
Tunable internal quantum well alignment in rationally designed oligomer-based perovskite films deposited by resonant infrared matrix-assisted pulsed laser evaporation†
Wiley A.
Dunlap-Shohl
 a,
E. Tomas
Barraza
b,
Andrew
Barrette
c,
Seyitliyev
Dovletgeldi
c,
Gamze
Findik
c,
David J.
Dirkes
d,
Chi
Liu
e,
Manoj K.
Jana
ae,
Volker
Blum
ae,
Wei
You
d,
Kenan
Gundogdu
c,
Adrienne D.
Stiff-Roberts
*b and
David B.
Mitzi
*ae
a,
E. Tomas
Barraza
b,
Andrew
Barrette
c,
Seyitliyev
Dovletgeldi
c,
Gamze
Findik
c,
David J.
Dirkes
d,
Chi
Liu
e,
Manoj K.
Jana
ae,
Volker
Blum
ae,
Wei
You
d,
Kenan
Gundogdu
c,
Adrienne D.
Stiff-Roberts
*b and
David B.
Mitzi
*ae
aDepartment of Mechanical Engineering and Materials Science, Duke University, Durham, North Carolina 27708, USA. E-mail: david.mitzi@duke.edu
bDepartment of Electrical and Computer Engineering, Duke University, Durham, North Carolina 27708, USA. E-mail: adrienne.stiffroberts@duke.edu
cDepartment of Physics, North Carolina State University, Raleigh, North Carolina 27695, USA
dDepartment of Chemistry, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA
eDepartment of Chemistry, Duke University, Durham, North Carolina 27708, USA
First published on 16th May 2019
Abstract
Hybrid perovskites incorporating conjugated organic cations enable unusual charge carrier interactions among organic and inorganic structural components, but are difficult to prepare as films due to disparate component chemical/physical characteristics (e.g., solubility, thermal stability). Here we demonstrate that resonant infrared matrix-assisted pulsed laser evaporation (RIR-MAPLE) mitigates these challenges, enabling facile deposition of lead-halide-based perovskite films incorporating variable-length oligothiophene cations. Density functional theory (DFT) predicts suitable organic and inorganic moieties that form quantum-well-like structures with targeted luminescence or exciton separation/quenching. RIR-MAPLE-deposited films enable confirmation of these predictions by optical measurements, which further display excited state behavior transcending traditional quantum-well models—i.e., with appropriate selection of specially synthesized organic/inorganic moieties, intercomponent carrier transfer efficiently converts excitons from singlet to triplet states in organics for which intersystem crossing cannot ordinarily compete with recombination. These observations demonstrate the uniquely versatile excited-state behavior in hybrid perovskite quantum wells, and the value of integrating DFT, organic synthesis, RIR-MAPLE and spectroscopy for screening/preparing rationally devised complex structures.
New conceptsHybrid organic–inorganic materials incorporating large, complex organic moieties can possess many interesting synergistic physical properties, such as targeted exciton migration and efficient organic singlet-to-triplet exciton conversion mediated by inorganic states. The vast diversity of organic and inorganic structural motifs possible for construction of such compounds suggests an unprecedented opportunity to tailor material properties. However, they are difficult to study systematically, in large part because the field lacks a thin film deposition method capable of reliably managing the disparate chemistries of the organic and inorganic components. Herein, we demonstrate that resonant infrared matrix-assisted pulsed laser evaporation (RIR-MAPLE) is an effective film deposition technique for this materials class, enabling detailed study of photophysical properties. Films deposited by RIR-MAPLE enable transient absorption spectroscopy measurements on oligothiophene-based lead halide perovskites, providing a critical window into excited state dynamics and validating hybrid density functional theory (DFT) predictions of quantum well alignment between the various inorganic/organic moieties. This work presents a paradigm for rational design and study of complex-organic hybrid materials, uniting the predictive capability of hybrid DFT, the versatile RIR-MAPLE film deposition method and sophisticated optical measurements, thereby facilitating investigation into the underexplored field of hybrid semiconductors with functional organic moieties. |
Introduction
Hybrid organic–inorganic perovskites have recently captured intense research attention due to their excellent optoelectronic and unique physical properties.5–10 However, the organic component generally plays only an indirect role in these properties through its effects on the inorganic lattice.11,12 The most widely studied hybrid perovskites incorporate relatively small organic cations, such as methyl-, phenethyl-, or butylammonium, for which the large energy gap between the organic component highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) straddles the gap between the inorganic valence band maximum (VBM) and conduction band minimum (CBM).13–15 Consequently, these materials are effectively inorganic from the perspective of optoelectronics, as features such as light absorption, luminescence and conductivity derive directly from this structural component. One of the most exciting yet underexplored features of hybrid perovskites is that the organic component need not be relegated to a supporting role. In structures incorporating large conjugated organic moieties, the HOMO–LUMO gap shrinks to a level that allows carrier or exciton transfer between the different structural components, enabling these materials to act as self-assembled quantum wells with tunable character as controlled by distinct organic/inorganic chemistries.3,18,19 As in traditional quantum wells, varying relative band positions can lead to control over exciton localization within the structure or carrier separation. However, involvement of the organic cation can introduce additional subtlety through participation of the organic triplet state(s), enabling behaviors unique to hybrid quantum well systems—i.e., band alignment may favor exciton transfer to singlet states, resulting in fluorescence from organic chromophores,16,17,20,21 or exciton transfer through inorganic bands to triplet states, potentially resulting in phosphorescence.18,22–25 An unprecedented degree of control over the excited state is therefore attainable in these perovskites, and by extension other prospective hybrid systems. The uniqueness of both the atomic structure and properties, as neither inorganic nor organic but possessing new characteristics derived from the synergy of these components, implies that these compounds represent a new and distinct class of materials.Although the properties of these materials are intriguing, a major challenge complicating the study of “complex-organic perovskites” (COPs) is thin film deposition. Most investigations into the properties of perovskites or related hybrids incorporating truly large cations, particularly those large enough for the organic singlet state to lie within the inorganic band gap and exhibit fluorescence,20,21,26,27 resort to single crystals or powders. However, bulk samples are often inadequate for sensitive and powerful techniques such as transient absorption spectroscopy (TAS), which can provide a wealth of information about excited state dynamics, but require films thin enough to provide a meaningful signal. Traditional solution processing methods are hindered by the disparate affinities of the organic and inorganic precursors for non-polar and polar solvents, respectively. Meanwhile, vapor deposition methods are hampered by risk of thermal damage and difficulty in controlling evaporation rates of organic precursors. Other techniques developed for film deposition of COPs also have major shortcomings. Film deposition of oligothiophene-derived COPs was achieved using single-source thermal ablation (SSTA),16,17 but this approach cannot reliably control film thickness and also entails significant risk of thermally decomposing the precursor materials. Era et al.28 have demonstrated formation of COP thin films via a Langmuir–Blodgett monolayer formation process, but this method also entails considerable challenges in developing compatible precursor–solvent–substrate systems due to immersion of partially-formed films in one or more solutions. The difficulty in producing COP films impedes not only device fabrication but also the detailed investigation of physical properties. There is thus a need for a deposition technique that unites the compositional precision of solution processing with the flexibility of vapor deposition, allowing COPs to be fabricated on any substrate.
Here, we tackle the challenges of depositing COP films using the RIR-MAPLE technique.29–32 The evaporation target in RIR-MAPLE is composed of a frozen precursor solution wherein the hydroxyl bonds of a “matrix” co-solvent are resonant with the incident infrared laser.32 This resonant absorption of laser energy avoids damaging sensitive precursor species, instead only evaporating the frozen solvent matrix surrounding them. Solubility problems are mitigated by the ability to use low concentration (∼10 mM or less) precursor solutions to create contiguous thin films. Most of the evaporated solvent is continuously removed by vacuum throughout the deposition, avoiding possible adverse solvent interactions. We therefore anticipate that RIR-MAPLE offers a maximally flexible approach for depositing a wide range of high-quality COP films in a substrate-agnostic manner. Some of us have previously deposited the small-cation perovskite CH3NH3PbI3 using RIR-MAPLE,29,30 but we see the technique filling a more prominent gap in available technologies when considering the challenging class of COPs.
Results and discussion
In this study, we focus on lead halide COPs incorporating oligothiophene chromophores terminated on either end by protonated aminoethyl groups to form divalent organic cations (AEnT2+, where n signifies the number of thiophene rings in the chain). These materials adopt the generalized chemical formula AEnTPbX4, for arbitrary halide X−. In an early experimental study by Mitzi et al.17 and a more recent hybrid density functional theory (DFT) study by Liu et al.,3 varying n and X can result in hybrid quantum wells with tunable behavior. Possible band alignments (Fig. 1) are Type 1A (inorganic bands straddle organic HOMO–LUMO), Type 1B (HOMO–LUMO straddle inorganic bands), or Type 2A/B (HOMO–LUMO and inorganic bands are staggered). For Type 1A (1B), excitons transfer to the organic (inorganic) part, leading to luminescence of that moiety. For Type 2, the excitons should dissociate to separate parts of the structure, quenching the luminescence. In the latter case, for appropriately selected organic/inorganic components, it is possible to subsequently reform excitons in the organic triplet states. The singlet–triplet transition is forbidden in the neat organic chromophore, but can be enabled in the hybrid material if the carriers cascade through appropriate inorganic states.18,22,25 In this sense, resulting hybrid quantum well structures may transcend the traditional inorganic quantum well model and involve synergistic organic–inorganic interactions.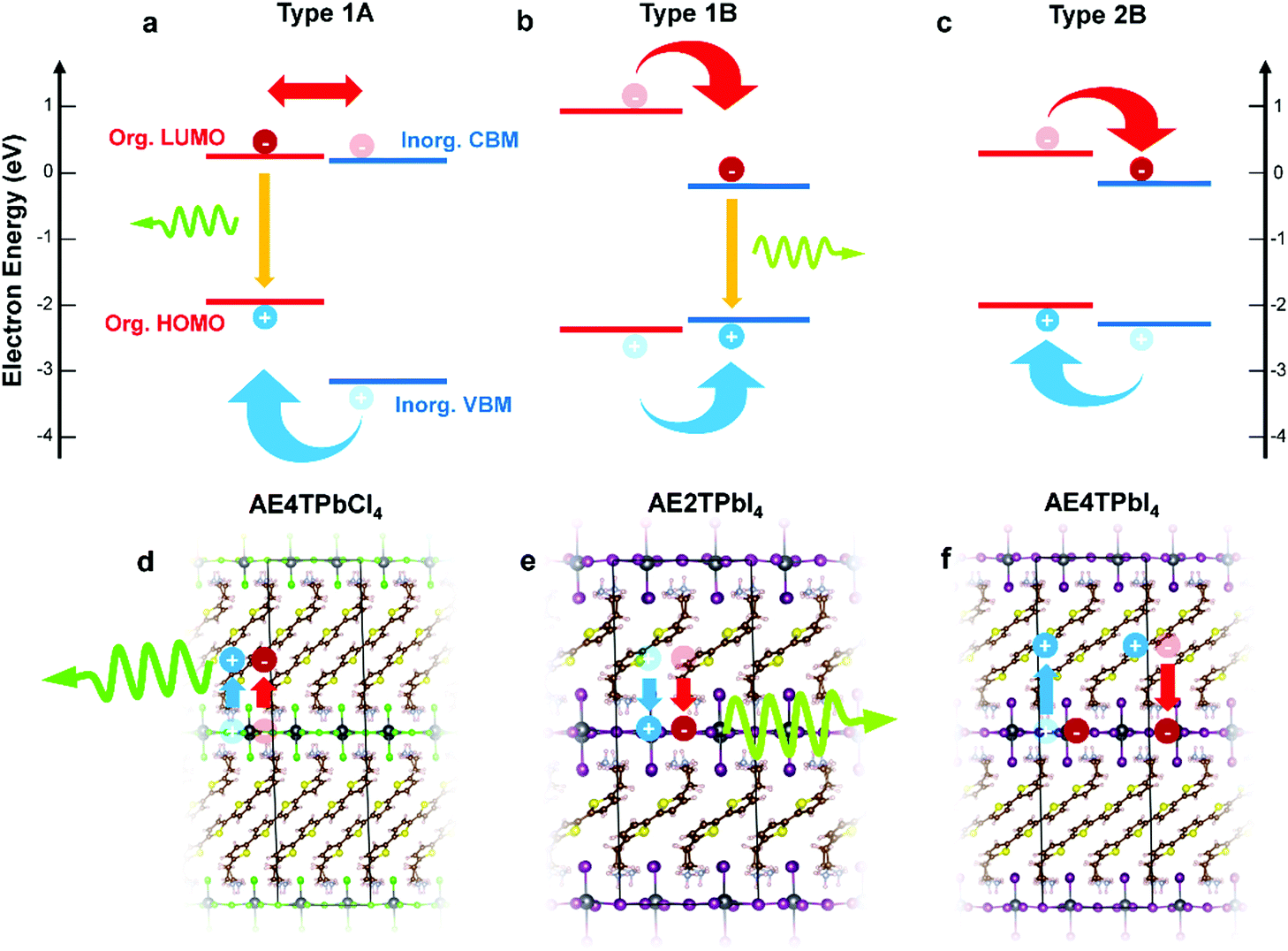 | ||
| Fig. 1 Schematics of hybrid spin–orbit coupled DFT predicted internal band/molecular orbital alignments (a–c) and anticipated exciton/luminescence behavior (d–f) in AE4TPbCl4 (a and d), AE2TPbI4 (b and e) and AE4TPbI4 (c and f). AE4TPbCl4 is expected to exhibit a Type 1A quantum well structure, wherein excitons generated in the inorganic part migrate to the organic part, leading to luminescence characteristic of the quaterthiophene. AE2TPbI4 is predicted to exhibit Type 1B behavior, wherein excitons generated in the organic part migrate to the inorganic part, leading to luminescence from the lead halide sheet. AE4TPbI4 is expected to exhibit Type 2B behavior, where excitons generated in either part dissociate across the organic–inorganic interface, inhibiting radiative recombination and quenching luminescence. Band positions are drawn to scale based on the calculations of Liu et al.3 for AE4TPbI4 and AE4TPbCl4, and on the calculations for AE2TPbI4 reported in this work. Note that, within the accuracy limits of currently applicable first-principles calculations, the organic LUMO of AE4TPbCl4 is essentially indistinguishable from the inorganic VBM; prior experimental results,16,17 as well as those described in this work, support its assignment as a Type 1A structure. | ||
All three band alignments are expected to exist within the AEnTPbX4 family. To facilitate rational selection of COP compounds exhibiting the three distinct behaviors, we extend the demanding hybrid spin–orbit-coupled DFT33–41 calculations of Liu et al.3 with newly-predicted band alignments in the AEnTPbX4 family with n = 1–5, X = Cl, Br, I (Fig. S1 and Table S1, ESI†). Among the 15 AEnTPbX4 compounds considered, AE4TPbCl4, AE2TPbI4, and AE4TPbI4 are expected to form Type 1A, 1B, and 2B structures, respectively, and were therefore selected for further study. Prior reports place the triplet state of the quaterthiophene (4T) molecule at ∼1.7 eV above the ground state,42 just below the ∼1.8 eV gap between the organic HOMO and the inorganic CBM predicted by our calculations, supporting the possibility that the triplet state may play a role in AE4TPbI4 photophysics.17 While intriguing, oligothiophene-based perovskites are challenging to work with, particularly those incorporating quaterthiophene; single crystals of AE4TPbI4 have only recently been reported,3 and those of AE4TPbCl4 have never been, emphasizing the difficulty posed by their low solubility. By providing a straightforward means of depositing films, RIR-MAPLE allows us to broadly explore excited-state behavior in these systems through steady-state and transient optical measurements, using targeted compositions selected for their theory-predicted properties. This work thereby furnishes a paradigm for rational design and investigation of COPs exhibiting exotic properties—i.e., tunable quantum wells with accessible triplet exciton states—by employing hybrid spin–orbit coupled DFT to select promising organic/inorganic components, organic synthesis to obtain targeted molecules, and RIR-MAPLE to prepare hybrid quantum well films for spectroscopic study (which in turn provides feedback for subsequent compound design).
To fabricate oligothiophene perovskite films, we use a similar procedure to that developed for CH3NH3PbI3,29,30 but using custom-synthesized AEnT molecular cations (Scheme 1), as described in the Methods section/ESI† and shown schematically in Fig. 2a. X-ray diffraction (XRD) patterns of the resulting COP thin films are shown in Fig. 2b–d, along with expected patterns simulated from the computationally optimized atomic positions (van der Waals corrected semilocal DFT43,44) used to calculate the band structures. In each pattern, the peaks are weak relative to the background and fairly broad, and do not appear to favor a particular crystallographic orientation. These features are unusual for 2D halide perovskite films, which normally comprise a single series of strong and sharp peaks, indicating films consisting of large grains with their layers parallel to the substrate. These patterns are instead characteristic of films composed of small, randomly oriented grains, a more common microstructure for 3D perovskites such as CH3NH3PbI3, especially those prepared by vapor-deposition methods.45–47 Film morphology in SEM images (Fig. S3, ESI†) is consistent with XRD, showing conformal substrate coverage with ∼50–100 nm grains. Visual examination of the films (Fig. S4, ESI†) indicates that they are smooth and homogeneous across the entire surface, roughly 1–4 cm2. When placed under a 365 nm blacklight, film fluorescence (Fig. S4, ESI† and Fig. 3 inset) is uniform in color and intensity, and consistent with the expected photoluminescence emission for each composition, as discussed below. Atomic force microscopy (AFM) measurements (Fig. S5, ESI†) of the films are broadly consistent with those previously performed on MAPLE-deposited CH3NH3PbI3,30 exhibiting a conformal topography and root-mean-square roughnesses in the range of 30–50 nm.
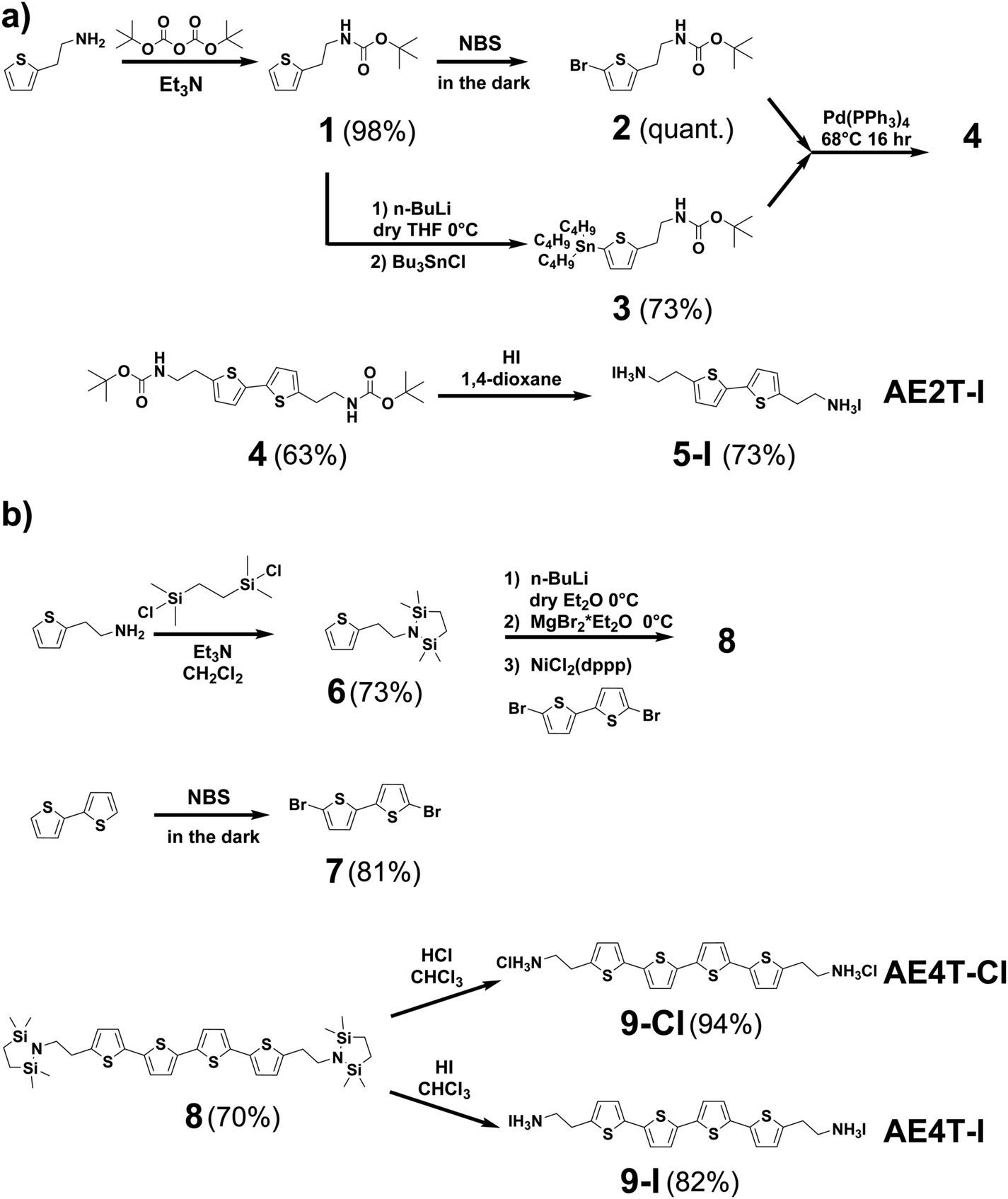 | ||
| Scheme 1 Synthesis pathways for (a) bi- and (b) quaterthiophene halide salts used in this study. Detailed discussion of the synthesis of each compound shown in the scheme is given in the Methods section and ESI.† Briefly, AE2T·2HI is synthesized starting from a thiophene-2-ethylamine precursor via a method reported by Harvey and Tovar.1 The amine is first protected using a tert-butyloxycarbonyl group to form tert-butyl (2-(thiophen-2-yl)ethyl)carbamate (1). The bithiophene diamine building blocks are prepared by treating 1 with N-bromosuccinimide (NBS) to form tert-butyl (2-(5-bromothiophen-2-yl)ethyl)carbamate (2) or tributyl tin chloride to form tert-butyl (2-(5-(tributylstannyl)thiophen-2-yl)ethyl)carbamate (3). 2 and 3 are then coupled via Stille reaction to form di-tert-butyl ([2,2′-bithiophene]-5,5′-diylbis(ethane-2,1-diyl))dicarbamate (4), which is thereafter deprotected using hydriodic acid, yielding the desired AE2T·HI product (5-I). AE4T·2HX salts are synthesized starting from bithiophene and thiophene-2-ethylamine precursors via a method reported by Muguruma et al.4 The amine is protected to form 2,2,5,5-tetramethyl-1-(2-(thiophen-2-yl)ethyl)-1,2,5-azadisilolidine (6), while bithiophene is brominated with NBS to form 5,5′-dibromo-2,2′-bithiophene (7). 7 and lithiated 6 are thereafter linked via Kumada coupling reaction to yield the protected quaterthiophene diamine 5,5′′′-bis(2-(2,2,5,5-tetramethyl-1,2,5-azadisilolidin-1-yl)ethyl)-2,2′:5′,2′′:5′′,2′′′-quaterthiophene (8), which is thereafter deprotected using the appropriate hydrohalic acids, yielding the desired ammonium salts AE4T·2HCl (9-Cl) and AE4T·2HI (9-I). | ||
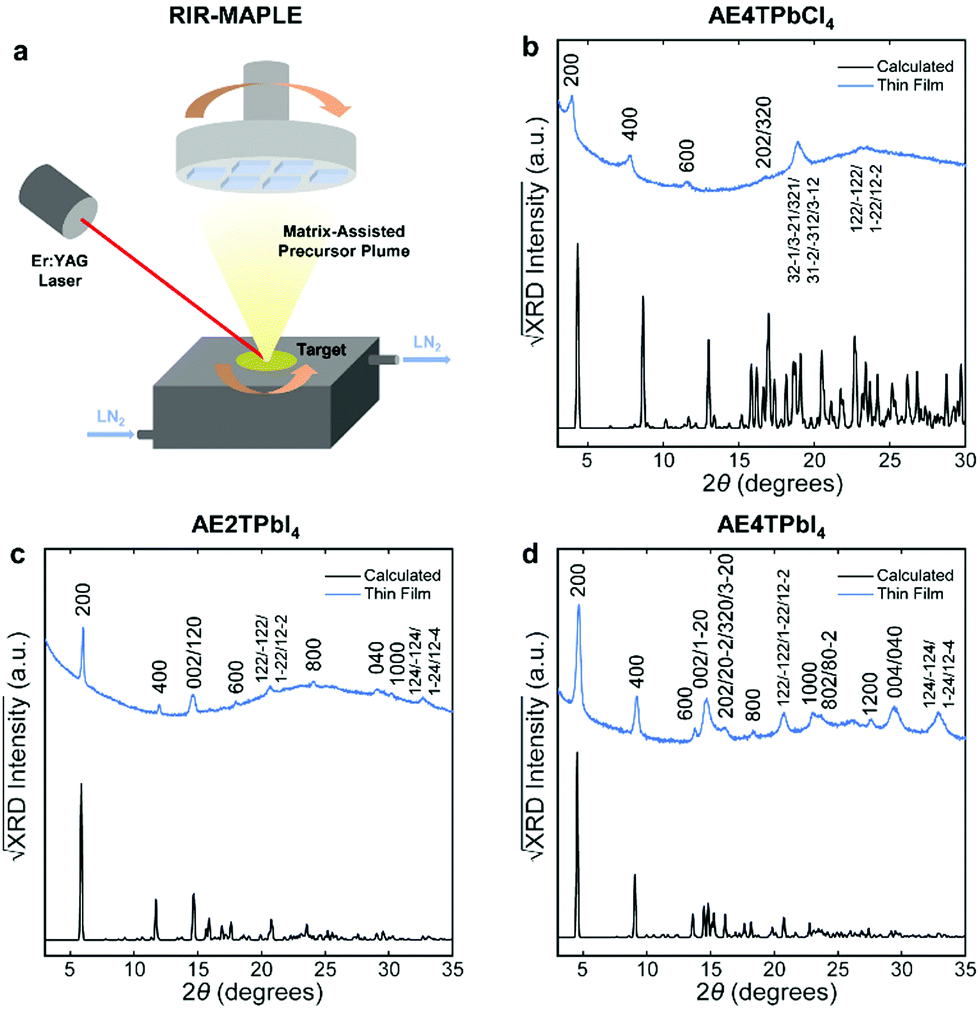 | ||
| Fig. 2 Schematic of the RIR-MAPLE process (a), and Cu Kα XRD patterns of oligothiophene perovskite films deposited by RIR-MAPLE, referenced against patterns calculated using computationally optimized structures from van der Waals corrected semilocal DFT: (b) AE4TPbCl4, (c) AE2TPbI4 and (d) AE4TPbI4. Comparison of the film XRD patterns against the calculated patterns indicates that all of the peaks in the former may be assigned to Miller indices corresponding to reasonably prominent peaks in the latter. Similar sets of reflections (e.g., h00, 0k0, 122) appear in the patterns of both (AE2T/AE4T)PbI4, indicating similar texture in these closely related materials. The AE4TPbCl4 pattern contains fewer and less-well-defined peaks. However, features resembling those in the other patterns are evident, such as the first three h00 reflections, which decrease in intensity in a manner consistent with the predicted pattern and the other two perovskites. The theory-derived AE4TPbCl4 pattern slightly underestimates the lattice constant along the a axis; however, the inferred values are still broadly consistent with the expected structure. Note that another recent computational study on AE4TPbCl4 predicts a similar value for the interlayer lattice parameter as our DFT calculations;2 however, also note that relatively subtle changes in the configuration of the quaterthiophene cations can have substantial impact on the lattice parameters in the DFT-predicted structures, as explored further in the ESI† (Fig. S2 and Table S2). Accurate determination of all subtleties of the structure would require the growth and XRD refinement of single crystals, which is beyond the scope of this work. Importantly, the key optoelectronic properties reported below are experimentally derived and thus not affected by small structural deviations between the computationally assumed model and the experimental material. | ||
 | ||
| Fig. 3 UV-vis (solid green lines), PL (solid pink lines) and PLE (dashed pink lines) spectra of oligothiophene COP and precursor salt thin films deposited by RIR-MAPLE: (a) AE4TPbCl4, (b) AE2TPbI4, (c) AE4TPbI4, (d) AE2T·2HI and (e) AE4T·2HI. PL spectra are collected using excitation by a monochromated Xe lamp at 280 nm (a) or 335 nm (b–e), at energies high enough to generate excitons in both parts of the perovskite structure. PLE spectra are collected using a monochromated Xe lamp at emission wavelengths of 600 nm (a and e), 560 nm (b), 570 nm (c) or 430 nm (d)—i.e., near the maximum intensity of the PL spectrum. In (b–d), the increase in the PLE signal at longer wavelengths is due to collection of scattered excitation light. All optical data are scaled as noted in the figure in order to be plotted on the same axes. Insets: Photographs of perovskite and thiophene salt films under a 365 nm UV lamp, displaying the characteristic luminescence of each composition (or lack thereof, in the case of AE4TPbI4). | ||
Successful formation of the 2D perovskite structure is also evident in the film optical properties. Excitons in the inorganic sheets cause a characteristic peak in the absorption spectrum to appear near 330 and 520 nm for lead chloride and iodide perovskites, respectively.17,48 The absorption spectrum of each COP film (Fig. 3) demonstrates the exciton peak in the expected location. These spectra also contain features assignable to the organic component, evidenced by their similarity to those in the corresponding spectra of the isolated organic precursor salts, which serve as proxies for the neat organics. Photoluminescence emission (PL) and excitation (PLE) spectroscopies were also performed on COP and precursor salt films (Fig. 3). The AE4TPbCl4 PL spectrum consists of a quaterthiophene feature resembling that of pristine AE4T·2HI, without a peak corresponding to excitons in the PbCl4 sheets near 330 nm. In the PLE spectrum of AE4TPbCl4, a short-wavelength shoulder lies off the main quaterthiophene absorption peak, closely aligned with the 330 nm exciton absorption peak. This feature is absent from the PLE spectrum of AE4T·2HI, demonstrating that excitons generated in the inorganic layer indeed contribute to the characteristic quaterthiophene luminescence of AE4TPbCl4, validating the proposed Type 1A quantum well structure. Note also that the presence of characteristic luminescence from the quaterthiophene shows that the organic molecule is transferred intact from the precursor solution to the final films, both in the perovskite and the pristine salt.
The AE2TPbI4 PL spectrum comprises a single peak that largely overlaps the exciton peak in the absorption spectrum. Excitation at 335 nm should generate excitons in both organic and inorganic moieties, but no PL from AE2T·2HI is evident in the AE2TPbI4 spectrum, signifying that excitons migrate to the lead iodide sheets and radiatively recombine there, consistent with the predicted Type 1B structure. Furthermore, the PLE spectrum does not exactly match the absorption spectrum; reduction of PLE at shorter wavelengths may signify that excitons generated in the organic component undergo some nonradiative recombination, competing with transfer to the inorganic component and leading to less efficient PL than if those excitons were generated directly within the inorganic part. The PLE spectrum also shows a feature at 470 nm. The corresponding absorption spectrum mostly appears featureless at that wavelength, but any absorption at that wavelength results in PL nearly as efficient as that generated by wavelengths resonant with the inorganic excitons. The origin of this feature is not completely clear, but its energetic position relative to the inorganic exciton peak suggests a possible interfacial state with the hole and electron in the organic and inorganic parts, respectively, a configuration only slightly higher in energy than that with both carriers in the inorganic bands (0.14 eV) (see Fig. 1b and Table S1, ESI†). This difference is reasonably close to the experimentally observed 0.25 eV difference between the 470 nm (2.64 eV) and 518 nm (2.39 eV) peaks in the PLE spectrum. For AE4TPbI4, PL and PLE spectra resemble those of AE4TPbCl4 but are considerably weaker. This difference is especially clear from the PLE spectra (collected for wavelengths near the PL peak for both perovskites to best represent their maximum emissive strength), as the AE4TPbI4 spectrum must be enhanced by a factor of 100 to appear on the same scale as that of AE4TPbCl4. The observed PL quenching is in general agreement with observations of Mitzi et al.17 for films prepared by SSTA, and consistent with the Type 2B quantum well structure.
To better understand excited state behavior, TAS was performed on the COP films. Fig. 4a depicts TAS spectra at 15 ps delay time of the three different samples excited at 400 nm (AE4TPbI4, AE2TPbI4) or 330 nm (AE4TPbCl4), chosen such that both organic and inorganic components can be excited. The resulting dynamics are traced by measuring the differential absorption of a broadband 400–800 nm probe pulse at various time delays. For AE2TPbI4, the TAS spectrum exhibits a strong exciton photobleach corresponding to the inorganic component (labeled PB2) and a weaker photobleach at higher energies (PB1). For AE4TPbI4, the TAS spectrum shows similar shortwave bleaching, inorganic exciton bleaching, and additionally two broad photoinduced absorption (PIA) features at 640 nm (PIA1) and 700 nm (PIA2). These peaks are the dominant spectral features observed in AE4TPbCl4 and also in AE4T·2HI films (Fig. 4d and Fig. S6, ESI†), implying an intrinsic connection to the quaterthiophene chromophore. For AE2TPbI4, the spectrum mostly shows response from the inorganic moiety, as predicted by the band alignment (Fig. 1). Similarly, for AE4TPbCl4, the response is predominantly from the organic moiety due to the lower energy of excitons in the organic layer. In AE4TPbI4, the band alignment favors charge separation; accordingly, the TAS spectrum shows prominent features from both components of the structure.
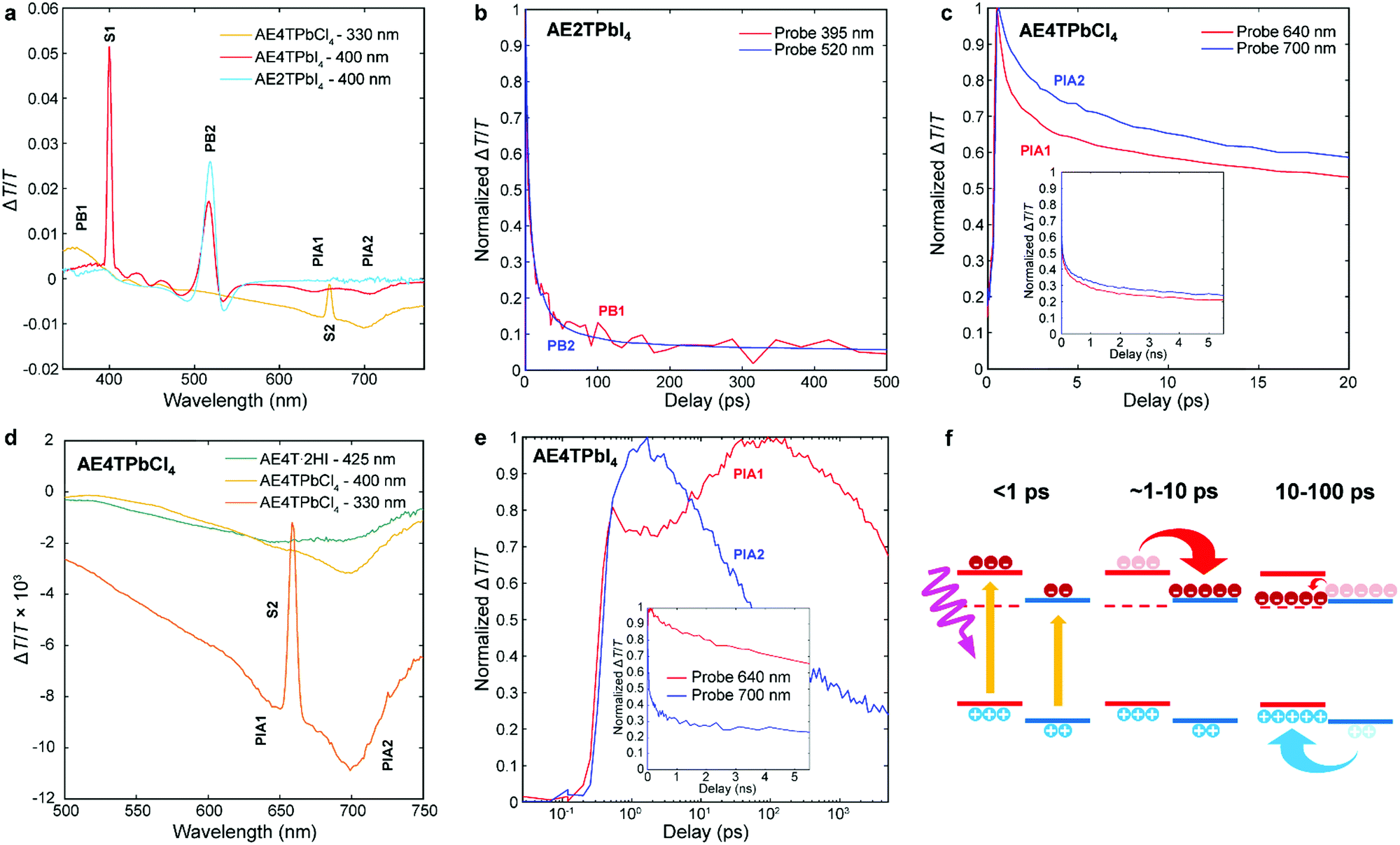 | ||
| Fig. 4 Transient absorption spectra of the AE4TPbI4, AE2TPbI4, and AE4TPbCl4 samples after a pump–probe delay time of 15 ps (a); decay kinetics of selected features in AE2TPbI4 (b), AE4TPbCl4 (c and d), and AE4TPbI4 (e); schematic of carrier transfer in AE4TPbI4 (f), wherein the triplet state is represented by the dashed line and the band positions are the same as in Fig. 1. The features labeled S1 and S2 in (a) and (d) correspond to scattered light from the pump beam at 400 nm and 660 nm, respectively. | ||
We further elucidate carrier population dynamics by analyzing time evolution of important spectral features. For AE2TPbI4 (Fig. 4b), the material is excited at 330 nm and the probe is traced at 395 nm (PB1) and 520 nm (PB2), aimed at tracking populations in the organic and inorganic parts, respectively. The time evolution of both features is very similar, suggesting that optical excitations quickly move to the PbI4 sheets and that the response is mostly determined by the inorganic component. For AE4TPbCl4 (Fig. 4c), we trace the dynamics at 640 (PIA1) and 700 nm (PIA2). Comparison of these features in AE4T·2HI with AE4TPbCl4 excited with 330 and 400 nm pump beams at 15 ps delay (Fig. 4d) indicates that PIA1 is due to excitons and PIA2 is due to hole polarons, as discussed further in the ESI† and briefly summarized below. Similar features are often used to distinguish excitons from free carriers in other organic compounds.49,50 When AE4TPbCl4 is excited at 400 nm, PIA2 strengthens because of a higher driving force for holes to concentrate in the organic part compared to electrons. When AE4TPbCl4 is excited at 330 nm, both PIA1 and PIA2 are increased further because carriers generated in the inorganic part now contribute to these populations as well, providing further evidence for the Type 1B band alignment. After photoexcitation, both features abruptly rise and decay, with similar decay dynamics over the first 10 ps. Afterward, the decay slows, suggesting that excitons accumulate in the organic part, consistent with the observed PL and PLE spectra.
In AE4TPbI4 (Fig. 4e), PIA2 rises for the first few ps, then decays, while PIA1 rises very sharply during excitation, first decays for about 3 ps, then rises again to a maximum intensity maintained over the first 100 ps before its final decay, which occurs on a time scale of ns. The initial decay of PIA1 coincides with the rise of PIA2 in the first 3 ps, which indicates separation of charges due to the band alignment. The second rise of PIA1 beyond its original intensity is unexpected from the perspective of traditional quantum well systems but perfectly consistent with an energy cascade from singlet to triplet excitons through the inorganic states, as illustrated schematically in Fig. 4f. Since both organic and inorganic components are initially excited, the total number of triplet excitons collected in the organic is more than the original singlet exciton population, explaining why PIA1 increases above its initial peak value due to this electron cascade. The long subsequent decay of PIA1 (Fig. 4e) also suggests that these triplet excitons survive much longer than the total time window of the experiment, with apparent lifetime of ∼10 ns. Note that exciton transfer occurs rapidly, with the triplet population in the organic reaching its maximum value within 30–40 ps, a reasonable value when compared to the results of Ema et al.,18 who report from time-resolved PL measurements that an analogous process in naphthylethylammonium lead bromide takes place with a time constant of ∼50–100 ps. Because triplet states in oligothiophenes generally do not lead to efficient phosphorescence,42 PL measurements are inferior to TAS for tracking excitations in oligothiophene-derived perovskites or other hybrid materials with weak triplet emission. This feature underscores the importance of the ability to produce high-quality COP thin films, which enable detailed study of their photophysical dynamics, and consequently tuning and control of exciton behavior.
Conclusions
In this work, we have combined the predictive capabilities of hybrid spin–orbit coupled DFT, the versatility of organic chemistry, the unique film deposition strengths of RIR-MAPLE and the detailed information available from TAS to design, fabricate and investigate hybrid perovskites incorporating specifically selected oligothiophene-derived cations to span a wide range of targeted excited state behaviors. While COPs provide the opportunity to rationally design (employing hybrid DFT and organic synthesis) quantum well structures that behave nominally like strictly inorganic analogs (e.g., AE4TPbCl4 and AE2TPbI4), hybrids based on oligothiophene-derived cations have been shown to provide an additional degree of freedom to tailor excited state properties involving organic triplet states (e.g., AE4TPbI4), extending beyond traditional quantum well models. RIR-MAPLE yields a convenient pathway for versatile deposition of COP films that span this range of possibilities, and TAS enables a detailed accounting and validation of the excited state dynamics. In particular, for AE4TPbI4, we see that favorable alignment of inorganic CBM and organic states leads to population of excited state carriers in the otherwise inaccessible triplet state, demonstrating an important synergistic effect involving both organic/inorganic COP components. The powerful combination of hybrid DFT, organic synthesis, RIR-MAPLE and TAS is expected to provide a more general framework for rationally exploring the broader range of possibilities provided by the vast COP family and further provides the opportunity to design new hybrid quantum wells (even beyond perovskites) that enable unique synergistic properties and useful hybrid configurations for optoelectronics and beyond.Experimental methods
Hybrid DFT calculations
All first-principles calculations are performed using the all-electron numeric atom-centered orbital code FHI-aims.33,34,36–38 For the relaxation of crystal geometries, we perform a full relaxation of unit cell parameters and cell-internal atomic coordinates on the level of semi-local DFT with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional,43 plus the Tkatchenko–Scheffler (TS) pairwise dispersion scheme44 to account for van der Waals (vdW) interactions. In the geometry relaxations, FHI-aims “tight” numerical defaults were employed. Unit cell sizes of up to 480 atoms (AE5TPbX4 with X = Cl, Br, I) were considered. k-Point grids of 1 × 2 × 2 were used to sample the Brillouin zones in the geometry relaxations for all the COPs investigated. The initial computational models for all the COPs investigated were built by substitution of inorganic or organic component of the experimental structure of AE4TPbBr4.17 Details of how the models were constructed can be found in the ESI Section IX of ref. 3 and analogous choices were made for new structures in the present work. Importantly, while these structures are thus directly derived from an experimentally known progenitor, small structural deviations from actual experimental compounds would not be critical due to the important synergy/division of labor between theory and experiment in our work. Specifically, we employ computational results as indicators for expected types of behavior (particularly, level alignments). These results are subsequently validated and extended by dedicated experiments for compounds of outstanding interest, completing the picture unambiguously. For predictions of frontier orbital and band structure properties, we rely on the Heyd–Scuseria–Ernzerhof (HSE06) hybrid density functional40,41 plus perturbative spin–orbit coupling (SOC).35 FHI-aims “Intermediate” settings and dense k-point grids setting of 3 × 3 × 3 were used in the HSE06 calculations for obtaining electronic properties. The internal parameters of the HSE06 functional were fixed at α = 0.25 for the exchange mixing parameter and ω = 0.11 Bohr−1 for the screening parameter, following ref. 40, avoiding any further materials-specific parameterization. In traditional inorganic semiconductors with similar band gaps, we would expect this choice to systematically underestimate the fundamental gap by approximately 0.3 eV.39Synthesis of oligothiophene salts
Synthesis of the quaterthiophene salts was adapted from the procedure described by Muguruma et al.;4 synthesis of bithiophene salts was adapted from that described by Harvey and Tovar.1 All chemicals were purchased from commercial sources (e.g., Sigma-Aldrich, Fisher, Matrix) and used as received except when specified. Tetrahydrofuran (THF) was distilled over sodium and benzophenone before use. Anhydrous dimethylformamide (DMF) and dichloromethane (DCM) were purchased in a sealed bottle from Fisher. For reactions performed under an inert atmosphere, the reaction flask was flame dried under vacuum and cycled between vacuum and argon three times before any reagents were added. 1H and 13C nuclear magnetic resonance (NMR) measurements (Fig. S7–S16, ESI†) were recorded with Bruker DRX spectrometers (400 MHz, 500 MHz, or 600 MHz). Details of the synthesis reactions are provided in the ESI† and are depicted in Scheme 1.Thin-film deposition by RIR-MAPLE
Solutions were prepared by dissolving equimolar amounts of the oligothiophene and lead halide salts in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume mixture of dimethyl sulfoxide (DMSO) and monoethylene glycol (MEG). All solutions were loaded into the stainless steel RIR-MAPLE target cup within the evaporation chamber at room temperature and the chamber was then evacuated. Once chamber pressure reached 10 Torr the target cup began to be cooled using liquid nitrogen. Chamber pressure was then cycled between 10 and 100 Torr using N2 gas until target temperature reached −196 °C. At this point, chamber pressure was raised to 250–275 Torr and held there for 5 minutes to allow the target solution to freeze. Afterwards, the chamber was once again evacuated until base pressure (1 × 10−4 Torr) was established, allowing target laser evaporation to begin. Unless otherwise noted, the target-to-substrate distance was 7 cm, and the deposition duration was 3 h.
:1 by volume mixture of dimethyl sulfoxide (DMSO) and monoethylene glycol (MEG). All solutions were loaded into the stainless steel RIR-MAPLE target cup within the evaporation chamber at room temperature and the chamber was then evacuated. Once chamber pressure reached 10 Torr the target cup began to be cooled using liquid nitrogen. Chamber pressure was then cycled between 10 and 100 Torr using N2 gas until target temperature reached −196 °C. At this point, chamber pressure was raised to 250–275 Torr and held there for 5 minutes to allow the target solution to freeze. Afterwards, the chamber was once again evacuated until base pressure (1 × 10−4 Torr) was established, allowing target laser evaporation to begin. Unless otherwise noted, the target-to-substrate distance was 7 cm, and the deposition duration was 3 h.
Films for optical and XRD measurements were deposited on soda-lime glass (Electron Microscopy Sciences, 1.2 mm thick) or quartz (GM Associates, 1 mm thick) substrates; films for SEM measurements were deposited on indium tin oxide (ITO) coated soda-lime glass substrates (10 Ω sq−1, 1.1 mm thick). Before deposition, substrates were cleaned by sonication in acetone, then isopropanol for 10 minutes each, and thereafter plasma cleaned in a mixture of Ar and O2 for 10 min. Specific growth considerations regarding each perovskite composition are listed below. AE4TPbI4: films were prepared using 4 mM and 8 mM solutions; the latter was used in conjunction with a 4 cm target-to-substrate distance and 6 h deposition time to produce thick samples for XRD and PL measurements. After deposition, the films were annealed in a nitrogen-filled glovebox containing <0.1 ppm H2O and O2 on a ceramic hot plate at 200 °C for 30 min. AE2TPbI4: films were prepared using 4 mM and 6 mM solutions. After deposition, they were annealed on a ceramic hot plate in a nitrogen-filled glovebox at 150 °C for 10 min. AE4TPbCl4: films were prepared using a 2 mM solution due to solubility restrictions, but the target-to-substrate distance was reduced to 4 cm and the deposition time increased to 6 h to compensate for the reduction in film thickness resulting from the lower concentration. After deposition, the films were annealed on a ceramic hot plate in a nitrogen-filled glovebox at 125 °C for 5 min.
Film characterization
XRD patterns were collected using a Panalytical Empyrean powder X-ray diffractometer with Cu Kα radiation and 45 kV/40 mA operating voltage/current. Reference XRD patterns were calculated from the computationally relaxed structures using CrystalDiffract software. SEM images were collected from an FEI Apreo high-resolution scanning electron microscope using an accelerating voltage of 2 kV. AFM measurements were collected in tapping mode using a Digital Instruments Dimension 3100 instrument, using a tapping frequency of 1 Hz, a voltage ranging from 0.9–1.1 V, and resolution of 512 samples/line and 512 lines/image. UV-vis spectra were collected using a Shimadzu UV-3600 spectrophotometer using a blank substrate as reference. Steady-state PL/PLE spectra were obtained from an Edinburgh Instruments FS920 fluorimeter that was equipped with a 450 W xenon arc lamp as the excitation source, and a Peltier-cooled Hamamatsu R2658P photomultiplier tube. TAS experiments were performed in a Helios system. Pump and probe were produced from output of a Libra regenerative amplifier, with a Coherent OPerA Solo for tuning pump, and CaF2 crystal generation for white light probe. Newport mechanical delay stages were used to control pump–probe delay.Author contributions
W. A. D.-S., E. T. B., A. D. S.-R. and D. B. M. conceived, designed, and directed the project. W. A. D.-S. was executive author of the manuscript, prepared target solutions, performed XRD, SEM, and UV-vis measurements, and analyzed resulting data. E. T. B. and A. D. S.-R. deposited the perovskite films and performed AFM measurements. D. D. and W. Y. synthesized organic precursor salts. C. L. and V. B. performed DFT calculations. A. B., S. D. G. F., and K. G. performed and analyzed PL, PLE, and TAS measurements. All authors contributed to the writing of the manuscript and discussion of the results.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Yosuke Kanai for useful discussion. W. A. D.-S., D. B. M., M. K. J., C. L., V. B., D. D., W. Y., A. B., S. D., and K. G. acknowledge support from the NSF DMREF program under grant numbers DMR-1729297, DMR-1728921 and DMR-1729383. W. A. D.-S. and E. T. B. also gratefully acknowledge support from the Fitzpatrick Institute for Photonics John T. Chambers Scholarship. A. D. S.-R. and E. T. B. acknowledge research supported as part of the Center for Hybrid Organic Inorganic Semiconductors for Energy (CHOISE), an Energy Frontier Research Center funded by the U. S. Department of Energy, Office of Science, Basic Energy Sciences (BES) (RIR-MAPLE film deposition). An award of computer time was provided by the INCITE program. This research used resources of the Argonne Leadership Computing Facility, which is a DOE Office of Science User Facility supported under Contract DE-AC02-06CH11357. This work was performed in part at the Duke University Shared Materials Characterization Facility (SMIF), a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), which is supported by the National Science Foundation (Grant ECCS-1542015) as part of the National Nanotechnology Coordinated Infrastructure (NNCI).References
- C. P. Harvey and J. D. Tovar, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4861–4874 CrossRef CAS.
- Z. Wang, A. M. Ganose, C. Niu and D. O. Scanlon, J. Mater. Chem. C, 2019, 7, 5139–5147 RSC.
- C. Liu, W. Huhn, K. Z. Du, A. Vazquez-Mayagoitia, D. Dirkes, W. You, Y. Kanai, D. B. Mitzi and V. Blum, Phys. Rev. Lett., 2018, 121, 146401 CrossRef CAS.
- H. Muguruma, T. Saito, S. Sasaki, S. Hotta and I. Karube, J. Heterocyclic Chem., 1996, 33, 173–178 CrossRef CAS.
- N. J. Jeon, H. Na, E. H. Jung, T.-Y. Yang, Y. G. Lee, G. Kim, H.-W. Shin, S. Il Seok, J. Lee and J. Seo, Nat. Energy, 2018, 3, 682–689 CrossRef CAS.
- A. Kojima, K. Teshima, Y. Shirai and T. Miyasaka, J. Am. Chem. Soc., 2009, 131, 6050–6051 CrossRef CAS PubMed.
- J. S. Manser, J. A. Christians and P. V. Kamat, Chem. Rev., 2016, 116, 12956–13008 CrossRef CAS PubMed.
- L. N. Quan, F. P. Garcia de Arquer, R. P. Sabatini and E. H. Sargent, Adv. Mater., 2018, 30, e1801996 CrossRef PubMed.
- M. D. Smith, B. A. Connor and H. I. Karunadasa, Chem. Rev., 2019, 119, 3104–3139 CrossRef CAS.
- D. A. Egger, A. Bera, D. Cahen, G. Hodes, T. Kirchartz, L. Kronik, R. Lovrincic, A. M. Rappe, D. R. Reichman and O. Yaffe, Adv. Mater., 2018, 30, e1800691 CrossRef PubMed.
- J. Knutson, J. D. Martin and D. B. Mitzi, Inorg. Chem., 2005, 44, 4699–4705 CrossRef CAS.
- K. Z. Du, Q. Tu, X. Zhang, Q. Han, J. Liu, S. Zauscher and D. B. Mitzi, Inorg. Chem., 2017, 56, 9291–9302 CrossRef CAS PubMed.
- S. Silver, J. Yin, H. Li, J.-L. Brédas and A. Kahn, Adv. Energy Mater., 2018, 8, 1703468 CrossRef.
- P. Umari, E. Mosconi and F. De Angelis, Sci. Rep., 2014, 4, 4467 CrossRef PubMed.
- W.-J. Yin, T. Shi and Y. Yan, Adv. Mater., 2014, 26, 4653–4658 CrossRef CAS PubMed.
- K. Chondroudis and D. B. Mitzi, Chem. Mater., 1999, 11, 3028–3030 CrossRef CAS.
- D. B. Mitzi, K. Chondroudis and C. R. Kagan, Inorg. Chem., 1999, 38, 6246–6256 CrossRef CAS.
- K. Ema, M. Inomata, Y. Kato, H. Kunugita and M. Era, Phys. Rev. Lett., 2008, 100, 257401 CrossRef CAS PubMed.
- D. B. Mitzi, K. Chondroudis and C. R. Kagan, IBM J. Res. Dev., 2001, 45, 29–45 CAS.
- M. Braun, W. Tuffentsammer, H. Wachtel and H. C. Wolf, Chem. Phys. Lett., 1999, 303, 157–164 CrossRef CAS.
- D. Cortecchia, C. Soci, M. Cametti, A. Petrozza and J. Martí-Rujas, ChemPlusChem, 2017, 82, 681–685 CrossRef CAS.
- M. Era, K. Maeda and T. Tsutsui, Chem. Phys. Lett., 1998, 296, 417–420 CrossRef CAS.
- G. C. Papavassiliou, G. A. Mousdis, G. Pagona, N. Karousis and M.-S. Vidali, J. Lumin., 2014, 149, 287–291 CrossRef CAS.
- G. C. Papavassiliou, G. Pagona, G. A. Mousdis and N. Karousis, Chem. Phys. Lett., 2013, 570, 80–84 CrossRef CAS.
- S. Yang, D. Wu, W. Gong, Q. Huang, H. Zhen, Q. Ling and Z. Lin, Chem. Sci., 2018, 9, 8975–8981 RSC.
- A. C. Véron, A. Linden, N. A. Leclaire, E. Roedern, S. Hu, W. Ren, D. Rentsch and F. A. Nüesch, J. Phys. Chem. Lett., 2018, 9, 2438–2442 CrossRef.
- H. Barkaoui, H. Abid, A. Yangui, S. Triki, K. Boukheddaden and Y. Abid, J. Phys. Chem. C, 2018, 122, 24253–24261 CrossRef CAS.
- M. Era, Y. Shironita and K. Soda, Jpn. J. Appl. Phys., 2018, 57, 03EG07 CrossRef.
- E. T. Barraza, W. A. Dunlap-Shohl, D. B. Mitzi and A. D. Stiff-Roberts, J. Electron. Mater., 2017, 47, 917–926 CrossRef.
- W. A. Dunlap-Shohl, E. T. Barraza, A. Barrette, K. Gundogdu, A. D. Stiff-Roberts and D. B. Mitzi, ACS Energy Lett., 2018, 3, 270–275 CrossRef CAS.
- R. Pate and A. D. Stiff-Roberts, Chem. Phys. Lett., 2009, 477, 406–410 CrossRef CAS.
- A. D. Stiff-Roberts and W. Ge, Appl. Phys. Rev., 2017, 4, 041303 Search PubMed.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- V. Havu, V. Blum, P. Havu and M. Scheffler, J. Comput. Phys., 2009, 228, 8367–8379 CrossRef CAS.
- W. Huhn and V. Blum, Phys. Rev. Mater., 2017, 1, 033803 CrossRef.
- A. C. Ihrig, J. Wieferink, I. Y. Zhang, M. Ropo, X. Ren, P. Rinke, M. Scheffler and V. Blum, New J. Phys., 2015, 17, 093020 CrossRef.
- S. V. Levchenko, X. Ren, J. Wieferink, R. Johanni, P. Rinke, V. Blum and M. Scheffler, Comput. Phys. Commun., 2015, 192, 60–69 CrossRef CAS.
- X. Ren, P. Rinke, V. Blum, J. Wieferink, A. Tkatchenko, A. Sanfilippo, K. Reuter and M. Scheffler, New J. Phys., 2012, 14, 053020 CrossRef.
- T. Zhu, W. P. Huhn, G. C. Wessler, D. Shin, B. Saparov, D. B. Mitzi and V. Blum, Chem. Mater., 2017, 29, 7868–7879 CrossRef CAS.
- A. V. Krukau, O. A. Vydrov, A. F. Izmaylov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 224106 CrossRef PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- S. Rentsch, J. P. Yang, W. Paa, E. Birckner, J. Scheidt and R. Weinkauf, Phys. Chem. Chem. Phys., 1999, 1, 1707–1714 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- M. Liu, M. B. Johnston and H. J. Snaith, Nature, 2013, 501, 395–398 CrossRef CAS PubMed.
- C. Momblona, L. Gil-Escrig, E. Bandiello, E. M. Hutter, M. Sessolo, K. Lederer, J. Blochwitz-Nimoth and H. J. Bolink, Energy Environ. Sci., 2016, 9, 3456–3463 RSC.
- W. A. Dunlap-Shohl, Y. Zhou, N. P. Padture and D. B. Mitzi, Chem. Rev., 2018, 119, 3193–3295 CrossRef PubMed.
- X. Hong, T. Ishihara and A. V. Nurmikko, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 6961–6964 CrossRef CAS.
- B. R. Gautam, C. Lee, R. Younts, W. Lee, E. Danilov, B. J. Kim and K. Gundogdu, ACS Appl. Mater. Interfaces, 2015, 7, 27586–27591 CrossRef CAS PubMed.
- R. Shivanna, S. Shoaee, S. Dimitrov, S. K. Kandappa, S. Rajaram, J. R. Durrant and K. S. Narayan, Energy Environ. Sci., 2014, 7, 435–441 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9mh00366e |
| This journal is © The Royal Society of Chemistry 2019 |