
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA continuing legend: the Brookhart-type α-diimine nickel and palladium catalysts
Fuzhou
Wang
 a and
Changle
Chen
*b
a and
Changle
Chen
*b
aInstitute of Physical Science and Information Technology, Anhui University, Hefei 230601, Anhui, China
bCAS Key Laboratory of Soft Matter Chemistry, Department of Polymer Science and Engineering, University of Science and Technology of China, Hefei 230026, China. E-mail: changle@ustc.edu.cn
First published on 17th April 2019
Abstract
The Brookhart-type α-diimine nickel and palladium catalysts are one of the most examined systems in the field of olefin polymerization. Even after more than twenty years of extensive research, they continue to attract a lot of attention. In addition to their ease of synthesis and structural versatility, the chain walking capability and great polar group tolerance represent some of their most fascinating features. Followed by some brief descriptions of the chain walking mechanism, this review summarizes some recent advances in α-diimine ligand modifications. Specifically, it is categorized into three aspects: modifications of N-aryl substituents, modifications of ligand backbone structures, and the utilization of dinucleating ligands. Influences of steric and electronic effects on catalytic properties are demonstrated. Some recent developments in copolymerizations of olefins with polar comonomers to produce functionalized polyolefin materials are also discussed.
 Fuzhou Wang | Fuzhou Wang received his MS degree in 2013 from Northwest Normal University. He carried out his doctoral work in polymer chemistry at Hiroshima University under the direction of Prof. Takeshi Shiono from 2013 to 2017. Subsequently, he joined Prof. Changle Chen's group at the University of Science and Technology of China as a research assistant. In 2018, he joined Anhui University as a special associate researcher. His current research interest is focused on olefin polymerization by late transition metal catalysts. |
 Changle Chen | Changle Chen obtained his B.S. degree from the University of Science and Technology of China (2005), and PhD from the University of Chicago (2010). After postdoctoral studies at Northwestern University and some time at Celanese Corporation, he started his independent career as a professor at USTC in 2013. His current research focuses on the development of new catalysts and new strategies for olefin polymerization and copolymerization. His notable awards include American Chemical Society DIC Young Investigator Award, IUPAC Prizes-Honorable Mention, Chinese Chemical Society Award for Outstanding Young Chemist, Society of Polymer Science Japan International Leading Young Scientist and NSFC Excellent Young Scholar Fellowship. |
1. Introduction
Polyolefins have been playing pivotal roles in the polymer industry as well as in our daily lives.1 After two decades of research, late-transition metal catalysts based on Ni and Pd for olefin polymerization have received extensive attention owing to their low oxophilicity, and correspondingly the potential in copolymerizing olefins with polar comonomers.2,3 Specifically, the seminal studies by Brookhart and co-workers on the discovery of cationic α-diimine-based Ni and Pd catalysts (Fig. 1) really marked the start of this new era in olefin polymerization studies.4,5 Branched polyolefins are generally produced by early transition metal catalyzed ethylene copolymerization with α-olefins. In contrast, this type of catalyst (C1a and C1b) can produce branched polyolefins using only ethylene as the feedstock. This is achieved through their characteristic chain walking process, which has been extensively studied both experimentally and theoretically.5,6 More importantly, the palladium catalysts are able to copolymerize olefins with polar comonomers to afford copolymers containing functional groups without the pre-protection of the polar groups.7,8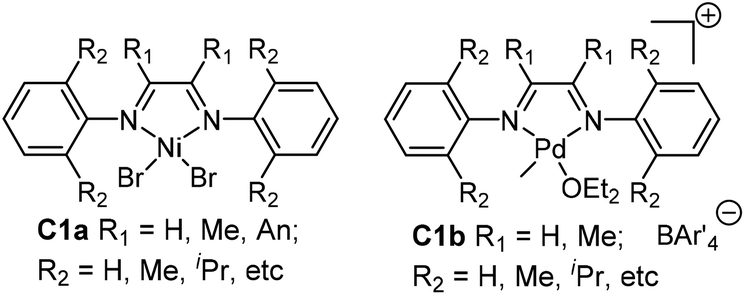 | ||
| Fig. 1 Brookhart's Ni and Pd α-diimine-based precatalysts.4,5 | ||
Recently, phosphine-containing palladium catalysts have emerged as powerful alternatives for ethylene copolymerization with polar comonomers.9–21 Some of these catalysts possessed comparably even better properties than α-diimine palladium catalysts in copolymerization reactions. However, the Brookhart-type α-diimine-based precatalysts continue to be of great research interest for the reasons that include: (i) the ease of synthesis for both the ligands and the metal complexes, (ii) the versatility of the ligand structures, (iii) the ability to tune the polymer microstructures such as branching and topologies, etc.22 As a matter of fact, it is truly amazing that great efforts are still being made nowadays concerning Brookhart catalysts from all around the world, even after twenty years of intensive research.
Over the past few years, there have been numerous review articles concerning α-diimine Ni and Pd catalysts, with aspects including the chain walking mechanism, ligand structures, different monomers, and polymer properties.23–32 However, there have been some major advances recently concerning catalyst developments,33 which allowed far greater control over the polymerization process as well as polymer microstructures. Most importantly, the suitable polar monomer substrate scope has been significantly expanded through these catalyst developments. Here we will summarize some of the advances in α-diimine ligand developments since 2013, as well as some new and challenging monomers that this class of catalysts can address through these ligand improvements. First, the chain walking mechanism is briefly discussed for readers who are not familiar with this field. The α-diimine ligand family has continued to grow rapidly. It seems that this is only limited by the chemists’ imagination and their synthesis capabilities. We do not intend to provide a comprehensive review of all the α-diimine ligands in this field. Instead, only selected examples in recent years will be discussed. Special attention is paid to the cases with unique catalytic properties. Finally, α-diimine Ni and Pd mediated copolymerization of olefins with polar comonomers will be described. The polar monomer substrate scope with Brookhart type catalysts has been greatly expanded in recent years; much of the success has been enabled through ligand modifications.
2. Chain walking mechanism
2.1. Chain walking ethylene polymerization
In ethylene polymerization, these Brookhart catalysts demonstrated good thermal stability and high activities to yield high molecular weight polyolefins with unique branched structures because of the chain walking process. Fink and co-workers34 originally presented a possible chain walking mechanism which was subsequently developed by Brookhart and co-workers.4,5Scheme 1 shows the mechanism of insertion and chain walking for ethylene polymerization. The chain propagation, chain transfer, and chain walking or isomerization are the major steps. During controlled chain walking polymerization, the cationic metal center migrated along the polymer backbone through a rapid β-H elimination reaction and reinsertion with opposite regio-chemistry. This can occur many times before ethylene trapping. Subsequently, methyl and longer chain branches were generated from ethylene trapping and insertion of the secondary metal-alky species. As such, alkyl chains included Me, Et, n-Pr, n-Bu, and sec-Bu branches and longer chains can be generated in ethylene polymerization.3,35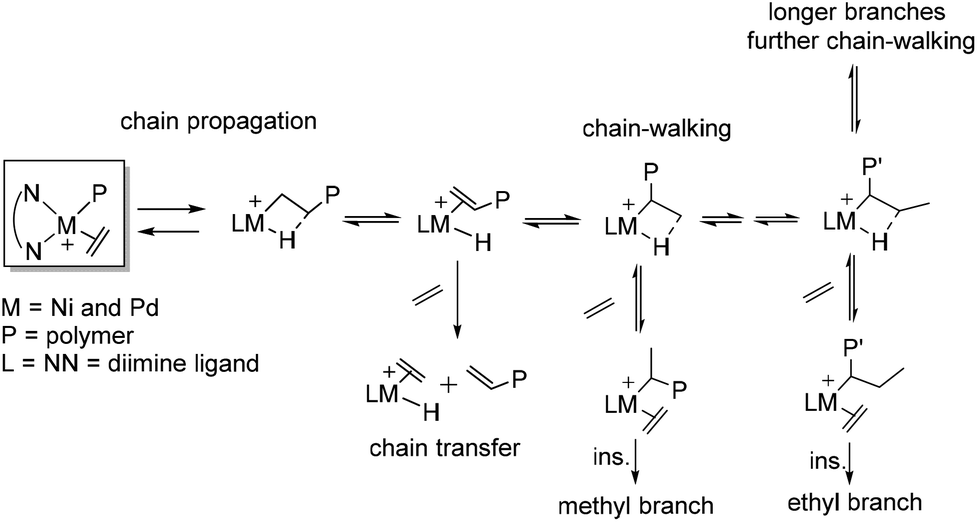 | ||
| Scheme 1 Mechanism of chain walking ethylene polymerization. | ||
Recently, Gao and co-workers have proposed a different mechanistic model for the α-diimine Ni catalyzed ethylene chain walking process.36 One-step chain walking followed by ethylene insertion gives a methyl branch. Meanwhile, long-chain branching (LCB) is obtained by ethylene insertion into the primary Ni-alkyl species originating from nickel migration to the methyl terminal, because of restricted ethylene insertion into the secondary Ni-alkyl species with an α-ethyl or a bulkier alkyl.
Different ligand backbone structures and their N-aryl substituents (Scheme 2) can provide various coordination environments and electronic and steric influences over the metal center, which correspondingly modulate the chain walking behavior and control the polymer microstructures.8,37 For example, it has been demonstrated that the substituents on the backbone of α-diimine ligands can significantly influence the molecular weights and branching densities of the polyethylene products.38,39 The ligand electronic effect is also highly important.40–43 For example, Guan and co-workers showed that the electron withdrawing substituent can increase the electrophilicity of the metal center, which favors chain propagation to yield higher molecular weight polyethylenes (PEs).43 Furthermore, polymer topology can be controlled via polymerization conditions (Scheme 2).8,29 For example, Guan and co-workers demonstrated that polyethylene topology can be changed from a hyperbranched structure at low ethylene pressures to a relatively linear structure at high pressures.37,44 Interestingly, Long and co-workers have recently demonstrated that the polyethylene microstructure can be tuned by visible light irradiation.45
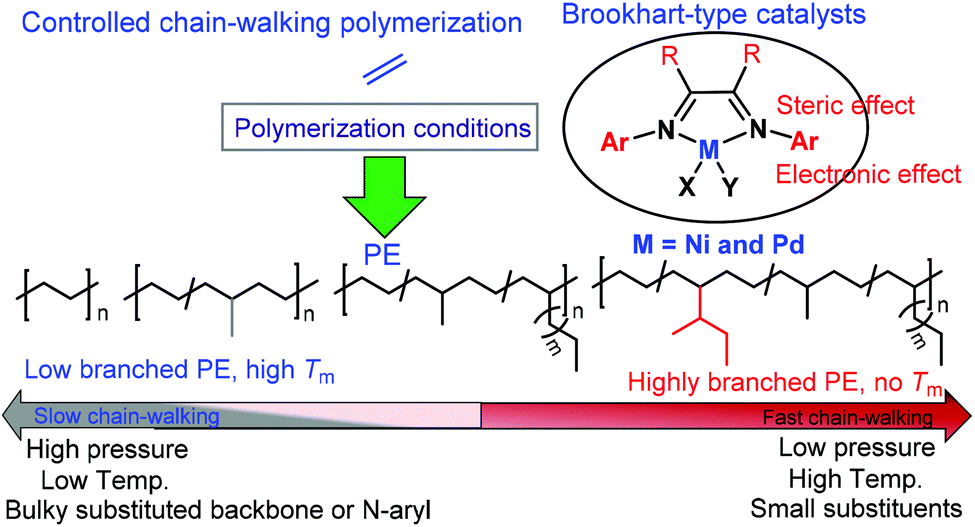 | ||
| Scheme 2 Influence of the chain walking process by ligand structures and polymerization conditions. | ||
2.2. Chain walking α-olefin polymerization
The mechanism for chain walking α-olefin polymerization is outlined in Scheme 3.46,47 Consecutive 1,2-insertion of α-olefins gives an n-alkyl (Cω-2) branch, while 1,2-insertion followed by chain walking and insertion gives a methyl branch (ω,2-enchainment).48 In contrast, 2,1-insertion and complete chain walking lead to chain-straightening with the formation of a long methylene sequence (ω,1-enchainment).44 Coates and co-workers have recently revised and updated the mechanistic picture of α-olefin insertion pathways to analyze and explain the overall branching microstructure (Scheme 3).46 The 13C NMR analyses of poly(α-olefins) produced from monomers with 13C-labeled carbons showed 2,1- or 1,2-insertion from the primary chain end position (1°), the penultimate chain end position (2p°), secondary positions on the polymer backbone (2°), and previously installed methyl groups (1m°). Therefore, various branched polyolefins containing methyl, different alkyl branches and long methylene sequences can be obtained.46,47 In a similar situation to ethylene polymerization, the chain walking process is influenced by ligand structures and polymerization conditions.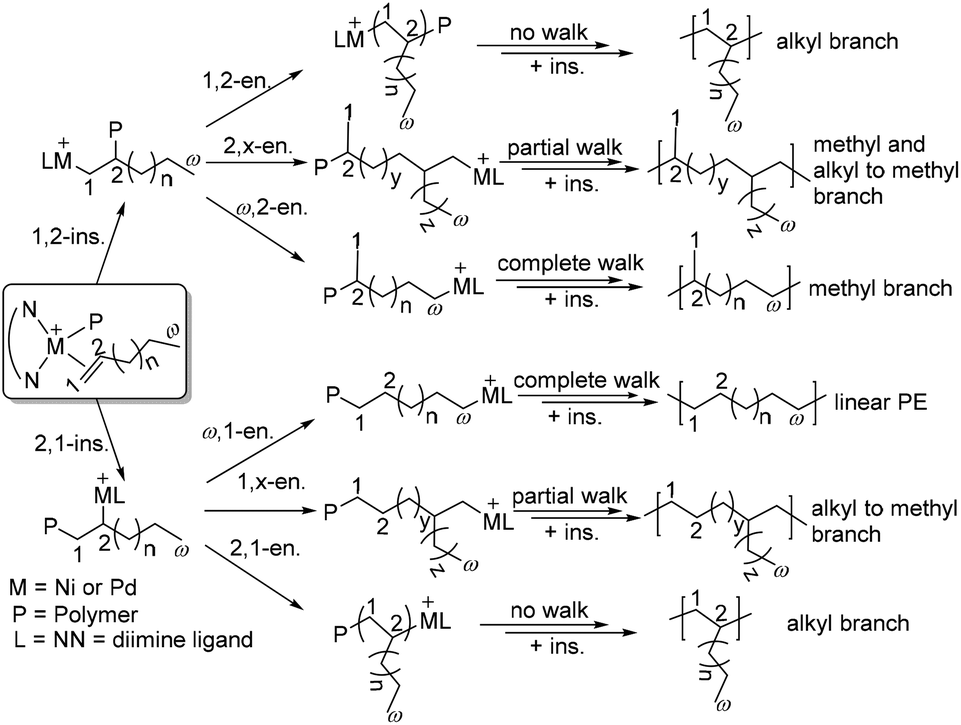 | ||
| Scheme 3 Mechanism of chain walking α-olefin polymerization.46 | ||
Living α-olefin polymerization is an important technique to obtain predictable polymer molecular weights as well as block copolymers.49 In addition, the control of the chain walking process to achieved regio- and stereo-selectivity in α-olefin polymerization represents a great challenge.49–53 Coates and co-workers reported living polymerization of propylene and higher α-olefins catalyzed by a C2-symmetric chiral sec-phenethyl substituted α-diimine nickel complex.50,51 The regioselectivity of these catalysts varied significantly with reaction temperatures: regioregular polypropylene (PP) with a high isotactic content was generated at −60 °C, but regioirregular PP was formed at elevated temperature.50 Therefore, regio-block PP can be generated by changing the polymerization temperature. Guan and co-workers showed that some cyclophane α-diimine nickel catalysts can realize living propylene polymerization at 50–75 °C.53–56 Recently, Coates and co-workers have demonstrated that α-diimine-nickel complexes catalyzed 1-butene polymerization in a stereo-retentive fashion to yield semicrystalline polyolefin, i.e., poly(1-propylpentan-1,5-diyl).53
3. Modified Brookhart-type α-diimine catalysts
Ligand structures are crucial in determining the properties of Brookhart catalysts. Significant advances have been made in the design and modifications of the ligand backbones and the N-aryl substituents.24,28 It is well established that bulky N,N-diaryl-substituents were required to enable high stability, high activity, high polymer molecular weights and high melting temperatures (Tm). In this section, we categorize the studies on ligand modifications into three classes: (1) modifications of the N-aryl substituents (Fig. 2–14), (2) modifications of the ligand backbone (Fig. 15–17), and (3) utilization of dinucleating ligands (Fig. 18–21).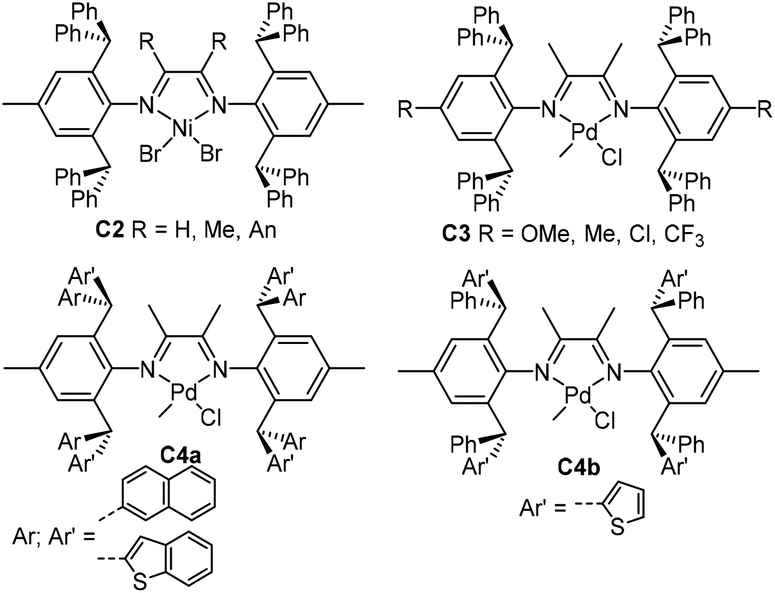 | ||
| Fig. 2 2,6-Diarylhydryl-based α-diimine Ni and Pd catalysts.57–62 | ||
 | ||
| Fig. 3 2,6-Dibenzhydryl-based α-diimine Ni catalysts.63,64 | ||
 | ||
| Fig. 4 2,6-Dibenzhydryl-based unsymmetrical α-diimine Ni and Pd complexes.65–67 | ||
 | ||
| Fig. 5 2,6-Dibenzhydryl-based unsymmetrical acenaphthene Ni- and Pd-diamine complexes.68–75 | ||
 | ||
| Fig. 6 ortho-Menthyl substituted α-diimine-based Ni and Pd complexes.76 | ||
 | ||
| Fig. 7 Unsymmetrical ortho-dibenzhydryl-substituted α-diimine-based Ni and Pd complexes.77–80 | ||
 | ||
| Fig. 8 Xanthene-bridged α-diimine-based Pd and Ni complexes.81,82 | ||
 | ||
| Fig. 9 para-Ferrocene-based α-diimine Pd catalysts.83 | ||
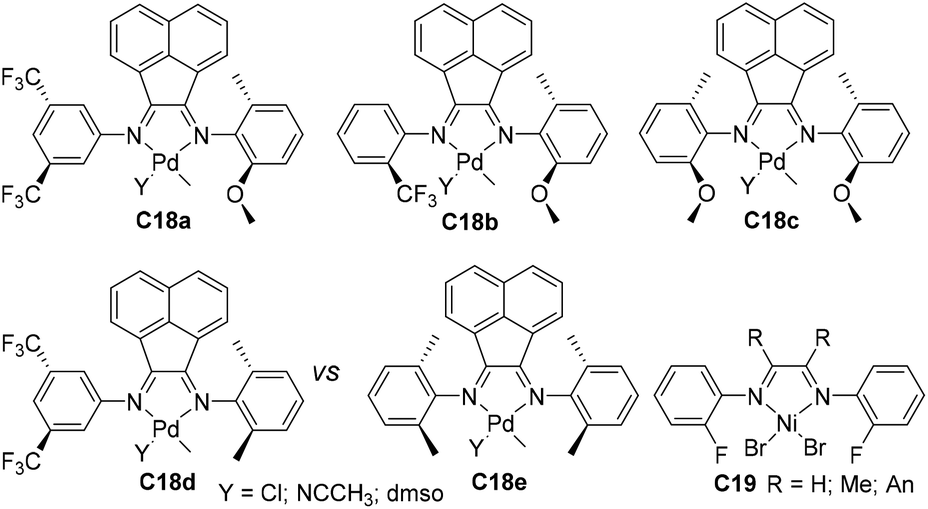 | ||
| Fig. 10 Structures of the CF3, methoxy substituted and fluorinated α-diimine-based Pd and Ni complexes.88–90 | ||
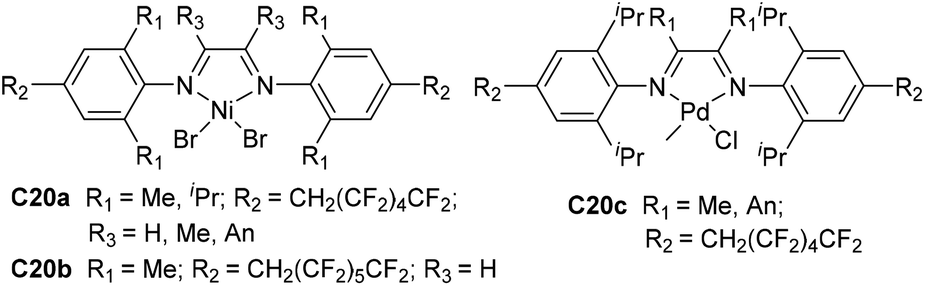 | ||
| Fig. 11 para-Fluorinated alkyl substituted α-diimine Ni- and Pd-based complexes.91 | ||
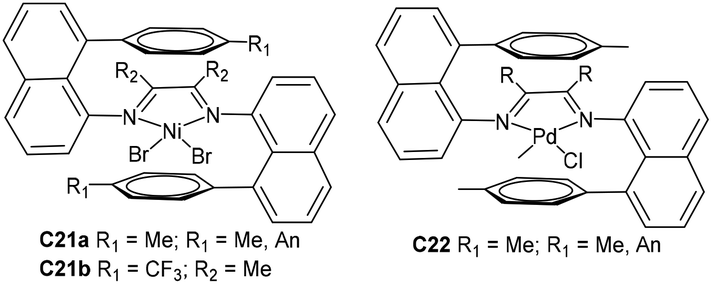 | ||
| Fig. 12 “Sandwich”-type naphthyl α-diimine Ni- and Pd-based catalysts.91–95 | ||
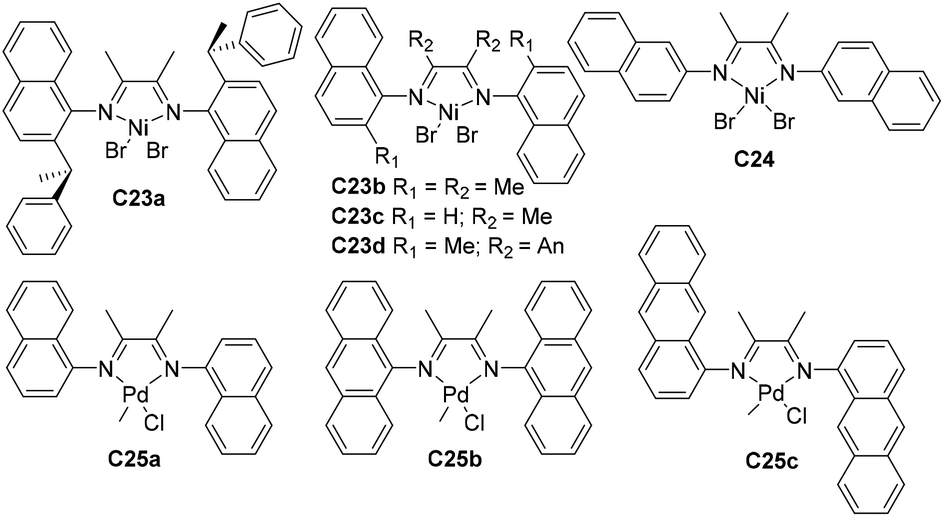 | ||
| Fig. 13 Structure of N-aryl α-diimine Ni- and Pd-based catalysts.96–98 | ||
 | ||
| Fig. 14 Unsymmetrical naphthyl-imine Ni catalysts.99,100 | ||
 | ||
| Fig. 15 Skeletal modifications in α-diimine Ni-based catalysts.101,102 | ||
 | ||
| Fig. 16 Modified acenaphthyl (An) backbone in α-diimine Ni- and Pd-based catalysts.103,104 | ||
 | ||
| Fig. 17 Three-dimensional geometry Ni- and Pd-based α-diimine complexes with a bulky bridged backbone.105–111 | ||
 | ||
| Fig. 18 N,N-Alkyl-bridged and aryl-linked dinculear α-diimine Ni and Pd precatalysts.113–117 | ||
 | ||
| Fig. 19 Conjugated-backbone α-diimine binuclear Ni and Pd catalysts.118,119 | ||
 | ||
| Fig. 20 Xanthene-, naphthalene- and biphenylene-bridged dinuclear Ni and Pd α-diimine precatalysts.120,121 | ||
 | ||
| Fig. 21 Double-decker structure of dinuclear α-diimine Pd catalysts.122–124 | ||
3.1. Modifications of the N-aryl substituents
In 2013, Long and co-workers reported studies of some cationic ortho-dibenzhydryl substituted α-diimine nickel complexes C2 (Fig. 2).57–59 These complexes displayed remarkable thermal stability, maintaining high activity of up to 2.85 × 106 g PE (mol Ni h)−1 (100 °C, 10 min).57 In addition, moderately branched polyethylenes (63–75 branches/1000 C) with high Mn values (>6 × 105 g mol−1) and narrow polydispersities (Mw/Mn ≤ 1.31) were generated. This type of catalyst showed the characteristics of living polymerization even at 75 °C. It is well known that sterically bulky substituents in α-diimine systems can improve catalyst stability and polymer molecular weights. However, the great performances of catalysts C2 exceeded general expectations, and inspired numerous research groups to further study related ligands and catalysts.Because of the sterically bulky nature, it is quite challenging to prepare these α-diimine ligands. In the initial reports, quite low yields (ca. 10%) were reported for the ligand synthesis.57–59 In 2015, Chen and co-workers developed a modified procedure, which can synthesize this type of ligand at yields of above 90%.60 Utilizing the modified procedure, some ligands containing either electron-donating or electro-withdrawing groups (OMe, Me, Cl and CF3) were prepared. The corresponding α-diimine palladium catalysts C3 (Fig. 2) displayed high thermal stability, high activities of up to 3.2 × 106 g PE (mol Pd h)−1 (60 °C, 15 min), and high polymer Mn (up to 538![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) in ethylene polymerization. More interestingly, semicrystalline low-density branched (23–29/1000 C) PEs with high Tm of up to 99 °C were obtained. Semicrystalline ethylene/methyl acrylate copolymers can also be generated through copolymerizations. This represents a very unique property for these and subsequently developed diarylhydryl based α-diimine palladium precatalysts. The majority of the previously reported α-diimine based palladium precatalysts led to the formation of highly branched (usually >80/1000 C) polyethylenes or copolymers. As such, totally amorphous polymeric materials with very poor mechanical properties were obtained. The ability to suppress the chain walking process and produce semicrystalline polar functionalized polyolefins is highly fascinating.
000) in ethylene polymerization. More interestingly, semicrystalline low-density branched (23–29/1000 C) PEs with high Tm of up to 99 °C were obtained. Semicrystalline ethylene/methyl acrylate copolymers can also be generated through copolymerizations. This represents a very unique property for these and subsequently developed diarylhydryl based α-diimine palladium precatalysts. The majority of the previously reported α-diimine based palladium precatalysts led to the formation of highly branched (usually >80/1000 C) polyethylenes or copolymers. As such, totally amorphous polymeric materials with very poor mechanical properties were obtained. The ability to suppress the chain walking process and produce semicrystalline polar functionalized polyolefins is highly fascinating.
Subsequently, the polyethylene branching density can be further lowered to 6/1000 C by two ortho-diarylhydryl-based α-diimine palladium catalysts C4a and C4b (Fig. 2) containing naphthalene or (benzo)thiophene groups.61,62 Most importantly, very high-molecular-weight polar functionalized copolymers possessing relatively linear microstructures and high Tm values were directly synthesized by copolymerization of ethylene with polar monomers. The ligand electronic effect was investigated in a class of dibenzhydryl-ligated nickel α-diimine catalysts C5a and C6 (Fig. 3).63 The CF3-substituted nickel catalyst displayed exceptionally high thermal stability and catalytic activity at elevated polymerization temperatures. The corresponding sterically hindered di(4-X-phenyl)methyl-ligated acenaphthene-nickel catalysts C5b (Fig. 3) possessing different X substituents (OMe, Me, tBu and Ph) were also synthesized.64 All of these catalysts were highly catalytically active at the level of 106 g PE (mol Ni h)−1 toward ethylene polymerization, and yielded semicrystalline polyethylenes with Mn values larger than one million. In addition, these catalytic systems maintained high catalytic activity and high Mn values at temperatures of up to 100 °C.
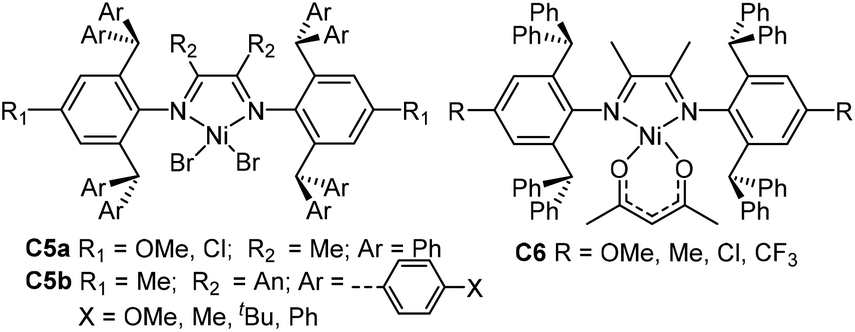
Based on these studies, Chen and co-workers have recently explored ethylene and 1-hexene polymerization studies using a class of ortho-dibenzhydryl-based unsymmetric Ni and Pd α-diimine catalysts (C7 and C8a–c, Fig. 4) containing systematically varied ligand sterics.65,66 The branching structures and Mn values of the polymers obtained by these catalysts can be regulated via catalyst structures as well as polymerization conditions. The total branching degrees of the polyethylenes obtained were decreased with increasing α-diimine ligand sterics and decreasing reaction temperatures.66 The 1-hexene polymerization results indicated that increasing ligand sterics led to enhancement of the chain straightening phenomenon.65 The comonomer incorporations and copolymer microstructures can also be tuned in C8a–c catalyzed ethylene copolymerization of methyl acrylate. These palladium catalysts also generated various branched carboxylic acid-functionalized polyolefins with better control over material properties.67
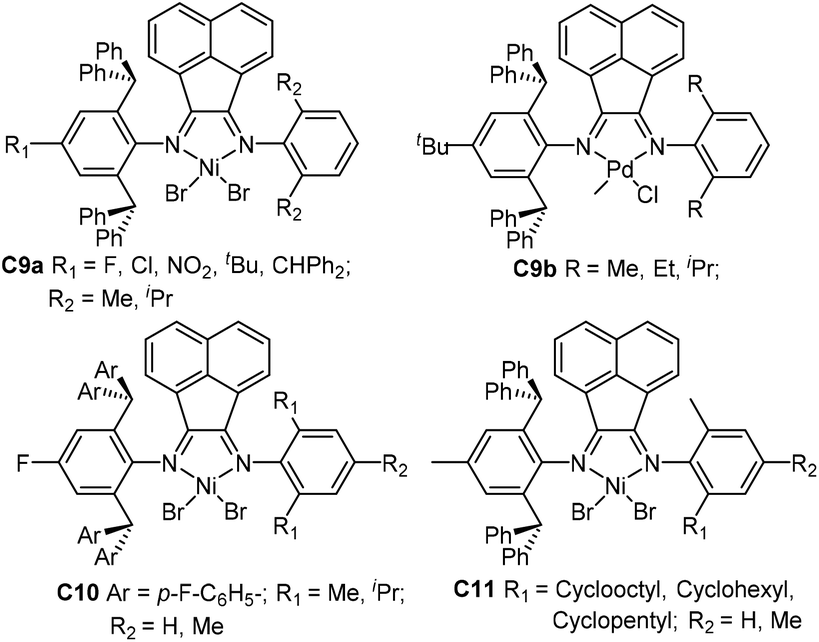
Sun and co-workers synthesized a family of 2,6-dibenzhydryl-based unsymmetrical diiminoacenaphthene nickel catalysts (C9a and C10, Fig. 5) bearing different substituents for ethylene polymerization.68–72 Electron-withdrawing groups (F, Cl, NO2) led to great thermal stability and high activities of up to 107 g PE (mol Ni h)−1. They also synthesized a class of 2,6-dibenzhydryl-based unsymmetrical nickel catalysts C11 (Fig. 5) containing one ring-size variable ortho-cycloalkyl substituted N-aryl groups.73 Ethylene polymerization using these nickel complexes displayed high activities of up to 1.75 × 107 g PE (mol Ni h)−1 (30 °C, 5 min), and generated hyperbranched and moderately branched polyethylene elastomers with Mn values in the range of 2.14–6.68 × 105 g mol−1 and Tm values as low as 44.2 °C. They recently synthesized some dibenzhydryl-based unsymmetrical α-diimine palladium complexes C9b (Fig. 5) bearing different substituents.74,75 These palladium complexes activated by methylalminoxanes (MAO) showed high activities for norbornene (NB) homopolymerization, producing insoluble polymers. E-NB copolymerization can also be achieved using these palladium catalysts.
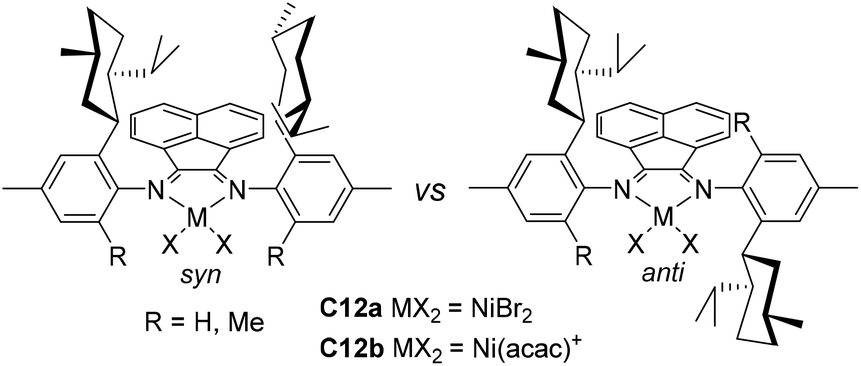
Recently, Jordan and co-workers have synthesized some ortho-menthyl-substituted α-diimine-based nickel precatalysts (C12a and C12b, Fig. 6) with both the syn and anti conformers for ethylene polymerization.76ortho-Menthyl-substituted C12b has a preference for the syn conformation, and displayed high activities of (2.5–6.6) × 106 g PE (mol Ni h)−1 (22 °C, 15 min). However, these syn–anti conformers of menthyl-substituted C12b exhibited substantially different polymerization behaviors. The Mn values and branching densities of the polyethylenes generated by the syn conformer were higher than those generated by the anti conformer. The 1-hexene polymerization using the syn conformer showed a greater preference for 6,1-enchainment to afford poly(1-hexene) with a high chain-straightening content (syn 50% vs. anti 41%, Scheme 3).
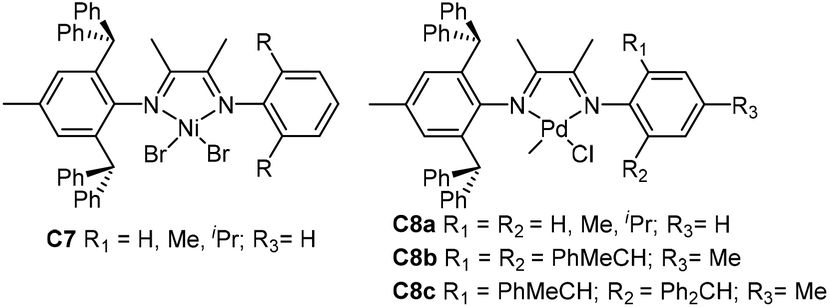
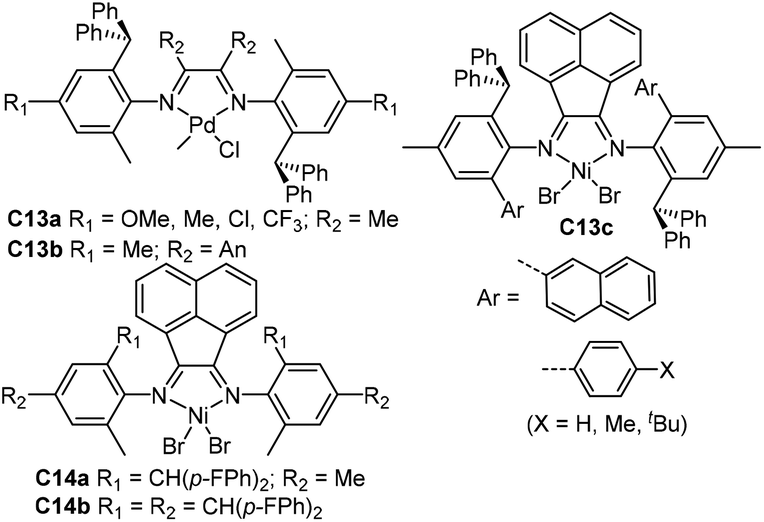
Chen and co-workers have recently synthesized some ortho-dibenzhydryl substituted unsymmetrical α-diimine-based palladium complexes (C13a and C13b, Fig. 7) with different backbone structures and para-substituents (OMe, Me, Cl and CF3).77 The ethylene and 1-hexene homopolymerization and ethylene/methyl acrylate (MA) copolymerizations were investigated by these catalysts. The electron-donating para-substituted C13a generated higher molecular weight polymers, and exhibited higher thermal stability. Interestingly, these catalysts produced monodisperse (co)polymers with narrow Mw/Mn values of 1.30–2.05. The corresponding acenaphthene-nickel catalysts containing both dibenzyhydryl and aryl moieties showed great properties to offer polyethylenes with great tensile and elastic properties.78 In α-olefin polymerizations, this type of catalyst led to a linear monomer unit via the chain walking process. In addition, polar functionalized polyolefins were synthesized by the propylene copolymerizations with 10-undecen-1-ol and methyl 10-undecenoate. Sun and co-workers also developed some unsymmetrical α-diimine nickel complexes (C14a79 and C14b,80Fig. 7) bearing ortho-difluorobenzhydryl moieties. These nickel catalysts exhibited high thermal stability and activities (106 g PE (mol Ni h)−1), generating high molecular weight PEs with relatively low branching density and narrow Mw/Mn values. It is possible that the sterically bulky dibenzhydryl-substituent prevents syn–anti isomerization in these two systems, or both isomers possess similar properties in ethylene polymerization reactions.
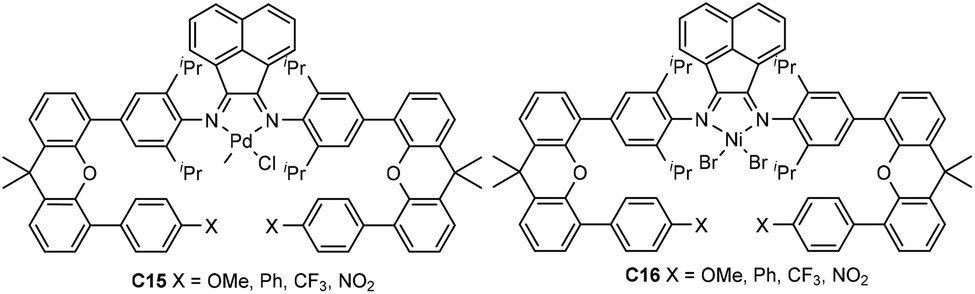
Recently, Chen and co-workers have explored a family of xanthene-containing α-diimine nickel and palladium complexes (C15 and C16, Fig. 8) bearing different substituents (OMe, Ph, CF3 and NO2) at a remote position.81,82 The steric and electronic factors of the metal center remain largely similar to each other. These palladium complexes C15 demonstrated enhanced thermal stability and polymer molecular weights in comparison with the classic Brookhart-type palladium complex.81 The palladium catalyst containing a phenyl group exhibited a much better catalytic activity of up to 2.5 × 104 g PE (mol Pd h)−1 (20 °C, 12 h) than the rest of the palladium complexes in ethylene polymerization, and produced higher Mn (up to 1.21 × 105 g mol−1) PEs with slightly lower branching density. Additionally, E-MA and E-NB copolymerizations were achieved using these palladium catalysts to produce copolymers with high molecular weights. Their thermostable nickel complexes C16 are very active of up to 6.9 × 106 g PE (mol Ni h)−1 (20 °C, 30 min), producing moderately branched PEs with very high Mn values of up to 1.53 × 106 g mol−1.82 Most importantly, the polyethylene products generated by the Ph-substituted nickel catalyst using only ethylene as feedstock possess excellent elastic properties. This provides an alternative strategy to prepare thermoplastic polyolefin elastomers (TPEs).
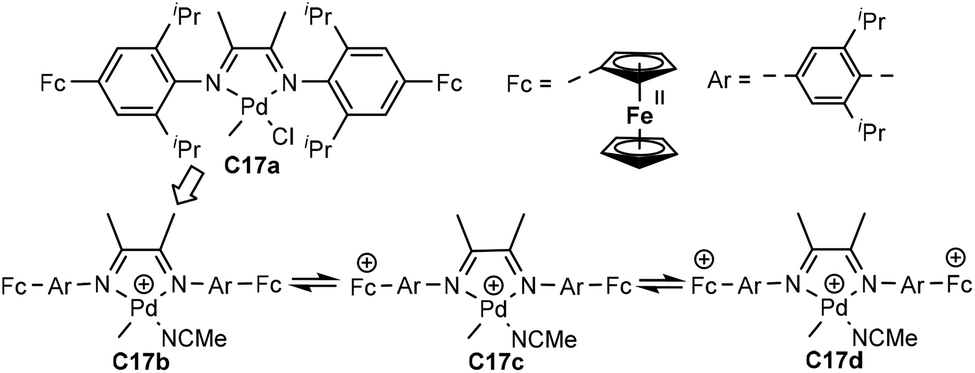
Chen and co-workers have recently demonstrated that the pre-installed two ferrocenyl units on the para-aryl position of the palladium complex based α-diimine ligand can be oxidized sequentially (C17a–d, Fig. 9).83 The catalyst properties were modulated via this stepwise redox process in ethylene and 1-hexene homopolymerization, as well as ethylene copolymerization with methyl acrylate, 5-norbornene-2-yl acetate (NB-Ac) and norbornene. Most interestingly, the microstructures and polydispersities of the produced polymers could be controlled in this stepwise oxidation processes. The concept of redox controlled olefin polymerization has also been realized in α-diimine nickel systems as well as some palladium-based catalysts.84–87
Milani and co-workers have recently designed a family of sterically open α-diimine palladium complexes C18a–e (Fig. 10) bearing trifluoromethyl or methoxy substituents.88 These complexes catalyzed the cooligomerization of ethylene with methyl acrylate to generate cooligomers under mild conditions. The ligand structures can significantly affect the catalytic properties. Ahmadjo and co-workers designed some fluorinated nickel α-diimine precatalysts C19 (Fig. 10) possessing various backbone structures.89,90 The acenaphthoquinone-based catalyst in ethylene polymerization exhibited a moderate activity of 1.4 × 104 g PE (mol Ni h)−1 (30 °C, 30 min).89 Despite the steric openness of these ligands, molecular weights of up to 2.48 × 105 g mol−1 with good crystallinity (58%) were generated. The introduction of ortho-fluorine into the ligand's N-aryl can effectively retard the β-hydride elimination through the influence of F–H bonds, leading to semicrystalline polymers.
Merna and co-workers designed some α-diimine-based Pd and Ni complexes C20a–c (Fig. 11) bearing fluorinated alkyl substituents at the para-aryl position.91 Propene and 1-hexene were polymerized in a controlled manner by nickel catalysts C20a and C20b at −10 °C, whereas palladium C20c allowed controlled ethylene polymerization at 0 °C. The effect of para-fluoroalkyl groups on the polymerization activities, thermal stability and olefin chain walking process was not significant. The branching densities of the polyolefins obtained by these catalysts can be controlled by the ligand backbones and ortho-aryl substituents.
Brookhart and co-workers have recently designed two “sandwich” type naphthyl α-diimine Ni-based precatalysts C21a (Fig. 12) containing two 8-p-tolylnaphthylimino moieties, which provided strong shielding in the Ni axial positions.92 These catalysts were shown to produce unique PEs with very high branching densities of up to 152 branches/1000 C and high molecular weights. Most importantly, some corresponding derivative nickel C21b (Fig. 12) was reported by Coates and co-workers and catalyzed higher α-olefin polymerizations by precision chain walking with a preference for the ω,1-enchainment (Scheme 3).93 Relatively low branched poly(α-olefin)s that are highly “chain-straightened” semicrystalline (Tm > 100 °C) were generated by these “sandwich” type catalysts. In addition, they also reported the synthesis of TPEs from the block copolymerization of 1-decene and ethylene using C21b.94 Polyolefin-based block copolymers with good crystallinity hard blocks were generated from 1-decene polymerization by high 2,1-insertion. Recently, Brookhart and co-workers have investigated some “sandwich”-type α-diimine palladium catalysts C22 (Fig. 12)95 in ethylene polymerization. These catalysts were used to conduct living ethylene polymerization at 25 °C and produced hyperbranched PEs with narrow Mw/Mn values (ca. 1.1). Comonomer incorporation of up to 14% was achieved in ethylene/methyl acrylate copolymerization.
Yuan and co-workers prepared some naphthyl-α-diimine Ni-based catalysts C23a–d and C24 (Fig. 13) bearing different substituents (sec-phenethyl, methyl and H) at the o-naphthyl position for ethylene polymerization.96C23a yielded PEs with highly branched structures and up to 2.8 × 106 g PE (mol Ni h bar)−1 (40 °C, 10 min) catalytic activities. Chen and co-workers also conducted higher α-olefin polymerization using these nickel catalysts C23a–d to afford poly(α-olefin)s with high molecular weight.97 The polymerization results showed the possibility of polymer structure control, via reaction temperatures, ligand structures, and the types and concentration of monomers. Specifically, increasing steric hindrance led to enhanced 2,1-insertion. Carfagna and co-workers synthesized some cationic palladium complexes C25a–c (Fig. 13) bearing α-diimine ligands with fused aromatic rings,98 and studied their properties in the stereo-controlled copolymerization of CO with vinyl arene.
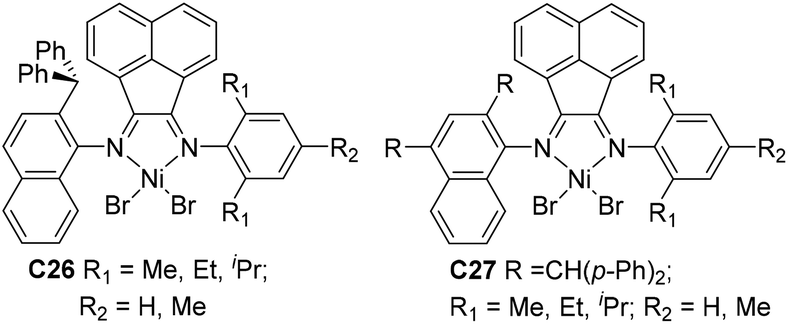
Sun and co-workers studied that some 2-benzhydrylnaphthyl-imino unsymmetrical nickel C26 (Fig. 14) catalyzed ethylene polymerization at high activities,99 and generated PEs with low branching densities of up to 12/1000 C and high Tm (up to 131 °C). The corresponding difluorobenzhydryl substituents in C27 (Fig. 14) led to high activities and high polyethylene molecular weights.100
3.2. Modifications of the α-diimine backbone structures
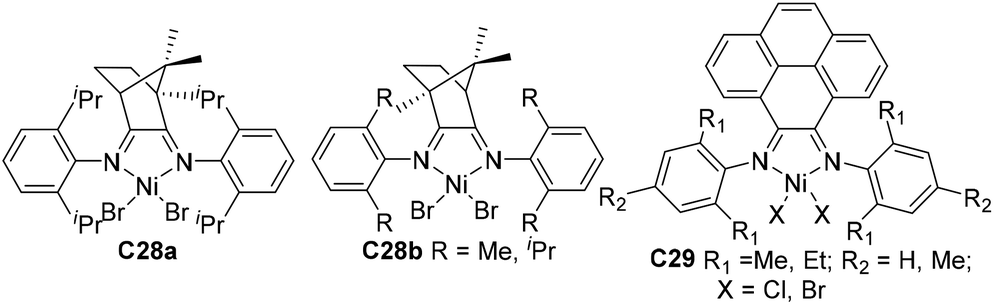
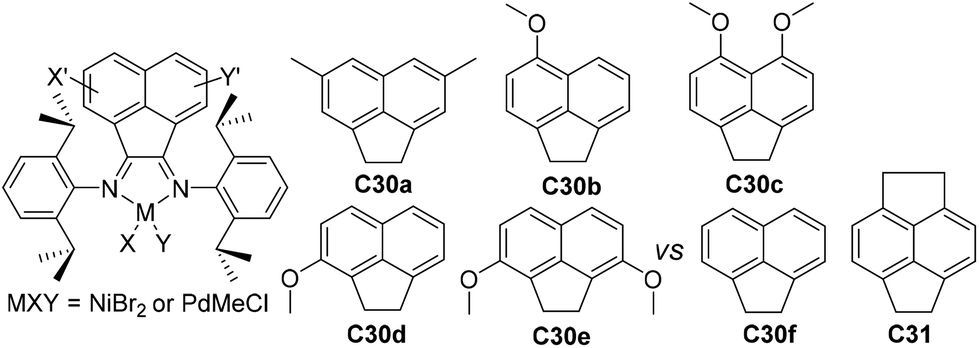
Compared with modifications of the N-aryl substituents, there have been far fewer studies related to the modifications of the α-diimine backbone structures. Wu and co-workers designed two chiral α-diimine based nickel catalysts C28a and C28b containing (1R)- and (1S)-camphyl backbones (Fig. 15) for olefin polymerization.101 Interestingly, the structure chirality had no obvious influence on the regioselectivity and catalytic activity. Living chain walking polymerization of 1-hexene and propylene in a wide range of temperatures was achieved. This camphyl type nickel catalyzed propylene homopolymerization at −60 °C in 45% 1,3-enchainment fraction to produce polypropylene with an obvious melting point. Sun and co-workers explored some 4,5-bis(arylimino)pyrenylidene based dihalide nickel C29 (Fig. 15) which catalyzed ethylene polymerization with high activities of up to 4.4 × 106 g PE (mol Ni h)−1 (40 °C, 15 min).102Recently, Chen and co-workers have designed some Ni- and Pd-based α-diimine precatalysts C30a–e (Fig. 16) possessing various groups on the acenaphthene-based backbone,103 which demonstrated activities of up to 1.6 × 107 g PE (mol Ni h)−1 (20 °C, 10 min), and yielded branched PEs with molecular weights of up to 4.2 × 105 g mol−1. The sterically bulky nickel complex C30e produced polymers with much lower branching density and higher Mn values than the other nickel complexes and the classic complex C30f. Significant effects of the backbone substituents were observed on related palladium catalyzed ethylene or E/MA (co)polymerizations. Once more, the palladium catalyst C30e produced a PE and E/MA copolymer with much higher Mn values than the rest of the palladium catalysts. Fu and co-workers also reported a similar α-diimine nickel catalyst C31 (Fig. 16) for ethylene polymerization, which demonstrated higher thermal stability and higher catalytic activity than classic Brookhart-type α-diimine catalyst C30f at elevated temperatures.104
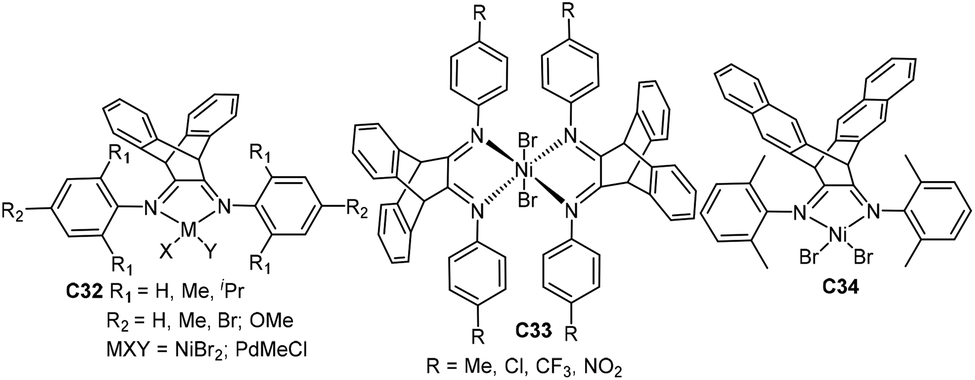
Gao and He groups studied a family of three-dimensional geometry Ni- and Pd-based α-diimine precatalysts C32 (Fig. 17) bearing a dibenzobarrelene backbone.105–108 The precision synthesis of functionalized polyolefins was achieved by Pd-catalyzed living coordination copolymerization of ethylene with various acrylate comonomers.105 In this system, the molecular weight, polydispersity, composition and branching topology of the E/MA copolymers can be tuned by changing ethylene pressure. These catalysts also showed high stability and high activity in NB polymerization and copolymerization with 5-norbornene-2-carboxylic acid methyl ester when activated by B(C6F5)3.107,108 They also synthesized some similar nickel catalysts C33 (Fig. 17) containing a bulky bis(α-diimine) ligand.109,110 These catalysts demonstrated high thermal stability and high catalytic activity toward NB polymerization. They also studied a related 6,13-dihydro-6,13-ethanopentacene-based nickel complex C34 (Fig. 17).111
3.3. Dinuclear α-diimine nickel and palladium catalysts
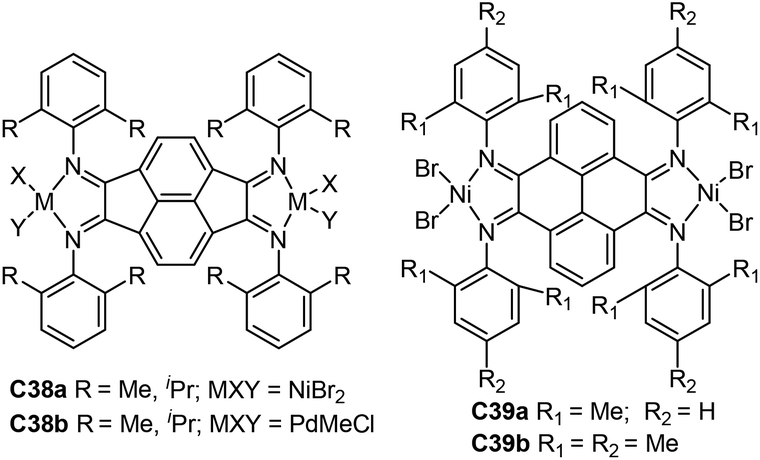
A large number of dinuclear Ni and Pd complexes as catalyst precursors have been explored, and many of them have been applied for olefin polymerization and copolymerization reactions.112 However, only a limited number of Brookhart-type dinuclear Ni and Pd precatalysts ligated by α-diimine were reporeted.113–116 For example, Revankar and co-workers developed a family of dinuclear Ni and Pd complexes (C35a and 35b, Fig. 18) with different groups at the ortho-position of aniline.113,114 These complexes efficiently oligomerized ethylene up to 106 g oligomer (mol M h bar)−1 high activities at 30 °C. In addition, dinuclear nickel complexes C35a exhibited high selectivity towards 1-butene polymerization. Sun and co-workers prepared a series of dinuclear nickel α-diimine complexes (C36a–c, Fig. 18) with methylene bridged for ethylene polymerization.115 Zohuri and co-workers reported some bridged dinuclear nickel α-diimine precatalysts C37a and C37b–f (Fig. 18) with alkyl- and 1,4-phenyl-linked based ligands.116,117 The ethylene polymerization results using C37b–f suggested that these steric and electronic substituent effects of α-diimine ligands have significant influences on polymerization behaviors.116C37a and C37f polymerized 1-hexene to obtain low to high molecular weight poly(1-hexene) with Mw of up to 1.7 × 106 g mol−1.117 The higher activities and lower level of branching densities were observed for C37f in comparison with C37a.Chen and co-workers reported the synthesis of some binuclear nickel and palladium catalysts (C38a and 38b, Fig. 19) bearing conjugated α-diimine ligands.118 Under similar conditions, C38a showed high activities of up to 1.05 × 106 g PE (mol Ni h)−1 (22 °C, 30 min) in comparison with the corresponding mononuclear catalyst. Interestingly, two separate peaks were observed in the GPC curves of the PEs obtained by these palladium catalysts C38b. Sun and co-workers also designed two dinuclear nickel catalysts (C39a and 39b, Fig. 19) supported by 4,5,9,10-tetra(arylimino)pyrenylidenes.119 These nickel catalysts demonstrated high stability and activities of 4.20 × 106 g PE (mol Ni h)−1 (30 °C, 15 min) in ethylene polymerization, and produced relatively linear PEs (7–14 branches/1000 C) with high Tm (up to 132 °C).
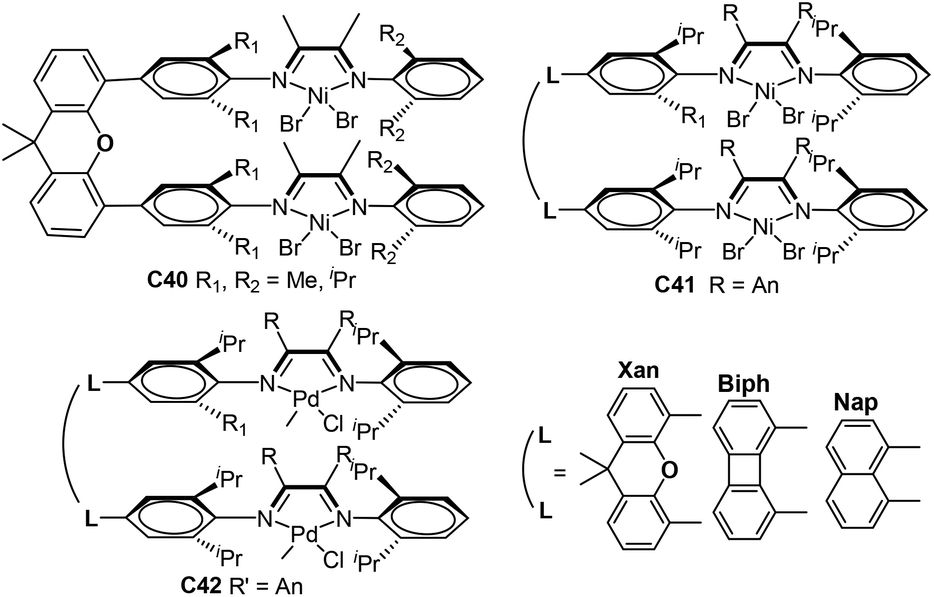
Chen and co-workers further synthesized some xanthene-, naphthalene- and biphenylene-bridged α-diimine dinuclear nickel complexes C40 and C41 (Fig. 20) bearing different substituents and backbones.120,121 These dinuclear nickel complexes showed good thermal stability in the polymerization of ethylene and high catalytic activity of up to 106 g PE (mol Ni h)−1. Interestingly, these Ni catalyst systems led to semicrystalline PEs with higher Mn values and relatively linear structures compared with their mononuclear analogues. The polymerization results indicated that the Ni–Ni cooperativity effect invoked by these dinucleating ligands may be able to suppress the β-H elimination reaction and retard the process of chain walking. Interesting differences were observed by dinuclear palladium complexes C42 (Fig. 20) in ethylene (co)polymerizations versus their monomer nuclear analogue in terms of polymer Mn values and activity.121 In ethylene-methyl acrylate copolymerizations, the mononuclear palladium complex can incorporate appreciable amounts of comonomer, whereas C42 cannot incorporate any comonomer.
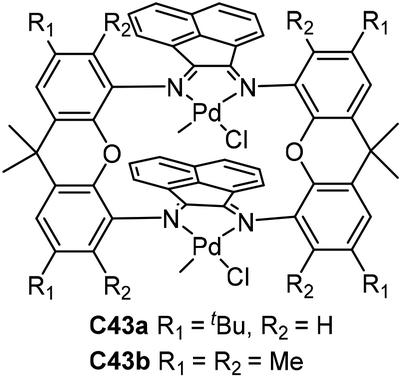
Takeuchi and co-workers developed two double-decker α-diimine dinuclear catalysts C43a and C43b (Fig. 21) bearing a macrocyclic ligand.122–124 In ethylene polymerization, dinuclear complex C43b displayed higher activity compared with the corresponding mononuclear catalyst and C43a. The polyethylenes obtained by C43a and C43b were less branched than those made with a mononuclear one. C43a and C43b catalyzed copolymerization to afford branched E-MA copolymers containing some acrylate units incorporated on the polymer backbone.122 The polymerization of α-olefins by C43b occurred with high selectivity “chain-straightened” in a ω,1-enchainment (>80%, Scheme 3) and generated high-linearity solid polymers,123 while the mononuclear analogues produced oily oligomers as a result of low selectivity in 1,2- or 2,1-insertion. In addition, C43b catalyzed copolymerization of E with acrylic anhydride to afford the copolymer containing the repeating unit from acrylic anhydride.124 These studies clearly indicated that structures of α-diimine ligands can strongly influence the polymerization processes.
4. Copolymerizations of olefins with polar monomers
In the process of exploring various Brookhart catalysts, various olefinic monomers have been polymerized and copolymerized.27,31,125–127 The applicable monomers have also been rapidly growing to include linear128,129 and branched130,131 α-olefins, cycloalkyl substituted α-olefins,132–136 cycloolefins,137 dienes,138 trienes,139,140 internal olefins,141–144 norbonene type monomers,101–104 and different polar comonomers.3,27,28 It should be noted that the continuous interest in Brookhart-type catalysts largely originates from their capabilities in copolymerizations of olefins with polar functionalized comonomers. The introduction of some polar functional substituents into polyolefins can significantly improve their properties and broaden their applications. Concerning industrially relevant polar comonomers, or the so called fundamental polar monomers, α-diimine palladium catalysts only worked for acrylates, vinyl ketones, silyl vinyl ethers, etc.145–147 A lot of specially designed polar monomers,27,28 such as the ones bearing long spacers between the double bond and acrylate group, are also suitable polar comonomers. However, many other fundamental polar monomers (such as vinyl ether, vinyl acetate, vinyl halide, acrylonitrile, styrene, etc.) completely shut down ethylene polymerization due to various reasons, such as cationic polymerization, β-X elimination, and/or formation of a stable metal chelate.148–150 The expansion of the polar monomer substrate scope through catalyst improvements represents a major challenge in this field. Compared with palladium, nickel is much more oxophillic and less tolerant towards polar monomers. In the presence of large amounts of cocatalysts or additives, and under harsh conditions, α-diimine nickel catalysts were demonstrated to copolymerize ethylene with methyl acrylate.151 The utilization of low-cost nickel α-diimine catalysts to address the polar monomer problem represents another fascinating challenge in this field.Chen and co-workers showed that ortho-dibenzhydryl-based palladium catalysts C3 led to the formation of semicrystalline E/MA copolymers with Mn of up to 1.9 × 104 g mol−1,60 and low branching density (25/1000 C). The sterically bulky catalysts C3 mediated efficient copolymerization of ethylene with acrylate or acrylic comonomers (Group B, Scheme 4) with relatively linear microstructures (as low as 9/1000 C) and high melting temperatures (Tm close to 120 °C). These “slow chain walking” palladium catalysts still cannot copolymerize ethylene with many fundamental polar monomers (Group A, Scheme 4). However, with some CH2 spacers between the polar groups and the double bond, the polar monomers of group C are suitable comonomers for catalysts C4a.61 Notably, the conventional Brookhart α-diimine palladium catalysts will undergo fast chain walking, which brings the metal center close to the polar groups and the subsequent termination reactions will completely shut down copolymerizations. The slow chain walking feature of C4 is the key factor for the successful expansion of the polar monomer substrate scope.
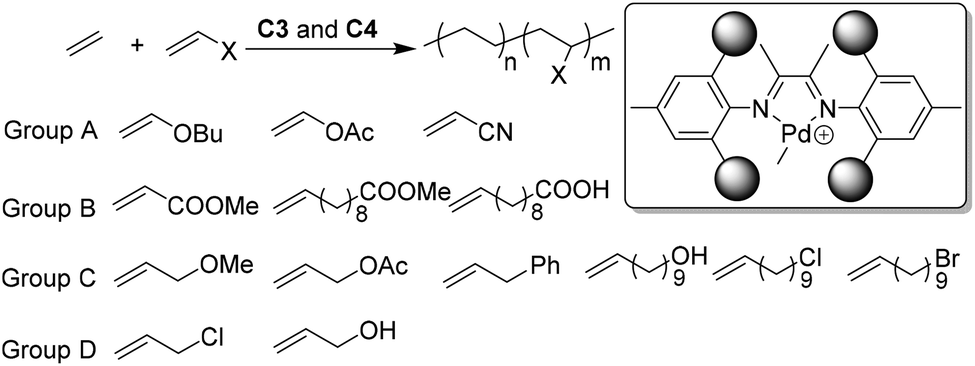 | ||
| Scheme 4 Direct synthesis of polar functionalized semicrystalline copolymers using slow chain walking palladium catalysts.60,61 | ||
During the above-mentioned copolymerization studies, it was found that polar monomers of group D (Scheme 4) quenched the C4a mediated ethylene polymerization.61 Although the chain walking process is efficiently slowed down, the occasional chain walking can still bring the metal center close to the Cl/OH groups and induce termination reactions. Strategies to address this issue are currently being explored in order to further expand the polar monomer substrate scope. Recently, Chen and co-workers have designed a family of Ni and Pd α-diimine complexes C44a–d (Fig. 22) with nitrogen-containing second coordination spheres.152 For the nickel catalysts, PEs with highly linear structures (branching < 1/1000 C) and high Tm (>130 °C) were obtained. For the palladium catalyst C44d, polyethylenes with moderately branched structures (ca. 70/1000 C) were generated. This suggested that the chain walking tendency of C44d is lower than that of classic Brookhart α-diimine palladium catalysts (ca. 100/1000 C), but much higher than that of the previously reported “slow chain walking” catalysts (C2 and C3, 6–25/1000 C). However, C44d can mediate efficient ethylene copolymerizations with the group D polar comonomers. Control experiments and computational results revealed the critical roles of the metal–nitrogen interactions in these (co)polymerization reactions.
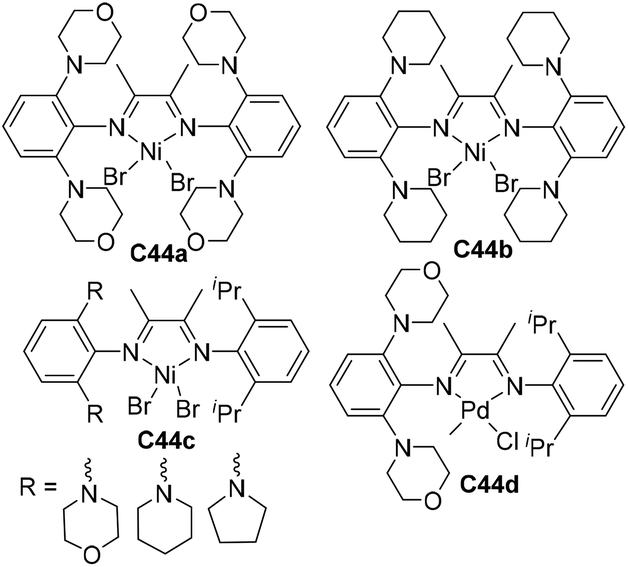 | ||
| Fig. 22 Structures of Ni and Pd α-diimine catalysts bearing nitrogen-containing second coordination spheres.152 | ||
Jordan and co-workers developed some amide-functionalized α-diimine palladium catalysts C45a–d (Fig. 23) containing –CONHMe or –CONMe2 groups on the N-phenyl rings.153 In ethylene polymerization, iPr-substituted C45c and C45d showed a higher activity of 107 g PE (mol Ni h)−1 than C45a and C45b with ortho-dibenzhydryl groups, and yielded branched (77–81 branches/1000 C) PEs with Mn values in the range of 2.6–60 × 104 g mol−1. In ethylene copolymerization, the incorporated levels of MA (Scheme 5) and acrylic acid (AA) using C45a were higher than 45b and iPr-substituted C8a.
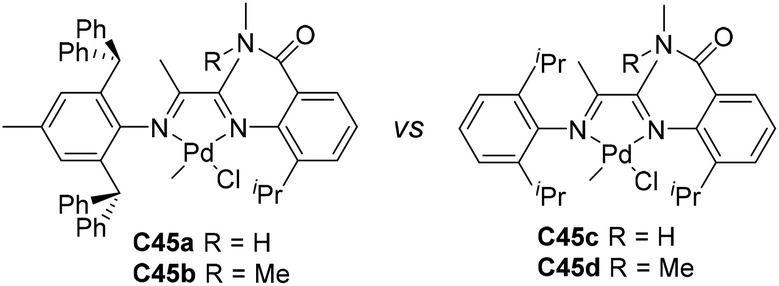 | ||
| Fig. 23 Amide-functionalized α-diimine Pd catalysts.153 | ||
 | ||
| Scheme 5 Copolymerizations of ethylene and norbornene derivatives or some nitrogen-containing monomers.81 | ||
Kaminsky and co-workers first investigated the E-NB copolymerization studies using classic Brookhart-type palladium catalysts,154 which displayed significantly higher NB incorporation ability than metallocene catalysts.155 Clearly, α-diimine palladium catalysts possess great reactivity towards norbornene monomers and great tolerance towards the ester type polar groups. However, the reactivity of α-diimine palladium catalysts towards polar functionalized norbornene monomers has not been explored over a very long period of time. Only recently, Chen and co-workers have studied the ethylene copolymerizations with a family of polar functionalized norbornene type comonomers or nitrogen-containing monomers (NB-NP, NB-OAc and NP, Scheme 5) using para-methoxy substituted α-diimine palladium catalyst C13a.156 Branched high molecular weight copolymers (55–89/1000 C; Mn up to 125000) with good comonomer incorporations (1.1–9.1%) were obtained. In addition, the same group reported the properties of C15 (Fig. 8) in ethylene copolymerizations with NB and NB-Ac (Scheme 5).81 The phenyl-substituted catalyst showed much higher activity, and produced a much higher Mn (up to 132500) copolymer with slightly lower NB (14%) and NB-Ac (1.67%) incorporation ratios than the rest of the catalysts.
Acrylic acid (AA) is a quite challenging polar monomer to be incorporated during olefin polymerization, due to the presence of both oxygen and hydroxyl polar groups. Successful copolymerizations have been achieved using phosphine-sulfonate palladium catalysts.157–159 Recently, Chen and co-workers have investigated the copolymerization of 1-octene with AA (Scheme 6)160 catalyzed by sterically bulky α-diimine palladium catalysts (C4a and C8a–c, Fig. 2 and 4), producing branched copolymers (58–98/1000 C) with high comonomer incorporation (1.07–15.7%) and high Mn values of up to 40400. This copolymerization behavior provides an alternative strategy to prepare carboxylic acid functionalized branched polyolefins.
 | ||
| Scheme 6 Copolymerization of 1-octene with acrylic acid catalyzed by α-diimine Pd catalysts.160 | ||
Recently, a major breakthrough in polar monomer substrate scope expansion has been achieved by Brookhart and co-workers (Scheme 7).161,162 They studied α-diimine Pd-catalyzed ethylene copolymerization of vinyltrialkoxysilanes (VTEoS) to afford copolymers with highly branched structures (ca. 100 branches/1000 C) and low molecular weights.161 Incorporation ratios from 0.25 to 2.0 mol% were generated with little rate retardation relative to ethylene polymerization. Compared with traditional catalytic systems, the sandwich-type catalyst produced polar functionalized copolymers with higher Mn values and narrow Mw/Mn values of 1.2–1.4. More importantly, multiple trialkoxysilyl groups can be incorporated per polymer chain.
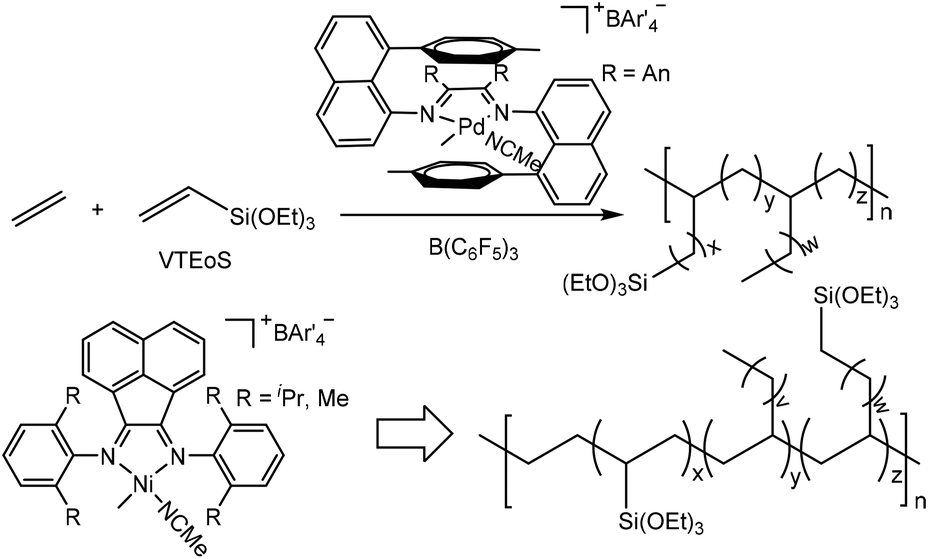 | ||
| Scheme 7 Cationic α-diimine-based Pd- and Ni-catalyzed ethylene copolymerization with vinyltrialkoxysilanes.161,162 | ||
There have also been some recent advances in α-diimine Ni-catalyzed ethylene copolymerization with polar monomers. For instance, Brookhart and co-workers achieved copolymerization of ethylene with VTEoS (Scheme 7) using cationic (α-diimine)NiMe(CH3CN)+ catalysts/B(C6F5)3 at 60 °C to give variously branched (5–60 branches/1000 C) copolymers with high molecular weights.162 Incorporation ratios ranging between 0.23 and 10.0 mol% with multiple –Si(OEt)3 groups incorporated per polymer chain were achieved.
Recently, Chen and co-workers have reported the preparation of silicon-functionalized polyolefin materials through subsequent dehydrogenative silylation catalysed by the pyridine-diimine cobalt catalyst and ethylene copolymerization using the classic α-diimine nickel catalyst (Scheme 8(I)).163 The allylsilanes with both the silanes functional groups and internal double bond were first prepared from dehydrogenative silylations of different terminal olefins and alkylsilanes using a pyridine-diimine cobalt-catalyst.164 Subsequently, an α-diimine traditional nickel catalyst [(2,4,6-Me3C6H2)N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C(Me)]2NiBr2 can mediate efficient ethylene copolymerizations with these allylsilanes to produce silicon-functionalized polyolefins. This provides an alternative strategy to utilize inexpensive and widely available starting materials such as olefins and silanes. Guironnet and co-workers demonstrated that the synthesis of semitelechelic polyethylene via the cationic α-diimine Pd-catalyzed chain-transfer ethylene (co)polymerization by various silanes as chain-transfer agents (Scheme 8(II)).165 The polymer end-group structure and molecular weight were effectively tuned via varying the types of silanes and their concentration.
C(Me)]2NiBr2 can mediate efficient ethylene copolymerizations with these allylsilanes to produce silicon-functionalized polyolefins. This provides an alternative strategy to utilize inexpensive and widely available starting materials such as olefins and silanes. Guironnet and co-workers demonstrated that the synthesis of semitelechelic polyethylene via the cationic α-diimine Pd-catalyzed chain-transfer ethylene (co)polymerization by various silanes as chain-transfer agents (Scheme 8(II)).165 The polymer end-group structure and molecular weight were effectively tuned via varying the types of silanes and their concentration.
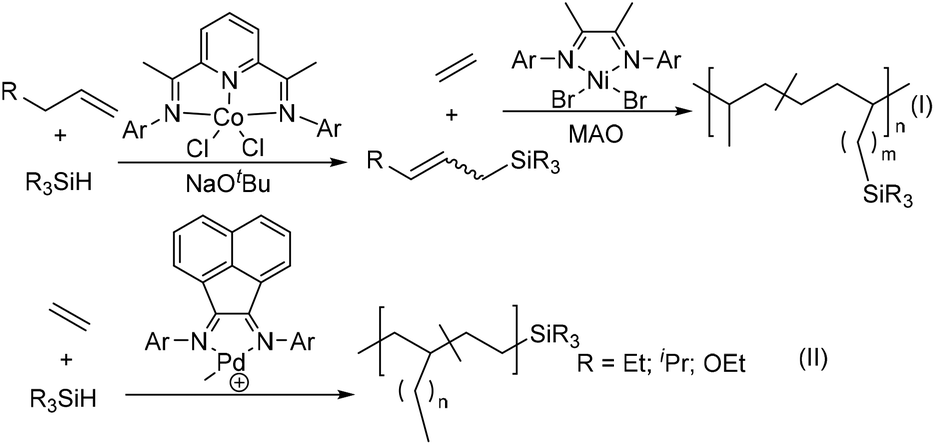 | ||
| Scheme 8 Preparation of polyolefins containing silicon-functionalities by ethylene (co)polymerization.163–165 | ||
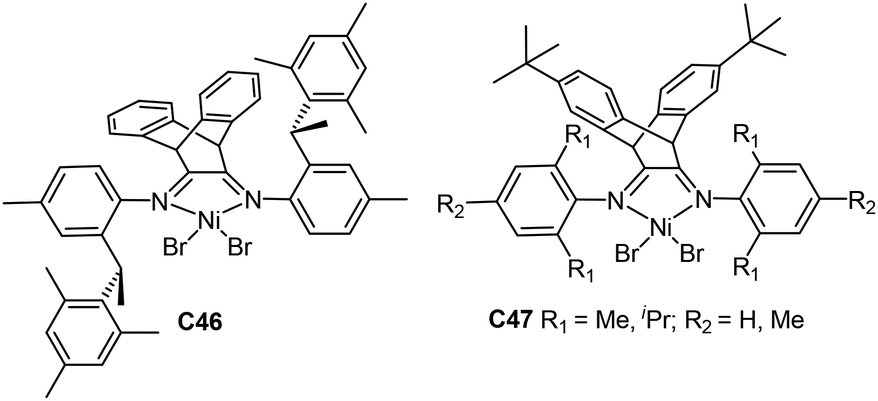
Coates and co-workers prepared a chiral sec-phenethyl based α-diimine nickel catalyst C46 (Fig. 24) containing a dibenzobarrelene-bridged backbone,166 which showed great properties in ethylene polymerization. The catalyst system yielded highly linear PEs with higher Mn values and narrow Mw/Mn values. Catalyst C46 showed living polymerization behavior at room temperature and generated PE with high Tm of up to 135 °C and a highly linear structure at −20 °C. Besides, copolymerization of ethylene with methyl 10-undecenoate (Scheme 4) was realized to afford high molecular weight (up to 1.05 × 105 g mol−1) ester-functionalized copolymers with very low branching densities.
 | ||
| Fig. 24 Dibenzobarrelene-bridged α-diimine nickel complexes.166–169 | ||
Wu and co-workers also demonstrated that a family of dibenzobarrelene-bridged α-diimine nickel C47 (Fig. 24) catalysed ethylene polymerization with activities of up to 107 g PE (mol Ni h)−1.167 The bulky ligand backbone led to high thermal stability and improved tolerance towards polar groups. These catalysts were stable at temperatures of up to 100 °C. Moreover, ethylene copolymerization with methyl 10-undecenoate was conducted in a living manner (Scheme 4). Conley and co-workers prepared some sulfated zirconia supported α-diimine nickel catalysts and applied them in ethylene copolymerization with methyl 10-undecenoate.168 Plenio and co-workers designed some bowl-shaped diimine nickel catalysts with two pentiptycenyl-substituents, and studied their properties in ethylene copolymerization with various comonomers including methyl 10-undecenoate.169
5. Conclusion and outlook
The Brookhart-type nickel and palladium complexes ligated by α-diimine have attracted considerable attention for olefin polymerization during the past two decades. Their catalytic properties are strongly influenced by ligand structures as well as polymerization conditions (temperature, types and concentration of monomers), which subsequently effectively influence the properties of the resulting polymers and copolymers. Most importantly, their chain walking capabilities provide great potential for the control of polymer microstructures.In this review, we first briefly discussed the chain walking mechanism of Brookhart-type catalysts in ethylene polymerization and α-olefin polymerization. Subsequently, some recent advances in catalyst developments were summarized. Specifically, the modifications of N-aryl substituents and of the ligand backbone structure, as well as the utilization of dinucleating ligands, were described. Finally, the applications of some of the newly developed α-diimine Ni and Pd catalysts in olefin copolymerizations with polar comonomers were demonstrated. Special attention was paid to the examples where a new polar comonomer substrate scope was enabled through the utilization of new catalysts. The Brookhart-type Ni and Pd catalysts ligated by α-diimine are easy to synthesize, extremely versatile, tolerant towards various functional groups, and capable of generating numerous polymer microstructures that are difficult to access otherwise. Because of these superior and unique properties, they continue to be of great research interest even after twenty years of extensive research. It is believed that this research enthusiasm will continue, and we will see more novel catalysts as well as novel polyolefin materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC, 21690071 and 21801002) and the Fundamental Research Funds for the Central Universities.Notes and references
- D. B. Malpass, Introduction to Industrial Polyethylene, Wiley, Hoboken, NJ, USA, 2010, pp. 85–97 Search PubMed.
- A. Nakamura, S. Ito and K. Nozaki, Chem. Rev., 2009, 109, 5215–5244 CrossRef CAS PubMed.
- Z. Chen and M. Brookhart, Acc. Chem. Res., 2018, 51, 1831–1839 CrossRef CAS PubMed.
- L. K. Johnson, C. M. Killian and M. Brookhart, J. Am. Chem. Soc., 1995, 117, 6414–6415 CrossRef CAS.
- L. K. Johnson, S. Mecking and M. Brookhart, J. Am. Chem. Soc., 1996, 118, 267–268 CrossRef CAS.
- S. D. Ittel, L. K. Johnson and M. Brookhart, Chem. Rev., 2000, 100, 1169–1204 CrossRef CAS PubMed.
- Z. Dong and Z. Ye, Polym. Chem., 2012, 3, 286–301 RSC.
- L. H. Guo, S. Y. Dai, X. L. Sui and C. L. Chen, ACS Catal., 2016, 6, 428–441 CrossRef CAS.
- Z. Jian, L. Falivene, G. Boffa, S. O. Sánchez, L. Caporaso, A. Grassi and S. Mecking, Angew. Chem., Int. Ed., 2016, 55, 14378–14383 CrossRef CAS PubMed.
- M. Chen and C. L. Chen, ACS Catal., 2017, 7, 1308–1312 CrossRef CAS.
- G. Song, W. M. Pang, W. Li, M. Chen and C. L. Chen, Polym. Chem., 2017, 8, 7400–7405 RSC.
- B. P. Yang, S. Y. Xiong and C. L. Chen, Polym. Chem., 2017, 8, 6272–6276 RSC.
- J. X. Gao, W. Cai, Y. Hu and C. L. Chen, Polym. Chem., 2019, 10, 1416–1422 RSC.
- S. Ito, Bull. Chem. Soc. Jpn., 2018, 91, 251–261 CrossRef CAS.
- Z. Cai and L. H. Do, Organometallics, 2018, 37, 3874–3882 CrossRef CAS.
- W. Zhang, P. M. Waddell, M. A. Tiedemann, C. E. Padilla, J. J. Mei, L. Y. Chen and B. P. Carrow, J. Am. Chem. Soc., 2018, 140, 8841–8850 CrossRef CAS PubMed.
- Y. Mitsushige, H. Yasuda, B. Carrow, S. Ito, M. Kobayashi, T. Tayano, Y. Watanabe, Y. Okuno, S. Hayashi, J. Kuroda, Y. Okumura and K. Nozaki, ACS Macro Lett., 2018, 7, 305–311 CrossRef CAS.
- X. Wang and K. Nozaki, J. Am. Chem. Soc., 2018, 140, 15635–15640 CrossRef CAS PubMed.
- T. Liang and C. L. Chen, Inorg. Chem., 2018, 57, 14913–14919 CrossRef CAS PubMed.
- M. Chen and C. L. Chen, Angew. Chem., Int. Ed., 2018, 57, 3094–3098 CrossRef CAS PubMed.
- J. X. Gao, B. P. Yang and C. L. Chen, J. Catal., 2019, 369, 233–238 CrossRef CAS.
- Z. Guan and C. S. Popeney, Top. Organomet. Chem., 2009, 2, 179–220 CrossRef.
- C. Tan and C. L. Chen, Angew. Chem., Int. Ed., 2019, 58 DOI:10.1002/anie.201814634.
- J. M. Kaiser and B. K. Long, Coord. Chem. Rev., 2018, 372, 141–152 CrossRef CAS.
- L. H. Guo and C. L. Chen, Sci. China: Chem., 2015, 58, 1663–1673 CrossRef CAS.
- Y. S. Chen, L. Wang, H. J. Yu, Y. L. Zhao, R. L. Sun, G. H. Jing, J. Huang, H. Khalid, N. M. Abbasi and M. Akram, Prog. Polym. Sci., 2015, 45, 23–43 CrossRef CAS.
- C. L. Chen, ACS Catal., 2018, 8, 5506–5514 CrossRef CAS.
- L. H. Guo, W. Liu and C. L. Chen, Mater. Chem. Front., 2017, 1, 2487–2494 RSC.
- D. Takeuchi, Polym. J., 2018, 50, 573–578 CrossRef CAS.
- H. Suo, G. A. Solan, Y. Ma and W. H. Sun, Coord. Chem. Rev., 2018, 372, 101–106 CrossRef CAS.
- C. L. Chen, Nat. Rev. Chem., 2018, 2, 6–14 CrossRef CAS.
- J. Wang, L. Wang, H. Yu, R. S. Ullah, M. Haroon, Z. Abdin, X. Xia and R. U. Khan, Eur. J. Inorg. Chem., 2018, 1450–1468 CrossRef CAS.
- N. E. Mitchell and B. K. Long, Polym. Int., 2019, 68, 14–26 CrossRef CAS.
- V. M. Mohring and G. Fink, Angew. Chem., Int. Ed. Engl., 1985, 24, 1001–1003 CrossRef.
- G. J. Domski, J. M. Rose, G. W. Coates, A. D. Bolig and M. Brookhart, Prog. Polym. Sci., 2007, 32, 30–92 CrossRef CAS.
- L. Pei, F. Liu, H. Liao, J. Gao, L. Zhong, H. Gao and Q. Wu, ACS Catal., 2018, 8, 1104–1113 CrossRef CAS.
- Z. Guan, P. M. Cotts, E. F. McCord and S. J. McLain, Science, 1999, 283, 2059–2062 CrossRef CAS PubMed.
- D. P. Gates, S. A. Svejda, E. Oñate, C. M. Killian, L. K. Johnson, P. S. White and M. Brookhart, Macromolecules, 2000, 33, 2320–2334 CrossRef CAS.
- F. S. Liu, H. B. Hu, Y. Xu, L. H. Guo, S. B. Zai, K. M. Song, H. Y. Gao, L. Zhang, F. M. Zhu and Q. Wu, Macromolecules, 2009, 42, 789–7796 Search PubMed.
- C. S. Popeney and Z. Guan, Organometallics, 2005, 24, 1145–1155 CrossRef CAS.
- C. S. Popeney and Z. Guan, Macromolecules, 2010, 43, 4091–4097 CrossRef CAS.
- J. Y. Liu, Y. G. Li, Y. S. Li and N. H. Hu, J. Appl. Polym. Sci., 2008, 109, 700–707 CrossRef CAS.
- C. S. Popeney, C. M. Levins and Z. Guan, Organometallics, 2011, 30, 2432–2452 CrossRef CAS.
- Z. Guan, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 3680–3692 CrossRef CAS.
- J. M. Kaiser, W. C. Anderson Jr. and B. K. Long, Polym. Chem., 2018, 9, 1567–1570 RSC.
- K. S. O'Connor, J. R. Lamb, T. Vaidya, I. Keresztes, K. Klimovica, A. M. LaPointe, O. Daugulis and G. W. Coates, Macromolecules, 2017, 50, 7010–7027 CrossRef.
- I. Pierro, G. Zanchin, E. Parisini, J. Martí-Rujas, M. Canetti, G. Ricci, F. Bertini and G. Leone, Macromolecules, 2018, 51, 801–814 CrossRef CAS.
- E. F. McCord, S. J. McLain, L. T. J. Nelson, S. D. Ittel, D. Tempel, C. M. Killian, L. K. Johnson and M. Brookhart, Macromolecules, 2007, 40, 410–420 CrossRef CAS.
- G. J. Domski, J. M. Rose, G. W. Coates, A. D. Bolig and M. Brookhart, Prog. Polym. Sci., 2007, 32, 30–92 CrossRef CAS.
- A. E. Cherian, J. M. Rose, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 13770–13771 CrossRef CAS PubMed.
- J. M. Rose, A. E. Cherian and G. W. Coates, J. Am. Chem. Soc., 2006, 128, 4186–4187 CrossRef CAS PubMed.
- D. N. Vaccarello, K. S. O'Connor, P. Iacono, J. M. Rose, A. E. Cherian and G. W. Coates, J. Am. Chem. Soc., 2018, 140, 6208–6211 CrossRef CAS PubMed.
- D. H. Camacho, E. V. Salo, J. W. Ziller and Z. Guan, Angew. Chem., Int. Ed., 2004, 43, 1821–1825 CrossRef CAS PubMed.
- D. H. Camacho and Z. Guan, Macromolecules, 2005, 38, 2544–2546 CrossRef CAS.
- C. S. Popeney, D. H. Camacho and Z. Guan, J. Am. Chem. Soc., 2007, 129, 10062–10063 CrossRef CAS PubMed.
- C. S. Popeney, C. M. Levins and Z. Guan, Organometallics, 2011, 30, 2432–2452 CrossRef CAS.
- J. L. Rhinehart, L. A. Brown and B. K. Long, J. Am. Chem. Soc., 2013, 135, 16316–16319 CrossRef CAS PubMed.
- J. L. Rhinehart, N. E. Mitchell and B. K. Long, ACS Catal., 2014, 4, 2501–2504 CrossRef CAS.
- L. A. Brown, W. C. J. Anderson, N. E. Mitchell, K. R. Gmernicki and B. K. Long, Polymers, 2018, 10, 41 CrossRef PubMed.
- S. Y. Dai, X. L. Sui and C. L. Chen, Angew. Chem., Int. Ed., 2015, 54, 9948–9953 CrossRef CAS PubMed.
- S. Y. Dai and C. L. Chen, Angew. Chem., Int. Ed., 2016, 55, 13281–13285 CrossRef CAS PubMed.
- Y. N. Na, S. Y. Dai and C. L. Chen, Macromolecules, 2018, 51, 4040–4048 CrossRef CAS.
- L. H. Guo, S. Y. Dai and C. L. Chen, Polymers, 2016, 8, 37 CrossRef PubMed.
- L. H. Guo, K. Lian, W. Kong, S. Xu, G. Jiang and S. Y. Dai, Organometallics, 2018, 37, 2442–2449 CrossRef CAS.
- S. Y. Dai, S. X. Zhou, W. Zhang and C. L. Chen, Macromolecules, 2016, 49, 8855–8862 CrossRef CAS.
- J. L. Sun, F. Z. Wang, W. M. Li and M. Chen, RSC Adv., 2017, 7, 55051–55059 RSC.
- S. Y. Dai and C. L. Chen, Macromolecules, 2018, 51, 6818–6824 CrossRef CAS.
- X. X. Wang, L. L. Fan, Y. C. Yuan, S. Z. Du, Y. Sun, G. A. Solan, C. Y. Guo and W. H. Sun, Dalton Trans., 2016, 45, 18313–18323 RSC.
- L. L. Fan, E. Yue, S. Z. Du, C. Y. Guo, X. Hao and W. H. Sun, RSC Adv., 2015, 5, 93274–93282 RSC.
- L. L. Fan, S. Z. Du, C. Y. Guo, X. Hao and W. H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2015, 53, 1369–1378 CrossRef CAS.
- S. Z. Du, Q. F. Xing, Z. Flisak, E. Yue, Y. Sun and W. H. Sun, Dalton Trans., 2015, 44, 12282–12291 RSC.
- S. Z. Du, S. L. Kong, Q. S. Shi, J. Mao, C. Y. Guo, J. J. Yi, T. L. Liang and W. H. Sun, Organometallics, 2015, 34, 582–590 CrossRef CAS.
- X. X. Wang, L. L. Fan, Y. P. Ma, C. Y. Guo, G. A. Solan, Y. Sun and W. H. Sun, Polym. Chem., 2017, 8, 2785–2795 RSC.
- H. Y. Suo, I. V. Oleynik, C. B. Huang, I. I. Oleynik, G. A. Solan, Y. P. Ma, T. L. Liang and W. H. Sun, Dalton Trans., 2017, 46, 15684–15697 RSC.
- Y. N. Zeng, Q. Mahmood, Q. Y. Zhang, T. L. Liang and W. H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2018, 56, 922–930 CrossRef CAS.
- F. Zhai and R. F. Jordan, Organometallics, 2017, 36, 2784–2799 CrossRef CAS.
- X. L. Sui, C. W. Hong, W. M. Pang and C. L. Chen, Mater. Chem. Front., 2017, 1, 967–972 RSC.
- J. Fang, X. L. Sui, Y. G. Li and C. L. Chen, Polym. Chem., 2018, 9, 4143–4149 RSC.
- S. Du, Q. Xing, Z. Flisak, E. Yue, Y. Sun and W. H. Sun, Dalton Trans., 2015, 44, 12282–12291 RSC.
- Y. Chen, S. Du, C. Huang, G. A. Solan, X. Hao and W. H. Sun, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 1971–1983 CrossRef CAS.
- R. K. Wang, M. H. Zhao and C. L. Chen, Polym. Chem., 2016, 7, 3933–3938 RSC.
- K. Lian, Y. Zhu, W. Li, S. Y. Dai and C. L. Chen, Macromolecules, 2017, 50, 6074–6080 CrossRef CAS.
- M. H. Zhao and C. L. Chen, ACS Catal., 2017, 7, 7490–7494 CrossRef CAS.
- M. Chen, B. P. Yang and C. L. Chen, Angew. Chem., Int. Ed., 2015, 54, 15520–15524 CrossRef CAS PubMed.
- W. C. Anderson Jr., J. L. Rhinehart, A. G. Tennyson and B. K. Long, J. Am. Chem. Soc., 2016, 138, 774–777 CrossRef PubMed.
- W. C. Anderson Jr., S. H. Park, L. A. Brown, J. M. Kaiser and B. K. Long, Inorg. Chem. Front., 2017, 4, 1108–1112 RSC.
- W. P. Zou, W. M. Pang and C. L. Chen, Inorg. Chem. Front., 2017, 4, 795–800 RSC.
- V. Rosar, A. Meduri, T. Montini, P. Fornasiero, E. Zangrando and B. Milani, Dalton Trans., 2018, 47, 2778–2790 RSC.
- S. Ahmadjo, S. Damavandi, G. H. Zohuri, A. Farhadipour, N. Samadieh and Z. Etemadinia, J. Organomet. Chem., 2017, 835, 43–51 CrossRef CAS.
- S. Ahmadjo, S. Damavandi, G. H. Zohuri, A. Farhadipour, N. Samadieh and Z. Etemadinia, Polym. Bull., 2017, 74, 3819–3832 CrossRef CAS.
- R. Mundil, A. Sokolohorskyj, J. Hošek, J. Cvačka, I. Císařová, J. Kvíčala and J. Merna, Polym. Chem., 2018, 9, 1234–1248 RSC.
- D. Zhang, E. T. Nadres, M. Brookhart and O. Daugulis, Organometallics, 2013, 32, 5136–5143 CrossRef CAS.
- T. Vaidya, K. Klimovica, A. M. LaPointe, I. Keresztes, E. B. Lobkovsky, O. Daugulis and G. W. Coates, J. Am. Chem. Soc., 2014, 136, 7213–7216 CrossRef CAS PubMed.
- K. S. O'Connor, A. Watts, T. Vaidya, A. M. LaPointe, M. A. Hillmyer and G. W. Coates, Macromolecules, 2016, 49, 6743–6751 CrossRef.
- K. E. Allen, J. Campos, O. Daugulis and M. Brookhart, ACS Catal., 2015, 5, 456–464 CrossRef CAS.
- J. C. Yuan, F. Z. Wang, W. B. Xu, T. J. Mei, J. Li, B. N. Yuan, F. Y. Song and Z. Jia, Organometallics, 2013, 32, 3960–3968 CrossRef CAS.
- F. Z. Wang, S. S. Tian, R. P. Li, W. M. Li and C. L. Chen, Chin. J. Polym. Sci., 2018, 36, 157–162 CrossRef CAS.
- C. Carfagna, G. Gatti, P. Paoli, B. Binotti, F. Fini, A. Passeri, P. Rossi and B. Gabriele, Organometallics, 2014, 33, 129–144 CrossRef CAS.
- C. Wen, S. Yuan, Q. Shi, E. Yue, D. Liu and W. H. Sun, Organometallics, 2014, 33, 7223–7231 CrossRef CAS.
- S. Yuan, E. Yue, C. Wen and W. H. Sun, RSC Adv., 2016, 6, 7431–7438 RSC.
- J. Liu, D. R. Chen, H. Wu, Z. F. Xiao, H. Y. Gao, F. M. Zhu and Q. Wu, Macromolecules, 2014, 47, 3325–3331 CrossRef CAS.
- K. Song, W. Yang, B. Li, Q. Liu, C. Redshaw, Y. Li and W. H. Sun, Dalton Trans., 2013, 42, 9166–9175 RSC.
- W. Zou and C. Chen, Organometallics, 2016, 35, 1794–1801 CrossRef CAS.
- L. Zhu, D. Zang, Y. Wang, Y. Guo, B. Jiang, F. He, Z. Fu, Z. Fan, M. A. Hickner, Z. K. Liu and L. Q. Chen, Organometallics, 2017, 36, 1196–1203 CrossRef CAS.
- S. Zhong, Y. Tan, L. Zhong, J. Gao, H. Liao, L. Jiang, H. Gao and Q. Wu, Macromolecules, 2017, 50, 5661–5669 CrossRef CAS.
- Z. Xiao, H. Zheng, C. Du, L. Zhong, H. Liao, J. Gao, H. Gao and Q. Wu, Macromolecules, 2018, 51, 9110–9121 CrossRef CAS.
- P. Huo, W. Y. Liu, X. H. He, H. M. Wang and Y. W. Chen, Organometallics, 2013, 32, 2291–2299 CrossRef CAS.
- P. Huo, W. Y. Liu, X. H. He, Z. Wei and Y. W. Chen, Polym. Chem., 2014, 5, 1210–1218 RSC.
- X. H. He, Y. J. Deng, Z. L. Han, Y. Yang and D. F. Chen, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3495–3505 CrossRef CAS.
- X. H. He, Y. J. Deng, X. Jiang, Z. J. Wang, Y. P. Yang, Z. L. Hana and D. F. Chen, Polym. Chem., 2017, 8, 2390–2396 RSC.
- P. Huo, J. B. Li, W. Y. Liu, G. Q. Mei and X. H. He, RSC Adv., 2017, 7, 51858–51863 RSC.
- M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed.
- S. P. Netalkar, P. P. Netalkar, M. P. Sathisha, S. Budagumpi and V. K. Revankar, Catal. Lett., 2014, 144, 181–191 CrossRef CAS.
- S. P. Netalkar, S. Budagumpi, H. H. Abdallah, P. P. Netalkar and V. K. Revankar, J. Mol. Struct., 2014, 1075, 559–565 CrossRef CAS.
- S. Kong, K. Song, T. Liang, C. Y. Guo, W. H. Sun and C. Redshaw, Dalton Trans., 2013, 42, 9176–9187 RSC.
- M. Khoshsefat, G. H. Zohuri, N. Ramezanian, S. Ahmadjo and M. Haghpanah, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 3000–3011 CrossRef CAS.
- M. Khoshsefat, S. Ahmadjo, S. M. M. Mortazavi, G. H. Zohuri and J. B. P. Soares, New J. Chem., 2018, 42, 8334–8337 RSC.
- L. Zhu, Z. S. Fu, H. J. Pan, W. Feng, C. Chen and Z. Q. Fan, Dalton Trans., 2014, 43, 2900–2906 RSC.
- Q. Xing, K. Song, T. Liang, Q. Liu, W. H. Sun and C. Redshaw, Dalton Trans., 2014, 43, 7830–7837 RSC.
- R. K. Wang, X. L. Sui, W. Pang and C. L. Chen, ChemCatChem, 2016, 8, 434–440 CrossRef CAS.
- Y. Na, X. Wang, K. Lian, Y. Zhu, W. Li, Y. Luo and C. L. Chen, ChemCatChem, 2017, 9, 1062–1066 CrossRef CAS.
- S. Takano, D. Takeuchi, K. Osakada, N. Akamatsu and A. Shishido, Angew. Chem., Int. Ed., 2014, 53, 9246–9250 CrossRef CAS PubMed.
- S. Takano, D. Takeuchi and K. Osakada, Chem. – Eur. J., 2015, 21, 16209–16218 CrossRef CAS PubMed.
- D. Takeuchi, T. Iwasawa and K. Osakada, Macromolecules, 2018, 51, 5048–5054 CrossRef CAS.
- L. Ding, H. Cheng, Y. Li, R. Tanaka, T. Shiono and Z. Cai, Polym. Chem., 2018, 9, 5476–5482 RSC.
- X. Fu, L. Zhang, R. Tanaka, T. Shiono and Z. Cai, Macromolecules, 2017, 50, 9216–9221 CrossRef CAS.
- D. B. Culver, H. Tafazolian and M. P. Conley, Organometallics, 2018, 37, 1001–1006 CrossRef CAS.
- F. Liu, H. Gao, Z. Hu, H. Hu, F. Zhu and Q. Wu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3859–3866 CrossRef CAS.
- A. Keyes, H. E. Basbug Alhan, U. Ha, Y. S. Liu, S. K. Smith, T. S. Teets, D. B. Beezer and E. Harth, Macromolecules, 2018, 51, 7224–7232 CrossRef CAS.
- H. Gao, X. Liu, Y. Tang, J. Pan and Q. Wu, Polym. Chem., 2011, 2, 1398–1403 RSC.
- H. Gao, J. Pan, L. Guo, D. Xiao and Q. Wu, Polymer, 2011, 52, 130–137 CrossRef CAS.
- T. Okada, D. Takeuchi, A. Shishido, T. Ikeda and K. Osakada, J. Am. Chem. Soc., 2009, 131, 10852–10853 CrossRef CAS PubMed.
- D. Takeuchi, J. Am. Chem. Soc., 2011, 133, 11106–11109 CrossRef CAS PubMed.
- J. Gao, Z. H. Ying, L. Zhong, H. Liao, H. Y. Gao and Q. Wu, Polym. Chem., 2018, 9, 1109–1115 RSC.
- J. Gao, L. Zhang, L. Zhong, C. Du, H. Liao, H. Gao and Q. Wu, Polymer, 2019, 164, 26–32 CrossRef CAS.
- D. Takeuchi and K. Osakada, Polymer, 2016, 82, 392–405 CrossRef CAS.
- D. Takeuchi, T. Iwasawa and K. Osakada, Macromolecules, 2018, 51, 5048–5054 CrossRef CAS.
- T. Okada, S. Park, D. Takeuchi and K. Osakada, Angew. Chem., Int. Ed., 2007, 46, 6141–6143 CrossRef CAS PubMed.
- K. Motokuni, D. Takeuchi and K. Osakada, Macromolecules, 2014, 47, 6522–6526 CrossRef CAS.
- K. Motokuni, D. Takeuchi and K. Osakada, Polym. Chem., 2015, 6, 1248–1254 RSC.
- A. E. Cherian, E. B. Lobkovsky and G. W. Coates, Chem. Commun., 2003, 20, 2566–2567 RSC.
- F. Z. Wang, R. Tanaka, Z. G. Cai, Y. Nakayama and T. Shiono, Polymer, 2017, 127, 88–100 CrossRef CAS.
- F. Z. Wang, R. Tanaka, Z. G. Cai, Y. Nakayama and T. Shiono, Macromol. Rapid Commun., 2016, 37, 1375–1381 CrossRef CAS PubMed.
- F. Z. Wang, R. Tanaka, Q. S. Li, Y. Nakayama and T. Shiono, Organometallics, 2018, 37, 1358–1367 CrossRef CAS.
- C. L. Chen, S. J. Luo and R. F. Jordan, J. Am. Chem. Soc., 2008, 130, 12892–12893 CrossRef CAS PubMed.
- C. Chen and R. F. Jordan, J. Am. Chem. Soc., 2010, 132, 10254–10255 CrossRef CAS PubMed.
- C. Chen, S. J. Luo and R. F. Jordan, J. Am. Chem. Soc., 2010, 132, 5273–5284 CrossRef CAS PubMed.
- M. Kang, A. Sen, L. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2002, 124, 12080–12081 CrossRef CAS PubMed.
- S. R. Foley Jr., R. A. Stockland, H. Shen and R. F. Jordan, J. Am. Chem. Soc., 2003, 125, 4350–4361 CrossRef PubMed.
- S. Li and Z. Ye, Macromol. Chem. Phys., 2010, 211, 1917–1924 CrossRef CAS.
- L. K. Johnson, L. Wang, S. McLain, A. Bennett, K. Dobbs, E. Hauptman, A. Ionkin, S. D. Ittel, K. Kutnisky, W. Marshall, E. McCord, C. Radzewich, A. Rinehart, K. J. Sweetman, Y. Wang, Z. Yin and M. Brookhart, ACS Symp. Ser., 2003, 857, 131–142 CrossRef CAS.
- M. Li, X. Wang, Y. Luo and C. L. Chen, Angew. Chem., Int. Ed., 2017, 56, 11604–11609 CrossRef CAS PubMed.
- F. Zhai, J. B. Solomon and R. F. Jordan, Organometallics, 2017, 36, 1873–1879 CrossRef CAS.
- J. Kiesewetter and W. Kaminsky, Chem. – Eur. J., 2003, 9, 1750–1758 CrossRef CAS PubMed.
- P. Xiang and Z. Ye, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 672–686 CrossRef CAS.
- Y. N. Na, D. Zhang and C. L. Chen, Polym. Chem., 2017, 8, 2405–2409 RSC.
- T. Runzi, D. Frohlich and S. Mecking, J. Am. Chem. Soc., 2010, 132, 17690–17691 CrossRef PubMed.
- J. C. Daigle, L. Piche and J. P. Claverie, Macromolecules, 2011, 44, 1760–1762 CrossRef CAS.
- D. Zhang and C. L. Chen, Angew. Chem., Int. Ed., 2017, 56, 14672–14676 CrossRef CAS PubMed.
- L. H. Guo, C. Zou, S. Y. Dai and C. L. Chen, Polymers, 2017, 9, 122 CrossRef PubMed.
- Z. Chen, W. J. Liu, O. Daugulis and M. Brookhart, J. Am. Chem. Soc., 2016, 138, 16120–16129 CrossRef CAS PubMed.
- Z. Chen, M. D. Leatherman, O. Daugulis and M. Brookhart, J. Am. Chem. Soc., 2017, 139, 16013–16022 CrossRef CAS PubMed.
- S. X. Zhou and C. L. Chen, Sci. Bull., 2018, 63, 441–445 CrossRef CAS.
- C. C. H. Atienza, T. Diao, K. J. Weller, S. A. Nye, K. M. Lewis, J. G. P. Delis, J. L. Boyer, A. K. Roy and P. J. Chirik, J. Am. Chem. Soc., 2014, 136, 12108–12118 CrossRef CAS PubMed.
- G. H. Michael and D. Guironnet, ACS Catal., 2017, 7, 5717–5720 CrossRef.
- B. K. Long, J. M. Eagan, M. Mulzer and G. W. Coates, Angew. Chem., Int. Ed., 2016, 55, 7222–7226 CrossRef.
- L. Zhong, G. L. Li, G. D. Liang, H. Y. Gao and Q. Wu, Macromolecules, 2017, 50, 2675–2682 CrossRef CAS.
- H. Tafazolian, D. B. Culver and M. P. Conley, Organometallics, 2017, 36, 2385–2388 CrossRef CAS.
- Y. Kanai, S. Foro and H. Plenio, Organometallics, 2019, 38, 544–551 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |