
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMatters of class: coming of age of class III and IV lanthipeptides
Julian D.
Hegemann
 * and
Roderich D.
Süssmuth
*
* and
Roderich D.
Süssmuth
*
Institute of Chemistry, Technische Universität Berlin, Straße des 17. Juni 124, 10623, Berlin, Germany. E-mail: roderich.suessmuth@tu-berlin.de; jdhegemann@googlemail.com
First published on 16th July 2020
Abstract
Lanthipeptides belong to the superfamily of ribosomally-synthesized and posttranslationally-modified peptides (RiPPs). Despite the fact that they represent one of the longest known RiPP subfamilies, their youngest members, classes III and IV, have only been described more recently. Since then, a plethora of studies furthered the understanding of their biosynthesis. While there are commonalities between classes III and IV due to the similar domain architectures of their processing enzymes, there are also striking differences that allow their discrimination. In this concise review article, we summarize what is known about the underlying biosynthetic principles of these lanthipeptides and discuss open questions for future research.
 Julian D. Hegemann | During his Diploma and PhD theses, Julian D. Hegemann investigated the biosynthesis and characteristic features of lasso peptides in the research group of Professor Mohamed Marahiel at the Philipps-Universität Marburg. For his postdoctoral research on the biochemistry of lanthipeptides, he joined the laboratory of Professor Wilfred van der Donk at the University of Illinois at Urbana-Champaign, where he worked from 2016–2019. Since Mid-2019, he is active at the Technische Universität Berlin. |
 Roderich D. Süssmuth | Roderich D. Süssmuth is Professor of the Department of Chemistry at the Technische Universität Berlin. He received his Diploma (1995) and PhD in Chemistry (1998) from the Eberhard Karls Universität Tübingen (Germany). This was followed by a postdoctoral stay at The Scripps Research Institute, La Jolla (2000–2001) and an Assistant Professor position as Emmy-Noether Fellow at the Eberhard Karls Universität Tübingen (2002–2004). He was appointed to the position of Associate Professor at the Technische Universität Berlin in 2004, and Full Professor in 2009. His research interests encompass medicinal and peptide chemistry, peptide drugs, and the biosynthesis of natural products. |
Introduction
Lanthipeptides belong to the superfamily of ribosomally synthesized and posttranslationally-modified peptide (RiPP) natural products. RiPPs are defined through a shared biosynthetic origin.1 A genetically encoded precursor peptide is synthesized at the ribosome and then matured into the final natural product by action of one or more processing enzymes. The precursors themselves can be subdivided into an N-terminal leader region, which is needed for enzymatic recognition, and a C-terminal core peptide, which becomes modified by the processing enzymes and yields the mature natural product after eventual removal of the leader peptide by proteolysis. A common nomenclature has been agreed on for RiPP precursors,1 where the N-terminal residue of the core peptide is designated as residue 1 and numbering increases towards the C-terminus, whereas the C-terminal residue of the leader peptide is designated as residue −1 and numbering decreases towards the N-terminus.The defining feature of lanthipeptides is the presence of β-thioether crosslinks, which are installed through posttranslational modifications of Ser/Thr and Cys residues (Fig. 1A).1,2 Depending on characteristics of their biosynthetic enzymes, lanthipeptides can be subdivided into four classes. It is worth mentioning that the biosynthesis of class I and class II lanthipeptides has been very well studied and more recently structures of the corresponding biosynthetic enzymes have been reported.1–7 Less is known about class III and class IV enzymes, although lately more details about their underlying mechanistic principles have been uncovered. In general, the biosynthetic enzymes of the different classes are defined as follows (Fig. 1B):
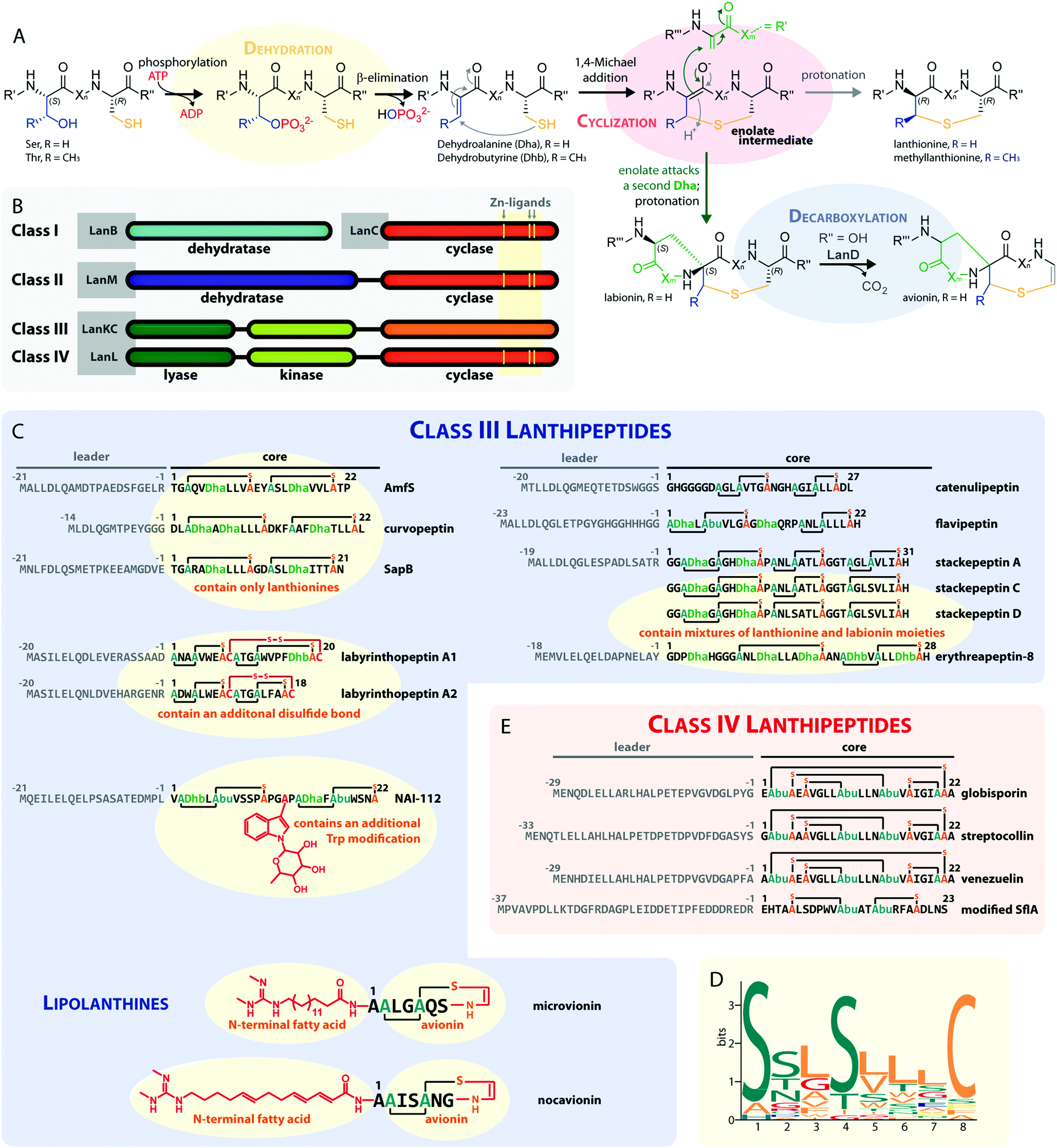 | ||
| Fig. 1 (A) Underlying principles of class III and IV lanthipeptide biosynthesis. (B) Overview of the four known lanthipeptide classes. Schematic showing the primary structures of important (C) class III and (D) class IV lanthipeptides. The color coding is as follows: leader peptide residues are grey, unmodified core peptide residues are black, cysteines involved in (methyl)lanthionines and labionins are orange, other residues in (methyl)lanthionines and labionins are teal, Dha/Dhb residues are green, additional posttranslational modifications are red. (E) MEME motif24 predicted on basis of all Sx2Sx2–5C motifs from the peptides in (C) highlighting the most common residues and arrangements of these motifs in studied class III lanthipeptides. Dha = Dehydroalanine. Dhb = Dehydrobutyrine. Abu = Aminobutyric acid. | ||
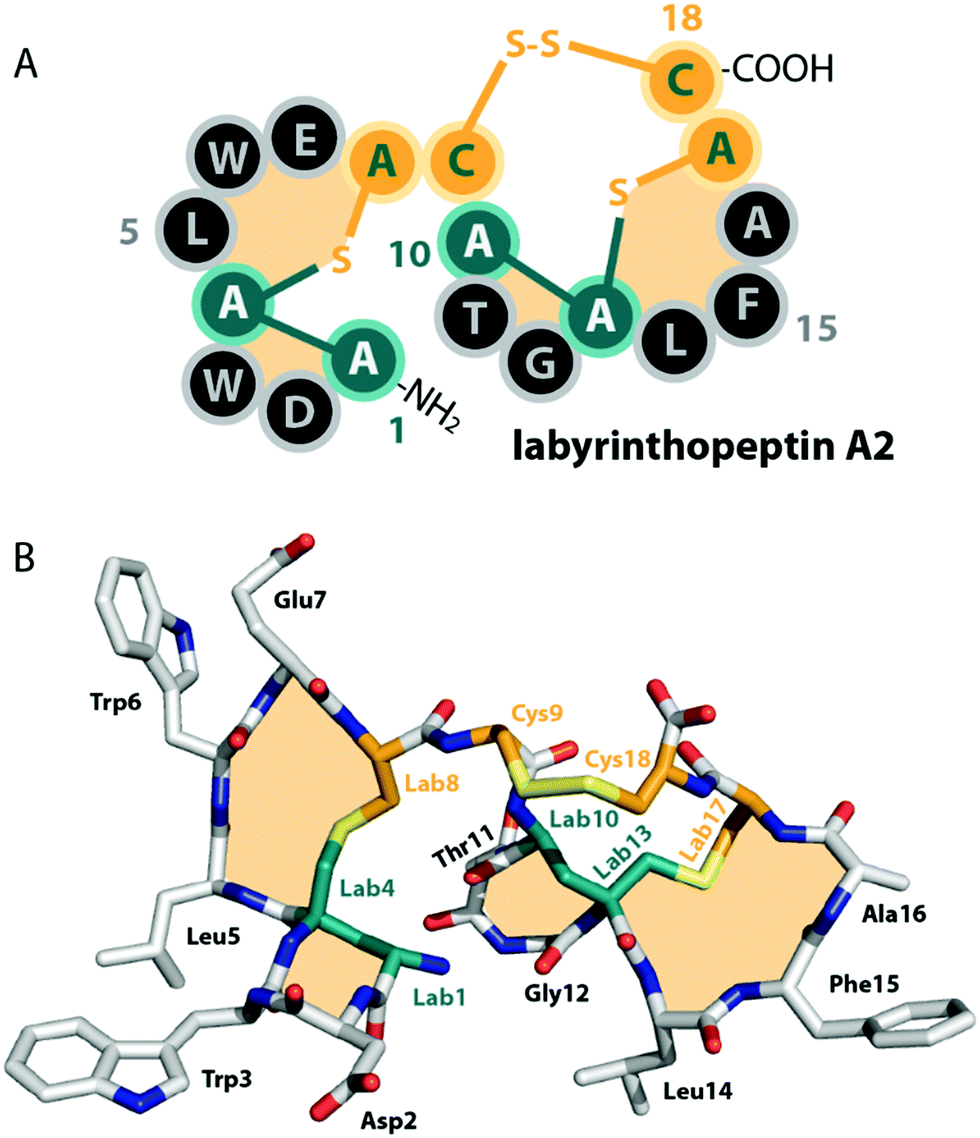 | ||
| Fig. 2 (A) Schematic representation and (B) crystal structure (CCDC number 721326) of labyrinthopeptin A2.13 The labionin macrocycles are highlighted by orange shading in the background. | ||
– Class I uses a combination of discrete aminoacyl-tRNA-dependent dehydratases (LanBs)3,8 and cyclases (LanCs).5
– Class II uses proteins with N-terminal ATP-dependent dehydratase and C-terminal cyclase domains (LanMs).6,7 In these enzymes, the phosphate transfer from ATP and the β-elimination of the phosphorylated Ser/Thr residues take place at the same active site in the dehydratase domain.2,6
– Class III and IV enzymes are also NTP-dependent and share a three-domain structure, featuring an N-terminal lyase, a central kinase, and a C-terminal cyclase domain.9–15 Thus, unlike in class II dehydratase domains, two distinct catalytic centers catalyze the phosphorylation and subsequent β-elimination reactions in these enzymes. Whereas the lyase and kinase domains share high homology, the differentiation of the classes is based on unique characteristics of their cyclase domains. While class IV enzymes (LanL) feature cyclase domains with the conserved zinc-binding motif also found in class I and class II cyclases,1,2,5,6 these are lacking in class III enzymes (LanKCs).
Class III and IV lanthipeptides have been described comparatively recently in 2010.13,14 Although the lanthipeptides SapB (Fig. 1C)16–18 and AmfS (Fig. 1C)19,20 were known since the late 1980s18 and early 1990s,20 respectively, they were only retrospectively recognized as class III representatives when the discovery of the labyrinthopeptins led to the classification of this new lanthipeptide subgroup.13,21
Unlike the mostly antibacterial activities observed for class I and II lanthipeptides, e.g. through inhibition of cell wall biosynthesis by binding to lipid II,1,2 the biological functions of many of the known representatives of class III and IV lanthipeptides have remained elusive, since they mostly showed no or only weak antibacterial activities. Initially, morphogenetic functions involved in aerial hyphae formation of their respective Streptomyces hosts have been described for SapB and AmfS, which likely represent their natural functions.1,2,16–20 However, amongst the class III lanthipeptides several other interesting bioactivities have been reported, including the antiallodynic properties of labyrinthopeptins and NAI-112 (Fig. 1C and 2)13,22,23 and the strong anti-MRSA activities of lipolanthines (Fig. 1C).25 Labyrinthopeptins additionally display remarkable broad-spectrum antiviral properties against Herpes simplex virus (HSV), HIV, dengue virus, and Zika virus.13,22,26–28
The underlying biosynthetic principles are basically the same for class III and IV lanthipeptides (Fig. 1A). First, the kinase domain activates the hydroxy groups of Ser/Thr side chains in the core peptide by NTP-dependent phosphorylation.9,10,29,30
Next, the lyase domain catalyzes a phosphate elimination reaction, which yields dehydroalanine (Dha) and dehydrobutyrine (Dhb) moieties.10–12,29,30 Finally, the cyclase domain mediates the nucleophilic attack by the Cys thiol of these unsaturated double bonds in fashion of a 1,4-Michael-addition.10,11,13,14,30 The nucleophilic attack yields an enolate intermediate that through protonation is converted into a (methyl)lanthionine β-thioether. In some cases, the enolate intermediate can undergo a second nucleophilic attack of another Dha moiety, which yields entwined, bicyclic structures that are called labionins (Fig. 1 and 2).11,13,23,29,31,32 So far, labionins have exclusively been found in class III lanthipeptides and are derived from a shared Sx2Sx2–5C motif. Compared to class IV lanthipeptides, the ring topologies in class III are less diverse and rather restricted. Particularly, the two serines in the Sx2Sx2–5C motif are always separated by two amino acids (Fig. 1D).33 Most commonly, the core peptides of class III precursors contain two iterations of adjacent Sx2Sx2–5C motifs. However, these motifs are not always converted into labionins but instead into lanthionines containing an internal Dha residue.
Remarkably, even mixed macrocycle topologies have been observed, e.g. for the erythreapeptins and stackepeptins31,32,34 (Fig. 1C). It should also be noted that the stackepeptins carry a third iteration of the Sx2Sx2–5C motif. This feature was hypothesized to have originated from motif multiplication,32 which is similarly known from cyanobactins like patellamides, trunkamides, and tenuecyclamides.35 Interestingly, with the discovery of the lipolanthines a monomer labionin motif was identified as well (Fig. 1C),25,36 which might have served as the basis for such multiplication events. Conversely, most of the characterized class IV lanthipeptides10,14,37,38 are closely related to each other, feature an identical, overlapping ring conformation, and only differ in some core peptide residues that are not further modified after the ribosomal assembly of the precursor (Fig. 1E). The only so far known class IV lanthipeptide with a different ring pattern features two non-overlapping methyllanthioine rings15 (Fig. 1E).
In this article, we will give a concise overview of what is known about the biosynthesis of class III and IV lanthipeptides with a particular focus on the similarities and differences of the LanKC and LanL processing enzymes and the role of each individual catalytic domain of these enzymes will be discussed in detail in the upcoming sections.
Directionality of the enzymatic processing
With regard to the directionality of core peptide processing, only limited information is available, as this subject was so far only studied in detail for the class III representatives LabKC13,29 (Fig. 1C and 2, from labyrinthopeptin biosynthesis) and CurKC30,34 (Fig. 1C, from curvopeptin biosynthesis), while for class IV enzymes, only SgbL9,10 (Fig. 1E, from globisporin biosynthesis) and SflL15 (Fig. 1E, from the maturation of an unnamed lanthipeptide from Streptomyces sp. NRRL S-1022) were investigated.For the class III systems, chemical synthesis of precursor peptide variants provided the advantage of selective labelling to directly elucidate directionality of peptide processing. Hence, Ser residues of the core peptides where sequentially replaced with α-deuterated Ser. Subsequently, these peptides were incubated with the respective class III enzyme and the assays were quenched before completion, which allowed detection of the completely dehydrated product as well as all dehydration intermediates by high-resolution mass spectrometry (MS).29,30 Identification of a dehydration event at an α-deuterated serine is possible as the elimination of HDO causes a 1 Da higher mass shift compared to the elimination of H2O. Remarkably, a general C-to-N-terminal directionality of the dehydration events was observed for the modifications introduced by LabKC and CurKC.29,30 The only exception to this rule was that Ser1 phosphorylation in CurA actually preceded modification of the most C-terminal Ser (Ser15).30 However, modification of the other CurA Ser residues generally followed the C-to-N-terminal order (Fig. 3).30 It should also be pointed out that the in-depth analysis of the observed isotopic distributions of the MS signals revealed some deviations from simulated distribution patterns.29,30 This observation, which at least holds true for in vitro experiments, demonstrates that there is a main route of modification but also a variety of minor modification sidetracks (Fig. 3). Without the usage of the sophisticated deuterium labeling strategy, the presence of such minor modification routes is hard to detect.
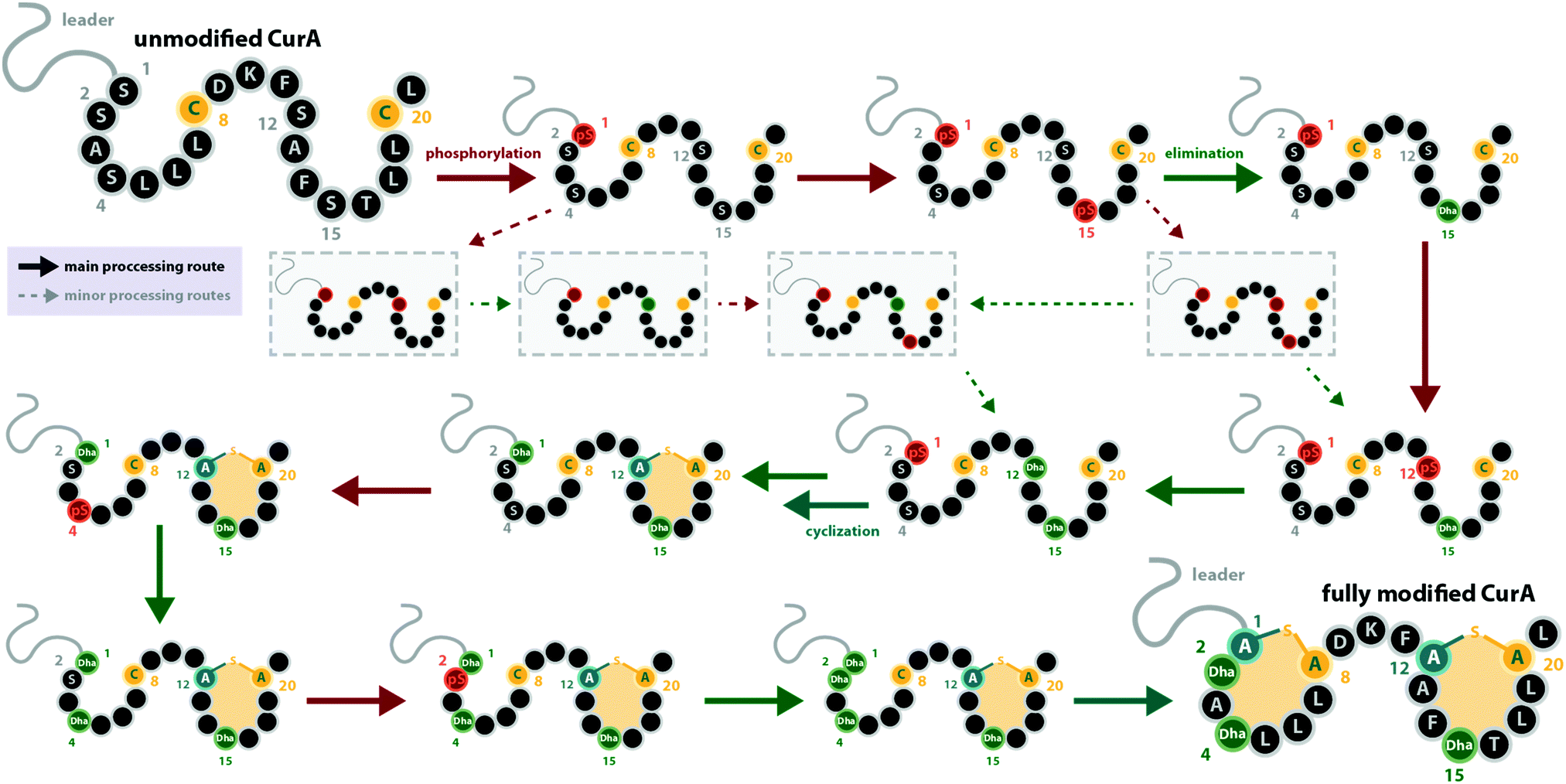 | ||
| Fig. 3 Processing order of the CurA precursor peptide by the CurKC processing enzyme, which was investigated in-depth using the deuterium-labeling approach as described in the main text.30 This case exemplifies that while a general C-to-N-terminal directionality is observed, there can be exceptions to this order (here, Ser1 is phosphorylated first) and that besides the major processing route, minor routes can be detected with sufficient scrutiny (note that only a few exemplary early minor processing routes are shown and not all that were described in the original publication30). This example also emphasizes that while usually the same residue becomes phosphorylated and eliminated before the next residue is processed, there are also exceptions to this rule. pS = phosphoserine. | ||
To determine the sequence of dehydration events in a class IV system, a SgbL variant lacking the C-terminal cyclase domain was incubated with the SgbA precursor and again the reaction was quenched before completion, thus allowing detection of all dehydration intermediates.10 Instead of using isotopic labelling, the order of modification was in this case determined by tandem MS analysis of the linear, partially dehydrated intermediates. In contrast to the class III enzymes, the in vitro experiments with SgbL revealed a preferred N-to-C-terminal directionality of the dehydration events in SgbA,10 which is similarly occurring for some representatives of class I and II maturation enzymes.2,39 Furthermore, in vitro assays with both SgbL and VenL variants lacking the C-terminal cyclase domain demonstrated that full dehydration can be accomplished in absence of macrocycle formation.7,10,14 Surprisingly, experiments performed with the SflA precursor peptide and the SflL processing enzyme did not show a clear order of modification events.15 Instead, these in vitro assays revealed only a weak preference of SflL for incorporating the modifications in a C-to-N-terminal direction, yet intermediates arising from a different order of modifications were very pronounced.
It is interesting to note that all of these experiments point towards a sequential order of phosphorylation and elimination steps, i.e. the enzymes have a pronounced preference for eliminating a phosphate group before the sequentially following side chain is phosphorylated. The exception to this rule is, again, the curvopeptin system. Whereas Ser1 in CurA is phosphorylated first, it only undergoes phosphate elimination after the most C-terminal (Ser15) and then the penultimate (Ser12) serine both become phosphorylated and eliminated (Fig. 3).30
It has not been elucidated in which order the macrocycles are installed by LabKC (class III) and SgbL (class IV) and if cyclization already occurs during the process of dehydration or only after complete dehydration of the core peptide is accomplished. However, in vitro experiments with CurA and CurKC showed that cyclization follows a C-to-N-terminal order and that the formation of the C-terminal lanthionine ring precedes both dehydration and cyclization of the N-terminal part of the core peptide (Fig. 3).30 Furthermore, if the formation of the C-terminal lanthionine ring in CurA is inhibited by a S12A exchange, formation of the N-terminal lanthionine ring is abrogated, whereas full dehydration can nonetheless be accomplished.30 The cyclization order by the class IV enzyme SflL for the non-overlapping methyllanthionine rings in the SflA precursor was also investigated.15 While a weak preference for the formation of the C-terminal before the N-terminal methyllanthionine was observed, a significant portion of SflA seemed to be modified in the opposite order. Yet, Ala exchange of the Thr residue involved in the C-terminal ring decreased the overall efficiency of the formation of the N-terminal methyllanthionine, but not completely abolished it.15 Conversely, Ala substitution of the Thr yielding the N-terminal β-thioether crosslink did not interfere with the formation of the C-terminal methyllanthionine.15 Hence, the presence of the C-terminal macrocycle in SflA might indeed facilitate the modification of the N-terminal part of the core peptide. Interestingly, in vitro assays with wild type SflA also demonstrated that dehydrations and cyclization reactions happen in parallel in this class IV system, as some observed intermediates lacked β-thioether crosslinks, but already featured both Dhb residues, whereas others had one of the methyllanthionine rings already formed while the second Thr was not yet dehydrated.15
While the directionality of the macrocycle incorporation by the class III enzyme AciKC during catenulipeptin (Fig. 1C) biosynthesis was not directly assessed, indirect evidence obtained from in vitro modification assays using AciKC and precursor peptide variants suggests a C-to-N-terminal cyclization order as well.11 Here, AciA variants were employed that had either of the labionin-forming Cys residues in the core peptide replaced with Ala. While replacement of the N-terminal Cys15 yielded a fully dehydrated precursor peptide with the C-terminal labionin still formed, the Ala substitution of the C-terminal Cys25 yielded only a linear, dehydrated peptide.11 This result suggests that in AciA the formation of the C-terminal labionin is a prerequisite for the formation of the N-terminal labionin and thus implies the C-to-N-terminal directionality of cyclization events.
When the two Cys thiol groups in AciA were protected by oxidation to a disulfide bond, AciKC still fully dehydrated the core peptide, yet could not install the labionin crosslinks.11 Subsequent reduction of the disulfide bond yielded linear, dehydrated AciA, which in turn could be cyclized by AciKC, whereas no cyclization was observed when AciKC was absent.11 These experiments demonstrate that cyclization is possible after the core peptide region of AciA is fully dehydrated, yet it remains unclear if labionin formation could already occur on a partially dehydrated peptide.
Similar in vitro experiments were carried out to investigate the maturation of the class III lanthipeptide stackepeptin (Fig. 1C). If the formation of the C-terminal labionin ring is disturbed by exchange of either of the corresponding ring-forming Ser in StaA (Ser23 and Ser26) to Ala, the resulting StaA variant is barely modified.32 Furthermore, a StaA variant with Ala substitutions of all three Cys residues of the core peptide (Cys10, Cys19, Cys30) was also only partially dehydrated.32 Hence, the formation of the C-terminal labionin is not only a prerequisite for the formation of the other two rings in this system but also crucial for the preceding modification of the StaA precursor peptide by StaKC. Interestingly, this is contrasted by Ala exchanges of the Ser residues in StaA that are needed for the formation of either the N-terminal (Ser3, Ser6) or central ring (Ser12, Ser15), as either exchange did not affect the formation of the remaining two other rings nor interfered with the dehydration of the remaining Ser residues.32
In the FlaKC-mediated maturation of the flavipeptin precursor FlaA (Fig. 1C), both labionin rings can form independently from one another. Exchange of all Ser/Thr residues in either the N- or C-terminal half of the core peptide abolished the formation of the labionin ring in the respective region, while dehydration and cyclization of the residues in the other region were not disturbed.40 Furthermore, a FlaA variant with replacements of both ring-forming Cys residues (Cys8, Cys21) to Ala was still fully dehydrated.40 Thus, it remains unclear if FlaKC has a preferred order of macrocyclization events.
For the class IV lanthipeptide synthetase VenL that is involved in venezuelin (Fig. 1E) maturation, the Ala scan of the Cys residues in its precursor VenA did not yield a clear conclusion regarding the order of cyclization,14 as the effects of the Cys substitutions did not yield uniform results.
In summary, it seems that the processing through the class III LanKC enzymes follows a mostly C-to-N-terminal directionality for both the dehydration and the cyclization reactions.29,30,32 This directionality is however counterintuitive as one would assume that recognition and immediate start of processing would be spatially located in close proximity. In fact, C-to-N-terminal processing occurs to a lesser extent than the N-to-C-terminal processing that is often observed for class I and II lanthipeptides.1,2,39 A generalizable statement cannot be made for class IV enzymes, as this was only investigated for two class IV systems so far. SgbL introduced dehydrations in a sequential N-to-C-terminal order into the SgbA precursor peptide.10 The cyclization order in this and other closely related class IV systems has not been studied yet, since their overlapping four (methyl)lanthionine rings (Fig. 1E) make this a very challenging task.10,14,37,38 However, the investigation of the incorporation of the methyllanthionine crosslinks in the SflA precursor by SflL did not yield a clear picture either.15 Although SflL seems to prefer to install both dehydrations and macrocycles in a C-to-N-terminal order, this preference is only weakly pronounced.
For class I and II lanthipeptide enzymes, full dehydration can usually be accomplished in the absence of the cyclase (domain).2 However, if both the dehydratase and the cyclase active sites are present under in vitro conditions, it is often observed that some cyclization events happen before full dehydration of the core peptide is achieved and that the enzymes introduce dehydrations and macrocycles following a more or less strict reaction sequence.2 Even though this was not explicitly tested for many of the investigated class III and IV systems, it is therefore assumed that under native conditions the first macrocycles are usually installed before the core peptide is fully dehydrated.
Often the sequence and/or maturation state of the core peptide will also affect the (further) processing by the biosynthetic enzymes as was shown by utilizing precursor peptide variants for in vitro modification assays. In general, it seems hard to make a generalizable statement about the processing orders for class III and IV enzymes, as different representative synthetases can behave contrarily to each other. Additionally, it has to be considered that there is a sampling bias for the clusters discovered and hence for the enzymes that were tested so far.
However, this non-uniformity emphasizes the importance of such studies, as only the examination of enough systems will allow the recognition of general principles and the underlying logic of these processes.
Removal of the leader peptide
Until recently, it was unknown how leader peptide removal is realized for class III and IV lanthipeptides as nearly all known biosynthetic gene clusters lack a gene encoding a potential peptidase that could fulfill such a function. Based on the observation that sometimes native producers would not yield a single, distinct class III or IV lanthipeptide but rather microheterogenic mixtures of two or more peptide species that differed merely by single amino acids present or missing at the N-terminus, the involvement of unspecific aminopeptidases in leader peptide removal was hypothesized.10,13,31,32,34,38,41 Considering that the cyclic (methyl)lanthione and labionin groups in the modified core peptides provide resistance against degradation by amino-, carboxy- and many endopeptidases, such a route seemed plausible.9,10,15,41In the flavipeptin (Fig. 1C) biosynthetic gene cluster, a prolyl oligopeptidase (POP)-family enzyme is encoded that was shown to cleave C-terminally of the Pro−12 residue in the leader region of the modified precursor peptide.40 However, after FlaP-mediated proteolysis, a portion of the leader peptide still remains attached to the suggested core peptide. Thus, further removal of the remaining leader peptide residues through activity of an aminopeptidase would be needed to obtain the final natural product. Processing of the class III lanthipeptide flavipeptin was therefore hypothesized to indeed utilize a combination of a site-specific, dedicated leader peptidase with subsequent unspecific aminopeptidase-mediated hydrolysis.
Remarkably, FlaP only acts on the modified precursor peptide (in particular the presence of the C-terminal labionin ring was shown to be important for FlaP activity), a feature that ensures that leader removal is only initiated after the core peptide has been processed.40 Another interesting observation concerning FlaP is that this protease is still able to cleave the leader of modified FlaA variants carrying Gly, Ala, Val, or Lys substitutions of Pro−12 in vitro.40 These findings imply that FlaP does not primarily use Pro−12 for recognition of the cleavage site. As mature flavipeptin was never isolated from its native producer, it seems also possible that the leader/core peptide border in FlaA was wrongly annotated. Between Pro−12 and the currently suggested start of the core peptide is a glycine-rich sequence, which is reminiscent to the linear glycine-rich N-terminus of the class III lanthipeptide catenulipeptin11 (Fig. 1C). Therefore, the modified peptide released from FlaP-mediated proteolysis might actually already be the mature natural product. As most class III and IV biosynthetic gene clusters lack a gene encoding a FlaP-homolog, this particular mode of leader peptide removal is however not ubiquitous.
Insight into a potentially more general approach to leader peptide removal in class III and IV lanthipeptide maturation came from study of the biosynthetic gene cluster producing the class III lanthipeptide NAI-112 (Fig. 1C). This gene cluster contains a gene coding for the Zn-dependent peptidase AlpP.23,41 It was demonstrated that AlpP exhibits both endopeptidase and aminopeptidase activities, when incubated with the corresponding precursor AlpA as well as with precursor or leader peptides of a selection of other class III lanthipeptides.41 Unlike the dedicated proteases involved in the maturation of class I and II lanthipeptides,2,42–45 AlpP does not cleave at a single specific site at the C-terminal end of the leader region (i.e. in between residues −1 and 1 of the precursor) but was shown to exhibit its endopeptidase activity at several different sites in the leader peptide.41 Prolonged incubation of AlpP with the AlpA precursor revealed the additional aminopeptidase activity inherent to AlpP, which is needed to remove the leader peptide residues that remain after the initial endopeptidase cleavage.41 Unlike FlaP, AlpP and tested homologs of AlpP from other class III lanthipeptide-producing strains do not differentiate between modified and unmodified precursor peptides.41
It seems likely that AlpP-homologs are commonly used for leader removal in class III and IV lanthipeptide biosynthesis. However, as the corresponding class III and class IV biosynthetic gene clusters usually do not encode such a peptidase, the respective endopeptidases are expected to be encoded at different loci in the respective genomes. Bioinformatic analysis indeed demonstrated that all known class III lanthipeptide producing strains carry at least one AlpP homolog in their genomes, as do the Streptomyces strains producing the class IV lanthipeptides venezuelin and streptocollin.41 The Streptomyces strain harboring the SflA/SflL class IV system encodes a total of four AlpP homologs in its genome, which were tested for in vitro activity against modified and unmodified SflA precursor peptide.15 While one AlpP homolog did not show any activity in these assays, two homologs exhibited the same combination of endo- and aminopeptidase activity as described for AlpP against both modified and unmodified SflA, while the fourth homolog was only active on the unmodified precursor.15 Two other homologs of AlpP were identified in the genome of the strain producing the class III lanthipeptide microvionin of the lipolanthine subclade.36 Both homologs were able to remove the leader peptide of the modified MicA precursor in vitro, although the observed cleavage sites differed slightly from the ones described for AlpP.36,41 When incubated with unmodified MicA, cleavage was even observed inside the core peptide region,36 which emphasizes the importance of a proteolytically stable macrocyclic structure resulting from core peptide processing for such leader removal strategies.
The fact that these endopeptidases are not necessarily located in the genetic vicinity of the respective lanthipeptide biosynthetic gene clusters and the observation that their preferred cleavage sites can differ15,36,41 suggest that these peptidases do fulfill more general functions in their native hosts and are merely borrowed by the lanthipeptide systems for completing the precursor modification. Combined with the lack of a single specific cleavage site in AlpA for AlpP, this observation could mean that class III and IV systems evolved towards utilizing a generally accessible proteolytic machinery for leader peptide removal. As expression in E. coli (which contains the ePepN aminopeptidase) revealed inherent stability issues of some class IV precursors in vivo,10,15 it seems feasible that lanthipeptides of these families might have adopted the usage of a proteolytically sensitive leader peptide, which facilitates utilization of generally available aminopeptidases for processing instead of employing dedicated proteases as reported for other RiPPs.1,42–49 In summary, the question how leader peptide removal is accomplished during maturation of many of the known class III and class IV lanthipeptides cannot be fully answered. However, the recent investigation of the role of AlpP in NAI-112 maturation,41 the experimental evidence that modified SflA precursor peptide can be processed by two out of four AlpP homologs from the same host strain,15 and the observation that for many class III/IV lanthipeptides their native producers yield series of congeners differing only by few amino acids at the N-terminus,10,13,31,32,34,38,41 strongly suggest a general involvement of aminopeptidases in processing, partially in combination with some more or less specific endopeptidases (Fig. 4).
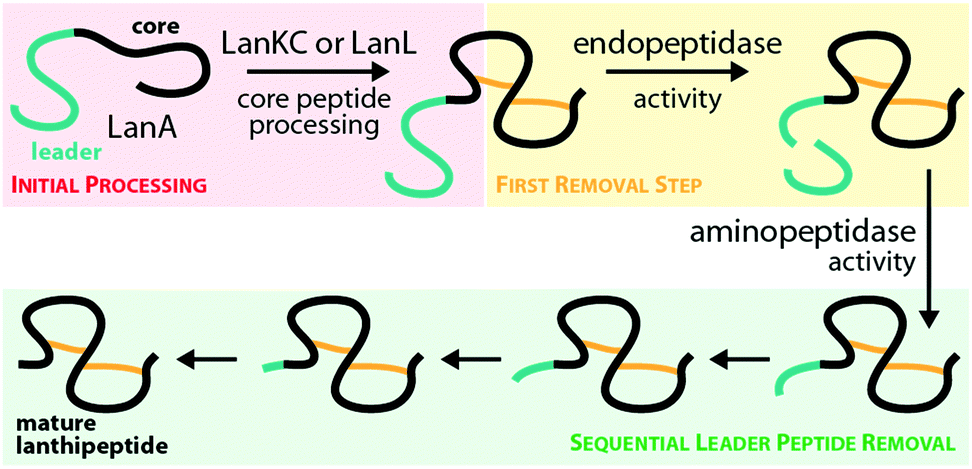 | ||
| Fig. 4 Hypothesized10,31,34,40,41 general route for leader peptide removal in class III and IV lanthipeptide systems. It should be noted that the endopeptidase step might not be required in all cases. | ||
Role of the kinase domain
The central kinase domains of class III and IV lanthipeptide synthetases initiate core peptide processing through activation of the Ser/Thr side chains for the subsequent β-elimination reactions by the lyase domain. For the biosynthesis of class III/IV lanthipeptides, this activation is accomplished by phosphorylation of the Ser/Thr hydroxy moieties by transfer of a phosphate group from, most commonly, ATP and subsequent release of ADP. All in vitro modification assays using class IV synthetases have been successfully carried out with ATP as phosphate donor and it has not yet been investigated if other (d)NTPs are accepted as co-substrates as well.7,10,12,14,15 This is contrasted by the initial finding that no precursor modification was observed in in vitro assays with LabKC and LabA2 in presence of ATP.50 However, when such assays were performed in the presence of GTP or dGTP as phosphate donor, full conversion of the LabA2 precursor peptide was accomplished.50 While LabKC has a strict requirement for (d)GTP, other class III enzymes were shown to have different preferences. While CurKC and StaKC show at least some activity with any (d)NTP in vitro, purine nucleotides seemed to be their preferred co-substrates.32,34 Conversely, EryKC31 and MicKC25 cannot utilize GTP and are specific towards ATP, whereas AciKC does not show a preference and was demonstrated to fully convert the AciA precursor peptide regardless of which NTP was present.11 FlaKC also accomplished full modification of the corresponding FlaA core peptide with either ATP or GTP but was not tested with TTP or CTP. Thus, LabKC is currently the only example of a class III/IV lanthipeptide synthetase that cannot utilize ATP.40 In general, there is a high homology between the kinase domains from class III and IV enzymes and thus it is assumed that they operate in a likewise manner.1,2,9,12 They further share homology with certain structurally characterized human Ser/Thr kinases and the mycobacterial protein kinases B (PknB) and G (PknG).9 The homology with PknB/PknG was used to identify the catalytic residues of the SgbL kinase domain.9 Indeed, an alignment of either class III or class IV enzymes with PknB and PknG shows that almost all catalytic residues reported for the mycobacterial proteins are fully conserved (Fig. 5A) in class III/IV kinase domains. In addition, an Ala scan of these residues in SgbL demonstrated that their presence was critical for core peptide processing.9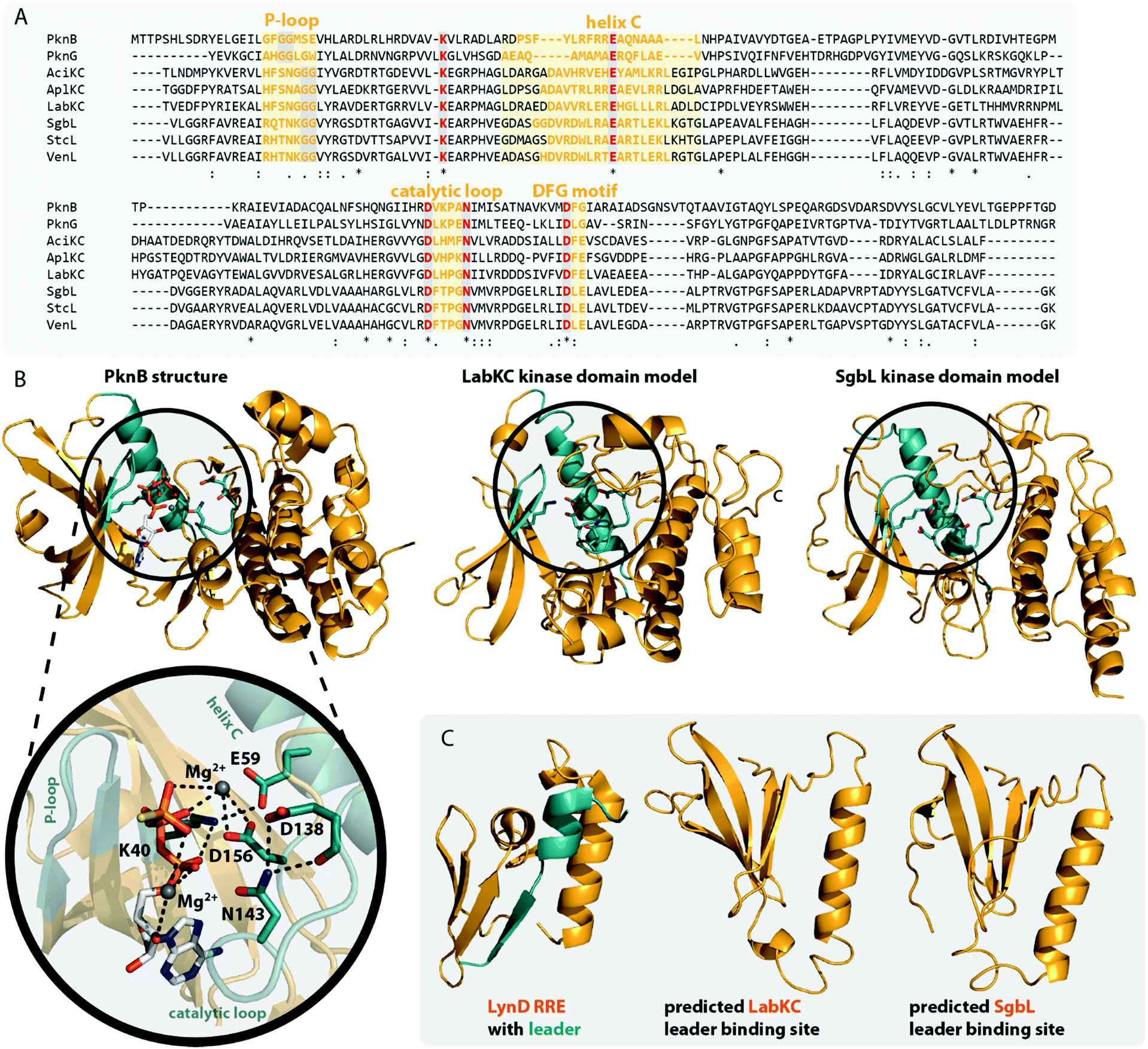 | ||
| Fig. 5 (A) Excerpt of an alignment of the mycobacterial protein kinases PknB and PknG with the kinase domains of exemplary class III (AciKC, AplKC, LabKC) and class IV (SgbL, StcL, VenL) lanthipeptide synthetases. Conserved catalytic residues are shown in red and structural features important for activity are highlighted. (B) Comparison of the crystal structure of PknB binding the ATP-mimic adenosine 5′-[γ-thio]triphosphate (PDB code 1MRU) with the Phyre2 models of the LabKC and SgbL kinase domains. A close up of the active site of PknB is shown. Catalytic residues and regions of importance are highlighted in teal. (C) Structure of the RRE domain of the cyanobactin heterocyclase LynD from Lyngbya aestuarii with bound leader (PDB code 4V1T) compared with the segments of the LabKC and SgbL structure models that are implied9 in leader binding. | ||
In more detail, the exchange of the conserved Lys and Glu residues in SgbL significantly decreased catalytic turnover.9 In PknB and PknG, these residues form a salt bridge that is needed for structural integrity of the active conformation.51,52 Furthermore, the side chain amine of this Lys is involved in the coordination of the α- and β-phosphates of ATP.51,52 The conserved Glu is located in a structural motif that is called helix C. Other residues found in helix C show little to no conservation between the different enzyme classes, yet the PsiPred webtool53 (Fig. 5A) indeed predicts with high confidence that the regions surrounding the conserved Glu residues always adopt an α-helical fold.
The so-called catalytic loop contains two other conserved residues that are needed for catalysis.51,52 The conserved Asp acts as an acceptor during proton transfer, while the conserved Asn residue forms a canonical contact with this Asp and also plays a role in Mg2+ coordination.51,52 Yet again, little conservation of other residues in the catalytic loop are observed between the different enzyme classes (Fig. 5A). The last catalytic residue identified is a second conserved Asp located in the so-called DFG motif (Fig. 5A), which is also required for Mg2+ coordination.51,52 The DFG motif varies between enzymes. PknB actually has a DFG sequence, while it differs in PknG (DLG), and e.g. in SgbL (DLE) and LabKC (DFE).9,51,52 Replacement of either of the two conserved Asp residues or the conserved Asn fully abolished enzymatic activity of SgbL.9 While the roles of these catalytic residues have not yet been confirmed experimentally for a class III enzyme, the high conservation and high homology to class IV kinase domains makes it likely that these residues share the same functions in catalysis.
However, one feature important for activity of PknB/PknG is not conserved in the class III/IV kinase domains. In PknB/PknG the backbones of conserved Gly residues in the so-called P-loop are involved in the coordination of the ATP phosphates.51,52 Yet, alignments demonstrate that these Gly residues are not conserved in class III and IV kinase domains (Fig. 5A).9 Interestingly, an alignment of only class III and IV enzymes show two conserved Gly residues in vicinity to the predicted P-loop region, thus it might be that the P-loop motif is still present in class III/IV lanthipeptide synthetases but is shifted compared to the P-loop regions in PknB/PknG. Experimental proof for this hypothesis is however still missing.
Homology of class III/IV kinase domains with structurally studied Ser/Thr kinases and PknB/PknG9,51,52 is sufficiently strong to predict a high confidence structure model for the class III/IV enzymes utilizing the Phyre2 webtool54 (Fig. 5B). Indeed, the catalytic residues in the predicted models for the LabKC and SgbL kinase domains are oriented towards the suggested active site and would be able to fulfill their functions in catalysis.
The kinase domains of class IV enzymes were proven to have significance beyond the phosphorylation activity though. It was observed that an SgbA variant lacking the part of the leader region needed for enzymatic recognition is not modified by SgbL in vivo,10 thus demonstrating that leader peptide recognition and binding is a prerequisite for efficient core peptide modification by SgbL. As the isolated kinase domains of VenL and SgbL were shown to be able to phosphorylate their precursors (VenA/SgbA),9,10,14 it was implied that the kinase domain would also contain the leader peptide binding site.
Indeed, in vitro binding studies with the SgbA leader peptide and the different separated domains of SgbL clearly demonstrated that the kinase domain is binding the leader peptide almost as tightly as full length SgbL,9,10 thus confirming the role of the kinase domain in substrate recognition. Recent studies of the class IV enzyme SgbL utilizing HDX-MS further revealed that it is the N-terminal part of its kinase domain (Fig. 5C) that is binding the leader.9 There is yet no experimental proof that the same is true for class III enzymes, but this is likely given due to the high similarity of the kinase domains between classes.
It was furthermore demonstrated that Ala substitutions of either of the two catalytic Asp or the catalytic Asn in the SgbL kinase domain did not affect the leader peptide binding.9 Conversely, Ala exchanges of the conserved catalytically important Lys and Glu residues significantly reduced the leader binding affinity.9 However, it remains unclear if these residues are directly involved in interactions with the leader peptide or if their Ala substitution just causes an overall destabilization of the active protein fold and thus indirectly affects the propensity of these SgbL variants to bind the SgbA leader.
The presence of a leader peptide binding site in class IV and potentially class III kinase domains also implies that for these RiPP subfamilies, leader recognition is independent of the common RiPP recognition element (RRE) motif.55 RREs consist of three N-terminal β-strands, followed by three C-terminal α-helices (Fig. 5C).3,46–49,56–60 Corresponding leaders are bound in between the β3-strand and the α3-helix and often form a β-strand fold themselves upon binding that aligns with the antiparallel β-sheet of the RRE. RREs can be found in a large variety of RiPP subfamilies, including for example the processing enzymes of lasso peptides,49,56 cyanobactins,59 sactipeptides,48,57 streptides,61 pyrroloquinoline quinone (PQQ),58,62 mycofactocin,63 and even class I lanthipeptides.3 Interestingly, the N-terminal region of the SgbL kinase domain model, which was suggested as leader binding region based on the HDX-MS results,9 is somewhat reminiscent of the RRE motif, as it contains an antiparallel β-sheet and the adjacent helix C. Yet, a co-crystal structure will be required to answer, if indeed a leader peptide would be bound in between a β-strand and the α-helix in this region similar to an RRE-leader interaction or rather by a completely different mode of coordination.
In summary, class III and IV kinase domains phosphorylate Ser/Thr hydroxy groups through phosphate transfer from a (d)NTP co-substrate (usually ATP), share high homology to structurally characterized Ser/Thr protein kinases, and are suggested to act as the central leader peptide binding site of these lanthipeptide synthetases.
Role of the lyase domain
The N-terminal lyase domains in class III/IV lanthipeptide synthetases catalyze the β-eliminations of the Ser/Thr side chains after hydroxyl phosphorylation. These reactions yield the Dha/Dhb moieties needed as electrophiles for the subsequent macrocyclizations.The lyase domains share homology to type III effector proteins of the OspF family, which are phosphothreonine (pThr) lyases that are produced by pathogens.64–67 They act as virulence factors by catalyzing the conversion of pThr to Dhb in mitogen-activated protein kinases (MAPK) through phosphate elimination. This activity in turn diminishes the immune response of the host because it is inhibiting signaling pathways that would otherwise activate proinflammatory genes.
The homology of the VenL lyase domain with members of the OspF protein family was utilized to identify residues important for catalyzing the pSer/pThr β-elimination in class IV lanthipeptide synthetases.12 Indeed, an alignment reveals that the lyase domains of both class III and IV enzymes share many of the conserved catalytic residues with the pThr lyases OspF and SpvC (Fig. 6A). However, mutational analysis studies demonstrated that not all of them were also important for the catalytic activity of VenL and that residues conserved only amongst class III and IV lyase domains are indeed required for catalysis (Fig. 6A).12
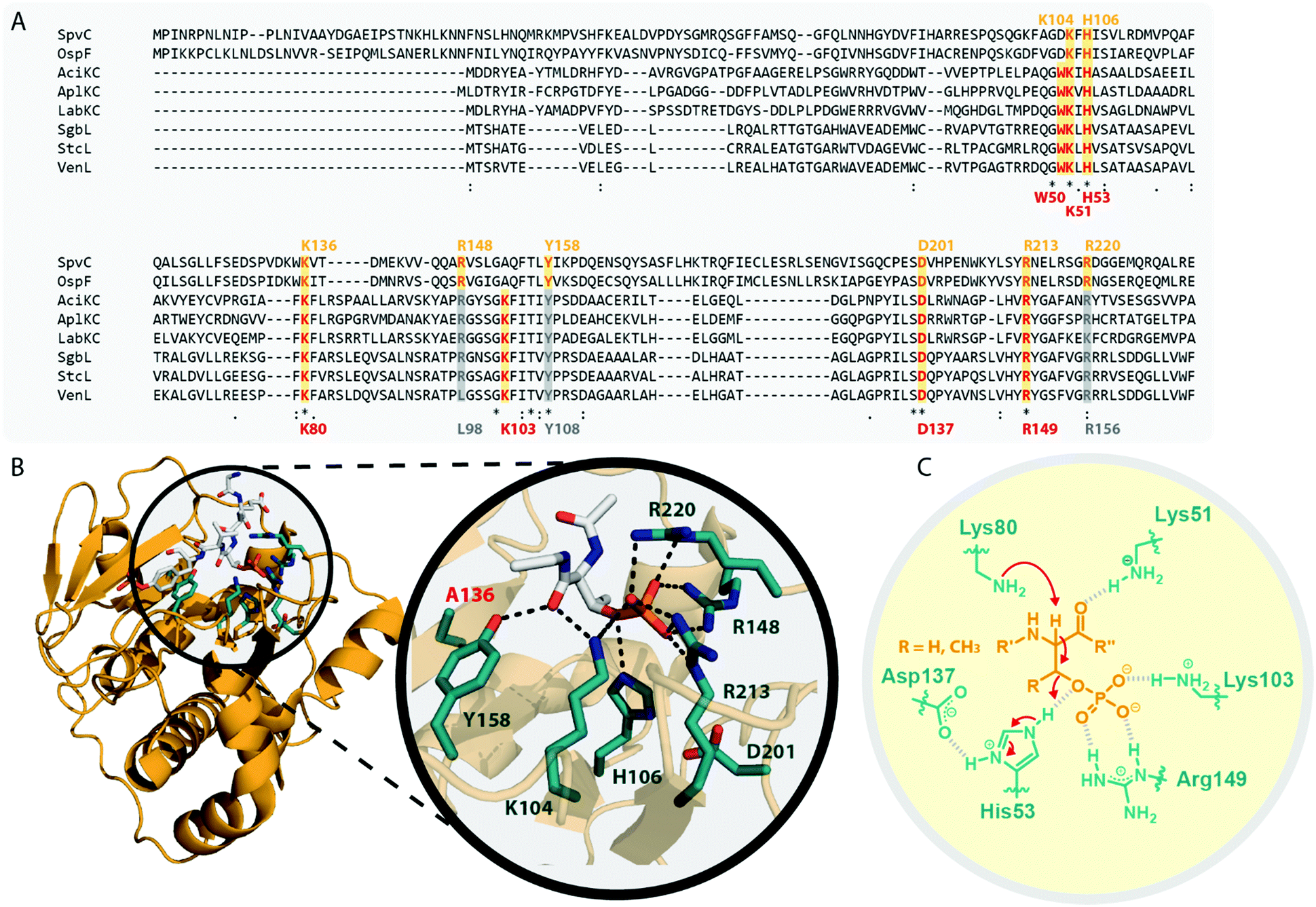 | ||
| Fig. 6 (A) Excerpt of an alignment of OspF from Shigella dysenteriae and SpvC from Salmonella paratyphi with the lyase domains of exemplary class III (AciKC, AplKC, LabKC) and class IV (SgbL, StcL, VenL) lanthipeptide synthetases. Catalytic residues of the OspF protein family pThr lyases are highlighted in orange, catalytic residues of class III/IV lyase domains in red. Above the alignment, numbering of the catalytic residues of SpvC are shown. Below the alignment, numbering of the catalytic residues of VenL are shown. (B) Crystal structure of a K136A variant of SpvC (PDB code 2Q8Y) with bound substrate zoomed in on the active site. For clarity, only a fragment of the pThr-containing peptide substrate is shown. The K136 residue of WT SpvC acts as catalytic base during the β-elimination reaction. (C) Schematic showing the catalyzed phosphate elimination reaction with the hypothesized12 functions for the catalytically important residues in VenL. | ||
Concretely, two Lys (Lys51 and Lys80), one Asp (Asp137), and one His (His53) residue that was conserved in both OspF family lyases and VenL were shown to be required for activity, whereas the substitution of a conserved Tyr (Tyr108) did not affect the ability of VenL to perform elimination reactions.12 In addition, only two of the three Arg residues important for the activity of SpvC64,65,67 were conserved in VenL. Of these two (Arg149 and Arg156), only one (Arg149) proved to be required for catalysis. Substitutions of the Trp50 and Lys103 residues, which were only conserved in class III/IV synthetases, did also reduce VenL activity.12 However, how strongly the substitution of these residues affected VenL varied. Whereas the D137N substitution reduced catalytic turnover but still allowed for the production of some amount of fully dehydrated product, only limited dehydrations were observed for the K51M and R149M variants, and no lyase activity was detected for variants carrying a W50D, H53F, K80M, or K103A exchange.12
The functions of these residues have been deduced based on their roles in the structurally characterized SpvC lyase (Fig. 6B).64,65 In this model for VenL12 (Fig. 6C), Lys80 would act as the catalytic base to deprotonate the α-proton of the pSer/pThr residues to initiate the elimination reaction. The acidity of this proton would be enhanced by interaction of the primary amine of the Lys51 side chain with the pSer/pThr backbone carbonyl.12 In SpvC, the conserved Tyr forms an additional hydrogen bond with the same backbone carbonyl and thus assists in deprotonation.64,65 As exchange of the corresponding Tyr108 in VenL did not reduce catalytic activity,12 this interaction is apparently not formed between class IV lyase domains and their substrates or, if present after all, at least does not contribute significantly to the overall catalytic efficacy.
His53 would act as catalytic acid for protonation of the bridging oxygen of the phosphate leaving group and an interaction with Asp137 would stabilize the protonated form of the His53 side chain for this function.12,67 In SpvC, a series of three Arg residues are furthermore forming contacts to the non-bridging phosphate oxygens and thereby are stabilizing the negatively charged phosphate group in the active site of the enzyme.64,65,67 Of these three Arg, two are conserved in VenL, but only the substitution of Arg149 decreased catalytic activity. Hence, it was hypothesized that instead of using three Arg for phosphate stabilization like in SpvC, VenL would utilize Lys103 and Arg149 instead.12 However, the role in catalysis of the Trp50 residue, which is like Lys103 unique to class III/IV lyase domains, remains unclear.
In contrast to the kinase and cyclase domains, protein structure databases do not contain structures of proteins with high enough similarity to the lyase domains of class III/IV lanthipeptide synthetases to allow for the prediction of a proper structure model, highlighting their somewhat unique status amongst so far characterized proteins.
In summary, the lyase domains in class III and IV enzymes catalyze the β-elimination of pSer/pThr residues that were generated through NTP-dependent phosphorylation carried out by the respective kinase domains.9–12,14 Experiments revealed the importance of several residues for the elimination reaction; which are in part shared with lyases of the OspF protein family, in part exclusive to class III/IV lanthipeptide synthetases.12 As for the kinase domain, experiments demonstrating the importance of these conserved lyase residues have only been performed with a class IV, but not yet with a class III enzyme. Because of the high similarity between class III and IV lyase domains, it is however likely that the roles of these conserved residues transcend class designation.
Role of the cyclase domain
The final processing step that class III/IV lanthipeptide synthetases accomplish is the thioether ring formation through 1,4-Michael-type additions of Cys thiol groups to free Dha or Dhb moieties. In general, the cyclases catalyze the formation of (methyl)lanthionines by lowering the pKa of Cys thiols, which increases their nucleophilicity and thus facilitates the attack on the dehydrated residues. The resulting enolate intermediate is then protonated to yield the (methyl)lanthionine moiety.1,2,5,6,68,69 In contrast, labionin formation requires that instead of being protonated, the enolate intermediate is positioned in a manner that enables a nucleophilic attack on another Dha residue.11,13 The labionins that are introduced into the labyrinthopeptins by LabKC were shown to have an LDL-stereochemistry.13 The (methyl)lanthionines introduced by VenL and SflL in their respective precursor peptides all had a DL-stereochemistry.15,37Class III and class IV cyclase domains share sequence homology to each other but differ in two key elements. Class III cyclase domains lack the conserved residues of other lanthipeptide cyclases and are so far the only enzymes observed to install labionin moieties.2,11,13,50 Interestingly, there are also some class III lanthipeptides that despite the presence of the typical Sx2Sx2–5C motif contain lanthionines and not labionins (Fig. 1C). For example, the CurA precursor peptides has two potential Sx2Sx3C labionin motifs, yet CurKC has been reported to mainly generate lanthionines and at most trace amounts of labionin rings have been detected from in vitro modification assays.30,34 EryKC and StaKC, the enzymes involved in the maturation of the lanthipeptides erythreapeptin and stackepeptin, respectively, were even shown to incorporate mixtures of labionins and lanthionines in their precursor peptides.31,32 It is however still unknown what criteria decide if a lanthionine or a labionin is formed, especially when the corresponding processing enzymes were shown to be able to generate both lanthionine and labionin crosslinks.
In the model class I cyclase NisC, the conserved residues Asp141 and His212 along with the zinc-binding residues Cys284, Cys330 and His331 are essential for activity.2,5,68–70 Sequence alignments (Fig. 7A) show that these residues are also conserved in other class I, II and IV cyclases. Conversely, the catalytic His and the zinc-binding residues are absent in class III cyclase domains and only the Asp residue is somewhat conserved (it is missing e.g. in LabKC). Due to the homology shared between all lanthipeptide cyclases, it is possible to generate high confidence models for the exemplary cyclase domains of LabKC and SgbL based on the NisC5 and CylM6 structures using the Phyre2 webtool54 (Fig. 7B). The resulting structure models thus also share the typical α-toroidal structure of the stand-alone NisC enzyme and the CylM cyclase domain. For the SgbL cyclase domain, Asp642, His710, Cys769, Cys814, and His815 are at the positions corresponding to the sites of the catalytic and zinc-binding residues in NisC and CylM (Fig. 7A and B). For lanthipeptide cyclases with the Cys–Cys–His zinc-binding motif, it is suggested that the coordinated Zn2+ ion would act as Lewis acid and thereby accomplish the activation of the thiol nucleophile (Fig. 7C).2,5,6,69,70 Since this binding motif is missing in class III cyclase domains, it is unclear how thiol activation is achieved in these enzymes.
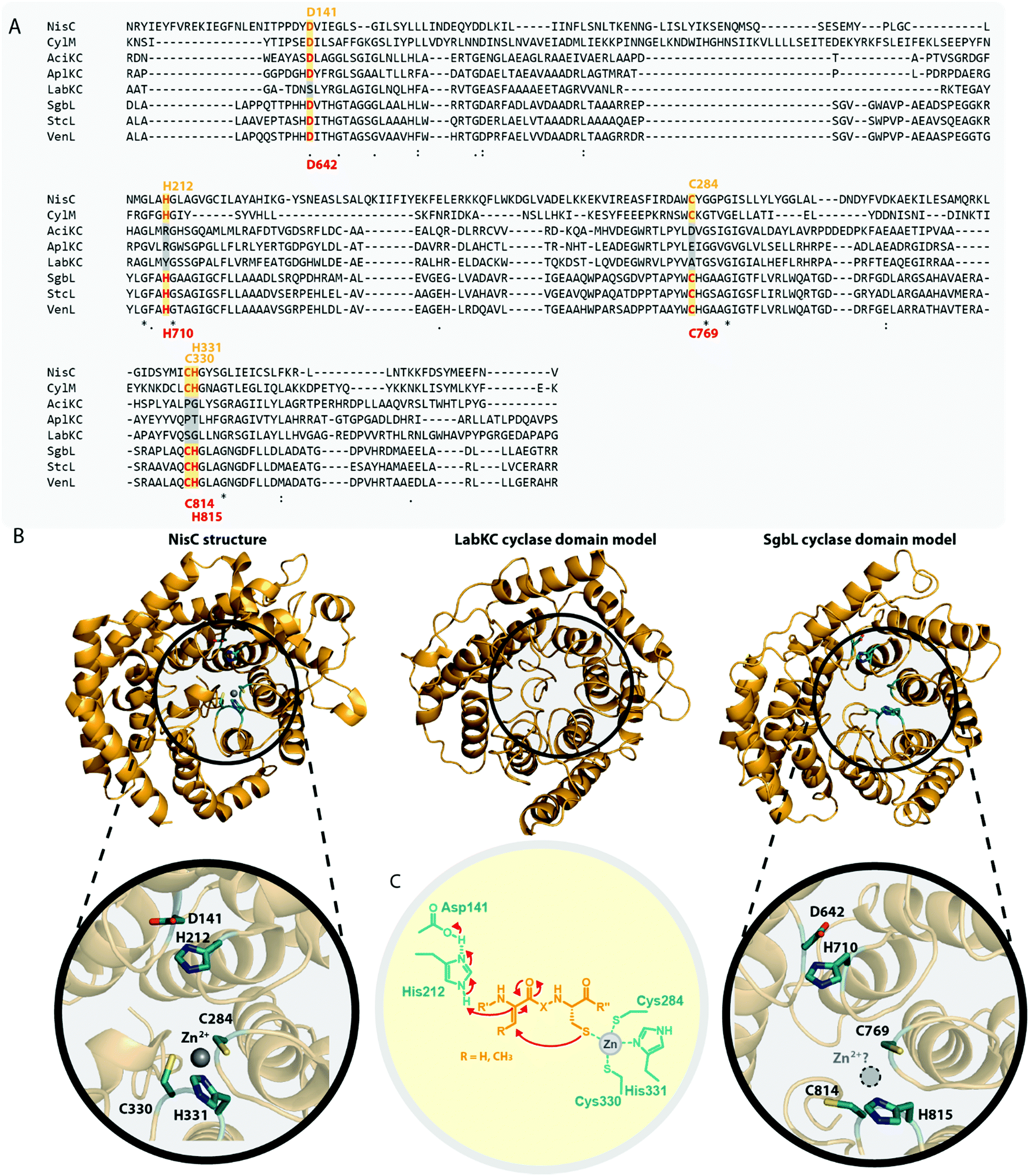 | ||
| Fig. 7 (A) Excerpt of an alignment of the NisC cyclase (involved in the biosynthesis of the class I lanthipeptide nisin) and the CylM cyclase domain (involved in the biosynthesis of the class II cytolysin lanthipeptides) with the cyclase domains of exemplary class III (AciKC, AplKC, LabKC) and class IV (SgbL, StcL, VenL) lanthipeptide synthetases. Catalytic residues of class I and II cyclases are highlighted in orange, catalytic residues of the class IV cyclase domains in red. Above the alignment, numbering of the catalytic residues of NisC are shown. Below the alignment, numbering of the predicted catalytic residues of SgbL are shown. (B) Comparison of the crystal structure of NisC (PDB code 2G0D) with the Phyre2 models of the LabKC and SgbL cyclase domains. Close ups of the active site of NisC and the predicted active site of the SgbL cyclase domain are shown. Catalytic residues are highlighted in teal. (C) Schematic showing the catalyzed cyclization reaction with the hypothesized2,5,68,69 functions for the catalytically important residues in NisC. | ||
The NisC crystal structure (Fig. 7B) furthermore shows that Asp141 forms a hydrogen bond with His212 and it was reported that substitution of either residue with Ala abolished catalytic activity.2,5,68–70 It is suggested that the conserved His residue acts as the catalytic acid for enolate protonation in class I and II enzymes and in turn that the hydrogen bonding with Asp141 facilitates this activity (Fig. 7C). Due to the conservation of these residues, a likewise mechanism of (methyl)lanthionine formation is anticipated for class IV enzymes. Conversely, the complete absence of the catalytic His residue and only partial conservation of the Asp residue in class III cyclase domains requires different, yet unidentified means of catalysis.
Interestingly, the lack of the conserved catalytic His residue might explain why labionin formation was so far only observed in class III lanthipeptides, as the absence of the catalytic acid protonating the intermediate would facilitate a second nucleophilic attack by this enolate moiety. Another intriguing observation in this regard is that in all so far characterized labionin-containing lanthipeptides, there are always two residues in between the Dha electrophile and the enolate nucleophile (Fig. 1C and D). Therefore, a fixed distance between these two reaction partners appears to be mandatory for labionin formation.33 However, it remains unclear how the eventual protonation of the labionin-yielding enolate is accomplished and why some class III enzymes are specific for labionins, while others preferentially form lanthionines.
Class III and IV cyclase domains have not been very well studied in vitro, as there are problems with solubility and stability when removing the domains from their respective synthetases.10,11,14 It was however confirmed through ICP-MS analysis that the class III enzyme AciKC did not only lack the conserved zinc-binding residues but indeed did not bind significant amounts of zinc or other metal ions.11 Similar experiments to prove zinc-binding for class IV enzymes have not yet been reported.
In vitro studies on AciKC furthermore confirmed that the cyclization reaction is not ATP-dependent as it was shown that AciKC was able to correctly install both labionin moieties in the absence of ATP into a previously dehydrated AciA precursor peptide.11 Moreover, no ring formation was observed in a similar experiment with AciKC using only the dehydrated core peptide of AciA.11 These experiments thereby demonstrated that the cyclization through class III lanthipeptide synthetases is leader-dependent and ATP-independent. Again, likewise experiments have not been done with class IV enzymes so far.
In summary, high homology is observed when comparing the cyclases of all lanthipeptide families on the level of predicted secondary structure elements. For class IV cyclase domains, this homology extends to the conservation of the catalytic Asp, His and the zinc-binding residues. However, experimental confirmation of the role of these residues are still missing for class IV enzymes. The fact that these residues are not conserved in class III cyclase domains points to a different cyclization mechanism, which remains elusive, yet might potentially explain how some class III enzymes are able to also generate labionins.
Leader peptide recognition
Experiments for both class III and IV systems have demonstrated the general importance of the leader peptide for enzymatic processing of the precursor peptide.10,11,32,71 The interactions of a leader peptide with its processing enzyme were further investigated for the class III LabA2/LabKC71 and the class IV SgbA/SgbL10 systems. Due to the lack of structural information, the interactions between leader peptides and processing enzymes are not understood on a molecular level, but underlying principles can be derived from the experimental data.For the class III system, in vitro modification assays using LabKC and LabA2 variants were performed.71 Precursor variants featuring truncations in the N- or C-terminal sections of the leader region quickly revealed that an ILELQ sequence close the N-terminus is essential for LabKC processing. However, C-terminal truncations also demonstrated that although this part of the leader is not involved in substrate recognition by the enzyme, it is acting as a spacer between the recognition site and the core peptide residues that get modified. When this spacer was shortened too much, the core peptide was not modified anymore, while spacers of intermediate length only allowed processing of the most C-terminal residues of the core peptide. For these variants, it was hypothesized that the core peptide cannot reach the active sites any longer and thus could not be processed properly.
Point mutations of residues in and surrounding the ILELQ motif confirmed the overall importance of the hydrophobic amino acids in this sequence.71 Ala exchanges of the five amino acids following the ILELQ motif did not affect processing through LabKC and neither did the Ala substitutions of the Glu or Gln residues. In contrast, an exchange of either of the Ile or Leu residues with Ala significantly reduced processing efficiency, while exchanges of either one of these hydrophobic amino acids to a charged residue abrogated enzymatic activity altogether. Hence, it was suggested that the hydrophobic interactions between the leader recognition site and the LabKC enzyme are the main driving force for substrate recognition.
Sequences with high similarity to ILELQ are found throughout the known class III lanthipeptide precursors (Fig. 8A), which implies that substrate recognition is accomplished by similar means in these systems.
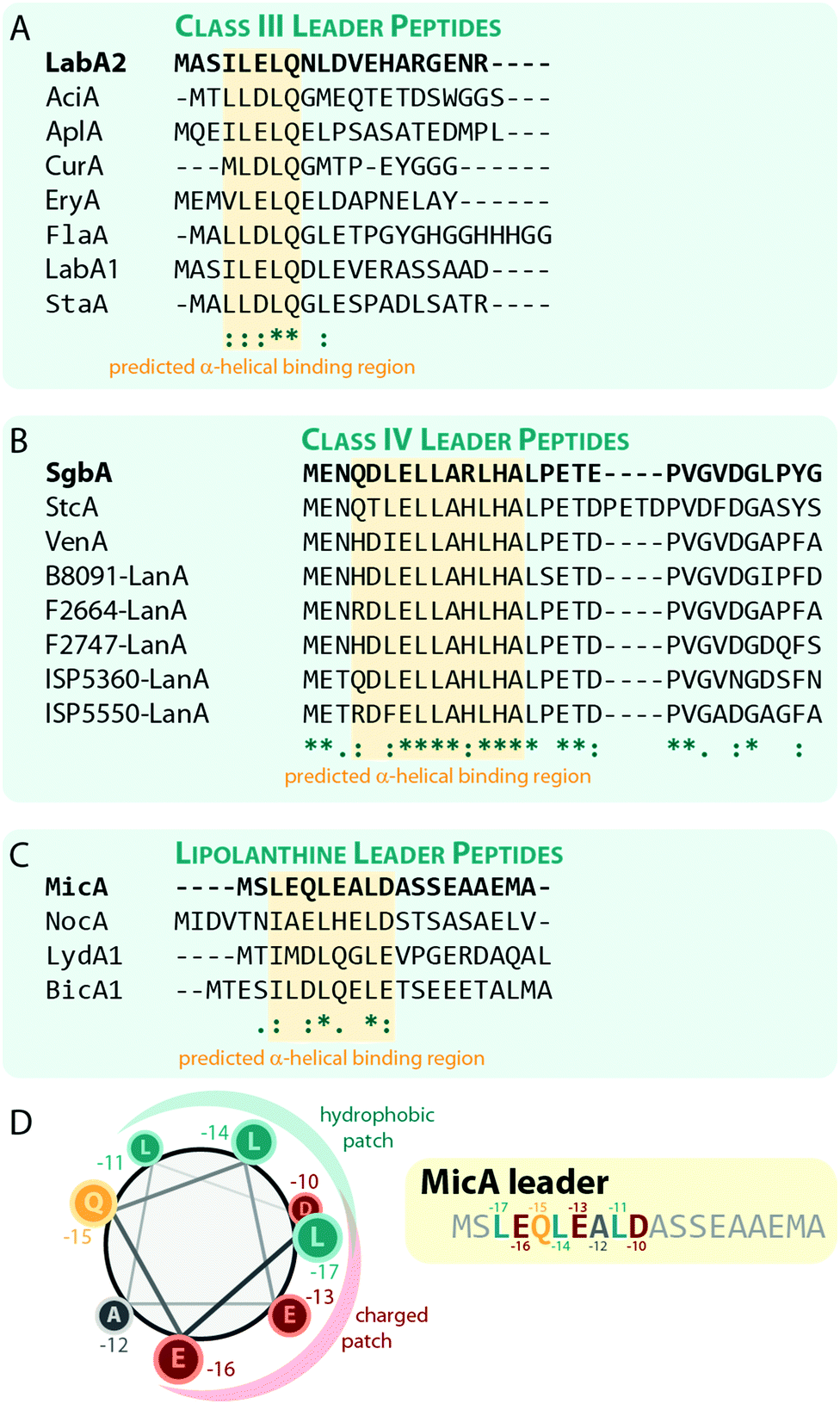 | ||
| Fig. 8 Alignments of a selection of (A) class III, (B) class IV, and (C) lipolanthine leader peptides highlighting the predicted α-helical regions that are suggested to be involved in substrate recognition by the respective processing enzymes. The peptides LabA2, SgbA, and MicA, which were used for the experimental validation of the importance of these regions, are shown on the top of each alignment and are highlighted in bold. (D) Helical-wheel representation of the MicA leader peptide highlighting the hydrophobic and charged patches on the helix surface. | ||
Interestingly, secondary structure predictions suggested that the binding motif adopts an α-helical fold. Yet, circular dichroism experiments with the LabA2 leader peptide in water did only confirm the presence of an α-helix upon addition of the helix-inducer trifluoroethanol.71
For class IV lanthipeptides, the interactions between the SgbA precursor peptide and the SgbL processing enzyme were investigated by in vivo modification studies and in vitro binding assays.10 Again, N-terminal truncations of the leader peptide beyond a certain point abolished the ability of SgbL to process the core peptide, while neither the deletion of the last five residues nor the addition of five Ala residues to the C-terminus of the leader affected processing. Furthermore, an experiment was conducted where portions of the leader spanning four to five adjacent residues were replaced with Ala residues. Surprisingly, all these Ala exchange variants were fully processed during co-expression with SgbL in E. coli, which implies the presence of an extended binding motif that is somewhat tolerant to substitutions.
In vitro binding studies between the SgbL enzyme and variants of the SgbA leader peptide helped to pinpoint the binding site to an N-terminal to central portion of the leader which is highly conserved in class IV precursor peptides (Fig. 8B).10 Intriguingly, this region was also predicted to adopt an α-helical fold, but this has not yet been confirmed experimentally.
The lipolanthines belong to the class III lanthipeptides but are a structurally distinct subclade due to their unique additional modifications that are introduced by specific tailoring enzymes. Recently, their leader peptides (Fig. 8C) were representatively investigated more closely for class III lanthipeptides.36 Indeed, analysis of the MicA leader by circular dichroism and NMR experiments demonstrated the presence of an α-helix ranging from Leu-17 to Asp-10 (Fig. 8C), which is most stable at lower temperatures and, unlike the helix formation in the LabA2 leader, is observed in absence of a helix-inducer like trifluoroethanol.36 However, the data also suggest that the MicA leader peptide is rather flexible and alternates between an unfolded and the α-helical structure, which means that at any given time, only a subpopulation of MicA inhabits the folded state. This flexible nature of the leader might in turn facilitate the eventual removal of the leader peptide by mixed endo- and aminopeptidase activity as described before and it is furthermore suggested that the folded state might be stabilized upon a binding interaction with MicKC. This hypothesis however still needs experimental validation.
The NMR data also shows that in the LEQLEALD helix region, the Leu residues form a distinct hydrophobic, whereas the Glu/Asp residues form a distinct negatively charged patch on the helix surface, thus rendering an amphipathic character (Fig. 8D).36 While no direct binding studies were performed, in vitro modification assays demonstrated that the substitution of either Leu with Ala in MicA fully prevented enzymatic turnover of the substrate by MicKC, whereas Ala exchange of any other residue in this region did not.36 These experiments therefore imply that hydrophobic residues in an α-helical region of the leader are essential for substrate recognition by the processing enzymes. As similar patterns of hydrophobic residues interspaced by two other amino acids are present in the predicted α-helical regions important for substrate recognition in other class III and IV leader peptides, a similar mechanism for leader-enzyme interaction seems likely but this hypothesis still needs to be validated experimentally.
In summary, processing through LanKC and LanL enzymes is leader-dependent and the regions of the leader peptides that are essential for substrate recognition by the processing enzymes have been identified. In both classes, these regions are predicted to adopt α-helices. However, this prediction needs further experimental validation and it has to be considered that the secondary structures of the precursors might change upon binding to a processing enzyme. A better understanding of the interactions between precursor peptides and processing enzymes demands the solution of co-crystal structures and/or NMR-based binding experiments in the future.
Additional tailoring enzymes and emerging subclasses
Additional enzymatic modifications are found in a few class III lanthipeptides. NAI-112 contains a 6-deoxyhexose moiety, which is N-linked to a central Trp side chain in the core peptide (Fig. 1C).23 This modification is introduced by a glycosyltransferase that is encoded in the NAI-112 biosynthetic gene cluster.23,41 Heterologous co-expression studies demonstrated that glycosylation through this transferase is possible in E. coli, which implies either the presence of the right activated sugar in both the native host and in E. coli or suggests that the enzyme is promiscuous enough to also use other activated (isobaric) sugar substrates.41 Furthermore, co-expression of only the precursor peptide with the glycosyltransferase did not yield glycosylated but merely unmodified peptide, thus revealing that labionin formation must precede the glycosylation reaction.41The probably most exciting novel biosynthetic features are contributed by the lipolanthines (Fig. 1C). These are class III lanthipeptides that have two additional modifications: a guanidino fatty acid attached to the N-terminus of the core peptide and the labionin ring at the C-terminus that is decarboxylated to render an avionin25,36 (Fig. 1A). Interestingly, the FAD-dependent cysteine decarboxylase MicD and the class III lanthipeptide synthetases MicKC were shown to be interdependent in vitro.25,36 On its own, MicKC only introduced one single Dha moiety into the core peptide. Conversely, co-incubation of the precursor with both MicKC and MicD enabled full conversion of the Sx2Sx2C motif into the avionin.
Since class III enzymes are leader-dependent,36,71 the sequence of modifications must start with the avionin formation, followed by the removal of the leader peptide to allow the subsequent coupling of the N-terminal α-amine with the guanidino fatty acid. However, this coupling step of the biosynthesis has not been investigated so far. For the known representatives of the lipolanthines, it is suggested that this modification is introduced by other enzymes encoded in the respective biosynthetic gene clusters.25,36
Interestingly, genome mining reveals that there seem to be at least four distinct lipolanthine subclades that are differentiated by the proposed enzymatic machineries installing the fatty acid moieties. The putative machinery synthesizing and incorporating the fatty acid in subtype I features a single standing PKS protein and a type I PKS synthase with an A-PCP loading domain. For subtype II, the genetic surroundings of the biosynthetic gene cluster imply involvement of only a single standing PKS protein. Subtype IV is similar to subtype I but its type I PKS enzyme features an N-terminal ACP instead of the A-PCP loading domain and an additional fatty-acyl AMP ligase is encoded that is suggested to carry out the ACP-loading step. Finally, subtype III features a PKS machinery similar to the one of subtype II, but does not encode a decarboxylase. Hence, lipolanthines of subtype III would feature the defining N-terminal guanidino fatty acid moiety, but would contain a simple labionin instead of the avionin crosslink.25,36
Taken together, lipolanthines represent the first reported case of a RiPP/PKS hybrid natural product. While this is not surprising as hybrids amongst other, longer known natural product superfamilies, e.g. mixed PKS/NRPS systems,72 have been known for a while now, the experimental validation of a mixed RiPP/PKS natural product enables the more targeted discovery of novel RiPP hybrid systems in the future.
Conclusion and outlook
A decade has passed since class III and IV lanthipeptides have been identified as separate entities amongst this RiPP subfamily. Since then, a vast body of work has helped to comprehend underlying principles of the biosynthesis in these classes (Fig. 9) and key features of the corresponding processing enzymes have been elucidated. Some representatives of these emerging lanthipeptide subclades exhibit truly remarkable bioactivities, but the associated mode-of-actions have not been well understood.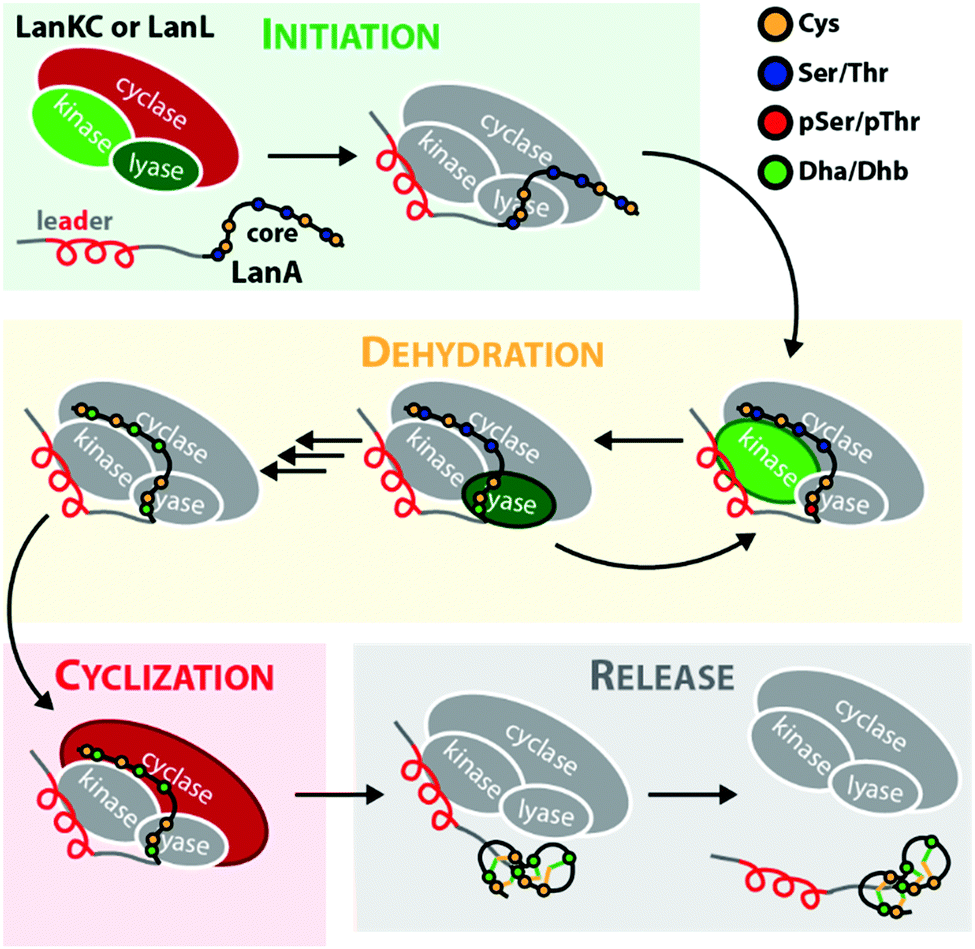 | ||
| Fig. 9 Simplified mechanism of the precursor processing through LanKC and LanL enzymes. Domains active in the respective steps are highlighted. Note that the directionality of the dehydration reactions was chosen arbitrarily to demonstrate that a specific Ser/Thr residue often undergoes first phosphorylation and then β-elimination, before the next residue is modified in the peptide.9,10,29,30 As discussed earlier, cyclization can sometimes be initiated before the complete dehydration of the core peptide is accomplished. In some systems, the formation of a thioether crosslink is even a prerequisite for further dehydrations to occur,30,32 while in other instances, complete dehydration in absence of any macrocycles is possible.9–12,14,15,40 | ||
In this review, we gave a concise overview of the corresponding studies and summarized their findings. Yet, we are still far from a complete understanding of the biosynthetic processes and the roles in nature of many of the characterized class III and IV representatives remain enigmatic. Future research will therefore have to address two major goals. First, obtaining structural information of the enzymes will be paramount for a molecular level understanding of the biosynthetic mechanisms and to get a deeper insight into the interactions between precursor peptides and processing enzymes. Second, additional focus must be directed towards studying the native functions of class III and IV lanthipeptides and it has to be established how far these compounds are distributed in nature. As the majority of so far studied class III and IV lanthipeptides did not show antimicrobial activity, this, however, is a challenging objective. It is furthermore of importance to investigate the mode-of-actions of compounds with known bioactivities in more detail.
Likewise, it is important to further assess the possibilities for in vivo engineering of the lanthipeptide structures. An initial attempt was made with labyrinthopeptin A1 by establishing a heterologous production system in Actinomadura namibiensis and performing a mutational analysis of the labA1 precursor gene.33 These experiments demonstrated that labionin formation is still possible, if a residue is deleted or inserted into the region between the Cys and the first Dha residue. However, the length of the highly conserved two amino acid linker region between the two Dha residues could not be altered.33
Recently, it was demonstrated that class I and II lanthipeptides can be used for generation of compound libraries through precursor diversification.7,73–75 Such libraries can furthermore be screened by using yeast- and phage-display techniques recently adopted for lanthipeptides.74,75 It would be worthwhile to extend these approaches to class III and IV lanthipeptides to allow access to a wider diversity of scaffolds and ring topologies. Class III systems in particular could be of great promise as they would also allow the use of compact labionin macrocycles that cannot be generated by the other three classes.
Further efforts should be directed towards a more complete study of the commonalities and differences of class III and IV enzymes. The high homology between their lyase and kinase domains suggests that they follow the same biosynthetic logic, yet experimental proof to substantiate this generalization is still missing. Of the three catalytic domains of class III and IV enzymes, the cyclase domains remain the least studied, although they are the key feature that allows the differentiation of these lanthipeptide classes. Here, experiments are required to confirm that the similarities of the cyclase domains of classes I, II, and IV hold true beyond the bioinformatic level. Furthermore, the enigmatic cyclase domains of class III enzymes need to be studied in-depth. Particular relevant questions that need to be addressed in this context are how the formation of labionins is accomplished compared to (methyl)lanthionines and which catalytic residues compensate for the lack of zinc in the active site and by what mechanism.
In conclusion, significant progress was made over the last decade towards the elucidation of underlying biosynthetic principles of class III and IV lanthipeptides, but the remaining questions about their biosynthesis and their roles in nature hold a manifold of new challenges for the next one.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. D. S. acknowledges support from the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – EXC 2008 – 390540038 – UniSysCat and the research training group “Bioactive Peptides” RTG 2473, No. 392923329. We would also like to thank Arnar Sigurdsson for constructive discussions during the writing of this review article.Notes and references
- P. G. Arnison, M. J. Bibb, G. Bierbaum, A. A. Bowers, T. S. Bugni, G. Bulaj, J. A. Camarero, D. J. Campopiano, G. L. Challis, J. Clardy, P. D. Cotter, D. J. Craik, M. Dawson, E. Dittmann, S. Donadio, P. C. Dorrestein, K. D. Entian, M. A. Fischbach, J. S. Garavelli, U. Goransson, C. W. Gruber, D. H. Haft, T. K. Hemscheidt, C. Hertweck, C. Hill, A. R. Horswill, M. Jaspars, W. L. Kelly, J. P. Klinman, O. P. Kuipers, A. J. Link, W. Liu, M. A. Marahiel, D. A. Mitchell, G. N. Moll, B. S. Moore, R. Muller, S. K. Nair, I. F. Nes, G. E. Norris, B. M. Olivera, H. Onaka, M. L. Patchett, J. Piel, M. J. Reaney, S. Rebuffat, R. P. Ross, H. G. Sahl, E. W. Schmidt, M. E. Selsted, K. Severinov, B. Shen, K. Sivonen, L. Smith, T. Stein, R. D. Süssmuth, J. R. Tagg, G. L. Tang, A. W. Truman, J. C. Vederas, C. T. Walsh, J. D. Walton, S. C. Wenzel, J. M. Willey and W. A. van der Donk, Ribosomally synthesized and post-translationally modified peptide natural products: overview and recommendations for a universal nomenclature, Nat. Prod. Rep., 2012, 30(1), 108 RSC.
- L. M. Repka, J. R. Chekan, S. K. Nair and W. A. van der Donk, Mechanistic Understanding of Lanthipeptide Biosynthetic Enzymes, Chem. Rev., 2017, 117(8), 5457 CrossRef CAS PubMed.
- M. A. Ortega, Y. Hao, Q. Zhang, M. C. Walker, W. A. van der Donk and S. K. Nair, Structure and mechanism of the tRNA-dependent lantibiotic dehydratase NisB, Nature, 2015, 517(7535), 509 CrossRef CAS PubMed.
- W. A. van der Donk and S. K. Nair, Structure and mechanism of lanthipeptide biosynthetic enzymes, Curr. Opin. Struct. Biol., 2014, 29, 58 CrossRef CAS PubMed.
- B. Li, J. P. Yu, J. S. Brunzelle, G. N. Moll, W. A. van der Donk and S. K. Nair, Structure and mechanism of the lantibiotic cyclase involved in nisin biosynthesis, Science, 2006, 311(5766), 1464 CrossRef CAS PubMed.
- S. H. Dong, W. Tang, T. Lukk, Y. Yu, S. K. Nair and W. A. van der Donk, The enterrococcal cytolysin synthetase has an unanticipated lipid kinase fold, eLife, 2015, 4, e07607 CrossRef PubMed.
- J. D. Hegemann, S. C. Bobeica, M. C. Walker, I. R. Bothwell and W. A. van der Donk, Assessing the Flexibility of the Prochlorosin 2.8 Scaffold for Bioengineering Applications, ACS Synth. Biol., 2019, 8(5), 1204 CrossRef CAS PubMed.
- C. P. Ting, M. A. Funk, S. L. Halaby, Z. Zhang, T. Gonen and W. A. van der Donk, Use of a scaffold peptide in the biosynthesis of amino acid-derived natural products, Science, 2019, 365(6450), 280 CrossRef CAS PubMed.
- J. D. Hegemann, L. Shi, M. L. Gross and W. A. van der Donk, Mechanistic Studies of the Kinase Domains of Class IV Lanthipeptide Synthetases, ACS Chem. Biol., 2019, 14(7), 1583 CrossRef CAS PubMed.
- J. D. Hegemann and W. A. van der Donk, Investigation of Substrate Recognition and Biosynthesis in Class IV Lanthipeptide Systems, J. Am. Chem. Soc., 2018, 140(17), 5743 CrossRef CAS PubMed.
- H. Wang and W. A. van der Donk, Biosynthesis of the class III lantipeptide catenulipeptin, ACS Chem. Biol., 2012, 7(9), 1529 CrossRef CAS PubMed.
- Y. Goto, A. Okesli and W. A. van der Donk, Mechanistic studies of Ser/Thr dehydration catalyzed by a member of the LanL lanthionine synthetase family, Biochemistry, 2011, 50(5), 891 CrossRef CAS PubMed.
- K. Meindl, T. Schmiederer, K. Schneider, A. Reicke, D. Butz, S. Keller, H. Guhring, L. Vertesy, J. Wink, H. Hoffmann, M. Bronstrup, G. M. Sheldrick and R. D. Süssmuth, Labyrinthopeptins: a new class of carbacyclic lantibiotics, Angew. Chem., Int. Ed., 2010, 49(6), 1151 CrossRef CAS PubMed.
- Y. Goto, B. Li, J. Claesen, Y. Shi, M. J. Bibb and W. A. van der Donk, Discovery of unique lanthionine synthetases reveals new mechanistic and evolutionary insights, PLoS Biol., 2010, 8(3), e1000339 CrossRef PubMed.
- H. Ren, C. Shi, I. R. Bothwell, W. A. van der Donk and H. Zhao, Discovery and Characterization of a Class IV Lanthipeptide with a Non-overlapping Ring Pattern, ACS Chem. Biol., 2020, 15(6), 1642–1649 CrossRef CAS PubMed.
- S. Kodani, M. E. Hudson, M. C. Durrant, M. J. Buttner, J. R. Nodwell and J. M. Willey, The SapB morphogen is a lantibiotic-like peptide derived from the product of the developmental gene ramS in Streptomyces coelicolor, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(31), 11448 CrossRef CAS PubMed.
- J. Willey, R. Santamaria, J. Guijarro, M. Geistlich and R. Losick, Extracellular complementation of a developmental mutation implicates a small sporulation protein in aerial mycelium formation by S. coelicolor, Cell, 1991, 65(4), 641 CrossRef CAS PubMed.
- J. Guijarro, R. Santamaria, A. Schauer and R. Losick, Promoter determining the timing and spatial localization of transcription of a cloned Streptomyces coelicolor gene encoding a spore-associated polypeptide, J. Bacteriol., 1988, 170(4), 1895 CrossRef CAS PubMed.
- K. Ueda, K. Oinuma, G. Ikeda, K. Hosono, Y. Ohnishi, S. Horinouchi and T. Beppu, AmfS, an extracellular peptidic morphogen in Streptomyces griseus, J. Bacteriol., 2002, 184(5), 1488 CrossRef CAS PubMed.
- K. Ueda, K. Miyake, S. Horinouchi and T. Beppu, A gene cluster involved in aerial mycelium formation in Streptomyces griseus encodes proteins similar to the response regulators of two-component regulatory systems and membrane translocators, J. Bacteriol., 1993, 175(7), 2006 CrossRef CAS PubMed.
- R. D. Kersten, Y. L. Yang, Y. Xu, P. Cimermancic, S. J. Nam, W. Fenical, M. A. Fischbach, B. S. Moore and P. C. Dorrestein, A mass spectrometry-guided genome mining approach for natural product peptidogenomics, Nat. Chem. Biol., 2011, 7(11), 794 CrossRef CAS PubMed.
- T. Dang and R. D. Süssmuth, Bioactive Peptide Natural Products as Lead Structures for Medicinal Use, Acc. Chem. Res., 2017, 50(7), 1566 CrossRef CAS PubMed.
- M. Iorio, O. Sasso, S. I. Maffioli, R. Bertorelli, P. Monciardini, M. Sosio, F. Bonezzi, M. Summa, C. Brunati, R. Bordoni, G. Corti, G. Tarozzo, D. Piomelli, A. Reggiani and S. Donadio, A glycosylated, labionin-containing lanthipeptide with marked antinociceptive activity, ACS Chem. Biol., 2014, 9(2), 398 CrossRef CAS PubMed.
- T. L. Bailey and C. Elkan, Fitting a mixture model by expectation maximization to discover motifs in biopolymers, Proc. Int. Conf. Intell. Syst. Mol. Biol., 1994, 2, 28 CAS.
- V. Wiebach, A. Mainz, M. J. Siegert, N. A. Jungmann, G. Lesquame, S. Tirat, A. Dreux-Zigha, J. Aszodi, D. Le Beller and R. D. Süssmuth, The anti-staphylococcal lipolanthines are ribosomally synthesized lipopeptides, Nat. Chem. Biol., 2018, 14(7), 652 CrossRef CAS PubMed.
- H. Prochnow, K. Rox, N. V. S. Birudukota, L. Weichert, S. K. Hotop, P. Klahn, K. Mohr, S. Franz, D. H. Banda, S. Blockus, J. Schreiber, S. Haid, M. Oeyen, J. P. Martinez, R. D. Süssmuth, J. Wink, A. Meyerhans, C. Goffinet, M. Messerle, T. F. Schulz, A. Kroger, D. Schols, T. Pietschmann and M. Bronstrup, Labyrinthopeptins Exert Broad-Spectrum Antiviral Activity through Lipid-Binding-Mediated Virolysis, J. Virol., 2020, 94(2), e01471 CAS.
- S. Blockus, S. M. Sake, M. Wetzke, C. Grethe, T. Graalmann, M. Pils, R. Le Goffic, M. Galloux, H. Prochnow, K. Rox, S. Huttel, Z. Rupcic, B. Wiegmann, R. Dijkman, M. A. Rameix-Welti, J. F. Eleouet, W. P. Duprex, V. Thiel, G. Hansen, M. Bronstrup, S. Haid and T. Pietschmann, Labyrinthopeptins as virolytic inhibitors of respiratory syncytial virus cell entry, Antiviral Res., 2020, 177, 104774 CrossRef CAS PubMed.
- G. Ferir, M. I. Petrova, G. Andrei, D. Huskens, B. Hoorelbeke, R. Snoeck, J. Vanderleyden, J. Balzarini, S. Bartoschek, M. Bronstrup, R. D. Süssmuth and D. Schols, The lantibiotic peptide labyrinthopeptin A1 demonstrates broad anti-HIV and anti-HSV activity with potential for microbicidal applications, PLoS One, 2013, 8(5), e64010 CrossRef CAS PubMed.
- B. Krawczyk, P. Ensle, W. M. Muller and R. D. Süssmuth, Deuterium labeled peptides give insights into the directionality of class III lantibiotic synthetase LabKC, J. Am. Chem. Soc., 2012, 134(24), 9922 CrossRef CAS PubMed.
- N. A. Jungmann, B. Krawczyk, M. Tietzmann, P. Ensle and R. D. Süssmuth, Dissecting reactions of nonlinear precursor peptide processing of the class III lanthipeptide curvopeptin, J. Am. Chem. Soc., 2014, 136(43), 15222 CrossRef CAS PubMed.
- G. H. Voller, J. M. Krawczyk, A. Pesic, B. Krawczyk, J. Nachtigall and R. D. Süssmuth, Characterization of new class III lantibiotics–erythreapeptin, avermipeptin and griseopeptin from Saccharopolyspora erythraea, Streptomyces avermitilis and Streptomyces griseus demonstrates stepwise N-terminal leader processing, ChemBioChem, 2012, 13(8), 1174 CrossRef PubMed.
- N. A. Jungmann, E. F. van Herwerden, M. Hugelland and R. D. Süssmuth, The Supersized Class III Lanthipeptide Stackepeptin Displays Motif Multiplication in the Core Peptide, ACS Chem. Biol., 2016, 11(1), 69 CrossRef CAS PubMed.
- J. M. Krawczyk, G. H. Völler, B. Krawczyk, J. Kretz, M. Brönstrup and R. D. Süssmuth, Heterologous expression and engineering studies of labyrinthopeptins, class III lantibiotics from Actinomadura namibiensis, Chem. Biol., 2013, 20(1), 111 CrossRef CAS PubMed.
- B. Krawczyk, G. H. Voller, J. Voller, P. Ensle and R. D. Süssmuth, Curvopeptin: a new lanthionine-containing class III lantibiotic and its co-substrate promiscuous synthetase, ChemBioChem, 2012, 13(14), 2065 CrossRef CAS PubMed.
- M. S. Donia, J. Ravel and E. W. Schmidt, A global assembly line for cyanobactins, Nat. Chem. Biol., 2008, 4(6), 341 CrossRef CAS PubMed.
- V. Wiebach, A. Mainz, R. Schnegotzki, M. J. Siegert, M. Hügelland, N. Pliszka and R. Süssmuth, An amphipathic alpha-helix guides maturation of the ribosomally-synthesized lipolanthines, Angew. Chem., Int. Ed., 2020 DOI:10.1002/anie.202003804.
- Q. Zhang, J. R. Doroghazi, X. Zhao, M. C. Walker and W. A. van der Donk, Expanded natural product diversity revealed by analysis of lanthipeptide-like gene clusters in actinobacteria, Appl. Environ. Microbiol., 2015, 81(13), 4339 CrossRef CAS PubMed.
- D. Iftime, M. Jasyk, A. Kulik, J. F. Imhoff, E. Stegmann, W. Wohlleben, R. D. Süssmuth and T. Weber, Streptocollin, a Type IV Lanthipeptide Produced by Streptomyces collinus Tu 365, ChemBioChem, 2015, 16(18), 2615 CrossRef CAS PubMed.
- Q. Zhang, M. Ortega, Y. Shi, H. Wang, J. O. Melby, W. Tang, D. A. Mitchell and W. A. van der Donk, Structural investigation of ribosomally synthesized natural products by hypothetical structure enumeration and evaluation using tandem MS, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(33), 12031 CrossRef CAS PubMed.
- G. H. Voller, B. Krawczyk, P. Ensle and R. D. Süssmuth, Involvement and unusual substrate specificity of a prolyl oligopeptidase in class III lanthipeptide maturation, J. Am. Chem. Soc., 2013, 135(20), 7426 CrossRef PubMed.
- S. Chen, B. Xu, E. Chen, J. Wang, J. Lu, S. Donadio, H. Ge and H. Wang, Zn-dependent bifunctional proteases are responsible for leader peptide processing of class III lanthipeptides, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(7), 2533 CrossRef CAS PubMed.
- S. C. Bobeica, S. H. Dong, L. Huo, N. Mazo, M. I. McLaughlin, G. Jimenez-Oses, S. K. Nair and W. A. van der Donk, Insights into AMS/PCAT transporters from biochemical and structural characterization of a double Glycine motif protease, eLife, 2019, 8, e42305 CrossRef PubMed.
- M. Montalban-Lopez, J. Deng, A. J. van Heel and O. P. Kuipers, Specificity and Application of the Lantibiotic Protease NisP, Front. Microbiol., 2018, 9, 160 CrossRef PubMed.
- M. Lagedroste, S. H. J. Smits and L. Schmitt, Substrate Specificity of the Secreted Nisin Leader Peptidase NisP, Biochemistry, 2017, 56(30), 4005 CrossRef PubMed.
- Y. Xu, X. Li, R. Li, S. Li, H. Ni, H. Wang, H. Xu, W. Zhou, P. E. Saris, W. Yang, M. Qiao and Z. Rao, Structure of the nisin leader peptidase NisP revealing a C-terminal autocleavage activity, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70(Pt 6), 1499 CrossRef CAS PubMed.
- J. D. Hegemann, C. J. Schwalen, D. A. Mitchell and W. A. van der Donk, Elucidation of the roles of conserved residues in the biosynthesis of the lasso peptide paeninodin, Chem. Commun., 2018, 54(65), 9007 RSC.
- S. Zhu, C. D. Fage, J. D. Hegemann, A. Mielcarek, D. Yan, U. Linne and M. A. Marahiel, The B1 Protein Guides the Biosynthesis of a Lasso Peptide, Sci. Rep., 2016, 6, 35604 CrossRef CAS PubMed.
- B. M. Wieckowski, J. D. Hegemann, A. Mielcarek, L. Boss, O. Burghaus and M. A. Marahiel, The PqqD homologous domain of the radical SAM enzyme ThnB is required for thioether bond formation during thurincin H maturation, FEBS Lett., 2015, 589(15), 1802 CrossRef CAS PubMed.
- J. D. Hegemann, M. Zimmermann, X. Xie and M. A. Marahiel, Lasso Peptides: An Intriguing Class of Bacterial Natural Products, Acc. Chem. Res., 2015, 48(7), 1909 CrossRef CAS PubMed.
- W. M. Muller, T. Schmiederer, P. Ensle and R. D. Süssmuth, In vitro biosynthesis of the prepeptide of type-III lantibiotic labyrinthopeptin A2 including formation of a C-C bond as a post-translational modification, Angew. Chem., Int. Ed., 2010, 49(13), 2436 CrossRef PubMed.
- N. Scherr, S. Honnappa, G. Kunz, P. Mueller, R. Jayachandran, F. Winkler, J. Pieters and M. O. Steinmetz, Structural basis for the specific inhibition of protein kinase G, a virulence factor of Mycobacterium tuberculosis, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(29), 12151 CrossRef CAS PubMed.
- T. A. Young, B. Delagoutte, J. A. Endrizzi, A. M. Falick and T. Alber, Structure of Mycobacterium tuberculosis PknB supports a universal activation mechanism for Ser/Thr protein kinases, Nat. Struct. Biol., 2003, 10(3), 168 CrossRef CAS PubMed.
- D. W. Buchan, F. Minneci, T. C. Nugent, K. Bryson and D. T. Jones, Scalable web services for the PSIPRED Protein Analysis Workbench, Nucleic Acids Res., 2013, 41(Web Server issue), W349 CrossRef PubMed.
- L. A. Kelley, S. Mezulis, C. M. Yates, M. N. Wass and M. J. Sternberg, The Phyre2 web portal for protein modeling, prediction and analysis, Nat. Protoc., 2015, 10(6), 845 CrossRef CAS PubMed.
- B. J. Burkhart, G. A. Hudson, K. L. Dunbar and D. A. Mitchell, A prevalent peptide-binding domain guides ribosomal natural product biosynthesis, Nat. Chem. Biol., 2015, 11(8), 564 CrossRef CAS PubMed.
- T. Sumida, S. Dubiley, B. Wilcox, K. Severinov and S. Tagami, Structural basis of leader peptide recognition in lasso peptide biosynthesis pathway, ACS Chem. Biol., 2019, 14(7), 1619 CrossRef CAS PubMed.
- T. L. Grove, P. M. Himes, S. Hwang, H. Yumerefendi, J. B. Bonanno, B. Kuhlman, S. C. Almo and A. A. Bowers, Structural Insights into Thioether Bond Formation in the Biosynthesis of Sactipeptides, J. Am. Chem. Soc., 2017, 139(34), 11734 CrossRef CAS PubMed.
- R. L. Evans, 3rd, J. A. Latham, Y. Xia, J. P. Klinman and C. M. Wilmot, Nuclear Magnetic Resonance Structure and Binding Studies of PqqD, a Chaperone Required in the Biosynthesis of the Bacterial Dehydrogenase Cofactor Pyrroloquinoline Quinone, Biochemistry, 2017, 56(21), 2735 CrossRef PubMed.
- J. Koehnke, G. Mann, A. F. Bent, H. Ludewig, S. Shirran, C. Botting, T. Lebl, W. Houssen, M. Jaspars and J. H. Naismith, Structural analysis of leader peptide binding enables leader-free cyanobactin processing, Nat. Chem. Biol., 2015, 11(8), 558 CrossRef CAS PubMed.
- H. Martin-Gomez, U. Linne, F. Albericio, J. Tulla-Puche and J. D. Hegemann, Investigation of the Biosynthesis of the Lasso Peptide Chaxapeptin Using an E. coli-Based Production System, J. Nat. Prod., 2018, 81(9), 2050 CrossRef CAS PubMed.
- K. M. Davis, K. R. Schramma, W. A. Hansen, J. P. Bacik, S. D. Khare, M. R. Seyedsayamdost and N. Ando, Structures of the peptide-modifying radical SAM enzyme SuiB elucidate the basis of substrate recognition, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(39), 10420 CrossRef CAS PubMed.
- J. A. Latham, A. T. Iavarone, I. Barr, P. V. Juthani and J. P. Klinman, PqqD is a novel peptide chaperone that forms a ternary complex with the radical S-adenosylmethionine protein PqqE in the pyrroloquinoline quinone biosynthetic pathway, J. Biol. Chem., 2015, 290(20), 12908 CrossRef CAS PubMed.
- R. Ayikpoe, V. Govindarajan and J. A. Latham, Occurrence, function, and biosynthesis of mycofactocin, Appl. Microbiol. Biotechnol., 2019, 103(7), 2903 CrossRef CAS PubMed.
- L. Chen, H. Wang, J. Zhang, L. Gu, N. Huang, J. M. Zhou and J. Chai, Structural basis for the catalytic mechanism of phosphothreonine lyase, Nat. Struct. Mol. Biol., 2008, 15(1), 101 CrossRef CAS PubMed.
- Y. Zhu, H. Li, C. Long, L. Hu, H. Xu, L. Liu, S. Chen, D. C. Wang and F. Shao, Structural insights into the enzymatic mechanism of the pathogenic MAPK phosphothreonine lyase, Mol. Cell, 2007, 28(5), 899 CrossRef CAS PubMed.
- H. Li, H. Xu, Y. Zhou, J. Zhang, C. Long, S. Li, S. Chen, J. M. Zhou and F. Shao, The phosphothreonine lyase activity of a bacterial type III effector family, Science, 2007, 315(5814), 1000 CrossRef CAS PubMed.
- G. K. Smith, Z. Ke, A. C. Hengge, D. Xu, D. Xie and H. Guo, Active-site dynamics of SpvC virulence factor from Salmonella typhimurium and density functional theory study of phosphothreonine lyase catalysis, J. Phys. Chem. B, 2009, 113(46), 15327 CrossRef CAS PubMed.
- X. Yang and W. A. van der Donk, Michael-type cyclizations in lantibiotic biosynthesis are reversible, ACS Chem. Biol., 2015, 10(5), 1234 CrossRef CAS PubMed.
- B. Li and W. A. van der Donk, Identification of essential catalytic residues of the cyclase NisC involved in the biosynthesis of nisin, J. Biol. Chem., 2007, 282(29), 21169 CrossRef CAS PubMed.
- M. Helfrich, K. D. Entian and T. Stein, Structure-function relationships of the lanthionine cyclase SpaC involved in biosynthesis of the Bacillus subtilis peptide antibiotic subtilin, Biochemistry, 2007, 46(11), 3224 CrossRef CAS PubMed.
- W. M. Muller, P. Ensle, B. Krawczyk and R. D. Süssmuth, Leader peptide-directed processing of labyrinthopeptin A2 precursor peptide by the modifying enzyme LabKC, Biochemistry, 2011, 50(39), 8362 CrossRef PubMed.
- J. Masschelein, M. Jenner and G. L. Challis, Antibiotics from Gram-negative bacteria: a comprehensive overview and selected biosynthetic highlights, Nat. Prod. Rep., 2017, 34(7), 712 RSC.
- X. Yang, K. R. Lennard, C. He, M. C. Walker, A. T. Ball, C. Doigneaux, A. Tavassoli and W. A. van der Donk, A lanthipeptide library used to identify a protein-protein interaction inhibitor, Nat. Chem. Biol., 2018, 14(4), 375 CrossRef CAS PubMed.
- K. J. Hetrick, M. C. Walker and W. A. van der Donk, Development and Application of Yeast and Phage Display of Diverse Lanthipeptides, ACS Cent. Sci., 2018, 4(4), 458 CrossRef CAS PubMed.
- J. H. Urban, M. A. Moosmeier, T. Aumuller, M. Thein, T. Bosma, R. Rink, K. Groth, M. Zulley, K. Siegers, K. Tissot, G. N. Moll and J. Prassler, Phage display and selection of lanthipeptides on the carboxy-terminus of the gene-3 minor coat protein, Nat. Commun., 2017, 8(1), 1500 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2020 |