
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The solid state forms of the sex hormone 17-β-estradiol†
Erin L.
Stevenson
a,
Robert W.
Lancaster
a,
Asma B. M.
Buanz
 b,
Louise S.
Price
a,
Derek A.
Tocher
a and
Sarah L.
Price
*a
b,
Louise S.
Price
a,
Derek A.
Tocher
a and
Sarah L.
Price
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: s.l.price@ucl.ac.uk
bUCL School of Pharmacy, 29-39 Brunswick Square, Bloomsbury, London WC1N 1AX, UK
First published on 7th December 2018
Abstract
The crystal structure of the single component form of the primary female sex hormone, 17-β-estradiol (BES), is reported, solved from single crystals obtained by sublimation. The Z′ = 2 P212121 structure was computationally predicted as one of the thermodynamically plausible structures. It appears that the dehydration process for the very stable hemihydrate structure is a complex process, strongly affected by particle size and conditions. An experimental polymorph screen has produced six solid forms of BES, including novel acetonitrile and highly labile ethylene dichloride solvates, and reproduced previously reported methanol and propanol solvates. These have been characterized, as far as possible given the metastability relative to the hemihydrate (BES·0.5H2O), by single-crystal X-ray diffraction (SCXRD), powder X-ray diffraction (PXRD), differential scanning calorimetry (DSC), thermogravimetric analysis (TGA), hot-stage microscopy and Fourier transform infrared spectroscopy (FT-IR), sorting out some of the confusion in the earlier literature.
Introduction
Solvent molecules (including water) commonly satisfy the hydrogen bonding sites of an organic molecule better than the molecule itself. Therefore, organic molecules often crystallize as solvated or hydrated solid forms, in which solvent or water molecules occupy a regular position within the crystal structure.1–3 The presence of a solvent molecule in the crystal packing arrangement plays a critical role in the activity of a drug by altering the intermolecular interactions within the solid. This can modify the internal energy and enthalpy and therefore affect pharmaceutically important properties such as solubility and bioavailability.1,4 Most frequently, hydrates are less soluble than the corresponding anhydrate leading to a possibly undesirable decrease in bioavailability.4 Therefore, it is of great interest to the pharmaceutical industry to identify and understand any interconversions between solid forms. This can be particularly demanding for hydrates, as water is difficult to exclude from a process but simple hydrate classifications can be inappropriate for pharmaceutical development.5 The control of production of crystalline products, which may be very sensitive to water activity in crystallization, filtration and drying processes, is essential in order to manufacture pharmaceutical material fit for purpose.6The studied estrogen, 17-β-estradiol (BES), is a white, odourless powder that is a primary female sex hormone associated with the female reproductive system. It is widely used in hormone replacement therapy for the treatment of post-menopausal symptoms, hormonal contraception and the treatment of hormone sensitive cancers.7,8 It is marketed as the hemihydrate form which in terms of pharmacodynamics is identical to the anhydrous form.9 However, the hemihydrate is significantly less soluble and has a lower oral bioavailability.10 It therefore needs to be micronized or esterified in order to be bioavailable to any significant extent.11 Remarkably for such an important pharmaceutical, there are no crystal structures containing just BES in the Cambridge Structural Database (CSD).12
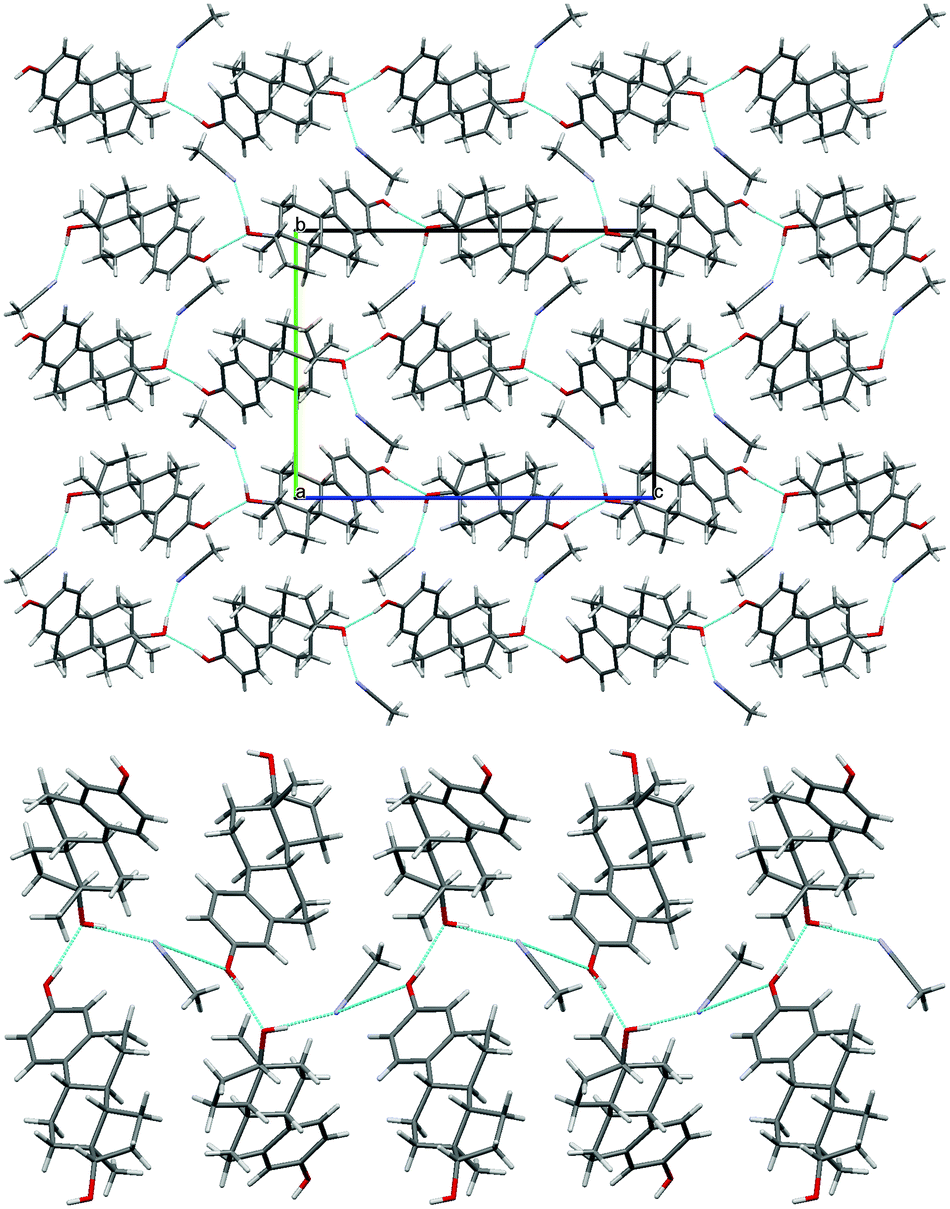
Busetta et al.13 determined the full crystal structure of BES·0.5H2O in 1972 (CSD Refcode ESTDOL10). The water molecules are located on the 2-fold axis and participate in hydrogen bonding to the hydroxyl groups on the A- and D-rings (Fig. 2). Differential thermal analysis (DTA) by Florence et al.,14 in 1975, shows that BES·0.5H2O exhibits endothermic peaks around 112 °C and 174 °C prior to the melting endotherm at 179 °C. Analysis of the effluent gas of DSC samples by Kuhnert-Brandstätter et al.,15 in 1976, showed that these pre-melting endotherms are associated with water loss. Thermomicroscopic examination by the same group suggests that there is some structural rearrangement of BES·0.5H2O before melting. It was therefore concluded by Salole,16 in 1987, that BES·0.5H2O dehydrates in two stages. The first stage is around 112 °C, and the second at ca. 174 °C, which results in the complete loss of water and transformation to an anhydrous form before melting. There appears to be no obvious explanation from examining the crystal structure (Fig. 2) as to why water is lost in two stages. Florence et al.14 also observed that after grinding BES·0.5H2O there is no change in the IR spectrum, however the DTA curve contains only a single enlarged pre-melting endotherm around 120 °C. This change suggests that BES·0.5H2O is partially dehydrated upon grinding and an anhydrous form can be obtained at temperatures about 60 °C lower than a sample that has not been ground. In 2000, Variankaval et al.17 investigated the release of water from BES·0.5H2O near the melting point by DSC to find the compete removal of water only occurred after the sample had melted, apparently contradicting the work mentioned above.
Smakula et al.18 first identified four different crystalline forms and one amorphous form of BES (BES(am)) in 1957, by IR absorption spectroscopy and X-ray powder diffraction. The IR spectra of the two anhydrous crystalline forms (BES Form I and BES Form II) suggested that BES Form I contained an O–H group not involved in hydrogen bonding and that in BES Form II there was ketonization of the steroid A-ring. The other two crystalline forms that Smakula described as anhydrous have been identified by Salole16 as BES·0.5H2O and the methanol hemisolvate (BES·0.5MeOH). Heating of any of the four forms between two rock salt plates produces a common glassy amorphous form, BES(am).
About the same time, Kuhnert-Brandstätter et al.19 independently concluded that 17-β-estradiol anhydrate is dimorphic from the thermomicroscopy of cooled melts and postulated that BES Form I is the more stable. They also showed that desolvation of BES·0.5H2O usually transforms to BES Form I. Variankaval et al.17 prepared BES Form I from slow cooling a melt of BES·0.5H2O, and prepared BES Form II from dissolving BES·0.5H2O in ethyl acetate followed by evaporation to dryness from boiling. The DSC analysis of these two forms showed that BES Form I had one melting endotherm around 178 °C, and BES Form II showed two endotherms, one at a lower temperature appearing as a shoulder on the other at 178 °C. This suggests that BES Form I and BES Form II are often found in mixtures or BES Form II converts to BES Form I prior to melting. The difficulty in obtaining BES Form II is cited in all three publications and there is little crystallographic evidence to prove that BES Form II is a polymorph of 17-β-estradiol, with the molecular structure shown in Fig. 1.
 | ||
| Fig. 1 17-β-estradiol (BES) with the conventional steroid atomic numbering scheme. | ||
 | ||
| Fig. 2 Packing diagram (projection down the a axis) for BES·0.5H2O. Location of water on a two-fold axis is illustrated. Blue lines indicate hydrogen bonding, with the through-molecule Chain 1 horizontal and a hydroxyl Chain 2 (ESI† Fig. S17) coming out of the page. | ||
The ability of 17-β-estradiol to form solvates with organic solvents has been shown by Busetta et al.20 who published an orthorhombic propanol monosolvate (BES·PrOH, refcode ESTRDP) in 1972. In 1999, Parrish et al.21 published a triclinic methanol hemisolvate (BES·0.5MeOH, refcode BEQJIQ). The presence of an ethanol solvate was proposed by Resetarits et al.22 in 1978. Variankaval et al.17 performed thermal analysis on BES·0.5MeOH showing two weight loss peaks in the TGA and two pre-melting endotherms in the DSC. This suggests that BES·0.5MeOH loses methanol from the crystal structure in two stages, similar to the behaviour of BES·0.5H2O and the proposed ethanol monosolvate.22 A mixed solvate (BES)3(MeOH)2(H2O) (WURWEL) has also been reported.23
The most characteristic property of 17-β-estradiol is its tendency to crystallize as BES·0.5H2O, in which it precipitates not only from aqueous solution, but also from ethyl acetate, chloroform, absolute ethanol and other supposedly anhydrous solvents.16 This may explain why the PXRD patterns presented24 for four different solvate forms of 17-β-estradiol are so extremely similar to that for BES·0.5H2O that it is likely that only BES·0.5H2O was obtained in these cases.
This history inspired us to perform a polymorph screen on BES, particularly seeking to characterize the anhydrous BES forms. Anticipating that the structure(s) would have to be solved from PXRD, a simultaneous crystal structure prediction (CSP) study was performed.
Experimental
Materials
Enantiopure 17-β-estradiol hemihydrate (BES·0.5H2O) was obtained as a white microcrystalline powder from Sigma Co. 17-β-estradiol anhydrate (BES Form I) was prepared by heating the hemihydrate up to 150 °C for four hours.Methods
Slow evaporation crystallization experiments were carried out by dissolving either microcrystalline BES·0.5H2O (∼10 mg or ∼5 mg) or anhydrous BES (∼10 mg or ∼5 mg) in various solvents (∼3 mL) in small sample tubes and left at ambient temperature or 5 °C. The vessels were either uncapped or had 4 holes punched into the lids. Samples prepared from anhydrous BES were placed in a P2O5 filled desiccator. Experiments involving evaporation to dryness from boiling solvent were carried out by dissolving microcrystalline BES·0.5H2O (∼30 mg) in ethyl acetate (∼6 mL) and heating on a hot plate until dryness was achieved. Cooling crystallization experiments were undertaken by heating microcrystalline BES·0.5H2O (∼10 mg) to a temperature above the melting point of 178 °C or to 150 °C for an extended period and subsequent slow cooling at various rates to RT. Sublimation experiments were performed by sealing a small amount of microcrystalline BES Form I (∼100 mg) in Pyrex tubing at approximately 5 × 10−2 mmHg and heating in a tube furnace with a temperature gradient of 150 to 25 °C. Further details of all crystallization techniques attempted and solid forms produced are provided in the ESI† (Part A section 1).
The stability of anhydrous BES was compared to that of BES·0.5H2O and other solvates by minimizing the solvates with the same CrystalOptimizer computational model and comparing the lattice energies of Utot (BES) and Utot (ice/solvent) with Utot (BES·0.5H2O/solvate) in the appropriate ratios relative to one mole of BES.36 Full details of the computational methods are in Part B of the ESI.†
Results
Structural studies
 | ||
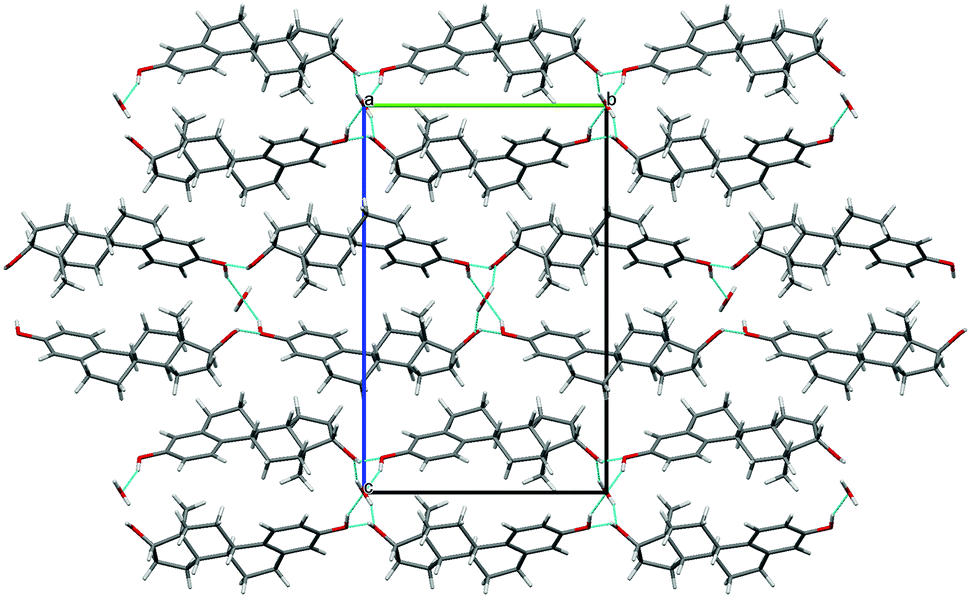
| Fig. 3 Packing diagram of BES Form I, molecules coloured by symmetry equivalence. Chain 1 through-molecule hydrogen bonding runs vertically through the green molecules, and the hydroxyl Chain 2 is perpendicular to the page linking each blue molecule with two adjacent green molecules (ESI† Part B Fig. S17). | ||
BES Form I crystallizes in the orthorhombic space group P212121 with Z′ = 2. Layers of estradiol molecules of the same symmetry equivalence are formed by translation along the a and b axes (green in Fig. 3). Between these layers, estradiol molecules of the other symmetry equivalence (blue in Fig. 3) are hydrogen bonded to the layers through the hydroxyl group on the D-ring forming ladder-like packing.
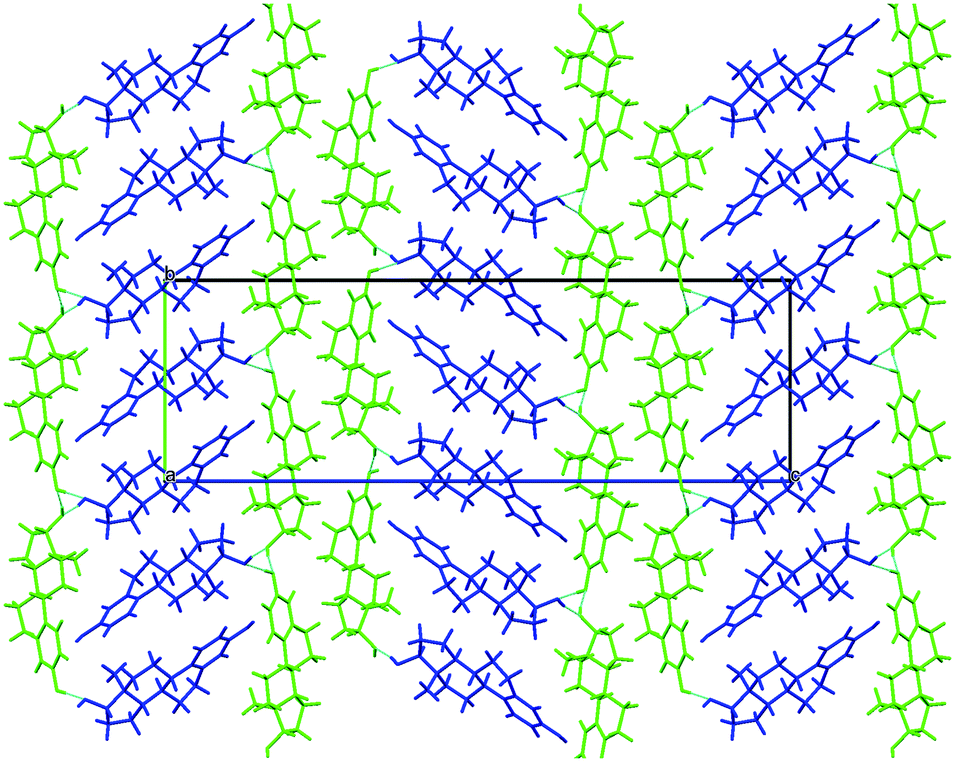
 | ||
| Fig. 4 (top) Packing diagram of BES·ACN, viewed down the a axis. (bottom) Hydrogen bonding between the estradiol hydroxyl groups and acetonitrile molecules (c.f. Chain 2 ESI† Fig. S17). Light blue lines indicate hydrogen bonding. | ||
Thermal analysis
Heating a microcrystalline sample of BES·0.5H2O on a hot stage microscope led to droplets of water appearing around 168–170 °C associated with the onset of melting which was completed at 180 °C. On cooling, amorphous BES was observed on the microscope slide. Sublimation occurred onto the cover slide to give a white crystalline powder with the same IR spectrum as BES Form I. In single crystal form, heating BES·0.5H2O did not show any droplets of water appearing before melting began at 175 °C.
DSC analysis of BES·0.5H2O as a microcrystalline powder shows two pre-melting endotherms, at 114–140 °C and 173.5 °C, before the melt at 179 °C (Fig. 5a). TGA analysis indicated that the first endothermic peak corresponds to a partial loss of water (1.2% of mass) and the second completes a total weight loss of 3.5% (Fig. 5b). DSC analysis of Form BES·0.5H2O in single crystal form gives rise to a single pre-melting endotherm at 173.5 °C and a melting endotherm at 178.5 °C (Fig. 5a). The TGA analysis indicates that the there is a gradual loss of 0.7% of mass, probably reflecting sublimation, before the pre-melting endotherm corresponding to complete loss of water (Fig. 5c). Thus, the loss of water is very different between the crystalline powder and as a single crystal.
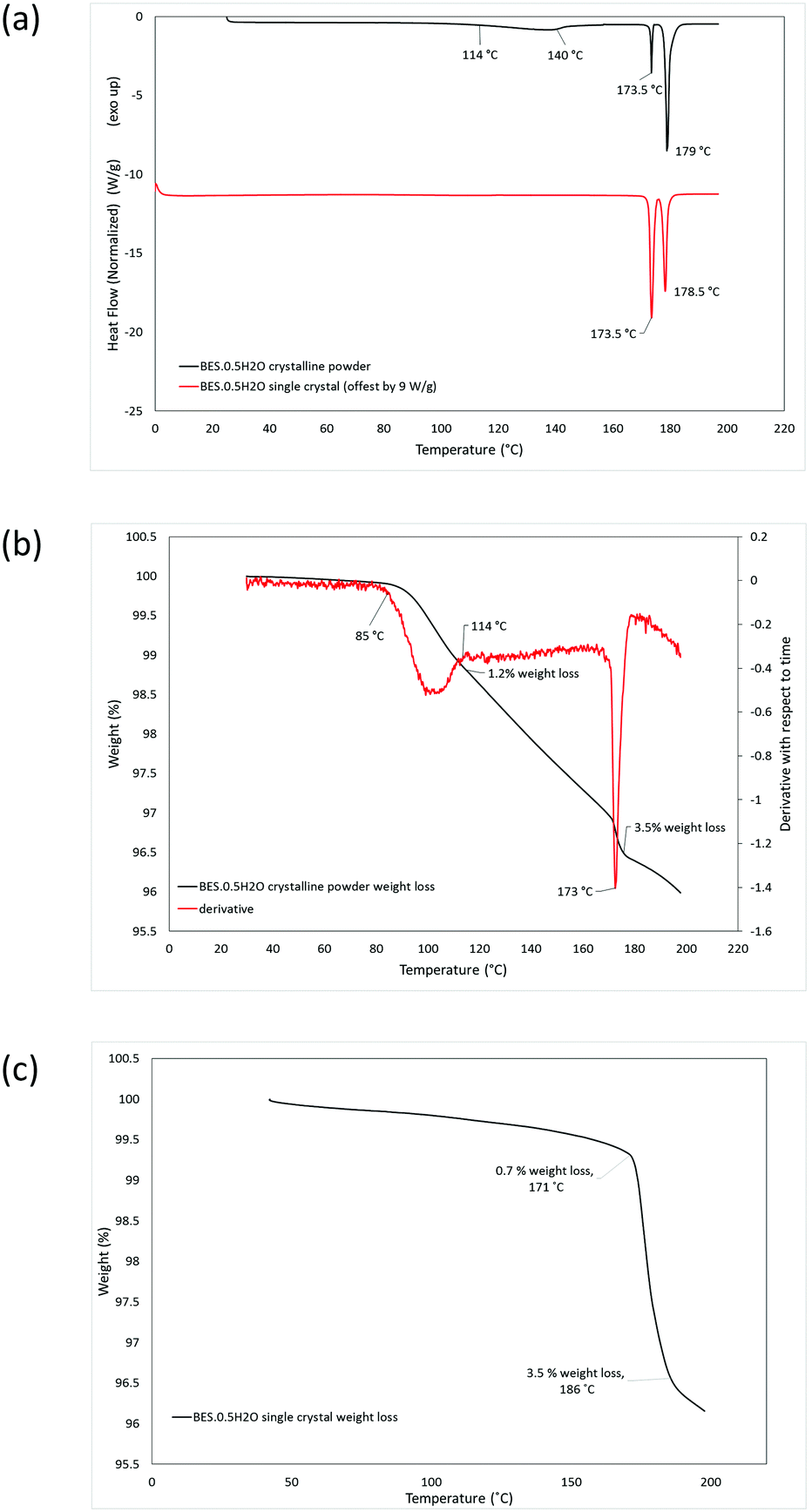 | ||
| Fig. 5 DSC and TGA traces of BES·0.5H2O. (a) DSC curve of a crystalline powder (black) and a single crystal (red). (b) TGA curve of a crystalline powder (black) including derivative with respect to time (red). (c) TGA curve of a single crystal form. | ||
The stability of BES·0.5H2O was also examined in the presence of the dehydrating agent, P2O5, in a sealed desiccator. After 6 weeks, the sample remained as a white microcrystalline powder. The IR spectrum (ESI† Fig. S1) gives rise to a small additional peak around 3600 cm−1 which might correspond to a slight rearrangement in the solid resulting in some non-hydrogen bonded O–H groups, however the PXRD analysis (ESI† Fig. S4) shows no significant structural change. The DSC analysis of this material shows one main pre-melting endothermic peak at 173.5 °C, (ESI† Fig. S7) suggesting that the water that would normally be released around 113–142 °C has been removed by the drying agent. There is a small broad endothermic peak between ∼80 and 120 °C (ESI† Fig. S7) indicating that part of the water has been removed by the drying over the desiccant but it was not completely removed. This is consistent with the TGA analysis (ESI† Fig. S7) showing two stages of water loss, the first at 80–94 °C, comprising 1.3% weight loss, and the second at 171–176 °C, with the total loss of water being 3.5% weight. The first loss of water occurs at a lower temperature than for the hemihydrate starting material, suggesting that the P2O5 drying agent affects the water binding. Throughout the TGA analyses of the desolvation, the final mass loss was somewhat greater than the stoichiometric 3.2%, in the range 3.5–4.0%, probably due to a mixture of sublimation and surface water loss. Hence, overall the dehydration of BES·0.5H2O is very dependent on the conditions.
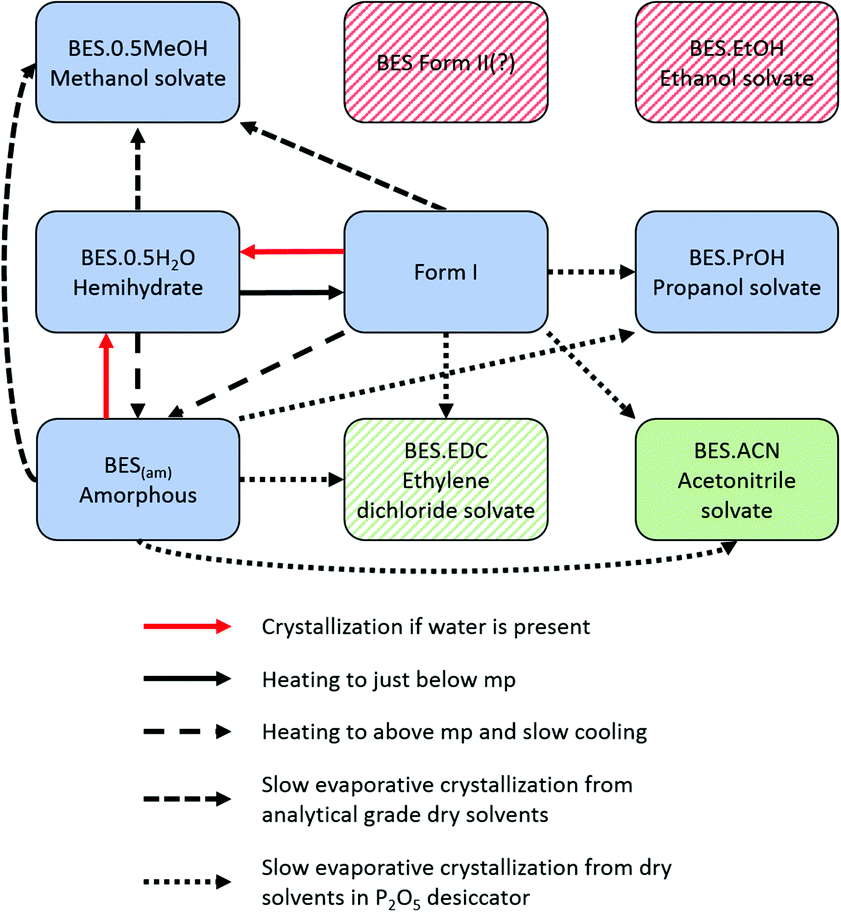 | ||
| Fig. 6 Summary of methods of interconversion between solid forms found in this project. Green denotes solid forms newly discovered in this work, blue denotes previously identified solid forms which we have been able to verify, and red denotes those in the literature which we have not verified. Shaded forms are those for which there is evidence of their existence but no crystal structure has been determined. | ||
Computational results
The CSP search generated a range of low energy structures (Fig. 7), which have been categorized by the conformation of the C3–O1–H hydroxyl group and the hydrogen bonding motif. The experimentally observed anhydrous BES Form I structure was found about 4 kJ mol−1 above the global minimum. This relative stability is predominantly from the steroid backbone packing as the structure has unsatisfied hydrogen bonding of half the O1 hydroxyl groups. The OH⋯OH⋯OH chain structure (Chain 2) does appear in the water and alcohol solvates with the solvent OH in every 3rd or 5th place.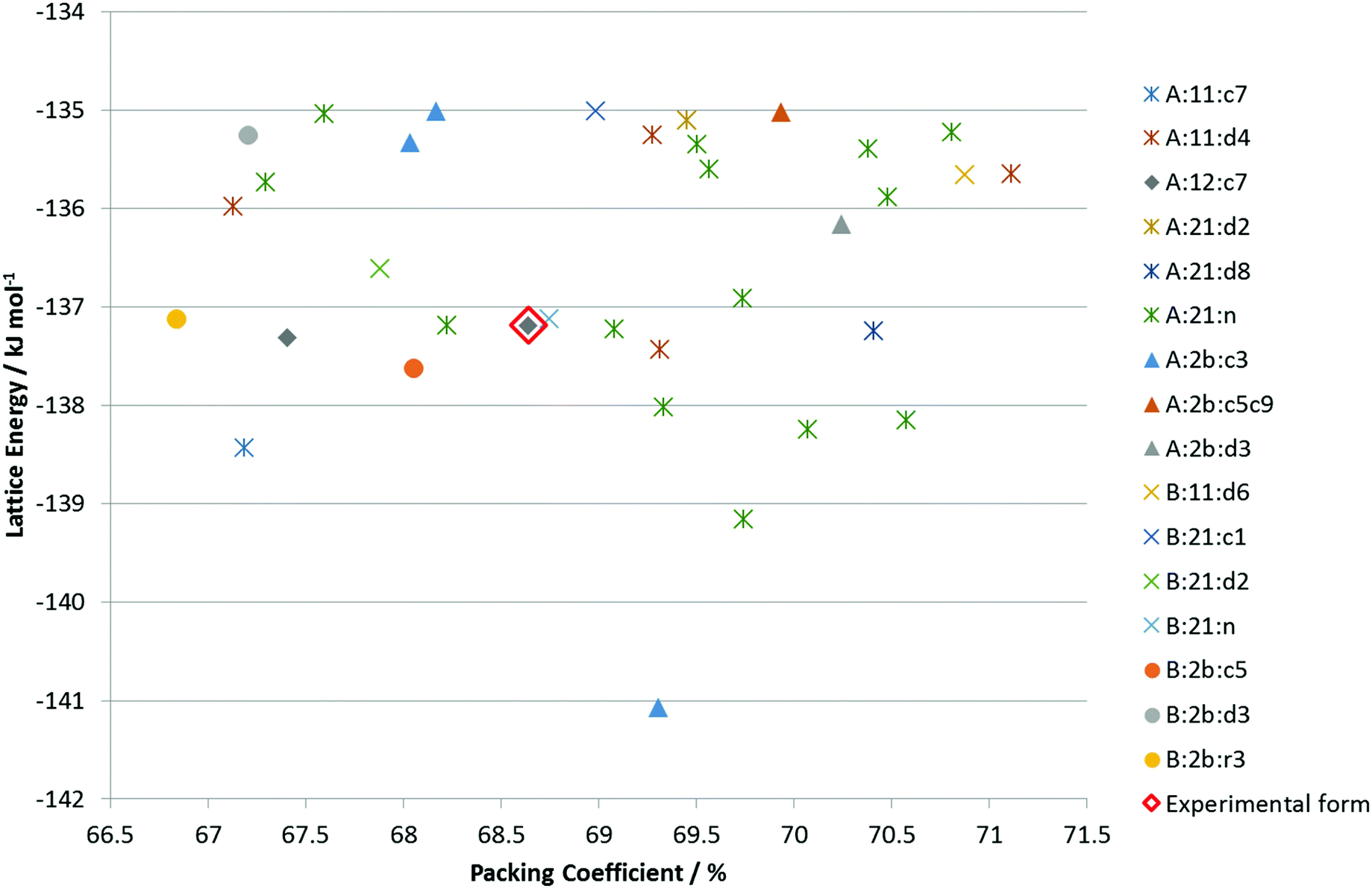 | ||
Fig. 7 Summary of the CSP study of BES. Each point corresponds to a structure which is a lattice energy minimum, calculated with the Ψmol method with intramolecular energy and charge density evaluated at the PBE0/6-31G(d,p) level and the FIT repulsion–dispersion model being used for the other terms. The structures have been categorized by motif X:nm:al where X![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) A or B denotes the O1H23 proton conformation; nm denotes the Chain 1 through-molecule hydrogen bonding type, with n denoting whether 1 or 2 molecules (of the Z′ = 2 molecules in the structure) are involved in through molecule hydrogen bonding and m denoting whether O1, O2 or both are acting as hydrogen bond donor; al denotes the Chain 2 hydroxyl-only hydrogen bonding type, n denotes that this is not present, c is that it is a chain, d a discrete interaction and r that it is a ring, the number l describes the order of the donor atoms along the hydroxyl only hydrogen bond motif as defined in ESI† Fig. S17. A or B denotes the O1H23 proton conformation; nm denotes the Chain 1 through-molecule hydrogen bonding type, with n denoting whether 1 or 2 molecules (of the Z′ = 2 molecules in the structure) are involved in through molecule hydrogen bonding and m denoting whether O1, O2 or both are acting as hydrogen bond donor; al denotes the Chain 2 hydroxyl-only hydrogen bonding type, n denotes that this is not present, c is that it is a chain, d a discrete interaction and r that it is a ring, the number l describes the order of the donor atoms along the hydroxyl only hydrogen bond motif as defined in ESI† Fig. S17. | ||
Given the size of the molecule and unit cells, the differences in the hydrogen bonding and range of densities, and the neglect of thermal effects by considering lattice rather than free energy, the observed Form I could be the thermodynamically most stable within computational error. However, it is unlikely that none of the other structures are thermodynamically plausible as polymorphs.
All the potential polymorphs of BES were calculated to be very unstable relative to the hemihydrate (Fig. 8).
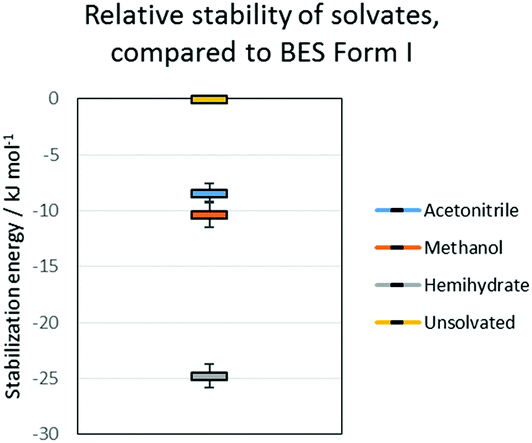 | ||
| Fig. 8 Relative stability of solvates, compared to BES Form I. The error bars on the solvates are the variation due to the energies of the pure solvent polymorphs. | ||
Discussion
17-β-estradiol has an experimentally diverse solid form landscape composed of one anhydrous form, one hemihydrate, one methanol hemisolvate, two monosolvates (ACN, PrOH) and an amorphous form. We found a highly labile, metastable ethylene dichloride solvate but no evidence of the previously proposed ethanol solvate.22 This may be a limitation of our range of experiments, as different alcohols could occupy the interlayer spacing (Fig. 9), as in the family of alcohol solvates of a pharmaceutical salt.37 The formation of the highly elusive Form II probably involves a transformation of some molecules to the keto-tautomer. It has been established that BES·0.5H2O is the most commonly found crystal form and BES Form I will only crystallize when water or other solvent molecules are rigorously excluded. The structure of this anhydrous form has been unambiguously established by single crystal X-ray diffraction. While a good deal of crystallization space has been explored, it nevertheless remains likely that many other undiscovered solid forms may still be found, such as mixed solvates exemplified by (BES)3(MeOH)2(H2O) (WURWEL).23 There are many more possibilities for multi-component complexes such as that formed with urea (ESOURE10)38 and recently, formamide.39,40 The extension to co-crystallization with larger molecules has been partially investigated, with cocrystals with isonicotinamide (ULUFIS) and piperazine dihydrate (ULUFOY) being reported from a search for increased solubility,41 and with pyrene (CUTBEZ), phenanthrene (RUGYID), benzo[h]quinoline (RUFYIC) and 1,2-dimethylnapthalene (RUFOY) in investigations of the molecular recognition of steroids.42,43 The crystal energy landscape (Fig. 7) suggests that other polymorphs of BES are highly likely, although Form I may well be the most thermodynamically stable form given the accuracy of the calculations.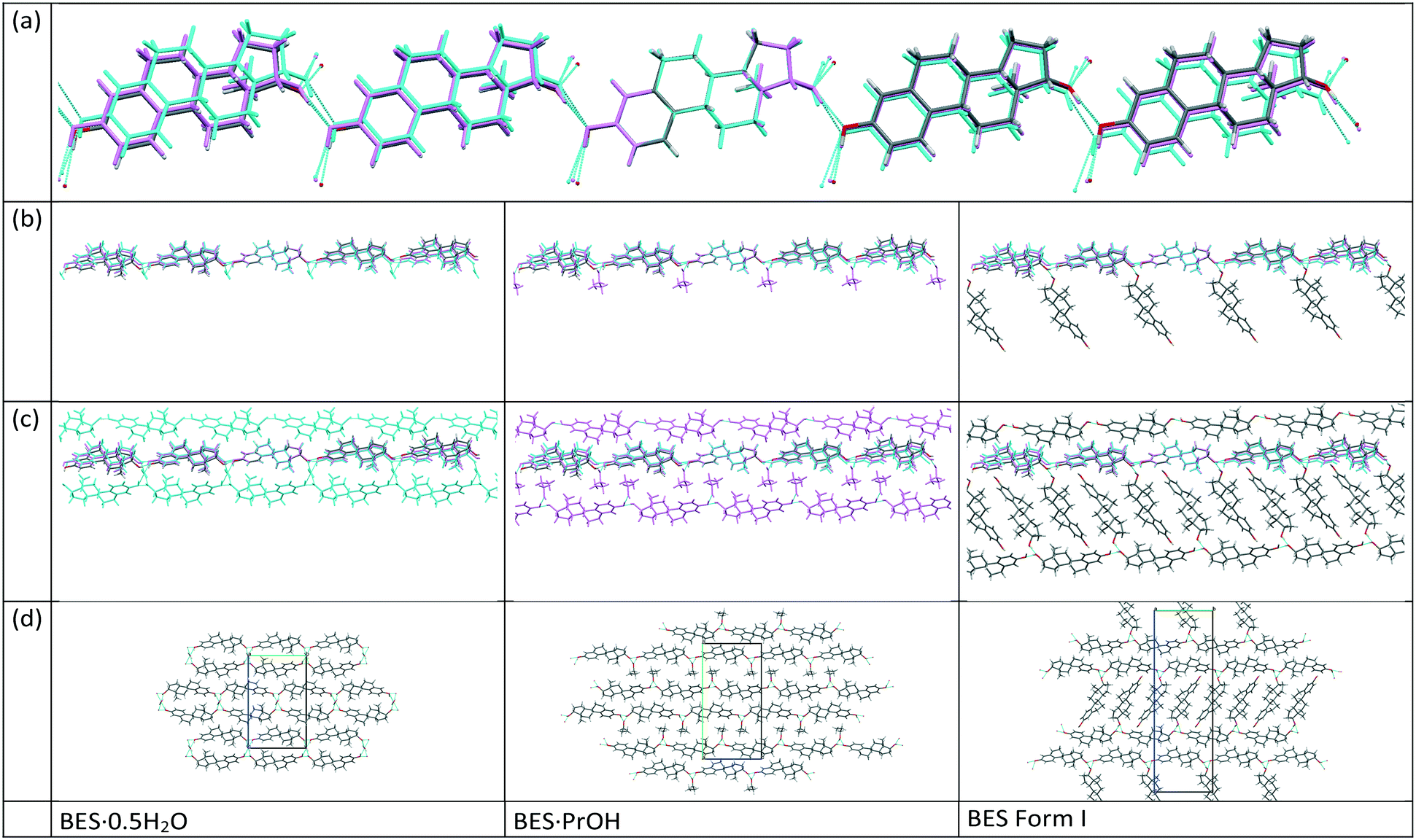 | ||
| Fig. 9 Comparison of the common packings of BES Form I (coloured by element), BES·0.5H2O (cyan) and BES·PrOH (violet). (a) Shows the overlaid chain, (b) shows the chain and hydrogen bonded solvent (second BES molecule in the case of BES Form I), (c) shows the surrounding chains and (d) shows the crystal packing of each form. | ||
Single crystal X-ray diffraction studies demonstrate that there are clear similarities in the arrangement of BES molecules in many of the structures. While the hemihydrate is not isomorphous with the propanol solvate, the water molecule occupies a similar position between the same hydrogen bonding chain (Chain 1) and plays a similar role in the crystal structures. The crystal structure of anhydrous BES Form I also contains a similar through-molecule hydrogen bonding Chain 1 arrangement of BES molecules, however the rows are now further apart as the sites previously occupied by solvent are occupied by a second set of BES molecules, approximately orthogonal and doubly hydrogen-bonded through a single oxygen atom (Chain 2), linking the through-molecule (Chain 1) motifs (Fig. 9).
Throughout the solid form screening experiments, it is evident that when water is available BES·0.5H2O will preferentially crystallize. The dominance of this form can be explained by the clear calculated relative stability of BES·0.5H2O over even the other stable solvates such as BES·0.5MeOH (Fig. 8). Experimentally, the crystallization of BES·0.5MeOH is significantly kinetically favoured over the remaining solid forms which take much longer to nucleate, thus BES·0.5MeOH can be isolated with a lesser degree of control than the other solvates. This highlights a key limitation of the computational model of Fig. 8 (ESI† Table S7) which is based purely on predicting thermodynamic stability of structures.44 The kinetics of crystallization are scarcely understood but are potentially the main factors in determining which solid forms are experimentally obtained,45 particularly in this case where the kinetically and thermodynamically favoured solid form is a hydrate. The prevalence of BES·0.5H2O is practically linked to the difficulty in removing water from organic solvents, and avoiding accidental seeding with the BES·0.5H2O starting material.46
All attempts to produce single crystals of the anhydrous form(s) from nominally anhydrous solvents using microcrystalline anhydrous BES were unsuccessful. In our hands each of these experiments led to a transformation to the corresponding solvate or the hydrate (presumably by the unintended inclusion of water from the solvent or atmosphere). It appears that it is more favourable for the 17-β-estradiol molecules to pack with the available solvents as opposed to just with themselves, despite Form I and other computer-generated structures having a reasonable packing density.
The mechanism of desolvation of BES·0.5H2O is unclear. Although there is a common chain that is preserved, the rearrangement of half the BES molecules to substitute for the solvent linkages between the chains requires a massive rearrangement of half the molecules (Fig. 9). This involves losing hydrogen bonding before gaining stabilization from the dispersion forces between the steroid backbones. Obtaining the anhydrate structure shows that desolvation involves such a substantial rearrangement of the molecules that it requires a nucleation and growth mechanism which will be very dependent on how the real crystals differ from the infinite perfect crystals used in the calculations. The desolvation studies show that when in microcrystalline form, water is often lost from BES·0.5H2O in two stages, though this is dependent on sample and conditions. When BES·0.5H2O is in single crystal form the water molecules appear to be bound more tightly and desolvation appears to occur in one step at 173.5 °C. The dissimilarity of the two structures could mean that intermediate forms, such as transient lower hydrates, BES(am) or BES Form II are also involved in a particle-size and structural-purity-dependent mechanism. It is also possible to rationalize the observation of an apparent two stage desolvation for BES·0.5H2O (ref. 14 and 16) (and BES·0.5MeOH (ref. 17)) as surface solvent being lost when there is a high surface area in microcrystalline form.
Conclusions
The crystal structure of anhydrous 17-β-estradiol has finally been established by SCXRD, along with those of an acetonitrile solvate and more modern determinations of the structures of the hemihydrate, and two alcohol solvates. 17-β-estradiol has a high affinity for water, making crystallization of the anhydrate challenging and the dehydration process dependent on the particle size and structural defects.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to Jeremy K Cockcroft and Andrea Sella (UCL Chemistry) for assistance with the crystallography and sublimation experiments, respectively, and EPSRC EP/K039229/1 for funding.Notes and references
- H. G. Brittain, Polymorphism in Pharmaceutical Solids, Informa Healthcare, New York, London, 2009 Search PubMed.
- T. L. Threlfall, Analyst, 1995, 120, 2435–2460 RSC.
- L. Infantes, J. Chisholm and S. Motherwell, CrystEngComm, 2003, 5, 480–486 RSC.
- R. K. Khankari and D. J. W. Grant, Thermochim. Acta, 1995, 248, 61–79 CrossRef CAS.
- D. E. Braun, L. H. Koztecki, J. A. McMahon, S. L. Price and S. M. Reutzel-Edens, Mol. Pharmaceutics, 2015, 12, 3069–3088 CrossRef CAS PubMed.
- A. Y. Lee, I. S. Lee, S. S. Dettet, J. Boerner and A. S. Myerson, J. Am. Chem. Soc., 2005, 127, 14982–14983 CrossRef CAS PubMed.
- G. R. Kraemer, R. R. Kraemer, B. W. Ogden, R. E. Kilpatrick, T. L. Gimpel and V. D. Castracane, Fertil. Steril., 2005, 79, 524–542 Search PubMed.
- G. Evans and E. L. Sutton, Med. Clin. North Am., 2015, 99, 479–503 CrossRef PubMed.
- IARC Working Group on the Evaluation of Carcinogenic Risk to Humans. Combined Estrogen–Progestogen Contraceptives and Combined Estrogen–Progestogen Menopausal Therapy, https://www.ncbi.nlm.nih.gov/books/NBK321672/, 2007.
- K. B. Lokind and F. H. Lorenzen, Int. J. Pharm. Anal., 1996, 127, 155–164 CrossRef CAS.
- Hormone Replacement Therapy, ed. W. A. Meikle, Springer, 1999 Search PubMed.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- B. Busetta and M. Hospital, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1972, 28, 560–567 CrossRef CAS.
- A. T. Florence and E. G. Salole, J. Pharm. Pharmacol., 1975, 28, 637–642 CrossRef.
- M. Kuhnert-Brandstätter and H. Winkler, Sci. Pharm., 1976, 44, 177–190 Search PubMed.
- E. G. Salole, J. Pharm. Biomed. Anal., 1987, 5, 635–648 CrossRef CAS.
- N. E. Variankaval, K. I. Jacob and S. M. Dinh, J. Cryst. Growth, 2000, 217, 320–331 CrossRef CAS.
- E. Smakula, A. Gori and H. H. Wotiz, Spectrochim. Acta, 1957, 9, 346–356 CrossRef CAS.
- M. Kuhnert-Brandstaetter and A. Kofler, Microchim. Acta, 1959, 47, 847–853 CrossRef.
- P. B. Busetta, C. Courseille, S. Geoffre and M. Hospital, Acta Crystallogr., 1971, 28, 1349–1351 CrossRef.
- D. A. Parrish and A. A. Pinkerton, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, IUC9900100 CrossRef.
- D. E. Resetarits, K. C. Cheng, B. A. Bolton, P. N. Prasad, E. Shefter and T. R. Bates, Int. J. Pharm., 1979, 2, 113–123 CrossRef CAS.
- D. A. Parrish and A. A. Pinkerton, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2003, 59, O80–O82 CrossRef.
- J.-S. Park, H. W. Kang, S. J. Park and C.-K. Kim, Eur. J. Pharm. Biopharm., 2005, 60, 407–412 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, SHELXS97, 2008 Search PubMed.
- G. M. Sheldrick, SHELXL-2013, 2013 Search PubMed.
- S. L. Price, Proc. R. Soc. A, 2018, 474, 20180351 CrossRef PubMed.
- M. Habgood, I. J. Sugden, A. V. Kazantsev, C. S. Adjiman and C. Pantelides, J. Chem. Theory Comput., 2015, 11, 1957–1969 CrossRef CAS PubMed.
- K. M. Anderson, A. E. Goeta and J. W. Steed, Cryst. Growth Des., 2008, 8, 2517–2524 CrossRef CAS.
- A. V. Kazantsev, P. G. Karamertzanis, C. S. Adjiman and C. C. Pantelides, in Molecular System Engineering, ed. C. S. Adjiman and A. Galindo, WILEY-VCH Verlag GmbH & Co., Weinheim, 2010, vol. 6, pp. 1–42 Search PubMed.
- A. J. Stone, GDMA2.2, 2010 Search PubMed.
- D. E. Williams and S. R. Cox, Acta Crystallogr., Sect. B: Struct. Sci., 1984, 40, 404–417 CrossRef.
- D. S. Coombes, S. L. Price, D. J. Willock and M. Leslie, J. Phys. Chem., 1996, 100, 7352–7360 CrossRef CAS.
- S. L. Price, M. Leslie, G. W. A. Welch, M. Habgood, L. S. Price, P. G. Karamertzanis and G. M. Day, Phys. Chem. Chem. Phys., 2010, 12, 8478–8490 RSC.
- N. Issa, P. G. Karamertzanis, G. W. A. Welch and S. L. Price, Cryst. Growth Des., 2009, 9, 442–453 CrossRef CAS.
- D. E. Braun, S. R. Lingireddy, M. D. Beidelschies, R. Guo, P. Muller, S. L. Price and S. M. Reutzel-Edens, Cryst. Growth Des., 2017, 17, 5349–5365 CrossRef CAS PubMed.
- D. Parrish, E. A. Zhurova, K. Kirschbaum and A. A. Pinkerton, J. Phys. Chem. B, 2006, 110, 26442–26447 CrossRef CAS PubMed.
- H.-P. Wang, J. Xu, L.-F. Ning, P. Li and X.-F. Chen, Jiegou Huaxue, 2018, 37, 730 CAS.
- H. P. Wang, J. Xu, L. F. Ning, P. Li and X. F. Chen, Z. Kristallogr. - New Cryst. Struct., 2018, 233, 199–201 CAS.
- J. R. Wang, X. J. Wang, Y. Yang, X. Y. Chen and X. F. Mei, CrystEngComm, 2016, 18, 3498–3505 RSC.
- K. J. Ardila-Fierro, V. Andre, D. Tan, M. T. Duarte, R. W. Lancaster, P. G. Karamertzanis and T. Friščić, Cryst. Growth Des., 2015, 15, 1492–1501 CrossRef CAS.
- T. Friščić, R. W. Lancaster, L. Fabian and P. G. Karamertzanis, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13216–13221 CrossRef PubMed.
- S. L. Price, Faraday Discuss., 2018, 211, 9–30 RSC.
- S. L. Price, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 313–328 CrossRef CAS PubMed.
- F. Tian, H. Qu, A. Zimmermann, T. Munk, A. C. Jørgensen and J. Rantanen, J. Pharm. Pharmacol., 2010, 62, 1534–1546 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of experimental methods of crystallization, IR spectra, single crystal diffraction data, PXRD patterns, thermal analyses, computational methods. Crystal structures have been deposited with the CSD with deposition numbers CCDC 1875323 (BES·0.5MeOH), CCDC 1875324 (BES·ACN), CCDC 1875325 (BES·0.5H2O), CCDC 1875326 (BES·PrOH) and CCDC 1875327 (BES Form I). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ce01874j |
| This journal is © The Royal Society of Chemistry 2019 |