
Oxygen vacancy-rich MoO3−x nanobelts for photocatalytic N2 reduction to NH3 in pure water†
Yehuan
Li
a,
Xin
Chen
a,
Mingjian
Zhang
a,
Yuanmin
Zhu
b,
Wenju
Ren
a,
Zongwei
Mei
*a,
Meng
Gu
 *b and
Feng
Pan
*a
*b and
Feng
Pan
*a
aSchool of Advanced Materials, Peking University, Shenzhen Graduate School, China. E-mail: meizw@pkusz.edu.cn; panfeng@pkusz.edu.cn
bDepartment of Materials Science and Engineering, Southern University of Science and Technology, China. E-mail: gum@sustc.edu.cn
First published on 10th January 2019
Abstract
Photocatalytic nitrogen fixation is a promising sustainable and green strategy for NH3 synthesis. The development of an efficient photocatalyst is the key to make this approach applicable. In this work, MoO3−x nanobelts are synthesized by a hydrothermal method, and they are firstly utilized as photocatalysts for N2 fixation without using sacrificial reagents and precious-metal co-catalysts at room temperature and atmospheric pressure. It is found that oxygen vacancies (OVS) are located on the (001) and (100) planes by STEM analysis. The surface OVS can chemisorb the N2 molecules by a side-on model on (001) while an end-on model on the (100) plane and elongate their bond length, playing a critical role in the photocatalytic N2 fixation activity. This result provides a new insight into developing efficient photocatalysts with original oxygen vacancies.
Introduction
Nitrogen fixation such as N2 reduction to NH3 is an important artificial synthesis in the chemical industry.1 However, the industrial synthetic strategies consume a large amount of the world's annual energy supply and release large quantities of CO2 aggravating global climate change.2 After the first report by Schrauzer and Guth in 1977, the photocatalytic N2 reduction to NH3 has attracted extensive research interest and shown high potential as a sustainable and green solution compared with the industrial Haber–Bosch process.3 However, the photocatalytic activity of N2 fixation is still not satisfactory due to the high reaction activation energy barriers and hard cleavage of the strong N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bond energy (941 kJ mol−1) of N2 molecules.4
N triple bond energy (941 kJ mol−1) of N2 molecules.4
It is well-known that crystal defect engineering has shown positive effects on many kinds of photocatalytic reactions,5 and oxygen vacancies (OVS) are one of the most widely used defects.6–8 There are three main kinds of ways to generate OVS for photocatalytic experiments. Generally, OVS could be formed via hydrothermal or solvothermal treatment naturally. For example, Tian et al. reported oxygen vacancy-rich BiPO4 through a one-pot solvothermal treatment for MB degradation.9 Additionally, thermal treatment in hydrogen or inert gases is also a usual method to modulate the surface OVS of samples.10 Chen and co-workers enhanced the photocatalytic performances of TiO2 nanocrystals for methylene blue degradation and water splitting by generating OVS in a hydrogen atmosphere and proved that the generation of OVS was positive in enhancing light absorption.11 Moreover, Ding et al. demonstrated that a triple atom defect cluster (Br,Bi,O) on BiOBr could promote the generation of singlet oxygen through an energy transfer process. It resulted in an excellent conversion rate and selectivity in the sulfoxidation reaction.12
It has been demonstrated that the assistant electron-donating center can firstly capture the N2 molecules and weaken the NN triple bond, and then the photogenerated electrons accumulate in the capture sites to reduce the N2 into NH3.13,14 Many research results have revealed that oxygen vacancies can also act as electron trap sites and activate the inert N2. For example, Shiraishi et al. reported that the original OVS in TiO2 directly determined the efficiency of photocatalytic nitrogen fixation from water and N2.45 Zhang and co-workers found that the energy barrier of N2 reduction to ammonia could be remarkably decreased by elongating the NN triple bond via an end-on configuration between the adsorbed N2 and OVS on the BiOBr surface.2,15 It was also reported that the OVS in Bi2MoO6 played an important role in the NN triple bond splitting.16,17 The OVS in LDH nanosheets enhanced the adsorption and activation of N2 and H2O, which made them show excellent photocatalytic activity for N2 reduction.18
Molybdenum trioxide (MoO3) has been extensively explored as a photochromic and electrochromic material due to its unique energetic and electrical properties.19–21 Oxygen vacancy-rich MoO3−x has an unusual oxygen defect structure and novel properties in the nanometer regime.22,23 It has been successfully used to photocatalyze isopropyl alcohol to propylene under visible light.24 Besides, electrocatalytic nitrogen fixation of MoO3 has also shown good activity in a recent study.25 However, there are few reports about photocatalytic N2 reduction by MoO3−x. In this work, MoO3−x nanobelts were synthesized by a simple one-pot hydrothermal method as previously described.24 In comparison with the annealed sample in air, it was found that the OVS on the surface of MoO3−x can chemisorb and reduce the activation energy of N2 molecules, playing a critical role in determining the photocatalytic activity for N2 reduction.
Experimental
Materials and methods
In a typical synthesis, 0.36 g of molybdenum acetylacetonate was added into 40 ml of distilled water. Then the obtained solution was loaded into a stainless steel Teflon-lined autoclave and the sealed autoclave was kept at 180 °C for 20 h. After naturally cooling to room temperature, the blue product was washed with distilled water several times to remove any ions and possible remnants and subsequently dried at 50 °C for 12 h in a vacuum oven. For comparison, the as-synthesized products were annealed at 400 °C in a tube furnace in air atmosphere for x minutes (x = 0, 1, 5, and 30) to reduce surface oxygen vacancy concentration, and the final reactants are denoted as MoO3−x-x min.Characterization of the photocatalysts
X-ray diffraction (XRD, Bruker, D8 Advance X-ray diffractometer) was used to characterize the crystal structure of the samples with the scanning range of 2θ from 10° to 50°. The morphology of the samples was observed on a field emission scanning electron microscope (FE-SEM, ZEISS SUPRA-®-55). The STEM and EELS analyses were performed on a Double-Cs aberration-corrected Themis G2 microscope at 300 kV with a Quantum GIF detector. UV-vis absorption spectroscopy was carried out on a UV-2450 (Shimadzu). X-ray photoelectron spectroscopy (XPS) was performed on a Thermo Scientific ESCALAB 250 Xi system. The Raman spectra were measured using a Horiba iHR 320 with a 532.1 nm laser as the excitation light, and the accumulation time was 25 s. The specific surface areas of the samples were determined through N2 adsorption–desorption isotherms at 77 K using an accelerated surface area and porosimetry system (ASAP, 2020 HD88). Electron spin resonance (ESR) spectroscopy was carried out using a Bruker A300, German under a magnetic field from 3400 to 3600 G. The temperature-programmed desorption of N2 (N2-TPD) was tested with an Autosorb-iQ-C chemisorption analyser (Quantachrome, USA) with the temperature from 0 to 773 K.Photocatalytic N2 fixation
The photocatalytic performance of the sample for N2 reduction was evaluated by the production rate of NH4+ under full spectrum illumination without any sacrificial agents at room temperature and atmospheric pressure. The light source was a 300 W xenon lamp (CEL-HXF300, wavelength: 300–2500 nm, radiant output 50 W) and LEDs with wavelengths of 365 nm, 385 nm, 400 nm and 427 nm (UVEC-4II). A water circulation system was used to maintain the temperature at 25 °C. In each experiment, 50 mg of the photocatalyst was added and dispersed in 100 mL of distilled water, and then the mixture was kept under stirring and irradiated from the top-window with a N2 flow at a rate of 60 mL min−1. 3 mL of the solution was taken out after every 2 h during the illumination. The catalyst in the sample was removed by centrifugation with a rotation rate of 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 r min−1 for 5 min and then was filtered two times. Finally, the photocatalytic product was determined by ion chromatography (Metrohm IC). The photocatalyst was recycled after the photocatalytic experiment by centrifugation and then was filtered several times. And the recycling photocatalytic experiment was carried out with a 300 W xenon lamp.
000 r min−1 for 5 min and then was filtered two times. Finally, the photocatalytic product was determined by ion chromatography (Metrohm IC). The photocatalyst was recycled after the photocatalytic experiment by centrifugation and then was filtered several times. And the recycling photocatalytic experiment was carried out with a 300 W xenon lamp.
DFT calculations
All calculations were performed with the plane-wave projector-augmented wave method,26,27 as implemented in the CASTEP package. The Perdew–Burke–Ernzerhof (PBE) form of generalized gradient approximation (GGA) was chosen as the exchange–correlation potential.28,29 The PBE + U approach with spin polarization was employed to take account of the strong on-site Coulomb interaction (U), present in the localized 4d electrons of Mo with U values being set to 2 eV. The cutoff energy for the plane wave was 340 eV. The integration in the Brillouin zone was carried out on a set of k-points determined by the Monkhorst and Pack scheme, 4 × 1 × 4. Convergence was assumed when the total energy difference was less than 10−6 eV between cycles of self-consistent iterations. Based on the results of the STEM analysis, a unit cell with one O vacancy at the 2c site (Mo4O11) was used to perform the geometry-optimization calculations. The atomic populations (Mulliken) were analyzed after optimization. Furthermore, the binding energy of O-deficient Mo atoms in this structure model was further calculated by removing another coordinating O atom. In comparison, the perfect unit cell of MoO3 (Mo4O12) was also calculated with the same calculation configurations.The calculation of the N–N bond was performed using the first-principles density functional theory (DFT) + U calculations with the exchange–correlation energy functional, which were described by a generalized gradient approximation with the Perdew–Burke–Ernzerhof (PBE) exchange–correlation function.28 We also took van der Waals interactions into correction by the DFT-D2 method of Grimme.30 To simulate N2 activation on the MoO3 (001) and (100) surfaces, (2 × 1 × 3) and (3 × 1 × 2) supercells were used, respectively, enabled by a VASP code in which the projector-augmented wave (PAW) method presented the electron–ion interaction with a kinetic energy cutoff of 520 eV.27,31 The k-points were 8 × 4 × 1, 4 × 4 × 1 and 4 × 8 × 1 for each supercell. During optimizations, the energy and force converged to 10−4 eV per atom and 0.02 eV Å−1, respectively.
Results and discussion
The XRD patterns of the as-synthesized and annealed samples with different times are shown in Fig. 1a. All the peaks can be indexed to orthorhombic MoO3 (o-MoO3) whose 2θ values are located at 12.8°, 23.4°, 25.7°, 25.8°, 27.3° and 39.0°. The result is consistent with previous reports.32,33 After annealing, the XRD patterns of the annealed samples don't show an obvious shift as presented in Fig. 1a. The SEM images of the as-synthesized and annealed products are shown in Fig. S1.† It can be observed that the products are all composed of nanobelts, and the morphologies remain stable even after annealing. In addition, as shown in the HRTEM image, a MoO3−x nanobelt is single crystalline and the inserted diffraction pattern in Fig. 1c proves the single-crystalline nature of the MoO3−x nanobelt. Detailed analysis for fringing the formation of oxygen vacancies was performed using atomic scale HAADF Z-contrast images in Fig. 1d. Ordered oxygen vacancies residing at certain crystal plane surfaces often show up as dark-lines in a Z-contrast image due to the reduced average atomic density.34 As shown in Fig. 1d, the white arrows point to the ordered oxygen vacancies residing at the (001) & (100) surfaces of MoO3−x, where we observed such dark-lines along these surface planes. In addition, the red lines mark the dislocation formed in MoO3−x. The dislocations formed in the MoO3−x may partially release the internal strain induced by the formation of such oxygen vacancies.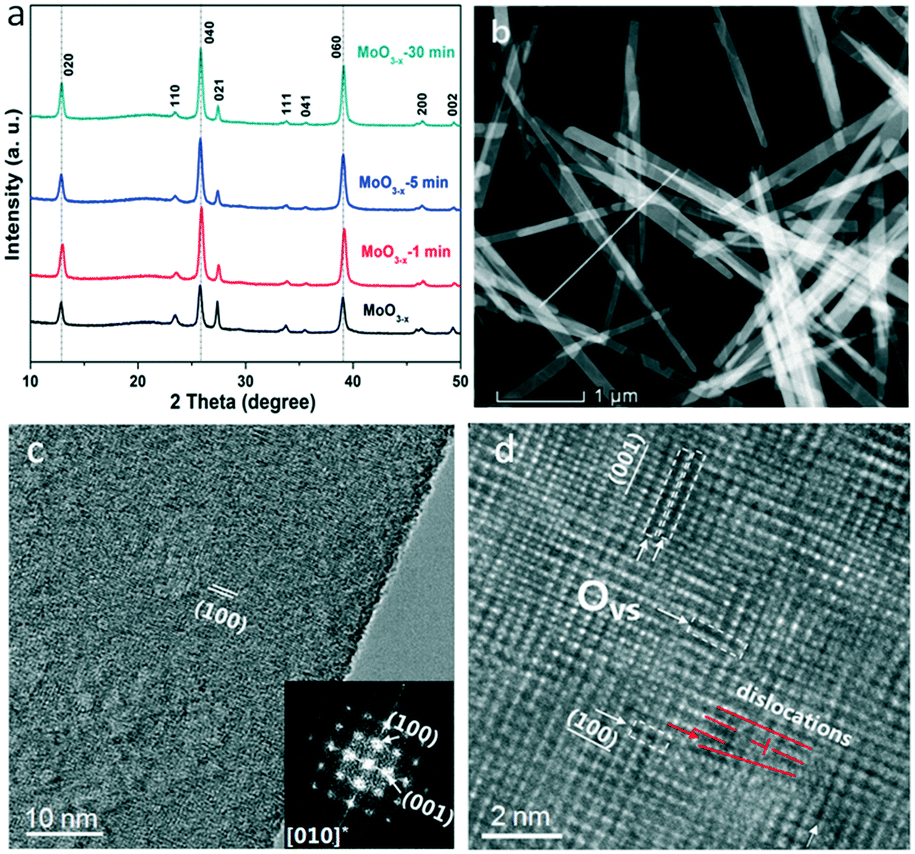 | ||
| Fig. 1 (a) XRD patterns of the as-synthesized and annealed MoO3−x nanobelts for various times; (b) HAADF STEM image of the overall morphology of the MoO3−x nanobelts; (c) HRTEM image of MoO3−x with the insert showing the electron diffraction pattern; (d) atomic scale HAADF image showing the ordered oxygen vacancies at the (001) & (100) planes. | ||
In order to prove the changes of OVS, the fine structures of the electron energy loss spectroscopy (EELS) edges were further measured. The EELS edges reflect the unoccupied local density of states and therefore, provide useful local oxidation states and coordination chemistry of materials.35 The Mo and O EELS results of the as-obtained MoO3−x and MoO3−x after 30 min annealing were acquired and plotted in Fig. 2 for comparison. The lattice distortion and reduced symmetry at the oxygen-deficient MoO3−x certainly caused changes in the oxygen K edges. As reported by previous literature studies, the enhancement of a peak labelled by the red arrow is a signature of the formation of oxygen vacancies.36 Another interesting feature to notice is the decreased intensity of the third peak as pointed by the purple arrow in the MoO3−x sample compared with the MoO3−x after 30 min annealing. Based on the multiple-scattering theory, the decreased intensity is caused by a decrease in the number of backscattering O2− ions at the first or second-nearest oxygen neighbours, further proving the presence of oxygen vacancies in the MoO3−x sample.37
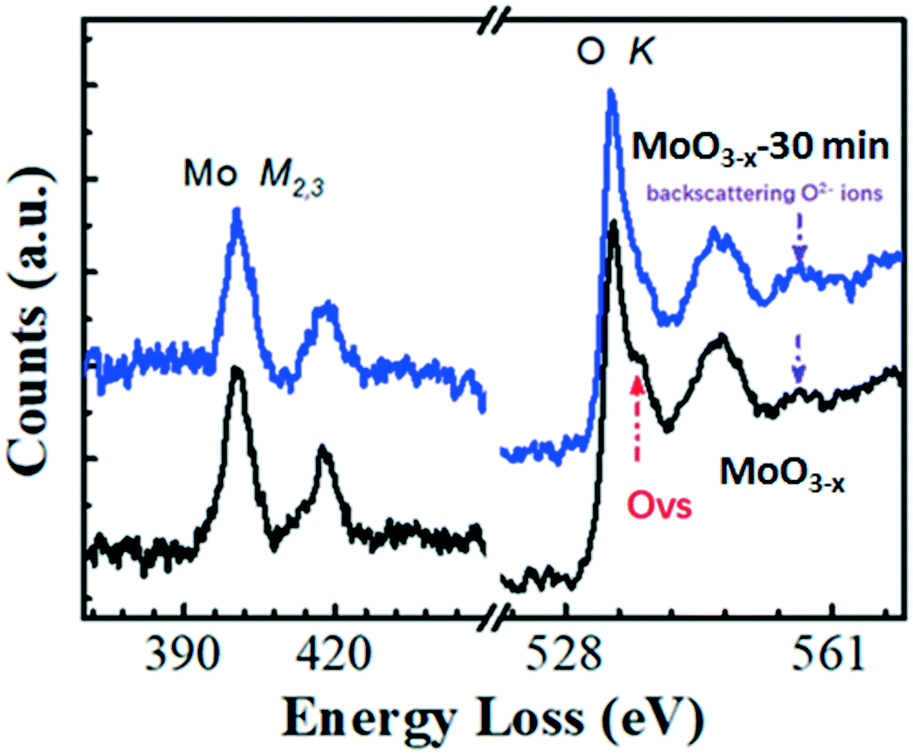 | ||
| Fig. 2 EELS analysis of the as-synthesized MoO3−x and MoO3−x after 30 min annealing in air atmosphere. | ||
The UV-vis absorption spectra of the samples in Fig. 3a were transformed from their reference spectra based on the Kubelka–Munk formula. There is a tail absorption for each sample, which results from the OVS,17 and decreases gradually as the annealing time increases. The colour of the sample gradually turns white after the heat treatment as shown in Fig. S2.† These results indicate that the annealing treatment can remove the OVS in the samples.
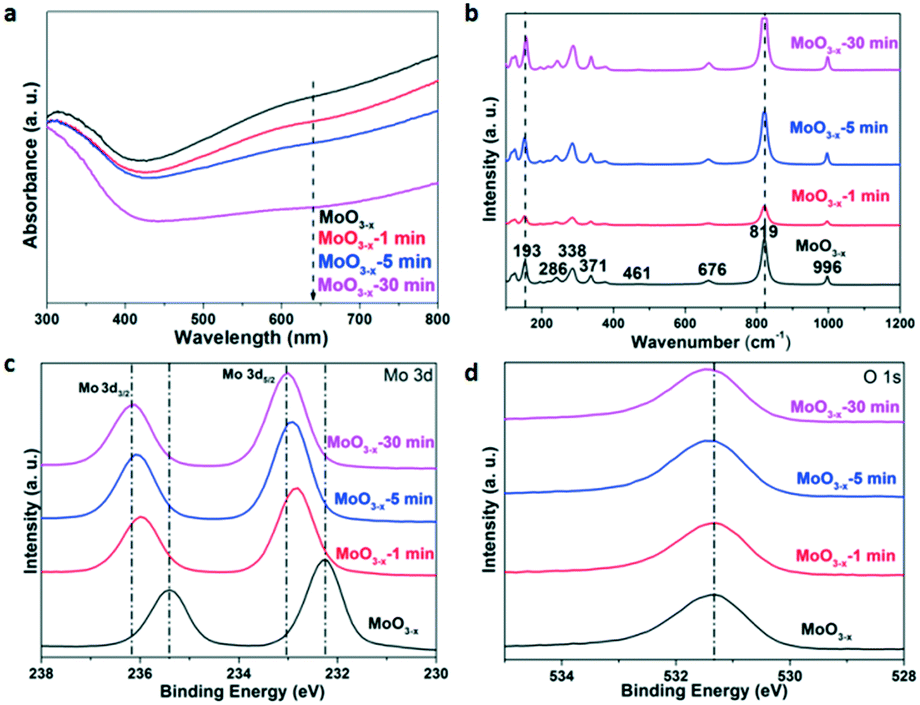 | ||
| Fig. 3 (a) UV-vis absorption spectra, (b) Raman spectra, (c) Mo 3d-related XPS spectra, and (d) O 1s-related XPS spectra of the as-synthesized and annealed MoO3−x for different times. | ||
The Raman spectroscopy characterization results are shown in Fig. 3b. The Raman peaks of the as-synthesized MoO3−x nanobelts are in agreement with those of previous research.38 The peak at 819 cm−1 shows a strong and well-defined line, assigned to the doubly coordinated bridging oxygen (Mo–O–Mo units). A couple of weak peaks at 461 and 676 cm−1 are attributed to the triply coordinated oxygen (Mo3–O). The peak at 996 cm−1 consists of the terminal oxygen (Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) stretching mode. Both the XRD patterns and Raman spectra indicate that the prepared MoO3−x nanobelts belong to an orthorhombic phase. However, there is no obvious shift in the Raman spectra of the annealed samples for different times, and it may result from the fact that the reduced OVS by annealing are not capable enough to change the Raman characteristics. Besides, we have tried to choose lower annealing temperatures (150 °C, 200 °C, and 300 °C) in our study before ensuring 400 °C as the final annealing temperature. And it is found that there was no obvious change in the sample colour although the annealing time was 1 h at 300 °C (Fig. S3†), indicating that the OVS do not decrease very much. On the other hand, the structure of the samples is stable after 400 °C annealing, because the XRD and Raman results are similar (Fig. 1a and 3b) for the as-synthesized and annealed samples. However, the absorption coefficient of the UV-vis tail absorption (Fig. 3a) decreases as the annealing time increases. This proves that OVS were drastically removed and the crystal structure remained stable after annealing at 400 °C.
O) stretching mode. Both the XRD patterns and Raman spectra indicate that the prepared MoO3−x nanobelts belong to an orthorhombic phase. However, there is no obvious shift in the Raman spectra of the annealed samples for different times, and it may result from the fact that the reduced OVS by annealing are not capable enough to change the Raman characteristics. Besides, we have tried to choose lower annealing temperatures (150 °C, 200 °C, and 300 °C) in our study before ensuring 400 °C as the final annealing temperature. And it is found that there was no obvious change in the sample colour although the annealing time was 1 h at 300 °C (Fig. S3†), indicating that the OVS do not decrease very much. On the other hand, the structure of the samples is stable after 400 °C annealing, because the XRD and Raman results are similar (Fig. 1a and 3b) for the as-synthesized and annealed samples. However, the absorption coefficient of the UV-vis tail absorption (Fig. 3a) decreases as the annealing time increases. This proves that OVS were drastically removed and the crystal structure remained stable after annealing at 400 °C.
The X-ray photoelectron spectra (XPS) are presented in Fig. 3c and d. For the as-synthesized MoO3−x nanobelts, the peaks at 232.3 eV and 235.6 eV could be ascribed to the 3d5/2 and 3d3/2 orbitals of Mo 3d, respectively.39,40 In Fig. 3d, the peaks at 531.4 eV originate from the lattice oxygen anions.41 After annealing, the peaks of Mo 3d gradually left-shift as the annealing time is increased. Similar phenomena are observed for the O 1s orbital as shown in Fig. 3d. In order to understand the deep reasons, the Mulliken electronegativity of the Mo and O elements of MoO3 with 25% or without OVS was calculated using CASTEP (see details in the Experimental section). Mo4O12 and Mo4O11 were chosen as the unit cells in this theoretical calculation, corresponding to the non-defect and defect MoO3, respectively. The calculation results are shown in Table S1.† The average charges for Mo and O in the non-defect MoO3 are 1.44 and −0.451, respectively. The corresponding values are 1.37 and −0.499 for the defect MoO3, respectively. Obviously, Mo and O in the defect MoO3 show more negative charges than those in the non-defect MoO3, which result in the increasing binding energy for Mo 3d and O 1s in the decreasing oxygen defect samples as shown in Fig. 3c and d.42 Besides, the results of binding energy for Mo4O12 and Mo4O11 have also been calculated. In comparison with Mo4O12, the binding energy of Mo4O11 (7.64 eV) becomes smaller than that of Mo4O12 (8.52 eV), which also confirms the weaker chemical bond of Mo–O in the sample with higher OVS concentration.
The photocatalytic performance was characterized under the irradiation of a 300 W xenon lamp with full spectra, and the quantity of the photo-fixed NH4+ was determined using an ion chromatograph. Compared with the traditional method (Nessler's reagent), the ion chromatograph could detect NH4+ more accurately and the measurement results would not be influenced by any organic reagents during the reaction.43 Pure water was used as the solvent and proton source in this work. Generally, the chemical reaction from N2 to NH3 in neutral solution can be described as the following equation:44
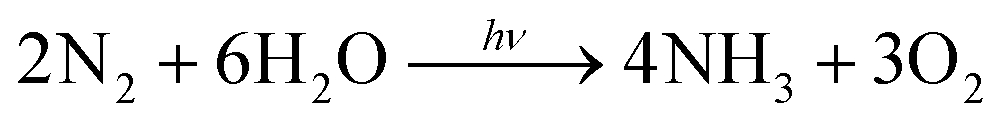 | (1) |
The photocatalytic nitrogen fixation activity is shown in Fig. 4 (the representative original data from ion chromatography are shown in Fig. S4†). The as-synthesized MoO3−x exhibits the highest NH3 production rate reaching nearly 11.1 μmol L−1 g−1 h−1, while the production rate is just a quarter remaining for the MoO3−x-1 min sample. In addition, there is no detectable N2 reduction product for the MoO3−x-5 min and MoO3−x-30 min samples. As we know, the BET surface area is also a significant parameter for the photocatalytic activity. However, the surface areas are all around 20 m2 g−1 (Fig. S5†). Accordingly, the surface area can't obviously affect the photocatalytic N2 reduction activity. Furthermore, the light absorption of MoO3−x-5 min doesn't show great difference in comparison with that of the as-synthesized sample, but its photocatalytic product is undetectable. To find out the reason, electron spin resonance (ESR) spectroscopy and temperature-programmed desorption of N2 (N2-TPD) were used to investigate the OVS and N2 adsorption on the surface OVS. In Fig. 5a, a signal exists at g = 2.001 caused by OVS in the as-synthesized and annealed samples.45 However, there are no obvious changes even if the samples are annealed for 30 minutes at 400 °C. This must result from the fact that the OVS have not been totally removed in the whole sample. In Fig. 5b, a single desorption peak of N2 begins at 180 °C and centers at 320 °C for the as-synthesized sample, which is ascribed to the chemisorbed N2.2 However, the peak becomes weaker obviously for sample MoO3−x-5 min and disappears for sample MoO3−x-30 min. These results indicate that the surface OVS were gradually removed but there were still OVS in the inner part of the annealed samples. Compared with the photocatalytic activities in Fig. 4, samples MoO3−x-5 min and MoO3−x-30 min don't show detectable products, which must be caused by the disappearance of enough surface OVS. In other words, the surface OVS are the key factor for the efficient photocatalytic N2 reduction.
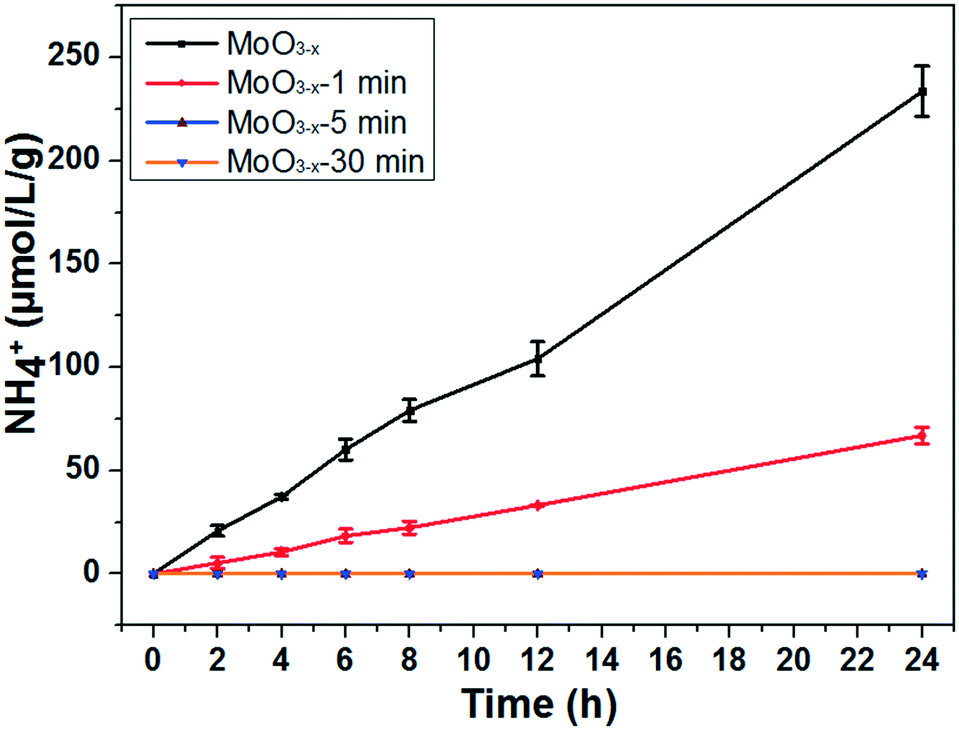 | ||
| Fig. 4 Photocatalytic experiments for nitrogen fixation of the as-synthesized and annealed MoO3−x nanobelts (light source: 300 W xenon lamp; photocatalyst: 0.05 g; solution: 100 ml of pure water. The error bar results came from photocatalytic experiments of the same samples performed three times.) | ||
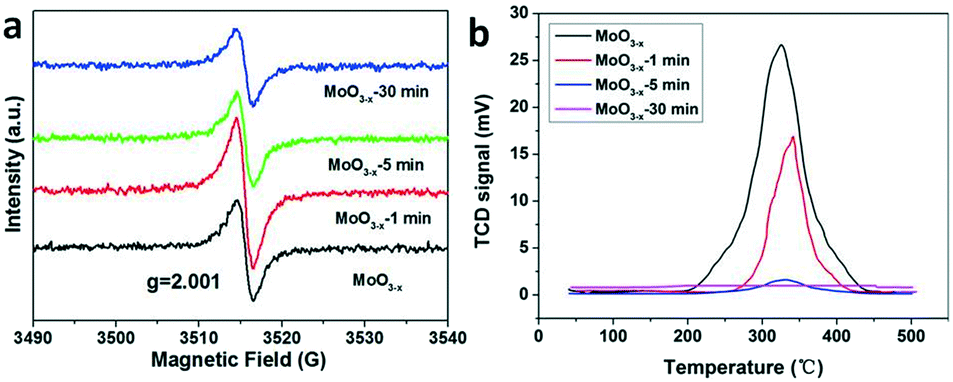 | ||
| Fig. 5 (a) ESR spectra and (b) N2-TPD profiles of the as-synthesized and annealed samples for different times. | ||
The reported photocatalytic activities of typical metal oxides were compared, which were characterized in pure water under UV-vis light irradiation (Table S2†). Hirakawa and co-workers created Ti3+ as active sites in TiO2 which exhibited an NH3 production rate of 35.0 μmol g−1 h−1.45 Zhang et al. reported that the NH3 production of Mo-doped W18O49 reached 61.9 μmol g−1 h−1.46 Most other reported metallic oxides exhibited production rates around 0.41–28.4 μmol g−1 h−1.47–51 The as-synthesized MoO3−x nanobelts show this production in the middle range.
It could be seen that there was strong IR absorption even at 850 nm from the UV-vis absorption (Fig. 3a). The photocatalytic performance was further characterized under LED irradiation with different wavelengths to find out the connection between the light wavelength and NH3 synthesis. LEDs with wavelengths of 365 nm, 384 nm, 400 nm, and 427 nm were used as the light source. The results have been normalized because of the different powers of LEDs. As shown in Fig. 6a, the LED with a wavelength of 365 nm exhibits the highest NH3 production rate reaching nearly 3.3 μmol L−1 g−1 h−1 W−1, while NH3 production is not detected in the reaction irradiated by 400 and 427 nm LEDs. This means that the sample could only fix N2 under ultraviolet light irradiation (λ < 400 nm) although there was still light absorption in the longer wavelength range (λ ≥ 400 nm). In addition, the quantum efficiency (QE) was also calculated by the following equation:52
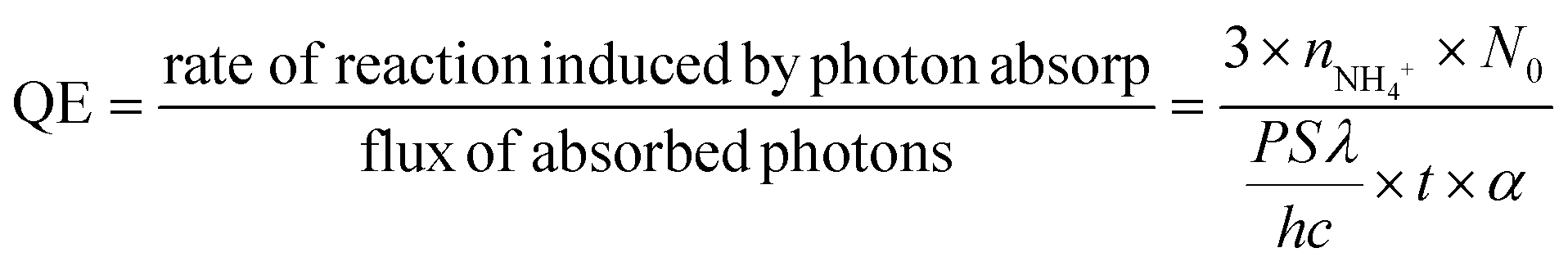 | (2) |
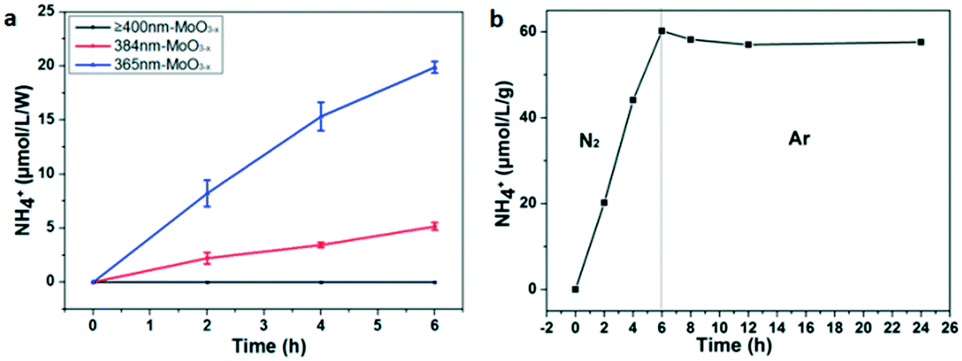 | ||
| Fig. 6 (a) Photocatalytic activities for nitrogen fixation of the as-synthesized MoO3−x nanobelts under LED light irradiation with various wavelengths; (b) control experiment in N2 and Ar gas in succession for photocatalytic nitrogen fixation (light source: 300 W xenon lamp). | ||
To ensure the source of the N element, argon was used to replace the N2 as the gas source after illumination for 6 h under a N2 flow. Obviously, the N2 fixation reaction hardly occurs under the Ar flow because the amount of NH4+ doesn't increase (Fig. 6b). This result indicates that the N atoms in NH3 come from N2. Besides, recycling photocatalytic experiments were also carried out to characterize the photochemical stability of MoO3−x. It has been found that the NH3 production is just 70% remaining during the first cycle, and 65% remaining for the second cycle (Fig. S6a†). The possible reason is that some OVS are inactive for the further photocatalytic N2 fixation after each cycle, because there are no obvious change for the XRD and TEM results of the sample before and after reaction (Fig. S7 and S8†). Moreover, we have also studied the effect of catalyst dosage on photocatalytic activities. As shown in Fig. S6b,† the rate of NH3 production would be enhanced obviously if the quantity of the catalyst is increased (nearly 1 μmol L−1 for 50 mg of catalysts, while 4 μmol L−1 for 1 g of catalysts). However, the rates of NH3 production decrease if the value is converted to one gram. It is reasonable that the larger dosage of the catalyst cannot be exposed enough to light for photocatalytic N2 fixation. Then a larger dosage of the catalyst exhibits a lower photocatalytic activity when converting to μmol L−1 g−1.
Based on the result of the electron diffraction pattern in Fig. 1c, the (100) and (001) planes exist in the MoO3−x nanobelts. Theoretical calculation was carried out to further study the formation energy of OVS in these two planes. As shown in Fig. 7, there are three and four kinds of O-sites on the (001) and (100) planes, respectively. The calculation results are shown in Table S3.† For the (001) plane, the OVS formation energy on the O1 site is equal to that on the O2 site (1.05 eV), which is more stable than that on the O3 site (1.29 eV). This means that it is easier to form OVS on the O1 (O2) site. For the (100) plane, the formation energy on the O1 and O2 sites is equal to −1.97 eV, and the value is 2.24 eV on the O3 and O4 sites. These results indicate that the OVS are easier to form on the O1 and O2 sites of the (001) plane, and the OVS can spontaneously form on the O1 and O2 sites of the (100) plane.
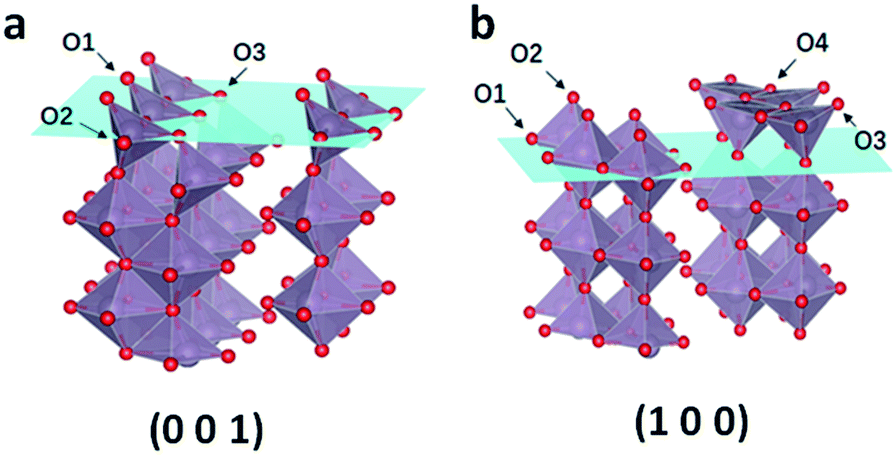 | ||
| Fig. 7 (a) Three kinds of O sites on the (001) plane and (b) four kinds of O sites on the (100) plane. | ||
The density of states of the (001) plane with or without OVS was calculated to explain the electron transfer processes during OVS-mediated photocatalytic nitrogen fixation (Fig. S9†). When there are OVS on the (001) plane, some defect levels appear between the valence and conduction bands. This also explains why there is tail absorption for defect MoO3−x (Fig. 3a and 8a). Due to the lower energy of the defect levels, the photoexcited electrons on the conduction band will move to the defect levels, and the OVS thus act as the trap sites of electrons. Subsequently, the photoexcited electrons with enough energy will reduce the chemically adsorbed N2 by OVS into NH3.
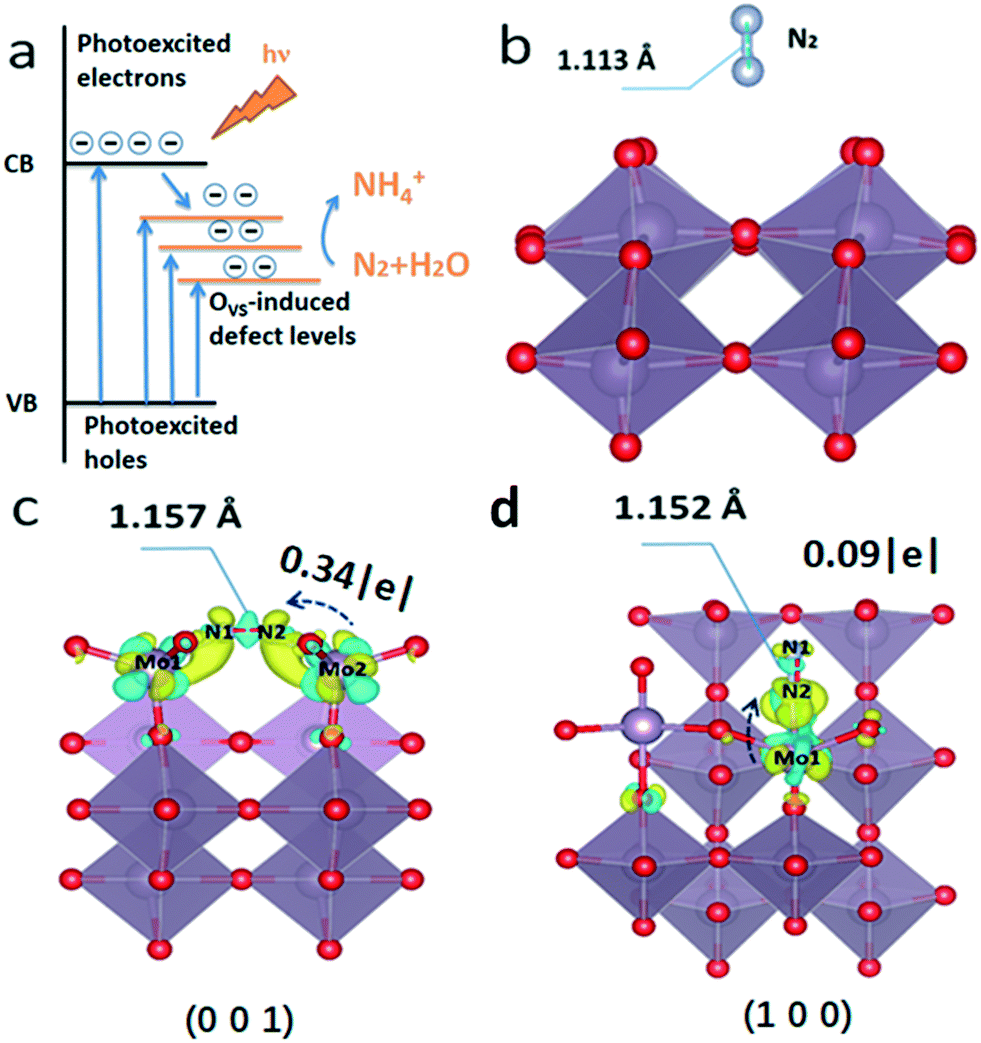 | ||
| Fig. 8 (a) Electron transfer processes during OVS-mediated nitrogen fixation. (b) N2 adsorbed on the pristine surface; (c) terminal side-on adsorption on (001) and (d) end-on bridging adsorption on (100) for N2 (yellow means increasing charge and blue means decreasing charge). | ||
Additionally, the theoretical calculation was carried out to further study the function of surface OVS for N2 photofixation. When there are no OVS on the surface of MoO3, the N2 molecule floats above the surface of the catalyst, and the NN triple bond length (1.113 Å) doesn't change (Fig. 8b). There are two models including end-on and side-on ways for nitrogen adsorption on the OVS on the (001) surface and (100) surface where the OVS are located as confirmed by atomic scale HAADF (Fig. 1d) and the above discussion. Based on the theoretical calculation results, there is little possibility for end-on ways on the (001) surface and side-on ways on the (100) surface for nitrogen adsorption, respectively, due to the reason that their adsorptions are too unstable to produce models for further calculation.
The bond length N1–N2 is 1.152 Å and 1.157 Å as observed on the (001) surface in Fig. 8c and on the (100) surface in Fig. 8d, respectively.
Obviously, the bond length of the N–N bond is elongated for the adsorbed N2 molecule. In other words, the N2 molecule chemisorbs on the OVS and its activation energy can be decreased, which is beneficial to photocatalytic N2 reduction to NH3. Fig. 8c and d show the charge distribution of nitrogen by side-on adsorption on the (001) plane and end-on adsorption on the (100) plane, respectively. It is well-known that N2 molecule reduction needed electrons to change into NH3. The Bader charge analysis in Fig. 8 demonstrates that there will be 0.34|e| electron on the (001) plane and 0.09|e| on the (100) plane transferring from the unsaturated Mo atom (OVS) to the N2 molecule, respectively. When the semiconductor is irradiated by light, the photoexcited electrons can be successfully injected from the OVS into the N2 molecule for its reduction reaction.
Conclusions
In summary, we have successfully synthesized o-MoO3−x by a simple one-pot hydrothermal method and tested its photocatalytic nitrogen fixation activity in pure water for the first time. We found that the OVS were located on the (001) plane and (100) plane by STEM measurement. The surface OVS play a critical role in the photocatalytic N2 reduction by chemisorption and the activation energy decrease of nitrogen molecules. The N2 molecule prefers to adsorb on the OVS by the side-on model on the (001) plane and the end-on model on the (100) plane, respectively. Our findings can provide useful information for further development of other efficient photocatalysts for N2 reduction.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was financially supported by Guangdong key lab Project (2017B030301013), Shenzhen Science (JCYJ20151015162256516), Technology Research Grant (JCYJ20160226105838578), National Key R&D Program of China (2016YFB0700600), and the Shenzhen Peacock Plan (KQTD2014062714543296).Notes and references
- D. E. Canfield, A. N. Glazer and P. G. Falkowski, Science, 2010, 330, 192–196 CrossRef CAS PubMed.
- H. Li, J. Shang, Z. Ai and L. Zhang, J. Am. Chem. Soc., 2015, 137, 6393–6399 CrossRef CAS PubMed.
- G. N. Schrauzer and T. D. Guth, J. Am. Chem. Soc., 1977, 99, 7189–7193 CrossRef CAS.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, ACS Catal., 2017, 7, 3713–3720 CrossRef CAS.
- Z. Dai, F. Qin, H. Zhao, J. Ding, Y. Liu and R. Chen, ACS Catal., 2016, 6, 3180–3192 CrossRef CAS.
- J. Ding, Z. Dai, F. Qin, H. Zhao, S. Zhao and R. Chen, Appl. Catal., B, 2017, 205, 281–291 CrossRef CAS.
- Q. Wang, W. Wang, L. Zhong, D. Liu, X. Cao and F. Cui, Appl. Catal., B, 2018, 220, 290–302 CrossRef CAS.
- Q. Wang, Z. Liu, D. Liu, G. Liu, M. Yang, F. Cui and W. Wang, Appl. Catal., B, 2018, 236, 222–232 CrossRef CAS.
- F. Tian, H. Zhao, G. Li, Z. Dai, H. Zhao, Y. Liu and R. Chen, ChemSusChem, 2016, 9, 1579–1585 CrossRef CAS PubMed.
- D. Tio, Nanoscale, 2013, 5, 3601–3614 RSC.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–751 CrossRef CAS PubMed.
- J. Ding, Z. Dai and F. Tian, J. Mater. Chem. A, 2017, 5, 23453–23459 RSC.
- K. C. MacLeod, D. J. Vinyard and P. L. Holland, J. Am. Chem. Soc., 2014, 136, 10226–10229 CrossRef CAS PubMed.
- S. Wang, X. Hai, X. Ding, K. Chang, Y. Xiang, X. Meng, Z. Yang, H. Chen and J. Ye, Adv. Mater., 2017, 29, 1–7 Search PubMed.
- J. Li, H. Li, G. Zhan and L. Zhang, Acc. Chem. Res., 2017, 50, 112–121 CrossRef CAS PubMed.
- C. Shi, X. Dong, Y. Hao, X. Wang, H. Ma and X. Zhang, RSC Adv., 2017, 7, 50040–50043 RSC.
- Y. Li, Y. Zhao, H. Cheng, Y. Hu, G. Shi, L. Dai and L. Qu, J. Am. Chem. Soc., 2012, 134, 15–18 CrossRef CAS PubMed.
- Y. Zhao, Y. Zhao, G. I. N. Waterhouse, L. Zheng, X. Cao, F. Teng, L. Z. Wu, C. H. Tung, D. O'Hare and T. Zhang, Adv. Mater., 2017, 29, 1–10 Search PubMed.
- H. Natori, K. Kobayashi and M. Takahashi, J. Oleo Sci., 2009, 58, 203–211 CrossRef CAS PubMed.
- Y. Yang, Y. Cao, B. Loo and J. Yao, J. Phys. Chem. B, 1998, 102, 9392–9396 CrossRef CAS.
- M. Greenblatt, Chem. Rev., 1988, 88, 31–53 CrossRef CAS.
- S. G. Processes, N. Pinna and M. Niederberger, Angew. Chem., Int. Ed., 2008, 47, 5292–5304 CrossRef PubMed.
- M. A. Ibrahem, F.-Y. Wu, A. Mengistie, C. Chang, D. A. Mengistie, L. Li, C. W. Chu and V. A. Online, Nanoscale, 2014, 6, 5484–5490 RSC.
- H. Bai, W. Yi, J. Li, G. Xi, Y. Li, H. Yang and J. Liu, J. Mater. Chem. A, 2016, 4, 1566–1571 RSC.
- J. Han, X. Ji, X. Ren, G. Cui, L. Li, F. Xie, H. Wang, B. Li and X. Sun, J. Mater. Chem. A, 2018, 6, 12974–12977 RSC.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef.
- D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- V. I. Anisimov, M. A. Korotin, J. Zaanen and O. K. Andersen, Phys. Rev. Lett., 1992, 68, 345–348 CrossRef CAS PubMed.
- S. Grimme, T. O. Chemie and O. I. D. U. Münster, J. Comput. Chem., 2006, 16, 1787–1799 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- N. Pinna and M. Niederberger, Angew. Chem., Int. Ed., 2008, 47, 5292–5304 CrossRef CAS PubMed.
- S. Hu and X. Wang, J. Am. Chem. Soc., 2008, 130, 8126–8127 CrossRef CAS PubMed.
- R. F. Klie and N. D. Browning, Microsc. Microanal., 2018, 8, 475–486 CrossRef.
- D. A. Muller, Nat. Mater., 2009, 8, 263–270 CrossRef CAS PubMed.
- D. Wang, D. S. Su and R. Schlögl, Z. Anorg. Allg. Chem., 2004, 630, 1007–1014 CrossRef CAS.
- N. D. Browning, H. O. Moltaji and J. P. Buban, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 8289–8300 CrossRef CAS.
- L. Seguin, M. Figlarz, R. Cavagnat and J. C. Lassegues, Spectrochim. Acta, Part A, 1995, 51, 1323–1344 CrossRef.
- M. A. Bica De Moraes, B. C. Trasferetti, F. P. Rouxinol, R. Landers, S. F. Durrant, J. Scarmínio and A. Urbano, Chem. Mater., 2004, 16, 513–520 CrossRef CAS.
- Q. Huang, S. Hu, J. Zhuang and X. Wang, Chem. – Eur. J., 2012, 18, 15283–15287 CrossRef CAS PubMed.
- J. Wang, Y. Xia, H. Zhao, G. Wang, L. Xiang, J. Xu and S. Komarneni, Appl. Catal., B, 2017, 206, 406–416 CrossRef CAS.
- Z. Mei, M. Zhang, J. Schneider, W. Wang, N. Zhang, Y. Su, B. Chen, S. Wang, A. L. Rogach and F. Pan, Catal. Sci. Technol., 2017, 7, 982–987 RSC.
- X. Gao, Y. Wen, D. Qu, L. An, S. Luan, W. Jiang, X. Zong, X. Liu and Z. Sun, ACS Sustainable Chem. Eng., 2018, 6, 5342–5348 CrossRef CAS.
- K. Tennakone, S. Punchihewa, S. Wickramanayake, C. A. N. A. N. Fernando, O. A. Illeperuma, O. A. Ileperuma and S. Punchihewa, J. Chem. Soc., Chem. Commun., 1987, 1078–1080 RSC.
- H. Hirakawa, M. Hashimoto, Y. Shiraishi and T. Hirai, J. Am. Chem. Soc., 2017, 139, 10929–10936 CrossRef CAS PubMed.
- N. Zhang, A. Jalil, D. Wu, S. Chen, Y. Liu, C. Gao, W. Ye, N. Zhang, A. Jalil, D. Wu, S. Chen, Y. Liu, C. Gao and W. Ye, J. Am. Chem. Soc., 2018, 140, 9434–9443 CrossRef CAS PubMed.
- Q. Li, K. Donmen, S. Naito, T. Onishi and K. Tamaru, Chem. Lett., 1983, 2, 321–324 CrossRef.
- M. M. Khader, N. N. Lichtin, G. H. Vurens, M. Salmeron and G. A. Somorjai, Langmuir, 1987, 3, 303–304 CrossRef CAS.
- J. Soria, J. C. Conesa, V. Augugliaro, L. Palmisano, M. Schiavello and A. Sclafani, J. Phys. Chem., 1991, 95, 274–282 CrossRef CAS.
- S. Sun, X. Li, W. Wang, L. Zhang and X. Sun, Appl. Catal., B, 2017, 200, 323–329 CrossRef CAS.
- K. T. Ranjit and B. Viswanathan, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 1996, 35, 443–453 Search PubMed.
- M. Liu, R. Inde, M. Nishikawa, X. Qiu, D. Atarashi, E. Sakai and Y. Nosaka, ACS Nano, 2014, 8, 7229–7238 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, BET, XRD, TEM, colour change of samples, density of states (DOS) of the (001) plane, ion chromatography images and tables of oxygen formation energy, atomic populations (Mulliken) of the Mo4O12 unit cell and Mo4O11 unit cell, and summary of some metal oxides. See DOI: 10.1039/c8cy02357c |
| This journal is © The Royal Society of Chemistry 2019 |