
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of glycopolymers with specificity for bacterial strains via bacteria-guided polymerization†
Yan
Luo
ab,
Yan
Gu
ab,
Ruyan
Feng
ab,
John
Brash
ac,
Ahmed M.
Eissa
 d,
David M.
Haddleton
*d,
Gaojian
Chen
*ab and
Hong
Chen
*a
d,
David M.
Haddleton
*d,
Gaojian
Chen
*ab and
Hong
Chen
*a
aThe Key Lab of Health Chemistry and Molecular Diagnosis of Suzhou, College of Chemistry, Chemical Engineering and Materials Science, Soochow University, 199 Ren-Ai Road, Suzhou, 215123, P. R. China. E-mail: chenh@suda.edu.cn
bCenter for Soft Condensed Matter Physics and Interdisciplinary Research & School of Physical Science and Technology, Soochow University, Suzhou, 215006, P. R. China. E-mail: gchen@suda.edu.cn
cSchool of Biomedical Engineering and Department of Chemical Engineering, McMaster University, Hamilton, Ontario L8S4L7, Canada. E-mail: brashjl@mcmaster.ca
dDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, CV4 7AL, UK. E-mail: d.m.haddleton@warwick.ac.uk
First published on 18th April 2019
Abstract
Identifying probiotics and pathogens is of great interest to the health of the human body. It is critical to develop microbiota-targeted therapies to have high specificity including strain specificity. In this study, we have utilized E. coli MG1655 bacteria as living templates to synthesize glycopolymers in situ with high selectivity. By this bacteria-sugar monomer-aptation-polymerization (BS-MAP) method, we have obtained glycopolymers from the surface of bacteria which can recognize template bacteria from two strains of E. coli and the specific bacteria-binding ability of glycopolymers was confirmed by both bacterial aggregation experiment and QCM-D measurements. Furthermore, the synthesized glycopolymers have shown a powerful inhibitory ability which can prevent bacteria from harming cells in both anti-infection and co-culture tests.
Introduction
Different types of bacteria populate the microbiota of the host in which they live. Probiotic bacteria protect the host from the external environment, while pathogenic bacteria may cause infectious diseases. Antibiotics treat infectious diseases by killing pathogens,1 however they may also kill non-pathogens thereby changing the composition of the microbiota and increasing susceptibility to secondary infections. Some non-lethal microbes may even become lethal.2 The bacterial strain is as important as the bacterial type because minor mutations may transform probiotic bacteria into pathogenic ones of the same type.3 Therefore, it is critical to develop microbiota-targeted therapies in which their agents have high specificity including strain specificity.4 Amongst the different selective reagents,5 sugars play an important role in biological processes involving recognition and signalling.6 Some sugars have high affinity for proteins, making them suitable for applications where such specificity is necessary.7 For example, lectins, sugar-binding cell surface receptor proteins,8 and a variety of different sugars have been utilized to recognize bacteria.9 Synthetic glycopolymers have been shown to be strongly selective based on specific binding between the sugar units and proteins.10 However, more work is still needed to obtain glycopolymers that can identify specific bacteria down to the level of the strain, which has not yet been reported to the best of our knowledge. Herein, we report the use of bacteria as “living” templates to synthesize glycopolymers in situ such that strain-specific affinity is realized. By choosing appropriate sugar monomer and polymerization method, we present a convenient approach to prepare glycopolymers with high specificity based on the optimized permutation of sugars using target bacteria as a live template. We refer to this approach as bacteria-sugar monomer-aptation-polymerization (BS-MAP).E. coli is a convenient system whose various genotypes can be easily found,11 therefore for this work, we chose E. coli as a model bacterium. 2-(Methacrylamido)glucopyranose (MAG) and 2-(N-3-m,sulfopropyl-N,N-dimethyl ammonium) ethyl methacrylate (MEDSA) were chosen as monomers for the synthesis of the glycopolymers. MAG is a sugar-containing monomer chosen for its binding ability and MEDSA is a non-sugar, non-binding monomer which can act as a spacer in the formed polymeric chain. The reducing properties of E. coli facilitate the activator regenerated electron transfer atom transfer radical polymerization (ARGET ATRP) of these monomers.12 Two polymers were obtained: one was formed in the solution phase (SP) and the other on the surface of the bacteria (BP) which was then released and collected from the bacterial surface by addition of mannose (1 mg mL−1, PBS), a strong competing binding agent to bacterial surfaces,13Scheme 1. The non-templated polymer (NP) was also prepared by living radical polymerization in the absence of bacteria for comparison.
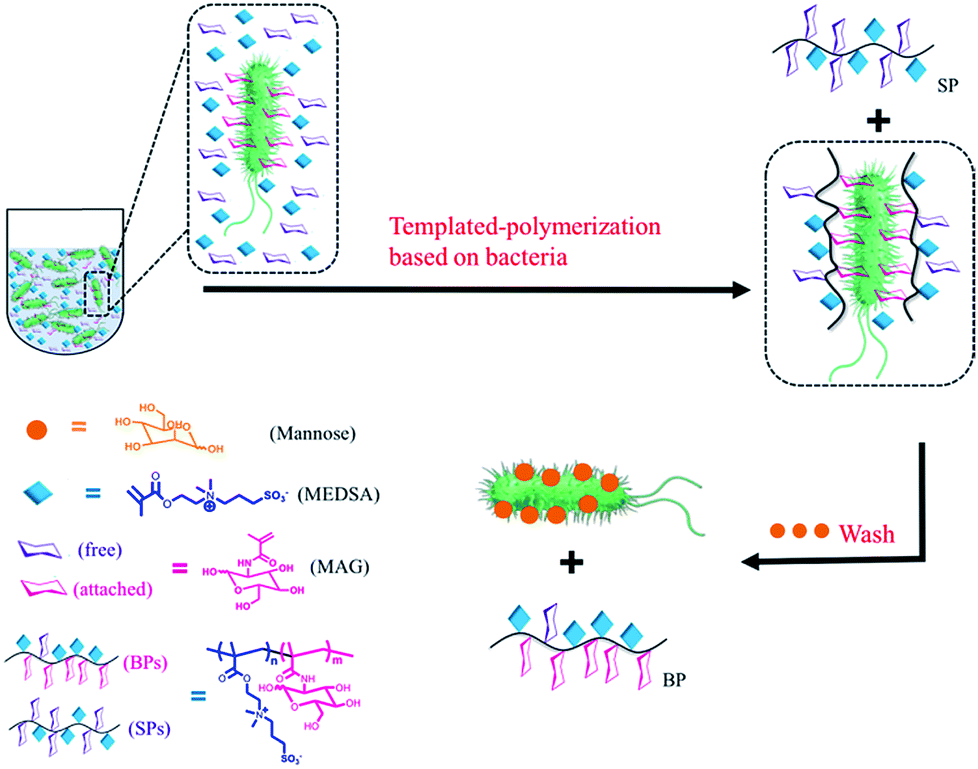 | ||
| Scheme 1 Scheme of the polymerization in the presence of bacteria: two different polymers were obtained: (1) in solution (SP), (2) on the surface of bacteria (BP). | ||
Results and discussion
The targeted molecular weights of all polymers were about 8 kDa, and Đ < 1.56. The polymers obtained from solution in the presence (SP) or absence (NP) of bacteria had similar sugar contents of about 30%, while the polymer washed from the surfaces of the bacteria (BP) had a sugar content as high as 50%. It has been previously established that the participation of bacteria in the polymerization can change the ratio of monomers in the copolymer.12 In our system, we believe that such change in monomer ratio is attributed to the preferential binding affinity of bacteria to sugar moieties, leading to greater incorporation of the MAG monomer in the polymer formed at the surface.We first used a bacterial aggregation experiment14 to investigate whether BP can bind templates specifically as expected (Fig. 1A). Besides the above three copolymers, homopolymers of MAG and MEDSA (pMAG, pMEDSA) were synthesized as controls (Table 1). To test the specificity of polymers, E. coli DH5α was chosen as a non-template bacteria, derived from the template bacteria E. coli MG1655 with slightly changed genome.11 As seen in Fig. 1A, the BP templated on E. coli MG1655, among all the polymers, had the greatest ability to cluster MG1655. pMEDSA caused very little MG1655 aggregation probably due to its antifouling properties as a zwitterion (Fig. S11†);15 pMAG caused some clustering of MG1655 but much less than BP, indicating that a higher content of sugar does not necessarily produce greater affinity. NP and the SP obtained from solution in the template reaction caused a little MG1655 clustering but less than either BP or the homopolymer pMAG. Compared to template bacteria, the ability of the polymers to cluster non-template bacteria DH5α did not change significantly except in the case of BP. The quantities of DH5α clusters in the solutions of BP decreased considerably and were even lower than those caused by NP. These data suggest that BP obtained from the surface of template bacteria possess strain-specific affinity.
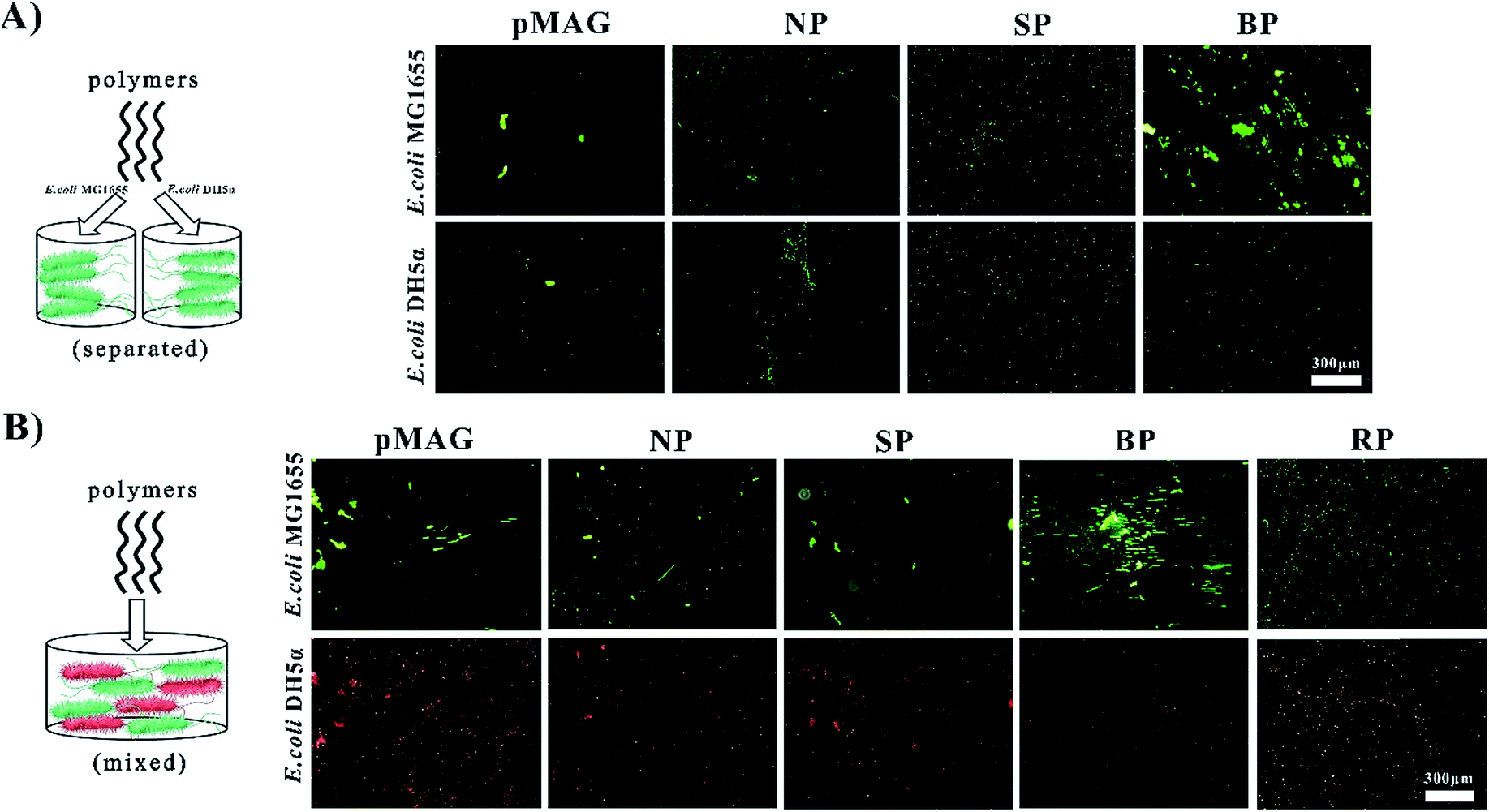 | ||
| Fig. 1 Clustering of bacteria by different polymers. (A) Aggregation of template bacteria E. coli MG1655 and non-template bacteria E. coli DH5α caused by different polymers (NP, SP, RP, BP), Both the strains were stained by STYO9 (green). (B) Separated and aggregation states in mixtures of template and non-template bacteria caused by the different polymers (NP, SP, RP, BP). The template bacteria were stained by STYO9 (green) and the non-template bacteria were modified with a target gene expressing red fluorescent protein. | ||
| Samples | pMAG | pMEDSA | NP | RP | SP | BP | SP2 | BP2 |
|---|---|---|---|---|---|---|---|---|
MAG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MEDSA (NMR) :MEDSA (NMR) |
1.00:0 |
0:1.00 |
0.55:1 |
0.98:1 |
0.51:1 |
0.91:1 |
0.02:1 |
1.00:1 |
| M n (g mol−1) | 8200 | 8700 | 9700 | 7000 | 8000 | 12800 |
8100 | 7600 |
| Đ | 1.15 | 1.16 | 1.35 | 1.13 | 1.56 | 1.40 | 1.14 | 1.20 |
As shown above, BP exhibited strong affinity and selectivity when interacting with bacteria alone. But in the “real world” involving more complex systems, it is desirable that the BP be able to recognize targets from flora bacteria of two or more types. With this in mind we investigated the ability of BP to identify template bacteria in mixtures of template and non-template bacteria. Non-template bacteria DH5α were modified with a target gene expressing red fluorescent protein to be distinguished from template bacteria which were stained by STYO9 (green). In addition to the control copolymers NP and SP, we synthesized a copolymer (RP) with virtually the same composition as BP to investigate whether a specific sugar content was required for specific affinity. In water solution without polymer, the bacteria did not aggregate (Fig. S13†) while in solutions of NP and SP, a few clusters showing both red and green fluorescence were observed (Fig. 1B), indicating that the interactions between the polymers and bacteria were weak and non-specific. RP also gave a few clusters of both bacteria. In contrast, in the solution of BP, large green clusters but no red clusters were observed, providing further confirmation that BP possesses specific affinity for green template bacteria MG1655 in mixtures with red non-template bacteria. These data for RP suggest that the selective binding property of BP does not require a specific monomer composition. It is, however, possible that the monomer sequence plays an important role in the strain-specific affinity. Thus in the case of the copolymer (BP), the monomer sequence may match the positioning of the receptor proteins so that the distance between adjacent MAG residues is the same as that between binding sites on the surface of the bacteria. We refer to this phenomenon as bacteria-sugar monomer-aptation-polymerization (BS-MAP).
In order to verify the process of template polymerization, we incubated the bacteria with the sugar-containing monomer MAG, and then re-suspended them in MEDSA so that there was no free MAG in the solution. After polymerization, two kinds of polymers were obtained as expected: one in solution (SP2) and the other on the surface of the bacteria (BP2). The sugar contents of these two polymers were calculated from 1H-NMR spectra. The data confirmed our hypothesis: that SP2 had only a few MAG moieties while BP2 had MAG content similar to that of BP. We believe that the process of BS-MAP has brought some sort of chain sequencing encoded with the information on bacterial surface. 13C-NMR spectroscopy was then used to explore the monomer sequence. Interestingly, some differences were observed in the 13C-NMR spectra of the produced polymers. In the region of –CH2– carbon (58–61 ppm), we noted that all spectra of copolymers show two broad peaks around 60.5 to 60.8 ppm except that of the copolymer BP shows two distinct peaks instead, assigned for carbons of two chemical environments of –CH2–O– on the two monomeric units of MAG and MEDSA (Fig. 2). This reveals the different chain sequence of synthesized glycopolymers which also explains the differences in specificity and binding ability. However, it is not possible to determine the total monomer sequence on account of the structural features of the BPs due to limitations of current analytical methods.
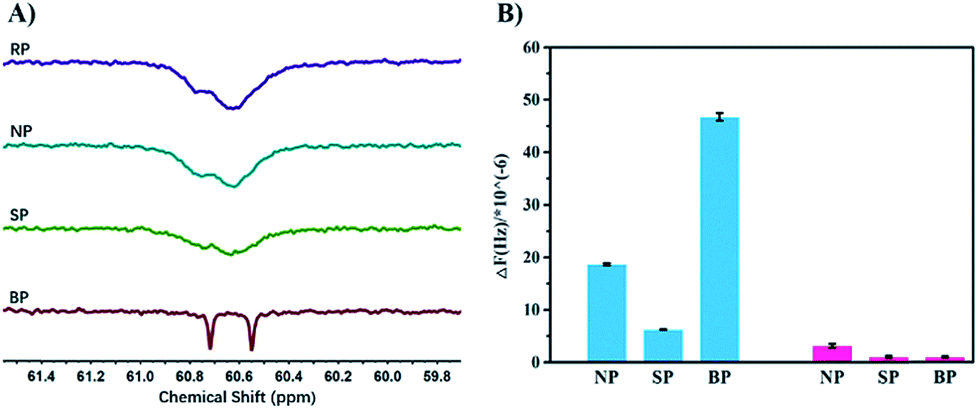 | ||
| Fig. 2 13C-NMR characterization of different copolymers and evaluation of bacteria-binding ability of glycopolymers from QCM-D measurements. (A) The typically different peaks of copolymers (RP, NP, SP, BP). (B) The average value of frequency change following the interaction between single bacterium and three polymers (NP, SP, BP). | ||
Quartz Crystal Microbalance with Dissipation monitoring (QCM-D) was used to further quantify the bacteria-binding ability of the different polymers. For the template bacteria, E. coli MG1655, BP showed the highest adsorption of all the polymers (Fig. S14A†), in agreement with the bacterial aggregation experiments. The adsorption of NP and SP was much lower, and NP showed higher adsorption than SP, possibly because as a random copolymer, it may contain a few short sugar sequences that match the binding sites of MG1655. SP, conversely, formed in solution and having low affinity, may be described as a polymer that is “less-liked” by the template bacteria. For the non-template bacteria, DH5α, the frequency changes caused by NP and SP were similar to those seen with the template bacteria (Fig. S14B†). However, the response to BP was quite different. A more rapid decrease of frequency was followed by a rapid increase upon injection of water, indicating that the interaction between BP and non-template bacteria is weak. This may seem surprising given that the two bacteria are highly similar and have the same major receptor proteins. However, the chain sequence in BP, matching the location of the receptor proteins in the template bacteria, presumably resulted in BP binding non-specifically and weakly to the non-template bacteria so that it could be washed off easily by water. The specificity of BP is more apparent by comparing the final frequency change which was about 47-fold greater for template bacteria than for non-template bacteria (Fig. S15†).
To obtain more detailed information on how the polymers interact with the template bacteria, they were attached to gold nano-particles (GNPs). The GNPs (diameter 13 nm by TEM, Fig. S18†) were added to a suspension of MG1655 and observed by TEM. It was found that SP-modified GNPs were distributed randomly around the bacteria, and only a few were adhered. For the BP-modified GNPs, in contrast, large quantities appeared near the surface of the bacteria indicating specific affinity between BP and MG1655 (Fig. 3).
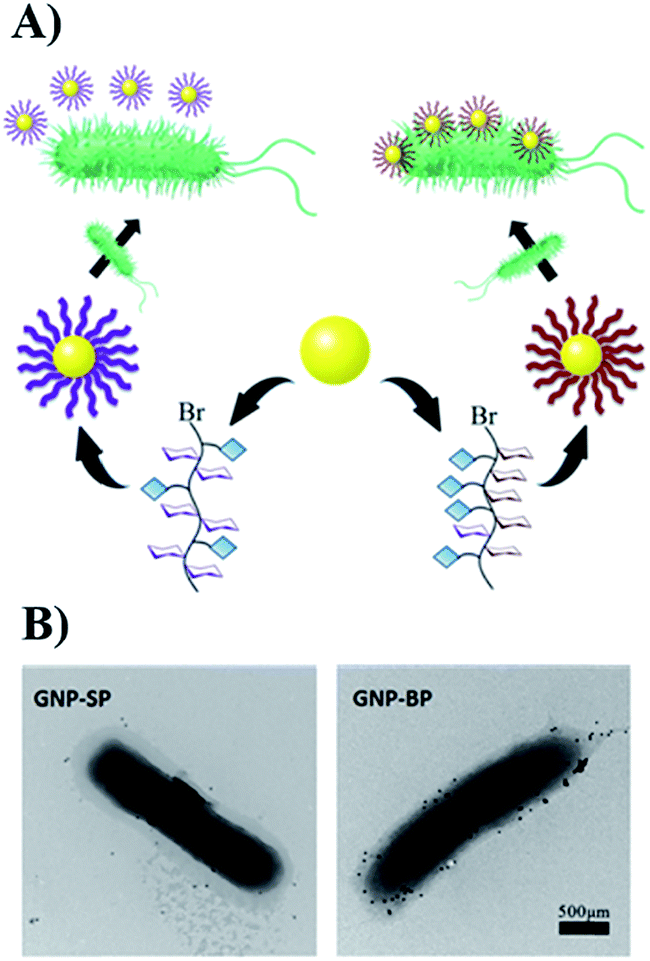 | ||
| Fig. 3 (A) The scheme of modification of GNPs with glycopolymers. (B) The interactions between modified-GNPs and template bacteria. | ||
Since this data shows that BP binds to the bacteria specifically, we carried out a preliminary test of its potential application as an anti-infection agent. BP is expected to occupy the binding sites of the bacteria, thereby preventing or weakening the interactions of the bacteria with cells.16 We used endothelial cells (ECs) as a model in these experiments. Two shapes of ECs are recognized: the spindle-shaped ECs which are associated with healthy functioning cells and the round-shaped ECs which are associated with poor activity (Fig. 4A). Upon exposure to bacteria, less than 20% of the cells remained active. Inclusion of SP and NP improved viability to about 50%, and with BP present, almost all of the cells were spindle-shaped, and presumably active, Fig. 4B. It is assumed that the glycopolymers prevented the bacteria from harming the cells, and that BP, with the highest affinity to MG1655, was therefore the most effective anti-bacterial agent.
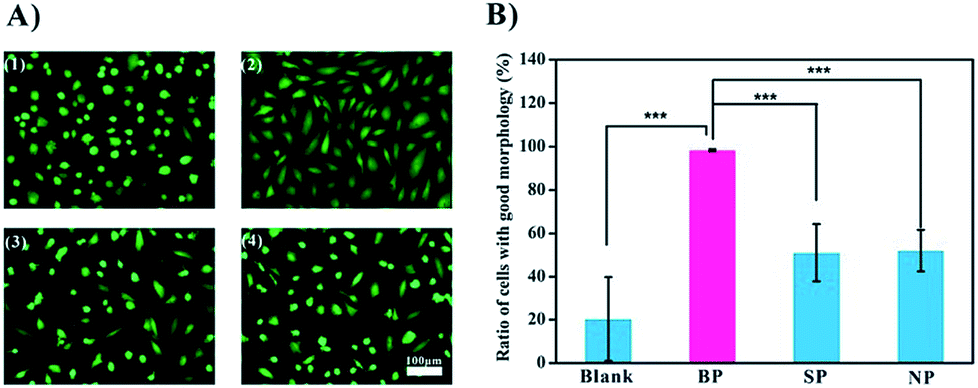 | ||
| Fig. 4 Inhibitory effect of glycopolymers in the anti-infection experiment. (A) Images of ECs after incubation with bacteria and different polymers for 12 h: (1) without polymers, (2) with addition of BP, (3) with addition of SP, (4) with addition of NP. (B) Percentage of spread ECs after exposure to different polymers. | ||
BP has been shown to have promise as an anti-infection agent against mature cells on the surface. A different situation occurs when bacteria are introduced in the early stages of adhesion, i.e. the growth of bacteria and cells on the surface of the implant is competitive.17 Co-culture experiments were carried out to test the possible beneficial effects of glycopolymer BP in this situation. Gold-coated slides were immersed in a mixed culture of MG1655 and ECs for 24 h at 37 °C. The slides were then treated with DAPI to stain the cell nuclei blue, and with FITC to stain the F-actin green. As seen in Fig. 5A, ECs were not competitive with MG1655, as there were no cells on the slides, only a few green dots which are probably cell debris. This result indicates that the bacteria hindered EC growth and even caused cell death. In the presence of BP, the cells were spindle-shaped and appeared healthier. The density of cells in the BP group was 8-fold greater than in the group without BP (Fig. 5B), suggesting that BP facilitates the adhesion and shape change of ECs in the presence of bacteria on gold surface. It should be noted that BP may not prevent bacterial colonization or eliminate negative effects on the cells completely. However, it seems likely that anti-bacterial performance can be greatly enhanced by introducing bactericidal groups like Ag+ or quarternary ammonium groups in other applications.
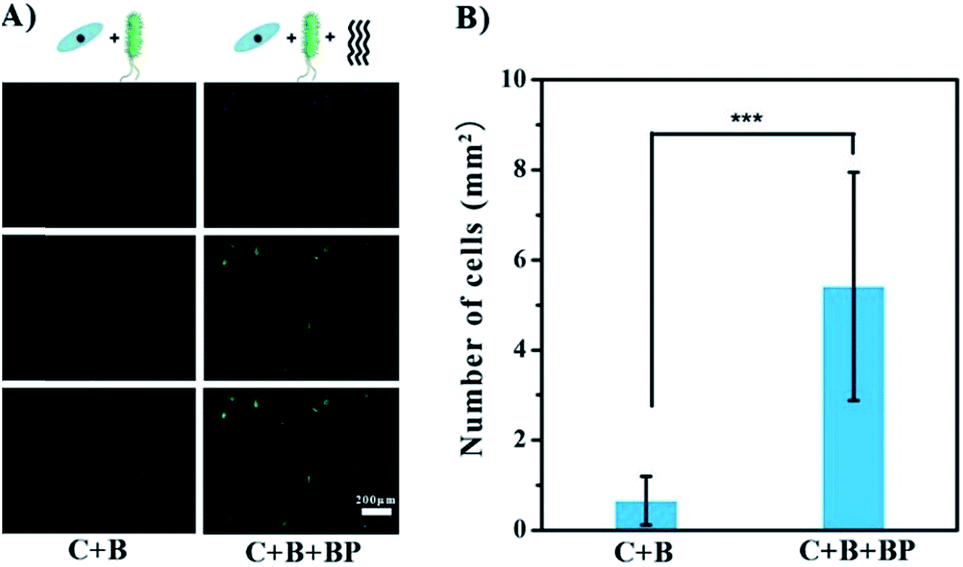 | ||
| Fig. 5 Inhibitory effect of BP in a co-culture experiment on gold surface. EC nuclei were stained with DAPI (blue) and F-actin were stained with FITC (green). (A) Surfaces without (C + B, left) and with (C + B + BP, right) glycopolymer BP. (B) Density of ECs on gold without (C + B) and with (C + B + BP) glycopolymer BP. | ||
Experimental section
Materials
Methacryloyl chloride (stabilized with hydroquinone monomethyl ether, 80%), tris(2-pyridylmethyl)amine (98%) and D-(+)-glucosamine hydrochloride (98%) were purchased from TCI. 2-(N-3-sulfopropyl-N,N-dimethyl ammonium)ethyl methacrylate (MEDSA, 97%), 2-hydroxyethyl 2-bromoisobutyrate (HEBIB, 95%), 2,2′-azoisobutyronitrile (AIBN, 98%), 4-(4-cyanopentanoic acid)dithiobenzoate (CTA, 98%) were purchased from Aldrich. Copper bromide (AR), potassium carbonate (AR), methanol (AR), and dichloromethane (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China). Nitrogen gas was of high purity grade. Dialysis membranes (1000 and 3500) were obtained from Shanghai Yuanye Bio-Technology Co., Ltd. LIVE/DEAD BacLight Bacterial Viability Kits were purchased from Invitrogen. β-Cysteamine (C2H7NS, 95%), trypsin, AM-calcein, 6-diamidino-2-phenylindole (DAPI), Actin-Tracker Green (Phalloidin-FITC) and the animal cell culture media were all purchased from Sigma-Aldrich Chemical Company. The bacteria growth media (LB media) were bought from Oxoid. E. coli MG1655 and E. coli DH5α were provided by the China General Microbiological Culture Collection Center (Beijing, China) and the red E. coli DH5α were given by Dr Ruiqing Yan from Institute of Biophysics, Chinese Academy of Sciences as a gift. Human umbilical vein endothelial cells (HUVECs) were supplied by ScienCell Research Laboratories (CA, USA). All aqueous solutions were prepared in 18.2 MΩ cm purified water from a Milli-Q water purification system (Millipore, Bedford, MA, USA).Instrumentation
1H-NMR spectra were measured with an Agilent 400 MHz NMR equipment using D2O as the solvent. 13C-NMR spectra were recorded on a Bruker Avance III HD 500 MHz (HD500) spectrometer with D2O as the solvent. Molecular weights and molecular weight distributions were obtained by a Waters 1515 gel permeation chromatography (GPC) system equipped with a PL aquagel-OHMIXED-M column (polymers were dissolved in the mobile phase consisted 0.2 M NaNO3 and 0.01 M NaH2PO4 (adjusted to pH = 9). The flow rate was 1.0 mL min−1. Calibration was with PEG standards). The fluorescent images of bacteria and cells were captured with an Inverted fluorescence Microscope (BX51, Olympus), Quartz crystal microbalance-dissipation (QCM-D) measurements were carried out by a Q-Sense-E4 instrument (Q-Sense, Sweden) with control software (Resonant Probes GmbH, Goslar, Germany). A transmission electron microscopy (TEM, Tecnai-G20, 120 kV, FEI) was used in this work. The particle size and zeta potential of GNPs were measured using a zetasizer nano ZS instrument (Malvern Inst. Ltd. Malvern, UK).Synthetic procedures
:4) and white solid (2.8 g) was obtained by rotary evaporation.
Bacterial aggregation experiment
:2. After incubation in 37 °C for 2 h, mixture (2 μL) of polymers and bacteria were dropped on a glass slide covered with glass slice. An optical microscope was used to examine the green clusters.
:1). Polymers were dissolved in water (1 mg mL−1) and then mixed with the suspension of two strains of E. coli (1:2). After incubation in 37 °C for 2 h, mixture (2 μL) of polymers and bacteria were dropped on a glass slide covered with glass slice. An optical microscope was used to examine the green and red clusters.
Quartz crystal microbalance with dissipation (QCM-D) measurements
The QCM chips were immersed separately in the suspension of two strains of bacteria (OD600 = 0.05) in 37 °C for 3 h, after which, the chips were washed by sterile water for three times. Then the chips were put into chambers and pumped with water at a flow rate of 50 μL min−1 until the base line keep stable. Polymers with water (1 mg mL−1) were injected at a speed of 10 μL min−1. 60 min later, the water was pumped again until no changes in the frequency detected by QCM-D to make sure that there were still bacteria after washing by water and calculate the frequency caused by per bacterium, the chips were treated with LIVE/DEAD BacLight kit and then imaged to get the number of bacteria on the chips.Interaction of bacteria and golden nano-particles (GNPs) decorated with polymers
000 rpm for 10 min). The GNPs were re-suspended with 500 μL water, 50 μL K2CO3 solution (10 mg mL−1) and 50 μL SP or BP solution (1 mg mL−1) to start the reaction, shaking at 55 °C for 18 h. The GNP-SP and GNP-BP were characterized by Transmission Electron Microscopy (TEM) and dynamic light scattering (DLS).
Conclusions
We have synthesized glycopolymers with high specificity for binding to bacteria using the bacteria as living templates via bacteria-sugar monomer-aptation-polymerization (BS-MAP). Specificity for bacteria strains was demonstrated via bacterial aggregation and QCM-D measurements. It was shown that the sugar content and ratio of MAG and MEDSA in the glycopolymers were not the main factors contributing to the specificity. Glycopolymers with optimized sugar content and chain sequence could be conveniently obtained by BS-MAP. BPs as potential inhibitors of bacterial adhesion and promoters of endothelial cell attachment and proliferation on gold substrate were tested in co-culture experiments both in the initial and later periods of culture. The effects of the glycopolymers were greater during the later period when the cells were growing well. This approach provides a convenient and general strategy for the preparation of synthetic glycopolymers with high specificity for bacteria used as templates in the synthesis. It is expected that this technique will provide the basis for many future applications such as cell labeling and capturing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by National Natural Science Foundation of China (21774084, 21674074). We thank Yanfei Luan and Ting Wei for the QCM-D measurements and Changming Hu for the TEM measurements.Notes and references
- T. J. Little, J. E. Allen, S. A. Babayan, K. R. Matthews and N. Colegrave, Nat. Med., 2012, 18, 217–220 CrossRef CAS PubMed.
- (a) M. Mondhe, A. Chessher, S. Goh, L. Good and J. E. M. Stach, PLoS One, 2014, 9, e89082 CrossRef PubMed; (b) M. Blaser, Nature, 2011, 476, 393–394 CrossRef CAS PubMed.
- M. Hartmann and T. K. Lindhorst, Eur. J. Org. Chem., 2011, 2011, 3583–3609 CrossRef CAS.
- K. P. Lemon, G. C. Armitage, D. A. Relman and M. A. Fischbach, Sci. Transl. Med., 2012, 4, 137rv5 Search PubMed.
- V. Templier, A. Roux, Y. Roupioz and T. Livache, TrAC, Trends Anal. Chem., 2016, 79, 71–79 CrossRef CAS.
- (a) F. Rolland, E. Baena-Gonzalez and J. Sheen, in Annual Review of Plant Biology, Annual Reviews, Palo Alto, 2006, vol. 57, pp. 675–709 Search PubMed; (b) C. R. Bertozzi and L. L. Kiessling, Science, 2001, 291, 2357–2364 CrossRef CAS PubMed.
- (a) L. L. Kiessling and J. C. Grim, Chem. Soc. Rev., 2013, 42, 4476–4491 RSC; (b) S. Goetze, N. Azzouz, Y.-H. Tsai, U. Gross, A. Reinhardt, C. Anish, P. H. Seeberger and D. V. Silva, Angew. Chem., Int. Ed., 2014, 53, 13701–13705 CrossRef CAS PubMed; (c) X. P. He, Y. L. Zeng, X. Y. Tang, N. Li, D. M. Zhou, G. R. Chen and H. Tian, Angew. Chem., Int. Ed., 2016, 55, 13995–13999 CrossRef CAS PubMed; (d) L. Xue, Z. Lv, Y. Luan, X. Xiong, J. Pan, G. Chen and H. Chen, Polym. Chem., 2015, 6, 3708–3715 RSC.
- H. Lis and N. Sharon, Chem. Rev., 1998, 98, 637–674 CrossRef CAS PubMed.
- N. Sharon, FEBS Lett., 1987, 217, 145–157 CrossRef CAS PubMed.
- (a) S. R. S. Ting, G. Chen and M. H. Stenzel, Polym. Chem., 2010, 1, 1392–1412 RSC; (b) Y. Miura, Y. Hoshino and H. Seto, Chem. Rev., 2016, 116, 1673–1692 CrossRef CAS PubMed; (c) C. R. Becer, Macromol. Rapid Commun., 2012, 33, 742–752 CrossRef CAS PubMed; (d) V. Ladmiral, E. Melia and D. M. Haddleton, Eur. Polym. J., 2004, 40, 431–449 CrossRef CAS.
- J. M. Monk, A. Koza, M. A. Campodonico, D. Machado, J. M. Seoane, B. O. Palsson, M. J. Herrgard and A. M. Feist, Cell Syst., 2016, 3, 238–251 CrossRef CAS PubMed.
- E. P. Magennis, F. Fernandez-Trillo, C. Sui, S. G. Spain, D. J. Bradshaw, D. Churchley, G. Mantovani, K. Winzer and C. Alexander, Nat. Mater., 2014, 13, 748–755 CrossRef CAS PubMed.
- J. Bouckaert, J. Berglund, M. Schembri, E. De Genst, L. Cools, M. Wuhrer, C. S. Hung, J. Pinkner, R. Slattegard, A. Zavialov, D. Choudhury, S. Langermann, S. J. Hultgren, L. Wyns, P. Klemm, S. Oscarson, S. D. Knight and H. De Greve, Mol. Microbiol., 2005, 55, 441–455 CrossRef CAS PubMed.
- G. Pasparakis, J. Am. Chem. Soc., 2007, 129, 11014–11015 CrossRef CAS PubMed.
- T. Goda, M. Tabata, M. Sanjoh, M. Uchimura, Y. Iwasaki and Y. Miyahara, Chem. Commun., 2013, 49, 8683–8685 RSC.
- (a) R. J. Pieters, Med. Res. Rev., 2007, 27, 796–816 CrossRef CAS PubMed; (b) S. Bhatia, L. C. Camacho and R. Haag, J. Am. Chem. Soc., 2016, 138, 8654–8666 CrossRef CAS PubMed.
- (a) A. G. Gristina, Science, 1987, 237, 1588–1595 CrossRef CAS PubMed; (b) A. G. Gristina, P. Naylor and Q. Myrvik, Med. Prog. Technol., 1988, 14, 205–224 Search PubMed.
- S. R. S. Ting, E. H. Min, P. B. Zetterlund and M. H. Stenzel, Macromolecules, 2010, 43, 5211–5221 CrossRef CAS.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc05561k |
| This journal is © The Royal Society of Chemistry 2019 |