
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox-responsive phosphonite gold complexes in hydroamination catalysis†
Eva
Deck
a,
Hanna E.
Wagner
a,
Jan
Paradies
 *b and
Frank
Breher
*a
*b and
Frank
Breher
*a
aInstitute of Inorganic Chemistry, Karlsruhe Institute of Technology (KIT), Engesserstr. 15, 76131 Karlsruhe, Germany. E-mail: breher@kit.edu
bDepartment of Chemistry, University of Paderborn, Warburger Str. 100, 33098 Paderborn, Germany. E-mail: jan.paradies@uni-paderborn.de
First published on 20th March 2019
Abstract
Very high activities were observed in the redox-induced hydroamination of alkynes by employing a redox-active gold(I) complex featuring an electron-deficient, terphenyl-substituted phosphonite-based ligand. The hydroamination proceeds roughly two-fold faster with the in situ oxidized catalysts than with their reduced form.
Tertiary phosphines (PR3) and phosphites (P{OR}3) represent some of the most important classes of ligands in transition metal chemistry and their steric and electronic properties can be easily adjusted.1 Although many variations of P-donor ligands have been described in the past, the development of new phosphines with exceptional donor2–4 or acceptor5 capacities or steric properties6 remain in the focus of current research activities. The driving force most often originates from the unsatisfied demand to modify and adjust the properties of phosphine metal complexes, e.g. to face activity, selectivity, or stability issues in the field of homogeneous catalysis. Landmark examples include, for instance, the well-known biaryl phosphines developed by Buchwald and co-workers (I, Chart 1), which found ubiquitous applications in homogeneous catalysis.6 One of the most important structural features of these ligands is the weak – for catalyst stability nonetheless crucial – arene π-interactions with the metal centre. In recent years, elegant variations of Buchwald-type biaryl phosphines have been reported by various groups.7–10 Our group could recently show that the biaryl structural motif can also be included in redox-active phosphine ligands, in our case [1]phosphaferrocenophanes (II, Chart 1).11 Palladium complexes thereof have been successfully applied in redox-induced12 Buchwald–Hartwig amination reactions. Furthermore, we have successfully applied these types of ligands in redox-induced rhodium-catalysed hydrosilylation reactions and found that it is possible to alter the activity and selectivity of the catalyst by using a stimulus-responsive phosphine ligand.13,14 Inspired by the recent developments in homogeneous gold(I) catalysis,15 in particular in catalytic hydroaminations,16 we became interested in studying redox-responsive P-donor ligands in this area.
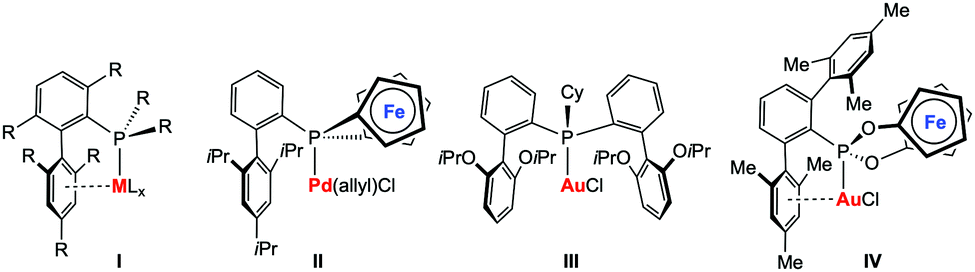 | ||
| Chart 1 Selected metal complexes with Buchwald-type ligand architectures (I–III) from the literature; target structure IV of this study. | ||
Gold-catalysed hydroamination reactions have been intensively studied in recent years. Xu and co-workers provided a comprehensive study on the influence of steric and electronic effects of phosphine ligands in the three major stages of the catalytic cycle in gold catalysis, also including catalytic hydroaminations of alkynes.17 The first stage consists of the initial electrophilic activation of the unsaturated bond, which appears to be favoured by electron-poor ligands. In contrast, the so-called protodeauration in the second stage of the catalytic cycle is accelerated by electron-rich ligands. Although the factors influencing the catalyst deactivation are more complex, biaryl-decorated phosphine ligands were found to be beneficial in terms of catalyst stability. To this end, a very active gold(I) complex (III, Chart 1)18 has been recently presented showing high efficiency at very low loadings (25 ppm) and relatively low temperatures (50 °C) in the gold-catalysed intermolecular hydroamination of alkynes. It is commonly accepted that for this type of catalysis, the protodeauration step19 is rate-determining, which should – in principle – be even more accelerated by increasing the donor-strength of the ligands. However, exceptions from this rather simple recipe have also been observed, such that most electron-rich phosphines are not very efficient in triggering the hydroamination reaction.20 We therefore became interested in studying electron-poor ligands in gold-catalysed hydroamination reactions and in elucidating the influence of a redox-switch on the catalyst performance. Based on our previous experiences in redox-switchable/-induced catalysis and our general interest in cooperative effects in multimetallic complexes,21 we targeted the ferrocene-based phosphonite ligand IV featuring both a redox-switchable entity and a Buchwald-type ligand architecture (Chart 1).
Although aryl- and alkyl-substituted 1,3-dioxa-2-phospha-[3]ferrocenophanes have already been described before by Herberhold and co-workers,22 their coordination chemistry or further substitution of the P-bound ligand has not been reported so far. The ligands of interest (1: R = Ter = 2,6-Mes2C6H3; 2: R = Ph;223: R = tBu22) were synthesised according to (or by adapting) the literature protocol from 1,1′-ferrocenediol and RPCl2 and obtained as orange crystals after work-up (ESI†). The ligands were fully characterised, also including the molecular structures of 1 and 3 (Fig. 1). The gold(I) complexes 4–6 were prepared in good yields (65–69%) by treating 1–3 with [Au(tht)Cl] in thf (Scheme 1). The successful coordination of the ligands was verified by 31P NMR spectroscopy. All 31P NMR chemical shifts show the expected upfield shift of ca. Δδ31P = 32–34 ppm upon κ1P-coordination (e.g. δ31P (1) = 183.8 ppm and δ31P (4) = 147.8 ppm). The structures of the complexes were determined by X-ray diffraction of suitable single crystals and confirmed the identities of the products (Fig. 1). All gold complexes display the expected linear coordination and typical Au–P bond lengths of ∼221 pm. The intermetallic distances between the Fe and the Au atoms vary from d(Fe⋯Au) = 418.6(1) pm in 4 to 423.1(1) in 5 and 426.4(1) pm in 6, respectively. As discussed above, low-coordinate gold complexes with Buchwald-type ligands show arene π-interactions.23 Also in our case, short Au⋯Caryl contacts of the gold atoms to one mesityl group of the terphenyl substituent were identified for 4 (d(Au1⋯C115) = 308.0(5) pm and d(Au1–C116) = 313.5(6) pm; ∢(dihedral angle) C100–C105–C115–C116 = 86.42°), which are shorter than typically found for gold(I) complexes of biaryl phosphonites.23 Hence, a stronger stabilising effect during catalysis may be anticipated. To determine the steric demand of the studied [3]ferrocenophanes 1, 2 and 3, their buried volumes (%Vbur)24 were calculated using the crystal structure data of the gold complexes and a fixed d(P–Au) of 228.0 pm. As expected, the terphenyl substituted ligand 1 (50.9%) is relatively bulky and similar to the known JohnPhos ligand (also 50.9%). For 2 and 3, %Vbur values of 34.8% and 33.8%, respectively, were calculated. Although we have also determined the Tolman electronic parameter (TEP) for 1 (2063.0 cm−1) from its iridium(I) complex [(κ1P-1)Ir(CO)2Cl],25 we refrain from a further discussion due to known problems in categorising phosphite-type ligands with TEP values.26
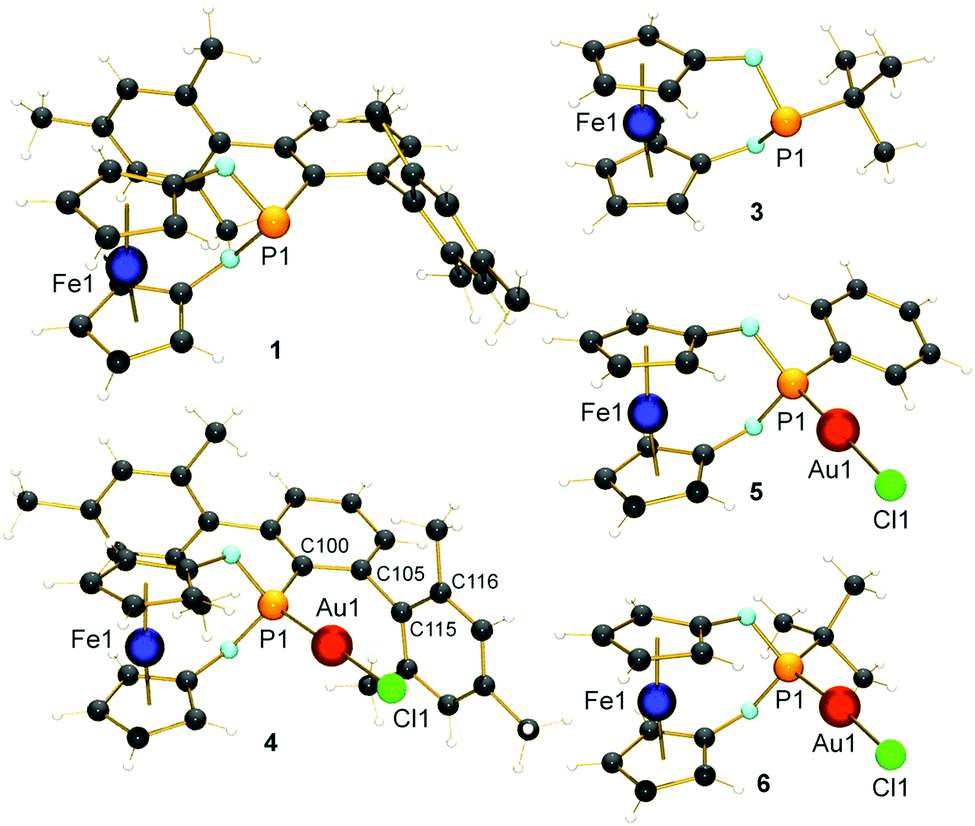 | ||
| Fig. 1 Molecular structures of 1 and 3–6. Selected distances (pm) and angles (°): 1: Fe1⋯P1 337.6(2); 3: Fe1⋯P1 330.8(1); 4: Fe1⋯P1 339.4(1), Fe1⋯Au1 418.6(1), P1–Au1 220.7(1), C100–C105–C115–C116 86.42; 5: Fe1⋯Au1 423.1(1), P1–Au1 220.3(1); and 6: Fe1⋯P1 338.4(2), Fe1⋯Au1 426.4(1), P1–Au1 221.4(1). | ||
 | ||
| Scheme 1 Synthesis of 1–6. | ||
The electrochemical properties of all title compounds (1–6) were probed with the aid of cyclic voltammetry (Fig. S3-1/2 and Table S3-1, ESI†). The E01/2 values for the (quasi)reversible iron-centred oxidations (ΔEp = 0.12–0.21 V; ipc/ipa = 1) depend on the coordinated transition metal fragment. Large anodic shifts of the iron-centred redox events are observed when the pure ligands (e.g.1: E01/2 = 0.03 V) are compared with their gold complex (e.g.4: E01/2 = 0.30 V). In line with our previous studies,11,13 this trend provides indications for a good electronic communication between the metal atoms, although slightly weaker than in the [1]ferrocenophane complexes reported earlier by our group.11,13 Nevertheless, it should be possible to alter the electronic properties of the gold(I) atom by simply changing the oxidation state of the ferrocene unit. The results prompted us to apply the stimulus-responsive metal complexes in redox-induced catalysis.
The application of complexes featuring [n]metallocenophanes as ligands in redox-switchable/-induced catalysis is limited to only some reports.11,13,27 In gold(I) catalysis, redox-responsive carbene complexes have been successfully applied by the groups of Sarkar,28 Peris29a and, more recently, Heinze.29b In order to investigate the applicability of the bimetallic complexes reported herein, we have studied the hydroamination of terminal alkynes using 4–6 and in situ generated 4+–6+30 as catalysts. All reactions were performed in C6D6 at room temperature with NaBArF as a chloride scavenger and were monitored by 1H NMR spectroscopy (referenced to an internal standard).
The Au(I) catalysts 4–6 were evaluated in the hydroamination of terminal and internal alkynes using primary amines as nucleophiles.31 First, we investigated the addition of aniline (7a) to phenylacetylene (8a) in the presence of 0.1 mol% gold catalyst in either the reduced or oxidized form (see Fig. 2a–c). The tBu-substituted complex 6 displayed medium activity in the hydroamination, nevertheless significant efficiency differences between the reduced and the oxidized form are apparent. The corresponding phenyl-derivative 5 was inferior to 6 and the reaction did not yield higher conversion than 50% (Fig. 2b), most probably a result of catalyst decomposition. Again, the oxidized form displayed higher activity than the reduced form. As a third system, we tested the terphenyl-substituted derivative 4 (Fig. 2c). This system displayed remarkable activity in the reduced and oxidized forms, giving 80% and 88% conversion, respectively, after 16 h at room temperature (25 °C). Notably, the reaction profiles of these catalytic reactions did not display catalyst degradation, as indicated by the second order rate plot up to 88% conversion (see Fig. S5-1, ESI†). The short Au-Mes contact, identified in the solid-state structure, has a stabilizing effect on the catalytically active cationic gold complex, which explains the poor performance of 5 and 6 in the hydroamination.
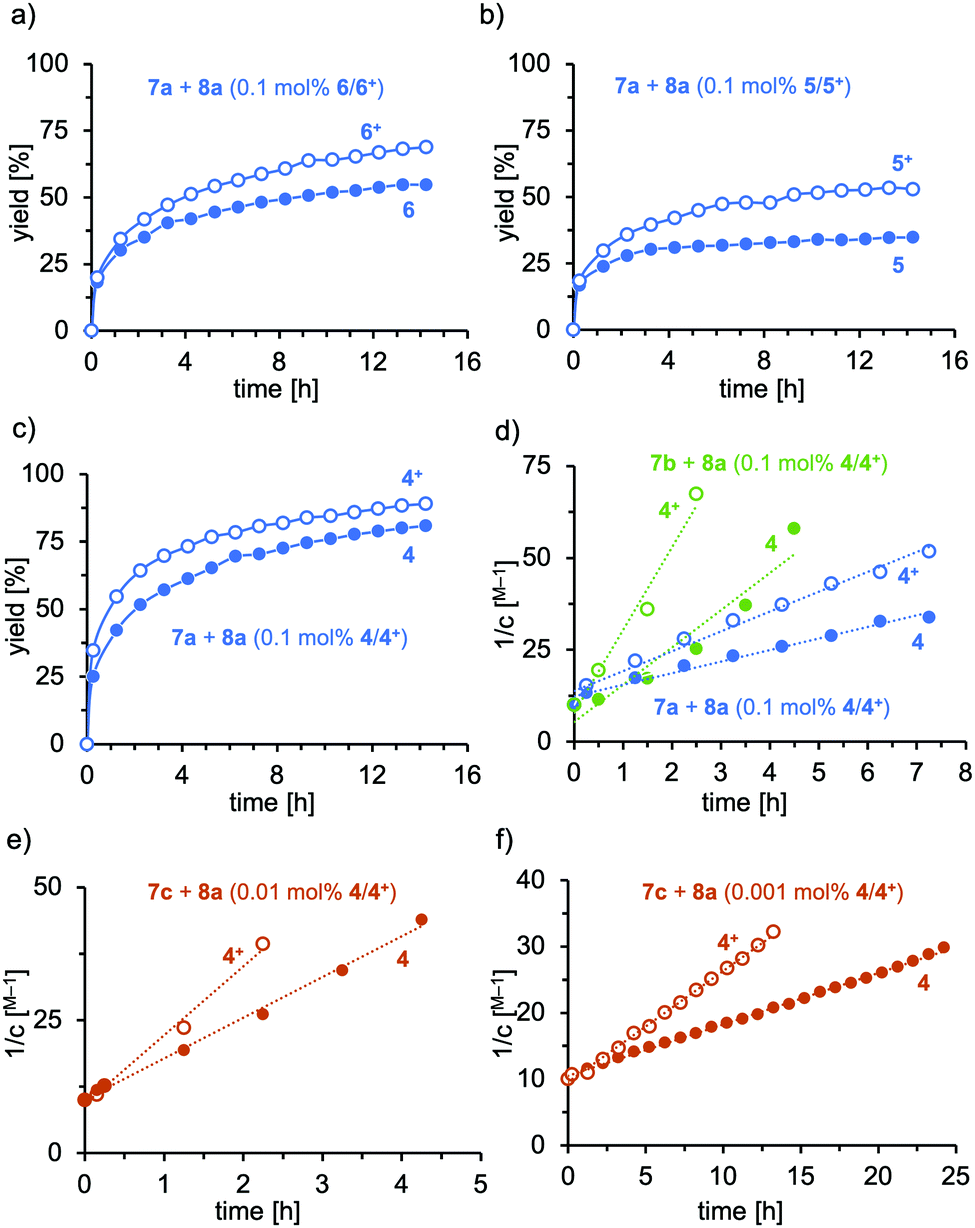 | ||
| Fig. 2 (a–c) Reaction profiles of the gold-catalyzed addition of aniline (7a, 1.1 equiv.) to phenylacetylene (8a, 1.0 equiv.) using 0.1 mol% of (a) 6/6+, (b) 5/5+ and (c) 4/4+ at room temperature (25 °C) in C6D6; (d) kinetic analysis and second order rate constants of the gold-catalyzed hydroamination (0.1 mol%) of phenylacetylene (8a) with Ph-NH2 (7a, blue): kred-7a = 3.2 M−1 h−1; kox-7a = 5.4 M−1 h−1 and Dipp-NH2 (7b, green): kred-7b = 10.2 M−1 h−1; kox-7a = 22.5 M−1 h−1; solid circles: 4, open circles: 4+; (e) hydroamination with Mes-NH2 (7c); 0.01 mol% 4 (kred-7c = 7.7 M−1 h−1) and 4+ (kox-7c = 12.9 M−1 h−1); (f) hydroamination with Mes-NH2 (7c), 0.001 mol% 4 (kred-7c = 0.8 M−1 h−1) and 4+ (kox-7c = 1.7 M−1 h−1). Control reactions without any Au complex but a pure oxidation reagent gave zero hydroamination. | ||
Also, the more sterically encumbered 2,6-diisopropylphenyl-amine (Dipp-NH2, 7b, green) was active in the gold-catalyzed hydroamination. Kinetic analysis of the Ph-NH2 and Dipp-NH2 addition to the alkyne 2a displayed second order behavior, suggesting the nucleophilic attack of the amine as the rate-determining step (see Fig. 2d). The hydroamination proceeds roughly two-fold faster with the oxidized catalyst 4+ than with its reduced form (4) for both amines (kox-7a/kred-7a = 1.7; kox-7b/kred-7b = 2.2). However, the hydroamination of 8a proceeds significantly faster with Dipp-NH2 (7b, green) than with aniline (7a, blue), probably as a result of the increased nucleophilicity. This picture is supported by the substantial rate increase of the hydroamination when Mes-NH2 (7c, red) was used as a nucleophile. In fact, the reaction became too fast to obtain reliable kinetic data and the catalyst loading needed to be reduced to 0.01 mol% and 0.001 mol% (Fig. 2e and f). The catalyst shows high stability under both conditions up to high conversions. Also for this reaction, app. two-fold rate increase was observed (0.01 mol% 4: kox-7c/kred-7c = 1.7; 0.001 mol% 4: kox-7b/kred-7b = 2.1). The turnover frequencies for this catalytic reaction at room temperature are 784 mmol s−1 and 1857 mmol s−1 for 4 and 4+, respectively. Thereby, impressive turnover numbers (TON) of 66.000 (4) and 82.000 (4+) were obtained in ca. 24 h at room temperature. It is important to note that only a few catalysts exist that allow for TON higher than 5.000 for the hydroamination of terminal alkynes. Lavallo and co-workers reported gold(I) complexes of a very bulky phosphine and obtained after 24 h a similar TON value of 67.000 at 10 ppm catalyst loading for the hydroamination of PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH (8a) with Mes-NH2 (7c), albeit at 50 °C.32 In our case, the reactions are performed at room temperature; furthermore, the TON can significantly be increased by in situ oxidation of catalyst 4.
CH (8a) with Mes-NH2 (7c), albeit at 50 °C.32 In our case, the reactions are performed at room temperature; furthermore, the TON can significantly be increased by in situ oxidation of catalyst 4.
These kinetic data strongly support the nucleophilic addition of the amine to the activated triple bond as the rate-determining step in these phosphinate ester-derived Au catalysts in contrast to the unimolecular protodeauration of electron-rich phosphine-derived Au catalysts.
Furthermore, the catalyst system 4 was employed in the hydroamination of electron-rich, electron-deficient and aliphatic alkynes (Scheme 2). The electronic nature of the alkyne has a significant impact on the efficiency. Electron-donating groups (compare 9c and 9g) slowed the reaction down, which supports the notion of the nucleophilic attack being the rate-determining step. Nevertheless, all reactions went to full conversion within 4–9 hours. However, when a strongly electron-deficient alkyne is applied, the reaction becomes unproductive. The fluorinated imine 9d is only formed in trace amounts (7%) as a result of inefficient alkyne activation by the catalyst, irrespective of the oxidation state of the iron. Aliphatic alkynes were only reluctantly hydroaminated within 24 h in medium yields. The hydroamination reactions were generally faster or gave higher yields when 4+ was applied, which gives rise to externally stimulated catalyst modification. Overall, we observed high activities and impressively high TON by employing a redox-active gold(I) complex featuring an electron-deficient phosphonite-based ligand with Buchwald-type architecture in the redox-induced hydroamination catalysis of alkynes. Very low catalyst loadings (down to 10 ppm) and very mild conditions (room temperature) were used. Second order rate kinetics were observed for the addition to phenyl acetylene, suggesting the nucleophilic attack of the amine as the rate-determining step for this catalyst system. In these studies, the hydroamination with the three amines 7a–c proceeds 1.7 to 2.1 times faster with the in situ oxidized catalysts than with their reduced form.
 | ||
| Scheme 2 Hydroamination of alkynes. Yields are given for 4 (upper value) and 4+ (lower value); numbers in parentheses correspond to reaction times in [h]. Reactions were conducted on a 0.1 mmol scale in d6-benzene with 0.1 mol% 4 and 0.1 mol% NaBArF as activating agents. 4+ was generated in situ prior to the reaction by addition of 0.1 mol% [Fc(OAc)][Al{OC(CF3)3}4]. The yields were determined using 1,3,5-(MeO)3C6H3 as an internal standard; areactions were conducted using a 1 mol% catalyst/activator. | ||
We acknowledge support from the DFG-funded transregional collaborative research centre SFB/TRR 88 “Cooperative effects in homo- and heterometallic complexes (3MET)” (project B4).
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. Hartwig, Organotransition Metal Chemistry, From bonding to catalysis, University Science Books, Sausalito, California, 2010 Search PubMed.
- M. A. Wünsche, P. Mehlmann, T. Witteler, F. Buß, P. Rathmann and F. Dielmann, Angew. Chem., Int. Ed., 2015, 54, 11857 CrossRef PubMed.
- T. Scherpf, C. Schwarz, L. T. Scharf, J.-A. Zur, A. Helbig and V. H. Gessner, Angew. Chem., Int. Ed., 2018, 57, 12859 CrossRef CAS PubMed.
- L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392 CrossRef CAS PubMed.
- (a) O. René and K. Fagnou, Adv. Synth. Catal., 2010, 352, 2116 CrossRef; (b) P. H. Lee, S. Kim, A. Park, B. C. Chary and S. Kim, Angew. Chem., Int. Ed., 2010, 49, 6806 CrossRef CAS PubMed; (c) T. Korenaga, A. Ko, K. Uotani, Y. Tanaka and T. Sakai, Angew. Chem., Int. Ed., 2011, 50, 10703 CrossRef CAS PubMed.
- (a) D. S. Surry and S. L. Buchwald, Angew. Chem., Int. Ed., 2008, 47, 6338 CrossRef CAS PubMed; (b) P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564 CrossRef CAS PubMed; (c) O. Diebolt, G. C. Fortman, H. Clavier, A. M. Z. Slawin, E. C. Escudero-Adan, J. Benet-Buchholz and S. P. Nolan, Organometallics, 2011, 30, 1668 CrossRef CAS.
- Y. Wang, Z. Wang, Y. Li, G. Wu, Z. Cao and L. Zhang, Nat. Commun., 2014, 5, 3470 CrossRef PubMed.
- X. Qiu, M. Wang, Y. Zhao and Z. Shi, Angew. Chem., Int. Ed., 2017, 56, 7233 CrossRef CAS PubMed.
- L. Noel-Duchesneau, N. Lugan, G. Lavigne, A. Labande and V. Ceśar, Organometallics, 2014, 33, 5085 CrossRef CAS.
- H. Tinnermann, C. Wille and M. Alcarazo, Angew. Chem., Int. Ed., 2014, 53, 8732 CrossRef CAS PubMed.
- A. Feyrer and F. Breher, Inorg. Chem. Front., 2017, 4, 1125 RSC.
- Selected examples: (a) I. M. Lorkovic, R. R. Duff and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 3617 CrossRef CAS; (b) C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410 CrossRef CAS PubMed; (c) X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264 CrossRef CAS PubMed.
- (a) A. Feyrer, M. K. Armbruster, K. Fink and F. Breher, Chem. – Eur. J., 2017, 23, 7402 CrossRef CAS PubMed; (b) F. Walz, E. Moos, D. Garnier, R. Köppe, C. E. Anson and F. Breher, Chem. – Eur. J., 2017, 23, 1173 CrossRef CAS PubMed.
- (a) A. M. Allgeier and C. A. Mirkin, Angew. Chem., Int. Ed., 1998, 37, 894 CrossRef; (b) V. Blanco, D. A. Leigh and V. Marcos, Chem. Soc. Rev., 2015, 44, 5341 RSC; (c) O. R. Luca and C. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440 RSC.
- Selected recent reviews: (a) A. M. Asiri and A. S. K. Hashmi, Chem. Soc. Rev., 2016, 45, 4471 RSC; (b) A. Fürstner, Angew. Chem., Int. Ed., 2018, 57, 4215 CrossRef PubMed; (c) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028 CrossRef CAS PubMed; (d) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2010, 49, 5232 CrossRef CAS PubMed.
- (a) L. Huang, M. Arndt, K. Gooßen, H. Heydt and L. J. Gooßen, Chem. Rev., 2015, 115, 2596 CrossRef CAS PubMed; (b) R. A. Widenhoefer and X. Han, Eur. J. Org. Chem., 2006, 4555 CrossRef CAS.
- W. Wang, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2012, 134, 5697 CrossRef CAS PubMed.
- D. Malhotra, M. S. Mashuta, G. B. Hammond and B. Xu, Angew. Chem., Int. Ed., 2014, 53, 4456 CrossRef CAS PubMed.
- C. A. Gaggioli, G. Ciancaleoni, D. Zuccaccia, G. Bistoni, L. Belpassi, F. Tarantelli and P. Belanzoni, Organometallics, 2016, 35, 2275 CrossRef CAS.
- (a) T. Witteler, H. Darmandeh, P. Mehlmann and F. Dielmann, Organometallics, 2018, 37, 3064 CrossRef CAS; (b) Footnote 33 of ref. 3.
- (a) S. González-Gallardo, I. Kuzu, P. Oña-Burgos, T. Wolfer, C. Wang, K. W. Klinkhammer, W. Klopper, S. Bräse and F. Breher, Organometallics, 2014, 33, 941 CrossRef; (b) J. M. Serrano-Becerra, A. F. G. Maier, S. González-Gallardo, E. Moos, C. Kaub, M. Gaffga, G. Niedner-Schatteburg, P. W. Roesky, F. Breher and J. Paradies, Eur. J. Org. Chem., 2014, 4515 CrossRef CAS.
- (a) M. Herberhold and H.-D. Brendel, J. Organomet. Chem., 1993, 458, 205 CrossRef CAS; (b) M. Herberhold, A. Hofmann and W. Milius, J. Organomet. Chem., 1998, 555, 187 CrossRef CAS.
- (a) P. Pérez-Galán, N. Delpont, E. Herrero-Gómez, F. Maseras and A. M. Echavarren, Chem. – Eur. J., 2010, 16, 5324 CrossRef PubMed; (b) A. S. K. Hashmi, B. Bechem, A. Loos, M. Hamzic, F. Rominger and H. Rabaa, Aust. J. Chem., 2014, 67, 481 CrossRef CAS; (c) J. T. Fleming, P. G. Waddell, M. R. Probert, W. Clegg and L. J. Higham, Eur. J. Inorg. Chem., 2017, 2837 CrossRef CAS.
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286 CrossRef CAS.
- E. Deck and F. Breher, unpublished results.
- A. R. Chianese, X. Li, M. C. Janzen, J. W. Faller and R. H. Crabtree, Organometallics, 2003, 22, 1663 CrossRef CAS.
- C. D. Varnado, Jr., E. L. Rosen, M. S. Collins, V. M. Lynch and C. W. Bielawski, Dalton Trans., 2013, 42, 13251 RSC.
- (a) L. Hettmanczyk, S. Manck, C. Hoyer, S. Hohloch and B. Sarkar, Chem. Commun., 2015, 51, 10949 RSC; (b) L. Hettmanczyk, L. Suntrup, S. Klenk, C. Hoyer and B. Sarkar, Chem. – Eur. J., 2017, 23, 576 CrossRef CAS PubMed; (c) S. Klenk, S. Rupf, L. Suntrup, M. van der Meer and B. Sarkar, Organometallics, 2017, 36, 2026 CrossRef CAS.
- (a) S. Ibáñez, M. Poyatos, L. N. Dawe, D. Gusev and E. Peris, Organometallics, 2016, 35, 2747 CrossRef; (b) P. Veit, C. Volkert, C. Förster, V. Ksenofontov, S. Schlicher, M. Bauer and K. Heinze, Chem. Commun., 2019 10.1039/c9cc00283a.
- All attempts to prepare the cationic complexes, e.g.4+, on a preparative scale have not yet been successful.
- E. Mizushima, T. Hayashi and M. Tanaka, Org. Lett., 2003, 5, 3349 CrossRef CAS PubMed.
- V. Lavallo, J. H. Wright, F. S. Tham and S. Quinlivan, Angew. Chem., Int. Ed., 2013, 52, 3172 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR data, cyclic voltammetry data, and X-ray crystal structure data. CCDC 1898143–1898147. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9cc01492f |
| This journal is © The Royal Society of Chemistry 2019 |