DOI:
10.1039/D0AN00304B
(Paper)
Analyst, 2020,
145, 3851-3856
An alternative approach for the preparation of a core–shell bimetallic central metal–organic framework as a hydrophilic interaction liquid chromatography stationary phase†
Received
11th February 2020
, Accepted 17th March 2020
First published on 18th March 2020
Abstract
A new type of core–shell composite material was prepared and applied as a hydrophilic interaction liquid chromatography (HILIC) stationary phase. In this work, silica spheres were first modified with a bimetallic central metal–organic framework (ZnCoMOF) by a new strategy of static self-assembled in situ growth. This strategy was beneficial for increasing the electrostatic interaction between the MOF ligand and silica via introducing a sodium dodecylbenzenesulfonate (SDBS) group. The ZnCoMOF@silica stationary phase was characterized and evaluated in comparison with amino-modified and bare silica columns in terms of various polar analytes including eight nucleosides and nucleobases, seven carbohydrates, and multiple sulfonamides and antibiotics. The effects of organic solvent concentration, water content, the concentration of the salt and the pH of the buffer solution on the retention time were studied, which demonstrated the typical retention behavior of HILIC on the ZnCoMOF@silica column. Compared with most reported MOF-based stationary phases, the new composite material showed excellent hydrophilic properties and separation efficiency for various polar analytes. Moreover, the obtained stationary phase showed good reproducibility and stability. The relative standard deviation (RSD) of the retention time for repeatability was found to range from 0.1% to 0.6%, and the RSD of the retention time for stability was found to range from 0.3% to 0.7%. Furthermore, the column batch-to-batch reproducibility showed excellent preparation reproducibility, which few reported in most previous MOF@silica composite materials. This specific preparation method offers an easy and novel way to manipulate the amount of MOF particles on silica, which extends a universal way to produce various MOF@silica stationary phases by the method of static self-assembled in situ growth.
1. Introduction
Metal–organic frameworks (MOFs) containing metal units and organic linkers are considered flexible materials for a wide range of applications due to their various advantages such as a large specific surface area, diverse structures, and excellent thermal stability.1–3 Furthermore, MOFs with unique features have been applied as chromatographic stationary phases. In comparison with more conventional stationary phases like silica-based materials such as saccharides,4–6 ionic liquids,7–9 amides,10–12 and polymers13,14 in high-performance liquid chromatography (HPLC), MOFs can be used in chromatographic separations based on molecular size and shape due to their uniform pore size distribution and ordered pore structure. Therefore, various MOF-based composites were also used as chromatography stationary phases.15–19
The earliest literature report on the use of MOFs as HPLC stationary phases was that the column was directly loaded with MOF crystals.20–22 Due to the obvious disadvantages such as high column pressure and low column efficiency of these columns, the application of core–shell composites has been increasing gradually. Yan et al. prepared ZIF-8@SiO2 shell–core microspheres to separate endocrine disruption chemicals and pesticides by HPLC.17 Ding et al. prepared core–shell UiO-66@SiO2 materials to separate structural isomers via the effect of molecular sieving and the reverse shape selectivity.23 Stefan Kaskel et al. used MIL-101 and UiO-67 to produce core–shell particles for separating C8 isomers and dichlorobenzene isomers.24 Alternatively, Qu et al. employed a kinetic controlling method to immerse MOF particles into silica pores and fabricated core–shell ZIF-8@SiO2 materials for separating positional isomers.25 To date, most of the above composites have been known for reverse phase liquid chromatography (RPLC) and few composites have been explored as HILIC stationary phases. In addition, the density of MOFs would be complicated and hardly controlled in most studies. Therefore, developing a facile synthesis method to prepare MOF@SiO2 shell–core microspheres as HILIC stationary phases and controlling the amount of MOF particles on silica are necessary.
ZnCoMOF is a well-known MOF built using Zn2+ and Co2+ ions and 2-methylimidazole ligands.26 ZnCoMOF particles were found to easily combine with the negative charge on the surface of silica on account of two metal centers, so they could be firmly fixed on the surface of silica compared to monometallic central metal–organic frameworks. Here, we first prepared ZnCoMOF@silica composite materials as the stationary phase by the static self-assembled in situ growth method. In this method, the negative charges were first introduced onto the surface of silica by the addition of a surfactant, followed by an electrostatic interaction with the MOF metal center ion. Chromatographic behaviors in HILIC mode were investigated with a set of polar compounds, including nucleosides and nucleobases, antibiotics, sulfonamides, alkaloids, carbohydrates, etc. In addition, chromatographic behavior in hydrophilic interaction mode was also studied. Moreover, the column batch-to-batch reproducibility was also evaluated, which demonstrated that the composite had superior stability and preparation reproducibility. The new ZnCoMOF@silica composite material was found to be superior to other MOF@silica stationary phases reported in terms of hydrophilic chromatographic separation performance, stability and preparation reproducibility. Finally, the static self-assembled in situ growth method offers a novel way to prepare core–shell composites on MOF@silica, which provides a feasible option for the analysis of polar compounds.
2. Experimental
2.1. Materials and instruments
Spherical silica (5 μm, 70 Å) and amino-modified spherical silica (5 μm, 70 Å) were obtained from Lanzhou Institute of Chemical Physics (Chinese Academy of Sciences, China). Zn(NO3)2·6H2O and Co(NO3)2·6H2O were from Energy Chemical (Shanghai, China). 2-Methylimidazole (98%) was obtained from InnoChem (Beijing, China). Nucleosides, nucleobases, antibiotics and sulfonamides were purchased from Aladdin Biochem Technology Co., Ltd (Shanghai, China). Carbohydrates, alkaloids, amino acid compounds and sodium dodecylbenzenesulfonate (SDBS) were supplied by Energy Chemical. Acetonitrile, ethanol and ammonium acetate were obtained from Yuwang Chemical (Shandong, China) and were of chromatographic grade. The details of the apparatus are the same as those given in ref. 27.
2.2. Mechanism of static self-assembled in situ growth
ZnCoMOF@silica was prepared by the static self-assembled in situ growth method onto the surface of silica, as shown in Fig. 1. The negative charges of SDBS solution were first carried onto the silica, and the zinc and cobalt ions were bonded with them by electrostatic interactions. Subsequently, 2-methylimidazole was coordinated with the metal ions on the surface of the silica for in situ growth.
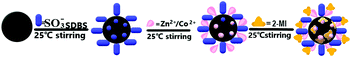 |
| | Fig. 1 Schematic diagram of the preparation route of ZnCoMOF@silica. | |
2.3. Preparation of ZnCoMOF nanocrystals, stationary phase synthesis, and column packing
The procedure of the synthesis of ZnCoMOF nanocrystals is as follows: a mixture of Co(NO3)2·6H2O (5 mmol) and Zn(NO3)2·6H2O (5 mmol) was dissolved in 4 mL of distilled water. The obtained solution was added to a solution containing 50 mmol 2-methylimidazole dissolved in 40 mL of distilled water. The mixture was stirred at 25 °C for 6 h; after this, the precipitate was separated by centrifugation (8000 rpm, 10 min), washed with ethanol 3 times, and dried at 60 °C for 24 h.
The ZnCoMOF@silica synthesis process is as follows: SDBS (0.1 g) was added to distilled water (100 ml), to which activated silica (5 g) was added and the mixture was stirred for 4 hours at 25 °C to obtain a uniform aqueous solution. Subsequently, the mixture was centrifuged 3 times with distilled water and dried at 80 °C under vacuum. Zn(NO3)2·6H2O (5 mmol) and Co(NO3)2·6H2O (5 mmol) were dissolved in 100 ml of water. The mixture of the above product was added to the first mixture and the mixture was stirred for 4 hours. The mixture was centrifuged 3 times with distilled water and dried under vacuum at 80 °C. The obtained product was added to a mixed solution containing 2-methylimidazole (25 mmol) and distilled water (20 ml), and then stirred at 25 °C for 6 hours. The obtained mixture was washed three times with deionized water, centrifuged and dried. Following the above steps, an electrostatically self-assembled ZnCoMOF@silica was obtained. Finally, a two-layer electrostatic self-assembled product was obtained by repeating the above steps. The details of column packing are the same as those given in ref. 27.
3. Results and discussion
3.1. Characterization of the prepared materials
Elemental analysis showed an obvious increase of the carbon content from bare silica (0.25%) to ZnCoMOF@silica (2.23%) (Table 1). The degree of surface modification of the silica spheres was determined by SEM, as shown in Fig. S1 (ESI†). It proved that a continuous layer of the MOF was there on the surface of the silica. The FT-IR spectra of ZnCoMOF and ZnCoMOF@silica (Fig. S1c†) showed some specific peaks for Zn/Co–N (425 cm−1 and 473 cm−1) and C–H (2922.7 and 2964.3 cm−1) bonds. The imidazole ring causes some peaks to appear in the range of 700 to 1500 cm−1. The peaks located at higher wavenumbers (>2900 cm−1) could be associated with the N–H vibrations.26 In the spectra of bare silica and the ZnCoMOF@silica stationary phase, a common band at 1100 cm−1 was observed, which was attributed to the SiO2 backbone stretching vibration. This further proved that ZnCoMOF@silica was successfully synthesized. XRD measurements showed the crystallinity of ZnCoMOF@silica (Fig. S1d†), which was consistent with ZnCoMOF. The characteristic signal of ZnCoMOF@silica and bare silica presented at 2θ = 20°–25° belongs to the silica backbone. Furthermore, the BET surface areas of all particle types are provided in Table S1.† The above results indicate that ZnCoMOF@silica has been successfully synthesized.
Table 1 Elemental analysis of the bare silica and ZnCoMOF@silica
| Sample |
Content of element (%) |
| N |
C |
H |
| Bare silica |
0.00 |
0.25 |
0.367 |
| ZnCoMOF@silica |
0.52 |
2.23 |
0.589 |
3.2. Chromatographic behavior of ZnCoMOF@silica vs. bare silica
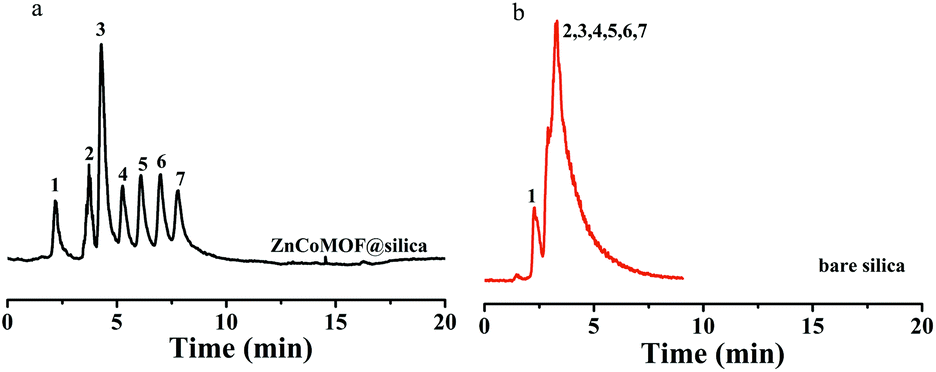
Seven carbohydrates were first selected to evaluate the separation performance of ZnCoMOF@silica composites. Fig. 2 shows the separation performance of carbohydrates on ZnCoMOF@silica and bare silica columns at a high acetonitrile concentration. Seven carbohydrates were successfully separated with good efficiency on the ZnCoMOF@silica stationary phase, which few studies had achieved on some MOF-based stationary phases. The peak sequence and elution order were consistent with the characteristics of hydrophilic chromatography. From the chromatogram, it can be observed that the stationary phases exhibited HILIC behavior.
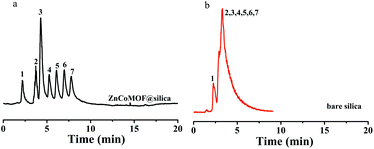 |
| | Fig. 2 Chromatograms for the separation of seven carbohydrates on ZnCoMOF@silica (a) and bare silica (b): L-rhamnose monohydrate (1), D-ribose (2), D-fructose (3), sucrose (4), lactose (5), melezitose (6) and raffinose (7) mobile phase: 90% acetonitrile, 10% water; flow rate = 0.8 mL min−1, T = 25 °C. ELS detector: gas flow: 4 L min−1, tube temperature 115 °C. | |
3.3. Batch-to-batch reproducibility
The reproducibility of the preparation of the stationary phase is also a crucial parameter. Therefore, three batches of ZnCoMOF@silica were prepared to characterize the reproducibility of the preparation of the stationary phase. Seven carbohydrates were separated on three batches of ZnCoMOF@silica columns. The chromatographic parameters can be reproduced well (see Fig. S2 and Table S2†), which proved the excellent preparation reproducibility of the new composite material and the feasibility of this method.
3.4. Mechanism description
3.4.1. Effect of water content and organic solvent concentration on retention for the separation of analytes.
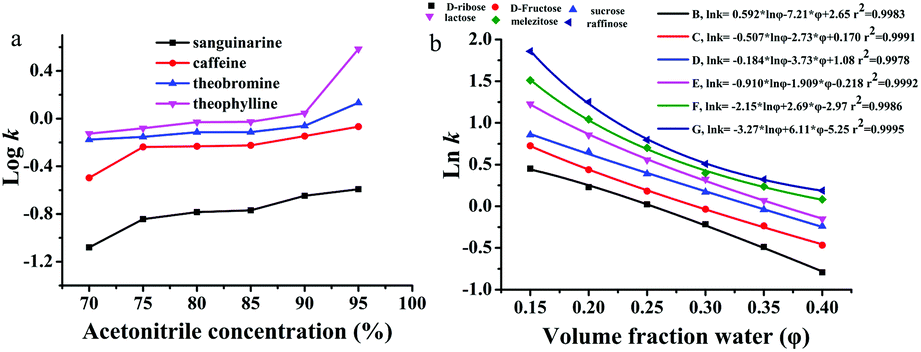
The retention mechanism of ZnCoMOF@silica was investigated by varying chromatography conditions, including water content, organic solvent concentration, buffer pH and buffer concentration. As shown in Fig. 3a, the retention factors of all alkaloids increased gradually with the increase of acetonitrile content. Thus, the typical HILIC mechanism was confirmed.28–30 The partitioning and adsorption mechanisms occur mainly in HILIC modes;31–33 therefore, the equation of combining the partitioning model and the adsorption model should be described by eqn (1):
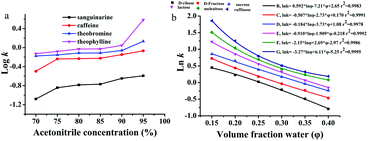 |
| | Fig. 3 Effect of water content and organic solvent concentration on retention for the separation of alkaloids (a) and carbohydrates (b), respectively. In a, mobile phase: acetonitrile and 100 mmol L−1 ammonium acetate; flow rate = 0.8 mL min−1, T = 25 °C, UV detection: 254 nm. The chromatography conditions in b are the same as in Fig. 2. | |
where a, b, and c are constants, k is the retention factor of the analytes, and φ is the water content in the mobile phase. The results of multiple regression analyses are presented in the ESI (Table S3†), and show excellent fits (r2 = 0.9884–0.9997) for carbohydrates at 6 different eluent compositions (φ = 0.14–0.30). The results of eqn (1) (r2 values) show that both adsorption and partitioning contribute to the retention mechanism in the separation system as observed in Fig. 3b.
3.4.2. Effect of buffer pH and buffer concentration on retention for the separation of compounds.
To better understand the retention mechanism of the ZnCoMOF@silica stationary phase, the effect of buffer pH and buffer concentration was also investigated. Four alkaloids were chosen as test compounds at pH values ranging from 4 to 9 on the ZnCoMOF@silica column. As shown in Fig. S3a,† the retention times of all compounds increased gradually with the increase of pH value. It was possible that the stationary phase obtained more negative charges, which enhanced the ion–exchange interaction with these compounds with the increasing pH value, leading to an increase of retention time. In addition, the reason may be that the ionization of silanol groups was suppressed at low pH, which weakens the ion–exchange interaction between the alkaloids and the residual silanol groups. Therefore, the changes in the charge state and density of stationary phases and compounds are also a factor that can affect the retention time. Ammonium salts with relatively high solubility are usually used to adjust the retention behaviors in HILIC mode. As shown in Fig. S3b,† the retention ability of ZnCoMOF@silica toward nucleosides and nucleobases increased with an increase in buffer concentration. It is probably due to that the water-rich layer on the stationary phase becomes thicker, as the buffer concentration increases.28 The thickening of the water-rich layer on the stationary phase was considered to promote partitioning.33
3.5. Chromatographic performance of the ZnCoMOF@silica composite
3.5.1. Separation performance for nucleosides and nucleobases.
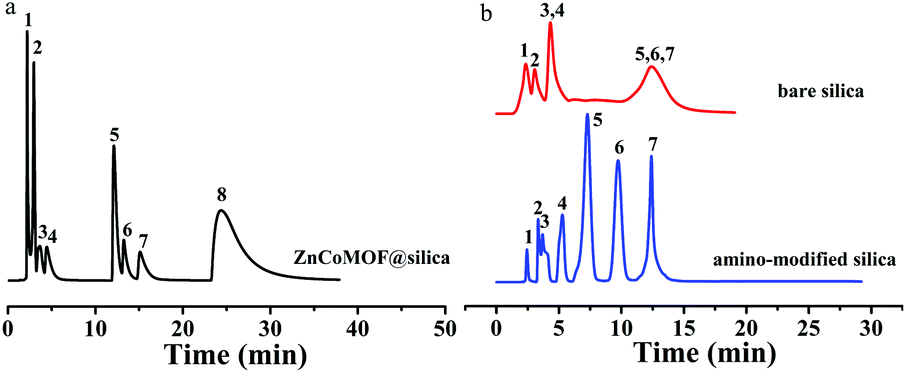
The application of the ZnCoMOF@silica stationary phase in HILIC mode was further investigated by the separation of some representative polar compounds. A test mixture of eight nucleosides and nucleobases (i.e., 6-chlorouracil, thymine, thymidine, uridine, inosine, guanosine, adenosine, and hypoxanthine) was separated in HILIC mode on the ZnCoMOF@silica, bare silica and amino-modified silica columns under the same elution conditions, respectively. Fig. 4 shows that the successful separation of eight nucleosides and nucleobases was achieved on the ZnCoMOF@silica stationary phase. Hypoxanthine formed a prominent tailing peak on the ZnCoMOF@silica column. This phenomenon was attributed to the residual Si–OH on the surface of ZnCoMOF@silica, which led to strong adsorption toward nucleobases. However, the quantity of Si–OH on the surface of ZnCoMOF@silica and the surface properties of the spheres were changed by ZnCoMOF particles. Besides, the ZnCoMOF@silica column presented better separation performance than the bare silica column under the same chromatography conditions, which explained that the modification of ZnCoMOF onto silica would improve the surface chemical properties of the silica sphere microparticles.
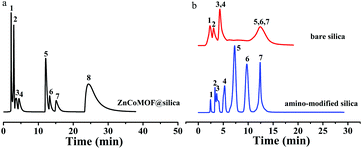 |
| | Fig. 4 Chromatograms for the separation of eight nucleosides and nucleobases with ZnCoMOF@silica (a), bare silica and amino-modified columns (b). 6-Chlorouracil (1), thymine (2), thymidine (3), uridine (4), inosine (5), guanosine (6), adenosine (7), hypoxanthine (8); mobile phase: 0–6 min 90% acetonitrile, 10% water, 6–8 min 70% acetonitrile, 30% water, 18–20 min 60% acetonitrile, 40% water, flow rate = 0.8 mL min−1, T = 25 °C, UV detection: 254 nm. | |
3.5.2. Separation performance for amino acid compounds and alkaloids.
Five amino acid compounds (i.e., cysteine, tryptophan, methionine, threonine, and glycine) were successfully separated on the column, as shown in Fig. S4.† The result showed that amino acid compounds were baseline separated only on ZnCoMOF@silica. Furthermore, seven alkaloids were also separated on the columns (Fig. S5†). Although the latter peaks had significant peak broadening effects, baseline separation can also be achieved on the ZnCoMOF@silica column compared to that on the amino silica and the bare silica columns. The chromatographic separation performance of the ZnCoMOF@silica column has indeed been greatly improved compared with those of the most reported MOF-based composites, but its column efficiency needs to be further improved. The reason behind this phenomenon may be that the size of the selected silica is large and the MOF particles grown on the surface of silica change the original pores of silica, resulting in the uniformity of the pores. In addition to the changes in the pore size and particle size of silica by MOF particles, there is also a problem of adsorption of analytes caused by the properties of MOFs themselves. The charged metal center shows a strong electrostatic interaction and strong adsorption of the analytes. In addition, the porous structure of MOFs can also cause strong adsorption of analytes. The strong adsorption of analytes results in a slow elution process and broadening of the peaks. Therefore, the strong adsorption of analytes caused by the structural properties of MOFs themselves results in chromatographic peak broadening and poor column efficiency. Reducing the particle size of silica or selecting MOF particles with a simpler structure compatible with the size of silica is a feasible improvement measure.
3.5.3. Separation performance for antibiotic compounds and sulfonamides.
In addition to the separation of the above-mentioned typical hydrophilic compounds, sulfonamides and antibiotics have also been further investigated. As shown in Fig. S6 and S7,† these compounds were capable of rapid separation on the ZnCoMOF@silica stationary phase compared to the amino-modified column or bare silica. At the same time, the ZnCoMOF@silica composite exhibited the advantage of separating such drugs, which could be close to the separation effect on some of the excellent MOF-based stationary phases reported. From the above results, it could be seen that the ZnCoMOF@silica stationary phase showed good selectivity in the separation of polar compounds in HILIC mode.
3.6. Reproducibility and stability of the ZnCoMOF@silica stationary phase
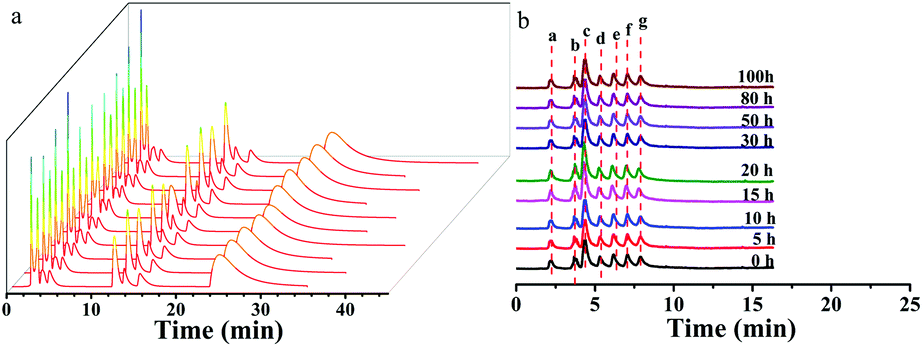
The ZnCoMOF@silica column demonstrated a high reproducibility of retention time and column stability. The repeatability of the ZnCoMOF@silica column was evaluated with a mixture of nucleosides and nucleobases through 10 continuous injections under the chromatography conditions of water/ACN as the mobile phase. Furthermore, the RSD of the retention time was found to range from 0.1% to 0.6% (Fig. 5a and Table S5†). The longevity of the column would also be obviously of concern. The retention of seven typical carbohydrates (D-ribose, L-rhamnose monohydrate, D-fructose, sucrose, D-lactose, melezitose, and raffinose) was studied over 100 h of continuous operation with a mobile phase containing 80% acetonitrile and 20% water. The RSD of the retention time for four model analytes was found to range from 0.3% to 0.7%, as shown in Fig. 5b (Table S4†). Finally, the above results fully demonstrate the superior reproducibility and stability of the material.
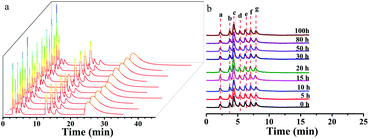 |
| | Fig. 5 Reproducibility and stability of the ZnCoMOF@silica stationary phase. a shows the reproducibility of eight nucleosides and nucleobases on the ZnCoMOF@silica column. b shows the stability of seven carbohydrates on the ZnCoMOF@silica column. | |
4. Conclusion
A facile static self-assembled in situ growth method for the synthesis of shell–core ZnCoMOF@silica microspheres as a novel HILIC stationary phase was reported. The new stationary phase was applied to the separation of diverse polar compounds. The retention mechanism was also confirmed, which is caused by a combination of partitioning and adsorption mechanisms. Furthermore, the new composite materials were found to be superior to most reported MOF@silica composites in terms of hydrophilic properties, separation efficiency, reproducibility, and stability. In summary, the proposed approach could effectively control the amount of MOF particles and is suitable for preparing MOF@silica composite materials, which could be an ideal choice for the synthesis of similar MOF-based stationary phases.
Conflicts of interest
The authors have declared no conflict of interest.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21575149 and 21575148) and the State Key Scientific Special Project (2016ZX05011-003).
References
- H. Furukawa, F. Gandara, Y. B. Zhang, J. Jiang, W. L. Queen, M. R. Hudson and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 4369–4381 CrossRef CAS PubMed.
- B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390–1393 CrossRef CAS.
- F. I. Pambudi, M. W. Anderson and M. P. Attfield, Chem. Sci., 2019, 10, 9571–9575 RSC.
- T. Chen, L. Zhu, H. Lu, G. Song, Y. Li, H. Zhou, P. Li, W. Zhu, H. Xu and L. Shao, Anal. Chim. Acta, 2017, 964, 195–202 CrossRef CAS PubMed.
- Q. Sheng, X. Su, X. Li, Y. Ke and X. Liang, J. Chromatogr. A, 2014, 1345, 57–67 CrossRef CAS PubMed.
- Z. Guo, A. Lei, Y. Zhang, Q. Xu, X. Xue, F. Zhang and X. Liang, Chem. Commun., 2007, 2491–2493, 10.1039/b701831b.
- L. Qiao, X. Shi, X. Lu and G. Xu, J. Chromatogr. A, 2015, 1396, 62–71 CrossRef CAS.
- L. Qiao, H. Li, Y. Shan, S. Wang, X. Shi, X. Lu and G. Xu, J. Chromatogr. A, 2014, 1330, 40–50 CrossRef CAS PubMed.
- H. Qiu, A. K. Mallik, M. Takafuji, S. Jiang and H. Ihara, Analyst, 2012, 137, 2553–2555 RSC.
- L. Qiao, W. Lv, M. Chang, X. Shi and G. Xu, J. Chromatogr. A, 2018, 1559, 141–148 CrossRef CAS PubMed.
- D. Kotoni, I. D'Acquarica, A. Ciogli, C. Villani, D. Capitani and F. Gasparrini, J. Chromatogr. A, 2012, 1232, 196–211 CrossRef CAS PubMed.
- G. Karlsson, S. Winge and H. Sandberg, J. Chromatogr. A, 2005, 1092, 246–249 CrossRef CAS PubMed.
- S. Ji, F. Zhang, S. Wu, B. Yang and X. Liang, Analyst, 2014, 139, 5594–5599 RSC.
- C. Zhang, E. Rodriguez, C. Bi, X. Zheng, D. Suresh, K. Suh, Z. Li, F. Elsebaei and D. S. Hage, Analyst, 2018, 143, 374–391 RSC.
- R. D. Arrua, A. Peristyy, P. N. Nesterenko, A. Das, D. M. D'Alessandro and E. F. Hilder, Analyst, 2017, 142, 517–524 RSC.
- L. Fan and X. P. Yan, Talanta, 2012, 99, 944–950 CrossRef CAS PubMed.
- Y. Y. Fu, C. X. Yang and X. P. Yan, Chemistry, 2013, 19, 13484–13491 CrossRef CAS PubMed.
- Y. Y. Wu, C. X. Yang and X. P. Yan, Analyst, 2015, 140, 3107–3112 RSC.
- K. Tanaka, T. Muraoka, Y. Otubo, H. Takahashi and A. Ohnishi, RSC Adv., 2016, 6, 21293–21301 RSC.
- X. Kuang, Y. Ma, H. Su, J. Zhang, Y. B. Dong and B. Tang, Anal. Chem., 2014, 86, 1277–1281 CrossRef CAS PubMed.
- W. W. Zhao, C. Y. Zhang, Z. G. Yan, L. P. Bai, X. Y. Wang, H. L. Huang, Y. Y. Zhou, Y. B. Xie, F. S. Li and J. R. Li, J. Chromatogr. A, 2014, 1370, 121–128 CrossRef CAS PubMed.
- S. Van der Perre, A. Liekens, B. Bueken, D. E. De Vos, G. V. Baron and J. F. M. Denayer, J. Chromatogr. A, 2016, 1469, 68–76 CrossRef CAS PubMed.
- X. Q. Zhang, Q. Han and M. Y. Ding, RSC Adv., 2015, 5, 1043–1050 RSC.
- S. Ehrling, C. Kutzscher, P. Freund, P. Müller, I. Senkovska and S. Kaskel, Microporous Mesoporous Mater., 2018, 263, 268–274 CrossRef CAS.
- Q. Qu, H. Xuan, K. Zhang, X. Chen, Y. Ding, S. Feng and Q. Xu, J. Chromatogr. A, 2017, 1505, 63–68 CrossRef CAS PubMed.
- N. Bagheri, B. Habibi, A. Khataee and J. Hassanzadeh, Talanta, 2019, 201, 286–294 CrossRef CAS PubMed.
- T. Si, X. Song, L. Wang, Y. Guo, X. Liang and S. Wang, Microchem. J., 2020, 152, 104330 CrossRef CAS.
- M. Taraji, P. R. Haddad, R. I. J. Amos, M. Talebi, R. Szucs, J. W. Dolan and C. A. Pohl, Anal. Chim. Acta, 2018, 1000, 20–40 CrossRef CAS PubMed.
- J. Wang, Z. Guo, A. Shen, L. Yu, Y. Xiao, X. Xue, X. Zhang and X. Liang, J. Chromatogr. A, 2015, 1398, 29–46 CrossRef CAS PubMed.
- G. Schuster and W. Lindner, J. Chromatogr. A, 2013, 1273, 73–94 CrossRef CAS PubMed.
- P. Hemström and K. Irgum, J. Sep. Sci., 2006, 29, 1784–1821 CrossRef PubMed.
- Y. Guo, Analyst, 2015, 140, 6452–6466 RSC.
- T. Cai, H. Zhang, J. Chen, Z. Li and H. Qiu, J. Chromatogr. A, 2019, 1597, 142–148 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0an00304b |
|
| This journal is © The Royal Society of Chemistry 2020 |

 a,
Xiaojing
Liang
a,
Xiaojing
Liang
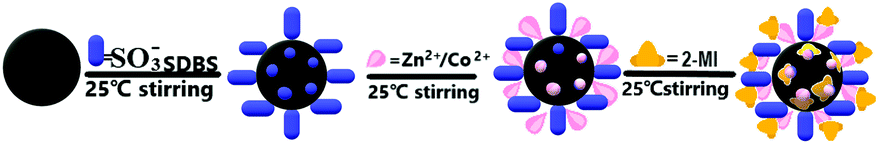
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k = a + b
k = a + b