
Photoelectrochemical solar fuels from carbon dioxide, water and sunlight
Jose Ramon
Galan-Mascaros
 *ab
*ab
aInstitute of Chemical Research of Catalonia (ICIQ), The Barcelona Institute of Science and Technology (BIST), Av. Països Catalans 16, 43007, Tarragona, Spain. E-mail: jrgalan@iciq.es; Tel: +34 977 920 808
bICREA, Passeig LLuís Companys 23, 08010, Barcelona, Spain
First published on 5th March 2020
Abstract
The production of fuels from renewable sources is one of the most promising alternatives to the current dominant position of fossil fuels in the energy market. Environmental, socio-economic, and geo-political issues make a fast and sustainable transition of high importance. One plausible opportunity for the generation of renewable fuels would be the capture of sunlight and the utilisation of this energy to reduce carbon dioxide, a pollutant, into energy rich molecules, mimicking natural photosynthesis. Here, we describe the challenge and the few successful examples able to produce fuels, exclusively from carbon dioxide, water and sunlight.
 Galán-Mascarós | Galán-Mascarós (aka JR) received his PhD from the University of Valencia, supervised by Prof. E. Coronado. After a post-doctoral stay with Prof. Kim R. Dunbar at Texas A&M University, he returned to Valencia in 2002 as a Research Fellow (Ramon y Cajal program). Currently, he is an ICREA Research Professor at the Institute of Chemical Research of Catalonia (ICIQ), where he joined in 2009. His current interests are centered in the design and development of affordable (photo)electrochemical schemes for the sustainable production of solar fuels. In this line of research, he coordinates the European project FET-PROACTIVE-A-LEAF (http://www.a-leaf.eu). |
Introduction
The search for solar fuels is becoming a major driving force in energy-related research.1,2 The excessive and continuous consumption of fossil fuels has caused high emissions of greenhouse gases, including CO2, into the atmosphere resulting in a global energy crisis. A sustainable, alternative, close energy cycle based on renewable resources is urgently needed. The opportunity to combine the most widely available renewable energy source (sunlight) with the most preferred energy storage vectors (fuels) offers unparalleled possibilities to meet the present and future energy needs,3,4 while offering highly attractive opportunities from a socio-economic perspective.5–7 The combustion of these fuels would return them to their starting materials, in a complete, circular and environmentally friendly process.Most of the promising strategies reported to date deal with water splitting, where the energy of the incident light is stored as molecular hydrogen, pointing towards a hydrogen economy.8,9 Despite their advantages,10 it is reasonable to state that liquid green fuels would be much preferred for a faster and easier transition from fossil fuels to a new environmentally friendly and carbon neutral energy cycle. If energy-rich molecules such as methane, ethanol, or small hydrocarbons could be obtained from a carbon source (CO2) and water, their implementation as fuels or fuel additives for industrial applications should be quite straightforward.11–13 Indeed, CO2 recycling into fuels is expected to become a key technology for the introduction of renewable sources in the energy cycle.14,15 However, intensive research is still needed to apply such technologies to viable and scalable processes.16
CO2 reduction is a complex reaction (see eqn (1)–(6)), where selectivity is difficult to achieve. The problem resides in the high number of different products that can be obtained, which are also prone to further reactivity. To make things worse, most of them require very similar energy input, precluding selectivity control by electrochemical potential, for example. Additionally, the simplest reduction products, such as carbon monoxide, are the most demanding in terms of energy. Meanwhile, the most interesting products, such as methane or alcohols, thermodynamically require less energy. However, the higher number of electrons and protons involved in those reactions generates a prohibiting activation energy. Such high overpotentials benefit the simpler reactions, that are not necessarily desired. This is particularly true for the hydrogen evolution reaction (HER), the most important side-reaction difficulting high CO2 selectivity, since protons are needed for CO2 reduction. The high complexity of the surface mechanisms for CO2 reduction makes the establishment of helpful computational models also difficult. Some recent developments in the theory of (photo)electrochemical CO2 fixation are shedding light on the problem, and bring appealing strategies to the table for future development.17,18 Good selectivity (>80%) has only been described for CO or HCOOH19 Probably, because of this reason, many strategies are targeting a mixture of H2 and CO as products, since this syngas can be converted to gasoline or diesel by the Fischer–Tropsch synthesis.20,21
| CO2 + 2H+ + 2e− → HCOOHE0 = −1.19 V | (1) |
| CO2 + 2H+ + 2e− → CO + H2OE0 = −0.11 V | (2) |
| CO2 + 4H+ + 4e− → HCHO + H2OE0 = −0.06 V | (3) |
| CO2 + 6H+ + 6e− → CH3OH + H2OE0 = +0.04 V | (4) |
| CO2 + 8H+ + 8e− → CH4 + 2H2OE0 = +0.18 V | (5) |
| CO2 + 1e− → CO2˙−E0 = −1.48 V | (6) |
Green plants and algae are extremely selective to reducing CO2 to sugars. Their enzymatic mechanism is an inspiration and valuable information can be extracted,22–25 despite their very low solar to biomass efficiency.26 One strategy would be to utilise solar H2 as feedstock for the reduction of CO2 as a second step, which could be attained by industrial methods or new approaches, including the use of microorganisms engineered to produce biomass and fine chemicals.27,28 Such a scheme, as any CO2 valorisation strategy, will need to be combined with CO2 producing processes (combustion, fermentation), needing additional concentration and purification steps.29 It is worthy to mention that CO2, despite its damaging effects on the climate, is not readily available in the atmosphere (still <500 ppm), and any strategies based on atmospheric CO2 purification would be cost-prohibitive.
Several comprehensive reviews have been published in recent years regarding the production of CO2-based fuels, including future perspectives.30–37 In this review, we will focus exclusively on photoelectrochemical (PEC) strategies, where fuels are obtained using sunlight energy, CO2 as feedstock and H2O as a source of protons and electrons, highlighting the light absorption requirements and schemes, along with catalyst integration. The combination of water oxidation with CO2 reduction is arguably the most appealing strategy, given its simplicity. It is also the most advantageous, since no additional power or sacrificial chemicals would be needed, allowing immediate product separation. In contrast, for example, in the so-called homogeneous Z-schemes, where products and reagents are present, oxidation and reduction occur in a one pot architecture.38,39
Light absorption by semiconductors with the right band alignment could drive both reactions, but they will need cocatalysts for high selectivity.40–45 This integrated approach that combines light absorbers and electrocatalysts allows for fine tuning their features and minimizing losses. The interface engineering is the key for the suitable function of these composite electrodes, since the properties of both semiconductors and catalysts can be severely affected by these surface modifications. Unfortunately, few absorbers can generate the required potential difference between cathodes and anodes. Thus, typical PEC devices need a combination of them, as a photocathode and photoanode, or addition of an external power supply (photovoltaic or not). This comes with a current limitation, since the current density is limited by the photocurrent of the lesser unit when connected in series. Because of this difficult implementation, from a technological perspective, a modular approach has been also proposed by simply wiring a photovoltaic device to a CO2 electrolyser in the dark. Despite being not so elegant, this approach has provided the highest solar to hydrogen efficiency in water splitting, reaching a spectacular 30% yield.46,47 The separation of the two modules avoids the complex interface engineering, and also the limitations of each component, since they can be individually scaled to match performance without the restrains of direct surface contact. The major disadvantage of this approach is the functional integration of both sub-systems that are currently individually optimised, whereas the integration of their respective function is still a must for successful and long-term application. For example, industrial electrolysers lose performance when working under dynamic operation conditions.48,49 The intermittent nature of sunlight will force the electrolyser part to follow continuous start/stop cycles, inducing random changes in potential, and favouring back-reactions at open circuit voltage causing fast deterioration of catalysts, supports and membranes. Systems engineering becomes a major requirement to mitigate these effects under intermittent operation conditions.
Photoelectrocatalytic CO2 conversion: photovoltaics plus electrolysers
As mentioned above, the combination of photovoltaics (PV) with electrolysers has become closer to technological development, since independent optimisation of each system appears to be an easier task. This modular approach allows taking advantage of highly efficient photovoltaic modules, encapsulated for long term stability and wired to an electrolyser. State-of-the-art PV devices easily reach over 20% solar to electricity efficiency,50–52 and new multi-junction technologies are reaching over 30%.53 One of the limitations comes from the voltage window, since CO2 electrolysers need well over a 2 volt difference, due to the large overpotential required by the catalysts. PV modules offering such power are formed by the combination of multiple cells, in series, with the corresponding limitations in current density. The second key parameter for this approach to be successful is the selectivity of the electrocatalysts that must minimise side-reactions, such as the HER.A record high solar to fuel (STF) efficiency was reported using a GaInP/GaInAs/Ge photovoltaic cell (Fig. 1a), reaching a 13.4% STF efficiency towards CO with bifunctional electrodes made of SnO2-modified CuO, which act as a cathode and anode.54 Bifunctionality and high selectivity of these electrodes was achieved by atomic layer deposition of the SnO2 decoration. Long term stability required each electrode to work at different pH levels.
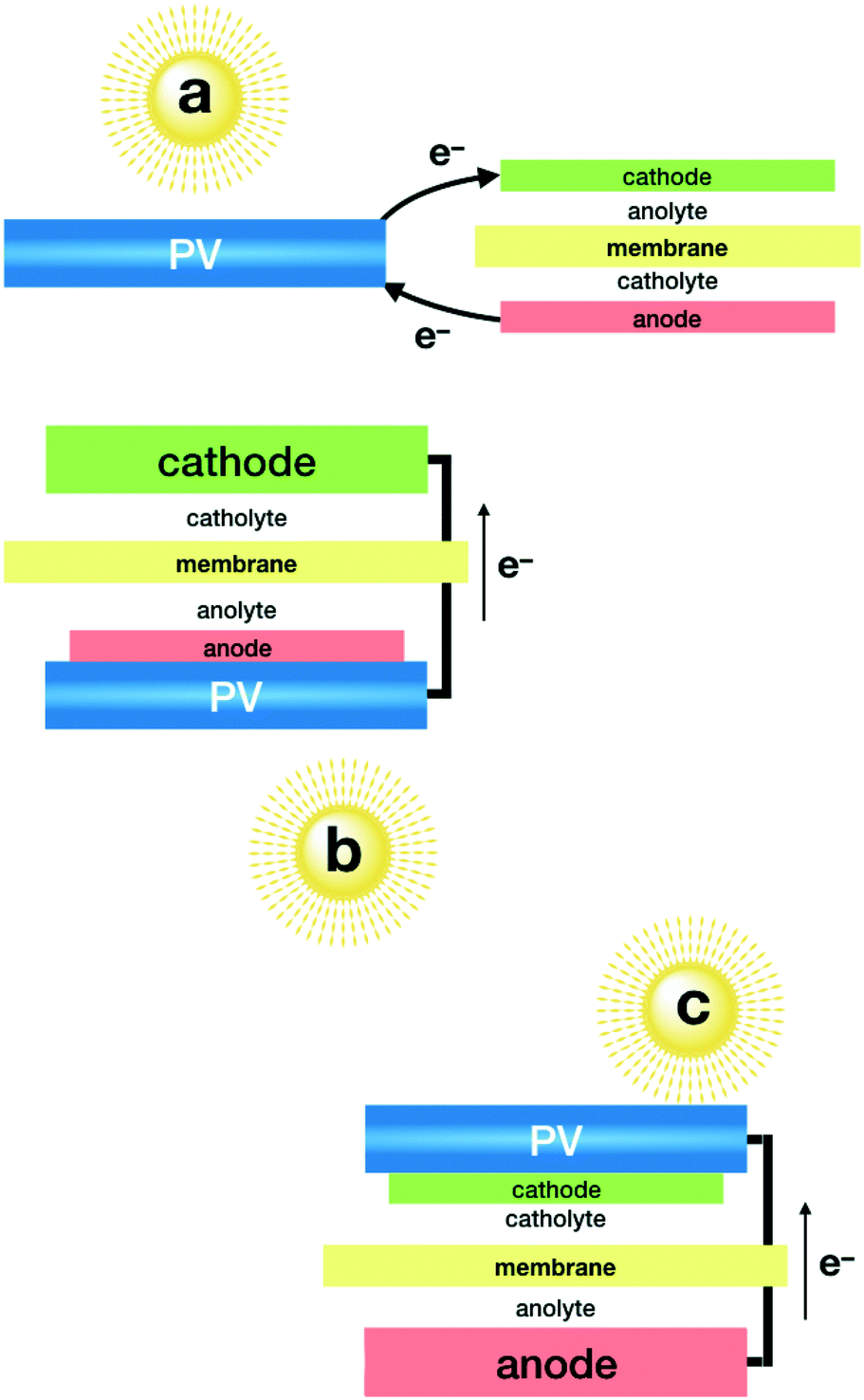 | ||
| Fig. 1 Plausible schemes for the conversion of sunlight, CO2 and H2O to fuels based on a PV plus unbiased electrolyser architecture: a) simple wired PV plus an electrolyser; b) and c) integration of the PV module as a support for a cathode or an anode, to drive both reactions through the wired counter electrode. | ||
A remarkable 6.5% efficiency towards CO was obtained using an electrolyser equipped with a nanostructured Au cathode and an IrO2 anode in bicarbonate neutral media, powered by three perovskite solar cells connected in series.55 The major drawback of this set-up (Fig. 1a) is the need for noble metal-based cathodes and anodes, both being key components. The gold cathode offers an excellent Faradaic efficiency of over 80%. The IrO2 anode offers good kinetics and, more importantly, good stability in neutral pH. Currently, no low cost alternatives are available to reach comparable performance under these conditions.
In a single compartment reactor, and exclusively using earth abundant elements, CO was obtained with a 3.4% STF efficiency.56 This was achieved by the combination of a homogeneous Mn catalyst on the cathode and a Ni–Fe oxo-hydroxide as an anode, both working in alkaline media. This study also included an interesting comparison of efficiencies when using different PV modules (Fig. 1a).
A first step towards integrated devices was reported by combination of four encapsulated silicon heterojunction solar cells (SHSCs)57 in series wired to a Ni foam electrode to build a self-standing photoanode (Fig. 1c). In combination with a cathode made from Cu foam decorated with Zn nanoflakes, a 16% Faradaic efficiency towards CO was reported, for a total of 4.3% STF production. As in previous examples, two different electrolytes were used to avoid noble metal catalysts as anodes.58
Beyond CO, only formate production has been reported with this modular configuration with the implementation of selective catalysts, such as Bi metal. Highly porous Bi electrodes were used to reach an impressive 8.5% STF efficiency for formate when combined with an IrO2 anode (Fig. 1a), both working in bicarbonate media.59 It is worthy to mention that when powered by a polycrystalline Si solar cell, this set-up works at currents over 10 mA cm−1, resulting in one of the highest STF production rates reported. Arguably, this probably comes from the relative high surface area of the cathode, and not necessarily from a higher electrocatalytic activity. The authors also present a computational analysis of the cathode selectivity, along with the influence of electrolytes.
Integrated photoelectrocatalytic CO2 conversion
Wireless decoration of PV cells appears to be the second step towards integrated systems. Following this strategy, two amorphous Si triple-junction cells in series were decorated with a WSe2 cathode and a CoOx anode to yield a 4.6% STF efficiency for CO.60 This system minimises the HER side-reaction, reaching a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO2/H2 ratio with the help of an ionic liquid catholyte, while the anode works in neutral phosphate buffer to stabilise the cobalt oxide catalyst.
:1 CO2/H2 ratio with the help of an ionic liquid catholyte, while the anode works in neutral phosphate buffer to stabilise the cobalt oxide catalyst.
Most examples from an integrated approach are built from monolithic semiconductors as photoelectrodes, decorated with catalysts, looking for optimum synergy by interface engineering. However, only a few common semiconductors are able to promote the right potential difference and band alignment to promote both the reactions, CO2RR and OER, such as TiO2, SiC, ZnO, or CdS.61 TiO2 has been the most studied material for photoelectrodes with inconsistent results during the last years.62 A photocathode built from a combination of CuFeO2 and CuO is another one of the very few examples of single absorber PEC electrode. This photocathode drives formate production and oxygen evolution at a Pt anode with a 1% efficiency.63
Given that single absorbers are very scarce, most integrated PEC devices need to combine two absorbers when working unbiased. This combination may come from heterostructured photoelectrodes; from two photoelectrodes in series: photoanode + photocathode; or by addition of an external photovoltaic device to provide the extra power needed to run both semi-reactions. An interesting compromise deals with the parallel combination of the two photoelectrodes, or one photoelectrode and a PV cell to absorb light from complementary regions in the solar light spectrum to improve photocurrent generation efficiency. All the possibilities are shown in Fig. 2.
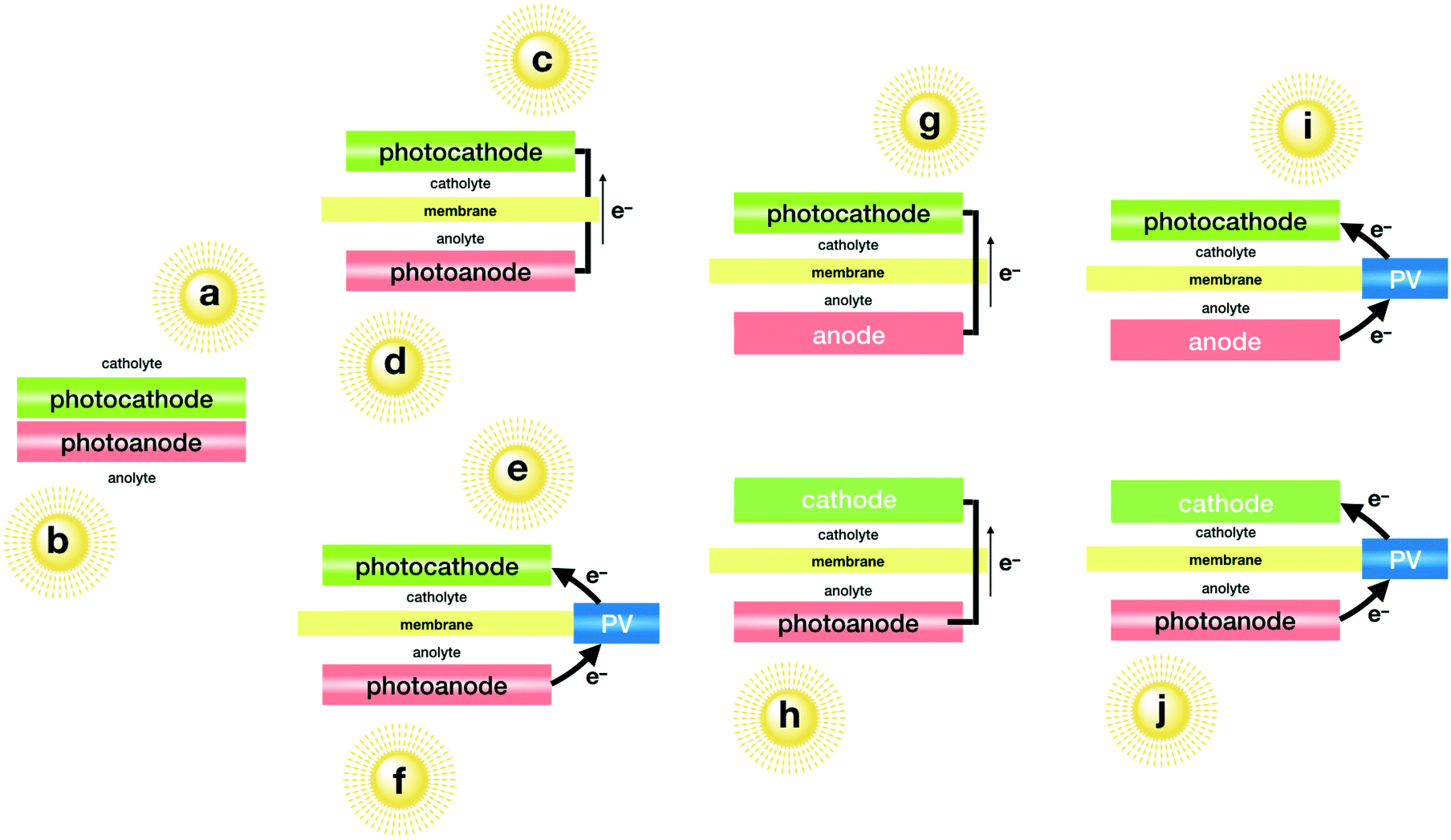 | ||
| Fig. 2 Plausible schemes for the conversion of sunlight, CO2 and H2O to fuels based on integrated, unbiased photoelectrochemical (PEC) architecture: a) and b) simple coupling of a photocathode and photoanode, irradiated from only one side (a or b) or from both sides (a and b). c) and d) Wired coupling of two complementary photoelectrodes, when irradiated from only one side (c or d) or from both sides (c and d); e) and f) wired coupling of two complementary photoelectrodes, assisted by an external PV cell, when irradiated from only one side (e or f) or from both sides (e and f); g) single photocathode architecture; h) single photoanode architecture; i) and j) single photoelectrode architectures assisted by an external PV cell. In the cases of two photoelectrodes irradiated only from one side, both should ideally absorb complementary regions of the solar spectrum to maximise solar to fuel efficiency. | ||
A single photoanode, built from a tandem Ni/TiO2/InGaP/GaAs architecture, delivered a record 10% STF efficiency for formate with the assistance of a highly selective Pd/C/Ti cathode. In this case, the implementation of a low resistance bipolar membrane allowed to minimise voltage losses and product crossover. This set-up works at a high current density under unbiased conditions, thanks to the nanostructuration and optimisation of all the parts, including the large pH difference between the anolyte and catholyte maintained with the membrane.64
Combination of a hybrid Ru–metal complex/zinc-doped InP photocathode with a reduced SrTiO3 photoanode yielded a maximum of 0.14% for formate when both photoelectrodes are connected with wires, working in different compartments.65 Taking advantage of the high selectivity of the photoanode towards water oxidation, the authors also explored a one-compartment cell, using back-to-back configuration (Fig. 2a). In this case, the yield was reduced to 0.08%. This difference was assigned to the pH difference that can be maintained in different compartments, which occurs because CO2 is bubbled only into the catholyte, decreasing the pH by about 1.5.
Previously, the same photocathodes were combined with TiO2 photoanodes in parallel (Fig. 2c).66 Since TiO2 only absorbs UV light, the visible light passing through the photoanode reached the photocathode. This light absorption scheme did not yield a better STF efficiency though, which was limited to 0.04%.
This parallel architecture also works with SrTiO3 photoanodes when combined with a more elaborated photocathode incorporating an analogous Ru complex.67 This combination with a Ru complex/TiO2/N,Zn–Fe2O3/Cr2O3 photocathode delivered a 0.15% STF efficiency, albeit at very low current densities.
A cobalt molecular catalyst has been used as a selective cathode catalyst to produce syngas (H2/CO2 = 3) from a perovskite–BiVO4 PEC tandem cell working in bicarbonate buffer, when irradiated from the photoanode side.68 Multiple protective layers on the photoabsorbers confer moderate stability to this architecture for at least 10 h when working completely immersed in the solution.
The rest of the examples incorporate an external PV cell (Fig. 2e–f and i–j) to reach the required potential for both reactions. This comes with a STF efficiency penalty, since this external PV cell is connected in series with the rest of the semiconductors. Such architecture results in voltage addition, but the current is limited by the photocurrent of the weakest photoabsorber, since it is constant throughout the whole circuit.69
A record high efficiency was achieved with this strategy with a tandem system of Cu/Ag/TiO2 photocathodes that yielded a 60% FE for C2+ products under illumination. These cathodes have been successfully combined with water oxidation when coupled to a series of perovskite solar cells, reaching a total of 3.5% STF efficiency.70 This also represents the only example of CO2-reduction PEC devices that produce carbon–carbon bonds.
An interesting combination of a photocathode/photovoltaic tandem assembly was designed to harvest a wide energy range of incident light and to power the CO2 to CO conversion at the photocathode (>0.35%), which was composed of gold-decorated ZnO/ZnTe/CdTe nanorods, while oxidising water into oxygen at the anode, powered by the tandem photocathode + CH3NH3PbI3 (MAPI) perovskite device.71
FeOOH-decorated BiVO4 photoanodes have been combined with a large surface area 3D TiN electrode able to transfer electrons to CO2-selective loaded enzymes to obtain formate in a very high FE. Reasonable currents need the additional power from a photovoltaic cell to deliver 0.08% STF conversions.72
Despite being less appealing from an application point of view, many attempts to combine metal complexes as chromophores with semiconductors to boost the performance of such hybrid photoelectrodes have been incorporated into CO2 reduction PEC devices.73 Again, few examples have been able to work unbiased.
Another molecular photocatalyst/semiconductor hybrid photocathode was reported from the impregnation of CuGaO2 with a mixed Ru–Re complex. This photocathode is able to produce CO and H2 operating at <75% selectivity when combined with an oxygen evolution CoOx/TaON photoanode.74 This architecture required an additional PV unit to achieve significant photocurrent.
Discussion
Despite the great technological and industrial interest on the direct storage of sunlight power into energy rich chemicals from CO2 reduction, this field is still in its infancy. Fortunately, many studies and reports on the optimisation of the needed parts are available. Surprisingly, the key step of implementation into working systems is missing. Unbiased transformation is scientifically demanding, but all problems derived from implementation can only be solved when tackled. Compared to the hundreds of results on photoelectrodes under biased conditions, and on photocatalysts with the help of ancillary chemicals (as an energy source), the focus of this review resulted in a very short list. Shorter than what we expected, indeed.Beyond the limited number, just looking at Tables 1 and 2, several additional conclusions can be derived from these promising results, obtained by a few research groups. Starting on the positive side, some research teams have already achieved over 10% STF efficiency, a common minimum required to receive industrial interest. All are based on different materials and architectures, with their own advantages and disadvantages. Most of them are fuelled by the authors' previous expertise. There has not been a sustained, rational stepwise progress. Given the importance of the challenge, a consistent, collaborative, multidisciplinary effort will be needed as soon as possible.
| Cathode | Anode | PV | j (mA cm−2) | FECO (%) | FECOOH (%) | STFa (%) | Ref. |
|---|---|---|---|---|---|---|---|
| a All efficiencies are quoted as reported, although not all the reports used identical methods for their estimation. | |||||||
| SnO2/CuO | SnO2/CuO | GaInP/GaInAs/Ge (2.4 V) | 0.1 | 81 | — | 13.8 | 54 |
| Anodised Au | IrO2 | Perovskite (3.1 V) | ? | 90 | — | 6.5 | 55 |
| Mn complex | FeOx/NiOx | Si (2.2 V) | 1 | 80 | — | 3.4 | 56 |
| Cu–Zn | Ni | Si (2.7 V) | 5 | 16 | — | 4.3 | 58 |
| Porous Bi | IrO2 | Si (2.7 V) | 10 | — | 95 | 8.5 | 59 |
| (photo)Cathode | (photo)Anode | j (mA cm−2) | FECO (%) | FECOOH (%) | FEothers (%) | STF (%)b/PVa | Ref. |
|---|---|---|---|---|---|---|---|
| a All efficiencies are quoted as reported, although not all the reports used identical methods for their estimation. b External photovoltaic cell, if any. | |||||||
| WSe2/PV-a-si-3jn | CoOx/PV-a-si-3jn | ? | 24 | — | — | 4.6 | 60 |
| CuFeO2/CuO | Pt | <0.2 | — | 90 | — | 0.7–1.2 | 63 |
| Pd/C/Ti | Ni/TiO2/InGaP/GaAs | 8.7 | — | >94 | — | >10 | 64 |
| Ru complex/InP | Reduced SrTiO3 | 0.1 | — | >71 | — | 0.14 | 65 |
| Ru-complex/InP | TiO2 | ? | — | >70 | — | 0.04 | 66 |
| Ru-complex/TiO2/Fe2O3/Cr2O3 | TiO2 | >0.1 | 16 | >79 | — | 0.15 | 67 |
| Co-complex/perovskite | CoOx/BiVO4 | >1 | 25 | — | — | 0.08 | 68 |
| Cu/Ag/TiO2 | IrO2 | — | — | — | 60 (C2/C3) | 3.5/perovskite | 70 |
| ZnO–ZnTe@CdTe-Au | Co-HCO3/Ni foam | 0.85 | 80 | — | — | 0.43/MAPI | 71 |
| Bio/TiN | FeOx/BiVO4 | <1 | — | 77 | — | 0.08/perovskite | 72 |
| RuRe-complex/CuGaO2 | CoOx/TaON | <0.05 | 41 | — | — | ? | 74 |
Another interesting conclusion is the clear superiority of the modular approach, combining a photovoltaic cell with an electrolyser. An over 3% STF efficiency has been reached with earth abundant, low-cost materials following this scheme, and a record high >13% using more complex, unscalable approaches. This approach is more promising to reach reasonable success in the near future, thanks to the better synergy between both parts. Photovoltaic cells can only deliver <30 mA cm−2,75–77 but industrial electrolysers can work at much higher currents, >0.5 A cm−2.78 The challenge here is the implementation of CO2RR cathodes, and the corresponding product separation and management into industry-ready systems. In the meantime, it does not make much sense to limit the electrolytic capabilities to low current densities offered by PV devices by matching the area and geometry of both parts, when a reduced-size electrolyser could efficiently do the same work. Furthermore, in the near future, when the grid becomes predominantly renewable, the “simple” development of appropriate electrolysers, associated with CO2 sources, will emerge as a plausible strategy, and an economically attractive one for industrial CO2 emitters.
On the integrated approach, too many examples yield marginal efficiencies (<0.5%), restricted by two major issues. On one end, the limited voltage offered by monolithic semiconductors combined with the very high overpotentials required by the catalysts hampers their productivity. This can be solved by using two photoelectrodes, but this limits the current and also STF efficiency. Ideally, both should absorb light from complementary regions of the solar spectrum to maximise efficiency, which is not a simple achievement. Using n external photovoltaic cells in series has been the most successful solution, and the only one yielding STF efficiencies over 1%. The other solution explored has been the decoration of heterojunction photovoltaics with catalysts to build photoelectrodes, but this approach does not improve the wired analog technology, although it could be preferred given some restrictions (space, costs, implementation, etc.). The second major issue comes from the interface that must protect the light absorber, while facilitating fast and energy efficient charge transfer. This complicates the processing and architecture, since the high efficiency of PV also depends on the perfect encapsulation and optimum definition of the current collector layers, that must be modified. Modifications on these key parts have necessarily a detrimental effect on PV efficiency. Finally, the same current density limitation applies to this approach, as fixed by the weakest photoabsorber.
With all these in mind, we can summarise the major challenges that prevent sunlight-to-CO2-based fuel technology. From a basic science perspective, the lack of selectivity towards the desired product is without a doubt the major drawback. With rare exceptions, only CO/H2 (syngas) and formate have been obtained in significant yields. These are interesting chemicals, but higher value products would be certainly preferred, like methane. Methane could be directly used as a fuel in current technologies, complementing natural gas, in a real sustainable cycle. Other products, like alcohols, would also be useful as additives for liquid fuels. There is a huge margin of improvement. On the development end, the field will need to follow the same route that water splitting electrolysers followed decades ago. The mitigation of losses in the electrolyser part (membrane, electrolyte, interface, reagent and product dynamics, etc.) must be optimised to exploit the excellent capabilities of electrode supports and catalysts. On the application end, the target should be adapted to industry standards. Scalability and economic feasibility should be taken into account, and not just scientific excellence. The latter is a must, but if we all, as a community, are discussing about contributing to solve a societal challenge, we better put our words into action. Finally, scientific cooperation will be fundamental to overcome all these hurdles. It is difficult to envision a successful solution coming from a single research team. Multiple disciplines and multiple points of view will be needed to deliver. This is why multi-institutional and multi-national programs are so important to move this field forward.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The author acknowledges the support from the European Union in the artificial photosynthesis project H2020 FET-PROACT A-LEAF (Grant Agreement No.732840).Notes and references
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- S. Chu, Y. Cui and N. Liu, Nat. Mater., 2017, 16, 16–22 CrossRef PubMed.
- D. Gust, T. A. Moore and A. L. Moore, Acc. Chem. Res., 2009, 42, 1890–1898 CrossRef CAS PubMed.
- H. B. Gray, Nat. Chem., 2009, 1, 7 CrossRef CAS PubMed.
- R. J. Detz, J. N. H. Reek and B. C. C. van der Zwaan, Energy Environ. Sci., 2018, 11, 1653–1669 RSC.
- J. Newman, P. G. Hoertz, C. A. Bonino and J. A. Trainham, J. Electrochem. Soc., 2012, 159, A1722–A1729 CrossRef CAS.
- A. Harriman, Philos. Trans. R. Soc., A, 2013, 20110415 CrossRef PubMed.
- I. Staffell, D. Scamman, A. V. Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- S. Ardo, D. F. Rivas, M. A. Modestino, V. S. Greiving, F. F. Abdi, E. Alarcon Llado, V. Artero, K. Ayers, C. Battaglia, J. P. Becker, D. Bederak, A. Berger, F. Buda, E. Chinello, B. Dam, V. Di Palma, T. Edvinsson, K. Fujii, H. Gardeniers, H. Geerlings, S. M. H. Hashemi, S. Haussener, F. Houle, J. Huskens, B. D. James, K. Konrad, A. Kudo, P. P. Kunturu, D. Lohse, B. Mei, E. L. Miller, G. F. Moore, J. Muller, K. L. Orchard, T. E. Rosser, F. H. Saadi, J. W. Schuttauf, B. Seger, S. W. Sheehan, W. A. Smith, J. Spurgeon, M. H. Tang, R. van de Krol, P. C. K. Vesborg and P. Westerik, Energy Environ. Sci., 2018, 11, 2768–2783 RSC.
- A. Saeedmanesh, M. A. Mac Kinnon and J. Brouwer, Curr. Opin. Electrochem., 2019, 12, 463–491 RSC.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef CAS.
- G. A. Olah, G. K. S. Pakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- A. Corma and H. Garcia, J. Catal., 2013, 308, 168–175 CrossRef CAS.
- G. Centi and S. Perathoner, Energy Environ. Sci., 2013, 6, 1711–1731 RSC.
- G. Centi and S. Perathoner, ChemSusChem, 2010, 3, 195–208 CrossRef CAS PubMed.
- H. L. Tuller, Mater. Renewable Sustainable Energy, 2017, 6, 3 CrossRef PubMed.
- S. Z. Xu and E. A. Carter, Chem. Rev., 2018, 119, 6631–6669 CrossRef PubMed.
- R. Garcia-Muelas, F. Dattila, A. J. Shinagawa, T. abd Martin, J. Pérez-Ramírez and N. López, J. Phys. Chem. Lett., 2019, 9, 7153–7159 CrossRef PubMed.
- G. O. Larrazábal, A. J. Martín and J. Pérez-Ramírez, J. Phys. Chem. Lett., 2017, 8, 3933–3944 CrossRef PubMed.
- S. Hernández, M. A. Farkhondehfal, F. Sastre, M. Makkee, G. Saracco and N. Russo, Green Chem., 2017, 19, 2326–2346 RSC.
- C. Graves, S. D. Ebbesen, M. Mogensen and K. S. Lackner, Renewable Sustainable Energy Rev., 2011, 15, 1–23 CrossRef CAS.
- G. D. Scholes, G. R. Fleming, A. Olaya-Castro and R. van Grondelle, Nat. Chem., 2011, 3, 763–774 CrossRef CAS PubMed.
- X. J. Xie and E. Bakker, Phys. Chem. Chem. Phys., 2014, 16, 19781–19789 RSC.
- Y. Kim, J. H. Lee, H. Ha, S. W. Im and K. T. Nam, Nano Convergence, 2014, 16, 19781–19789 RSC.
- N. Kornienmko, J. Z. Zhang, K. K. Sakimoto, P. D. Yang and E. Reisner, Nat. Nanotechnol., 2018, 13, 890–899 CrossRef PubMed.
- X. G. Zhu, S. P. Long and D. R. Ort, Curr. Opin. Biotechnol., 2008, 19, 153–159 CrossRef CAS PubMed.
- J. P. Torella, C. J. Gagliardi, J. S. Chen, D. K. Bediako, B. Colón, J. C. Way, P. A. Silver and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 2337–2342 CrossRef CAS PubMed.
- D. K. Dogutan and D. G. Nocera, Acc. Chem. Res., 2019, 52, 3143–3148 CrossRef CAS PubMed.
- P. C. Psarras, S. Comello, P. Bains, P. Charoensawadpong, S. Reichelstein and J. Wilcox, Environ. Sci. Technol., 2017, 51, 11440–11449 CrossRef CAS PubMed.
- E. V. Kondratenko, G. Mul, J. Baltrusaitis, G. O. Larrazábal and J. Pérez-Ramírez, Energy Environ. Sci., 2013, 6, 3112–3135 RSC.
- C. A. Trickett, A. Helal, B. A. Al-Maythalony, Z. H. Yamani, K. E. Cordova and O. M. Yaghi, Nat. Rev. Mater., 2017, 2, 17045 CrossRef CAS.
- S. C. Roy, O. K. Varghese, M. Paulose and C. A. Grimes, ACS Nano, 2010, 4, 1259–1278 CrossRef CAS PubMed.
- W. G. Tu, Y. Zhou and Z. G. Zou, Adv. Mater., 2014, 26, 4607–4626 CrossRef CAS PubMed.
- Y. Izumi, Coord. Chem. Rev., 2013, 257, 171–186 CrossRef CAS.
- M. K. Brennaman, R. J. Dillon, L. Alibabaei, M. K. Gish, C. J. Dares, D. L. Ashford, R. L. House, G. J. Meyer, J. M. Papanikolas and T. J. Meyer, J. Am. Chem. Soc., 2016, 138, 13085–13102 CrossRef CAS PubMed.
- A. U. Pawar, C. W. Kim, M. Nguyen-Le and Y. S. Kang, ACS Sustainable Chem. Eng., 2019, 7, 7431–7455 CrossRef CAS.
- N. Vu, S. Kaliaguine and T. Do, Adv. Funct. Mater., 2019, 29, 1901825 CrossRef.
- P. Zhou, J. G. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920–4935 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- X. Li, J. G. Yu, M. Jaroniec and X. B. Chen, Chem. Rev., 2019, 119, 3962–4179 CrossRef CAS PubMed.
- X. Chang, T. Wang, P. Yang, G. Zhang and J. Gong, Adv. Mater., 2019, 31, 1804710 CrossRef PubMed.
- J. R. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30, 1704649 CrossRef PubMed.
- J. X. Low, B. Cheng and J. G. Yu, Appl. Surf. Sci., 2017, 392, 658–686 CrossRef CAS.
- S. Nahar, M. F. M. Zain, A. A. H. Kadhum, H. A. Hasan and M. R. Hasan, Materials, 2017, 10, 629 CrossRef PubMed.
- J. L. White, M. F. Baruch, J. E. Pander III, Y. Hu, I. C. Fortmeyer, J. E. Park, T. Zhang, K. Liao, J. Gu, Y. Yan, T. W. Shaw, E. Abelev and A. B. Bocarsly, Chem. Rev., 2015, 115, 12888–12935 CrossRef CAS PubMed.
- J. Jia, L. C. Seitz, J. D. Benck, Y. Huo, Y. Chen, J. W. D. Ng, T. Bilir, J. S. Harris and T. F. Jaramillo, Nat. Commun., 2016, 7, 13237 CrossRef CAS PubMed.
- J. S. Luo, J. H. Im, M. T. Mayer, M. Schreier, M. K. Nazeeruddin, N. G. Park, S. D. Tilley, M. Fan and H. J. Grätzel, Science, 2014, 345, 1593–1596 CrossRef CAS PubMed.
- S. M. Alia, S. Stariha and R. L. Borup, J. Electrochem. Soc., 2019, 166, F1164–F1172 CrossRef CAS.
- A. Weiß, A. Siebel, M. Bernt, T.-H. Shen, V. Tileli and H. A. Gasteiger, J. Electrochem. Soc., 2019, 166, F487–F497 CrossRef.
- G. Grancini and M. K. Nazeeruddin, Nat. Rev. Mater., 2019, 4, 4–22 CrossRef CAS.
- J. P. Correa-Baena, M. Saliba, T. Buonassisi, A. Grätzel, W. Tress Abate and A. Hagfeldt, Science, 2017, 358, 739–744 CrossRef CAS.
- M. A. Green, Science, 2013, 371, 20110413 Search PubMed.
- S. Essig, C. Allebe, T. Remo, J. F. Geisz, M. A. Steiner, K. Horowitz, L. Barraud, J. S. Ward, M. Schnabel, A. Descoeudres, D. L. Young, M. Woodhouse, M. Despeisse, C. Ballif and A. Tamboli, Nat. Energy, 2017, 2, 17144 CrossRef CAS.
- M. Schreier, F. Héroguel, L. Steier, S. Ahmad, J. S. Luterbbacher, M. T. Mayer, J. Luo and M. Grätzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS.
- M. Schreier, L. Curvat, F. Giordano, L. Steier, A. Abate, S. M. Zakeeruddin, J. Luo, M. T. Mayer and M. Grätzel, Nat. Commun., 2015, 6, 7326 CrossRef CAS PubMed.
- T. Arai, S. Sato, K. Sekizawa, T. M. Suzuki and T. Morikawa, Chem. Commun., 2019, 55, 237–240 RSC.
- C. Battaglia, A. Cuevas and S. De Wolf, Energy Environ. Sci., 2016, 9, 1552–1576 RSC.
- F. Urbain, P. Tang, N. M. Carretero, T. Andreu, L. G. Gerling, C. Voz, J. Arbiol and J. R. Morante, Energy Environ. Sci., 2017, 10, 2256–2266 RSC.
- G. Pia, S. H. Yoon, D. S. Han and H. Park, ChemSusChem, 2020, 13, 698–706 CrossRef PubMed.
- M. Asadi, K. Kim, C. Liu, A. V. Addepalli, P. Abbasi, P. Yasaei, P. Phillips, A. Behranginia, J. M. Cerrato, R. Haasch, P. Zapol, B. Kumar, R. F. Klie, J. Abiade, L. A. Curtiss and A. Salehi-Khojin, Science, 2016, 353, 467–470 CrossRef CAS PubMed.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638 CrossRef CAS.
- S. Xie, Q. Zhang, G. Liu and Y. Wang, Chem. Commun., 2016, 52, 35–59 RSC.
- U. Kang, S. K. Choi, D. J. Ham, S. M. Ji, W. Choi, D. S. Han, A. Abdel-Wahab and H. Park, Energy Environ. Sci., 2015, 8, 2638–2643 RSC.
- X. Zhou, R. Liu, K. Sun, Y. Chen, E. Verlage, S. A. Francis, N. S. Lewis and C. Xiang, ACS Energy Lett., 2016, 1, 764–770 CrossRef CAS.
- T. Arai, S. Sato, T. Kajino and T. Morikawa, Energy Environ. Sci., 2013, 6, 1274–1282 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS.
- K. Sekizawa, S. Sato, T. Arai and T. Morikawa, ACS Catal., 2018, 8, 1405–1416 CrossRef CAS.
- V. Andrei, B. Reuillard and E. Reisner, Nat. Mater., 2020, 19, 189–194 CrossRef CAS PubMed.
- T. J. Jacobsson, V. Fjällström, M. Edoff and T. Edvinsson, Sol. Energy Mater. Sol. Cells, 2015, 138, 86–95 CrossRef CAS.
- J. W. Beeman Gurudayal, J. Bullock, J. Wang, H. nd Eichhorn, C. Towle, A. Javey, F. M. Toma, N. Mathews and J. W. Ager, Energy Environ. Sci., 2019, 12, 1068–1077 RSC.
- Y. J. Jang, I. Jeong, J. Lee, J. Lee, M. J. Ko and J. S. Lee, ACS Nano, 2016, 10, 6980–6987 CrossRef CAS.
- S. K. Kuk, Y. Ham, K. Gopinath, P. Boonmongkolras, Y. Lee, Y. W. Lee, S. Kondaveeti, C. Ahn, B. Shin, J. Lee, S. Jeon and C. B. Park, Adv. Energy Mater., 2019, 9, 1900029 CrossRef.
- K. Maeda, Adv. Mater., 2019, 31, 1808205 CrossRef PubMed.
- H. Kumagai, G. Sahara, K. Maeda, M. Higashi, R. Abe and O. Ishitani, Chem. Sci., 2017, 8, 4242–4249 RSC.
- P. Tonui, S. O. Oseni, G. Sharma, Q. Yan and G. Tessema Mola, Renewable Sustainable Energy Rev., 2018, 91, 1025–1044 CrossRef CAS.
- S. Q. Zhang, L. Ye and J. H. Hou, Renewable Sustainable Energy Rev., 2016, 6, 1502529 Search PubMed.
- H. P. Shen, D. Walter, Y. L. Wu, K. C. Fong, D. A. Jacobs, T. Duong, J. Peng, K. Weber, T. P. White and K. R. Catchpole, Adv. Energy Mater., 2019, 6, 1902840 CrossRef.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2019, 6, 1902840 Search PubMed.
| This journal is © The Royal Society of Chemistry 2020 |