
2D/2D heterojunction of Ti3C2/g-C3N4 nanosheets for enhanced photocatalytic hydrogen evolution†
Tongming
Su
 *ab,
Zachary D.
Hood
bd,
Michael
Naguib
c,
Lei
Bai
bf,
Si
Luo
b,
Christopher M.
Rouleau
b,
Ilia N.
Ivanov
b,
Hongbing
Ji
ae,
Zuzeng
Qin
*a and
Zili
Wu
*b
*ab,
Zachary D.
Hood
bd,
Michael
Naguib
c,
Lei
Bai
bf,
Si
Luo
b,
Christopher M.
Rouleau
b,
Ilia N.
Ivanov
b,
Hongbing
Ji
ae,
Zuzeng
Qin
*a and
Zili
Wu
*b
aSchool of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China. E-mail: sutm@gxu.edu.cn; qinzuzeng@gxu.edu.cn
bCenter for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA. E-mail: wuz1@ornl.gov
cDepartment of Physics and Engineering Physics, Tulane University, New Orleans, Louisiana 70118, USA
dElectrochemical Materials Laboratory, Department of Materials Science and Engineering, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA
eSchool of Chemistry, Sun Yat-sen University, Guangzhou 510275, China
fWest Virginia University, Department of Chemical and Biomedical Engineering, Morgantown, West Virginia 26506, USA
First published on 21st February 2019
Abstract
Photocatalytic hydrogen evolution from water has received enormous attention due to its ability to address a number of global environmental and energy-related issues. Here, we synthesize 2D/2D Ti3C2/g-C3N4 composites by electrostatic self-assembly technique and demonstrate their use as photocatalysts for hydrogen evolution under visible light irradiation. The optimized Ti3C2/g-C3N4 composite exhibited a 10 times higher photocatalytic hydrogen evolution performance (72.3 μmol h−1 gcat−1) than that of pristine g-C3N4 (7.1 μmol h−1 gcat−1). Such enhanced photocatalytic performance was due to the formation of 2D/2D heterojunctions in the Ti3C2/g-C3N4 composites. The intimate contact between the monolayer Ti3C2 and g-C3N4 nanosheets promotes the separation of photogenerated charge carriers at the Ti3C2/g-C3N4 interface. Furthermore, the ultrahigh conductivity of Ti3C2 and the Schottky junction formed between g-C3N4/MXene interfaces facilitate the photoinduced electron transfer and suppress the recombination with photogenerated holes. This work demonstrates that the 2D/2D Ti3C2/g-C3N4 composites are promising photocatalysts thanks to the ultrathin MXenes as efficient co-catalysts for photocatalytic hydrogen production.
Introduction
With the development of human society and the increasing consumption of fossil fuels, renewable chemical fuels have attracted intensive research attention over the past decades.1 Converting solar energy into chemical energy in the form of hydrogen is an ideal route to obtain clean renewable energy resources.2,3 Due to the fact that water and solar energy are inexhaustible, photocatalytic water splitting into hydrogen over a semiconductor with visible light represents a promising strategy to store solar energy as chemical energy. The key of photocatalytic water splitting lies in the development of cheap and highly-active photocatalysts that function under visible light irradiation. Over the past decades, various semiconductors, such as metal oxides,4 sulphides,5,6 metal oxynitrides,7 nitrides,8 amongst others, have been used as photocatalysts for hydrogen evolution. Among these photocatalysts, graphitic carbon nitride (g-C3N4) grabs considerable attention in the field of visible-light photocatalytic water splitting due to its suitable band structure, high thermal and chemical stability, low cost, non-toxicity, and high surface area.9,10Unfortunately, the rapid recombination of photogenerated electrons and holes on g-C3N4 largely hinders its practical application in photocatalytic water splitting.9 Various strategies have been developed to improve the separation of photoinduced electron–hole pairs on g-C3N4, such as structural engineering,11 increasing the crystallinity,12,13 doping with heteroatom,14 construction of heterojunction,15–18 and coupling with a co-catalyst.19,20 Among these approaches, co-catalyst loading is an effective method to promote the separation of photoinduced electron–hole pairs. For example, Pt, MoS2, and graphene can all be used as co-catalysts to capture the photo-induced electrons, thus inhibited the recombination of photoinduced charge carriers.21,22 Among them, Pt is the most efficient co-catalyst for photocatalytic hydrogen evolution. However, the high cost and scarcity of this noble metal limit its large-scale application. Therefore, the development of low-cost and effective co-catalysts is imperative to meet the requirements of industrial applications of semiconductor-based photocatalysts.
MXenes, a new family of two-dimensional (2D) materials of early transition metal carbides/carbonitrides, has attracted a great deal of research interest since it was discovered by Gogotsi and coworkers in 2011.23 Attributing to its excellent electrical conductivity, hydrophilicity, and stability, MXenes have been extensively investigated for various applications, such as electrochemical supercapacitors,24 batteries,25 biomedicine,26 catalysis,27 among other applications. The theoretical studies show that the Gibbs free energy for hydrogen adsorption (ΔGH) of several MXenes are close to zero, thus these MXenes are considered as effective electrocatalysts for the hydrogen evolution reaction.28,29 Furthermore, the electrocatalytic hydrogen evolution activity of MXenes was confirmed by experiment.30 With the near-zero ΔGH, electrical conductivity, hydrophilicity, and low Fermi level compared to semiconductors, MXene may be an effective co-catalyst for photocatalytic hydrogen production from water. Recently, MXenes, such as Ti3C2, Ti2C, and Nb2C have been investigated as efficient co-catalysts of photocatalysts (e.g. TiO2, CdS, g-C3N4) for water splitting.31–35 When MXene was coupled with semiconductors, a Schottky barrier will be presented at the MXene/semiconductor interface.33 The Schottky barrier can serve as the electron reservoir, thus facilitating the separation of photoinduced electrons and holes. For example, the Ti3C2 nanoparticles (0D) can be used as the co-catalyst to enhance the photocatalytic hydrogen production of g-C3N4.34 Nevertheless, the Ti3C2/g-C3N4 shows a 0D/2D type heterojunction, and this 0D/2D heterojunction possesses a limited interface between the Ti3C2 and g-C3N4. Furthermore, due to the multilayer nature of Ti3C2, the photogenerated electrons have to transfer a long distance to the surface of the catalyst, which lead to the poor separation of charge carriers. Therefore, an intimate interfacial contact between Ti3C2 and g-C3N4 is needed in order to improve charge separation efficiency.
Construction of 2D/2D heterojunction, with obvious advantages over the 0D/2D heterojunction, is considered as an effective route to enhance the charge separation efficiency and improve the photocatalytic hydrogen evolution.2 Firstly, the 2D/2D heterojunction has much more contact areas than that of 0D/2D. Secondly, the intimate 2D/2D heterojunction improves the electron transfer rate and shortens the migration distance and time of photogenerated electrons to the catalyst surface. Therefore, the photocatalytic hydrogen production can be greatly enhanced through the formation of a 2D/2D heterojunction. For example, the photocatalytic hydrogen production rate over 2D/2D g-C3N4/ZnIn2S4 is 8.2 times higher than that of 0D/2D g-C3N4/ZnIn2S4.36 Recently, d-Ti3C2/TiO2/g-C3N4 heterostructured composite was synthesized and used for photocatalytic hydrogen production. In this system, delaminated Ti3C2 (d-Ti3C2) served as the fast electron transfer channel to develop the separation efficiency of the photogenerated charge carriers, thus enhance the photocatalytic activity of g-C3N4. However, this d-Ti3C2 still exhibit multilayered structure, and the hydrogen evolution rate of the optimal d-Ti3C2/TiO2/g-C3N4 composites showed only 2.4 times higher than that of the pure g-C3N4 with 3.0 wt% Pt as the co-catalyst.37 To shed more light on the effect of the 2D MXene and the 2D/2D heterojunction on the photocatalytic activity of the photocatalyst, in this work, monolayer Ti3C2 and g-C3N4 nanosheets were successfully prepared and the 2D/2D Ti3C2/g-C3N4 composites were synthesized by electrostatic self-assembly technique. The hydrogen evolution rate of obtained 2D/2D Ti3C2/g-C3N4 nanosheets showed over 10 times higher than that of the pure g-C3N4, and also significantly higher than that of the 0D/2D Ti3C2/g-C3N4, and the 2D/2D graphene/g-C3N4 without any noble metal as the co-catalyst. Such enhanced activity is largely attributed to the intimate interfacial contact between the monolayer Ti3C2 and g-C3N4 nanosheets and the highly active sites on the monolayer Ti3C2.
Experimental section
Synthesis of bulk g-C3N4 and g-C3N4 nanosheets
Bulk g-C3N4 powder was synthesized by thermal polymerization of urea. Briefly, urea (25 g) was placed into a covered ceramic crucible, and then the crucible was heated to 600 °C at a ramp rate of 5 °C min−1 and maintained for 4 h. After cooling to room temperature, the resulting light-yellow solid was ground with the mortar to obtain the bulk g-C3N4 powder (denoted as B-g-C3N4). To obtain the g-C3N4 nanosheet, B-g-C3N4 (1.0 g) was placed in an open ceramic crucible and was heated to 550 °C with a ramp rate of 5 °C min−1 and held in an ambient air atmosphere for 2 h. Finally, a light yellow powder of g-C3N4 nanosheets was obtained, which is herein denoted as g-C3N4.Synthesis of multilayer Ti3C2 and monolayer Ti3C2
The Ti3AlC2 was prepared according to the previous reports.23 Briefly, commercial Ti2AlC and TiC were mixed evenly and heated to 1350 °C for 2 h under continuous flow of argon gas, Ar, to obtain Ti3AlC2. To synthesize the multilayer Ti3C2 (M-Ti3C2), Ti3AlC2 powder (2.0 g) was immersed into of a 48% HF aqueous solution (20 mL) and stirred at room temperature for 15 h. The obtained Ti3C2 suspension was washed with deionized (DI) water several times and centrifuged to separate the multilayer Ti3C2 from the solution. After that, the multilayer Ti3C2 was dried with a vacuum oven. Monolayer Ti3C2 was prepared via a previously reported method.38 In short, LiF (1.0 g) was dissolved completely in 6 M HCl (20 mL) under stirring at room temperature to obtain the LiF/HCl solution. Then, Ti3AlC2 (1.0 g) was slowly added to the obtained LiF/HCl solution over the course of 5 minutes. The temperature of the suspension was raised to 35 °C and maintained at this temperature for 24 h with vigorous stirring. The resulting suspension was washed with DI water repeatedly until the pH of the filtrate was ≥6. After that, the suspension was centrifuged at 8000 rpm to separate the un-delaminated Ti3AlC2 and the multilayer Ti3C2 from the monolayer Ti3C2 colloidal solution. The Ti3C2 colloidal solution was subsequently filtered through a PVDF membrane (0.25 μm pore size, Millipore) and dried in a vacuum oven at room temperature for 24 h to obtain the stacked monolayer of Ti3C2 (S-Ti3C2).Synthesis of 2D/2D Ti3C2/g-C3N4
The 2D/2D Ti3C2/g-C3N4 was synthesized via electrostatic self-assembly approach. Firstly, g-C3N4 (0.3 g) was added to a 0.5 M HCl aqueous solution (100 mL) and protonated at room temperature for 0.5 h via sonication. After that, the protonated g-C3N4 was repeatedly washed with DI water until the pH of the g-C3N4 suspension was equal to 4. Secondly, 80 mg S-Ti3C2 was re-dispersed in 40 mL deionized water by sonication for 1 h in Ar atmosphere to obtain the 2 mg mL−1 monolayer Ti3C2 solution. After that, a quantitative monolayer Ti3C2 solution (1.0, 2.0, 3.0, 4.0, and 5.0 wt%) was added dropwise to the above protonated g-C3N4 suspension and was stirred for 0.5 h. Then the suspension was centrifuged to obtain the Ti3C2/g-C3N4 precipitate, which was dried at 80 °C for 24 h. The Ti3C2/g-C3N4 composites with different amount of monolayer Ti3C2 (1.0, 2.0, 3.0, 4.0, and 5.0 wt%) were denoted as 1-TC/CN, 2-TC/CN, 3-TC/CN, 4-TC/CN, and 5-TC/CN. The protonated g-C3N4 (P-g-C3N4) powder was obtained by the same method without adding Ti3C2. We applied the same approach by replacing monolayer Ti3C2 with multilayer Ti3C2 or graphene in order to synthesize 3.0 wt% M-Ti3C2/g-C3N4 (3-MTC/CN) and 3.0 wt% graphene/g-C3N4 (3-G/CN), respectively. We also replaced g-C3N4 with B-g-C3N4 in order to prepare 3.0 wt% Ti3C2/B-g-C3N4 (3-TC/BCN).Photocatalytic hydrogen evolution reaction
Photocatalytic experiments were carried out in a side-irradiation quartz reactor. For comparison, the photocatalytic activity of the g-C3N4 by using 3.0 wt% Pt as the co-catalyst was investigated. The loading of 3.0 wt% Pt co-catalyst was conducted by directly dissolving H2PtCl6 into the suspension. Thereafter, the suspension was stirred and irradiated (200 W Hg lamp) for 30 min at room temperature to reduce the Pt species. For each experiment, the photocatalyst (30 mg) were dispersed in deionized water (40 mL) with 10% triethanolamine as the hole scavenger. Prior to the photocatalytic reaction, the system was degassed with ultrahigh pure argon for 30 min. The light input is provided by a 200 W Hg lamp equipped with a cutoff filter to allow the pass of light with wavelength higher than 400 nm. The solution was vigorously stirred during the photocatalytic reaction and the temperature was maintained at 25 °C by the circulating cooling water. The gas product was quantified by a gas chromatography (Model BUCK 910) equipped with a molecular sieve column and thermal conductivity detector (TCD) and with argon as the carrier gas.Characterizations
X-ray diffraction (XRD) measurements were performed on a PANalytical X'Pert MPD Pro powder diffractometer equipped with a Si-based position-sensitive one-dimensional detector and Ni-filtered Cu Kα radiation source, and the X-rays were generated at 45 kV/40 mA at a beam wavelength of λ = 1.5416 Å (Cu Kα radiation). A Zeiss Merlin scanning electron microscope with an acceleration voltage of 20.0 kV was used for scanning electron microscope (SEM) imaging. High-resolution transmission electron microscopy (HRTEM) imaging of the samples was performed on an aberration-corrected FEI Titan S 80-300 STEM/TEM microscope equipped with a Gatan OneView camera at an acceleration voltage of 300 kV. The BET surface area of the samples was measured with a Quantachrome system using N2 as the probe gas. An Acton Trivista 555 spectrometer (Princeton Instruments) with laser excitation at 325 nm was used to collect the Raman spectra. The UV–vis absorption spectra were recorded using a Cary 5000 UV/Vis spectrophotometer. Photoluminescence (PL) spectra were performed on a Horiba Jobin Yvon Fluorolog fluorescence spectrometer, and the excitation monochromator was set at 325 nm. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific K-Alpha spectrometer at an operating pressure under 3.0 × 10−7 Pa and a spot size of 400 μm using an Al-Kα microfused monochromatized source (1486.6 eV) with a resolution of 0.1 eV. All XPS data was processed using Avantage Data System, which is a software package provided by Thermo Scientific. Atomic force microscopy (AFM) was performed on a Dimension Icon scanning probe microscope with the contact and tapping modes.Photoelectrochemical measurements
Photoelectrochemical (PEC) measurements were performed on a BioLogic SP150 electrochemical workstation using a standard three-electrode cell with a working electrode, Ag/AgCl electrode as reference electrode and Pt mesh as the counter electrode. A 0.2 M Na2SO4 aqueous solution was utilized as the buffer solution. Electrochemical impedance spectroscopy (EIS) was collected in a range from 0.1 kHz to 100 kHz with an AC amplitude of 5 mV. The transient photocurrent was performed in the same three-electrode system, and the light source was a 200 W Hg lamp equipped with a cutoff filter (400 nm). The working electrodes were prepared as follows: 4 mg of the synthesized photocatalyst was first ultrasonically dispersed in the mixture of 250 μL of deionized water, 250 μL of ethanol and 20 μL of Nafion® solutions (Sigma Aldrich, 5 wt% in mixture of lower aliphatic alcohols and water, contains 45% water) for 1.0 h. After that, 20 μL of the catalyst suspension was transferred onto the indium tin oxide (ITO) substrate and dried at room temperature to obtain the working electrode.Results and discussion
The crystal structure of as-synthesized samples was studied by XRD analysis (Fig. S1A and B, ESI†). As shown in Fig. S1A,† the XRD patterns of original Ti3AlC2 exhibit intense peaks, which is in accordance with the previous reports.23 After reacting the MAX phase with a LiF/HCl solution, the most intense diffraction peak (2θ = 39°) of Ti3AlC2 disappears in the XRD patterns for stacked monolayer of Ti3C2 (S-Ti3C2) (see Fig. S2† for the photograph and SEM images of the cross-section of S-Ti3C2). Moreover, the (002) peak of Ti3C2 shifts to a lower angle due to the increased interlayer spacing of the Ti3C2, indicating the Ti3AlC2 was successfully transformed to Ti3C2. The XRD patterns of the g-C3N4 show two evident peaks (Fig. S1A†); the peak at 2θ = 13.3° is attributed to the in-plane structural packing motif, and the peak at 2θ = 27.7° is assigned to the interlayer stacking of aromatic segments.39 The XRD patterns of the protonated g-C3N4 (P-g-C3N4) show similar diffraction peaks to that of g-C3N4, indicating that the protonation did not change the crystal structure of the g-C3N4. The Ti3C2/g-C3N4 composites were fabricated via an electrostatic self-assembly strategy. According to the previous reports, the surface of g-C3N4 nanosheet is positively charged after the protonation treatment,40 while the surface of monolayer Ti3C2 is negatively charged.41 Therefore, when the g-C3N4 suspension was mixed with the Ti3C2 colloidal solution, g-C3N4 and Ti3C2 will first stick together via electrostatic interaction and then coagulate at the bottom of the beaker (Fig. S3, ESI†). The XRD patterns of the Ti3C2/g-C3N4 composites show similar diffraction peaks to that of g-C3N4, and no diffraction peaks of Ti3C2 was observed due to its low content and high dispersion (Fig. S1B†).The chemical structure of the as-prepared samples was investigated by Raman spectroscopy (Fig. S1C and D, ESI†). For the Raman spectra of the g-C3N4 and the Ti3C2/g-C3N4 composites, several characteristics peaks located at 480, 708, 771, 987, 1122, 1494, and 1629 cm−1 can be observed, confirming the formation of the polymeric structure of g-C3N4.42,43 In detail, two wide peaks located at 1355 cm−1 and 1587 cm−1 are assigned to the disordered sp3 carbon (D band) and graphitic sp2 carbon (G band) of a typical graphitic structure in the g-C3N4.44 The Raman shifts at 500–1300 cm−1 originate from the typical heptazine units. The two peaks at 708 cm−1 and 771 cm−1 correspond to the in-plane bending vibrations of the heptazine linkages, while the peaks at 987 cm−1 belonged to the symmetric N-breathing mode of heptazine units.36 The Raman spectra of all the samples are notably similar, suggesting that the protonation, thermal exfoliation, and loading processes during the preparation of Ti3C2/g-C3N4 composites did not change the chemical structure of the g-C3N4. In addition, no Raman peaks of Ti3C2 can be detected in the Ti3C2/g-C3N4 composites due to its low concentration, which is consistent with the XRD results.
g-C3N4 nanosheets and monolayer Ti3C2 were successfully prepared from bulk g-C3N4 and Ti3AlC2, respectively. The SEM images of the bulk g-C3N4 and Ti3AlC2 are shown in Fig. S4C and S6A,† and the SEM images of g-C3N4 nanosheets and monolayer Ti3C2 are shown in Fig. 1A and B, respectively. Fig. 1A shows the sheet structure of g-C3N4, which indicates that the bulk g-C3N4 was split into small nanosheets during thermal exfoliation at 550 °C. After the thermal exfoliation, the g-C3N4 nanosheets display a lighter yellow color if compared to that of bulk g-C3N4 (Fig. S4A and B, ESI†). The flake-shaped morphology of Ti3C2 is displayed in Fig. 1B; these Ti3C2 flakes show clean surfaces and sizes ranging from dozens of nanometers to hundreds of nanometers. The thickness of the Ti3C2 flakes was investigated by AFM (Fig. 1C). From the AFM height profile (Fig. S5, ESI†) measured along the red dashed line in Fig. 1C, the height of the folded region relative to the layer underneath was measured as 1.7 nm, which corresponds to a monolayer Ti3C2.38 The thickness of the Ti3C2 flakes relative to the Si substrate is 2.7 nm, which is possibly due to the surface adsorbates (such as water molecules) present between the Ti3C2 flakes and the Si substrate.38 The typical Tyndall effect was clearly observed in an aqueous suspension of Ti3C2 flakes (Fig. S6B, ESI†), demonstrating their excellent hydrophilicity and dispersity.45 In addition, the thickness of Ti3C2 flakes obtained from AFM range from 1.7 nm to 2.6 nm (Fig. S6C and D, ESI†), indicating that all the flakes are monolayer Ti3C2.
 | ||
| Fig. 1 SEM images of g-C3N4 nanosheets (A), monolayer Ti3C2 (B), P-g-C3N4 (D), 1-TC/CN (E), 2-TC/CN (F), 3-TC/CN (G), 4-TC/CN (H), and 5-TC/CN (I); AFM image (C) of a representative monolayer Ti3C2 deposited on Si wafe. | ||
The morphology of the protonated g-C3N4 and the 2D/2D Ti3C2/g-C3N4 composites was investigated by SEM (Fig. 1). After the ultrasonic treatment in an aqueous HCl solution, the sheet-like structure of g-C3N4 remains unchanged (Fig. 1D). However, these g-C3N4 nanosheets stacked together after drying at 80 °C, and the BET specific surface area of the g-C3N4 nanosheet decreased from 153.6 m2 g−1 to 79.0 m2 g−1 according to N2 adsorption–desorption analysis (Fig. S7 and Table S1, ESI†). In addition, the Ti3C2/g-C3N4 composites with different content of Ti3C2 are composed of thin nanosheets (Fig. 1E, F, G, H, and I). The BET specific surface area of the 1-TC/CN, 2-TC/CN, 3-TC/CN, 4-TC/CN, and 5-TC/CN samples are 73.8, 76.6, 82.2, 83.1, and 84.3 m2 g−1, respectively (Fig. S7 and Table S1, ESI†). Because Ti3C2 and g-C3N4 are both thin flakes, it is hard to distinguish the Ti3C2 and g-C3N4 from the SEM images of the Ti3C2/g-C3N4 composites. The EDS elemental mapping verifies the presence of C, N, and Ti element in the Ti3C2/g-C3N4 composites, which demonstrates that the composites are composed of Ti3C2 and g-C3N4 (Fig. S8, ESI†).
To better understand the morphology and microstructure of the as-synthesized photocatalysts, the g-C3N4 and 3-TC/CN samples were further investigated by TEM and HRTEM (Fig. 2). The TEM images of the g-C3N4 exhibit a 2D morphology (Fig. 2A), implying the ultrathin structures of g-C3N4 nanosheets. As shown in Fig. 2B and C, the g-C3N4 had low crystallinity and was mostly amorphous, which is consistent with previous reports.46,47 After the g-C3N4 nanosheets were combined with monolayer Ti3C2, the Ti3C2/g-C3N4 composites still displayed the sheet-shape morphology (Fig. 2D). The HRTEM images of the Ti3C2/g-C3N4 composites display large two-dimensional structures consisting of flat irregularly shaped nanosheets of 2D/2D structures. Additionally, an obvious composite structure can be seen in the HRTEM image of 3-TC/CN, as shown in Fig. 2E and F. The clear lattice fringes were assigned to Ti3C2 flakes, whereas the disordered areas were ascribed to the g-C3N4 nanosheets. Notably, an obvious intimate interface was observed between Ti3C2 and g-C3N4 suggesting the formation of 2D/2D Ti3C2/g-C3N4 heterojunctions, which is in accordance with the EDS analysis. This 2D/2D Ti3C2/g-C3N4 interface is beneficial to the transfer of photoinduced electrons from g-C3N4 to Ti3C2 due to the excellent electronic conductivity of the Ti3C2.
 | ||
| Fig. 2 TEM and HRTEM images of g-C3N4 (A, B, C) and 3-TC/CN (D, E, F). | ||
The chemical composition and oxidation state of the g-C3N4 and 3-TC/CN samples were characterized by XPS, as shown in Fig. 3. C and N elements were detected in the XPS survey spectrum of the g-C3N4 sample (Fig. S9A, ESI†), and the C and N elements were further investigated by the high resolution XPS (Fig. 3A and B). There are three carbon species in the C 1s spectra of the g-C3N4 sample. The 284.8 eV peak was assigned to the adsorbed carbon species (C–C) on the g-C3N4 surface, and the 286.8 eV and 288.0 eV peaks can be ascribed to the sp2-hybridized carbon in triazine rings (C–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) and N-contained aromatic ring (N–CN), respectively. The carbon in N–CN is considered as the major carbon species in g-C3N4 nanosheets.48 Three peaks were observed in the N 1s spectrum (Fig. 3B), and the peaks located at binding energies of 398.9, 400.6, and 401.5 eV can be attributed to the sp2-hybridized nitrogen involved in the triazine rings (C–NC), the tertiary nitrogen N–(C)3 groups, and the free amino groups (C–N–H), respectively.48,49 Ti, N, and C elements can be observed in the XPS survey spectrum of 3-TC/CN (Fig. S9B, ESI†), indicating that Ti3C2 and g-C3N4 are present in the composite. The high resolution spectra of Ti 2p, N 1s, and C 1s are shown in Fig. 3C, D and E, and the XPS peak fitting results are displayed in Table S2.† As shown in Fig. 3C, the peaks located at 454.9 (461.2), 455.9 (462.0), and 457.2 eV (463.4 eV) belong to the Ti (I, II, or IV), Ti2+(I, II, or IV), and Ti3+(I, II, or IV) species in the Ti3C2, respectively, indicating the existence of Ti3C2 in the 3-TC/CN sample. Furthermore, the peaks located at 459.4 eV (465.6 eV) correspond to the Ti in the TiO2−xFx species, which are likely originated from the slight oxidation of the Ti3C2.50 The C 1s spectrum exhibits four peaks at 281.4, 284.7, 286.8, and 288.0 eV. The peaks at 281.4 eV correspond to C–Ti–Tx(I, II, III or IV) species of Ti3C2, which further confirms the presence of the Ti3C2.50 In addition, the peaks at 284.8, 286.8, and 288.0 eV can be assigned to the carbon in C–C, C–NC, and N–CN, which is similar to the peaks in the XPS spectra of g-C3N4 sample. Moreover, the high resolution spectra of N 1s in the 3-TC/CN sample are in accordance with that of g-C3N4. The above analysis demonstrates that the loading of Ti3C2 did not change the structure and chemical composition of the g-C3N4, and the 3-TC/CN composite is consist of both Ti3C2 and g-C3N4. The XPS and TEM analyses indicate that the 2D/2D heterojunction of Ti3C2/g-C3N4 was successfully formed by the electrostatic self-assembly method.
C) and N-contained aromatic ring (N–CN), respectively. The carbon in N–CN is considered as the major carbon species in g-C3N4 nanosheets.48 Three peaks were observed in the N 1s spectrum (Fig. 3B), and the peaks located at binding energies of 398.9, 400.6, and 401.5 eV can be attributed to the sp2-hybridized nitrogen involved in the triazine rings (C–NC), the tertiary nitrogen N–(C)3 groups, and the free amino groups (C–N–H), respectively.48,49 Ti, N, and C elements can be observed in the XPS survey spectrum of 3-TC/CN (Fig. S9B, ESI†), indicating that Ti3C2 and g-C3N4 are present in the composite. The high resolution spectra of Ti 2p, N 1s, and C 1s are shown in Fig. 3C, D and E, and the XPS peak fitting results are displayed in Table S2.† As shown in Fig. 3C, the peaks located at 454.9 (461.2), 455.9 (462.0), and 457.2 eV (463.4 eV) belong to the Ti (I, II, or IV), Ti2+(I, II, or IV), and Ti3+(I, II, or IV) species in the Ti3C2, respectively, indicating the existence of Ti3C2 in the 3-TC/CN sample. Furthermore, the peaks located at 459.4 eV (465.6 eV) correspond to the Ti in the TiO2−xFx species, which are likely originated from the slight oxidation of the Ti3C2.50 The C 1s spectrum exhibits four peaks at 281.4, 284.7, 286.8, and 288.0 eV. The peaks at 281.4 eV correspond to C–Ti–Tx(I, II, III or IV) species of Ti3C2, which further confirms the presence of the Ti3C2.50 In addition, the peaks at 284.8, 286.8, and 288.0 eV can be assigned to the carbon in C–C, C–NC, and N–CN, which is similar to the peaks in the XPS spectra of g-C3N4 sample. Moreover, the high resolution spectra of N 1s in the 3-TC/CN sample are in accordance with that of g-C3N4. The above analysis demonstrates that the loading of Ti3C2 did not change the structure and chemical composition of the g-C3N4, and the 3-TC/CN composite is consist of both Ti3C2 and g-C3N4. The XPS and TEM analyses indicate that the 2D/2D heterojunction of Ti3C2/g-C3N4 was successfully formed by the electrostatic self-assembly method.
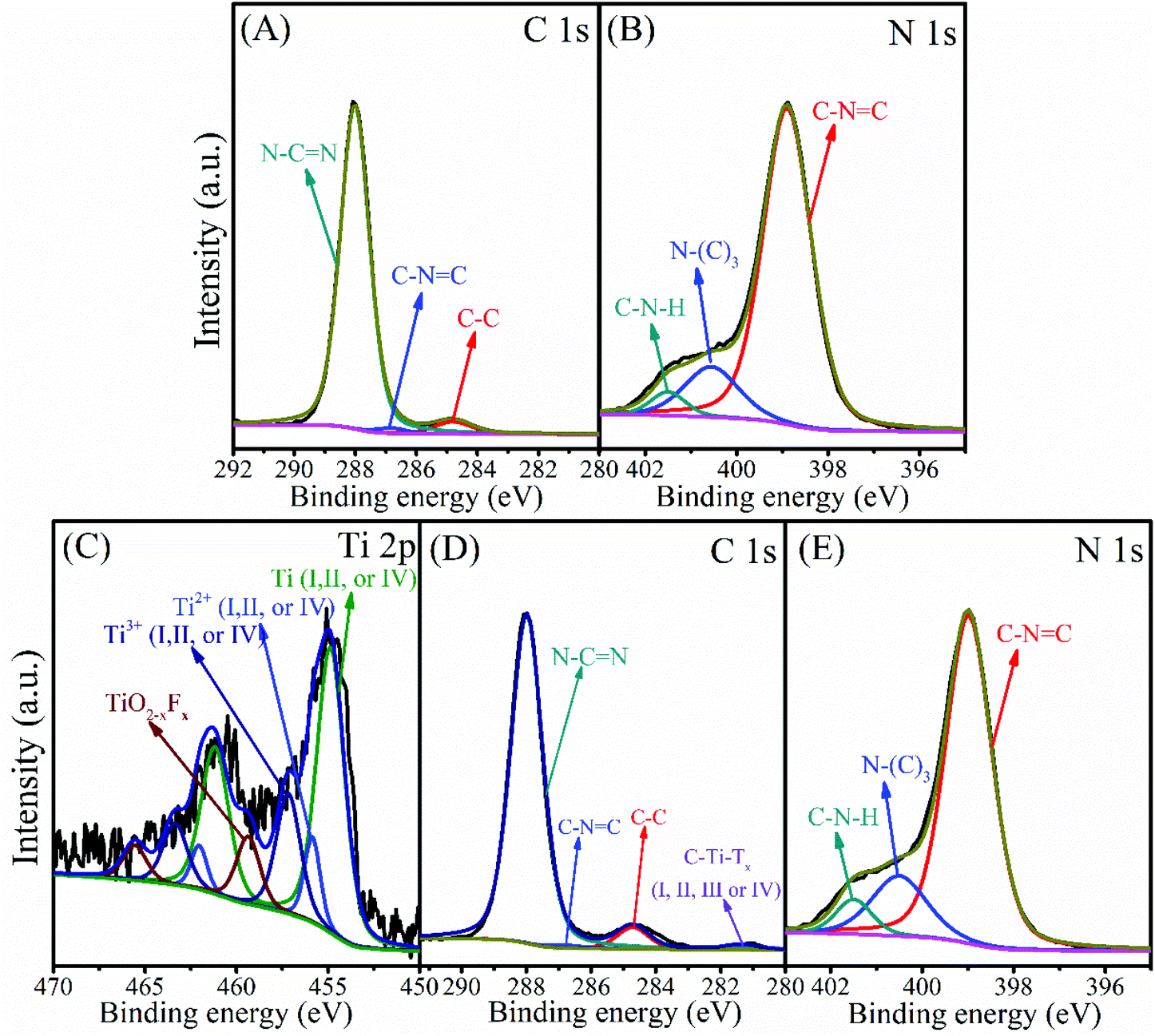 | ||
| Fig. 3 XPS spectra of C 1s (A) and N 1s (B) in g-C3N4, and Ti 2p (C), C 1s (D) and N 1s (E) in 3-TC/CN. | ||
The optical absorption properties of the photocatalyst play an important role in their activity. The UV-vis absorption spectra of as-synthesized photocatalysts are shown in Fig. 4A and Fig. S10.† The g-C3N4 nanosheets show a clear blue shift of the intrinsic absorption edge compared with that of bulk g-C3N4. Moreover, the band gap of bulk g-C3N4 (B-g-C3N4) and the g-C3N4 nanosheets is 2.57 and 2.77 eV, respectively, as determined from the (ahν)1/2versus photon-energy plots (Fig. S10, ESI†). When B-g-C3N4 is exfoliated to ultrathin g-C3N4 nanosheets, the band gap increases due to the quantum confinement effect by shifting the conduction and valence band edges in opposite directions, which is identical with the previous reports.51,52 It can be seen from Fig. 4A that all the Ti3C2/g-C3N4 composites exhibit enhanced light absorption compared to the pure g-C3N4 nanosheets. With the increase of Ti3C2 content, the absorption intensity of Ti3C2/g-C3N4 composites gradually increased within the range of 250–800 nm. Such increased light absorption is attributed to the full-spectrum absorption of dark Ti3C2. Although the enhanced light absorption might not be able to excite the photoelectrons and holes in Ti3C2/g-C3N4 composites, it is likely to facilitate the surface catalytic reactions by converting light energy to heat to activate the catalyst,53 due to the efficient photothermal conversion characteristics of Ti3C2.45,54
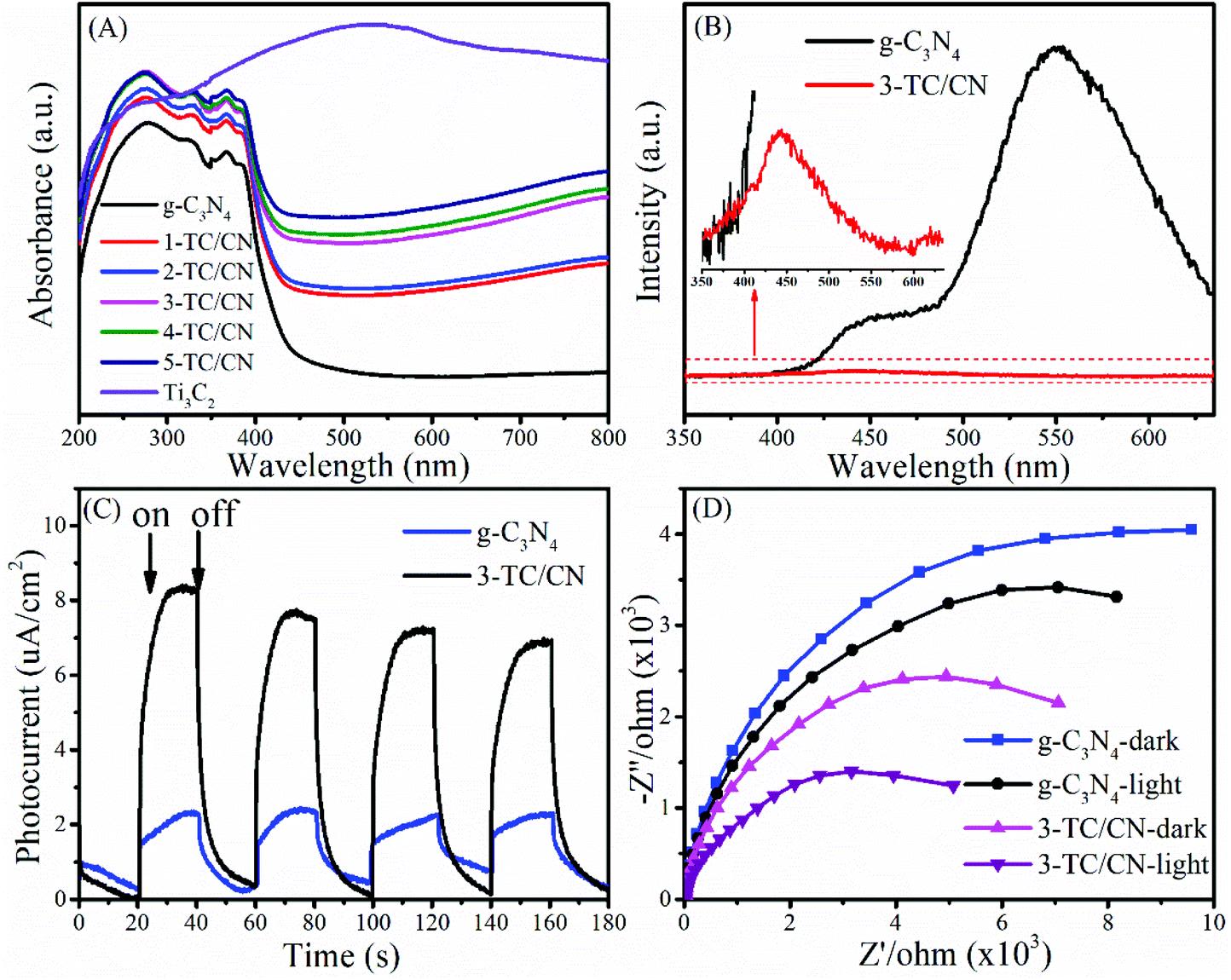 | ||
| Fig. 4 UV-vis absorption spectra of different photocatalysts (A), PL spectra of g-C3N4 and 3-TC/CN (B), transient photocurrent (C) and Nyquist plots (D) of g-C3N4 and 3-TC/CN in the darkness or under visible light irradiation. | ||
Photoluminescence (PL) spectroscopy represents a useful tool to understand the destiny of electron–hole pairs in photocatalysts, as PL emission is caused by the recombination of electrons and holes. Therefore, a low PL intensity generally indicates a high separation efficiency of electron–hole pairs.55,56 As shown in Fig. 4B, the PL intensity of the 3-TC/CN decreased significantly compared with that of pure g-C3N4 nanosheets, indicating that radiative recombination of charge carriers was suppressed. Notably, there are two obvious peaks located at around 445 and 550 nm in the PL spectrum of g-C3N4. The peak centred at 445 nm can be attributed to the band–band transition, which matches well with the absorption edge of the g-C3N4 from UV-Vis absorption measurements. While the peak at 550 nm may be due to the radiative recombination from the shallow trapped electrons, indicating the presence of sub-gap defects in the g-C3N4 nanosheets. Similar phenomenon was found in the previous reports.57–59 When the g-C3N4 nanosheets were combined with the Ti3C2 flakes, a 2D/2D intimate interface was formed, such a 2D/2D interface provides maximum contact surface between the g-C3N4 nanosheets and the Ti3C2 flakes. Furthermore, due to the high electrical conductivity of the metallic Ti3C2, the photogenerated electrons can easily transfer from the g-C3N4 nanosheets to the Ti3C2 flakes instead of to the defect states, resulted in the elimination of the main peak at 550 nm and the significantly lowered PL intensity. Such an electron transfer path is beneficial to the separation of photoelectrons and holes.
In order to shed more light on the charge transfer in the photocatalysts, transient photocurrent response and electrochemical impedance spectra (EIS) were collected for typical samples. As shown in Fig. 4C, the photocurrent intensity for 3-TC/CN is about four times stronger than that of pristine g-C3N4 when the light is on, indicating the better charge transfer efficiency within the ultrathin 2D/2D Ti3C2/g-C3N4 nanosheets. Fig. 4D shows the Nyquist plots of g-C3N4 and 3-TC/CN in the darkness or under visible light irradiation. It is well known that smaller arc radius of the Nyquist plot indicates lower electric charge transfer resistance in the samples.60 The arc radius of g-C3N4 and 3-TC/CN under visible light irradiation was obviously smaller than those in the darkness, which suggests the generation of photogenerated carriers in the samples. Moreover, the arc radius of 3-TC/CN sample was distinctly smaller than that of g-C3N4, further confirming the better charge transfer ability within the 3-TC/CN. Such enhanced charge transfer efficiency is likely due to the efficient interfacial charge transfer upon the 2D/2D heterojunction of ultrathin Ti3C2/g-C3N4 nanosheets. In the 2D/2D Ti3C2/g-C3N4 system, the metallic Ti3C2 is not able to generate the photoelectrons and holes under visible light irradiation. However, the Ti3C2 can be used as the electron acceptor to capture the photoelectrons generated by the g-C3N4 due to its excellent electrical conductivity, which can effectively suppress the recombination of photoinduced charge carriers.
The activity of all as-prepared samples was evaluated by photocatalytic hydrogen evolution from water under visible light irradiation (>400 nm), with triethanolamine (TEOA) as the hole scavenger. Control experiments indicate that no hydrogen was produced in the absence of photocatalyst or light irradiation. As displayed in Fig. 5A and B, the coupling of Ti3C2 flakes with g-C3N4 nanosheets leads to a remarkable enhancement in the photocatalytic hydrogen evolution rate. Pristine g-C3N4 nanosheets showed a very low hydrogen evolution rate of 7.1 μmol h−1 gcat−1, which is comparatively in consistent with the literature values.61,62 In contrast, when the g-C3N4 nanosheets combined with small content of Ti3C2 flakes (1.0 wt%), the photocatalytic activity of 1-TC/CN was obviously increased to 25.8 μmol h−1 gcat−1. With the increase of the content of Ti3C2 flakes, the photocatalytic hydrogen evolution rate of the Ti3C2/g-C3N4 composites is gradually improved.
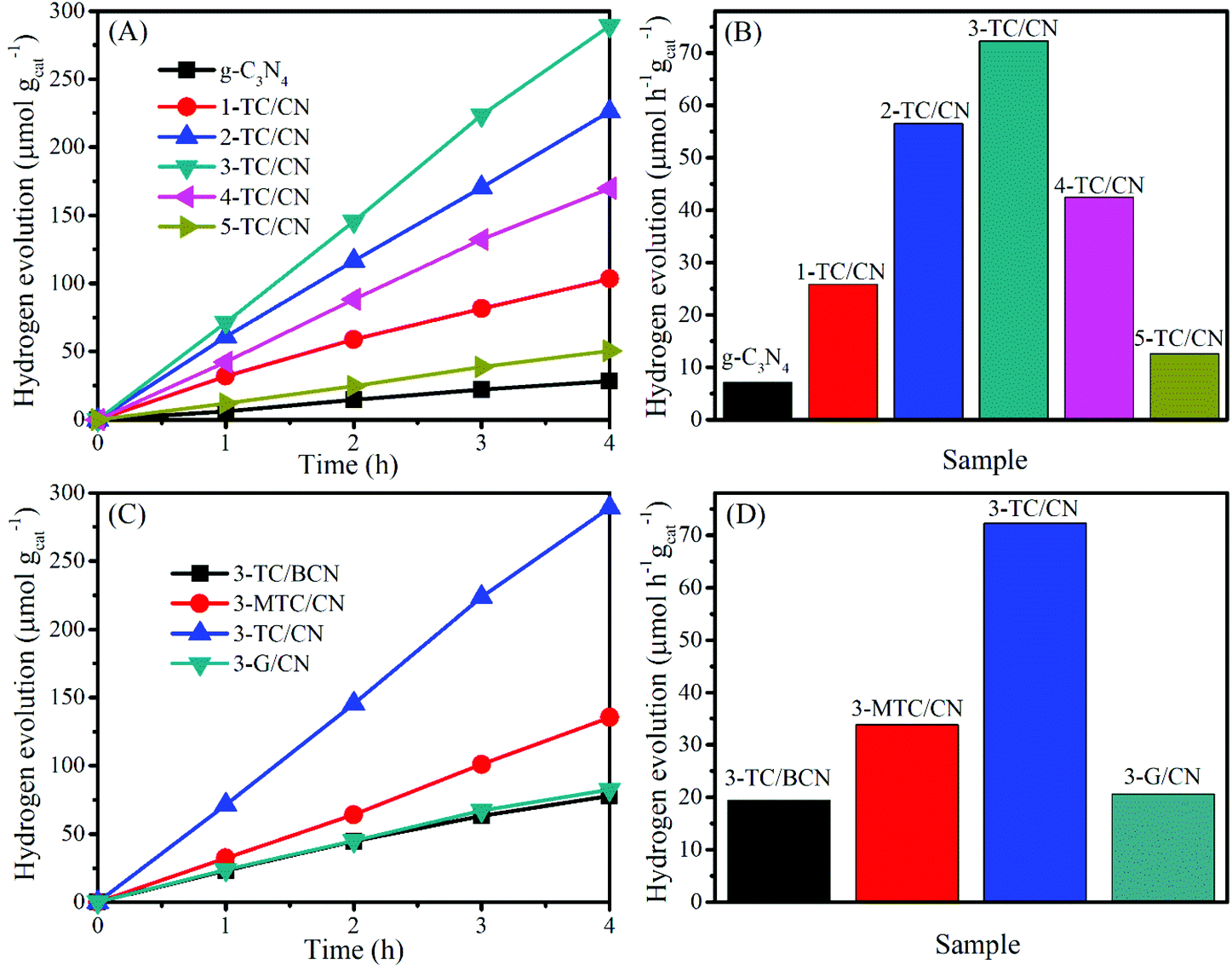 | ||
| Fig. 5 Photocatalytic hydrogen evolution over g-C3N4, 1-TC/CN, 2-TC/CN, 3-TC/CN, 4-TC/CN, 5-TC/CN, 3-TC/BCN, 3-MTC/CN, and 3-G/CN. | ||
The optimized sample 3-TC/CN shows the highest hydrogen production rate of 72.3 μmol h−1 gcat−1, which is more than 10 times higher than that of pristine g-C3N4. Moreover, the hydrogen production rate of 3-TC/CN is higher than that of the 3.0 wt% MWNTs/g-C3N4 (64.1 μmol h−1 gcat−1) and 1.0 wt% Pt/g-C3N4 (20.3 μmol h−1 gcat−1) under visible light (>400 nm).63 Such enhanced activity can be ascribed to the formation of 2D/2D compact interfaces in the ultrathin Ti3C2/g-C3N4 nanosheets. Under visible light irradiation, the photogenerated electrons produced on the conduction band of the g-C3N4 can effectively transfer via the 2D/2D interfaces and accumulate on the metallic Ti3C2, due to the short charge transfer distance and the large and compact contact surface. As a consequence, the recombination of the photogenerated electrons and holes was impeded and the photocatalytic activity of g-C3N4 was enhanced. However, the hydrogen production rate of the 3-TC/CN is still lower than that of the 3.0 wt% Pt/g-C3N4 (175.6 μmol h−1 gcat−1) under the same experimental conditions (Fig. S11, ESI†), which might be due to the less active sites on 3-TC/CN than the 3.0 wt% Pt/g-C3N4. Notably, increasing the content of Ti3C2 flakes past 3.0 wt% decreases the photocatalytic activity of g-C3N4. Such reduced activity is due to the excessive amount of Ti3C2 flakes hindering the light absorption of g-C3N4. Nevertheless, the 5-TC/CN sample still shows higher photocatalytic activity (12.6 μmol h−1 gcat−1) than that of the pristine g-C3N4. Moreover, no hydrogen can be detected when the Ti3C2 was used as the photocatalyst under visible light irradiation, indicating the photocatalytic reaction cannot be evoked by the Ti3C2 alone due to its metallic character. This result further confirms that the role of Ti3C2 is an electron acceptor and co-catalyst rather than a photocatalyst.
In comparison, under the same experimental conditions and with the same loading (3.0 wt%), 3-TC/BCN, 3-MTC/CN, and 3-G/CN were prepared and used in the photocatalytic hydrogen evolution reaction (Fig. 5C and D). The SEM images of the multilayer Ti3C2 (M-Ti3C2) and monolayer graphene can be found in Fig. S12.† In addition, the XRD patterns and the UV-vis absorption spectra of the 3-TC/BCN, 3-MTC/CN, and 3-G/CN samples can be seen in Fig. S13.† As shown in Fig. 5C and D, 3-G/CN shows higher photocatalytic activity (20.7 μmol h−1 gcat−1) than that of pristine g-C3N4 (7.1 μmol h−1 gcat−1) and shows much lower activity than that of 3-TC/CN (72.3 μmol h−1 gcat−1). Graphene has been widely used as an electron acceptor for photocatalysts to enhance their photocatalytic activity due to its good electrical conductivity. Herein, the lower activity of 3-G/CN than 3-TC/CN may be attributed to the following reasons: (1) O-terminated Ti3C2 with a low content of –OH and –F terminal groups was obtained by the LiF/HCl etching method,64 and the O-terminated MXenes have high work functions between 5.75 and 6.25 eV,65 which is much higher than that of graphene (4.5 eV).66,67 (2) The Gibbs free energy for hydrogen adsorption (ΔGH) on graphene is 0.79 eV,68 while the O-terminated Ti3C2 shows a near-zero value of ΔGH = 0.00283 eV at the same H* coverage (θ = 1/2), indicating that the O-terminated Ti3C2 is superior for H2 evolution reaction.33 (3) The hydrophobic nature of pure graphene is adverse to the adsorption of water molecules and is unfavorable for the hydrogen evolution from water.69,70 On the contrary, Ti3C2 possesses numerous hydrophilic functionalities (–OH and –O) on its surface, which can promote its strong interaction with water molecules.71 (4) Graphene is made of nothing but carbon. However, the Ti3C2 is consist of carbon and titanium, and the exposed terminal metal sites on the Ti3C2 might lead to stronger redox reactivity than that of the carbon materials.33 (5) The higher work functions of O-terminated Ti3C2 than that of graphene resulted in a higher Schottky barrier in 3-TC/CN than that in 3-G/CN.32 Thus the separation efficiency of the photogenerated electrons and holes in 3-TC/CN is higher than that of 3-G/CN, and this is confirmed by PL analysis (Fig. S14, ESI†).
In order to reveal whether the formation of 2D/2D heterojunction is the key factor for the enhanced photocatalytic activity or not, the photocatalytic activities of the 3-TC/BCN (2D/0D) and 3-MTC/CN (0D/2D) samples were investigated and compared with that of 3-TC/CN (2D/2D). Fig. 5C and D shows that the 3-TC/BCN and 3-MTC/CN samples exhibit much lower photocatalytic hydrogen production rate than that of 3-TC/CN sample, which suggests that the 2D/2D heterojunction of photocatalysts is more beneficial to suppress the recombination of photoinduced electrons and holes, thus enhancing the performance of photocatalysts. The advantage of the 2D/2D heterojunction has also been reported on the other 2D photocatalysts.1,46 In addition, the apparent quantum yield (400 nm) of the g-C3N4, 1-TC/CN, 2-TC/CN, 3-TC/CN, 4-TC/CN, 5-TC/CN, 3-TC/BCN, 3-MTC/CN and 3-G/CN was 0.08%, 0.29%, 0.63%, 0.81%, 0.47%, 0.14%, 0.22%, 0.38% and 0.23%, respectively.
The photochemical stability of a photocatalyst is very important for photocatalytic hydrogen evolution. Therefore, the photocatalytic stability of the optimal sample (3-TC/CN) was investigated (Fig. S15A, ESI†). After 3 cycles (reaction for 28 h), the photocatalytic hydrogen evolution rate of the 3-TC/CN did not show a remarkable decrease in activity, indicating its photochemical stability. To further study the stability of 3-TC/CN, XRD, SEM, UV-vis absorption and N2 adsorption–desorption was used to characterize the 3-TC/CN sample after photocatalysis. From the XRD pattern of the 3-TC/CN (Fig. S15B, ESI†), no obvious change can be observed, suggesting the composition and structure of the 3-TC/CN was maintained after reaction. Fig. S15C† shows that the light absorption of the 3-TC/CN decreased slightly after reaction, which is probably due to the separation of Ti3C2 flakes from the g-C3N4 nanosheets. Since the 2D/2D Ti3C2/g-C3N4 composites are prepared by the electrostatic self-assembly method, some Ti3C2 might not be able to bind with g-C3N4 tightly. Therefore, a long reaction with agitation might cause some Ti3C2 to separate from the Ti3C2/g-C3N4 composites, which leads to the deactivation of the photocatalysts. The BET specific surface area of the 3-TC/CN before and after reaction is 82.0 and 83.1 m2 g−1, respectively, which further confirms the stable nanosheet structure of the 3-TC/CN (Fig. S15D, ESI†). Moreover, the nanosheet morphology of the 3-TC/CN samples did not change according to the SEM images of 3-TC/CN (Fig. S16, ESI†). Besides, X-ray photoelectron spectra of the used 3-TC/CN indicate that no remarkable changes can be observed on the binding energies of Ti, C, and N, indicating the excellent chemical stability of the Ti3C2/g-C3N4 composites (Fig. S17 and Table S3, ESI†).
According to the above results and discussion, a mechanism of 2D/2D Ti3C2/g-C3N4 for photocatalytic hydrogen evolution under visible light irradiation is proposed and illustrated in Fig. 6. Under light irradiation, the electrons in the valence band (VB) of g-C3N4 can be excited to the conduction band (CB), generating photoinduced electrons and holes. Due to the excellent electronic conductivity of Ti3C2, the large intimate 2D/2D interface between the Ti3C2 and g-C3N4 and the short charge transfer distance, the migration rate of the photoinduced electrons can be greatly improved. In addition, the conduction band potential of the g-C3N4 was determined to be around −1.02 V vs. NHE using the Mott–Schottky plots.10,72–74 Besides, the Ti3C2 obtained from the LiF/HCl etching method should be O-terminated,65 and the Fermi level of the O-terminated Ti3C2 was theoretically determined to be 0.71 eV vs. NHE.53 Therefore, the most positive Fermi level of O-terminated Ti3C2 exhibits great ability to capture photoinduced electrons from the CB of the g-C3N4 and the holes was left on the VB of g-C3N4, which will significantly enhance the separation of electrons and holes.35 Moreover, a Schottky junction can be formed due to the close contact in 2D/2D Ti3C2/g-C3N4.35 In this case, the role of Ti3C2 as an electron receiver promotes the electrons transfer from g-C3N4 to Ti3C2. Therefore, the photogenerated electrons were accumulated on the Ti3C2 and the H+ is reduced to H2 on the surface of the Ti3C2. At the same time, the holes in the VB of g-C3N4 are used to oxidize TEOA into TEOA+. Thus, the photoinduced electrons and holes can be effectively separated and transferred in the 2D/2D Ti3C2/g-C3N4, which leads to remarkable enhancement of photocatalytic performance.
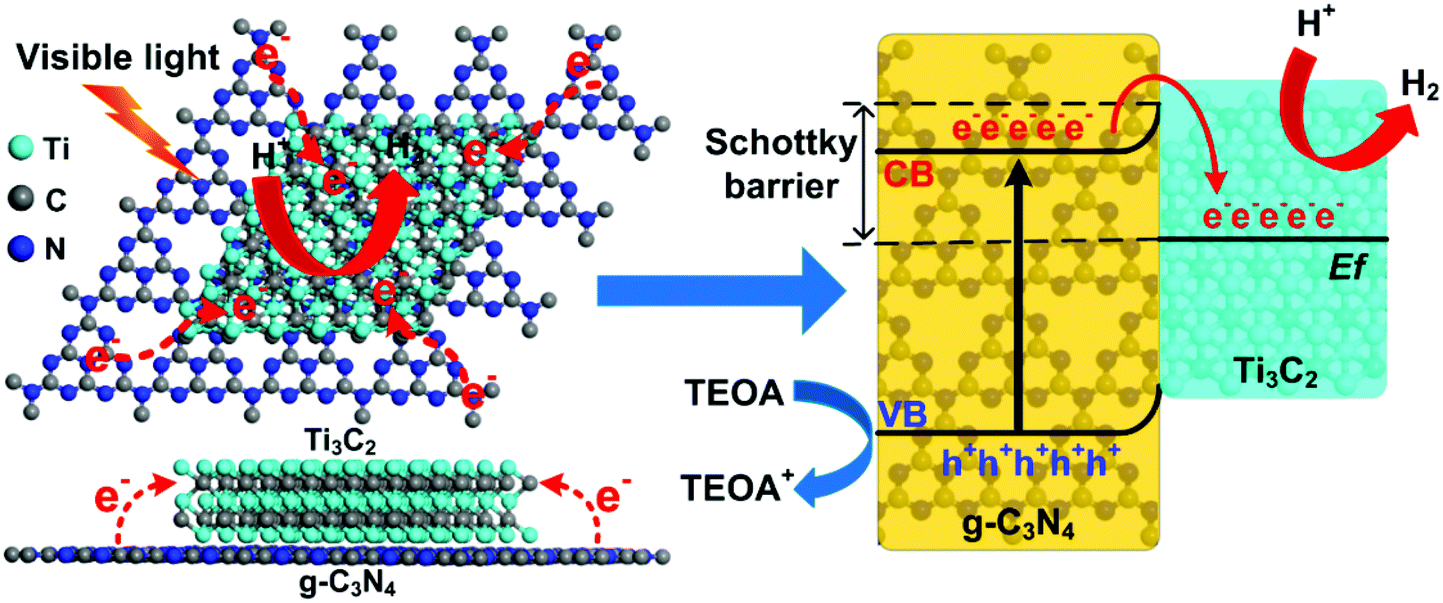 | ||
| Fig. 6 Proposed mechanism of photocatalytic hydrogen evolution over the Ti3C2/g-C3N4 system. | ||
Furthermore, the O-terminated Ti3C2 shows a near-zero value of Gibbs free energy for hydrogen adsorption (ΔGH = 0.00283 eV) at the H* coverage of 1/2 according to theoretical calculations,33 thus the H+ can be facilely reduced to H2 on the Ti3C2 surface. The hydrophilic functionalities and the exposed terminal metal sites on the Ti3C2 might also be beneficial to enhance the photocatalytic performance of the Ti3C2/g-C3N4. It is noted that the light adsorption of the Ti3C2/g-C3N4 composites is enhanced due to the dark color nature of Ti3C2. Although the enhanced light adsorption is unlikely to excite more photoinduced electrons and holes, the photothermal effect of Ti3C2 can convert light to heat.45,54 According to previous reports, the photocatalytic hydrogen production rate can be enhanced by heating of the reaction system, indicating the photothermal effect is related to the energy required for photocatalytic reaction.75 Moreover, the temperature of the photocatalyst can be increased and provide energy to activate reactant and boost the local photocatalytic reaction.53,76 In our current study, the reactor is kept at a constant temperature of 25 °C with a cooling recirculator. Yet the photothermal effect of Ti3C2 at local scale can still be potentially present and favorable for activating the photocatalysts and improving the photocatalytic hydrogen evolution efficiency.35
Conclusions
In summary, the 2D/2D Ti3C2/g-C3N4 nanosheets were successfully prepared by the electrostatic self-assembly approach. The formation of the 2D/2D heterojunctions in the Ti3C2/g-C3N4 composites was demonstrated. The photocatalytic hydrogen evolution rate of the optimized 2D/2D Ti3C2/g-C3N4 (3-TC/CN) was over 10 times higher than that of the pure g-C3N4. The superior activity of the 3-TC/CN was attributed to the 2D/2D intimate contact between the monolayer Ti3C2 and the g-C3N4 nanosheets. The 2D/2D interfaces provided a large charge transfer channel for accelerating the photoinduced electrons transfer from the conduction band of g-C3N4 to the metallic Ti3C2. Moreover, the construction of the Schottky junction further promoted the separation of the photogenerated electrons and holes, which will greatly enhance the photocatalytic activity of the g-C3N4. We believe that this work will stimulate ongoing interest in utilizing 2D MXene as co-catalysts to improve the performance of photocatalysts for hydrogen evolution as well as other reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported and conducted at the Center for Nanophase Materials Sciences, which is a DOE Office of Science User Facility. TMS acknowledges the support from China Scholarship Council. ZDH gratefully acknowledges a Graduate Research Fellowship award from the National Science Foundation (DGE-1650044). LB acknowledges financial support from National Science Foundation supplemental intern funding 1511818.References
- S. Liu, F. Chen, S. Li, X. Peng and Y. Xiong, Appl. Catal., B, 2017, 211, 1–10 CrossRef CAS.
- T. Su, Q. Shao, Z. Qin, Z. Guo and Z. Wu, ACS Catal., 2018, 8, 2253–2276 CrossRef CAS.
- X. Li, S. Liu, K. Fan, Z. Liu, B. Song and J. Yu, Adv. Energy Mater., 2018, 8, 1800101 CrossRef.
- Y. Jia, D. Zhao, M. Li, H. Han and C. Li, Chin. J. Catal., 2018, 39, 421–430 CrossRef CAS.
- R. Shi, H.-F. Ye, F. Liang, Z. Wang, K. Li, Y. Weng, Z. Lin, W.-F. Fu, C.-M. Che and Y. Chen, Adv. Mater., 2018, 30, 1705941 CrossRef PubMed.
- P.-Y. Kuang, P.-X. Zheng, Z.-Q. Liu, J.-L. Lei, H. Wu, N. Li and T.-Y. Ma, Small, 2016, 12, 6735–6744 CrossRef CAS PubMed.
- M. Ahmed and G. Xinxin, Inorg. Chem. Front., 2016, 3, 578–590 CAS.
- S. Zhao, Y. Zhang, Y. Zhou, Y. Wang, K. Qiu, C. Zhang, J. Fang and X. Sheng, Carbon, 2018, 126, 247–256 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- M. Zhu, S. Kim, L. Mao, M. Fujitsuka, J. Zhang, X. Wang and T. Majima, J. Am. Chem. Soc., 2017, 139, 13234–13242 CrossRef CAS PubMed.
- Y. Yu, Y. Wei, W. Xiaofang, L. Pei, G. Wenyu, Z. Haihan, W. Songmei and D. Kejian, Adv. Mater., 2018, 30, 1705060 CrossRef PubMed.
- L. Lin, W. Ren, C. Wang, A. M. Asiri, J. Zhang and X. Wang, Appl. Catal., B, 2018, 231, 234–241 CrossRef CAS.
- L. Lin, Z. Yu and X. Wang, Angew. Chem., Int. Ed., 2018 DOI:10.1002/anie.201809897.
- Y. Wang, S. Zhao, Y. Zhang, J. Fang, Y. Zhou, S. Yuan, C. Zhang and W. Chen, Appl. Surf. Sci., 2018, 440, 258–265 CrossRef CAS.
- R. Ma, L. Dong, B. Li, T. Su, X. Luo, Z. Qin and H. Ji, ChemistrySelect, 2018, 3, 5891–5899 CrossRef CAS.
- S. P. Adhikari, Z. D. Hood, V. W. Chen, K. L. More, K. Senevirathne and A. Lachgar, Sustainable Energy Fuels, 2018, 2, 2507–2515 RSC.
- S. P. Adhikari, Z. D. Hood, K. L. More, V. W. Chen and A. Lachgar, ChemSusChem, 2016, 9, 1869–1879 CAS.
- Y. Zheng, Z. Yu, H. Ou, A. M. Asiri, Y. Chen and X. Wang, Adv. Funct. Mater., 2018, 28, 1705407 CrossRef.
- L. Kong, Y. Ji, Z. Dang, J. Yan, P. Li, Y. Li and S. Liu, Adv. Funct. Mater., 2018, 28, 1800668 CrossRef.
- R.-B. Wei, Z.-L. Huang, G.-H. Gu, Z. Wang, L. Zeng, Y. Chen and Z.-Q. Liu, Appl. Catal., B, 2018, 231, 101–107 CrossRef CAS.
- Y.-J. Yuan, Y. Yang, Z. Li, D. Chen, S. Wu, G. Fang, W. Bai, M. Ding, L.-X. Yang, D.-P. Cao, Z.-T. Yu and Z.-G. Zou, ACS Appl. Energy Mater., 2018, 1, 1400–1407 CrossRef CAS.
- G. Zhang, Z.-A. Lan, L. Lin, S. Lin and X. Wang, Chem. Sci., 2016, 7, 3062–3066 RSC.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- Y. Xia, T. S. Mathis, M.-Q. Zhao, B. Anasori, A. Dang, Z. Zhou, H. Cho, Y. Gogotsi and S. Yang, Nature, 2018, 557, 409–412 CrossRef CAS PubMed.
- C. Chen, X. Xie, B. Anasori, A. Sarycheva, T. Makaryan, M. Zhao, P. Urbankowski, L. Miao, J. Jiang and Y. Gogotsi, Angew. Chem., Int. Ed., 2018, 57, 1846–1850 CrossRef CAS PubMed.
- K. Huang, Z. Li, J. Lin, G. Han and P. Huang, Chem. Soc. Rev., 2018, 47, 5109–5124 RSC.
- W. Yuan, L. Cheng, Y. An, H. Wu, N. Yao, X. Fan and X. Guo, ACS Sustainable Chem. Eng., 2018, 6, 8976–8982 CrossRef CAS.
- G. Gao, A. P. O'Mullane and A. Du, ACS Catal., 2017, 7, 494–500 CrossRef CAS.
- X. Yang, N. Gao, S. Zhou and J. Zhao, Phys. Chem. Chem. Phys., 2018, 20, 19390–19397 RSC.
- S. Li, P. Tuo, J. Xie, X. Zhang, J. Xu, J. Bao, B. Pan and Y. Xie, Nano Energy, 2018, 47, 512–518 CrossRef CAS.
- T. Su, R. Peng, Z. D. Hood, M. Naguib, I. N. Ivanov, J. K. Keum, Z. Qin, Z. Guo and Z. Wu, ChemSusChem, 2018, 11, 688–699 CrossRef CAS PubMed.
- H. Wang, R. Peng, Z. D. Hood, M. Naguib, S. P. Adhikari and Z. Wu, ChemSusChem, 2016, 9, 1490–1497 CrossRef CAS PubMed.
- J. Ran, G. Gao, F.-T. Li, T.-Y. Ma, A. Du and S.-Z. Qiao, Nat. Commun., 2017, 8, 13907 CrossRef CAS PubMed.
- Y. Sun, D. Jin, Y. Sun, X. Meng, Y. Gao, Y. Dall'Agnese, G. Chen and X.-F. Wang, J. Mater. Chem. A, 2018, 6, 9124–9131 RSC.
- X. An, W. Wang, J. Wang, H. Duan, J. Shi and X. Yu, Phys. Chem. Chem. Phys., 2018, 20, 11405–11411 RSC.
- B. Lin, H. Li, H. An, W. Hao, J. Wei, Y. Dai, C. Ma and G. Yang, Appl. Catal., B, 2018, 220, 542–552 CrossRef CAS.
- M. Zhang, J. Qin, S. Rajendran, X. Zhang and R. Liu, ChemSusChem, 2018, 11, 4226–4236 CAS.
- A. Lipatov, M. Alhabeb, M. R. Lukatskaya, A. Boson, Y. Gogotsi and A. Sinitskii, Adv. Electron. Mater., 2016, 2, 1600255 CrossRef.
- Y. Kang, Y. Yang, L.-C. Yin, X. Kang, G. Liu and H.-M. Cheng, Adv. Mater., 2015, 27, 4572–4577 CrossRef CAS PubMed.
- W.-J. Ong, L.-L. Tan, S.-P. Chai, S.-T. Yong and A. R. Mohamed, Nano Energy, 2015, 13, 757–770 CrossRef CAS.
- C. E. Ren, K. B. Hatzell, M. Alhabeb, Z. Ling, K. A. Mahmoud and Y. Gogotsi, J. Phys. Chem. Lett., 2015, 6, 4026–4031 CrossRef CAS PubMed.
- B. Tahir, M. Tahir and N. A. S. Amin, Appl. Surf. Sci., 2017, 419, 875–885 CrossRef CAS.
- Z. A. Lan, G. G. Zhang and X. C. Wang, Appl. Catal., B, 2016, 192, 116–125 CrossRef CAS.
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. N. Waterhouse, Y. Zhao, C. Zhou, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080–5086 CrossRef CAS PubMed.
- H. Lin, X. Wang, L. Yu, Y. Chen and J. Shi, Nano Lett., 2017, 17, 384–391 CrossRef CAS PubMed.
- Q. Wang, W. Wang, L. Zhong, D. Liu, X. Cao and F. Cui, Appl. Catal., B, 2018, 220, 290–302 CrossRef CAS.
- F. Cheng, H. Wang and X. Dong, Chem. Commun., 2015, 51, 7176–7179 RSC.
- Q. Y. Lin, L. Li, S. J. Liang, M. H. Liu, J. H. Bi and L. Wu, Appl. Catal., B, 2015, 163, 135–142 CrossRef CAS.
- S. Guo, Y. Tang, Y. Xie, C. Tian, Q. Feng, W. Zhou and B. Jiang, Appl. Catal., B, 2017, 218, 664–671 CrossRef CAS.
- J. Halim, K. M. Cook, M. Naguib, P. Eklund, Y. Gogotsi, J. Rosen and M. W. Barsoum, Appl. Surf. Sci., 2016, 362, 406–417 CrossRef CAS.
- P. Niu, L. Zhang, G. Liu and H.-M. Cheng, Adv. Funct. Mater., 2012, 22, 4763–4770 CrossRef CAS.
- Q. Han, B. Wang, J. Gao, Z. Cheng, Y. Zhao, Z. Zhang and L. Qu, ACS Nano, 2016, 10, 2745–2751 CrossRef CAS PubMed.
- C. Shaowen, S. Baojia, T. Tong, F. Junwei and Y. Jiaguo, Adv. Funct. Mater., 2018, 28, 1800136 CrossRef.
- R. Li, L. Zhang, L. Shi and P. Wang, ACS Nano, 2017, 11, 3752–3759 CrossRef CAS PubMed.
- T. Su, H. Tian, Z. Qin and H. Ji, Appl. Catal., B, 2017, 202, 364–373 CrossRef CAS.
- Y. Huang, H. Xu, H. Yang, Y. Lin, H. Liu and Y. Tong, ACS Sustainable Chem. Eng., 2018, 6, 2751–2757 CrossRef CAS.
- P. Wu, J. Wang, J. Zhao, L. Guo and F. E. Osterloh, J. Mater. Chem. A, 2014, 2, 20338–20344 RSC.
- G. Dong, D. L. Jacobs, L. Zang and C. Wang, Appl. Catal., B, 2017, 218, 515–524 CrossRef CAS.
- J. S. Kim, J. W. Oh and S. I. Woo, Catal. Today, 2017, 293–294, 8–14 CrossRef CAS.
- Y. Huang, K. Li, Y. Lin, Y. Tong and H. Liu, ChemCatChem, 2018, 10, 1982–1987 CrossRef CAS.
- Z. Sun, M. Zhu, M. Fujitsuka, A. Wang, C. Shi and T. Majima, ACS Appl. Mater. Interfaces, 2017, 9, 30583–30590 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- L. Ge and C. Han, Appl. Catal., B, 2012, 117–118, 268–274 CrossRef CAS.
- M. A. Hope, A. C. Forse, K. J. Griffith, M. R. Lukatskaya, M. Ghidiu, Y. Gogotsi and C. P. Grey, Phys. Chem. Chem. Phys., 2016, 18, 5099–5102 RSC.
- S. Chertopalov and V. N. Mochalin, ACS Nano, 2018, 12, 6109–6116 CrossRef CAS PubMed.
- E. J. H. Lee, K. Balasubramanian, R. T. Weitz, M. Burghard and K. Kern, Nat. Nanotechnol., 2008, 3, 486 CrossRef CAS PubMed.
- R. Maiti, S. Haldar, D. Majumdar, A. Singha and S. K. Ray, Nanotechnology, 2017, 28, 075707 CrossRef CAS PubMed.
- H.-J. Qiu, Y. Ito, W. Cong, Y. Tan, P. Liu, A. Hirata, T. Fujita, Z. Tang and M. Chen, Angew. Chem., Int. Ed., 2015, 54, 14031–14035 CrossRef CAS PubMed.
- I. Rahim, M. Shah, A. Khan, J. Luo, A. Zhong, M. Li, R. Ahmed, H. Li, Q. Wei and Y. Fu, Sens. Actuators, B, 2018, 267, 42–50 CrossRef CAS.
- Y. Liu, J. Liu, Q. Ding, J. Tan, Z. Chen, J. Chen, X. Zuo, A. Tang and K. Zeng, Macromol. Mater. Eng., 2018, 303, 1800053 CrossRef.
- M. Ghidiu, S. Kota, V. Drozd and M. W. Barsoum, Sci. Adv., 2018, 4, eaao6850 CrossRef PubMed.
- M. Wang, M. Shen, L. Zhang, J. Tian, X. Jin, Y. Zhou and J. Shi, Carbon, 2017, 120, 23–31 CrossRef CAS.
- R. Wang, L. Gu, J. Zhou, X. Liu, F. Teng, C. Li, Y. Shen and Y. Yuan, Adv. Mater. Interfaces, 2015, 2, 1500037 CrossRef.
- M. Yan, Y. Hua, F. Zhu, L. Sun, W. Gu and W. Shi, Appl. Catal., B, 2017, 206, 531–537 CrossRef CAS.
- S. I. Nikitenko, T. Chave, C. Cau, H.-P. Brau and V. Flaud, ACS Catal., 2015, 5, 4790–4795 CrossRef CAS.
- Z. Gan, X. Wu, M. Meng, X. Zhu, L. Yang and P. K. Chu, ACS Nano, 2014, 8, 9304–9310 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplemental characterization of photocatalysts by XRD, Raman, SEM, AFM, BET, EDS, XPS, UV-vis absorption, and PL, additional photocatalytic experiments, characterization of used photocatalysts by XRD, UV-vis absorption, BET, SEM, and XPS. See DOI: 10.1039/c9nr00168a |
| This journal is © The Royal Society of Chemistry 2019 |