DOI:
10.1039/C9SC01669D
(Edge Article)
Chem. Sci., 2019,
10, 5397-5404
Emergent supramolecular assembly properties of a recognition-encoded oligoester†
Received
4th April 2019
, Accepted 25th April 2019
First published on 2nd May 2019
Abstract
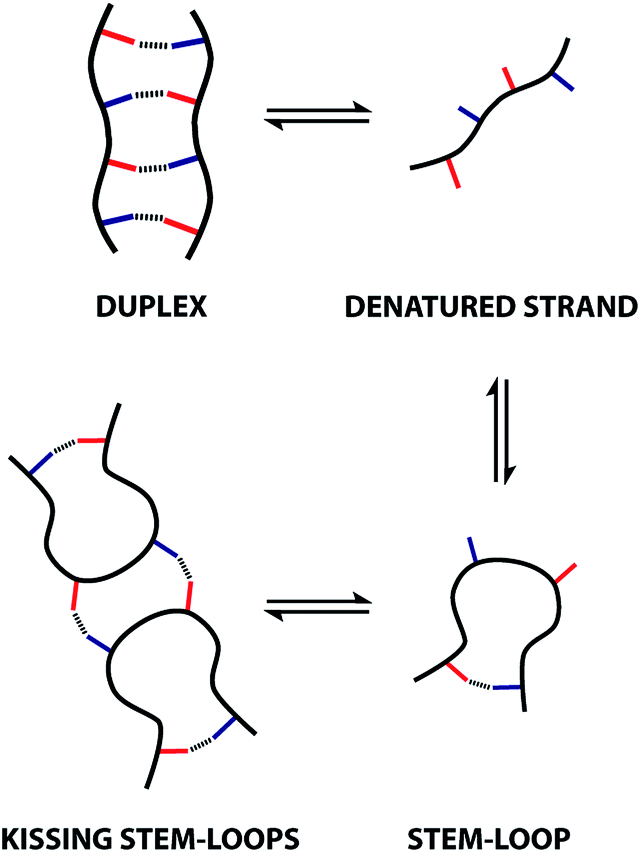
The sequences of oligomeric molecules equipped with interacting side-chains encode the three-dimensional structure, the supramolecular assembly properties, and ultimately function. In an attempt to replicate the duplex forming properties of nucleic acids, an oligoester containing an alternating sequence of hydrogen bonding donor (D) and acceptor (A) residues was synthesised. Characterisation of assembly properties of the ADAD oligomer revealed a supramolecular architecture that resembles the kissing stem-loops motif found in folded RNA. NMR dilution and melting experiments in chloroform and 1,1,2,2-tetrachloroethane show that intramolecular hydrogen bonding interactions between the terminal phenol and phosphine oxide recognition sites in the ADAD 4-mer lead to 1,4-folding. This folded stem-loop structure can be denatured to give the single strand by heating. At higher concentrations or lower temperatures, the stem-loop dimerises via intermolecular hydrogen bonding interactions between the two inner recognition sites, leading to a kissing stem-loops structure. The results suggest a rich supramolecular chemistry for these recognition-encoded oligoesters and lay strong foundations for the future development of new functional materials based on synthetic information molecules.
1 Introduction
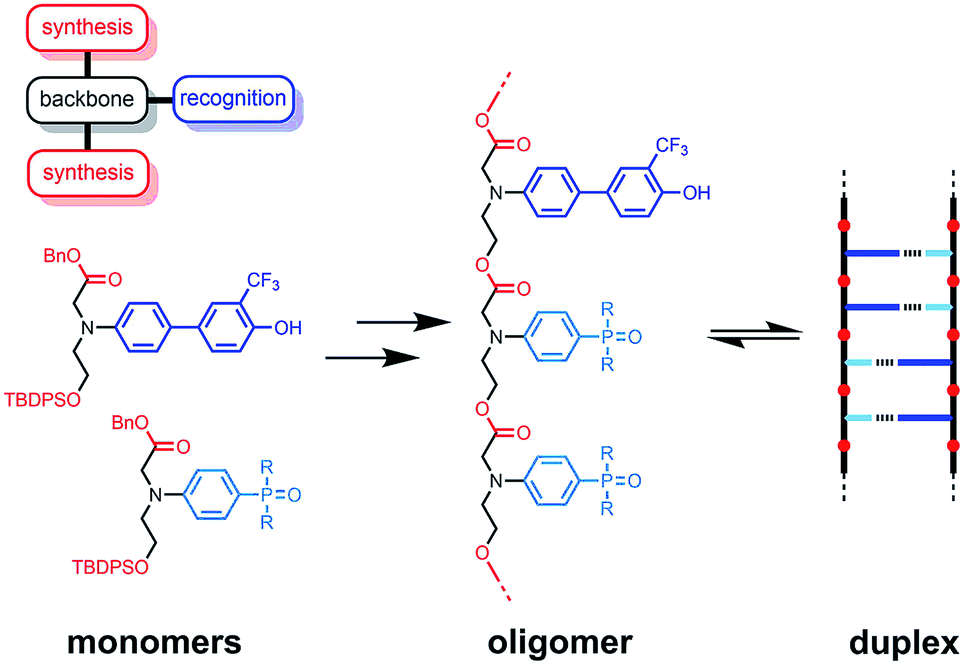
The functional properties of biomacromolecules are encoded in the sequence of building blocks that determine the three-dimensional structures that are adopted when the linear oligomers fold.1–8 The relationship between sequence, folding and function has been extensively explored by Huc in families of amide oligomers.9–11 These results suggest that sequence-encoded synthetic oligomers will be capable of showing the sophisticated recognition, self-assembly, catalysis and replication properties exhibited by biological oligomers.12 We have been investigating the properties of synthetic oligomers based on the nucleic acid blueprint: an oligomeric backbone equipped with side-chains containing complementary hydrogen bonding recognition sites (Fig. 1).13–21 Hydrogen bonding interactions between sequence-complementary oligomers leads to formation of stable duplexes in organic solvents for a wide range of molecular designs based on different synthesis, backbone and recognition modules.
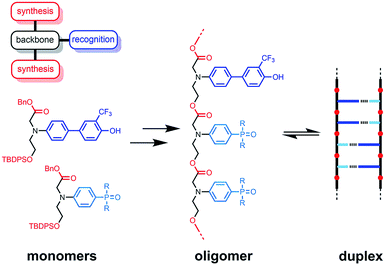 |
| | Fig. 1 A blueprint for duplex forming molecules. There are three key design elements: the coupling chemistry used for the synthesis of oligomers (red), the recognition module, which controls intermolecular binding (blue), and the backbone module, which links these components together (black). The protected monomers used for the synthesis of oligoesters equipped with phenol and phosphine oxide recognition modules are shown, along with a schematic representation of the proposed duplex. | |
One successful architecture is based on an oligoester backbone and phenol–phosphine oxide base-pairing interactions.21 Although the backbone ester groups could potentially compete for hydrogen bonding interactions with the phenol recognition groups, these interactions are very weak (β ≈ 5.5) compared with the very strong hydrogen bonds formed with phosphine oxide recognition sites (β ≈ 10.5), which leads to high fidelity base-pairing in this system.22 Backbone flexibility is another critical parameter in determining the duplex forming properties of recognition-encoded oligomers. If the backbone is too flexible, intramolecular interaction between adjacent complementary bases leads to 1,2-folding, which precludes duplex formation in mixed sequence oligomers.17 However, sufficient flexibility is required to allow the backbone to adapt to a conformation compatible with multiple base-pairing interactions in longer duplexes.20 The combination of the short–long base-pair geometry and the short flexible ester backbone shown in Fig. 1 provides the right balance of flexibility and geometric constraint to ensure cooperative formation of closed duplexes for the AA, DD, and AD 2-mers with no competition from 1,2-folding.21 Ester backbone chemistry is particularly attractive because it provides straightforward access to longer oligomers via stepwise synthesis from orthogonally protected monomers.23–30 Here we describe the synthesis of the ADAD 4-mer and show that there are emergent supramolecular assembly properties that resemble those found in nucleic acids.
2 Results and discussion
2.1 Synthesis
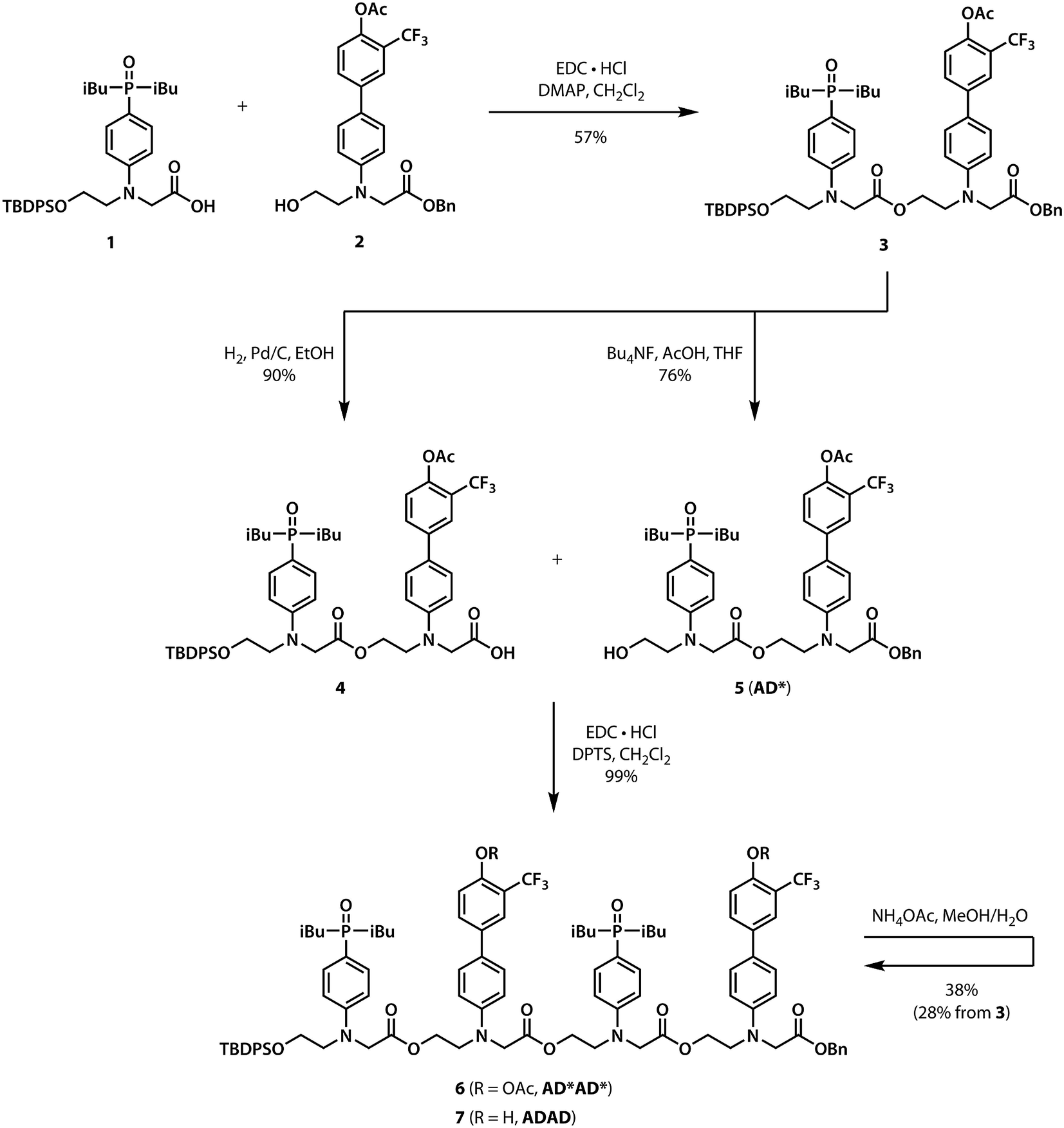
Scheme 1 shows the synthesis of the ADAD 4-mer. Synthesis of the two monomer building blocks (1 and 2) required for the preparation of oligomers was described previously.21 Coupling of 1 and 2 with EDC gave the acetyl-protected 2-mer 3. Hydrogenation of 3 selectively removed the carboxylic acid protecting group to give 4. The alcohol protecting group on 3 was selectively removed with n-tetrabutylammonium fluoride to give 5. Coupling 4 with 5 provided AD*AD*, the 4-mer with the phenol groups protected as acetate esters. The ester protecting groups were then hydrolysed to give ADAD. As with peptides, the backbone has a direction, so we write the sequence starting from the alcohol terminus and ending at the carboxylic acid terminus.
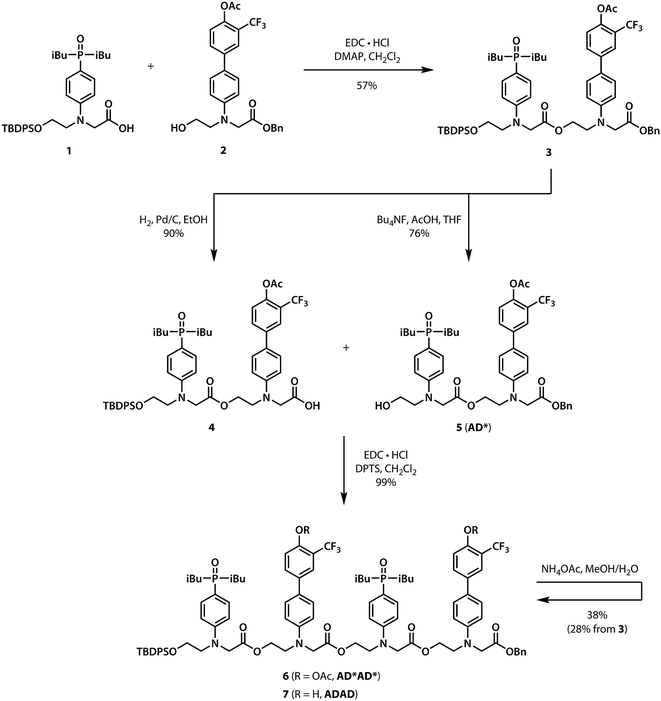 |
| | Scheme 1 Synthesis of the ADAD 4-mer. | |
2.2 NMR sequential assignment
The backbone of the ADAD 4-mer has a direction, so none of the residues are equivalent, and all of the 1H NMR signals were resolved in 500 MHz spectrum recorded in deuterochloroform. Two well-separated signals were observed in both the 19F and 31P NMR spectra, which indicates that the two donor residues and two acceptor residues are both in quite different environments, suggesting that the oligomer either folds or forms a higher order supramolecular structure in solution. In order to determine the three-dimensional structure, assignment of the signals due to different residues was required. The sequential assignment strategy is summarized below (see Fig. S3 to S8† for full details).
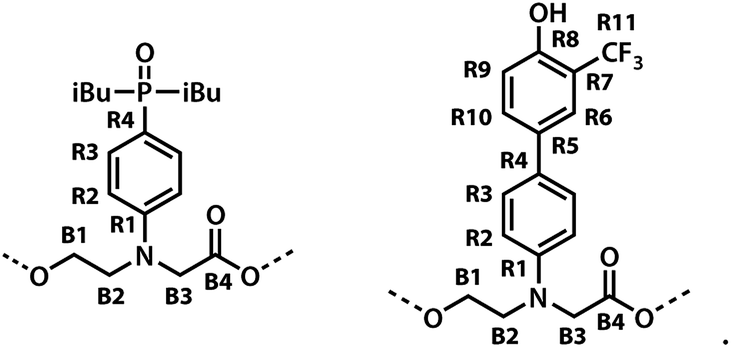
The labelling scheme used to assign the NMR spectra is illustrated in Fig. 2: B represents signals due to the backbone units and R represents signals due to the recognition units; carbon atoms are numbered sequentially for each unit (R1, R2, …, B1, B3, …) and the four different residues are distinguished using colours (red, blue, yellow and green). Protons have the same label as the carbon atom to which they are bonded; the signals due the phosphorus and fluorine atoms are labeled outer and inner to distinguish the two residues in the oligomer; the terminal protecting groups and the iso-butyl groups on the phosphine oxides were not assigned.
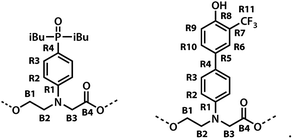 |
| | Fig. 2 Carbon labelling scheme for ADAD. | |
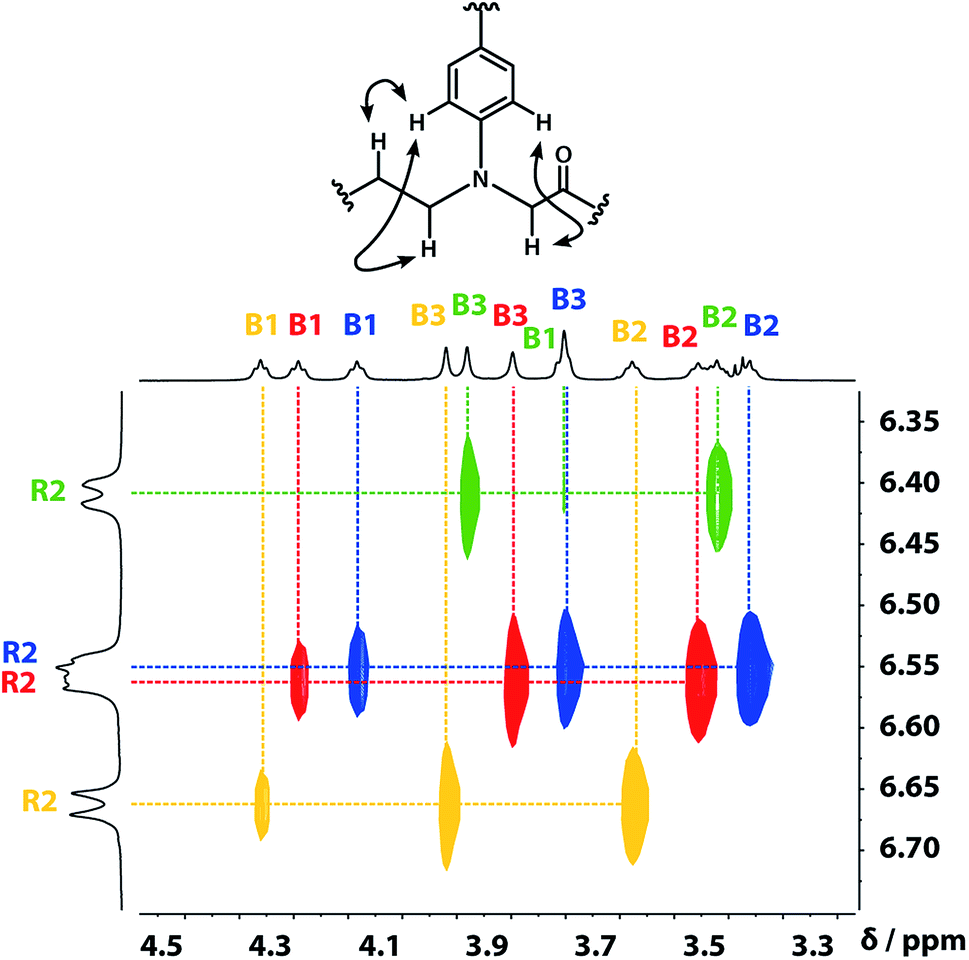
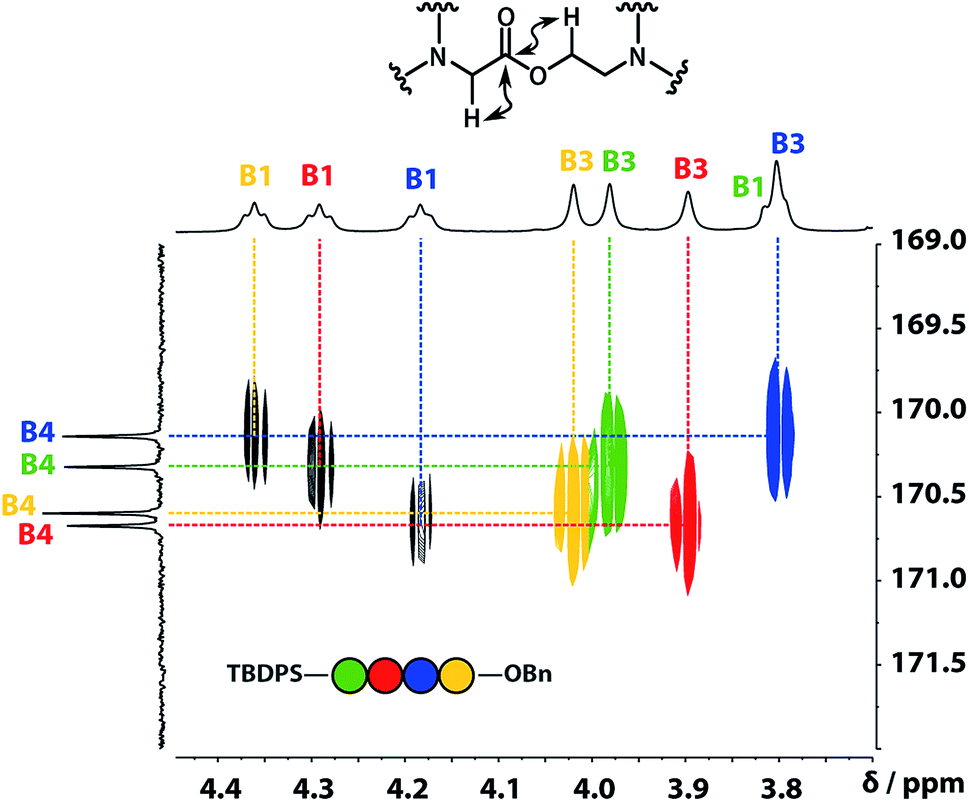
A COSY spectrum provided the connectivity within the different 1H spin systems present in the oligomer. A 1H–1H NOESY spectrum showed cross-peaks between the signals due to the aromatic protons of the recognition units and the aliphatic signals due to the backbone allowing unambiguous assignment of the R2 signals and the B3 signals, the only aliphatic singlets in the 1H NMR spectrum (Fig. 3). Cross peaks were also observed between the R2 signals and the B1 and B2 signals in the 1H–1H NOESY spectrum (Fig. 3). The B1 and B2 1H NMR signals were distinguished using heteronuclear multiple-bond correlations with the 13C signals due to the ester carbonyl groups B4. Fig. 4 shows the correlations observed between the B4 13C signals and the B3 and B1 1H NMR signals. These correlations also allow us to establish the connectivity of the four residues through the ester linkages. For example, the blue cross-peak highlighted in Fig. 4 is the correlation between B3 and B4 of the blue residue, but the B4 signal of the blue residue also shows a correlation with the B1 signal of the yellow residue, which means that blue and yellow residue are next to each other in the sequence. This methodology gives the color-coded sequence of residues illustrated in Fig. 4 green-red-blue-yellow in the alcohol terminus to carboxylic acid direction.
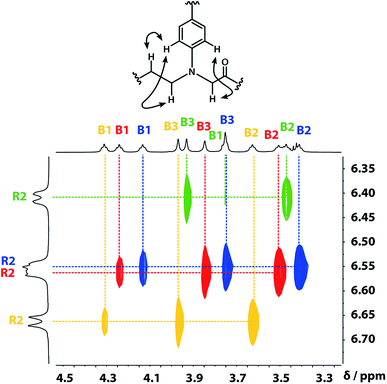 |
| | Fig. 3 Part of the 1H–1H NOESY (500 MHz, CDCl3, 298 K, ca. 5 mM) spectrum of the ADAD 4-mer showing cross-peaks with the R2 1H signal. Signals due to different residues are distinguished by colour. | |
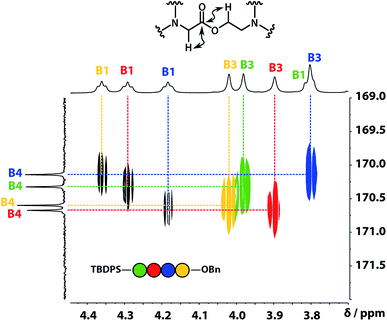 |
| | Fig. 4 Part of the 1H–13C HMBC (500 MHz, CDCl3, 298 K, ca. 5 mM) spectrum of the ADAD 4-mer showing cross-peaks with the B4 13C signal. This spectrum established the connectivity of the residues illustrated. | |
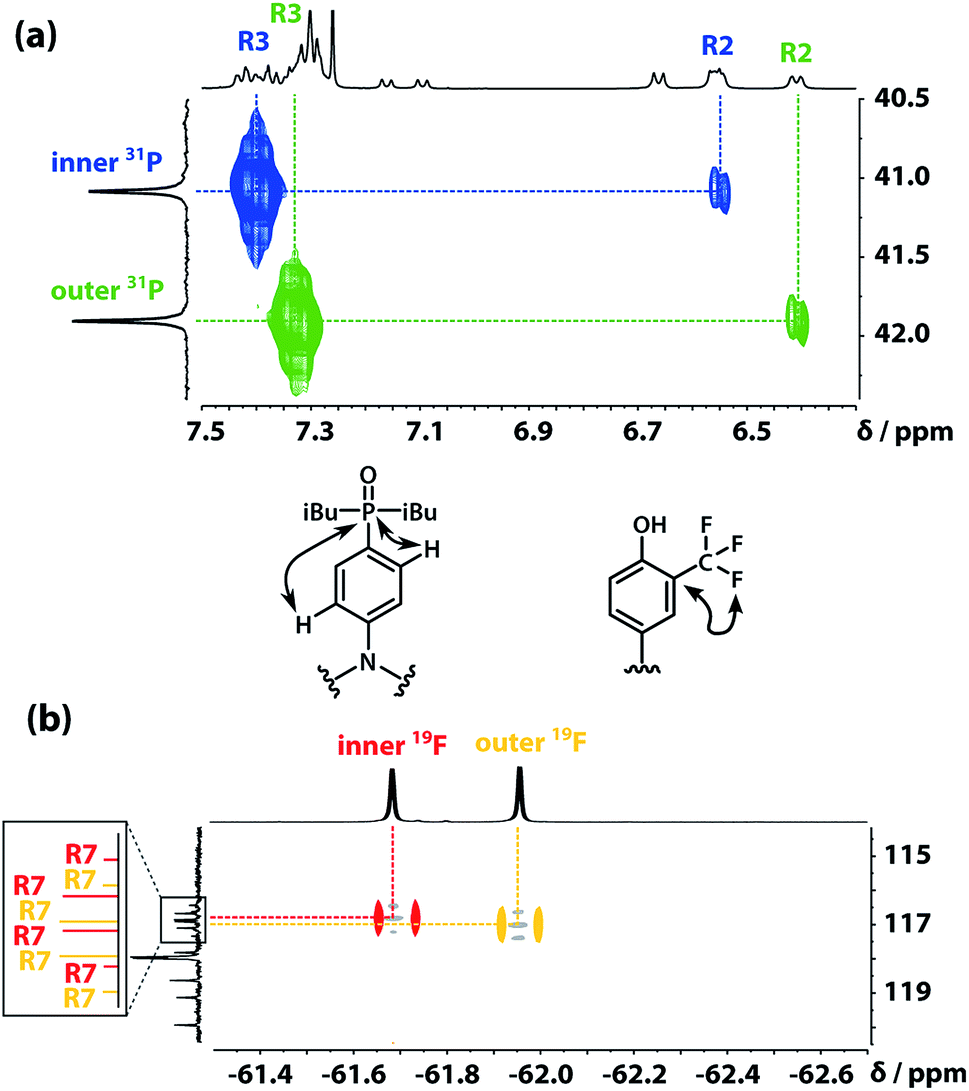
The 31P signals of the two acceptor residues were assigned using heteronuclear multiple-bond correlations observed in the 1H–31P HMBC spectrum shown in Fig. 5(a). Strong two-bond coupling was observed between the 31P signals and the 1H signals due to the R3 protons of the acceptor residues, and weaker cross-peaks were observed between the 31P signals and the 1H signals due to the R2 protons. The green 31P signal has a significantly lower chemical shift than the blue 31P signal, which suggests that the extent of hydrogen bond formation with the outer acceptor residue is greater than with the inner acceptor residue at this concentration.
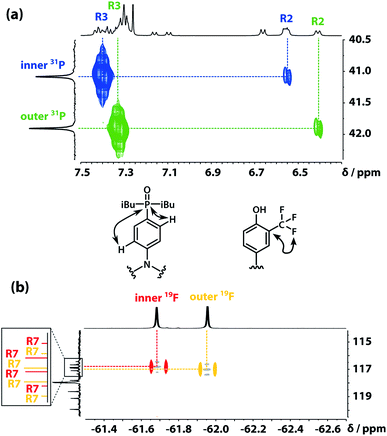 |
| | Fig. 5 (a) Part of the 1H–31P HMBC (400 MHz, CDCl3, 298 K, ca. 5 mM) spectrum of the ADAD 4-mer. (b) Overlay of the 19F–13C HMBC (400 MHz, CDCl3, 298 K, ca. 5 mM) and 19F–13C HSQC (CDCl3, 298 K, ca. 5 mM, light grey) spectra of the ADAD 4-mer. | |
The fluorine signals of the donor residues were assigned in a similar manner (Fig. 5(b)). Long range 1H–13C correlations observed in the HMBC spectrum connected the R3 1H signals of the donor residues to the R5 13C signals. Cross-peaks between the R9 1H signals and both the R5 and R7 13C signals were used to identify the signals due to the carbons ipso to the trifluoromethyl group. Fig. 5(b) shows long range 13C–19F correlations observed between the R7 13C signals and the 19F signals in the HMBC spectrum. Hence, the upfield 19F signal was identified as the yellow outer residue, and the downfield signal corresponds to the red inner residue.
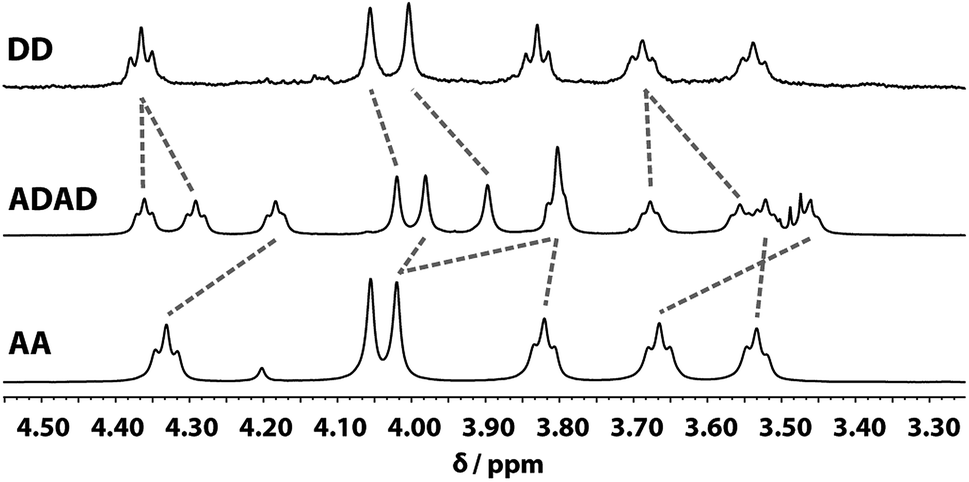
Despite the similar chemical environments of the backbone methylene groups, the signals span a large range of chemical shifts in the 1H NMR spectrum, which implies substantial differences in the local magnetic fields they experience. Fig. 6 compares the methylene region of 1H NMR spectrum of the ADAD 4-mer with the 1H NMR spectra of the previously reported DD and AA 2-mers. There are significant differences in the chemical shifts of chemically related signals. The 2-mers AA and DD 2-mers cannot form hydrogen bonds in the absence of the complementary strand, so the significant differences in chemical shift imply that hydrogen bonding interactions in ADAD lead to a different backbone conformation from DD and AA, which are likely to be unstructured. The differences in chemical shift are larger for the inner residues than for the outer residues, which is presumably related to the different extents of hydrogen bonding observed for the inner and outer residues in the 19F and 31P NMR spectra.
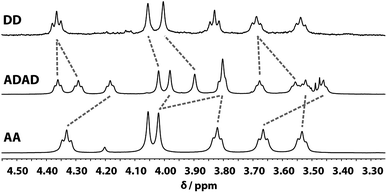 |
| | Fig. 6 Aliphatic region of the 1H NMR (CDCl3, 298 K) spectra of DD (400 MHz, 1 mM), ADAD (500 MHz, 5 mM), and AA (400 MHz, 5 mM). Chemically related signals are connected with dashed lines. | |
2.3 Diffusion ordered spectroscopy
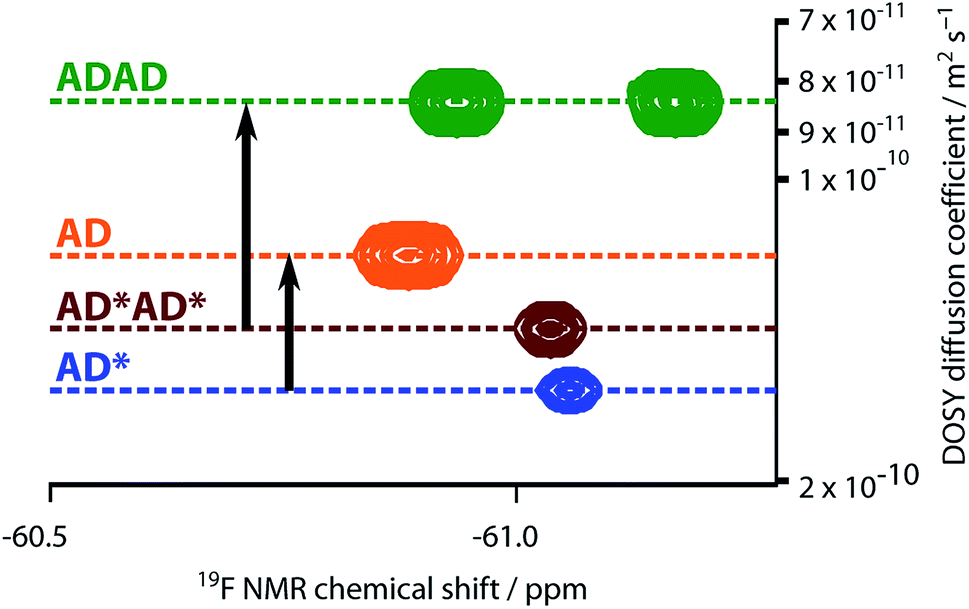
The NMR spectra suggest that there are hydrogen bonding interactions between the recognition groups in the ADAD 4-mer, and these interactions could be due to intramolecular folding or intermolecular interactions. Diffusion Order 19F NMR Spectroscopy was therefore used to determine whether intermolecular complexes are present in chloroform solution. The diffusion coefficient of the ADAD 4-mer was compared with the corresponding values for the AD 2-mer, which we have shown forms a duplex under these conditions,21 and the 4-mer and 2-mer, AD*AD* and AD*, in which the phenol groups are protected as acetate esters and so cannot form hydrogen bonds. Fig. 7 shows an overlay of the 19F DOSY spectra of samples prepared at comparable concentrations in 1,1,2,2-tetrachloroethane-d2. The diffusion coefficients for AD and ADAD were both significantly lower than the values for the corresponding non-hydrogen bonding analogues AD* and AD*AD*, which implies formation of intermolecular complexes through hydrogen bonding interactions involving the phenol groups in both AD and ADAD at a concentration of 5 mM in TCE-d2.31
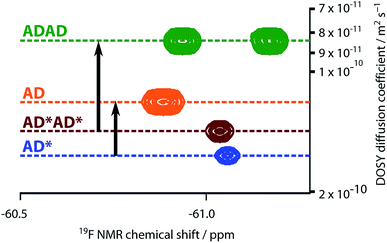 |
| | Fig. 7 Overlay of the 19F NMR DOSY (400 MHz, TCE-d2, 298 K, ca. 5 mM) spectra of AD, AD*, ADAD, and AD*AD*. Arrows highlight the decrease in the diffusion coefficient due to intermolecular hydrogen bonding interactions involving the phenols in AD and ADAD. In AD*AD*, the two 19F signals are not resolved. | |
2.4 NMR dilution studies
The self-association of ADAD was studied using 1H, 19F and 31P NMR dilutions experiment at 298 K in chloroform-d and in TCE-d2. Dilution resulted in a very small upfield shift of the outer 19F signal and a large downfield shift of the inner 19F signal. Similarly in the 31P experiment, dilution resulted in a very small downfield shift of the outer 31P signal and a large downfield shift of the inner 31P signal. The dilution data for both sets of 19F and 31P signals in both solvents fit well to dimerisation isotherms, and the results are shown in Table 1. The corresponding data for dilution of AD in chloroform are also shown for comparison. The bound chemical shifts are similar in all cases, which indicates that all of the phosphine oxides and all of the phenols form hydrogen bonds in the dimeric state of ADAD. The chemical shifts of the signals due to the inner donor and acceptor residues of ADAD in the monomeric state are practically identical to the chemical shifts of the corresponding signals for AD in the monomeric state. This result shows that the inner residues of ADAD are not involved in hydrogen bonding interactions, as we have previously shown that there are no intramolecular hydrogen bonds in the monomeric state of AD 2-mer. In contrast, the chemical shifts of the signals due to the outer donor and acceptor residues of ADAD in the monomeric state are practically identical to the chemical shifts in the dimeric states of both AD and ADAD. This result shows that the two outer residues of ADAD form a fully bound intramolecular bond in the monomeric state. The association constants for dimerization of AD and ADAD are similar, which suggests that the number of additional hydrogen bonding interactions formed on dimerization is similar for both oligomers. The chemical shift data show that it is only the two inner residues of ADAD that form new hydrogen bonds on dimerization, as the outer residues are already fully bound.
Table 1 Dimerisation constants (K, 95% CI) and limiting NMR chemical shifts from dilution experiments in CDCl3 and TCE-d2 at 298 K
| Oligomer |
Solvent |
K/M−1 |
Residue |
19F NMR δ/ppm |
31P NMR δ/ppm |
| Monomer |
Dimer |
Monomer |
Dimer |
|
AD
|
CDCl3 |
183 ± 23 |
— |
−60.7 |
−62.1 |
38.7 |
41.6 |
|
ADAD
|
CDCl3 |
487 ± 3 |
Inner |
−60.9 |
−62.1 |
38.9 |
42.1 |
| Outer |
−62.0 |
−61.9 |
41.7 |
41.9 |
|
AD
|
TCE-d2 |
211 ± 25 |
— |
−59.2 |
−60.4 |
|
|
|
ADAD
|
TCE-d2 |
340 ± 60 |
Inner |
−59.3 |
−60.3 |
|
|
| Outer |
−60.2 |
−60.2 |
These observations indicate that 1,4-folding in the ADAD 4-mer leads to formation of a stem-loop in the monomeric state, but it not possible to assign the structure of the dimeric state based on these experiments. A schematic representation of different possible species is shown in Fig. 8. The ADAD·ADAD complex could be assembled from two stem-loops by making two intermolecular hydrogen bonds between the inner residues without disruption of the intramolecular interaction between the outer residues. By analogy with nucleic acids, we call this structure kissing stem-loops.32 The other likely structure for the ADAD·ADAD complex is a duplex in which the intramolecular interactions between the two outer residues are broken to give four intermolecular hydrogen bonds.
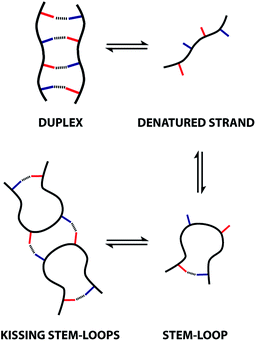 |
| | Fig. 8 Possible structures of the monomeric and dimeric ADAD. | |
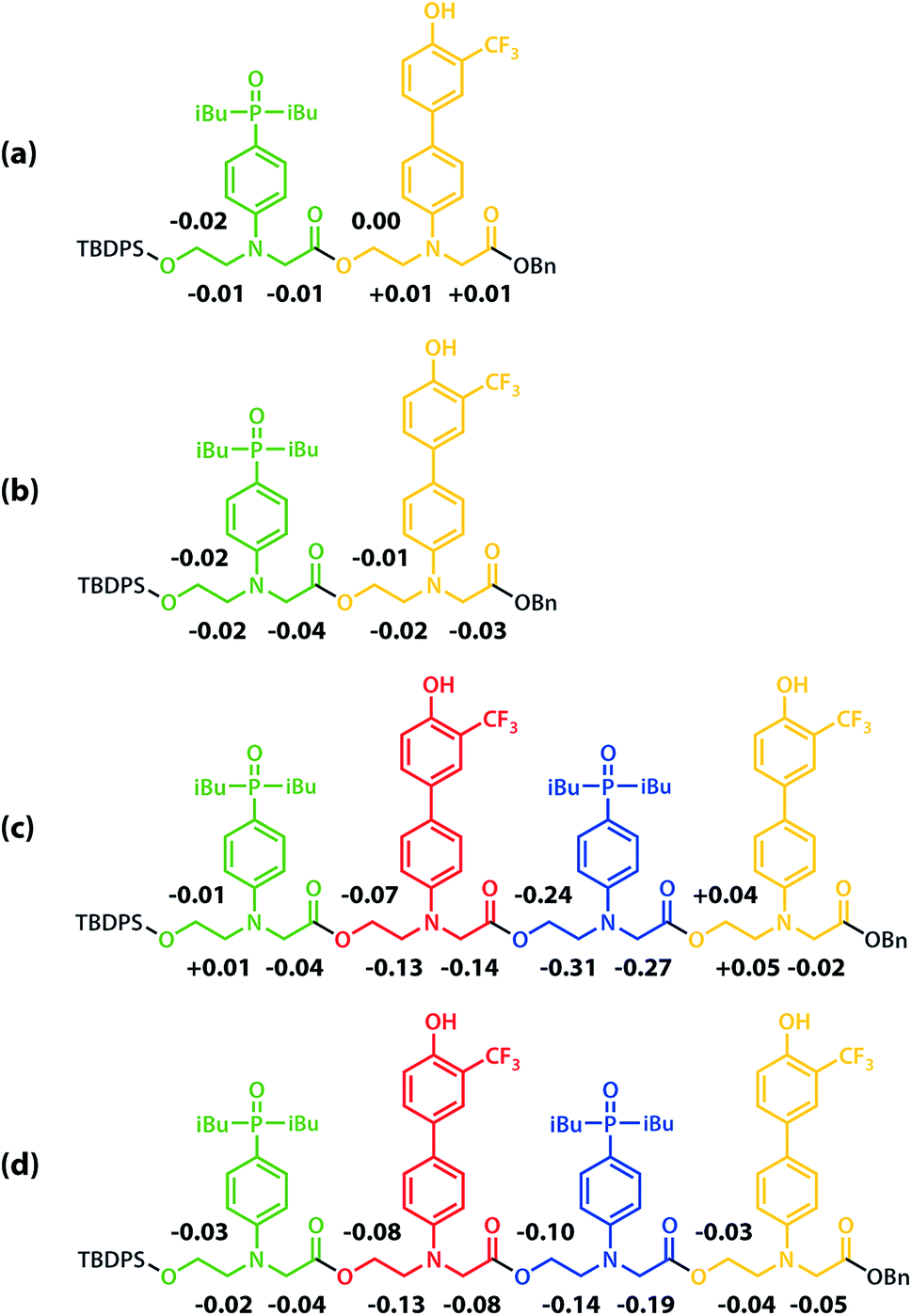
All of the structures in Fig. 8 are in fast exchange on the NMR timescale but the limiting 1H NMR chemical shifts provide some insights into the relative populations and the nature of the dimeric state of ADAD. Fig. 6 highlights the differences observed in the methylene region of 1H NMR spectra of the ADAD 4-mer compared with the AA 2-mer and the DD 2-mer in chloroform solution. Neither AA nor DD undergo self-association or intramolecular folding, because AA contains only hydrogen bond acceptors and DD contains only hydrogen bond donors. The AA and DD spectra therefore serve as reference points representing the chemical shifts of a denatured single strand (see Fig. S9 and S10† for details). 1H NMR dilution experiments were used to determine the limiting chemical shifts of the AD 2-mer and the ADAD 4-mer in the monomeric and dimeric states in chloroform solution. Fig. 9 shows the differences between these limiting chemical shifts and the corresponding reference values measured using AA and DD (Δδ). The AD 2-mer is a denatured strand in the monomeric state, and the Δδ values are all close to zero as expected (Fig. 9(a)). The AD 2-mer is a hydrogen-bonded duplex in the dimeric state, but there are no significant changes in the values of Δδ, which are all close to zero for the dimer (Fig. 9(b)). This result indicates that duplex formation does not cause a significant change in backbone conformation and is not associated with any changes in the backbone 1H NMR chemical shifts. The ADAD 4-mer is a hydrogen-bonded stem-loop in the monomeric state, and the relatively compact structure associated with 1,4-folding leads to large upfield shifts (up to 0.3 ppm) for all of the backbone signals due to the two inner residues (Fig. 9(c)). These large chemical shift differences persist in the dimeric state of ADAD (Fig. 9(d)), which indicates that the kissing stem-loops architecture predominates and that the duplex, which would have Δδ values close to zero, is not significantly populated.
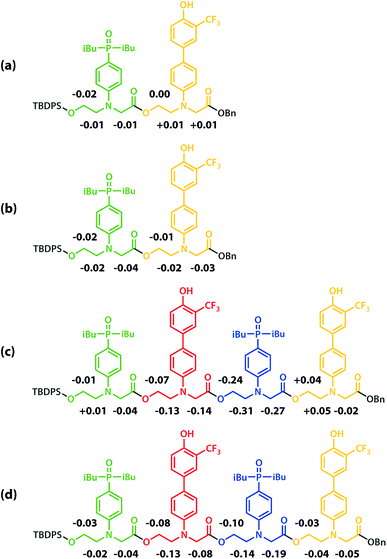 |
| | Fig. 9 Differences in the 1H NMR chemical shifts (Δδ/ppm) of the signals due to the backbone protons of (a) monomeric AD, (b) dimeric AD, (c) monomeric ADAD, and (d) dimeric ADAD compared with the signals due to the corresponding protons in AA or DD in CDCl3 at 298 K. | |
2.4.1 Molecular modelling.
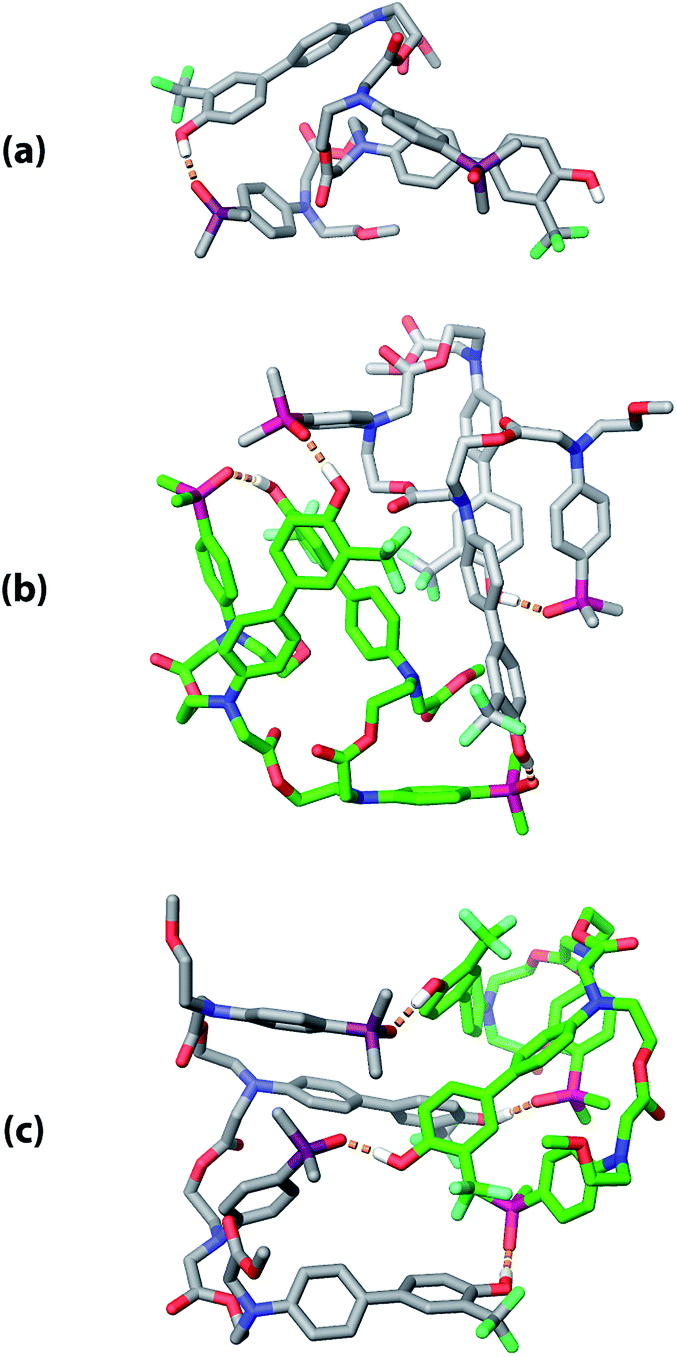
The three supramolecular structures shown in Fig. 4 were investigated using molecular mechanics calculations (see ESI† for further details). For the monomeric state of ADAD, a conformational search gave the 1,4-folded stem-loop with a hydrogen bond between the outer residues as the lowest energy structure, which is consistent with the NMR data. In order to reduce the size of the conformational search space for the dimeric state, constraints were used to make sure that the two inner residues formed the intermolecular hydrogen bonds, which are present in both the duplex and kissing stem-loop structures. Two distinct local minima corresponding to the duplex and kissing stem loops structures were found for dimeric ADAD (Fig. 10). While the calculated energy difference between the two structures (ΔEOPLS3 ≈ 10 kJ mol−1) is small, the calculations are consistent with the NMR data, which suggest predominant formation of the kissing stem-loops structure.
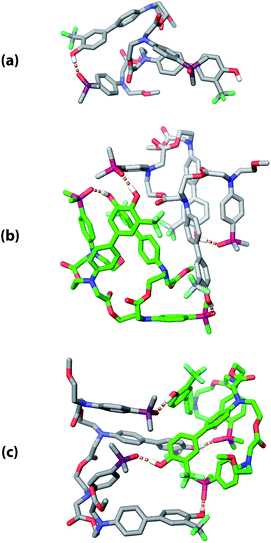 |
| | Fig. 10 Low energy structures obtained from molecular mechanics conformational searches. (a) ADAD stem-loop, (b) ADAD·ADAD kissing stem-loops, and (c) ADAD·ADAD duplex. Protecting groups and alkyl groups on phosphorus were replaced with methyl groups. Carbon is coloured grey or green for the two molecules in the dimeric structures. Hydrogen bonds are shown with orange dashed lines; non-polar hydrogen atoms are not shown for clarity. | |
2.5 NMR melting experiments
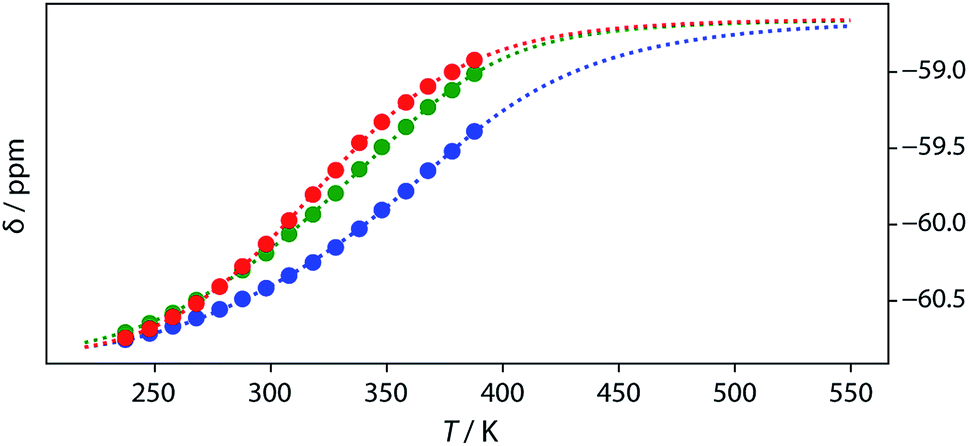
Thermal denaturation of the AD·AD and ADAD·ADAD complexes was studied by measuring the 19F NMR signals at regular temperature intervals between 238 K to 388 K in TCE-d2. Fig. 11 shows the best fit of the two-state melting isochore to the AD variable temperature 19F NMR data. The two-state model explains the behaviour well, giving a melting temperature of the duplex of 321 K and an enthalpy of melting of 39 kJ mol−1. The limiting 19F NMR chemical shifts of the AD 2-mer are in agreement with the corresponding values from the dilution experiments in Table 1 (δmonomer = −58.7 and δdimer = −60.9 ppm). However, a simple two-state melting equilibrium does not explain the behaviour of ADAD (see Fig. S11† for further details). In biochemistry, deviations from the enthalpies calculated using the van't Hoff method imply denaturation of proteins through an intermediate state.33 If we assume that the melting of the ADAD dimer proceeds through a highly populated intermediate and use a three-state equilibrium, it is possible to describe the experimental data well (Fig. 11).
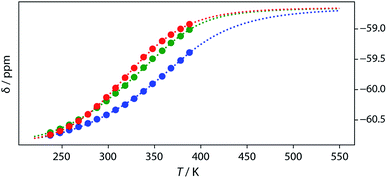 |
| | Fig. 11
19F NMR (500 MHz, TCE-d2, 7.4 mM) melting experiments. The lines show fitting of a two-state equilibrium isochore to the AD data (red) and a three-state isochores to the ADAD data (inner residue green, outer residue blue). | |
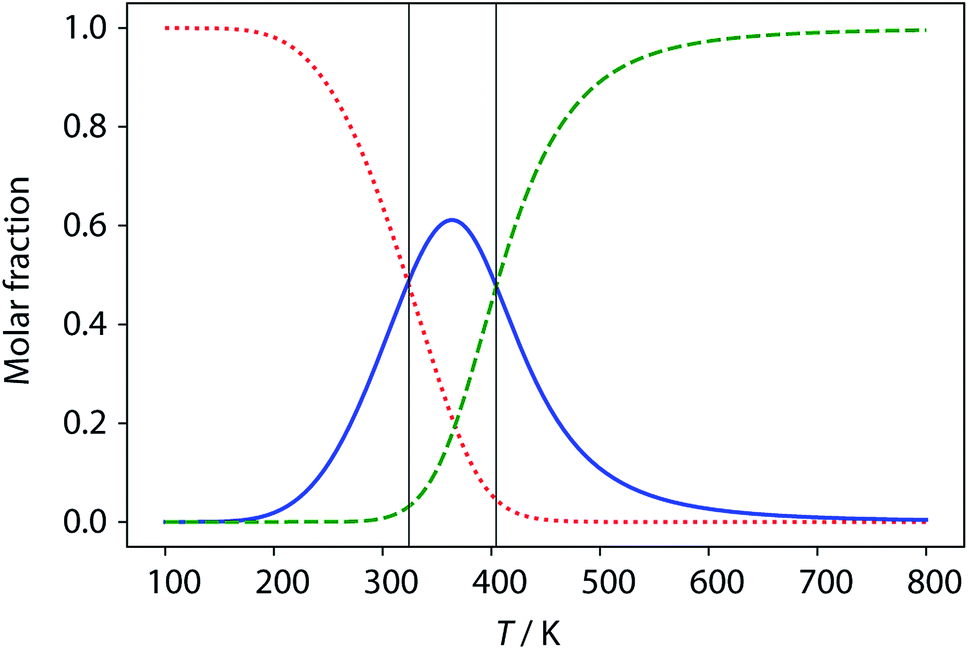
The limiting 19F chemical shifts resulting from fitting the three-state equilibrium model to the 19F NMR data provide information about the nature of the intermediate involved in the melting transition. The chemical shift of the signal due to the inner donor residue in the intermediate species is −58.9 ppm, which is similar to the chemical shift in the denatured single strand state implying that the inner residues are not involved in hydrogen bonding. In contrast, the chemical shift of the signal due to the outer donor residue in the intermediate is −59.6 ppm, indicating substantial hydrogen bonding between the outer residues. These results suggest that the intermediate form of ADAD observed in the melting experiment is the stem-loop, which assembles into kissing stem-loops upon cooling and opens to give the denatured single strand on heating. Calculated populations of the three forms of ADAD as a function of temperature are shown in Fig. 12.
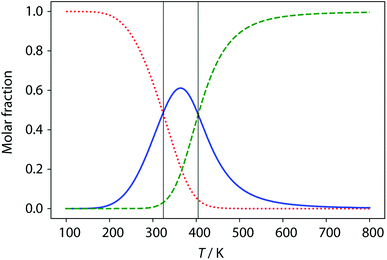 |
| | Fig. 12 Calculated populations of the three forms of ADAD as a function of temperature: kissing stem-loops (red dots), monomeric stem-loop (blue line), and fully denatured single strand (green dashes). Vertical lines represent the calculated transition temperatures (324 K and 404 K). | |
3 Conclusions
We have synthesised an oligoester bearing a sequence of four hydrogen bond recognition sites as side chains. The sequence of donor and acceptor recognition sites causes the ADAD 4-mer to fold via intramolecular hydrogen bonding interactions between the two terminal bases. This 1,4-folding leads to a large chemical shift dispersion of the backbone signals in the 1H NMR spectrum allowing complete sequential assignment and structure determination. The two inner bases in the sequence do not form hydrogen-bonds in the monomeric state, but are available for formation of intermolecular hydrogen-bonds. At higher concentrations or lower temperatures, intermolecular interactions between these inner recognition sites leads to dimerization of the ADAD 4-mer and the formation a kissing stem-loop architecture, which is reminiscent of one of the key structural elements of folded RNA. The equilibria between the denatured single strand, the stem-loop and the kissing stem-loops dimer were characterized in chloroform and TCE solution using a combination of 19F NMR dilution and melting experiments that show there are no other significant species present. The results suggest that longer oligoesters based on this architecture are likely to show interesting folding and self-assembly properties providing a new route to encoding functions, such a recognition or catalysis, in synthetic oligomers based on sequence.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the European Research Council (ERC-2012-AdG 320539-DUPLEX) for funding. L. G. thanks Marie Skłodowska-Curie Actions for Individual Fellowship (H2020-MSCA-IF-2016 number 745730).
References
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171, 737–738 CrossRef CAS PubMed.
-
W. Saenger, Principles of Nucleic Acid Structure, Springer-Verlag, New York, 1984 Search PubMed.
- M. L. Sipski and T. E. Wagner, Biopolymers, 1977, 16, 573–582 CrossRef CAS PubMed.
- R. D. Kornberg and Y. Lorch, Cell, 1999, 98, 285–294 CrossRef CAS PubMed.
- S. Lindsay, Biophys. J., 2007, 92, 1113 CrossRef CAS PubMed.
- A. Mujeeb, J. Clever, T. Billeci, T. James and T. Parslow, Nat. Struct. Mol. Biol., 1998, 5, 432–436 CrossRef CAS.
- J. Zhang, M. W. Lau and A. R. Ferre-D'Amare, Biochemistry, 2010, 49, 9123–9131 CrossRef CAS PubMed.
- W. A. Cantara, P. F. Crain, J. Rozenski, J. A. McCloskey, K. A. Harris, X. Zhang, F. A. P. Vendeix, D. Fabris and P. F. Agris, Nucleic Acids Res., 2011, 39, D195–D201 CrossRef CAS PubMed.
- C. Zhan, J. M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 4625–4628 CrossRef CAS PubMed.
- Q. Gan, Y. Ferrand, C. Bao, B. Kauffmann, A. Grélard, H. Jiang and I. Huc, Science, 2011, 331, 1172–1175 CrossRef CAS PubMed.
- M. L. Singleton, G. Pirotte, B. Kauffmann, Y. Ferrand and I. Huc, Angew. Chem., Int. Ed., 2014, 53, 13140–13144 CrossRef CAS PubMed.
- I. Huc, Eur. J. Org. Chem., 2004, 2004, 17–29 CrossRef.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016, 7, 94–101 RSC.
- A. E. Stross, G. Iadevaia and C. A. Hunter, Chem. Sci., 2016, 7, 5686–5691 RSC.
- G. Iadevaia, A. E. Stross, A. Neumann and C. A. Hunter, Chem. Sci., 2016, 7, 1760–1767 RSC.
- D. Núñez-Villanueva and C. A. Hunter, Chem. Sci., 2017, 8, 206–213 RSC.
- D. Núñez-Villanueva, G. Iadevaia, A. E. Stross, M. A. Jinks, J. A. Swain and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 6654–6662 CrossRef PubMed.
- A. E. Stross, G. Iadevaia, D. Núñez-Villanueva and C. A. Hunter, J. Am. Chem. Soc., 2017, 139, 12655–12663 CrossRef CAS PubMed.
- J. A. Swain, G. Iadevaia and C. A. Hunter, J. Am. Chem. Soc., 2018, 140, 11526–11536 CrossRef CAS PubMed.
- G. Iadevaia, D. Núñez-Villanueva, A. E. Stross and C. A. Hunter, Org. Biomol. Chem., 2018, 4183–4190 RSC.
- F. T. Szczypiński and C. A. Hunter, Chem. Sci., 2019, 10, 2444–2451 RSC.
- C. A. Hunter, Angew. Chem., Int. Ed., 2004, 43, 5310–5324 CrossRef CAS PubMed.
- B. Huang and M. E. Hermes, J. Polym. Sci., Part A: Polym. Chem., 1995, 33, 1419–1429 CrossRef CAS.
- U. D. Lengweiler, M. G. Fritz and D. Seebach, Helv. Chim. Acta, 1996, 79, 670–701 CrossRef CAS.
- C. M. Krell and D. Seebach, Eur. J. Org. Chem., 2000, 2000, 1207–1218 CrossRef.
- J. B. Williams, T. M. Chapman and D. M. Hercules, Macromolecules, 2003, 36, 3898–3908 CrossRef CAS.
- R. M. Weiss, E. M. Jones, D. E. Shafer, R. M. Stayshich and T. Y. Meyer, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1847–1855 CrossRef CAS.
- J. Li, R. M. Stayshich and T. Y. Meyer, J. Am. Chem. Soc., 2011, 133, 6910–6913 CrossRef CAS PubMed.
- R. M. Stayshich and T. Y. Meyer, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 4704–4711 CrossRef CAS.
- M. A. Washington, D. J. Swiner, K. R. Bell, M. V. Fedorchak, S. R. Little and T. Y. Meyer, Biomaterials, 2017, 117, 66–76 CrossRef CAS PubMed.
-
T. D. W. Claridge, High-Resolution NMR Techniques in Organic Chemistry, Elsevier, 2009, vol. 27, pp. 303–334 Search PubMed.
- J. Marino, R. Gregorian, G. Csankovszki and D. Crothers, Science, 1995, 268, 1448–1454 CrossRef CAS PubMed.
- Y. Zhou, C. K. Hall and M. Karplus, Protein Sci., 1999, 8, 1064–1074 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures with spectroscopic characterization data, 19F NMR titration spectra, binding isotherms, dimerisation isochores, limiting chemical shifts for free and bound states. See DOI: 10.1039/c9sc01669d |
| ‡ Current address: Department of Chemistry, Imperial College London, Molecular Sciences Research Hub, White City Campus, London W12 0BZ, UK. |
|
| This journal is © The Royal Society of Chemistry 2019 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Luca
Gabrielli
and
Christopher A.
Hunter
,
Luca
Gabrielli
and
Christopher A.
Hunter