
Direct bromocarboxylation of arynes using allyl bromides and carbon dioxide†
Yu
Zhang
,
Wenfang
Xiong
,
Jinghe
Cen
,
Wuxin
Yan
,
Yaodan
Wu
,
Chaorong
Qi
 ,
Wanqing
Wu
and
Huanfeng
Jiang
*
,
Wanqing
Wu
and
Huanfeng
Jiang
*
Key Lab of Functional Molecular Engineering of Guangdong Province, School of Chemistry and Chemical Engineering, South China University of Technology, Guangzhou 510640, P. R. China. E-mail: jianghf@scut.edu.cn
First published on 19th September 2019
Abstract
An unprecedented three-component coupling involving arynes, allyl bromides, and CO2 is described, providing efficient and facile access to structurally diverse ortho-brominated aryl esters. Unlike the conventional role played in organic synthesis as electrophiles, organic bromides served as nucleophiles in this reaction, affording a new approach to multicomponent reactions (MCRs) involving aryne intermediates. Additionally, Hammett analysis suggests that two reaction mechanisms exist, depending on the electronic nature of the cinnamyl bromides used.
Arynes have become versatile synthetic tools in organic synthesis, especially for the construction of diverse 1,2-functionalized aromatic rings.1 Due to their highly electrophilic strained triple bond, arynes can undergo different types of reactions such as cycloaddition reaction, multicomponent coupling, and insertion reaction to realize the double functionalization. Among them, the combination of organic halides and arynes with various nucleophiles provides an alternative way to preserve halogen atoms or organic parts in the final products. Typically, organic halides such as α-bromo ketones2 can undergo nucleophilic attack by in situ zwitterions or nucleophiles to afford the corresponding alkylated products (Scheme 1, Mode 1). On the other hand, the incorporation of halogen atoms into arynes, which greatly influences the pharmacological activities of bioactive molecules3 and is utilized for diverse functional group transformations,4 can be accomplished by using alkynyl or polyfluoroaryl bromides,5a,g perfluoroalkyl5b,c or alkynyl iodides,5d,e and carbon tetrachloride5f,g (Scheme 1, Mode 2).
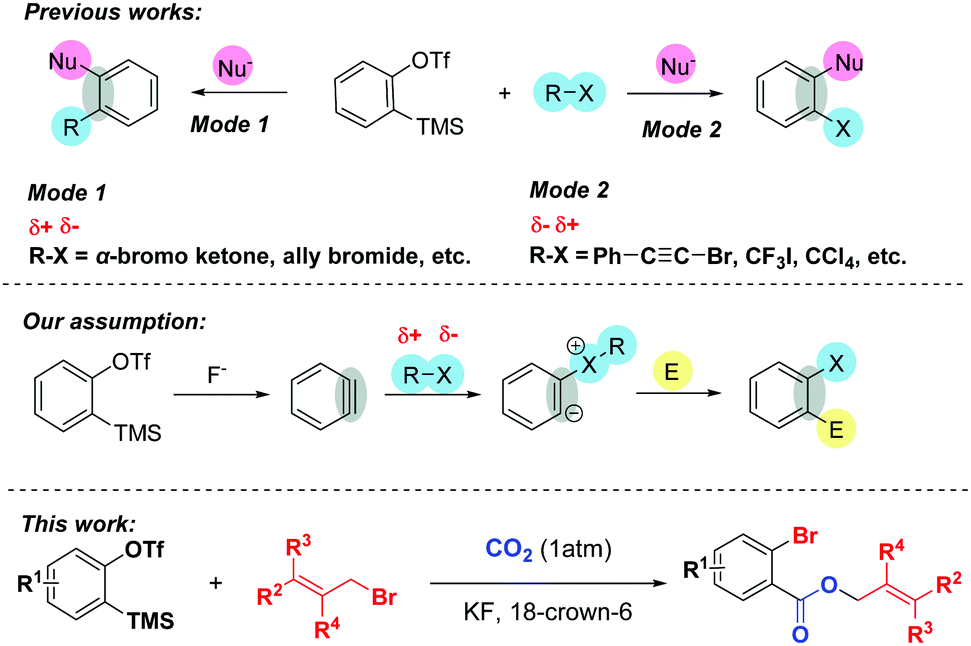 | ||
| Scheme 1 Background and our approach. | ||
Despite these elegant studies described, as far as we know, there has been no precedent utilizing organic halides as nucleophiles to trigger MCRs involving aryne intermediates. Inspired by Yoshida's brilliant work6 that arynes could be inserted into the C–X σ-bonds of acid halides and the fact that arynes could even be initiated with different solvents such as THF,5a,7 DMF8 and DMSO,9 we envisaged that the benzyne intermediate generated in situ might undergo nucleophilic attack by the halogen atoms of organic halides, and the corresponding zwitterions might capture another electrophilic reagent to furnish the functionalized halobenzenes.10 Additionally, given that CO2 is an inexpensive, safe and renewable C1 building block, the transformation of CO2 into high value-added products shows great potential in organic synthesis and has attracted numerous organic chemists’ attention.11 However, very limited success involving both CO2 and arynes has been achieved so far.12
Recently, we12g reported a 3P-4CR of arynes to afford diverse o-chlorobenzoates using KCl as a Cl source. Surprisingly, we found that the reaction could give a moderate yield of allyl 2-bromobenzoate 3aa when the solvent DCE (1,2-dichloroethane) was replaced with allyl bromide even without KBr. Therefore, as part of our continuous interest in the utilization of CO2,13 herein, we report an unprecedented three-component coupling reaction of arynes, allyl bromides and CO2. Compared with our previous studies, the reaction proceeded well without any additional inorganic salts and the incorporation of both Br and CO2 into the arynes made further facile functionalizations possible.
We initially investigated the reaction of aryne precursor 1a14 with allyl bromide 2a under 1 atm CO2 at 46 °C for 12 h, and observed that the three-component coupling product 3aa was obtained in 52% yield. After screening of temperatures, we were delighted to discover that the reaction yield could be optimized to 79%.15 The reaction system could be compatible with various benzyne precursors (Scheme 2), especially for those containing the methoxy group, affording allyl o-bromobenzoate derivatives in good to excellent yields. As expected, the reaction of 4-methoxybenzyne (generated from 1c) provided the corresponding product in 64% yield with poor regioselectivity (1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Moreover, both multisubstituted symmetrical and unsymmetrical silyl aryl triflates (1d–1g) reacted smoothly with complete regioselectivity16 under the reaction conditions. Furthermore, indanyl and 1,3-benzodioxole derivatives (1h and 1j) as well as 2,3-naphthalyne (1i) could also work well, furnishing the expected products in 67–74% yields. However, the reaction of the 4,5-difluoroaryne precursor with allyl bromide 2a resulted in a much lower yield.
:1). Moreover, both multisubstituted symmetrical and unsymmetrical silyl aryl triflates (1d–1g) reacted smoothly with complete regioselectivity16 under the reaction conditions. Furthermore, indanyl and 1,3-benzodioxole derivatives (1h and 1j) as well as 2,3-naphthalyne (1i) could also work well, furnishing the expected products in 67–74% yields. However, the reaction of the 4,5-difluoroaryne precursor with allyl bromide 2a resulted in a much lower yield.
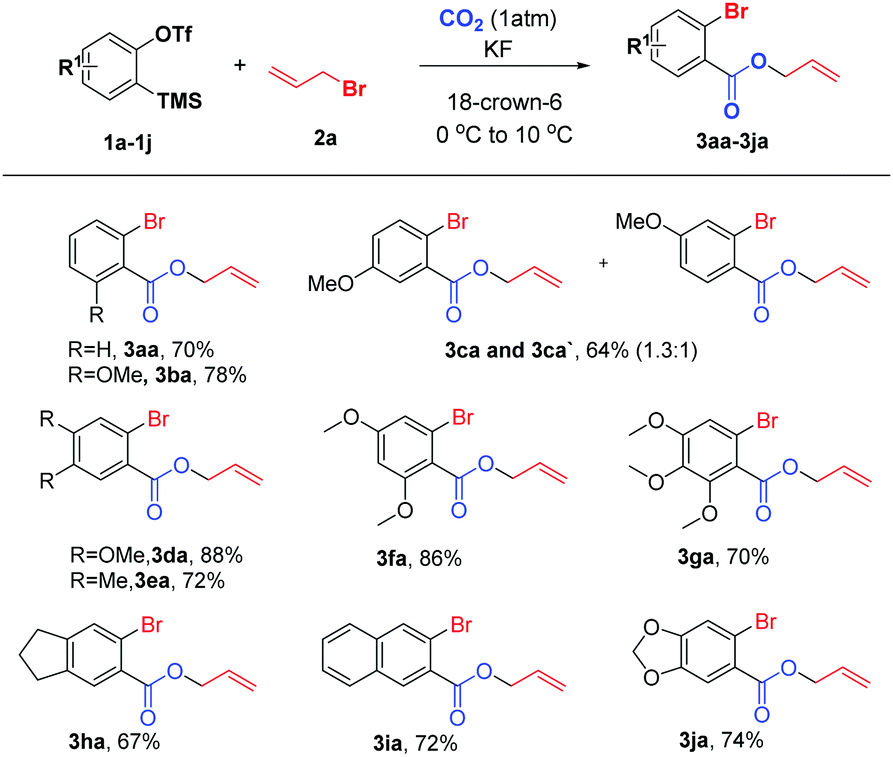 | ||
| Scheme 2 Reaction conditions: 1a (0.2 mmol), 2a (0.5 mL), CO2 (1 atm), KF (4 equiv.), 18-crown-6 (4 equiv.), 0 to 10 °C for 12 h. | ||
Encouraged by these preliminary results, we then directed our attention to the optimization of non-terminal allyl bromide 2b (Table 1). Among the different solvents examined, CF3-Ph was found to be the best solvent, whereas the other solvents afforded inferior results (entries 1–4). The reaction yield could be further improved to 56% by performing the reaction at −10 °C and gradually warming the reaction mixture to 10 °C (entry 5). Remarkably, when we proportionally decreased the dosages of both the aryne precursor and F source, the yield of 3ab increased to 66% (entries 5–7). To our delight, a similar result was achieved even when the amounts of KF and 18-crown-6 were reduced to 2.4 equivalents (entry 8). Finally, diluting the reaction mixture and altering the charging way of 2b gave the best result, with 3ab obtained in 76% yield (entry 10). The control experiments indicated that both 18-crown-6 and CO2 were essential for transformation (entries 11 and 12).
|
|
|||||
|---|---|---|---|---|---|
| Entry |
1a:2b |
F− source (equiv.) | Solvent (mL) | T [°C] | Yieldb [%] |
| a Reaction conditions: 2b (0.2 mmol), KF (4 equiv.) and 18-crown-6 (4 equiv.) in solvent (0.1 mL), CO2 (1 atm), 12 h. b Determined by 1H NMR analysis using CH3NO2 as an internal standard. c CF3-Ph (0.5 mL). d CF3-Ph (1.0 mL). e 2b was added in the N2 glove box f Without 18-crown-6. g Under a N2 atmosphere. | |||||
| 1 | 1.5:1 |
4 | THF | 0 | 21 |
| 2 | 1.5:1 |
4 | MeCN | 0 | n.d. |
| 3 | 1.5:1 |
4 | DCE | 0 | 12 |
| 4 | 1.5:1 |
4 | CF3-Ph | 0 | 41 |
| 5c | 1.5:1 |
4 | CF3-Ph | −10 to 10 | 56 |
| 6c | 1.2:1 |
3.2 | CF3-Ph | −10 to 10 | 66 |
| 7c | 1:1 |
2.6 | CF3-Ph | −10 to 10 | 64 |
| 8c | 1.2:1 |
2.4 | CF3-Ph | −10 to 10 | 66 |
| 9d | 1.2:1 |
2.4 | CF3-Ph | −10 to 10 | 69 |
| 10 , |
1.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1
|
2.4 | CF 3 -Ph | −10 to 10 | 76 |
| 11d,e,f | 1.2:1 |
2.4 | CF3-Ph | −10 to 10 | — |
| 12d,e,g | 1.2:1 |
2.4 | CF3-Ph | −10 to 10 | — |
Next, we examined various cinnamyl bromides17 under the optimized reaction conditions, and the results are shown in Scheme 3. To our delight, a broad range of cinnamyl bromides (3ab–3ai) reacted smoothly to afford the corresponding carboxylated products in moderate to good yields, allowing F, Cl, Br, NO2, and CF3 substituents on the aryl ring to be tolerated. Intriguingly, cinnamyl bromides with electron-neutral groups provided higher yields than those with electron-withdrawing or electron-donating groups, and the substrates with electron-donating groups (3af and 3ag) led to dramatic decreases in the yields of the products. In addition, both disubstituted and 1-naphthyl-substituted cinnamyl bromides (3aj–3al) were suitable reaction partners, leading to the desired products in 64–71% yields. Furthermore, the substrate scope was further expanded to the allyl groups at different positions with various substituents (3am–3ap), all of which were compatible with our reaction system. Moreover, when 1-bromo-5-phenyl-2,4-pentadiene (2q) was employed, 3aq could also be obtained in 65% yield, thus demonstrating the versatility of this new protocol.
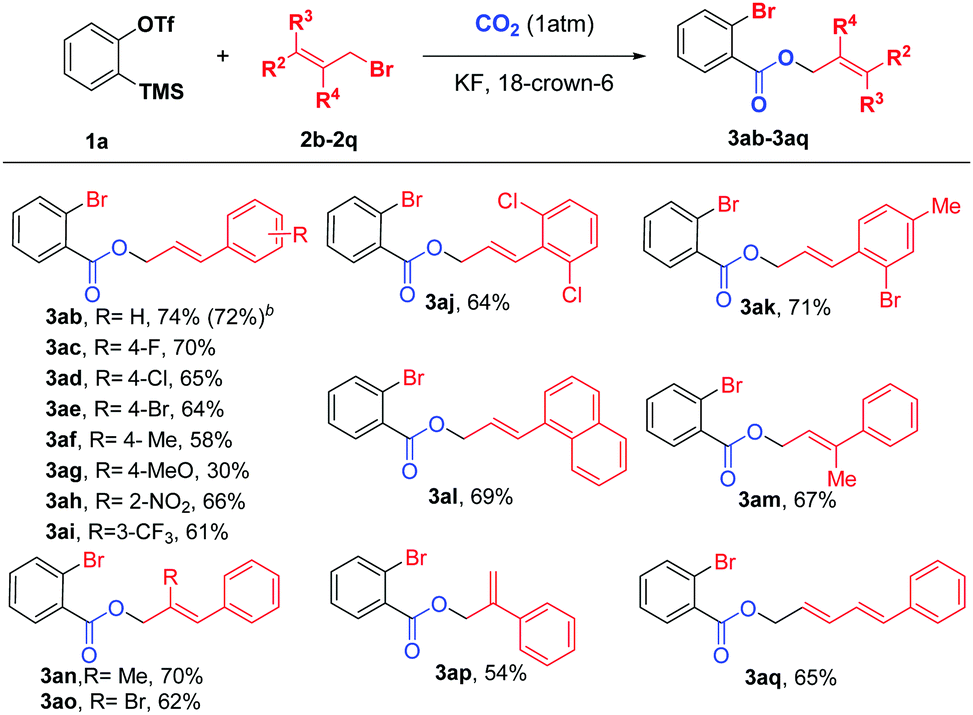 | ||
| Scheme 3 a Reaction conditions: 1a (0.24 mmol), 2 (0.2 mmol), CO2 (1 atm), KF (2.4 equiv.), 18-crown-6 (2.4 equiv.), CF3–Ph (1 mL), −10 to 10 °C for 12 h. bReaction conditions: 1a (1.2 mmol), 2 (1 mmol), CO2 (balloon), KF (2.4 equiv.), 18-crown-6 (2.4 equiv.), CF3-Ph (5 mL), −10 to 10 °C for 12 h. | ||
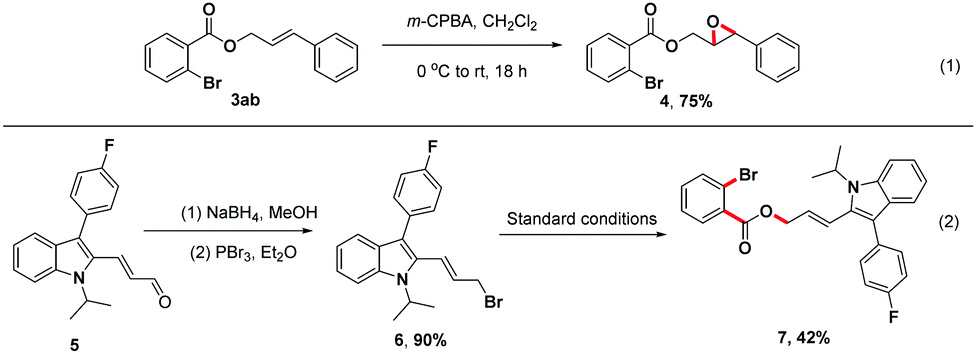
To further illustrate the utility of this unique three-component reaction, treatment of product 3ab with m-chloroperoxybenzoic acid (m-CPBA) in CH2Cl2 provided the desired epoxide derivative 4 in 75% isolated yield18a (Scheme 4, (1)). Moreover, the indole derivative 5, a synthetic precursor for the construction of Fluvastatin,18b,c underwent reduction and bromination efficiently to afford brominated compound 6, which could be transformed to the structurally complex product 7 in 42% isolated yield using this one-step strategy (Scheme 4, (2)).
 | ||
| Scheme 4 Synthetic applications. | ||
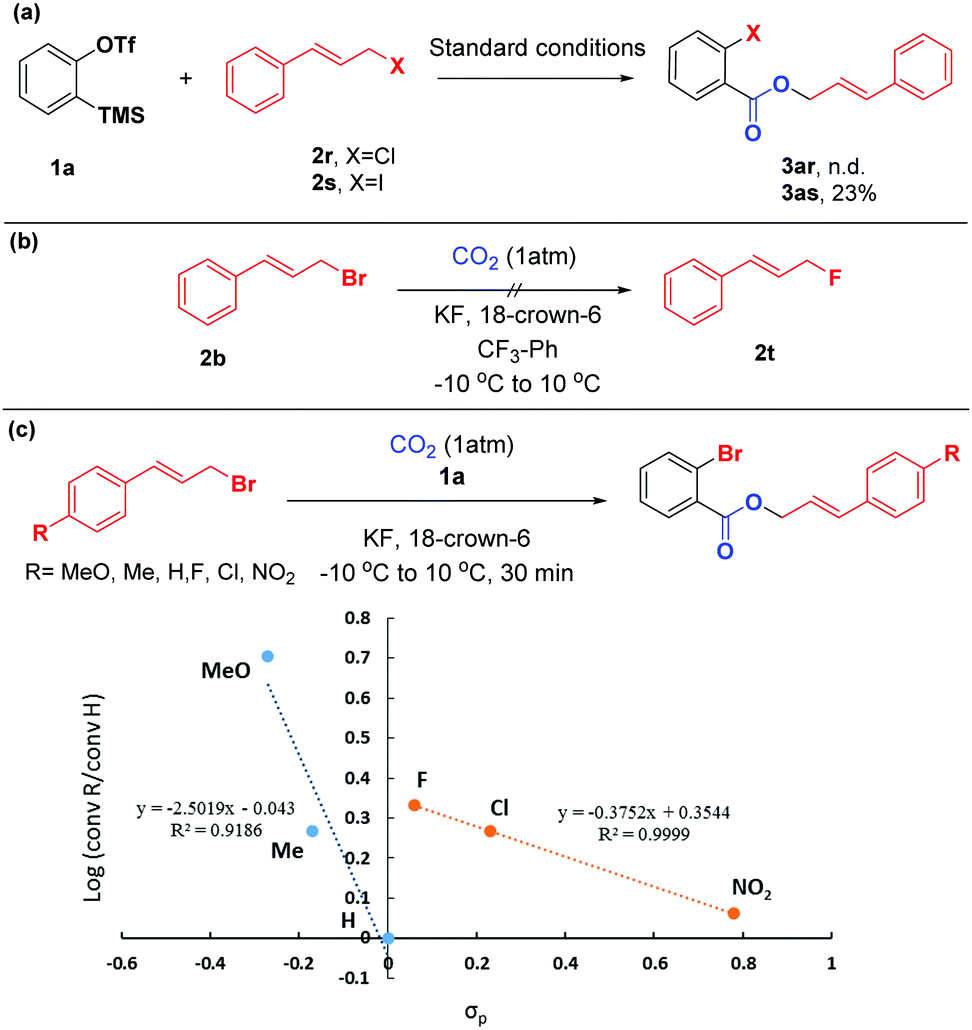
To shed light on the mechanism of this difunctionalization reaction of arynes, a series of control experiments were performed (Scheme 5). Compared with cinnamyl bromide 2b, the reaction of cinnamyl chloride 2r with aryne precursor 1a failed to give the target product 3ar, while cinnamyl iodine 2s could only afford the corresponding product 3as in 23% yield (based on 1H NMR), with iodobenzene and ortho-diiodobenzene as the main side products (detected by GC-MS). These observations indicated that cinnamyl bromide possessed a more appropriate nucleophilicity toward arynes. Additionally, given that the organic bromide might undergo nucleophilic fluorination by KF,19 we conducted the reaction without aryne precursor 1a. To our surprise, the formation of 2t was not detected by 19F NMR spectroscopy (Scheme 5(b)). We further evaluated the electronic effects of this reaction by Hammett analysis.20 The two lines with different negative ρ values indicated that a considerable build-up of positive charge in the transition state, however, with a change in the mechanism or rate-determining step, depending on the character of the substituents on the cinnamyl bromides. For substituents with σp < 0, the value ρ = −2.5 was consistent with an SN1-like mechanism.21 But in the case of electron deficient substrates, the sensitivity constant ρ was only −0.4, which might proceed in a different way (Scheme 5(c)).
 | ||
| Scheme 5 Mechanistic experiments. | ||
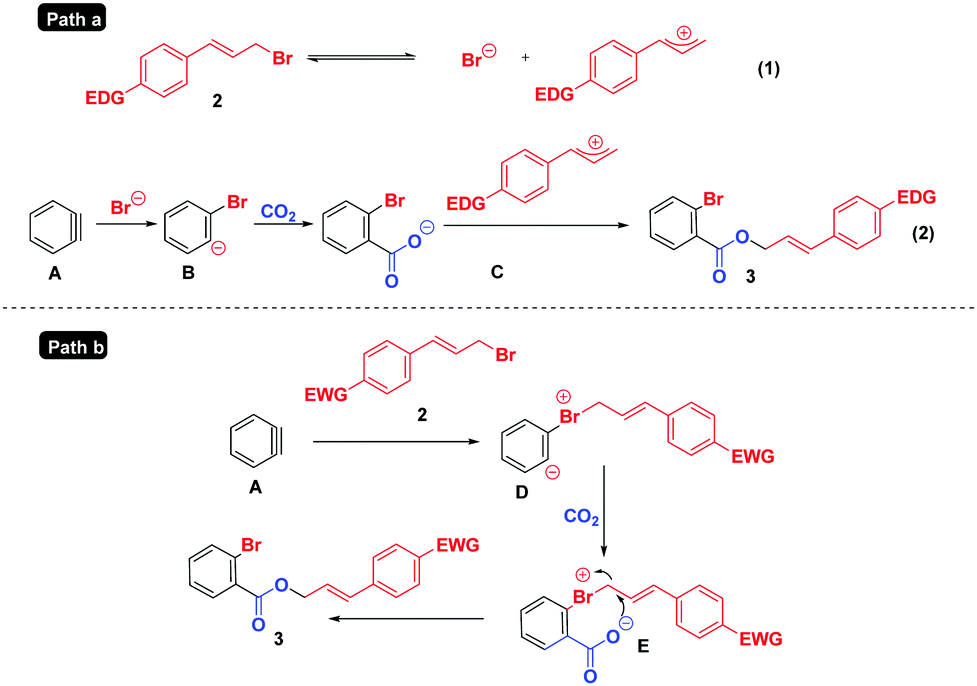
Based on the above results and previous studies,6,12 two tentative mechanisms are proposed in Scheme 6. For substrates containing electron-rich/-neutral groups (Path a), the ionization22 of 2 generates a cinnamyl cation and Br−; the latter reacts with aryne intermediate A, CO2 and the cinnamyl cation successively to deliver the final products 3. However, in the case of electron deficient substrates (Path b), which do not easily undergo the ionization process due to the instability of the cinnamyl cation, the reaction may proceed as follows: the nucleophilic attack of cinnamyl bromide 2 on the benzyne intermediate A affords the 1,3-zwitterion D, which is then intercepted by CO2 to form the 1,5-zwitterion E. Finally, a subsequent intramolecular nucleophilic attack of intermediate E yields cinnamyl 2-bromobenzoates 3.
 | ||
| Scheme 6 Plausible mechanisms. | ||
In summary, we have developed an efficient three-component reaction of CO2 with arynes and allyl bromides to construct various allyl o-bromobenzoate scaffolds. The reaction demonstrates the first example using cinnamyl bromide as a nucleophile in MCRs of arynes and helps deepen our understanding of the high electrophilicity of the aryne intermediate. Further studies focusing on the transformation of CO2 are ongoing in our laboratory.
We thank the National Program on the Key Research Project (2016YFA0602900), the National Natural Science Foundation of China (21420102003) and the Guangdong Natural Science Foundation (2018B030308007) for financial support.
Conflicts of interest
There are no conflicts to declare.Notes and references
- For reviews on arynes, see: (a) R. Sanz, Org. Prep. Proced. Int., 2008, 40, 215 CrossRef CAS; (b) H. Yoshida, J. Ohshita and A. Kunai, Bull. Chem. Soc. Jpn., 2010, 83, 199 CrossRef CAS; (c) P. M. Tadross and B. M. Stoltz, Chem. Rev., 2012, 112, 3550 CrossRef CAS PubMed; (d) A. Bhunia, S. R. Yetra and A. T. Biju, Chem. Soc. Rev., 2012, 41, 3140 RSC; (e) K. Okuma, Heterocycles, 2012, 85, 515 CrossRef CAS; (f) A. V. Dubrovskiy, N. A. Markina and R. C. Larock, Org. Biomol. Chem., 2013, 11, 191 RSC; (g) S. S. Bhojgude, A. Bhunia and A. T. Biju, Acc. Chem. Res., 2016, 49, 1658 CrossRef CAS PubMed; (h) J. A. García-López and M. F. Greaney, Chem. Soc. Rev., 2016, 45, 6766 RSC; (i) T. Roya and A. T. Biju, Chem. Commun., 2018, 54, 2580 RSC; (j) H. Takikawa, A. Nishii, T. Sa-kaib and K. Suzuki, Chem. Soc. Rev., 2018, 47, 8030 RSC.
- (a) W.-M. Shu, J.-R. Ma, K.-L. Zheng and A.-X. Wu, Org. Lett., 2016, 18, 196 CrossRef CAS PubMed; (b) Y. Li, D. Qiu, R. Gu, J. Wang, J. Shi and Y. Li, J. Am. Chem. Soc., 2016, 138, 10814 CrossRef CAS PubMed . Also see ref. 14g.
- M. Z. Hernandes, S. M. T. Cavalcanti, D. R. Moreira, W. F. A. Junior and A. C. L. Leite, Curr. Drug Targets, 2010, 11, 303 CrossRef CAS PubMed.
- (a) R. Martin and S. L. Buchwald, Acc. Chem. Res., 2008, 41, 1461 CrossRef CAS PubMed; (b) J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed; (c) J. Ruan and J. Xiao, Acc. Chem. Res., 2011, 44, 614 CrossRef CAS PubMed; (d) O. Baudoin, Acc. Chem. Res., 2017, 50, 1114 CrossRef CAS PubMed.
- (a) H. Yoshida, Y. Asatsu, Y. Mimura, Y. Ito, J. Ohshita and K. Takaki, Angew. Chem., 2011, 123, 9850 CrossRef; (b) Y. Zeng and J. Hu, Chem. – Eur. J., 2014, 20, 6866 CrossRef CAS PubMed; (c) Y. Zeng, G. Li and J. Hu, Angew. Chem., Int. Ed., 2015, 54, 10773 CrossRef CAS PubMed; (d) Y. Zeng, L. Zhang, Y. Zhao, C. Ni, J. Zhao and J. Hu, J. Am. Chem. Soc., 2013, 135, 2955 CrossRef CAS PubMed; (e) Y. Zeng and J. Hu, Org. Lett., 2016, 18, 856 CrossRef CAS PubMed; (f) S.-J. Li, Y. Wang, J.-K. Xu, D. Xie, S.-K. Tian and Z.-X. Yu, Org. Lett., 2018, 20, 4545 CrossRef CAS PubMed; (g) M. Zhou, C. Ni, Y. Zeng and J. Hu, J. Am. Chem. Soc., 2018, 140, 6801 CrossRef CAS PubMed.
- H. Yoshida, Y. Mimura, J. Ohshita and A. Kunai, Chem. Commun., 2007, 2405 RSC.
- (a) K. Okuma, Y. Fukuzaki, A. Nojima, A. Sou, H. Hino, N. Matsunaga, N. Nagahora, K. Shioji and Y. Yokomori, Bull. Chem. Soc. Jpn., 2010, 83, 1238 CrossRef CAS; (b) M. Thangaraj, S. S. Bhojgude, M. V. Maneb and A. T. Biju, Chem. Commun., 2016, 52, 1665 RSC.
- For selected reports, see: (a) E. Yoshioka, S. Kohtani and H. Miyabe, Org. Lett., 2010, 12, 1956 CrossRef CAS PubMed; (b) H. Yoshida, Y. Ito and J. Ohshita, Chem. Commun., 2011, 47, 8512 RSC; (c) E. Yoshioka, S. Kohtani and H. Miyabe, Angew. Chem., Int. Ed., 2011, 50, 6638 CrossRef CAS PubMed; (d) E. Yoshioka, H. Tanaka, S. Kohtani and H. Miyabe, Org. Lett., 2013, 15, 3938 CrossRef CAS PubMed; (e) F. Liu, H. Yang, X. Hu and G. Jiang, Org. Lett., 2014, 16, 6408 CrossRef CAS PubMed.
- (a) F.-L. Liu, J.-R. Chen, Y.-Q. Zou, Q. Wei and W.-J. Xiao, Org. Lett., 2014, 16, 3768 CrossRef CAS PubMed; (b) H.-Y. Li, L.-J. Xing, M.-M. Lou, H. Wang, R.-H. Liu and B. Wang, Org. Lett., 2015, 17, 1098 CrossRef CAS PubMed; (c) H. Hazarika, K. Neog, A. Sharma, B. Das and P. Gogoi, J. Org. Chem., 2019, 84, 5846 CrossRef CAS PubMed.
- Other methods for the incorporation of halogen atoms into arynes. (a) D. Rodríguez-Lojo, A. Cobas, D. Peña, D. Pérez and E. Guitián, Org. Lett., 2012, 14, 1363 CrossRef PubMed; (b) H. Yoshida, R. Yoshida and K. Takaki, Angew. Chem., Int. Ed., 2013, 52, 8629 CrossRef CAS PubMed; (c) C. E. Hendrick, S. L. McDonald and Q. Wang, Org. Lett., 2013, 15, 3444 CrossRef CAS PubMed; (d) C. E. Hendrick and Q. Wang, J. Org. Chem., 2015, 80, 1059 CrossRef CAS PubMed; (e) S. Bhattacharjee, A. Guin, R. N. Gaykar and A. T. Biju, Org. Lett., 2019, 21, 4383 CrossRef CAS PubMed ; also see ref. 6 and 14g.
- For recent reviews on CO2 chemistry, see: (a) L. Zhang and Z. Hou, Chem. Sci., 2013, 4, 3395 RSC; (b) Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed; (c) K. Sekine and T. Yamada, Chem. Soc. Rev., 2016, 45, 4524 RSC; (d) A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Angew. Chem., Int. Ed., 2018, 57, 15948 CrossRef CAS PubMed; (e) S. Wang and C. Xi, Chem. Soc. Rev., 2019, 48, 382 RSC; (f) C. S. Yeung, Angew. Chem., Int. Ed., 2019, 58, 5492 CrossRef CAS PubMed.
- (a) H. Yoshida, H. Fukushima, J. Ohshita and A. Kunai, J. Am. Chem. Soc., 2006, 128, 11040 CrossRef CAS PubMed; (b) H. Yoshida, T. Morishita and J. Ohshita, Org. Lett., 2008, 10, 3845 CrossRef CAS PubMed; (c) W. J. Yoo, T. V. Q. Nguyen and S. Kobayashi, Angew. Chem., Int. Ed., 2014, 53, 10213 CrossRef CAS PubMed; (d) T. Kaicharla, M. Thangaraj and A. T. Biju, Org. Lett., 2014, 16, 1728 CrossRef CAS PubMed; (e) S. S. Bhojgude, T. Roy, R. G. Gonnade and A. T. Biju, Org. Lett., 2016, 18, 5424 CrossRef CAS PubMed; (f) W. Xiong, C. Qi, R. Cheng, H. Zhang, L. Wang, D. Yan and H. Jiang, Chem. Commun., 2018, 54, 5835 RSC; (g) H. Jiang, Y. Zhang, W. Xiong, J. Cen, L. Wang, R. Cheng, C. Qi and W. Wu, Org. Lett., 2019, 21, 345 CrossRef CAS PubMed.
- (a) W. Xiong, C. Qi, H. He, L. Ouyang, M. Zhang and H. Jiang, Angew. Chem., Int. Ed., 2015, 54, 3084 CrossRef CAS PubMed; (b) W. Xiong, C. Qi, Y. Peng, T. Guo, M. Zhang and H. Jiang, Chem. – Eur. J., 2015, 21, 14314 CrossRef CAS PubMed; (c) Y. Peng, J. Liu, C. Qi, G. Yuan, J. Li and H. Jiang, Chem. Commun., 2017, 53, 2665 RSC; (d) W. Xiong, C. Qi, T. Guo, M. Zhang, K. Chen and H. Jiang, Green Chem., 2017, 19, 1642 RSC; (e) W. Xiong, D. Yan, C. Qi and H. Jiang, Org. Lett., 2018, 20, 672 CrossRef CAS PubMed; (f) H. Jiang, H. Zhang, W. Xiong, C. Qi, W. Wu, L. Wang and R. Cheng, Org. Lett., 2019, 21, 1125 CrossRef CAS PubMed.
- Y. Himeshima, T. Sonoda and H. Kobayashi, Chem. Lett., 1983, 1211 CrossRef CAS.
- See the ESI† for details.
- (a) J. M. Medina, J. L. Mackey, N. K. Garg and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 15798 CrossRef CAS PubMed; (b) E. Picazo, K. N. Houk and N. K. Garg, Tetrahedron Lett., 2015, 56, 3511 CrossRef CAS PubMed.
- Other organic bromides were also investigated. When (3-bromoprop-1-yn-1-yl)benzene, (bromomethyl)benzene or ethyl 2-bromoacetate was used instead of cinnamyl bromides, the reactions could provide the corresponding products in 44%, 52% and 42% yields, respectively.
- (a) Q. Yao, H. Yu, H. Zhang, S. Dong, F. Chang, L. Lin, X. Liu and X. Feng, Chem. Commun., 2018, 54, 3375 RSC; (b) J. T. Zacharia, T. Tanaka and M. Hayashi, J. Org. Chem., 2010, 75, 7514 CrossRef CAS PubMed; (c) V. K. A. Kalalbandi, J. Seetharamappa and U. Katrahalli, RSC Adv., 2015, 5, 38748 RSC.
- (a) E. Lee and D. V. Yandulov, J. Fluorine Chem., 2009, 130, 474 CrossRef CAS; (b) K. Dempsey, R. Mir, I. Smajlagic and T. Dudding, Tetrahedron, 2018, 74, 3507 CrossRef CAS.
- C. Hansch, A. Leo and R. W. Taft, Chem. Rev., 1991, 91, 165 CrossRef CAS.
- (a) K. A. Mix and R. T. Raines, Org. Lett., 2015, 17, 2358 CrossRef CAS PubMed; (b) E. V. Anslyn and D. A. Dougherty, Modern Physical Organic Chemistry, University Science Books, Sausalito, CA, 2006 Search PubMed.
- B. Denegri, A. R. Ofial, S. Jurić, A. Streiter, O. Kronja and H. Mayr, Chem. – Eur. J., 2006, 12, 1657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization of all compounds, and copies of 1H and 13C NMR spectra for selected compounds. See DOI: 10.1039/c9cc05495b |
| This journal is © The Royal Society of Chemistry 2019 |