
A new and different insight into the promotion mechanisms of Ga for the hydrogenation of carbon dioxide to methanol over a Ga-doped Ni(211) bimetallic catalyst†
Qingli
Tang
ab,
Wenchao
Ji
b,
Christopher K.
Russell
c,
Yulong
Zhang
d,
Maohong
Fan
 *ae and
Zhemin
Shen
*b
*ae and
Zhemin
Shen
*b
aSchool of Energy Resouces and Departments of Chemical and Petroleum Engineering, University of Wyoming, 1000 East University Avenue, Laramie, 82071, Wyoming, USA. E-mail: mfan@uwyo.edu
bSchool of Environmental Science and Engineering, Shanghai Jiao Tong University, 800 Dongchuan Road, Minhang District, Shanghai, 200240, P.R. China. E-mail: zmshen@sjtu.edu.cn
cDepartment of Civil and Environmental Engineering, Stanford University, Stanford 94305, CA, USA
dCollege of Chemistry and Chemical Engineering, Henan Polytechnic University, Jiaozuo, 454000, P. R. China
eSchool of Civil and Environmental Engineering, Georgia Institute of Technology, North Avenue, Atlanta 30332, Georgia, USA
First published on 22nd April 2019
Abstract
The hydrogenation of CO2 to CH3OH is one of the most promising technologies for the utilization of captured CO2 in the future. Nano Ni–Ga bimetallic catalysts have been proven to be excellent catalysts in the hydrogenation of CO2 to CH3OH. To investigate the promotion mechanisms of Ga for the hydrogenation of CO2 to CH3OH over Ga-doped Ni catalysts and the wide application of these promotion mechanisms in other catalysts and reactions, herein, density functional theory (DFT) was employed. The reaction mechanisms and the properties of Ni(211) and Ga–Ni(211) surfaces were comparatively studied. The results show that the Ni sites on both the Ni(211) and the Ga–Ni(211) surfaces are active sites, and the most stable structures of the intermediates are similar. Moreover, the Ga–Ni(211) surface is more favorable for the hydrogenation of CO2, whereas Ni(211) is more favorable for the dissociation of CO2. The activation barrier of the rate-limiting step in the CH3OH formation pathway on Ni(211) is 0.54 eV higher than that on Ga–Ni(211). According to the analyses of the projected density of states (PDOS) and Hirshfeld charge transfer, the addition of Ga atoms demonstrates the reactivity of the Ga-doped Ni(211) surfaces. Most importantly, the replacement of some secondary active sites of Ni atoms with the non-active Ga atoms may lower the activities of the secondary active sites and strengthen the activities of the active sites at the step edge. These results provide a new perspective for the reaction mechanism of the hydrogenation of CO2 to CH3OH over the state-of-the-art Ga-doped catalysts.
1. Introduction
Converting carbon dioxide (CO2) to value-added and highly demanded products1–3 in the future via electrocatalytic reduction,4,5 photocatalytic reduction,4,6,7 and thermocatalytic reduction8–13 is a practical way for dealing with the captured CO2;14,15 in the regard, one of the related strategies is the hydrogenation of CO2 to methanol (CH3OH), an easily marketable chemical. Catalysis is significant for the success of state-of-the-art conversion technologies. Conventional Cu/ZnO/Al2O3 and their modified catalysts16–18 used for the production of CH3OH are well known for their advantages. However, their disadvantages, including low stability resulting from CO accumulation, are obvious.In catalytic reactions, many heterogeneous catalysts have been proven to have an “ensemble effect”;19 moreover, promoters are very important for catalysts. For example, according to Studt et al.,10 Ga–Ni bimetallic catalysts are better for the formation of CH3OH than Cu/ZnO/Al2O3 although Ni-based catalysts have been widely used for CO/CO2 methanation,20,21 CO2 decomposition,22 and CO2 reforming with methane (CH4).23 They guessed that the reverse water–gas shift (rWGS) reaction and methanation (at Ni sties) and CH3OH formation (at Ga sites) occur in parallel or in series at different sites; however, they did not provide any sufficient evidence for this hypothesis. On the other hand, in our previous studies,24,25 we found that in the process of hydrogenation of CO2 to CH3OH on Ga–Ni bimetallic catalysts, the active sites were Ni sites, whereas the Ga sites had weak or no interaction with the intermediates. Obviously, the promotion mechanisms of Ga in the Ga–Ni bimetallic catalysts cannot be fully determined by the increase in the active sites. Moreover, there are a number of studies in which the promotion mechanisms of Ga have been investigated. For example, Collins and coworkers26 experimentally studied the hydrogenation of CO2 to CH3OH on a Pd/Ga2O3 catalyst and found that the presence of Ga was critical to the generation and evolution of atomic hydrogen. Ladera et al.27 studied the use of Ga as a promoter for Cu–ZnO–ZrO2 in the hydrogenation of CO2 to CH3OH, and their results showed that the presence of Ga increased the surface area of Cu and thus the active sites; this agreed very well with a previous study conducted by Toyir et al.28 Similarly, Li and coworkers29 have claimed that Ga3+ can increase the reduction of ZnO to Zn0 and further enhance the hydrogenation of CO2 to CH3OH. Medina et al.30 suggested that Ga, a promoter for the Cu/SiO2 catalysts, could create new active sites for the hydrogenation of CO2 to CH3OH. Theoretical investigations conducted by Santiago-Rodríguez et al.31 and Fiordaliso et al.32 concluded that Ga/Cu(111) and GaPd2 might be promising catalysts for the hydrogenation of CO2 to CH3OH. Thus, the possible promotion mechanisms of Ga vary, and the Ga atom is a promising promoter in the catalytic hydrogenation of CO2 to CH3OH.
This study was designed to make progress in the understanding of the function of Ga in Ga-doped Ni bimetallic catalysts towards the hydrogenation of CO2 to CH3OH. The research started with the investigation of the stability and surface segregation of Ga–Ni(211), followed by the adsorption characteristics of intermediates and active sites on Ni(211) and Ga–Ni(211) involved in the catalytic process. Then, the activation barriers and reaction energies of the elementary steps were calculated, and main pathways for Ni(211) and Ga–Ni(211) were obtained, followed by the study of micro-kinetic modeling. PDOS of the Ni atoms on Ni(211) and Ga–Ni(211) and the electron transfer abilities of Ni(211) and Ga–Ni(211) were determined for the clarification of the roles of Ga in the catalytic hydrogenation of CO2 to CH3OH. The findings obtained in this research can inspire scientists to further prepare new catalysts. Furthermore, it can have broad impacts on the enhancement of the performance of the existing catalysts.
2. Computational details
2.1 Calculation models
In previous studies,24,25 it has been found that the Ga3Ni5(111) step surface is more active than the Ga3Ni5(221) flat surface for the promotion of the hydrogenation of CO2 to CH3OH, and the structure of the Ni(211) surface is similar to that of the Ga3Ni5(111) surface. To investigate the promotion mechanisms of Ga, the first layer of Ni(211) was replaced with Ga atoms, similar to the case of the first layer of the Ga3Ni5(111) surface. Therefore, a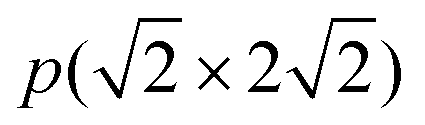 supercell of the Ni(211) slab with four layers was used, and eight Ni atoms on the first layer of the Ni(211) surface were replaced by Ga atoms; this led to the formation of a Ga–Ni(211) surface. A 15 Å vacuum was used to separate the slab to prevent periodic interactions. The top two layers and the adsorbates were relaxed, whereas the bottom two layers were fixed at their bulk positions. The structures of the Ni(211) and Ga–Ni(211) surfaces are shown in Fig. 1, and the corresponding bond lengths are listed in Table 1, in which the Ga3Ni5(111) surface has been used for comparison.
supercell of the Ni(211) slab with four layers was used, and eight Ni atoms on the first layer of the Ni(211) surface were replaced by Ga atoms; this led to the formation of a Ga–Ni(211) surface. A 15 Å vacuum was used to separate the slab to prevent periodic interactions. The top two layers and the adsorbates were relaxed, whereas the bottom two layers were fixed at their bulk positions. The structures of the Ni(211) and Ga–Ni(211) surfaces are shown in Fig. 1, and the corresponding bond lengths are listed in Table 1, in which the Ga3Ni5(111) surface has been used for comparison.
 | ||
| Fig. 1 Side views and top views of the optimized surface structures of the Ni(211), Ga–Ni(211) and Ga3Ni5(111) surfaces; the values in the figure are the corresponding bond lengths, which are in Å. | ||
| No. | Bond length/Å | ||
|---|---|---|---|
| Ni(211) | Ga–Ni(211) | Ga3Ni5(111) | |
| SE1 | 2.480 | 2.464 | 2.549 |
| SE2 | 2.480 | 2.500 | 2.517 |
| S1 | 2.423 | 2.404 | 2.447 |
| S2 | 2.423 | 2.458 | 2.536 |
| S3 | 2.423 | 2.400 | 2.395 |
| S4 | 2.423 | 2.391 | 2.423 |
| S5 | 2.480 | 2.441 | 2.452 |
| S6 | 2.480 | 2.534 | 2.613 |
| L1 | 2.447 | 2.426 | 2.394 |
| L2 | 2.447 | 2.554 | 2.618 |
| L3 | 2.447 | 2.428 | 2.480 |
| L4 | 2.447 | 2.687 | 2.394 |
| L5 | 2.481 | 2.478 | 2.558 |
| L6 | 2.481 | 2.498 | 2.537 |
2.2 Calculation methods
The 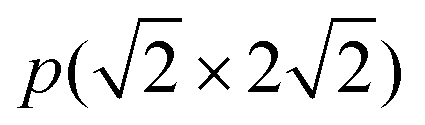 supercell of the four-layer Ni(211) slabs was used for the Ni(211) and Ga-doped Ni(211) surfaces. The k-points grid of 4 × 2 × 1 was used for the integration of the Brillouin zone. The chosen cutoff energy and Sigma value were tested. Moreoevr, 400 and 500 eV cutoff energies were employed for the Ni(211) and Ga-doped Ni(211) surfaces, respectively. In addition, 0.12 and 0.10 eV sigma values were obtained for the Ni(211) and Ga-doped Ni(211) surfaces, respectively. All calculations were conducted by considering spin polarization due to the magnetic properties of Ni.
supercell of the four-layer Ni(211) slabs was used for the Ni(211) and Ga-doped Ni(211) surfaces. The k-points grid of 4 × 2 × 1 was used for the integration of the Brillouin zone. The chosen cutoff energy and Sigma value were tested. Moreoevr, 400 and 500 eV cutoff energies were employed for the Ni(211) and Ga-doped Ni(211) surfaces, respectively. In addition, 0.12 and 0.10 eV sigma values were obtained for the Ni(211) and Ga-doped Ni(211) surfaces, respectively. All calculations were conducted by considering spin polarization due to the magnetic properties of Ni.
Surface stability can represent the degree of ease or difficulty of surface formation. The stability of Ga–Ni(211) was investigated by calculating the formation energy (Ef) of the surface. Surface segregation is a common phenomenon in alloys; thus, the surface segregation of Ga–Ni(211) has been determined. The energies of the configurations with one Ni atom of the first, second and fourth layer of the Ni(211) surfaces replaced by Ga were calculated. Note that the first and the second layers represent the surface and the subsurface, respectively, and the fourth layer represents the bulk Ni catalyst. Thus, three types of replaced sites, i.e. those at the step edge, slope and low edge, were examined.
The adsorption energies of the intermediates and the activation barriers and reaction energies of the elementary steps of CO2 hydrogenation to CH3OH on the Ni(211) and Ga–Ni(211) surfaces were calculated. A total energy below 1.0 × 10−5 eV was used to stop the geometric optimization. CI-NEB39 was used to search the transition states, where the maximum force of 0.05 eV Å−1 was used to stop the algorithm. Transition states were verified by frequency calculations.
3. Results and discussion
3.1 The adsorption behaviors of intermediates on the Ni(211) and Ga–Ni(211) surfaces
| Ef = (EGa–Ni(211) + nENi − ENi(211) − nEGa)/n | (1) |
The calculated formation energy for Ga–Ni(211) is −1.34 eV, indicating the ease of surface formation. Based on this definition, the positive Ef value suggests that the formation process is endothermic. The larger the value, the weaker the surface.46 Negative Ef value represents both exothermic and easy characteristics of surface formation.
| Esegr = Esurface1(or Esurface2) − Esurface4 | (2) |
The calculated segregation energies are displayed in Table 2. All the energy values are positive except that at the step edge of the first layer; this indicates that surface segregation cannot occur at the slope and low edge on both the first and second layers. Although slight surface segregation can take place at the step edge of the first layer, it does not affect the surface used in this study because only the slope and low edge Ni atoms have been replaced on the Ga–Ni(211) surface.
| Sites | E segr | ||
|---|---|---|---|
| Step edge | Slope | Low edge | |
| First layer | −0.37 | 0.01 | 0.39 |
| Second layer | 0.30 | 0.55 | 0.65 |
| Eads = Esys − (Eslab + Eadsorbate) + ΔZPE | (3) |
The intermediates involved in the hydrogenation of CO2 to CH3OH on the Ni(211) and Ga–Ni(211) surfaces and their adsorption energies are displayed in Fig. 2. For comparison, their adsorption energies on Ga3Ni5(111) are also listed. All the adsorption sites for these intermediates have been examined on the Ni(211) and Ga–Ni(211) surfaces, and the most stable structures are displayed in Fig. S1 and S2 of the ESI.†
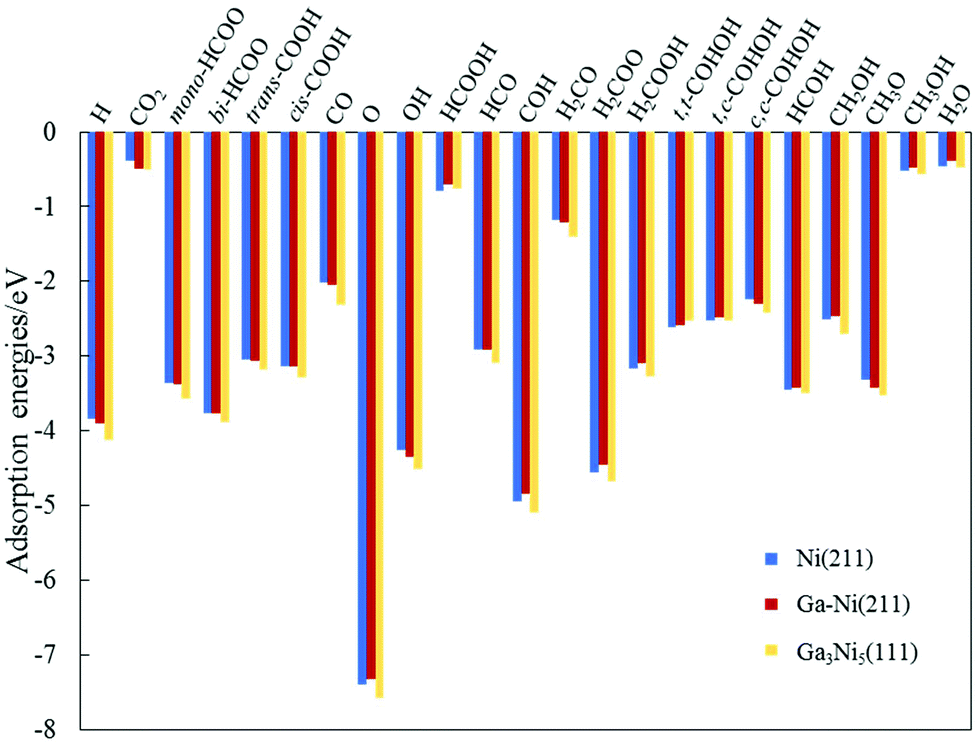 | ||
| Fig. 2 Values of the adsorption energies of the intermediates involved in the hydrogenation of CO2 to CH3OH on the Ni(211) and Ga–Ni(211) surfaces. The values obtained for the Ga3Ni5(111) surface are used for comparison.25 | ||
Considering the most stable configurations of the intermediates on Ni(211) and Ga–Ni(211), as shown in Fig. S1 and S2,† respectively, all the adsorption processes of the intermediates occur at the Ni sites along the step edge. CO2, OH, mono-HCOO, bi-HCOO, trans-COOH, cis-COOH, t,t-COHOH, t,c-COHOH, c,c-COHOH, HCO, HCOH, CH2OH and CH3O prefer to adsorb at the SE-bridge sites. On the other hand, O, H, CO, H2CO, H2COOH, and COH likely choose to adsorb at the SE-3-Fold hollow sites. HCOOH has high affinity for adsorption at the P-Bridge sites; H2COO prefers to adsorb at the SE-4-Fold hollow sites, whereas H2O and CH3OH have high selectivity for the SE-Atop sites. Fig. 2 shows that the adsorption energies of most of the intermediates on Ni(211) and Ga–Ni(211) are lower than the corresponding values on the Ga3Ni5(111) surface25 and higher than those on the Cu(111)49 and ZnO(0001)50 surfaces; this means that the Ga3Ni5(111) surface more strongly interacts with the intermediates. Note that the adsorption energy of CO2 on Ga–Ni(211) is −0.49 eV, which is very close to that on the Ga3Ni5(111) surface (−0.51 eV), whereas the corresponding value on the Ni(211) surface is −0.38 eV.
In addition, the Hirshfeld charges of CO2 and CO2 adsorbed on the Ni(211) and Ga–Ni(211) surfaces were calculated, and the results are displayed in Table 3. Table 3 shows that charge transfer to CO2 from Ni(211) (0.12 e) is less than that from Ga–Ni(211) (0.14e); this indicates that the interaction between CO2 and Ga–Ni(211) is stronger.51,52 Thus, the adsorption energy of CO2 on Ni(211) is 0.11 eV lower than that on Ga–Ni(211).
| Species | q/e | ||
|---|---|---|---|
| CO2 | CO2/Ni(211) | CO2/Ga–Ni(211) | |
| C | 0.30 | 0.16 | 0.15 |
| O1 | −0.15 | −0.14 | −0.15 |
| O2 | −0.15 | −0.14 | −0.14 |
| Sum | 0 | −0.12 | −0.14 |
3.2 The main reaction pathways of the hydrogenation of CO2 to CH3OH on the Ni(211) and Ga–Ni(211) surfaces
To investigate the main reaction pathways of the hydrogenation of CO2 to CH3OH on Ni(211) and Ga–Ni(211), activation barriers and reaction energies of the elementary steps were calculated, which are listed in Table S1 of the ESI.† The structures of the initial states (ISs), transition states (TSs), and final states (FSs) of all the elementary steps on Ni(211) and Ga–Ni(211) are displayed in Fig. S3 and S4 of the ESI.† The activation barrier (Ea) and reaction energy (ΔE) were calculated using the following equations:| EZPEa = ETS − EIS + ΔZPE | (4) |
| ΔE = EFS − EIS + ΔZPE | (5) |
The reaction rate constants were calculated using the equations reported in a previous study.25
3.2.1.1 The dissociation of H2. Table S1† indicates that the dissociation of H2 is spontaneous on all the considered surfaces; this suggests that the existing adsorption form of H2 is dissociative adsorption, and two dissociated H atoms adsorb at two adjacent SE-3-Fold-Hollow sites. The reaction energies on the Ni(211) and Ga–Ni(211) surfaces are −0.72 and −0.76 eV, respectively, which mean that H2 dissociation is highly exothermic. The calculated results for the dissociation of H2 on Ni(211) agree very well with those obtained by Zhi et al.53
3.2.1.2 The hydrogenation and dissociation of CO2. The hydrogenation or dissociation of CO2 is the first step of the CH3OH formation process. As previously reported,24,25 the first step may proceed via three possible ways: dissociation of CO2 to CO and O, hydrogenation of CO2 to COOH, and hydrogenation of CO2 to HCOO. Table S1† shows that the activation barriers of R2, R3, and R4 are 0.85, 0.88 and 0.74 eV on the Ni(211) surface, and they are 0.64, 0.61, and 0.95 eV on the Ga–Ni(211) surface, which are clearly displayed in Fig. 3(a) and (b), respectively. The results show that the easiest CO2 dissociation occurs on the Ni(211) surface, whereas CO2 prefers to undergo hydrogenation on the Ga–Ni(211) surface; this is consistent with the results obtained for the previously studied Ga3Ni5(111) surface.25 Furthermore, on the Ni(211) surface, the active sites will deactivate rapidly due to CO poisoning. Generally, CO is considered as an inevitable by product in the process of CO2 hydrogenation to CH3OH on Cu-based catalysts, resulting in low selectivity of CH3OH.54
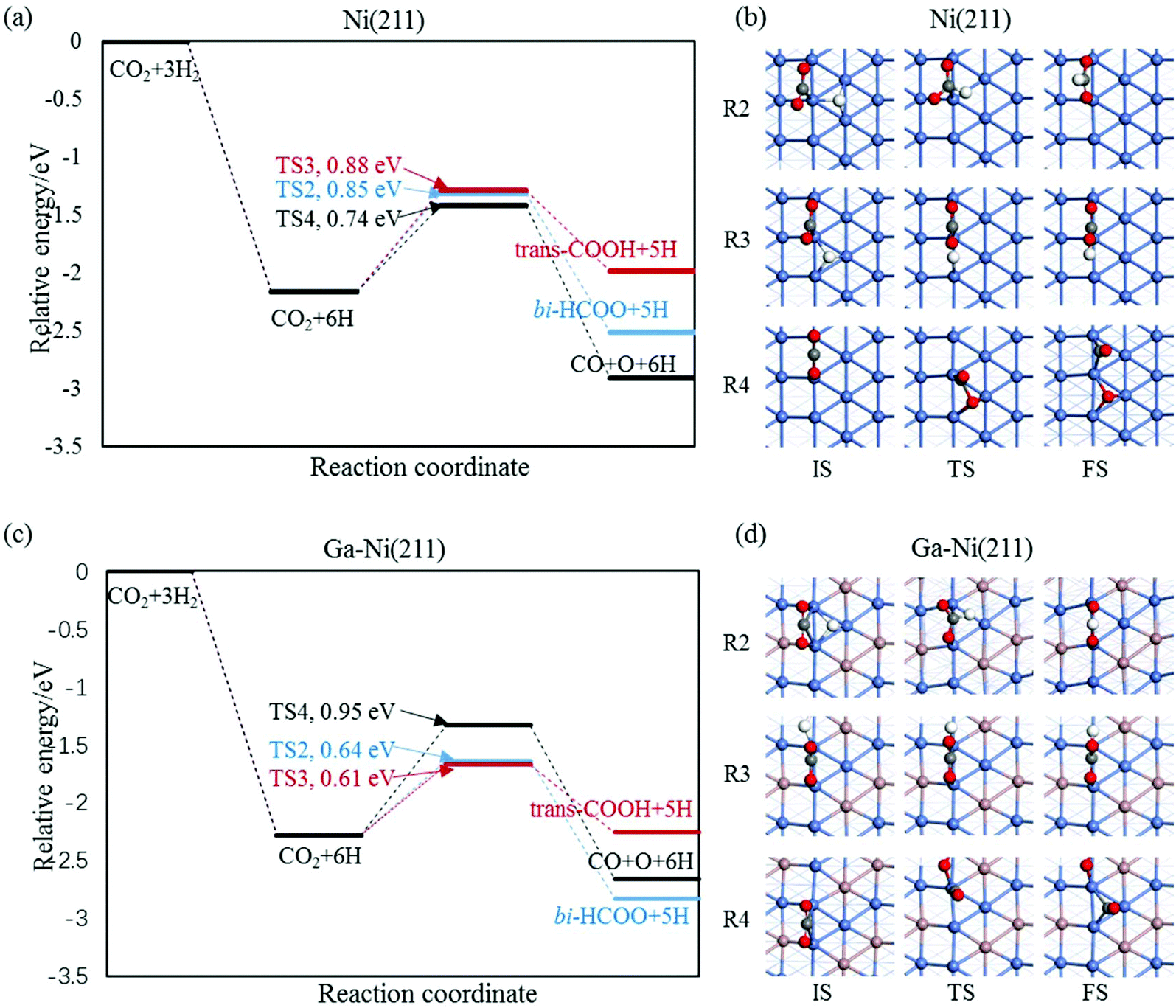 | ||
| Fig. 3 Reaction barriers of CO2 hydrogenation and dissociation on (a) the Ni(211) and (c) Ga–Ni(211) surfaces, and their elementary steps on (c) the Ni(211) and (d) Ga–Ni(211) surfaces. | ||
3.2.1.3 Successive reactions. During the reaction of bi-HCOO, the introduction of Ga increases the activation barriers for the dissociation of bi-HCOO into HCO and O and its hydrogenation to H2COO by 0.15 and 0.13 eV, respectively, whereas it lowers the activation barrier for its hydrogenation to HCOOH by 0.13 eV. Note that the hydrogenation of bi-HCOO is highly endothermic; this indicates that the reverse reaction readily occurs; this is highly consistent with the results reported by Vesselli et al.22 The activation barriers for the formation of H2COOH from HCOOH on the Ni(211) and Ga–Ni(211) surfaces are almost equal (0.88 and 0.90 eV, respectively), whereas the activation barrier for the formation of H2COO on Ga–Ni(211) is 0.17 eV lower than that on the Ni(211) surface; moreover, the activation barriers for the dissociation of HCOOH into HCO and OH and the hydrogenation of HCO to H2CO and H2CO to CH2OH and CH3O on the Ga–Ni(211) surface are lower than those on the Ni(211) surface. For the transition of isomers including trans-COOH and cis-COOH, mono-HCOO and bi-HCOO, and t,t-COHOH, t,c-COHOH and c,c-COHOH, their corresponding activation barriers on both Ni(211) and Ga–Ni(211) are almost equal. The activation barriers of the hydrogenation of trans-COOH and the dissociation of c,c-COHOH are higher on Ga–Ni(211) than those on the Ni(211) surface; however, they are low enough to make the corresponding hydrogenation and dissociation occur. On both the Ni(211) and the Ga–Ni(211) surfaces, the hydrogenation of CO has higher activation barrier. On the Ga–Ni(211) surface, the activation barriers of the hydrogenation of COH to HCOH and HCOH to CH2OH are 0.08 and 0.10 eV lower than those on the Ni(211) surface, respectively. Note that the activation barrier of H2O formation on the Ga–Ni(211) surface is 0.54 eV lower than that on the Ni(211) surface; this means that on the Ni(211) surface, OH has reached overcapacity status.
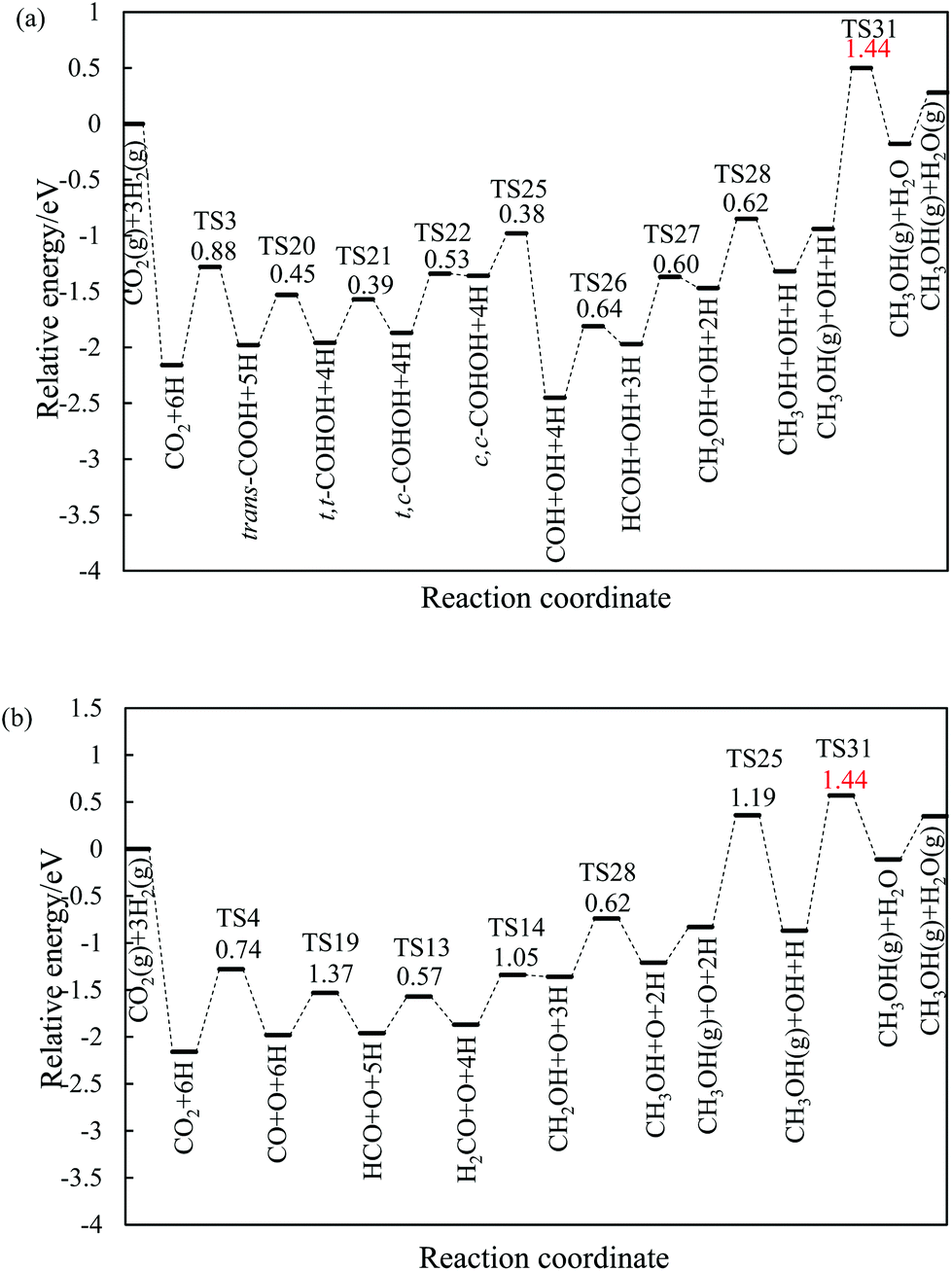 | ||
| Fig. 4 (a) COOH pathway and (b) rWGS + CO hydrogenation pathway of the CO2 hydrogenation to methanol on the stepped Ni(211) surface. The values in the figure are the corresponding reaction barriers, which are in eV. | ||
On the Ga–Ni(211) surface, the HCOO pathway starts with the hydrogenation of CO2 to bi-HCOO (EZPEa = 0.64 eV) followed by HCOOH (EZPEa = 0.95 eV). Compared with the hydrogenation (EZPEa = 0.90 eV) and desorption of HCOOH (EZPEa = 0.70 eV), the dissociation of HCOOH (EZPEa = 0.65 eV) is much easier. Then, the generated HCO is hydrogenated to CH3OH via H2CO (EZPEa = 0.84 eV) and CH2OH (EZPEa = 0.60 eV) intermediates. OH can be hydrogenated to H2O with the activation barrier of 0.90 eV. Thus, HCOOH formation is the rate-limiting step in this pathway with the reaction barrier of 0.95 eV. In the COOH pathway, CO2 is first hydrogenated to trans-COOH (EZPEa = 0.61 eV) and then to t,t-COHOH (EZPEa = 0.60 eV). t,t-COHOH is subsequently transformed into t,c-COHOH (EZPEa = 0.38 eV) and c,c-COHOH (EZPEa = 0.47 eV) isomers, followed by its dissociation into COH and OH (EZPEa = 0.75 eV). COH is further hydrogenated to CH3OH (EZPEa = 0.60 eV) via HCOH (EZPEa = 0.56 eV) and CH2OH (EZPEa = 0.50 eV). The rate-limiting step is the formation of H2O (EZPEa = 0.90 eV). The pathway of rWGS + CO hydrogenation has the same intermediates as those in the case of the Ni(211) surface, in which CO2 is first dissociated into CO and O (EZPEa = 0.95 eV), and then, hydrogenation of CO to HCO (EZPEa = 1.28 eV) occurs. HCO is consecutively hydrogenated to H2CO (EZPEa = 0.39 eV), CH2OH (EZPEa = 0.84 eV), and CH3OH (EZPEa = 0.60 eV). The activation barriers of the OH and H2O formations are 1.20 and 0.90 eV, respectively. The rate-limiting step of the rWGS + CO hydrogenation pathway is the hydrogenation of CO to HCO with the activation barrier of 1.28 eV, a 0.38 eV increase as compared to that in the COOH pathway.
In brief, on the Ga–Ni(211) surface, initial generation of COOH is the main pathway with the activation barrier of the rate-limiting step being 0.90 eV, as shown in Fig. 5. The value is 0.42 eV lower than that calculated by Behrens et al.56 on the Cu (211) surface. To the best of our knowledge, the COOH pathway as the main pathway has only been reported on the Cu(111) surfaces with the presence of H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 49 and the Ga3Ni5(221)24 and Ga3Ni5(111)25 surfaces. In addition, the dissociation of CO2 is more difficult than its hydrogenation because the activation energy of the former is 0.30 eV higher than that of the latter, and thus, the accumulation of CO is less likely to occur. Therefore, the doping of Ga into Ni can lower the activation barrier of the rate-limiting step in CH3OH synthesis.
49 and the Ga3Ni5(221)24 and Ga3Ni5(111)25 surfaces. In addition, the dissociation of CO2 is more difficult than its hydrogenation because the activation energy of the former is 0.30 eV higher than that of the latter, and thus, the accumulation of CO is less likely to occur. Therefore, the doping of Ga into Ni can lower the activation barrier of the rate-limiting step in CH3OH synthesis.
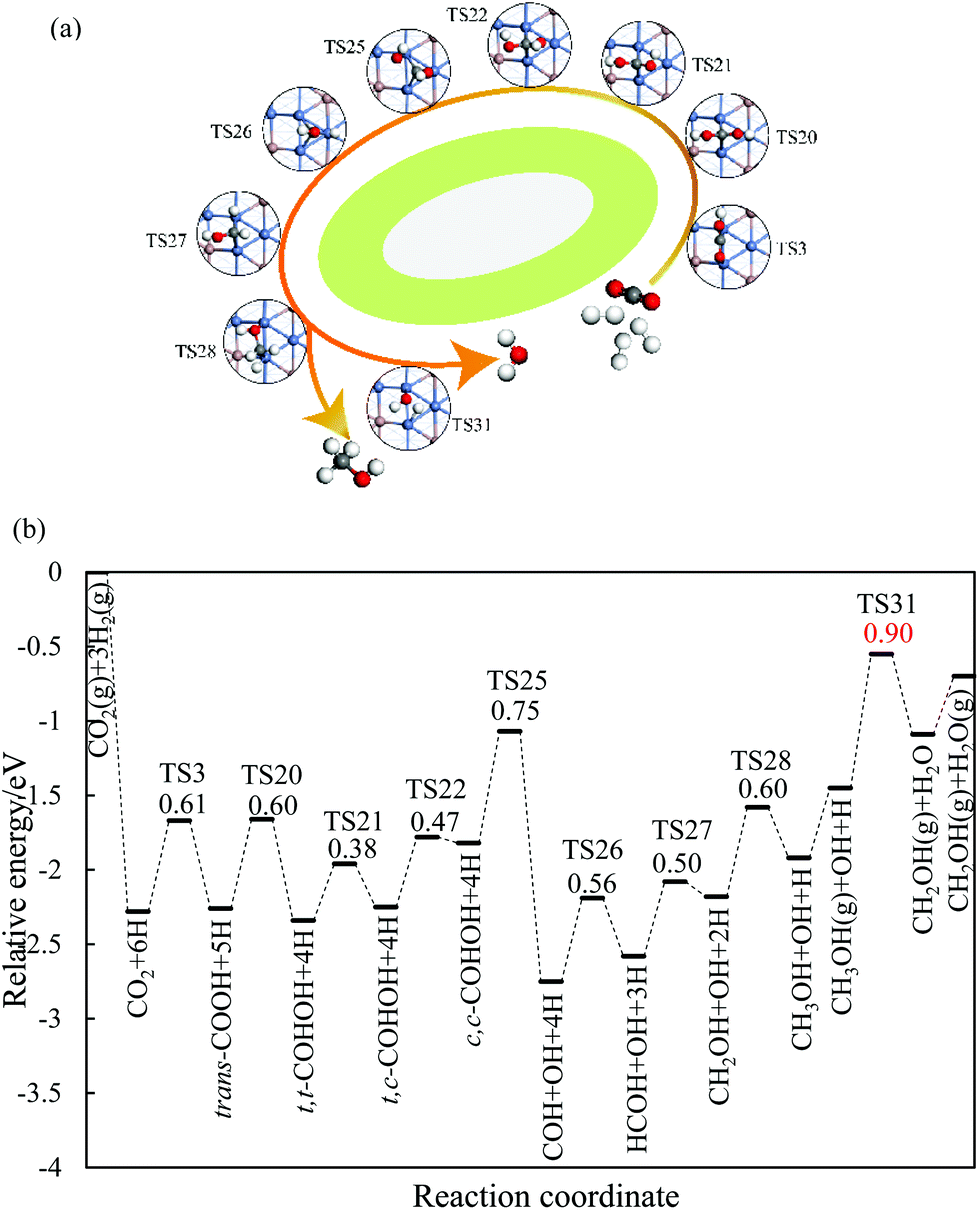 | ||
| Fig. 5 Most possible reaction pathway of CO2 hydrogenation to methanol on the Ga–Ni(211) surface. The values in the figure are the corresponding reaction barriers, which are in eV. | ||
Interestingly, many previous studies have indicated that the use of a second metal in addition to the abovementioned Ga can change the activities of the catalysts. For example, Ling et al.57 studied the role of Fe in Cu-based catalysts for the conversion of syngas to C2 oxygenates and ethanol. Their results show that both the Fe and the Fe–Cu sites are active, indicating that Fe can create new active sites. In addition, Zhang et al.58 studied C2 oxygenates and ethanol syntheses using Co-doped Cu bimetallic catalysts, and the results show that Co can promote C–C formation due to the synergetic effect between Cu and Co. Therefore, bimetallic catalysts are a promising perspective for the preparation of new catalysts.
:CO2 = 3:1) on the Ga–Ni(211) surface. The calculated reaction rate constants that will be used in the modeling are displayed in Table S2 of the ESI.† Under the estimation conditions, only the reactions related with the main pathway were considered, as presented in Table 4. The reaction rate for the hydrogenation of CO2 to trans-COOH is 5 orders of magnitude higher than that of CO2 dissociation into CO and O, indicating that Ga–Ni(211) prefers CO2 hydrogenation instead of dissociation.
| k/s−1 | |||||||
|---|---|---|---|---|---|---|---|
| Reactions | 500 K | 525 K | 550 K | 575 K | 600 K | ||
| 1 | H2 → H + H | k 1 | 1.59 × 1013 | 1.57 × 1013 | 1.56 × 1013 | 1.54 × 1013 | 1.53 × 1013 |
| 3 | CO2 + H → trans-COOH | k 3 | 2.53 × 108 | 5.23 × 108 | 1.02 × 109 | 1.86 × 109 | 3.26 × 109 |
| 4 | CO2 → CO + O | k 4 | 3.00 × 103 | 9.05 × 103 | 2.47 × 104 | 6.20 × 104 | 1.44 × 105 |
| 16 | trans-COOH → cis-COOH | k 16 | 2.08 × 108 | 3.60 × 108 | 5.95 × 108 | 9.43 × 108 | 1.44 × 109 |
| 17 | cis-COOH → CO + OH | k 17 | 6.23 × 103 | 1.81 × 104 | 4.78 × 104 | 1.16 × 105 | 2.63 × 105 |
| 20 | trans-COOH + H → t,t-COHOH | k 20 | 2.96 × 106 | 6.05 × 106 | 1.16 × 107 | 2.11 × 107 | 3.65 × 107 |
| 21 | t,t-COHOH → t,c-COHOH | k 21 | 3.27 × 109 | 5.21 × 109 | 7.98 × 109 | 1.18 × 1010 | 1.69 × 1010 |
| 22 | t,c-COHOH → c,c-COHOH | k 22 | 4.43 × 108 | 7.72 × 108 | 1.28 × 109 | 2.04 × 109 | 3.12 × 109 |
| 23 | t,t-COHOH → COH + OH | k 23 | 5.19 × 101 | 1.93 × 102 | 6.37 × 102 | 1.90 × 103 | 5.18 × 103 |
| 24 | t,c-COHOH → COH + OH | k 24 | 1.39 × 103 | 4.47 × 103 | 1.30 × 104 | 3.44 × 104 | 8.42 × 104 |
| 25 | c,c-COHOH → COH + OH | k 25 | 5.78 × 105 | 1.39 × 106 | 3.09 × 106 | 6.43 × 106 | 1.26 × 107 |
| 26 | COH + H → HCOH | k 26 | 2.41 × 107 | 4.70 × 107 | 8.65 × 107 | 1.51 × 108 | 2.52 × 108 |
| 27 | HCOH + H → CH2OH | k 27 | 3.07 × 107 | 5.62 × 107 | 9.75 × 107 | 1.62 × 108 | 2.57 × 108 |
| 28 | CH2OH + H → CH3OH | k 28 | 4.16 × 106 | 8.50 × 106 | 1.63 × 107 | 2.96 × 107 | 5.13 × 107 |
| 31 | OH + H → H2O | k 31 | 8.65 × 103 | 2.47 × 104 | 6.40 × 104 | 1.53 × 105 | 3.42 × 105 |
The detailed micro-kinetic modeling calculation process is presented in the ESI.† The formation rates of CH3OH and CO were calculated using the following equations:
| kCH3OH = k28 × θCH2OH × θH | (6) |
| kCO = k4 × θCO2 | (7) |
The productivities of CH3OH and CO are 0.097 and 0.20 per site at 500 K, and therefore, the relative selectivity values of CH3OH and CO are 32.36% and 67.64%, respectively, which are very close to the results obtained for Ga3Ni5 studied by Studt et al.10 Similarly, the productivities of CH3OH and CO at 525, 550, 575, and 600 K are obtained, and the relative selectivity of CH3OH increases with an increase in temperature.
3.3 General discussion
To analyze the promotion mechanism of Ga atom in Ga-doped Ni catalysts, PDOS of Ni in Ni(211) and Ga–Ni(211) was calculated. The calculated results for the d-band centers are displayed in Fig. 6(a). Both the d-band center and the peak of the Ni atom in the Ga–Ni(211) surface shift to Fermi level when compared with those in the Ni(211) surface, and the proximity to Fermi level is beneficial to the improvement of the activity of the catalyst according to previous studies.52,61,62 Hence, the Ga–Ni(211) surface is more active than Ni(211).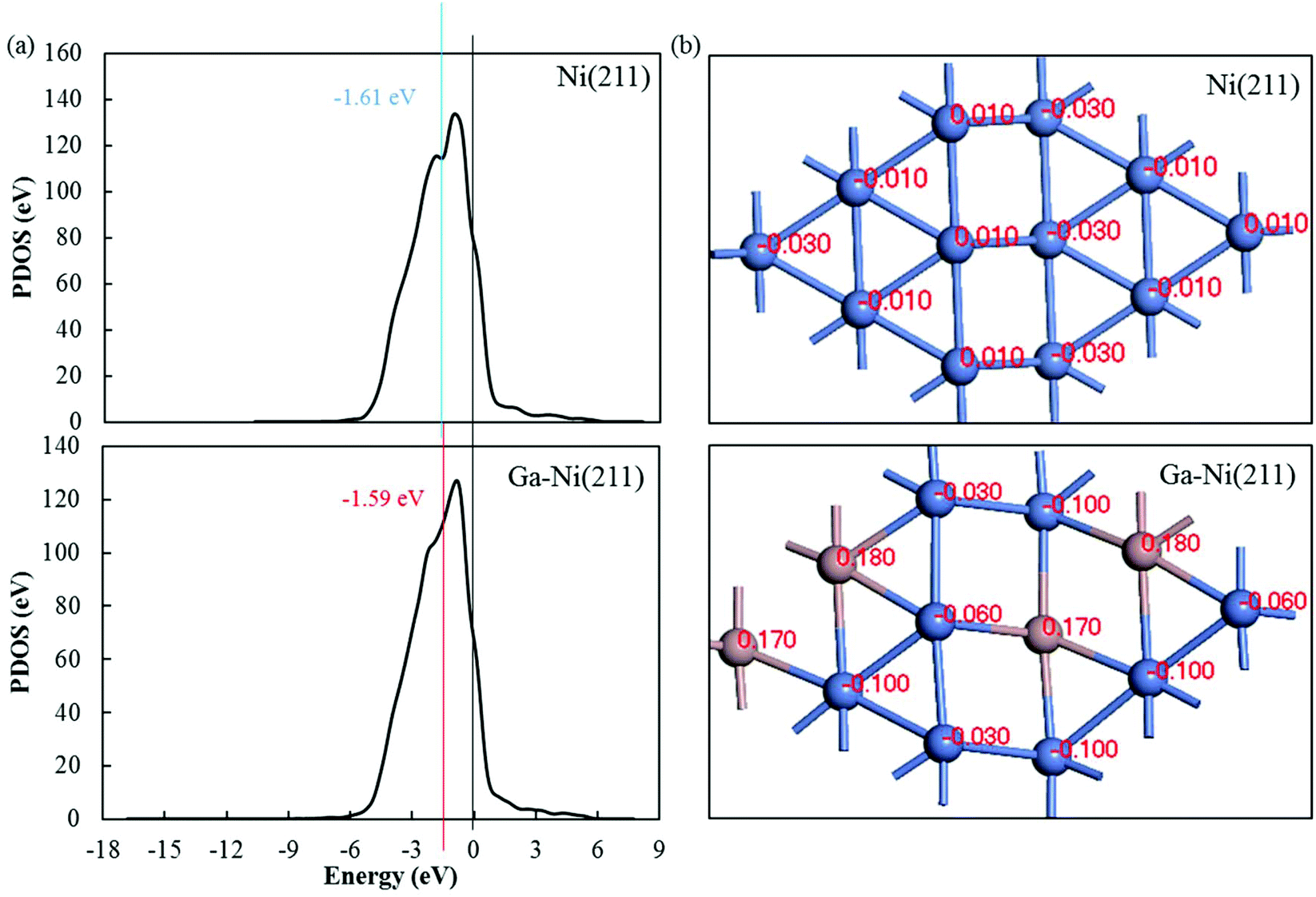 | ||
| Fig. 6 (a) Difference in the d-band center of the Ni atom in Ni(211) and Ga–Ni(211) and (b) Hirshfeld charge of the atoms on the first layer of the Ni(211) and Ga–Ni(211) surfaces. | ||
To further study the promotion mechanism of Ga, the Hirshfeld charges of the atoms on the first layer of the Ni(211) and Ga–Ni(211) surfaces were studied, and the results are shown in Fig. 6(b). As abovementioned, the Ni(211) surface has a step edge effect on CO2 hydrogenation; this means that the atoms at the step edge are more active than those at the slope and low edge. Replacements with Ga atoms were conducted to substitute some of the slope and low edge Ni atoms, that is, Ga atoms partially replace the second active sites. Fig. 6(b) shows that the Hirshfeld charge of each Ni atom at the step edge is 0.01 on Ni(211). After the introduction of Ga, two charges (−0.03 and −0.06) occurred. The charge of the slope Ni atoms changed from −0.01 to −0.10. The charge of the lower edge Ni atoms changed from −0.03 to −0.10. This demonstrates that the addition of Ga to the Ni(211) atoms leads to a decrease in the Hirshfeld charge of the Ni atoms; this indicates that the Ni atoms obtain electrons from the Ga atoms. It is generally known that chemical synthesis reactions highly depend on electron transfer processes, and many metal catalysts are electron donors, especially for reduction reactions. Therefore, the introduction of Ga atoms increases the electron losing or reduction abilities of Ni atoms that are the active sites for CH3OH formation on Ga–Ni(211), especially the atoms along the step edge. This is also demonstrated by the adsorption of CO2 and the Hirshfeld charge transfers between CO2 and Ni(211) and Ga–Ni(211), as shown in Table 3. Therefore, the promotion mechanism of Ga in Ga-doped Ni bimetallic catalysts involves the replacement of a part of the secondary active centers or sites and enhancement of the activity of the active centers or sites, which is different from the previously reported mechanisms.10 In summary, the conditions for the catalyst to have high-performance are as follows: the replaced atoms should not be the active sites and should transfer electrons to the active sites.
4. Conclusions
In this study, DFT was employed to study the promotion mechanisms of Ga over Ga-doped Ni bimetallic catalysts for the hydrogenation of CO2 to CH3OH. The adsorption behaviors of the intermediates on Ni(211) and Ga–Ni(211) were studied. The results show that the active sites of the intermediates on both surfaces are Ni sites along the step edge, and the differences in adsorption energies of most intermediates on both surfaces are lower than 0.10 eV except for CO2, COH and CH3O. From the aspect of electron transfers, the electrons transfer to CO2 from Ga–Ni(211) (0.14 e) is more than that (0.12 e) from the Ni(211) surface; this means that the interaction between CO2 and Ga–Ni(211) is stronger; this consistent with the calculated adsorption energies.In addition, the activation barriers and reaction energies of the elementary steps during the process on both Ni(211) and Ga–Ni(211) were calculated. The results show that on both surfaces, H2 dissociation is spontaneous. Moreover, the Ga–Ni(211) surface is favorable for CO2 hydrogenation, whereas the Ni(211) surface is advantageous for CO2 dissociation. The activation barriers of CO2 hydrogenation to COOH (0.61 eV) and HCOO (0.64 eV) on the Ga–Ni(211) surface are 0.34 and 0.31 eV lower than those for CO2 dissociation on the surface, respectively. On the Ni(211) surface, the activation barrier of CO2 dissociation (0.74 eV) is 0.14 eV and 0.11 eV higher than that of CO2 hydrogenation to COOH and HCOO, respectively. The introduction of Ga into the Ni(211) surface does not lower all the activation barriers of the elementary steps in the process of CO2 hydrogenation to CH3OH; however, it can change the reaction pathway of CO2 hydrogenation. Moreover, the PDOS and Hirshfeld charge of Ni in both the Ni(211) and the Ga–Ni(211) surfaces were calculated. The results show that the PDOS of Ga–Ni(211) is closer to the Fermi level; this means that the Ga–Ni(211) surface is more active. Moreover, the introduction of Ga into Ni increases the reducing ability of the active sites (along the step edge) of Ni via transfer of electrons; this further enhances the catalytic activity of the catalyst. This new finding may provide an insight into the theoretical guidance for the development of high-performance catalysts for the hydrogenation of CO2 to CH3OH.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The computational calculation in this study was performed using the resources of the Advanced Research Computing Center (2012. Mount Moran: IBM System X cluster. Laramie, WY: University of Wyoming) and the USNSF-sponsored NCAR-Wyoming Supercomputing Center (NWSC). This work was financially supported by the China Scholarship Council (CSC).References
- W. Umchoo, C. Sriakkarin, W. Donphai, C. Warakulwit, Y. Poo-arporn, P. Jantaratana, T. Witoon and M. Chareonpanich, Green and sustainable methanol production from CO2 over magnetized Fe-Cu/core-shell and infiltrate mesoporous silica-aluminosilicates, Energy Convers. Manage., 2018, 159, 342–352 CrossRef CAS.
- Y. Li, S. H. Chan and Q. Sun, Heterogeneous catalytic conversion of CO2: A comprehensive theoretical review, Nanoscale, 2015, 7, 8663–8683 RSC.
- J. Gao, Q. Jiang, Y. Liu, W. Liu, W. Chu and D. S. Su, Probing the enhanced catalytic activity of carbon nanotube supported Ni-LaOx hybrids for the CO2 reduction reaction, Nanoscale, 2018, 10, 14207–14219 RSC.
- W. Wang, J. X. Zhang, H. Wang, L. J. Chen and Z. Y. Bian, Photocatalytic and electrocatalytic reduction of CO2 to methanol by the homogeneous pyridine-based systems, Appl. Catal., A, 2016, 520, 1–6 CrossRef CAS.
- X. Cui, W. An, X. Liu, H. Wang, Y. Men and J. Wang, C2N-graphene supported single-atom catalysts for CO2 electrochemical reduction reaction: Mechanistic insight and catalyst screening, Nanoscale, 2018, 10, 15262 RSC.
- Z. Jiang, X. Liang, H. Zheng, Y. Liu, Z. Wang, P. Wang, X. Zhang, X. Qin, Y. Dai, M.-H. Whangbo and B. Huang, Photocatalytic reduction of CO2 to methanol by three-dimensional hollow structures of Bi2WO6 quantum dots, Appl. Catal., B, 2017, 219, 209–215 CrossRef CAS.
- Y.-H. Cheng, V.-H. Nguyen, H.-Y. Chan, J. C. S. Wu and W.-H. Wang, Photo-enhanced hydrogenation of CO2 to mimic photosynthesis by CO co-feed in a novel twin reactor, Appl. Energy, 2015, 147, 318–324 CrossRef CAS.
- F. Studt, I. Sharafutdinov, F. Abild-Pedersen, C. F. Elkjaer, J. S. Hummelshoj, S. Dahl, I. Chorkendorff and J. K. Norskov, Discovery of a Ni-Ga catalyst for carbon dioxide reduction to methanol, Nat. Chem., 2014, 6, 320–324 CrossRef CAS PubMed.
- M. Huš, V. D. B. C. Dasireddy, N. S. Štefančič and B. Likozar, Mechanism, kinetics and thermodynamics of carbon dioxide hydrogenation to methanol on Cu/ZnAl2O4 spinel-type heterogeneous catalysts, Appl. Catal., B, 2017, 207, 267–278 CrossRef.
- S. Kattel, P. J. Ramírez, J. G. Chen, J. A. Rodriguez and P. Liu, Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts, Science, 2017, 355, 1296–1299 CrossRef CAS.
- A. Ateka, P. Pérez-Uriarte, M. Gamero, J. Ereña, A. T. Aguayo and J. Bilbao, A comparative thermodynamic study on the CO2 conversion in the synthesis of methanol and of DME, Energy, 2017, 120, 796–804 CrossRef CAS.
- S. Saric, B. Biggs, M. Janbahan, R. Hamilton, H. K. Do, S. Mayoral and J. L. Haan, An integrated device to convert carbon dioxide to energy, Appl. Energy, 2016, 183, 1346–1350 CrossRef CAS.
- T. Witoon, N. Kachaban, W. Donphai, P. Kidkhunthod, K. Faungnawakij, M. Chareonpanich and J. Limtrakul, Tuning of catalytic CO2 hydrogenation by changing composition of CuO–ZnO–ZrO2 catalysts, Energy Convers. Manage., 2016, 118, 21–31 CrossRef CAS.
- N. Hedin, L. Andersson, L. Bergström and J. Yan, Adsorbents for the post-combustion capture of CO2 using rapid temperature swing or vacuum swing adsorption, Appl. Energy, 2013, 104, 418–433 CrossRef CAS.
- Q. Lai, S. Toan, M. A. Assiri, H. Cheng, A. G. Russell, H. Adidharma, M. Radosz and M. Fan, Catalyst-TiO(OH)2 could drastically reduce the energy consumption of CO2 capture, Nat. Commun., 2018, 9, 2672 CrossRef PubMed.
- J. D. Grunwaldt, A. M. Molenbroek, N. Y. Topsøe, H. Topsøe and B. S. Clausen, In situ investigations of structural changes in Cu/ZnO catalysts, J. Catal., 2000, 194, 452–460 CrossRef CAS.
- P. L. Hansen, J. B. Wagner, S. Helveg, J. R. Rostrup-Nielsen, B. S. Clausen and T. Henrik, Atom-resolved imaging of dynamic shape changes in supported copper nanocrystals, Science, 2002, 295, 2053–2055 CrossRef CAS PubMed.
- S. Bachu, CO2 storage in geological media: Role, means, status and barriers to deployment, Prog. Energy Combust. Sci., 2008, 34, 254–273 CrossRef CAS.
- R. G. Zhang, B. Zhao, L. X. Ling, A. J. Wang, C. K. Russell, B. J. Wang and M. H. Fan, Cost-Effective Palladium-Doped Cu Bimetallic Materials to Tune Selectivity and Activity by using Doped Atom Ensembles as Active Sites for Efficient Removal of Acetylene from Ethylene, ChemCatChem, 2018, 10, 2424–2432 CrossRef CAS.
- G. D. Weatgerbee and C. H. Bartholomew, Hydrogenation of CO2 on group Viii metals: II. Kinetics and mechanism of CO2 hydrogenation on nicklel, J. Catal., 1982, 77, 460–472 CrossRef.
- A. E. Aksoylu and Z. I. Onsan, Hydrogenation of carbon oxides using coprecipitated and impregnated Ni/A12O3 catalysts, Appl. Catal., A, 1997, 164, 1–11 CrossRef.
- E. Vesselli, M. Rizzi, L. De Rogatis, X. L. Ding, A. Baraldi, G. Comelli, L. Savio, L. Vattuone, M. Rocca, P. Fornasiero, A. Baldereschi and M. Peressi, Hydrogen-assisted transformation of CO2 on nickel: The role of formate and carbon monoxide, J. Phys. Chem. Lett., 2010, 1, 402–406 CrossRef CAS.
- Y. A. Zhu, D. Chen, X. G. Zhou and W. K. Yuan, DFT studies of dry reforming of methane on Ni catalyst, Catal. Today, 2009, 148, 260–267 CrossRef CAS.
- Q. Tang, Z. Shen, L. Huang, T. He, H. Adidharma, A. G. Russell and M. Fan, Synthesis of methanol from CO2 hydrogenation promoted by dissociative adsorption of hydrogen on a Ga3Ni5(221) surface, Phys. Chem. Chem. Phys., 2017, 19, 18539–18555 RSC.
- Q. Tang, Z. Shen, C. K. Russell and M. Fan, Thermodynamic and kinetic study on carbon dioxide hydrogenation to methanol over a Ga3Ni5(111) surface: The effects of step edge, J. Phys. Chem. C, 2018, 122, 315–330 CrossRef CAS.
- S. E. Collins, J. J. Delgado, C. Mira, J. J. Calvino, S. Bernal, D. L. Chiavassa, M. A. Baltanás and A. L. Bonivardi, The role of Pd-Ga bimetallic particles in the bifunctional mechanism of selective methanol synthesis via CO2 hydrogenation on a Pd/Ga2O3 catalyst, J. Catal., 2012, 292, 90–98 CrossRef CAS.
- R. Ladera, F. J. Pérez-Alonso, J. M. González-Carballo, M. Ojeda, S. Rojas and J. L. G. Fierro, Catalytic valorization of CO2 via methanol synthesis with Ga-promoted Cu-ZnO-ZrO2 catalysts, Appl. Catal., B, 2013, 142–143, 241–248 CrossRef CAS.
- J. Toyir, P. R. de la Piscina, J. L. G. Fierro and N. Homs, Catalytic performance for CO2 conversion to methanol of gallium-promoted copper-based catalysts: Influence of metallic precursors, Appl. Catal., B, 2001, 34, 255–266 CrossRef CAS.
- M. M. J. Li, Z. Y. Zeng, F. L. Liao, X. L. Hong and S. C. E. Tsang, Enhanced CO2 hydrogenation to methanol over CuZn nanoalloy in Ga modified Cu/ZnO catalysts, J. Catal., 2016, 343, 157–167 CrossRef CAS.
- J. C. Medina, M. Figueroa, R. Manrique, J. R. Pereira, P. D. Srinivasan, J. J. Bravo-Saurez, V. G. B. Medrano, R. Jimenez and A. Karelovic, Catalytic consequences of Ga promotion on Cu for CO2 hydrogenation to methanol, Catal. Sci. Technol., 2017, 7, 3375–3387 RSC.
- Y. Santiago-Rodríguez, E. Barreto-Rodríguez and M. C. Curet-Arana, Quantum mechanical study of CO2 and CO hydrogenation on Cu(111) surfaces doped with Ga, Mg, and Ti, J. Mol. Catal. A: Chem., 2016, 423, 319–332 CrossRef.
- E. M. Fiordaliso, I. Sharafutdinov, H. W. P. Carvalho, J. D. Grunwaldt, T. W. Hansen, I. Chorkendorff, J. B. Wagner and C. D. Damsgaard, Intermetallic GaPd2 nanoparticles on SiO2 for low-pressure CO2 hydrogenation to methanol: Catalytic performance and in situ characterization, ACS Catal., 2015, 5, 5827–5836 CrossRef CAS.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS.
- G. Kresse and J. Furthmiiller, Efficiency of ab initio total energy calculations for metals and semiconductors using a plane-wave basis set, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmuller, Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- G. Kresse and D. Joubert, From ultrasoft pseudopotentials to the projector augmented-wave method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A climbing image nudged elastic band method for finding saddle points and minimum energy paths, J. Chem. Phys., 2000, 113, 9901–9904 CrossRef CAS.
- F. L. Hirshfeld, Bonded-atom fragments for describing molecular charge densities, Theoret. Claim. Acta, 1977, 44, 129–138 CrossRef CAS.
- V. Milman, B. Winkler, J. A. White, C. J. Pickard, M. C. Payne, E. V. Akhmatskaya and R. H. Nobes, Electronic structure, properties, and phase stability of inorganic crystals: A pseudopotential plane-wave study, Int. J. Quantum Chem., 2000, 77, 895–910 CrossRef CAS.
- M. D. Segall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, First-principles simulation: Ideas, illustrations and the CASTEP code, J. Phys.: Condens. Matter, 2002, 14, 2717–2744 CrossRef CAS.
- B. Delley, An all electron numerical method for solving the local density functional for polyatomic molecules, J. Chem. Phys., 1990, 92, 508–517 CrossRef CAS.
- B. Delley, From molecules to solids with the DMol3 approach, J. Chem. Phys., 2000, 113, 7756–7764 CrossRef CAS.
- B. Delley, Hardness conserving semilocal pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155125 CrossRef.
- J. Li, H. Zheng, X. Zhang and Z. Li, First-principles investigation on Cu/ZnO catalyst precursor: Energetic, structural and electronic properties of Zn-doped Cu2(OH)2CO3, Comput. Mater. Sci., 2015, 96, 1–9 CrossRef CAS.
- T. Duan, R. Zhang, L. Ling and B. Wang, Insights into the effect of Pt atomic ensemble on HCOOH oxidation over Pt-decorated Au bimetallic catalyst to maximize Pt utilization, J. Phys. Chem. C, 2016, 120, 2234–2246 CrossRef CAS.
- O. M. Lovvik, Surface segregation in palladium based alloys from density-functional calculations, Surf. Sci., 2005, 583, 100–106 CrossRef CAS.
- Y. Zhao, Y. Yang, C. Mims, C. H. F. Peden, J. Li and D. Mei, Insight into methanol synthesis from CO2 hydrogenation on Cu(111): Complex reaction network and the effects of H2O, J. Catal., 2011, 281, 199–211 CrossRef CAS.
- Y. F. Zhao, R. Rousseau, J. Li and D. H. Mei, Theoretical study of syngas hydrogenation to methanol on the polar Zn-terminated ZnO(0001) surface, J. Phys. Chem. C, 2012, 116, 15952–15961 CrossRef CAS.
- H. Liu, B. Teng, M. Fan, B. Wang, Y. Zhang and H. Gordon Harris, CH4 dissociation on the perfect and defective MgO(001) supported Ni4, Fuel, 2014, 123, 285–292 CrossRef CAS.
- K. Li, H. Liu, R. Zhang, L. Ling and B. Wang, Interaction between bimetal cluster Ni2Co2 and MgO and its effect on H adsorption and H2 dissociation: A DFT study, Appl. Surf. Sci., 2016, 390, 7–16 CrossRef CAS.
- C. Zhi, R. Zhang and B. Wang, Comparative studies about CO methanation over Ni(211) and Zr-modified Ni(211) surfaces: Qualitative insight into the effect of surface structure and composition, Mol. Catal., 2017, 438, 1–14 CrossRef CAS.
- Y. Yang, J. Evans, J. A. Rodriguez, M. G. White and P. Liu, Fundamental studies of methanol synthesis from CO(2) hydrogenation on Cu(111), Cu clusters, and Cu/ZnO(0001), Phys. Chem. Chem. Phys., 2010, 12, 9909–9917 RSC.
- Z. W. Ulissi, A. J. Medford, T. Bligaard and J. K. Norskov, To address surface reaction network complexity using scaling relations machine learning and DFT calculations, Nat. Commun., 2017, 8, 14621 CrossRef PubMed.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B. L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts, Science, 2012, 336, 893–896 CrossRef CAS PubMed.
- L. Ling, Q. Wang, R. Zhang, D. Li and B. Wang, Formation of C2 oxygenates and ethanol from syngas on an Fe-decorated Cu-based catalyst: insight into the role of Fe as a promoter, Phys. Chem. Chem. Phys., 2017, 19, 30883–30894 RSC.
- R. G. Zhang, F. Liu and B. J. Wang, Co-decorated Cu alloy catalyst for C2 oxygenate and ethanol formation from syngas on Cu-based catalyst: Insight into the role of Co and Cu as well as the improved selectivity, Catal. Sci. Technol., 2016, 6, 8036–8054 RSC.
- P. Liu, A. Logadottir and J. K. Nørskov, Modeling the electro-oxidation of CO and H2/CO on Pt, Ru, PtRu and Pt3Sn, Electrochim. Acta, 2003, 48, 3731–3742 CrossRef CAS.
- H. Liu, R. Zhang, L. Ling, Q. Wang, B. Wang and D. Li, Insight into the preferred formation mechanism of long-chain hydrocarbons in Fischer-Tropsch synthesis on Hcp Co(10–11) surfaces from DFT and microkinetic modeling, Catal. Sci. Technol., 2017, 7, 3758–3776 RSC.
- B. Hammer and J. K. Norskov, Electronic factors determining the reactivity of metal surfaces, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- X. Z. Duan, G. Qian, Y. Liu, J. Ji, X. G. Zhou, D. Chen and W. K. Yuan, Structure sensitivity of ammonia decomposition over Ni catalysts: A computational and experimental study, Fuel Process. Technol., 2013, 108, 112–117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr01245a |
| This journal is © The Royal Society of Chemistry 2019 |