixing section of the reactor. Gas-phase AP (250 ppb) was generated by injection of the pure liquid (Sigma Aldrich, >99%) from an automated syringe pump (New Era Pump System Inc., Model NE-1000) into a 10 L min−1 flow of clean, dry air. Ozone was generated by flowing 0.4 L min−1 O2 gas (Praxair, 99.993%) through a Pen-Ray® mercury lamp (UVP, LLC), and was subsequently diluted with 9.6 L min−1 of air before being added to the system. An additional 14 L min−1 of air was introduced to create a total flow rate of 34 L min−1, and the resulting reactor concentrations were 250–300 ppb O3 measured using an ozone monitor (Teledyne Photometric O3 Analyzer 400E; Advanced Pollution Instrumentation, Inc. Photometric O3 Analyzer 400). For all the experiments, the dry clean air originated from a purge gas generator (Parker Balston, model 75-62) followed by a carbon/alumina media (PermaPure LLC) and an inline 0.1 µm filter (Headline Filter, DIF-N70). The reaction of AP with O3 produces OH radicals which, unless scavenged, will react with the organic nitrates.70 Thus in some experiments, cyclohexane (CH, Fisher Scientific, 99.9%), used as an OH scavenger, was evaporated into the flow of air to give a concentration of 2.5 × 1015 molecules per cm3 (100 ppm).Three organic nitrates (Fig. 1) were used as “tracer” compounds because of their ability to be detected via both FTIR and mass spectrometry. An alkyl nitrate, 2-ethylhexyl nitrate (2EHN, Sigma Aldrich, 97%) was used as purchased. Two multifunctional organic nitrates, β-hydroxyhexyl nitrate and β-hydroxypropyl nitrate (HHN and HPN, respectively, 82–93% purity in the liquid phase) were synthesized using the method of Cavdar and Saracoglu71 as described previously.67 In brief, epoxypropane (Sigma Aldrich, ≥99%) or epoxyhexane (Sigma Aldrich, 97%) were reacted with Bi(NO3)3·5H2O (Sigma Aldrich, 98%) in dichloromethane (Macron, ≥ 99.5%) for 16–24 hours under N2 gas (Praxair, 99.999%) at room temperature, after which the solvent was evaporated off. After synthesis, each organic nitrate was stored under an inert atmosphere of N2 in a freezer (T ∼ −20 °C). The purity of the resulting liquid organic nitrate was quantified by 1H NMR, with the major impurities identified as the corresponding di-alcohols. Compared to the impurities in the liquid phase, analysis of the gas-phase directly flowing out of a glass trap by direct analysis in real time mass spectrometry (DART-MS) did not detect impurities (Ionsense DART SVP source with Vapur Interface coupled to a Waters Xevo TQ-S mass spectrometer).67 The isomeric ratios of the hydroxy-terminated to nitrate-terminated isomer of the pure liquid estimated by 1H NMR were ∼2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 for HPN and ∼4:3 for HHN.
:1 for HPN and ∼4:3 for HHN.
For Series A experiments, each individual organic nitrate was introduced by flowing 0.06 L min−1 air through the glass trap to yield high gas-phase concentrations approaching the saturation vapor pressure before being introduced into the ATR-FTIR cell. The concentration of each gas-phase organic nitrate was controlled by keeping the trap at room temperature using a water bath, and the concentration of organic nitrate exiting the trap was measured by GC-MS as described below. For the limited Series B experiments, a reservoir of liquid 2EHN was placed inside the mini flow tube in order to expose particles on-the-fly to the saturation vapor pressure of 2EHN in a total flow of air of 0.4–1.5 L min−1. In a separate experiment for Series B, a glass trap containing the pure 2EHN liquid was used in place of the reservoir to flow 0.04–0.15 L min−1 into the mini flow tube to provide more dilute 2EHN concentrations. For Series C and D experiments, each organic nitrate was introduced by flowing 1 L min−1 air through the glass trap containing the pure liquid into a stream of air totaling 10 L min−1 simultaneously with AP, either with or without CH.
Experiments were performed under ambient temperature (295–298 K) and pressure, and dry conditions (RH < 5%), without seed particles. The particles were sampled along the length of the flow system at 7 min and 31 min reaction time. Particle size distributions were monitored using a scanning mobility particle sizer (SMPS, TSI), equipped with a model 3080 classifier, a 3081 long differential mobility analyzer, and a 3776 butanol-based CPC.
Gas phase measurements
Gas phase concentrations of AP and CH were measured using electron impact (EI) GC-MS (Agilent 7890A GC system with a 5975C MS detector) in a dual total ion/single ion monitoring (SIM) system (total ion monitoring was used for CH while m/z 93 was followed in SIM mode for AP), with the particles and ozone filtered out using a quartz filter (TissuQuartz®; 37 mm; PallFlex) and a KI (Fisher Chemical, 100.0%) ozone scrubber, respectively.The gas phase concentrations of all three organic nitrates were measured by flowing air at 1 L min−1 through the trap containing each nitrate into a 1 mL sampling loop on the GC-MS and comparing to a calibration using the synthesized liquid standards. The concentrations measured in this manner are shown in Table 1. Independently, the vapor pressures of all three organic nitrates were estimated using two group contribution methods72–74 and are also listed in Table 1. The estimated vapor pressures are in reasonable agreement with the measured values, indicating that these group contribution methods are good predictors of vapor pressure for these species. Hereafter, the concentration measured by GC-MS of each organic nitrate exiting the trap is used in all calculations, factoring in any additional dilution factors. For example, after the dilution factor of 34 in the flow reactor, the final gas phase concentrations are 1.4 × 1014 (2EHN), 1.2 × 1014 (HPN) and 5.0 × 1012 (HHN) molecules per cm3. Concentrations of 2EHN measured by GC-MS directly from the flow reactor were in good agreement with the calculated values. However, this comparison was not possible for HPN and HHN due to greater losses in the sampling line. Although some losses of the organic nitrate are expected to occur to the walls of the flow reactor, for some experiments the walls were conditioned with a flow of the organic nitrate overnight. There was no statistical difference in the partition coefficients for these experiments compared to experiments where the walls were not conditioned.
| Organic nitrates | Gas phase concentration at trap exit (1015 molecules per cm3 and Pa)b | Vapor pressure using Moller73,74 (Pa) | Vapor ressurec using SIMPOL.172 (Pa) |
|---|---|---|---|
| a Measured concentrations are from the average of triplicate measurements. b Error bars are ±1σ from the average of triplicate measurements. c SIMPOL.1 does not distinguish between isomers. d Hydroxy-terminated isomer. e Nitrate-terminated isomer. f Error bars are ±1σ. | |||
| 2EHN | 4.7 ± 0.2 (19 ± 1.0 Pa) | 14 | 18 |
| HPN | 3.9 ± 0.2 (16 ± 1.0 Pa) | 12d | 16 |
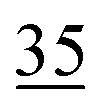
|
|||
| Average:f 24 ± 16 | |||
| HHN | 0.17 ± 0.05 (0.70 ± 0.21 Pa) | 0.35d | 0.85 |
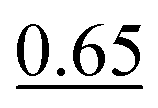
|
|||
| Average:f 0.50 ± 0.21 | |||
Incorporation into impacted particles
Previously, partition coefficients (K) and uptake coefficients (γ) were measured using ATR-FTIR for uptake of these organic nitrates into impacted particles from AP ozonolysis in the absence of an OH scavenger.67 For comparison, the partition coefficients for these organic nitrates into impacted particles from AP ozonolysis in the presence of CH as an OH scavenger have been measured in the same manner here (Series A). In brief, the polydisperse particles were collected onto a Ge ATR crystal using a custom-designed impactor with a 50% cut-off diameter of 240 nm.41 This resulted in >60% of the total mass concentration of the particles in the flow reactor being collected by the impactor under the reaction conditions used here. The particles were sampled at a total flow of 30 L min−1 for 10–30 min at the end of the reactor (∼20–60 µg total impacted mass), corresponding to a reaction time of 31 min. The amount impacted onto the crystal was varied to ensure the film thickness was smaller than the depth of penetration (dp) of the infrared evanescent wave, and thus ensuring that the entire film was probed by the IR beam. Using the wavelength of the IR beam and the refractive indices of the Ge crystal and air, the dp was calculated to be 0.35 µm at 1730 cm−1, 0.37 µm at 1630 cm−1, and 0.47 µm at 1280 cm−1.75 The path length (l) of the IR beam through the impacted particles can be estimated using dp and factoring in the 10 bounces of the beam within the ATR crystal, giving total path lengths of 3.5 µm at 1730 cm−1, 3.7 µm at 1630 cm−1, and 4.7 µm at 1280 cm−1.75 After impaction, the Ge crystal was placed in an ATR cell inside an FTIR spectrometer (Nicolet 6700), and the spectrum of the impacted SOA particles was acquired at 4 co-added scans and a resolution of 8 cm−1. The selected organic nitrate was introduced by flowing clean, dry air over the pure liquid and subsequently over the impacted particles at a constant flow of 0.060 ± 0.005 L min−1. The 1280 cm−1 peak was used for analysis of the organic nitrates since there was some overlap of the carbonyl peaks with the 1630 cm−1 peak of the –ONO2 group. The partition coefficients were calculated once the organic nitrate signal had reached steady-state (∼1000 seconds) based on the intensity of the infrared peaks for the –ONO2 symmetric stretch (1280 cm−1)48,76 and the carbonyl stretch (1733 cm−1),76 as follows: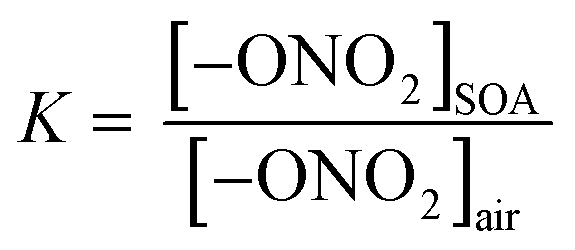 | (1) |
In eqn (1), [–ONO2]SOA is the concentration of organic nitrate in the impacted particles in units of moles per liter of SOA and [–ONO2]air is the concentration of organic nitrate in air in units of moles per liter of air. The [–ONO2]air is the gas phase concentration measured from the trap by GC-MS. The [–ONO2]SOA was calculated using eqn (2)–(4):
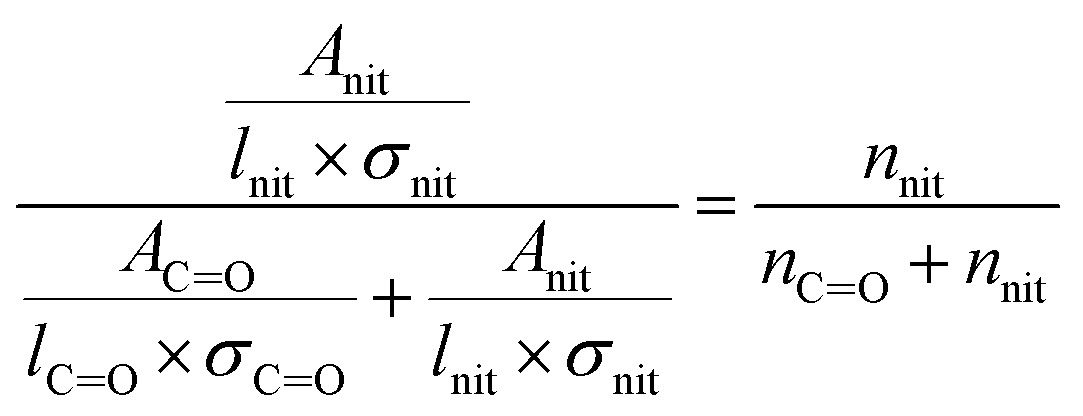 | (2) |
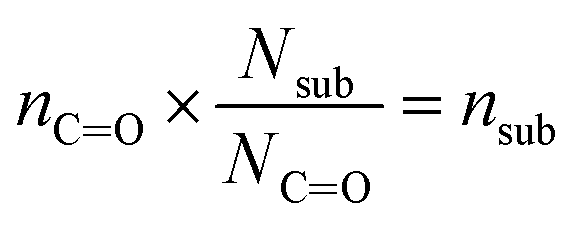 | (3) |
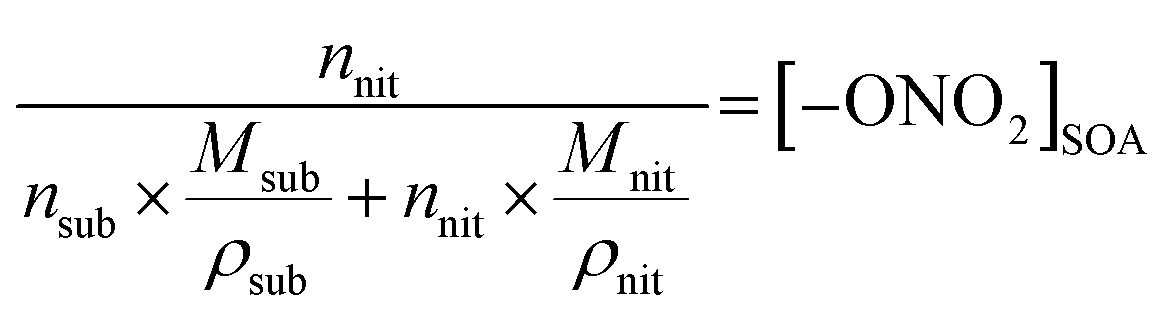 | (4) |
In eqn (2), Anit and AC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O are the absorbances for the –ONO2 and the CO, respectively. The absorption cross-section σ is in units of cm2 mole−1 (base 10), l is the pathlength of the IR beam through the film in cm at the selected wavenumbers, and nnit and nco concentrations are in moles –ONO2 cm−3 and moles CO cm−3 of film, respectively. The nco is converted into nsub (moles substrate cm−3) in eqn (3) using the number of CO groups (NCO) per substrate molecule. This includes any carbonyl, acid, anhydride, or ester functional groups that may be present in α-pinene SOA products. The value of NCO/Nsub is taken to be 2 for this SOA based on the literature.77,78 In eqn (4), the number of moles of substrate and nitrate in one cubic centimeter are converted to volume (in units of liters) using the molecular weights (M = 175 g mole−1 for 2EHN; 121 g mole−1 for HPN; 163 g mole−1 for HHN, assuming M = 200 g mole−1 for SOA),77–79 and the densities (ρ = 9.6 × 102 g L−1 for 2EHN; 1.2 × 103 g L−1 for HPN; 1.1 × 103 g L−1 for HHN, and using ρ = 1.2 × 103 g L−1 for SOA).67,80 The IR cross sections for all three organic nitrates and the proxy used for SOA, were previously reported.67
O are the absorbances for the –ONO2 and the CO, respectively. The absorption cross-section σ is in units of cm2 mole−1 (base 10), l is the pathlength of the IR beam through the film in cm at the selected wavenumbers, and nnit and nco concentrations are in moles –ONO2 cm−3 and moles CO cm−3 of film, respectively. The nco is converted into nsub (moles substrate cm−3) in eqn (3) using the number of CO groups (NCO) per substrate molecule. This includes any carbonyl, acid, anhydride, or ester functional groups that may be present in α-pinene SOA products. The value of NCO/Nsub is taken to be 2 for this SOA based on the literature.77,78 In eqn (4), the number of moles of substrate and nitrate in one cubic centimeter are converted to volume (in units of liters) using the molecular weights (M = 175 g mole−1 for 2EHN; 121 g mole−1 for HPN; 163 g mole−1 for HHN, assuming M = 200 g mole−1 for SOA),77–79 and the densities (ρ = 9.6 × 102 g L−1 for 2EHN; 1.2 × 103 g L−1 for HPN; 1.1 × 103 g L−1 for HHN, and using ρ = 1.2 × 103 g L−1 for SOA).67,80 The IR cross sections for all three organic nitrates and the proxy used for SOA, were previously reported.67
AMS measurements
An HR-ToF-AMS (Aerodyne)81–83 was used to characterize the particles formed in the absence or presence of the gas-phase organic nitrates and in the absence or presence of the OH scavenger. Particles were sampled at a flow rate of ∼0.082 L min−1 into the AMS and focused with an aerodynamic lens, vaporized at 600 °C, and ionized via EI (70 eV). The data presented were acquired in V-mode without HEPA-filter dilution. Measurements were taken with a particle filter at the beginning and end of each experiment to adjust the isotope ratio of 15N14N that interferes with the CHO+ fragment using the “Improved-Ambient” method of Canagaratna et al.84 Data were analyzed using Igor Pro v. 6.3 and 6.37 (Wavemetrics, Inc.) with SQUIRREL (v. 1.57I and 1.62A) and PIKA (v. 1.16I and 1.22A). Elemental analysis was carried out using the default calibration factors for O:C and H:C.Previous studies have shown that organic nitrates fragment in EI ionization to yield NO+ and NO2+ as major fragments,85–89 with small CHNO+ or CHN+ fragments.86,88 The ratio of NO+/NO2+ can be used to differentiate organic nitrates in the particles from inorganic nitrates or nitric acid.86,88,90 Details on this measurement and the measured ratios (Table S1†) are found in the electronic ESI.† The ratios indicate that the organic nitrate functional group remains unreacted once taken up into the particles. This is also supported by FTIR data showing the lack of detectable peaks due to inorganic NO3− in the infrared spectra (Fig. S1†) by comparison to NaNO3 (Fisher Scientific, 99.9%).
To quantify the amount of organic nitrate in the particles and to compare to the FTIR data, the AMS mass concentrations of NO+ and NO2+ were expressed as moles –ONO2 per liter of SOA. Thus, the mass loading (µg m−3) of NO+ and NO2+ were converted using eqn (5) into moles m−3 air of organic nitrate using the molecular weights of NO+ and NO2+ (30 and 46 g mole−1, respectively), assuming that each organic nitrate has only one nitrate group which will give either an NO+ or an NO2+ fragment. The mass concentration of SOA (µg m−3 air), represented by HROrg, is converted to volume concentration of SOA (L m−3 air) using its density (ρ = 1.2 × 103 g L−1)80 as shown in eqn (5):
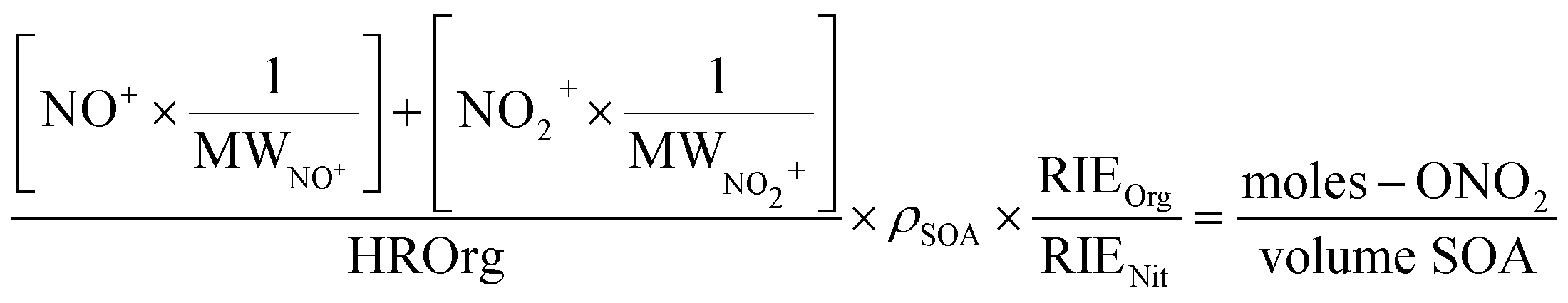 | (5) |
The default value for the relative ionization efficiency (RIE) of organics (1.4) was used for SOA, while an RIE of 1.0 was used for all organic nitrates, assuming their respective ionization efficiency is similar to that of inorganic nitrate as assumed by other researchers.51 To calculate the partition coefficient, K, the moles RONO2 per liter of SOA from eqn (5) was divided by the gas phase concentration of the organic nitrate in the flow reactor in moles –ONO2 per L air (eqn (1)). These concentrations were determined by measuring the concentration exiting the trap and factoring in the dilution into the flow reactor.
To examine changes in composition as a function of particle diameter, the high-resolution particle time-of-flight (HR-PToF) feature was used, which allows size-dependent composition analysis of specific fragments. The particle size distribution was separated into 12 evenly spaced bins between 157 and 822 nm Dva, and the high-resolution mass spectrum collected up to m/z 120. The data for diameters <157 nm and diameters >822 nm Dva were omitted due to the very small mass loading which caused large contributions from noise, and due to decreased lens transmission efficiency in these diameter ranges.91
FTIR quantification
For Series D (incorporation during growth), the polydisperse particles were impacted at a total flow of 30 L min−1 for 10–30 min at the end of the reactor. The partition coefficient was then quantified using the –ONO2 and carbonyl stretches as described above in eqn (1)–(4). Note that for this Series D, [–ONO2]SOA can include both the parent organic nitrate and a product from oxidation by OH.Results and discussion
(I) In the presence of an OH scavenger, cyclohexane
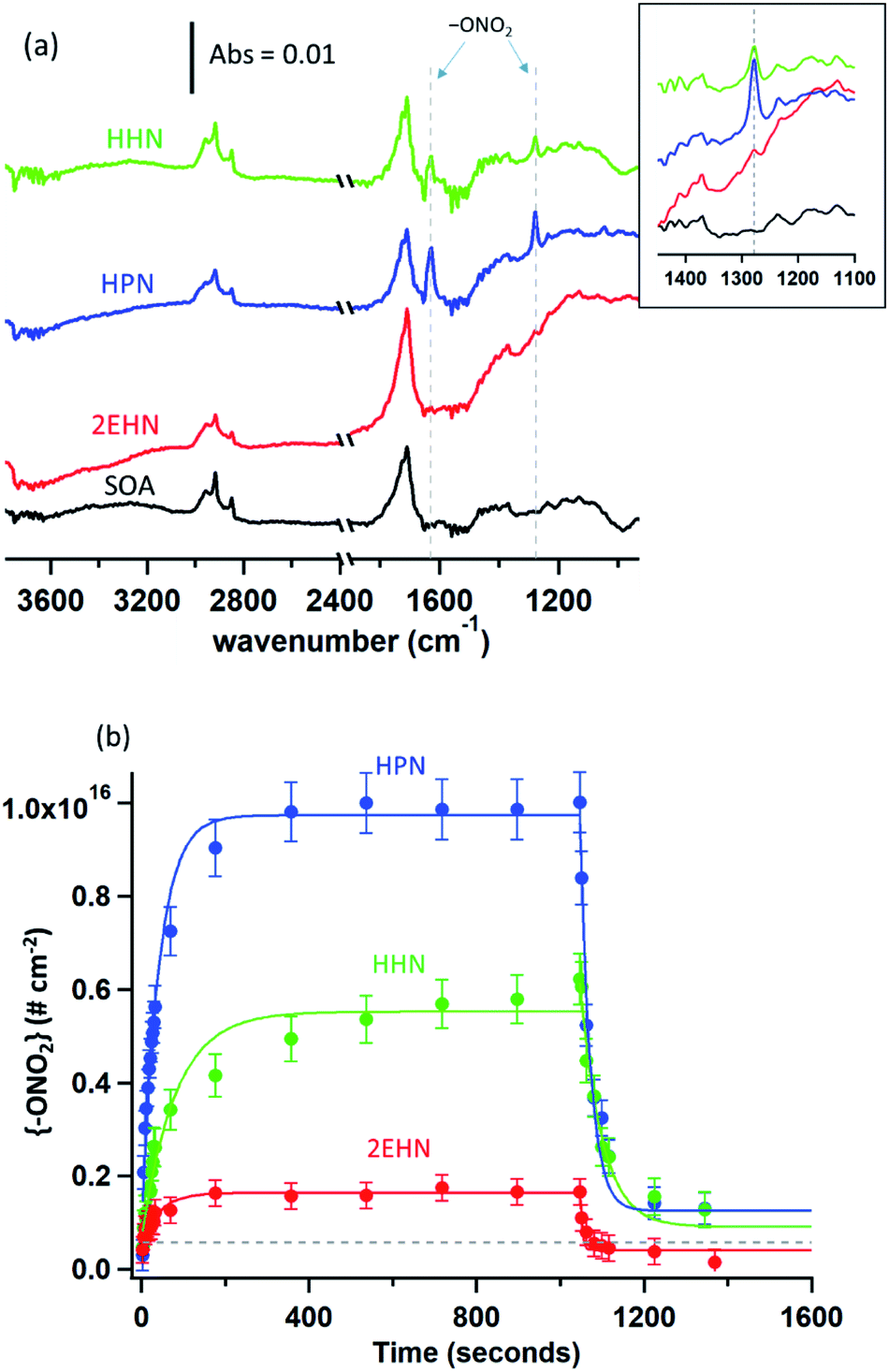 | ||
| Fig. 3 (a) ATR-FTIR spectra of the impacted particles formed in the presence of an OH scavenger, after exposure to 2EHN (4.7 × 1015 molecule per cm3, red), HPN (3.9 × 1015 molecule per cm3, blue), or HHN (1.7 × 1014 molecule per cm3, green) after equilibrium was reached for all organic nitrates (∼1000 seconds) (Series A). Also shown is the spectrum of SOA alone (black). The inset shows an expanded view of the 1280 cm−1 peak characteristic of the –ONO2 stretch. The region between 2500–2000 cm−1 is not shown due to variations in the CO2 in the sampling compartment. (b) Concentrations of the {–ONO2} functional group in molecules per cm2 after exposure of impacted particles (with total impacted mass of ∼30 µg) to the gaseous organic nitrates, and subsequent desorption by exposure to clean, dry air. The dashed black line indicates the experimentally-determined limit of detection for the nitrates. Solid lines are fits to guide the eye, and error bars are ± 2σ determined from the uncertainty in the measured absorption cross sections of 2EHN, HPN and HHN. | ||
To establish that the uptake into impacted particles is similar to particles suspended in air, experiments were carried out in which the SOA particles were denuded (to remove the α-pinene gas phase oxidation products), diverted to a mini glass flow tube and subsequently exposed to gas phase 2EHN (Series B). In this experiment, the measured 2EHN concentration (∼3 × 1015 molecules per cm3) was similar in magnitude to that used for the impacted particles in Series A. The partition coefficient for Series B was determined using AMS for times of exposure to 2EHN of approximately one to eight minutes. These exposure times encompass the timeframe for 2EHN to reach equilibrium in Series A experiments on impacted particles, which was approximately 2–3 min (Fig. 3b). The average partition coefficient (KB2EHN) is in excellent agreement with that for Series A (Table 2), confirming that suspended particles come to the same equilibrium as the thin film of impacted particles upon exposure to high concentrations of 2EHN. It also demonstrates that the AMS and the FTIR measurements are in excellent agreement with each other.
| Organic nitrate | K A (Series A)a,b | K B (Series B)c,d | K C (Series C)b,c,e | Ratio KC/KA |
|---|---|---|---|---|
| a Using ATR-FTIR. b Error bars are ±1σ from the average of three experiments. c Using AMS. d Error bars are ±1σ from the average of ∼1–8 minutes exposure time. e Values are taken at 31 minutes reaction time in the stainless steel flow reactor. f Series B was only done for 2EHN. | ||||
| 2EHN | (3.2 ± 1.5) × 104 | (2.9 ± 0.7) ×104 | (4.7 ± 1.0) × 105 | 15 ± 7.6 |
| HPN | (4.4 ± 2.0) × 105 | n/af | (1.7 ± 0.2) × 106 | 3.9 ± 1.8 |
| HHN | (4.9 ± 0.8) × 106 | n/af | (1.6 ± 0.3) × 107 | 3.3 ± 0.81 |
The concentration of 2EHN was reduced by removing the reservoir of 2EHN in Fig. 2b and instead introducing the 2EHN into the mini glass flow tube using a trap and flowing clean air over the pure liquid. This diluted the 2EHN by about a factor of 10, giving a concentration of ∼3 × 1014 molecules per cm3. Under these conditions, the organic nitrate signal in the particles is expected to approach the limit of detection, and in fact it was undetected. This smaller gas phase 2EHN concentration is comparable to the concentration used in Series C (discussed below) where a plasticizing effect was not observed. The lack of detection of the organic nitrate is thus consistent with earlier experiments that have shown that SOA from the ozonolysis of AP under dry conditions is a high viscosity semi-solid.35,37,41,92–96 In this case, net uptake of the organic nitrate into high viscosity SOA will be smaller than at equilibrium due to slow diffusion through the particles. Diffusion coefficients for particles from AP ozonolysis formed under dry conditions range from 10−14–10−17 cm2 s−1.40,42,92,97 Using the Stokes–Einstein relation26,37 and assuming a molecular radius of 1 nm, this results in viscosities ranging from 105–108 Pa s, consistent with measured viscosities for SOA from AP ozonolysis.37,98–102 The resulting characteristic timescale for diffusion26 through a semi-solid 200 nm particle is at least half an hour, much longer than the maximum residence time for Series B of ∼8 minutes.
The results show that the partition coefficients in Series C are much larger than those in Series A, which might seem surprising since they imply a larger than equilibrium concentration in the particles. As described above, the difference observed is not associated with bias in the two analytical techniques (AMS and ATR-FTIR) that were used. Additionally, Series C used much lower gas phase concentrations than for Series A, so the explanation cannot be a significant plasticizing effect. In support of this, Fig. S2† shows typical impaction patterns for SOA formed either with or without an organic nitrate or OH scavenger present. Also shown in Fig. S2† for comparison are the impaction patterns for deliquesced Na2SO4 particles, dry carboxylate-modified latex particles, and SOA particles formed at 87% relative humidity which is known to decrease viscosity.41,99,100,103 Upon impaction, particles hit and may stick to the substrate directly below the orifice plate to form spots, form midlines due to multi-orifice interactions, or they may bounce and either be re-entrained into the gas stream or subsequently be re-captured on the substrate to form a cloud or halo.104 The impaction patterns in Fig. S2† are distinctly different from both the deliquesced Na2SO4 and the SOA formed at high relative humidity, indicating the particles in the current study are highly viscous. The patterns do not change across the experimental conditions, suggesting there was no significant change in the viscosity upon addition of the organic nitrates or cyclohexane. One might therefore expect that the uptake into high viscosity semi-solid particles would be hindered; however, our results (Series C) show the opposite, which highlights that the incorporation of organic nitrates is driven by a different phenomenon than diffusion.
When the 2EHN concentration used in Series B was lowered to that used in Series C, the organic nitrate signal became undetectable, consistent with a higher viscosity limiting uptake into and diffusion through the pre-formed and denuded particles.38KC values from Series C taken at 7 min and 31 min are not significantly different, and thus the higher partition coefficients in Series C are not resulting from the longer time spent in the flow reactor. The presence of the gas-phase ozonolysis products in the large flow reactor in Series C must therefore play a central role in the incorporation of the organic nitrates during particle formation and growth that enhances organic nitrate uptake beyond the expected equilibrium established in Series A/B.
(II) In the absence of an OH scavenger
| Organic nitrate | Partition coefficient (KA)a for SOA with CH | Partition coefficient (KA)a,b for SOA without CH67 |
|---|---|---|
| a Error bars are ±1σ from the average of three experiments. b These values have been adjusted from previous concentrations using estimated vapor pressures to the newly measured gas phase concentrations in this study. | ||
| 2EHN | (3.2 ± 1.5) × 104 | (1.1 ± 0.1) × 105 |
| HPN | (4.4 ± 2.0) × 105 | (5.4 ± 2.0) × 105 |
| HHN | (4.9 ± 0.8) × 106 | (9.0 ± 1.0) × 106 |
For HPN, the partition coefficient on impacted particles formed without CH is not statistically different from that measured on impacted particles formed in the presence of CH, and is in excellent agreement with the air-octanol partition coefficient of (4.2 ± 0.3) ×105 reported by Treves et al.105 This suggests that the magnitude of the HPN partitioning into the particles is unaffected by composition changes in the SOA resulting from changes in the chemistry in the presence of the OH scavenger. It is interesting that the partition coefficients into SOA and octanol are similar and both larger than into water,106,107 suggesting that even with the hydrogen bonding possibility to the alcohol group of HPN, dispersion forces between HPN and SOA are important as well.
For 2EHN and HHN, the partition coefficients decrease when the SOA is pre-formed in the presence of the OH scavenger by factors of ∼3 and ∼2, respectively. This reduction in partitioning indicates decreased solubility of 2EHN or HHN into the bulk of the film, which could reflect differences in the SOA composition when CH is added to scavenge the OH. Table S2† shows the O:C and H:C ratios and the oxidation state of carbon (OSc) determined by AMS.108 Only very small changes in the bulk elemental composition were exhibited, consistent with previous reports for AP ozonolysis SOA formed with or without an OH scavenger.109 However, this may simply reflect that functional group changes important in determining solubility are not detected in these bulk measurements. The percent change of a few major fragments by AMS when OH scavenger is present (Fig. S3†) shows that there are some changes in the SOA bulk composition. Why this results in changes in the partition coefficients for 2EHN and HHN, and not for HPN, is not clear, but illustrates the need for a detailed molecular level understanding of the composition of SOA.110,111
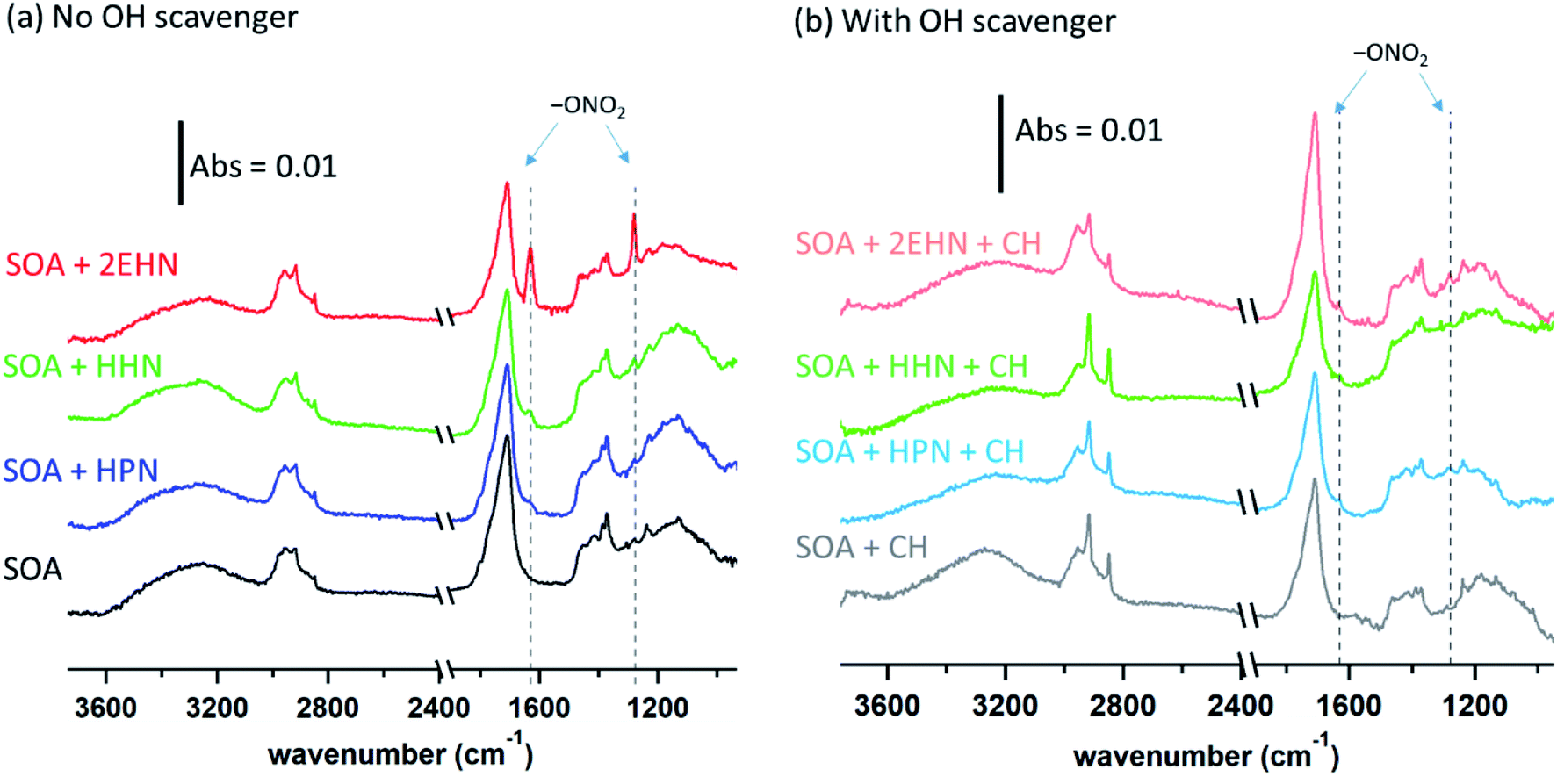 | ||
| Fig. 4 ATR-FTIR spectra for (a) impacted particles alone and formed in the presence of 2EHN (1.4 × 1014 molecules per cm3), HPN (1.2 × 1014 molecules per cm3), or HHN (5.0 × 1012 molecules per cm3) in the stainless steel flow reactor (Series D) without an OH scavenger, and (b) impacted particles alone and formed in the presence of 2EHN, HPN or HHN at the same concentrations in the stainless steel flow reactor in the presence of CH as an OH scavenger (Series D). The region between 2500–2000 cm−1 is not shown due to variations in the CO2 in the sampling compartment. | ||
When the gas-phase organic nitrate is present in the flow reactor in the absence of an OH scavenger (Series C and D, no CH), the organic nitrates can react in the gas phase with the OH radical generated in the ozonolysis. The calculated OH rate constants for 2EHN and HHN are similar, and are a factor of approximately three larger than that for HPN (Table S3†).115 This is due to the long alkyl chains in 2EHN and HHN which provide a number of potential sites for hydrogen abstraction by OH. Many of the products formed from OH oxidation of 2EHN and HHN are expected to be more functionalized and have lower volatility than the parent organic nitrate. Scheme 1 shows some of the likely routes for oxidation of those organic nitrates. Thus, the combination of higher rate constants and lower volatility OH oxidation products are such that the organic nitrate–OH oxidation products for 2EHN and HHN can contribute significantly to the nitrate content of the SOA in the absence of an OH scavenger. As a result, suppressing OH by the addition of cyclohexane significantly lowers the formation of the OH oxidation products of the organic nitrates.
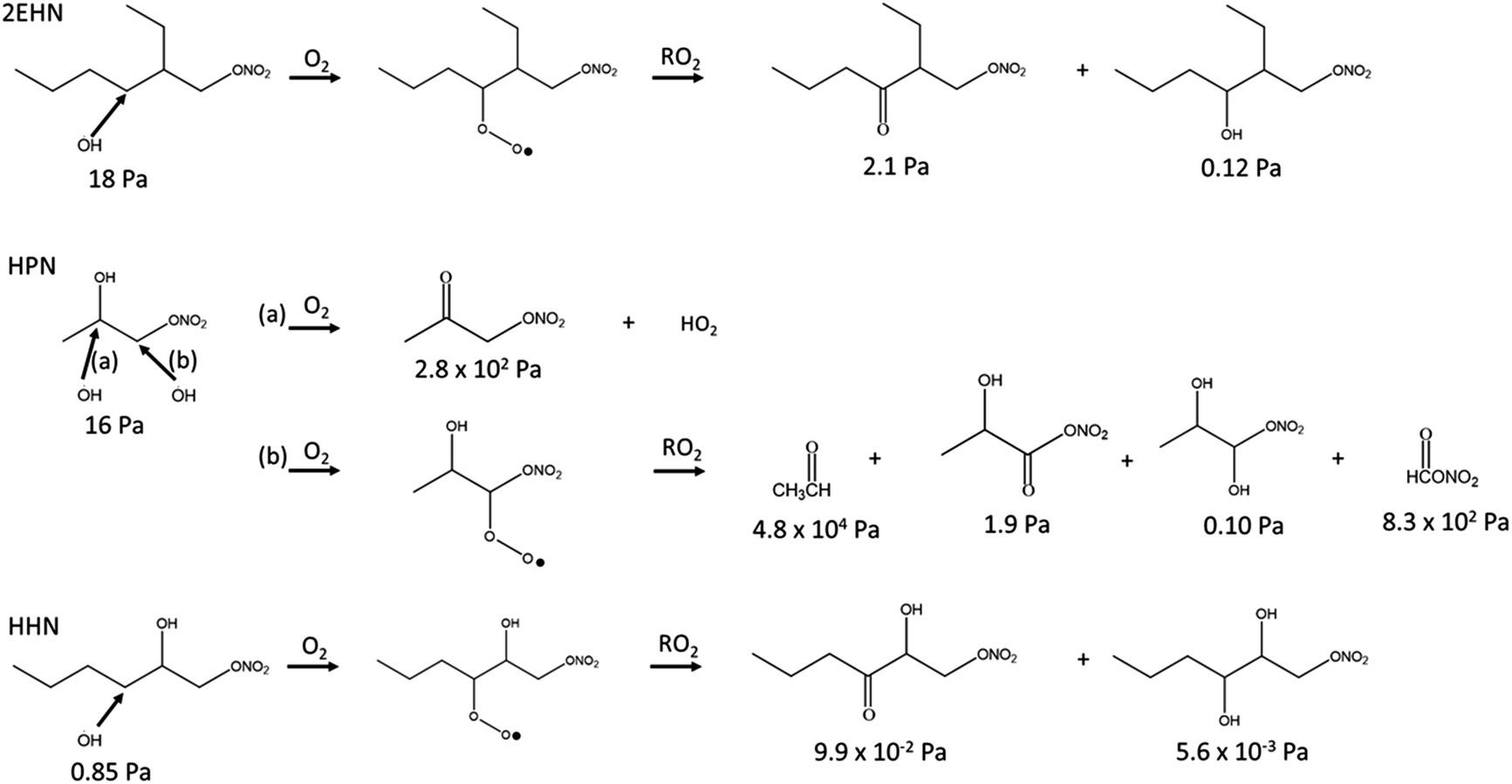 | ||
| Scheme 1 Simplified reaction scheme showing some pathways for OH oxidation of 2EHN, HPN and HHN. The vapor pressures for each compound were estimated using SIMPOL.1.72 Hydroxyl radical attack on a secondary carbon is shown for 2EHN and HHN due to the higher relative contribution of the sum of the secondary carbons to the total OH rate constant compared to that of the one tertiary carbon. | ||
The effect is more dramatic for 2EHN because its concentration in the experiments is about 28 times greater than that of HHN so that 2EHN itself is a more efficient OH scavenger. While HPN can also react with OH, oxidation at the tertiary carbon of the molecule is most likely and will lead to smaller, more volatile species which may not partition significantly into the particles (Scheme 1).
The trends seen in the organic nitrate signal measured by FTIR (Series D) in the presence or absence of the OH scavenger are also supported by the AMS data (Series C). Fig. 5 shows the number of moles RONO2 taken up per liter of SOA calculated from the AMS data (eqn (5)) at 31 min reaction time (there was no statistical difference between 7 min and 31 min reaction time). Consistent with the FTIR data, the signal for 2EHN increases significantly (by a factor of ∼14) when the SOA is formed in the absence of the OH scavenger. This factor is larger than that of Series A, indicating that it is the oxidation products of 2EHN that are incorporated into the SOA, and is not simply due to a difference in the SOA bulk composition. A similar but smaller trend is seen for HHN (a factor of ∼2.5), whereas the amount of HPN in the particles is unaffected by the presence of the OH scavenger. However, it should be noted that the detection methods used here (FTIR and AMS) focus on functional group analysis and are not direct measurements of the parent organic nitrate.
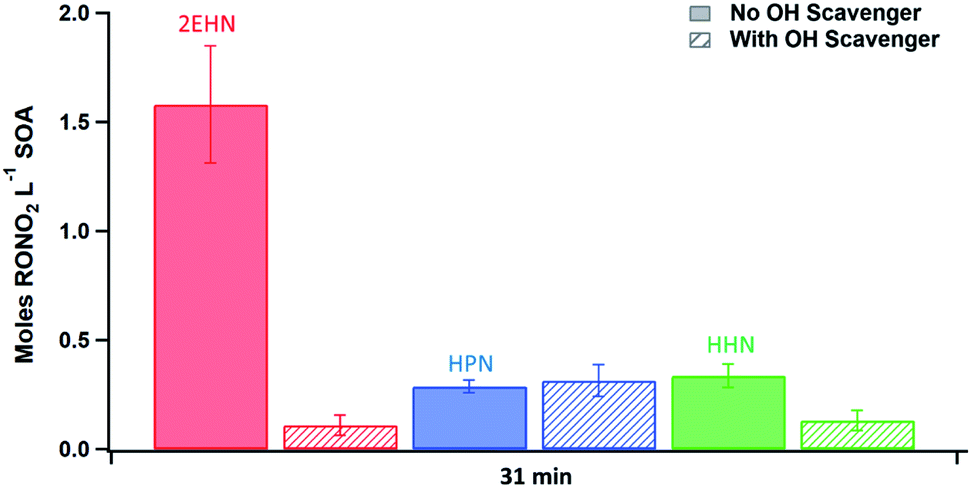 | ||
| Fig. 5 Quantification of moles of organic nitrate per liter of SOA (moles RONO2 per L SOA) for SOA formed in the presence of 2EHN (1.4 × 1014 molecules per cm3), HPN (1.2 × 1014 molecules per cm3), or HHN (5.0 × 1012 molecules per cm3), with or without OH scavenger at 31 minutes reaction time (Series C). Error bars are ± 2σ from the average of three experiments. | ||
The much smaller effect seen for HHN is due to the smaller gas phase concentrations that were able to be added to the flow system, which results in HHN not competing very effectively with the α-pinene for the OH radical generated in the ozonolysis. Thus, the initial first order rates of loss of OH, estimated using k[X]0, where X = AP or RONO2, are 3.3 × 102 s−1 for AP and 8.8 × 102 s−1 for 2EHN under the conditions shown in Fig. 5 (the rate for AP decreases with time due to reaction with O3). As a result, 2EHN competes with AP for OH, forming oxidized alkyl nitrate products that are incorporated into the SOA. On the other hand, k[X]0 for HHN is only 27 s−1 so that relatively small amounts are oxidized by OH whose removal is now mainly via reaction with AP, and incorporation of the unoxidized parent HHN contributes relatively more than its oxidation products to SOA growth. This is consistent with the magnitude of change in the incorporation of HHN into SOA formed with versus without CH being similar for Series A (uptake into preformed SOA) and Series C/D (incorporation during SOA formation). While the first order loss rate for HPN by OH is 1.9 × 102 s−1, its oxidation products are sufficiently small that they will not be efficiently incorporated into the SOA.
The contribution of OH radical chemistry is also manifested in the decrease in the particle number concentration in the presence of the OH scavenger, and in the presence of the organic nitrates which can also scavenge OH (Fig. 6). Note that the total particle number concentration is smaller in the presence of CH (Fig. 6a). This is consistent with previously reported work on the impact of the OH chemistry on SOA formation.116,117 For example, Berndt et al.118 showed that the OH radical plays an important role in the formation of highly oxidized multifunctional organic compounds (HOMs), and thus scavenging the OH suppresses the formation of these HOMs, lowering SOA yields. Fig. 6b, d, and 6f show the particle size distributions when organic nitrates are present during SOA formation without CH. A decrease in SOA is seen in the presence of 2EHN in a manner that is qualitatively similar to that due to addition of CH alone (Fig. 6a), while there is little impact for HPN, and no impact at all for HHN. This is consistent with the relative rate constants for OH reaction with the organic nitrates versus CH (Table S3†) and the initial concentrations of these compounds. As described above, 2EHN competes with AP for OH, but HHN does not. The rate of loss of OH by CH is 1.8 × 104 s−1, which overwhelms the reaction of OH with AP or the organic nitrates. Based on kinetic modeling using Kintecus®119 and a simplified reaction scheme (Table S3†), less than 0.1% of the organic nitrates reacts with OH after 31 min reaction time when 100 ppm CH is present. Additional experiments were done for 2EHN using 500 ppm CH, and there was no statistical difference in the amount of 2EHN incorporated, indicating that 100 ppm CH is enough to adequately scavenge the OH. At 31 min reaction time without CH present, approximately 1.3% of the 2EHN has reacted with OH compared to ∼0.8% of the HPN and ∼4.3% of the HHN. This is consistent with the trend in the impacts on SOA, given the much lower initial gas phase concentration of HHN compared to the other organic nitrates.
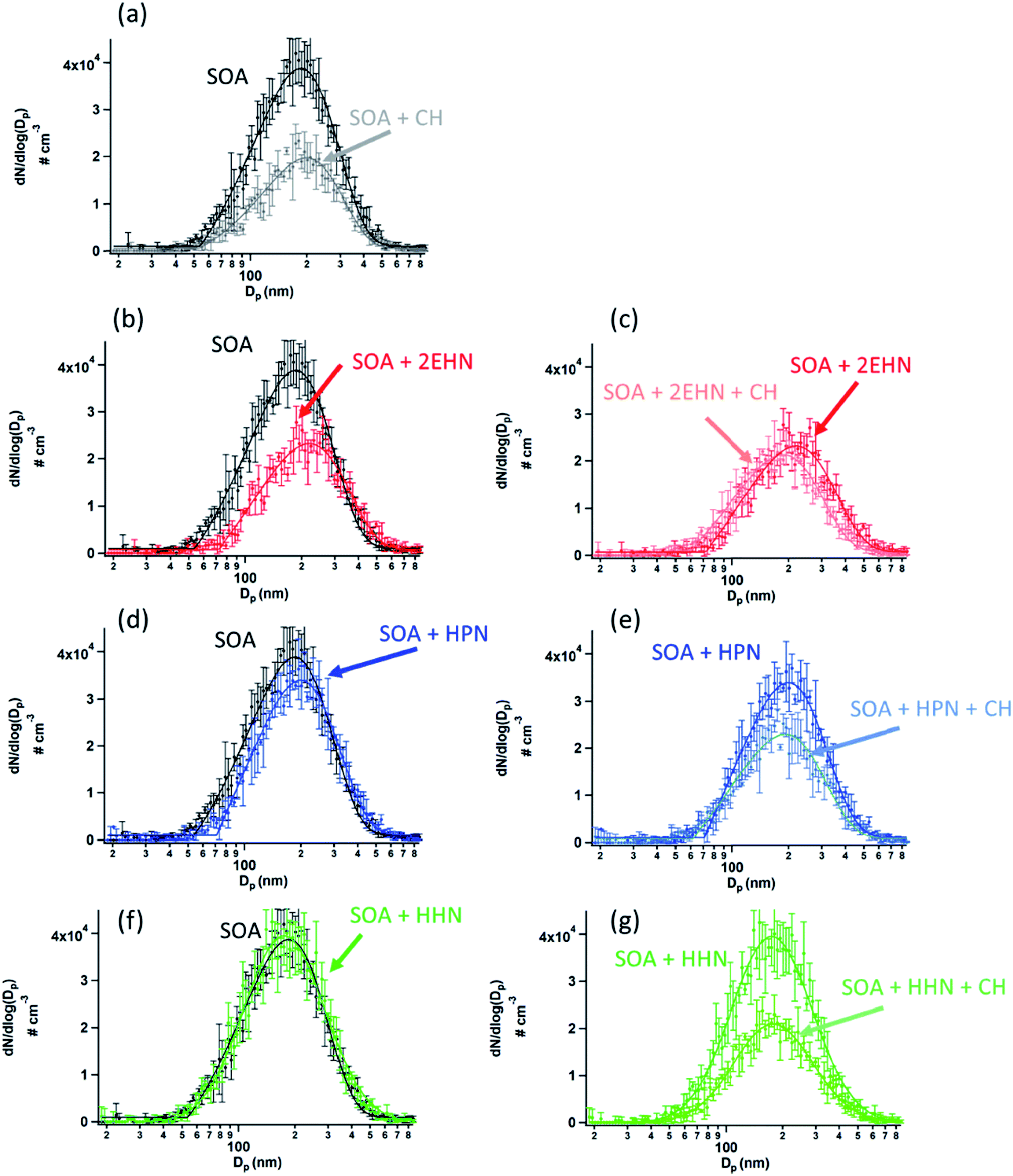 | ||
| Fig. 6 Particle number distributions (# cm−3) at 31 minutes reaction time for (a) SOA alone and formed in the presence of CH as an OH scavenger, (b) SOA alone and formed in the presence of 2EHN (1.4 × 1014 molecules per cm3), (c) SOA formed in the presence of 2EHN either with or without CH, (d) SOA alone and formed in the presence of HPN (1.2 × 1014 molecules per cm3), (e) SOA formed in the presence of HPN either with or without CH, (f) SOA alone and formed in the presence of HHN (5.0 × 1012 molecules per cm3), and (g) SOA formed in the presence of HHN either with or without CH. Error bars are ±1σ from the average of three scans, and solid lines are best fit distributions to guide the eye. | ||
In the absence of an OH scavenger, partition coefficients for 2EHN and HHN cannot be reliably quantified due to contributions from the RONO2 + OH oxidation products whose identity and gas phase concentrations are not known. Instead, the concentrations of RONO2 in SOA (moles of RONO2 per liter of SOA) were measured using both AMS (Fig. 5) and FTIR as described in the experimental (Series C and D). The values for 2EHN and HHN are summarized in Table 4, showing the two methods are in agreement within 50%. Only the AMS value (Series C) is reported for HPN, as it was below the detection limit by FTIR.
| RONO2 content (moles L−1 SOA) a Series C (AMS) | RONO2 content (moles L−1 SOA) a,b Series D (FTIR) | |
|---|---|---|
| a Error bars are ± 2σ from the average of three experiments. b FTIR quantification used the absorption cross section of the parent organic nitrate.67 c HPN was below the detection limit for FTIR. | ||
| 2EHN | 1.6 ± 0.3 | 1.6 ± 0.2 |
| HPN | 0.29 ± 0.03 | n/ac |
| HHN | 0.34 ± 0.05 | 0.50 ± 0.05 |
In short, OH oxidation of these organic nitrates forms more oxidized organic nitrates that, in the case of 2EHN and HHN, partition to a greater extent into the SOA. The rate constant for OH with HPN is smaller than for 2EHN and HHN, and in addition, its oxidation is expected to lead to smaller, higher volatility products, which will not be readily taken up into SOA (Scheme 1).
Physical mechanism for particle growth
As described previously, the growth of the SOA particles by ozonolysis products and the incorporation of the organic nitrates into the particles in the stainless steel flow reactor may be best described by a kinetically limited “burying” mechanism. Fig. 7 shows a simplified schematic that describes this mechanism, where RONO2 represents the organic nitrate, and P1 is a proxy low volatility organic from the ozonolysis of α-pinene. When the organic nitrate is present during particle growth in the flow reactor (Series C/D), condensing P1 molecules can bury the organic nitrate and hinder re-evaporation into the gas phase, resulting in larger partition coefficients than those measured after particle growth at equilibrium (Series A/B).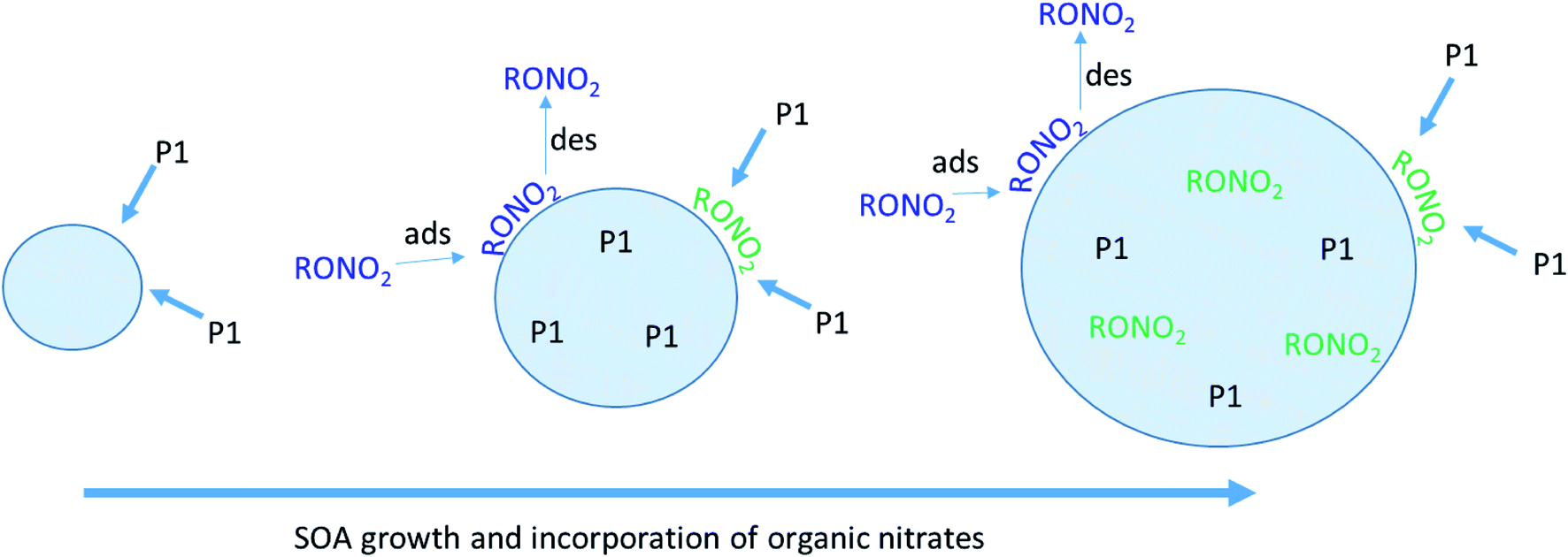 | ||
| Fig. 7 Schematic of “burying” mechanism for incorporation of organic nitrate tracers as SOA particles are forming. RONO2 represents the organic nitrate, and P1 is a proxy for low volatility organics from the ozonolysis of α-pinene. When the organic nitrate is present during particle growth, the P1 can bury the organic nitrate and hinder its re-evaporation into the gas-phase. | ||
Fig. 8 shows the HR-PToF data for both the total organic and the ratio of (NO+ + NO2+) to total organics as a function of particle size for SOA formed in the presence of 2EHN at 7 min reaction time, either with or without CH. The ratio (NO+ + NO2+)/HROrg is a measure of the relative concentrations of organic nitrate in the SOA. This ratio is approximately constant across the range of particle sizes, confirming that relative rates of incorporation of the organic nitrates and the organics that grow the particles do not vary significantly as the particles grow across this diameter range. The corresponding data for SOA formed in the presence of HPN and HHN are found in Fig. S5 and S6,† respectively, and show similar results. For comparison, Fig. S7† shows HR-PToF analysis for two major fragments, C2H3O+ (a marker for carbonyl groups) and CO2+ (a marker for carboxylic acid groups),120,121 and the ratio of these two fragments. In contrast to the uniform organic nitrate composition over all particle diameters, smaller diameter SOA particles are composed of more acid groups on average than the larger particles, either in the absence or presence of OH.
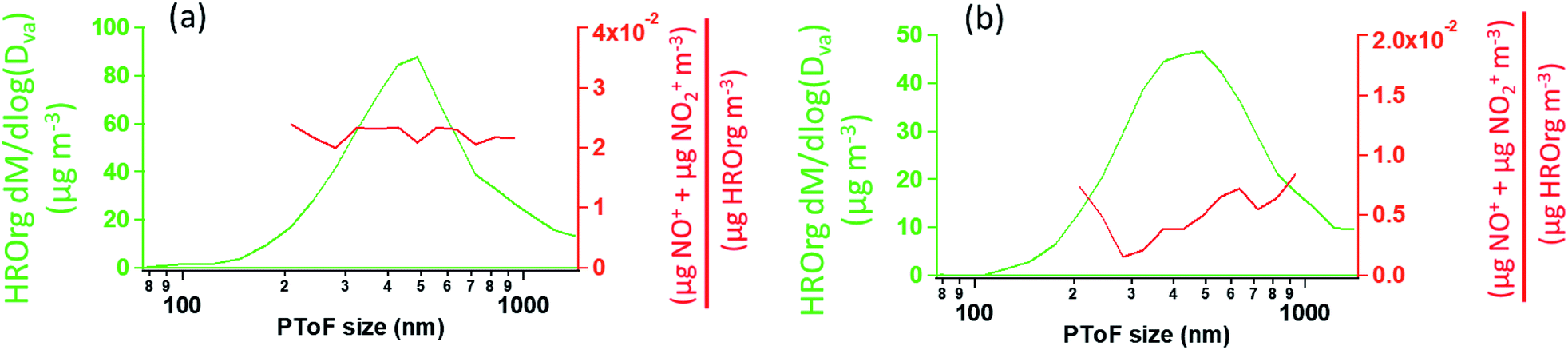 | ||
| Fig. 8 The HR-PToF mass distribution of total HROrg (green) and the HR-PToF mass ratio of (NO+ + NO2+) to HROrg (red) for (a) SOA formed in the presence of 2EHN (1.4 × 1014 molecules per cm3) at 7 min reaction time, and (b) SOA formed in the presence of 2EHN and CH (2.5 × 1015 molecules per cm3) at 7 min reaction time. Note the NO+ and NO2+ signals in the presence of CH have high uncertainty due to weak signal. | ||
Conclusions
Uptake of organic nitrate tracers into highly viscous, semi-solid SOA particles during their formation in the ozonolysis of AP offers new insights into the molecular interactions between gases and particles that ultimately lead to particle growth. The role of the OH radical resulting from the ozonolysis reaction is important not only for the bulk composition of the particles and overall SOA number concentration, but also plays an important role in the gas-phase chemistry of the organic nitrates. In the case of the smallest organic nitrate, HPN, the partitioning was unaffected both by the reaction of HPN with OH and by any changes in the particle composition by scavenging OH. However, the two long-chain organic nitrates were affected by both of these factors.The amount of organic nitrate taken up into growing particles relative to the gas phase concentration was found to be larger than expected based on the equilibrium partition coefficients into pre-existing impacted particles. This may be attributed to the evolution of particles during growth, such that continued uptake of organics leads to ‘burying’ of the organic nitrate, hindering their re-evaporation into the gas-phase. This is consistent with the HR-PToF analysis which shows that the organic nitrate was evenly distributed across all particle diameters. This could play a role in cases where mechanisms in addition to thermodynamic partitioning have been implicated.34,60,68,122 The results of these studies highlight the importance of a molecular level understanding of the interactions of gases with particle surfaces and their bulk as the foundation for accurately predicting their impacts on air quality and climate.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the NSF (Grant #1647386), and by the NSF Major Research Instrumentation (MRI) program (Grants #1337080 and #0923323), and the Army Research Office (Grant #W911NF1710105). We thank Dr Donna Sueper at Aerodyne Research, Inc. for her help with the HR-PToF analysis and Prof. David Soulsby of the University of Redlands for the NMR analysis of the organic nitrates.References
- C. A. Pope and D. W. Dockery, Health effects of fine particulate air pollution: lines that connect, J. Air Waste Manage. Assoc., 2006, 56, 709–742 CrossRef CAS PubMed.
- J. L. Mauderly and J. C. Chow, Health effects of organic aerosols, Inhalation Toxicol., 2008, 20, 257–288 CrossRef CAS.
- M. R. Heal, P. Kumar and R. M. Harrison, Particles, air quality, policy and health, Chem. Soc. Rev., 2012, 41, 6606–6630 RSC.
- A. Nel, Air pollution-related illness: effects of particles, Science, 2005, 308, 804–806 CrossRef CAS.
- P. M. Mannucci, S. Harari, I. Martinelli and M. Franchini, Effects on health of air pollution: a narrative review, Intern Emerg Med., 2015, 10, 657–662 CrossRef.
- P. J. Landrigan, R. Fuller, N. J. R. Acosta, O. Adeyi, R. Arnold, N. Basu, A. B. Baldé, R. Bertollini, S. Bose-O'Reilly, J. I. Boufford, P. N. Breysse, T. Chiles, C. Mahidol, A. M. Coll-Seck, M. L. Cropper, J. Fobil, V. Fuster, M. Greenstone, A. Haines, D. Hanrahan, D. Hunter, M. Khare, A. Krupnick, B. Lanphear, B. Lohani, K. Martin, K. V. Mathiasen, M. A. McTeer, C. J. L. Murray, J. D. Ndahimananjara, F. Perera, J. Potočnik, A. S. Preker, J. Ramesh, J. Rockström, C. Salinas, L. D. Samson, K. Sandilya, P. D. Sly, K. R. Smith, A. Steiner, R. B. Stewart, W. A. Suk, O. C. P. van Schayck, G. N. Yadama, K. Yumkella and M. Zhong, The Lancet Commission on pollution and health, Lancet, 2018, 391, 462–512 CrossRef.
- U. Poschl, Atmospheric aerosols: Composition, transformation, climate and health effects, Angew. Chem., Int. Ed., 2005, 44, 7520–7540 CrossRef.
- B. J. Finlayson-Pitts and J. N. Pitts, Chemistry of the Upper and Lower Atmosphere: Theory, Experiments, and Applications, Academic Press, 2000 Search PubMed.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, Wiley, 2006 Search PubMed.
- W. C. Hinds, Aerosol Technology: Properties, Behavior, and Measurement of Airborne Particles, John Wiley & Sons, 1982 Search PubMed.
- A. Singh, W. J. Bloss and F. D. Pope, 60 years of UK visibility measurements: impact of meteorology and atmospheric pollutants on visibility, Atmos. Chem. Phys., 2017, 17, 2085–2101 CrossRef CAS.
- O. Boucher, D. Randall, P. Artaxo, C. Bretherton, G. Feingold, P. Forster, V.-M. Kerminen, Y. Kondo, H. Liao, U. Lohmann, P. Rasch, S. K. Satheesh, S. Sherwood, B. Stevens and X. Y. Zhang, Clouds and Aerosols, in Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change, ed. T. F. Stocker, D. Qin, G.-K. Plattner, M. Tignor, S. K. Allen, J. Boschung, A. Nauels, Y. Xia, V. Bex and P. M. Midgley, Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA, 2013, DOI:10.1017/CBO9781107415324.
- M. Kanakidou, J. H. Seinfeld, S. N. Pandis, I. Barnes, F. J. Dentener, M. C. Facchini, R. Van Dingenen, B. Ervens, A. Nenes, C. J. Nielsen, E. Swietlicki, J. P. Putaud, Y. Balkanski, S. Fuzzi, J. Horth, G. K. Moortgat, R. Winterhalter, C. E. L. Myhre, K. Tsigaridis, E. Vignati, E. G. Stephanou and J. Wilson, Organic aerosol and global climate modelling: a review, Atmos. Chem. Phys., 2005, 5, 1053–1123 CrossRef CAS.
- M. Hallquist, J. C. Wenger, U. Baltensperger, Y. Rudich, D. Simpson, M. Claeys, J. Dommen, N. M. Donahue, C. George, A. H. Goldstein, J. F. Hamilton, H. Herrmann, T. Hoffmann, Y. Iinuma, M. Jang, M. E. Jenkin, J. L. Jimenez, A. Kiendler-Scharr, W. Maenhaut, G. McFiggans, T. F. Mentel, A. Monod, A. S. H. Prevot, J. H. Seinfeld, J. D. Surratt, R. Szmigielski and J. Wildt, The formation, properties and impact of secondary organic aerosol: current and emerging issues, Atmos. Chem. Phys., 2009, 9, 5155–5236 CrossRef CAS.
- J. P. D. Abbatt, A. K. Y. Lee and J. A. Thornton, Quantifying trace gas uptake to tropospheric aerosol: recent advances and remaining challenges, Chem. Soc. Rev., 2012, 41, 6555–6581 RSC.
- R. Y. Zhang, G. H. Wang, S. Guo, M. L. Zarnora, Q. Ying, Y. Lin, W. G. Wang, M. Hu and Y. Wang, Formation of urban fine particulate matter, Chem. Rev., 2015, 115, 3803–3855 CrossRef CAS PubMed.
- R. Y. Zhang, A. Khalizov, L. Wang, M. Hu and W. Xu, Nucleation and growth of nanoparticles in the atmosphere, Chem. Rev., 2012, 112, 1957–2011 CrossRef CAS PubMed.
- C. E. Kolb, R. A. Cox, J. P. D. Abbatt, M. Ammann, E. J. Davis, D. J. Donaldson, B. C. Garrett, C. George, P. T. Griffiths, D. R. Hanson, M. Kulmala, G. McFiggans, U. Pöschl, I. Riipinen, M. J. Rossi, Y. Rudich, P. E. Wagner, P. M. Winkler, D. R. Worsnop and C. D. O' Dowd, An overview of current issues in the uptake of atmospheric trace gases by aerosols and clouds, Atmos. Chem. Phys., 2010, 10, 10561–10605 CrossRef CAS.
- T. Berkemeier, A. J. Huisman, M. Ammann, M. Shiraiwa, T. Koop and U. Pöschl, Kinetic regimes and limiting cases of gas uptake and heterogeneous reactions in atmospheric aerosols and clouds: a general classification scheme, Atmos. Chem. Phys., 2013, 13, 6663–6686 CrossRef.
- N. M. Donahue, A. L. Robinson, C. O. Stanier and S. N. Pandis, Coupled partitioning, dilution, and chemical aging of semivolatile organics, Environ. Sci. Technol., 2006, 40, 2635–2643 CrossRef CAS PubMed.
- N. M. Donahue, E. R. Trump, J. R. Pierce and I. Riipinen, Theoretical constraints on pure vapor-pressure driven condensation of organics to ultrafine particles, Geophys. Res. Lett., 2011, 38, L16801 CrossRef.
- U. Pöschl, Y. Rudich and M. Ammann, Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions - Part 1: General equations, parameters, and terminology, Atmos. Chem. Phys., 2007, 7, 5989–6023 CrossRef.
- M. Ammann, U. Pöschl and Y. Rudich, Effects of reversible adsorption and Langmuir-Hinshelwood surface reactions on gas uptake by atmospheric particles, Phys. Chem. Chem. Phys., 2003, 5, 351–356 RSC.
- M. Ammann and U. Poschl, Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions - Part 2: Exemplary practical applications and numerical simulations, Atmos. Chem. Phys., 2007, 7, 6025–6045 CrossRef CAS.
- C. Pfrang, M. Shiraiwa and U. Poschl, Chemical ageing and transformation of diffusivity in semi-solid multi-component organic aerosol particles, Atmos. Chem. Phys., 2011, 11, 7343–7354 CrossRef CAS.
- M. Shiraiwa, M. Ammann, T. Koop and U. Pöschl, Gas uptake and chemical aging of semisolid organic aerosol particles, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 11003–11008 CrossRef CAS.
- J. F. Pankow, Further discussion of the octanol/air partition coefficient KOA as a correlating parameter for gas/particle partitioning coefficients, Atmos. Environ., 1998, 32, 1493–1497 CrossRef CAS.
- J. F. Pankow, Review and comparative-analysis of the theories on partitioning between the gas and aerosol particulate phases in the atmosphere, Atmos. Environ., 1987, 21, 2275–2283 CrossRef CAS.
- J. F. Pankow, Gas/particle partitioning of neutral and ionizing compounds to single and multi-phase aerosol particles. 1. Unified modeling framework, Atmos. Environ., 2003, 37, 3323–3333 CrossRef CAS.
- T. Salthammer and K.-U. Goss, Predicting the gas/particle distribution of SVOCs in the indoor environment using poly parameter linear free energy relationships, Environ. Sci. Technol., 2019, 53, 2491–2499 CrossRef CAS.
- F. Yu, A secondary organic aerosol formation model considering successive oxidation aging and kinetic condensation of organic compounds: global scale implications, Atmos. Chem. Phys., 2011, 11, 1083–1099 CrossRef CAS.
- L. I. Kleinman, S. R. Springston, J. Wang, P. H. Daum, Y. N. Lee, L. J. Nunnermacker, G. I. Senum, J. Weinstein-Lloyd, M. L. Alexander, J. Hubbe, J. Ortega, R. A. Zaveri, M. R. Canagaratna and J. Jayne, The time evolution of aerosol size distribution over the Mexico City plateau, Atmos. Chem. Phys., 2009, 9, 4261–4278 CrossRef CAS.
- J. Ye, P. Van Rooy, C. H. Adam, C.-H. Jeong, B. Urch, D. R. Cocker III, G. J. Evans and A. W. H. Chan, Predicting secondary organic aerosol enhancement in the presence of atmospherically relevant organic particles, ACS Earth Space Chem., 2018, 2, 1035–1046 CrossRef CAS.
- I. Riipinen, J. R. Pierce, T. Yli-Juuti, T. Nieminen, S. Häkkinen, M. Ehn, H. Junninen, K. Lehtipalo, T. Petäjä, J. Slowik, R. Chang, N. C. Shantz, J. Abbatt, W. R. Leaitch, V. M. Kerminen, D. R. Worsnop, S. N. Pandis, N. M. Donahue and M. Kulmala, Organic condensation: a vital link connecting aerosol formation to cloud condensation nuclei (CCN) concentrations, Atmos. Chem. Phys., 2011, 11, 3865–3878 CrossRef CAS.
- T. D. Vaden, D. Imre, J. Beránek, M. Shrivastava and A. Zelenyuk, Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2190–2195 CrossRef CAS.
- A. Virtanen, J. Kannosto, H. Kuuluvainen, A. Arffman, J. Joutsensaari, E. Saukko, L. Hao, P. Yli-Pirila, P. Tiitta, J. K. Holopainen, J. Keskinen, D. R. Worsnop, J. N. Smith and A. Laaksonen, Bounce behavior of freshly nucleated biogenic secondary organic aerosol particles, Atmos. Chem. Phys., 2011, 11, 8759–8766 CrossRef CAS.
- L. Renbaum-Wolff, J. W. Grayson, A. P. Bateman, M. Kuwata, M. Sellier, B. J. Murray, J. E. Shilling, S. T. Martin and A. K. Bertram, Viscosity of α-pinene secondary organic material and implications for particle growth and reactivity, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8014–8019 CrossRef CAS.
- T. Koop, J. Bookhold, M. Shiraiwa and U. Pöschl, Glass transition and phase state of organic compounds: dependency on molecular properties and implications for secondary organic aerosols in the atmosphere, Phys. Chem. Chem. Phys., 2011, 13, 19238–19255 RSC.
- M. Shiraiwa and J. H. Seinfeld, Equilibration timescale of atmospheric secondary organic aerosol partitioning, Geophys. Res. Lett., 2012, 39, L24801 CrossRef.
- S. M. Zhou, M. Shiraiwa, R. D. McWhinney, U. Pöschl and J. P. D. Abbatt, Kinetic limitations in gas-particle reactions arising from slow diffusion in secondary organic aerosol, Faraday Discuss., 2013, 165, 391–406 RSC.
- C. Kidd, V. Perraud, L. M. Wingen and B. J. Finlayson-Pitts, Integrating phase and composition of secondary organic aerosol from the ozonolysis of alpha-pinene, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7552–7557 CrossRef CAS PubMed.
- V. Perraud, E. A. Bruns, M. J. Ezell, S. N. Johnson, Y. Yu, M. L. Alexander, A. Zelenyuk, D. Imre, W. L. Chang, D. Dabdub, J. F. Pankow and B. J. Finlayson-Pitts, Nonequilibrium atmospheric secondary organic aerosol formation and growth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2836–2841 CrossRef CAS.
- A. Virtanen, J. Joutsensaari, T. Koop, J. Kannosto, P. Yli-Pirila, J. Leskinen, J. M. Makela, J. K. Holopainen, U. Pöschl, M. Kulmala, D. R. Worsnop and A. Laaksonen, An amorphous solid state of biogenic secondary organic aerosol particles, Nature, 2010, 467, 824–827 CrossRef CAS.
- C. D. Cappa and K. R. Wilson, Evolution of organic aerosol mass spectra upon heating: implications for OA phase and partitioning behavior, Atmos. Chem. Phys., 2011, 11, 1895–1911 CrossRef CAS.
- P. J. Ziemann, Atmospheric chemistry phase matters for aerosols, Nature, 2010, 467, 797–798 CrossRef CAS.
- F. H. Marshall, R. E. H. Miles, Y. C. Song, P. B. Ohm, R. M. Power, J. P. Reid and C. S. Dutcher, Diffusion and reactivity in ultraviscous aerosol and the correlation with particle viscosity, Chem. Sci., 2016, 7, 1298–1308 RSC.
- J. P. Reid, A. K. Bertram, D. O. Topping, A. Laskin, S. T. Martin, M. D. Petters, F. D. Pope and G. Rovelli, The viscosity of atmospherically relevant organic particles, Nat. Commun., 2018, 9, 956 CrossRef PubMed.
- J. M. Roberts, The atmospheric chemistry of organic nitrates, Atmos. Environ. A Gen. Top., 1990, 24, 243–287 CrossRef.
- N. Sobanski, J. Thieser, J. Schuladen, C. Sauvage, W. Song, J. Williams, J. Lelieveld and J. N. Crowley, Day and night-time formation of organic nitrates at a forested mountain site in south-west Germany, Atmos. Chem. Phys., 2017, 17, 4115–4130 CrossRef CAS.
- R. Atkinson, S. M. Aschmann, W. P. Carter, A. M. Winer and J. N. Pitts Jr, Alkyl nitrate formation from the nitrogen oxide (NOx)-air photooxidations of C2–C8 n-alkanes, J. Phys. Chem., 1982, 86, 4563–4569 CrossRef CAS.
- J. L. Fry, A. Kiendler-Scharr, A. W. Rollins, T. Brauers, S. S. Brown, H. P. Dorn, W. P. Dubé, H. Fuchs, A. Mensah, F. Rohrer, R. Tillmann, A. Wahner, P. J. Wooldridge and R. C. Cohen, SOA from limonene: role of NO3 in its generation and degradation, Atmos. Chem. Phys., 2011, 11, 3879–3894 CrossRef CAS.
- J. L. Fry, D. C. Draper, K. C. Barsanti, J. N. Smith, J. Ortega, P. M. Winkler, M. J. Lawler, S. S. Brown, P. M. Edwards, R. C. Cohen and L. Lee, Secondary organic aerosol formation and organic nitrate yield from NO3 oxidation of biogenic hydrocarbons, Environ. Sci. Technol., 2014, 48, 11944–11953 CrossRef CAS PubMed.
- J. H. Slade, C. de Perre, L. Lee and P. B. Shepson, Nitrate radical oxidation of γ-terpinene: hydroxy nitrate, total organic nitrate, and secondary organic aerosol yields, Atmos. Chem. Phys., 2017, 17, 8635–8650 CrossRef CAS.
- T. Berkemeier, M. Ammann, T. F. Mentel, U. Pöschl and M. Shiraiwa, Organic nitrate contribution to new particle formation and growth in secondary organic aerosols from α-pinene ozonolysis, Environ. Sci. Technol., 2016, 50, 6334–6342 CrossRef CAS PubMed.
- J. M. O'Brien, P. B. Shepson, K. Muthuramu, C. Hao, H. Niki, D. R. Hastie, R. Taylor and P. B. Roussel, Measurements of alkyl and multifunctional organic nitrates at a rural site in Ontario, J. Geophys. Res.: Atmos., 1995, 100, 22795–22804 CrossRef.
- R. G. Fischer, J. Kastler and K. Ballschmiter, Levels and pattern of alkyl nitrates, multifunctional alkyl nitrates, and halocarbons in the air over the Atlantic Ocean, J. Geophys. Res.: Atmos., 2000, 105, 14473–14494 CrossRef CAS.
- J. Kastler and K. Ballschmiter, Bifunctional alkyl nitrates – trace constituents of the atmosphere, Fresenius. J. Anal. Chem., 1998, 360, 812–816 CrossRef CAS.
- J. M. O'Brien, P. B. Shepson, Q. Wu, T. Biesenthal, J. W. Bottenheim, H. A. Wiebe, K. G. Anlauf and P. Brickell, Production and distribution of organic nitrates, and their relationship to carbonyl compounds in an urban environment, Atmos. Environ., 1997, 31, 2059–2069 CrossRef.
- J. Kastler, W. Jarman and K. Ballschmiter, Multifunctional organic nitrates as constituents in European and US urban photo-smog, Fresenius. J. Anal. Chem., 2000, 368, 244–249 CrossRef CAS PubMed.
- J. L. Fry, D. C. Draper, K. J. Zarzana, P. Campuzano-Jost, D. A. Day, J. L. Jimenez, S. S. Brown, R. C. Cohen, L. Kaser, A. Hansel, L. Cappellin, T. Karl, A. Hodzic Roux, A. Turnipseed, C. Cantrell, B. L. Lefer and N. Grossberg, Observations of gas- and aerosol-phase organic nitrates at BEACHON-RoMBAS 2011, Atmos. Chem. Phys., 2013, 13, 8585–8605 CrossRef.
- A. W. Rollins, S. Pusede, P. Wooldridge, K. E. Min, D. R. Gentner, A. H. Goldstein, S. Liu, D. A. Day, L. M. Russell, C. L. Rubitschun, J. D. Surratt and R. C. Cohen, Gas/particle partitioning of total alkyl nitrates observed with TD-LIF in Bakersfield, J. Geophys. Res.: Atmos., 2013, 118, 6651–6662 CAS.
- B. H. Lee, C. Mohr, F. D. Lopez-Hilfiker, A. Lutz, M. Hallquist, L. Lee, P. Romer, R. C. Cohen, S. Iyer, T. Kurten, W. W. Hu, D. A. Day, P. Campuzano-Jost, J. L. Jimenez, L. Xu, N. L. Ng, H. Y. Guo, R. J. Weber, R. J. Wild, S. S. Brown, A. Koss, J. de Gouw, K. Olson, A. H. Goldstein, R. Seco, S. Kim, K. McAvey, P. B. Shepson, T. Starn, K. Baumann, E. S. Edgerton, J. M. Liu, J. E. Shilling, D. O. Miller, W. Brune, S. Schobesberger, E. L. D'Ambro and J. A. Thornton, Highly functionalized organic nitrates in the southeast United States: contribution to secondary organic aerosol and reactive nitrogen budgets, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 1516–1521 CrossRef CAS PubMed.
- D. T. Allen, E. J. Palen, M. I. Haimov, S. V. Hering and J. R. Young, Fourier transform infrared spectroscopy of aerosol collected in a low pressure impactor (LPI/FTIR): method development and field calibration, Aerosol Sci. Technol., 1994, 21, 325–342 CrossRef CAS.
- W. Huang, H. Saathoff, X. Shen, R. Ramisetty, T. Leisner and C. Mohr, Chemical characterization of highly functionalized organonitrates contributing to night-time organic aerosol mass loadings and particle growth, Environ. Sci. Technol., 2019, 53, 1165–1174 CrossRef CAS PubMed.
- A. K. Y. Lee, M. G. Adam, J. Liggio, S. M. Li, K. Li, M. D. Willis, J. P. D. Abbatt, T. W. Tokarek, C. A. Odame-Ankrah, H. D. Osthoff, K. Strawbridge and J. R. Brook, A large contribution of anthropogenic organo-nitrates to secondary organic aerosol in the Alberta oil sands, Atmos. Chem. Phys., 2019, 19, 12209–12219 CrossRef CAS.
- D. A. Day, S. Liu, L. M. Russell and P. J. Ziemann, Organonitrate group concentrations in submicron particles with high nitrate and organic fractions in coastal southern California, Atmos. Environ., 2010, 44, 1970–1979 CrossRef CAS.
- A. C. Vander Wall, P. S. J. Lakey, E. Rossich Molina, V. Perraud, L. M. Wingen, J. Xu, D. Soulsby, R. B. Gerber, M. Shiraiwa and B. J. Finlayson-Pitts, Understanding interactions of organic nitrates with the surface and bulk of organic films: implications for particle growth in the atmosphere, Environ. Sci.: Processes Impacts, 2018, 20, 1593–1610 RSC.
- A. Zelenyuk, D. Imre, J. Beránek, E. Abramson, J. Wilson and M. Shrivastava, Synergy between secondary organic aerosols and long-range transport of polycyclic aromatic hydrocarbons, Environ. Sci. Technol., 2012, 46, 12459–12466 CrossRef CAS PubMed.
- M. J. Ezell, S. N. Johnson, Y. Yu, V. Perraud, E. A. Bruns, M. L. Alexander, A. Zelenyuk, D. Dabdub and B. J. Finlayson-Pitts, A new aerosol flow system for photochemical and thermal studies of tropospheric aerosols, Aerosol Sci. Technol., 2010, 44, 329–338 CrossRef CAS.
- R. Atkinson and J. Arey, Atmospheric degradation of volatile organic compounds, Chem. Rev., 2003, 103, 4605–4638 CrossRef CAS PubMed.
- H. Cavdar and N. Saracoglu, Synthesis of new β-hydroxy nitrate esters as potential glycomimetics or vasodilators, Eur. J. Org. Chem., 2008, 2008, 4615–4621 CrossRef.
- J. F. Pankow and W. E. Asher, SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds, Atmos. Chem. Phys., 2008, 8, 2773–2796 CrossRef CAS.
- B. Moller, J. Rarey and D. Ramjugernath, Estimation of the vapour pressure of non-electrolyte organic compounds via group contributions and group interactions, J. Mol. Liq., 2008, 143, 52–63 CrossRef CAS.
- Y. Nannoolal, J. Rarey, D. Ramjugernath and W. Cordes, Estimation of pure component properties: Part 1. Estimation of the normal boiling point of non-electrolyte organic compounds via group contributions and group interactions, Fluid Phase Equilib., 2004, 226, 45–63 CrossRef CAS.
- N. J. Harrick, Internal Reflection Spectroscopy, Interscience Publishers, 1967 Search PubMed.
- G. Socrates, Infrared and Raman Characteristic Group Frequencies, John Wiley & Sons, New York, 2001 Search PubMed.
- X. Zhang, R. C. Mcvay, D. D. Huang, N. F. Dalleska, B. Aumont, R. C. Flagan and J. H. Seinfeld, Formation and evolution of molecular products in alpha-pinene secondary organic aerosol, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 14168–14173 CrossRef CAS PubMed.
- R. Winterhalter, R. Van Dingenen, B. R. Larsen, N. R. Jensen and J. Hjorth, LC-MS analysis of aerosol particles from the oxidation of α-pinene by ozone and OH-radicals, Atmos. Chem. Phys., 2003, 3, 1–39 CrossRef.
- B. Witkowski and T. Gierczak, Early stage composition of SOA produced by α-pinene/ozone reaction: α-acyloxyhydroperoxy aldehydes and acidic dimers, Atmos. Environ., 2014, 95, 59–70 CrossRef CAS.
- A. Zelenyuk, J. Yang, C. Song, R. A. Zaveri and D. Imre, A new real-time method for determining particles' sphericity and density: application to secondary organic aerosol formed by ozonolysis of α-pinene, Environ. Sci. Technol., 2008, 42, 8033–8038 CrossRef CAS PubMed.
- J. T. Jayne, D. C. Leard, X. F. Zhang, P. Davidovits, K. A. Smith, C. E. Kolb and D. R. Worsnop, Development of an aerosol mass spectrometer for size and composition analysis of submicron particles, Aerosol Sci. Technol., 2000, 33, 49–70 CrossRef CAS.
- P. F. DeCarlo, J. R. Kimmel, A. Trimborn, M. J. Northway, J. T. Jayne, A. C. Aiken, M. Gonin, K. Fuhrer, T. Horvath, K. S. Docherty, D. R. Worsnop and J. L. Jimenez, Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer, Anal. Chem., 2006, 78, 8281–8289 CrossRef CAS PubMed.
- M. R. Canagaratna, J. T. Jayne, J. L. Jimenez, J. D. Allan, M. R. Alfarra, Q. Zhang, T. B. Onasch, F. Drewnick, H. Coe, A. Middlebrook, A. Delia, L. R. Williams, A. M. Trimborn, M. J. Northway, P. F. DeCarlo, C. E. Kolb, P. Davidovits and D. R. Worsnop, Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer, Mass Spectrom. Rev., 2007, 26, 185–222 CrossRef CAS PubMed.
- M. R. Canagaratna, J. L. Jimenez, J. H. Kroll, Q. Chen, S. H. Kessler, P. Massoli, L. Hildebrandt Ruiz, E. Fortner, L. R. Williams, K. R. Wilson, J. D. Surratt, N. M. Donahue, J. T. Jayne and D. R. Worsnop, Elemental ratio measurements of organic compounds using aerosol mass spectrometry: characterization, improved calibration, and implications, Atmos. Chem. Phys., 2015, 15, 253–272 CrossRef.
- R. T. M. Fraser and N. C. Paul, The mass spectrometry of nitrate esters and related compounds. Part II, J. Chem. Soc. B, 1968, 140, 1407–1410 RSC.
- E. A. Bruns, V. Perraud, A. Zelenyuk, M. J. Ezell, S. N. Johnson, Y. Yu, D. Imre, B. J. Finlayson-Pitts and M. L. Alexander, Comparison of FTIR and particle mass spectrometry for the measurement of particulate organic nitrates, Environ. Sci. Technol., 2010, 44, 1056–1061 CrossRef CAS PubMed.
- A. W. Rollins, J. L. Fry, J. F. Hunter, J. H. Kroll, D. R. Worsnop, S. W. Singaram and R. C. Cohen, Elemental analysis of aerosol organic nitrates with electron ionization high-resolution mass spectrometry, Atmos. Meas. Tech., 2010, 3, 301–310 CrossRef CAS.
- D. K. Farmer, A. Matsunaga, K. S. Docherty, J. D. Surratt, J. H. Seinfeld, P. J. Ziemann and J. L. Jimenez, Response of an aerosol mass spectrometer to organonitrates and organosulfates and implications for atmospheric chemistry, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6670–6675 CrossRef CAS PubMed.
- R. T. M. Fraser and N. C. Paul, The mass spectrometry of nitrate esters and related compounds. Part I, J. Chem. Soc. B, 1968, 6, 659–663 RSC.
- J. L. Fry, A. Kiendler-Scharr, A. W. Rollins, T. Brauers, S. S. Brown, H. P. Dorn, W. P. Dube, H. Fuchs, A. Mensah, F. Rohrer, R. Tillmann, A. Wahner, P. J. Wooldridge and R. C. Cohen, SOA from limonene: role of NO3 in its generation and degradation, Atmos. Chem. Phys., 2011, 11, 3879–3894 CrossRef CAS.
- P. S. K. Liu, R. Deng, K. A. Smith, L. R. Williams, J. T. Jayne, M. R. Canagaratna, K. Moore, T. B. Onasch, D. R. Worsnop and T. Deshler, Transmission efficiency of an aerodynamic focusing lens system: comparison of model calculations and laboratory measurements for the aerodyne aerosol mass spectrometer, Aerosol Sci. Technol., 2007, 41, 721–733 CrossRef CAS.
- E. Abramson, D. Imre, J. Beránek, J. Wilson and A. Zelenyuk, Experimental determination of chemical diffusion within secondary organic aerosol particles, Phys. Chem. Chem. Phys., 2013, 15, 2983–2991 RSC.
- E. Saukko, A. T. Lambe, P. Massoli, T. Koop, J. P. Wright, D. R. Croasdale, D. A. Pedernera, T. B. Onasch, A. Laaksonen, P. Davidovits, D. R. Worsnop and A. Virtanen, Humidity-dependent phase state of SOA particles from biogenic and anthropogenic precursors, Atmos. Chem. Phys., 2012, 12, 7517–7529 CrossRef CAS.
- A. P. Bateman, A. K. Bertram and S. T. Martin, Hygroscopic influence on the semisolid-to-liquid transition of secondary organic materials, J. Phys. Chem. A, 2015, 119, 4386–4395 CrossRef CAS PubMed.
- M. Kuwata and S. T. Martin, Phase of atmospheric secondary organic material affects its reactivity, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17354–17359 CrossRef CAS PubMed.
- D. M. Bell, D. Imre, S. T. Martin and A. Zelenyuk, The properties and behavior of α-pinene secondary organic aerosol particles exposed to ammonia under dry conditions, Phys. Chem. Chem. Phys., 2017, 19, 6497–6507 RSC.
- C. Kidd, V. Perraud and B. J. Finlayson-Pitts, New insights into secondary organic aerosol from the ozonolysis of α-pinene from combined infrared spectroscopy and mass spectrometry measurements, Phys. Chem. Chem. Phys., 2014, 16, 22706–22716 RSC.
- J. W. Grayson, Y. Zhang, A. Mutzel, L. Renbaum-Wolff, O. Böge, S. Kamal, H. Herrmann, S. T. Martin and A. K. Bertram, Effect of varying experimental conditions on the viscosity of α-pinene derived secondary organic material, Atmos. Chem. Phys., 2016, 16, 6027–6040 CrossRef CAS.
- M. L. Hinks, M. V. Brady, H. Lignell, M. Song, J. W. Grayson, A. K. Bertram, P. Lin, A. Laskin, J. Laskin and S. A. Nizkorodov, Effect of viscosity on photodegradation rates in complex secondary organic aerosol materials, Phys. Chem. Chem. Phys., 2016, 18, 8785–8793 RSC.
- Y. Zhang, M. S. Sanchez, C. Douet, Y. Wang, A. P. Bateman, Z. Gong, M. Kuwata, L. Renbaum-Wolff, B. B. Sato, P. F. Liu, A. K. Bertram, F. M. Geiger and S. T. Martin, Changing shapes and implied viscosities of suspended submicron particles, Atmos. Chem. Phys., 2015, 15, 7819–7829 CrossRef CAS.
- A. Pajunoja, J. Malila, L. Hao, J. Joutsensaari, K. E. J. Lehtinen and A. Virtanen, Estimating the viscosity range of SOA particles based on their coalescence time, Aerosol Sci. Technol., 2014, 48, i–iv CrossRef CAS.
- T. Yli-Juuti, A. Pajunoja, O.-P. Tikkanen, A. Buchholz, C. Faiola, O. Väisänen, L. Hao, E. Kari, O. Peräkylä, O. Garmash, M. Shiraiwa, M. Ehn, K. Lehtinen and A. Virtanen, Factors controlling the evaporation of secondary organic aerosol from α-pinene ozonolysis, Geophys. Res. Lett., 2017, 44, 2562–2570 CrossRef CAS PubMed.
- S. S. Petters, S. M. Kreidenweis, A. P. Grieshop, P. J. Ziemann and M. D. Petters, Temperature- and humidity-dependent phase states of secondary organic aerosols, Geophys. Res. Lett., 2019, 46, 1005–1013 CrossRef.
- J. M. Rocklage, V. A. Marple and B. A. Olson, Study of secondary deposits in multiple round nozzle impactors, Aerosol Sci. Technol., 2013, 47, 1144–1151 CrossRef CAS.
- K. Treves, L. Shragina and Y. Rudich, Measurement of octanol–air partition coefficients using solid-phase microextraction (SPME)—application to hydroxy alkyl nitrates, Atmos. Environ., 2001, 35, 5843–5854 CrossRef CAS.
- P. B. Shepson, E. Mackay and K. Muthuramu, Henry's law constants and removal processes for several atmospheric β-hydroxy alkyl nitrates, Environ. Sci. Technol., 1996, 30, 3618–3623 CrossRef CAS.
- J. Kames and U. Schurath, Alkyl nitrates and bifunctional nitrates of atmospheric interest: Henry's law constants and their temperature dependencies, J. Atmos. Chem., 1992, 15, 79–95 CrossRef CAS.
- J. H. Kroll, N. M. Donahue, J. L. Jimenez, S. H. Kessler, M. R. Canagaratna, K. R. Wilson, K. E. Altieri, L. R. Mazzoleni, A. S. Wozniak, H. Bluhm, E. R. Mysak, J. D. Smith, C. E. Kolb and D. R. Worsnop, Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol, Nat. Chem., 2011, 3, 133–139 CrossRef CAS PubMed.
- A. L. Putman, J. H. Offenberg, R. Fisseha, S. Kundu, T. A. Rahn and L. R. Mazzoleni, Ultrahigh-resolution FT-ICR mass spectrometry characterization of α-pinene ozonolysis SOA, Atmos. Environ., 2012, 46, 164–172 CrossRef CAS.
- A. Laskin, M. K. Gilles, D. A. Knopf, B. Wang and S. China, Progress in the analysis of complex atmospheric particles, Annu. Rev. Anal. Chem., 2016, 9, 117–143 CrossRef PubMed.
- B. Nozière, M. Kalberer, M. Claeys, J. Allan, B. D'Anna, S. Decesari, E. Finessi, M. Glasius, I. Grgić, J. F. Hamilton, T. Hoffmann, Y. Iinuma, M. Jaoui, A. Kahnt, C. J. Kampf, I. Kourtchev, W. Maenhaut, N. Marsden, S. Saarikoski, J. Schnelle-Kreis, J. D. Surratt, S. Szidat, R. Szmigielski and A. Wisthaler, The molecular identification of organic compounds in the atmosphere: state of the art and challenges, Chem. Rev., 2015, 115, 3919–3983 CrossRef PubMed.
- A. A. Chew and R. Atkinson, OH radical formation yields from the gas-phase reactions of O3 with alkenes and monoterpenes, J. Geophys. Res.: Atmos., 1996, 101, 28649–28653 CrossRef CAS.
- M. S. Alam, M. Camredon, A. R. Rickard, T. Carr, K. P. Wyche, K. E. Hornsby, P. S. Monks and W. J. Bloss, Total radical yields from tropospheric ethene ozonolysis, Phys. Chem. Chem. Phys., 2011, 13, 11002–11015 RSC.
- S. M. Aschmann, A. A. Chew, J. Arey and R. Atkinson, Products of the gas-phase reaction of OH radicals with cyclohexane: reactions of the cyclohexoxy radical, J. Phys. Chem. A, 1997, 101, 8042–8048 CrossRef CAS.
- E. S. C. Kwok and R. Atkinson, Estimation of hydroxyl radical reaction rate constants for gas-phase organic compounds using a structure-reactivity relationship: An update, Atmos. Environ., 1995, 29, 1685–1695 CrossRef CAS.
- Y. Iinuma, O. Böge, Y. Miao, B. Sierau, T. Gnauk and H. Herrmann, Laboratory studies on secondary organic aerosol formation from terpenes, Faraday Discuss., 2005, 130, 279–294 RSC.
- Å. M. Jonsson, M. Hallquist and E. Ljungström, Influence of OH scavenger on the water effect on secondary organic aerosol formation from ozonolysis of limonene, Δ3-carene, and α-pinene, Environ. Sci. Technol., 2008, 42, 5938–5944 CrossRef PubMed.
- T. Berndt, S. Richters, T. Jokinen, N. Hyttinen, T. Kurtén, R. V. Otkjær, H. G. Kjaergaard, F. Stratmann, H. Herrmann, M. Sipilä, M. Kulmala and M. Ehn, Hydroxyl radical-induced formation of highly oxidized organic compounds, Nat. Commun., 2016, 7, 13677–13684 CrossRef CAS PubMed.
- J. C. Ianni, Kintecus, www.kintecus.com Search PubMed.
- A. C. Aiken, P. F. DeCarlo and J. L. Jimenez, Elemental analysis of organic species with electron ionization high-resolution mass spectrometry, Anal. Chem., 2007, 79, 8350–8358 CrossRef CAS PubMed.
- J. Duplissy, P. F. DeCarlo, J. Dommen, M. R. Alfarra, A. Metzger, I. Barmpadimos, A. S. H. Prevot, E. Weingartner, T. Tritscher, M. Gysel, A. C. Aiken, J. L. Jimenez, M. R. Canagaratna, D. R. Worsnop, D. R. Collins, J. Tomlinson and U. Baltensperger, Relating hygroscopicity and composition of organic aerosol particulate matter, Atmos. Chem. Phys., 2011, 11, 1155–1165 CrossRef CAS.
- Y. Zhao, N. M. Kreisberg, D. R. Worton, G. Isaacman, R. J. Weber, S. Liu, D. A. Day, L. M. Russell, M. Z. Markovic, T. C. VandenBoer, J. G. Murphy, S. V. Hering and A. H. Goldstein, Insights into secondary organic aerosol formation mechanisms from measured gas/particle partitioning of specific organic tracer compounds, Environ. Sci. Technol., 2013, 47, 3781–3787 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9em00379g |
| This journal is © The Royal Society of Chemistry 2020 |