
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReversible ON–OFF switching of single-molecule-magnetism associated with single-crystal-to-single-crystal structural transformation of a decanuclear dysprosium phosphonate†
Haiquan
Tian
ad,
Jing-Bu
Su
b,
Song-Song
Bao
a,
Mohamedally
Kurmoo
c,
Xin-Da
Huang
a,
Yi-Quan
Zhang
 *b and
Li-Min
Zheng
*a
*b and
Li-Min
Zheng
*a
aState Key Laboratory of Coordination Chemistry, Coordination Chemistry Institute, School of Chemistry and Chemical Engineering, Collaborative Innovation Center of Advanced Microstructures, Nanjing University, Nanjing 210023, P. R. China. E-mail: lmzheng@nju.edu.cn
bJiangsu Key Laboratory for NSLSCS, School of Physical Science and Technology, Nanjing Normal University, Nanjing 210023, P. R. China. E-mail: zhangyiquan@njnu.edu.cn
cUniversite de Strasbourg, Institut de Chimie, CNRS-UMR7177, 4 rue Blaise Pascal, Strasbourg Cedex 67007, France
dShandong Provincial Key Laboratory of Chemical Energy Storage and Novel Cell Technology, School of Chemistry and Chemical Engineering, Liaocheng University, Liaocheng 252000, P. R. China
First published on 2nd July 2018
Abstract
{Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)(μ4-C15H11PO3)(μ2-AcO)2(AcO)2(H2O)(CH3OH)2}2(μ4-C2O4)·xH2O (I), where H2EDDC is N′,N′′,E,N′,N′′,E-N′,N′′-(ethane-1,2-diylidene)dipyrazine-2-carbohydrazide and C15H11PO3H2 is 9-anthrylmethylphosphonic acid, is found to undergo two consecutive single-crystal-to-single-crystal transformations. The first is under UV irradiation (λ = 365 nm for 3 d in air) to {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)2(AcO)2(H2O)3}2(μ4-C2O4)·xH2O (I-UV) where the two CH3OH are replaced by two H2O and the second by annealing under N2 at 100 °C on a diffractometer or under Ar in a glovebox to {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)4(H2O)}2(μ4-C2O4) (I-A-N2 or I-A-Ar) where it has lost two H2O molecules. The second transformation is reversible by exposure to air at room temperature (I-A-N2-cool). While the overall structures are the same (retaining the space group P21/c), there is a considerable expansion of the unit cell from I (8171 Å3) to I-UV (8609 Å3) and I-A-N2 (8610 Å3) and the coordinations of the Dy atoms undergo major reconstructions. This is associated with switching the single-molecule-magnetism (SMM) from OFF for I to ON for I-UV and to OFF again for I-A-Ar in air. Such a switching mechanism associated with the retention of crystallinity is unique in the chemistry of dysprosium. The structure of the molecule is formed from two symmetry related pentamers joined by an oxalate. A related compound containing two isolated neutral pentamers {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)3(AcO)2(H2O)2}{Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)(μ4-C15H11PO3)(μ2-AcO)3(AcO)2(CH3OH)2}·2CHCl3 (II) has also been isolated with closely related Dy coordination and it exhibits similar SMM behaviour in zero field.
Introduction
Molecular compounds that can have their physical properties switched from one state to another reversibly and retain their crystallinity are quite rare.1 And clusters that can switch their single-molecule-magnetism (SMM) ON and OFF are even fewer. In general, numerous compounds are known where the parameters defining their SMM are altered but are not switched ON and OFF reversibly. Since the discovery of SMM in the early nineties,2 there is a general wish to use these SMMs as ON–OFF switches which will endow them with a bi-stable magnetic ground state that has been coined as a great attribute in applications for information storage and quantum computing.3–5 Consequently, there is major interest in realising these properties by design using both d- and f-elements.6–8 Lanthanide complexes are becoming more promising candidates for SMMs because of their significant single-ion magnetic anisotropy arising from the large unquenched orbital angular momentum and strong spin–orbit coupling, and thus may lead to high energy barriers for spin reversal.9–12 Additionally, the very weak intermolecular interaction compared to those made up of d-elements makes them very appealing for single-molecule magnetism. However, the magnetization dynamics of lanthanide based SMMs (Ln-SMMs) is still far from being fully understood, because they are influenced by different factors such as the symmetry and the charge distribution of the coordination sphere around the lanthanide ions, the hyperfine couplings, dipolar spin–spin interactions, etc.9–13 For polynuclear clusters the magnetic behaviour can be further complicated by the different moment vector directions.While great efforts have been devoted to the design and syntheses of new Ln-SMMs with high energy barriers and blocking temperatures,14–18 the manipulation of the magnetic properties of Ln-SMMs through external stimuli has rarely been explored. Such manipulations can be realized by imposing chemical influences such as ligand redox19–21 and pH control with a base or acid.22 Guest solvent exchange was also found to induce a reversible structural transformation but only tunable magnetization relaxation.23,24 Reversible switching of SMM behaviors can also be achieved by using physical stimuli. Light-induced switching of magnetic relaxation has been realized in Dy compounds containing optically active diarylethene25 or 1,2-bis(4-pyridyl)ethane linkages.26 Thermal treatment can remove lattice or coordinated solvent molecules, switching the magnetic behavior of Ln-SMMs.27,28 But only in one case, as far as we are aware, ON–OFF SMM switching coupled with reversible single-crystal-to-single-crystal (SC–SC) structural transformation was achieved for Ln-SMMs.29
Cyclic lanthanide rings belong to a specific subclass of Ln-based clusters. These rings not only have structural aesthetics but also are magnetically interesting. In particular, the observation of a toroidal arrangement of local magnetization vectors in a Dy3 triangular cluster30,31 stimulates the enthusiasm for the exploration of new cyclic Ln-based clusters.32 Unfortunately, there are a very few examples of cyclic lanthanide rings reported so far, and most contain even-numbered lanthanide ions.32–37 The syntheses of odd-numbered lanthanide rings larger than three have therefore been a great challenge.38 Recently we developed a new synthetic strategy by combining pre-designed trimeric and dimeric building blocks and a phosphonate ligand as an adhesive co-ligand.39 The present cyclic heptanuclear Dy7 clusters were successfully obtained, which turn out to be the first with seven Dy on the ring and none inside.
In this paper, we report the syntheses, structures and magnetic properties of a novel series containing oxalate bridged pentamers, {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)(μ4-C15H11PO3)(μ2-AcO)2(AcO)2(H2O)(CH3OH)2}2(μ4-C2O4)·xH2O (I) and its SC–SC transformations to two other forms, {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)2(AcO)2(H2O)3}2(μ4-C2O4)·xH2O (I-UV) and {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)4(H2O)}2(μ4-C2O4) (I-A-N2), where H2EDDC is N′,N′′,E,N′,N′′,E-N′,N′′-(ethane-1,2-diylidene)dipyrazine-2-carbohydrazide, and C15H11PO3H2 is 9-anthrylmethylphosphonic acid (Scheme 1), consisting of a fused pair of a cyclic pentamer by oxalate. I undergoes two consecutive SC–SC transformations accompanied by switching ON and OFF of the single-molecule-magnetism reversibly. Another compound consisting of two independent structurally related pentamers, {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)3(AcO)2(H2O)2}{Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)(μ4-C15H11PO3)(μ2-AcO)3(AcO)2(CH3OH)2}·2CHCl3 (II), has also been isolated and characterised. It displays similar SMM behaviour. These phases are the first examples of odd-numbered rings with cyclic Ln-pentamers displaying the unique ON–OFF switching of SMM properties.
 | ||
| Scheme 1 Molecular structures of N′,N′′,E,N′,N′′,E-N′,N′′-(ethane-1,2-diylidene)dipyrazine-2-carbohydrazide (H2EDDC, left) and 9-anthrylmethylphosphonic acid (C15H11PO3H2, right). | ||
Results and discussion
Structures of I and II
I was synthesized as dark red crystals from a water–methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) mixture containing H2EDDC neutralised with triethylamine, Dy(OAc)3·4H2O, sodium oxalate and C15H11PO3H2. As shown in Scheme 2, the crystals undergo two SC–SC transformations, the first under UV irradiation to form I-UV and the second by annealing at 100 °C under N2 on a diffractometer (or under Ar in a glovebox) to form I-A-N2 (or I-A-Ar). Upon exposure to air, I-A-N2 (or I-A-Ar) transforms into I-A-N2-cool (or I-A-Ar-cool). Therefore, we have determined the structure in each state (I, I-UV, I-A-N2 and I-A-N2-cool). A summary of the crystallographic data is given in Tables 1 and S1.† We therefore describe the five structures with enough details for a thorough understanding. Further details can be seen in the ESI (Tables S2 and S3†) and the crystallographic information files. We note that there are three different molecular contents for the five structures where those of I-UV, I-A-N2-cool and I-A-Ar-cool are the same. Therefore, only the structure of I-UV is described.
:3) mixture containing H2EDDC neutralised with triethylamine, Dy(OAc)3·4H2O, sodium oxalate and C15H11PO3H2. As shown in Scheme 2, the crystals undergo two SC–SC transformations, the first under UV irradiation to form I-UV and the second by annealing at 100 °C under N2 on a diffractometer (or under Ar in a glovebox) to form I-A-N2 (or I-A-Ar). Upon exposure to air, I-A-N2 (or I-A-Ar) transforms into I-A-N2-cool (or I-A-Ar-cool). Therefore, we have determined the structure in each state (I, I-UV, I-A-N2 and I-A-N2-cool). A summary of the crystallographic data is given in Tables 1 and S1.† We therefore describe the five structures with enough details for a thorough understanding. Further details can be seen in the ESI (Tables S2 and S3†) and the crystallographic information files. We note that there are three different molecular contents for the five structures where those of I-UV, I-A-N2-cool and I-A-Ar-cool are the same. Therefore, only the structure of I-UV is described.
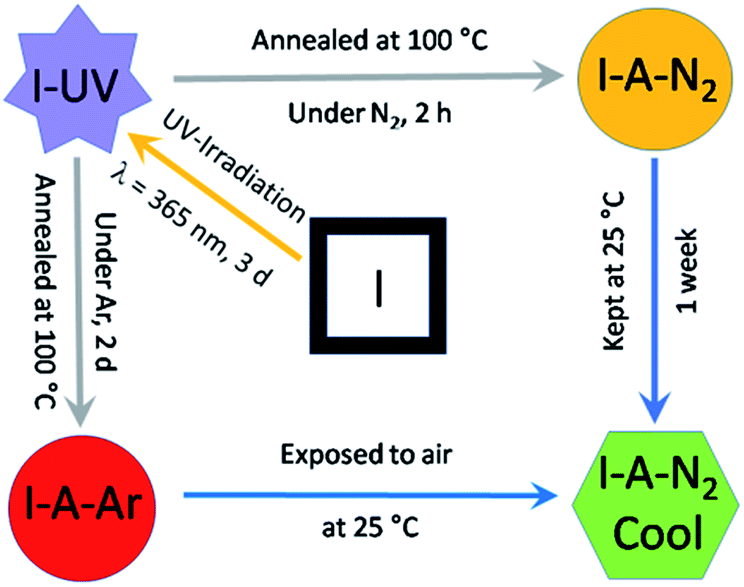 | ||
| Scheme 2 Protocol used for the transformations of I. | ||
| Compound | I | I-UV | I-A-N2 | I-A-N2-cool | II |
|---|---|---|---|---|---|
| a R 1 = Σ‖Fo| − |Fc‖/Σ|Fo|. b wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2. | |||||
| Formula | C138H132Dy10N32O54P4 | C134H148Dy10N32O66P4 | C134H116Dy10N32O50P4 | C134H148Dy10N32O66P4 | C140H132Dy10N32O52P4Cl6 |
| M r | 4851.63 | 5011.71 | 4719.43 | 5011.71 | 5056.35 |
| Cryst size [mm3] | 0.05 × 0.15 × 0.15 | 0.08 × 0.2 × 0.3 | 0.2 × 0.2 × 0.3 | 0.2 × 0.2 × 0.3 | 0.3 × 0.4 × 0.4 |
| T [K] | 123(2) | 123(2) | 373(2) | 123(2) | 123(2) |
| Crystal system | Monoclinic | Monoclinic | Monoclinic | Monoclinic | Trigonal |
| Space group | P21/c #14 | P21/c #14 | P21/c #14 | P21/c #14 | R32 #155 |
| a [Å] | 24.731(5) | 24.653(3) | 25.525(6) | 24.536(3) | 26.7135(7) |
| b [Å] | 14.057(3) | 14.565(2) | 14.312(4) | 14.4433(18) | 26.7135(7) |
| c [Å] | 25.496(5) | 26.067(3) | 25.318(6) | 26.078(3) | 71.325(4) |
| α [°] | 90 | 90 | 90 | 90 | 90 |
| β [°] | 112.797(4) | 112.969(3) | 111.413(5) | 112.936(2) | 90 |
| γ [°] | 90 | 90 | 90 | 90 | 120 |
| V [Å3] | 8171(3) | 8617(2) | 8611(4) | 8510.9(18) | 44079(4) |
| Z | 2 | 2 | 2 | 2 | 9 |
| ρ [g cm−3] | 1.972 | 1.931 | 1.822 | 1.956 | 1.714 |
| 2ϑ [deg] | 2.2–26.0 | 2.0–26.0 | 2.0–26.0 | 2.0–26.0 | 2.2–27.6 |
| F(000) | 4672 | 4848 | 4528 | 4848 | 21906 |
| Reflns collected | 45959 |
55815 |
52072 |
56468 |
127761 |
| Unique reflns | 15879 |
16870 |
16839 |
16708 |
22630 |
| R int | 0.059 | 0.082 | 0.149 | 0.168 | 0.037 |
| GOF | 1.090 | 1.004 | 1.040 | 1.060 | 0.987 |
| R1 [I > 2σ(I)]a | 0.0935 | 0.1021 | 0.1482 | 0.1148 | 0.0617 |
| wR2 (all data)b | 0.2675 | 0.2725 | 0.3499 | 0.2714 | 0.1459 |
| (Δρ)max, (Δρ)min/[e Å−3] | 3.10, −3.02 | 3.82, −2.61 | 3.43, −1.90 | 2.22, −2.15 | 4.54, −2.28 |
| CCDC number | 1819929 | 1819930 | 1819931 | — | 1819932 |
The crystals adopt the monoclinic space group P21/c (Z = 2) in all the forms. The key feature of the structures is two symmetry-related cyclic [Dy5] units, looking like a distorted star, fused by a bis-chelating oxalate ligand (Fig. 1 and 2). Each Dy5 unit contains a common fragment consisting of five independent Dy atoms, two EDDC2− and two μ3-acetate (shown in grey in Fig. 2). The oxygen atoms of one EDDC2− ligand form two bridges between three Dy atoms (Dy1, Dy2 and Dy3) in a pseudo-linear array. It also provides five chelate units (4 N–O and 1 N–N) to hold the three Dy atoms. The other EDDC2− is hinged at the apical Dy atom (Dy1) and hold two other Dy atoms (Dy4 and Dy5) by the chelating N–N and N–O. The two μ3-acetate groups, adopting a syn–syn–anti mode, bridge Dy3 and Dy4 into a rigid pair and form an irregular star-shaped ring. The structure of this base unit is common to all the clusters of the two compounds.
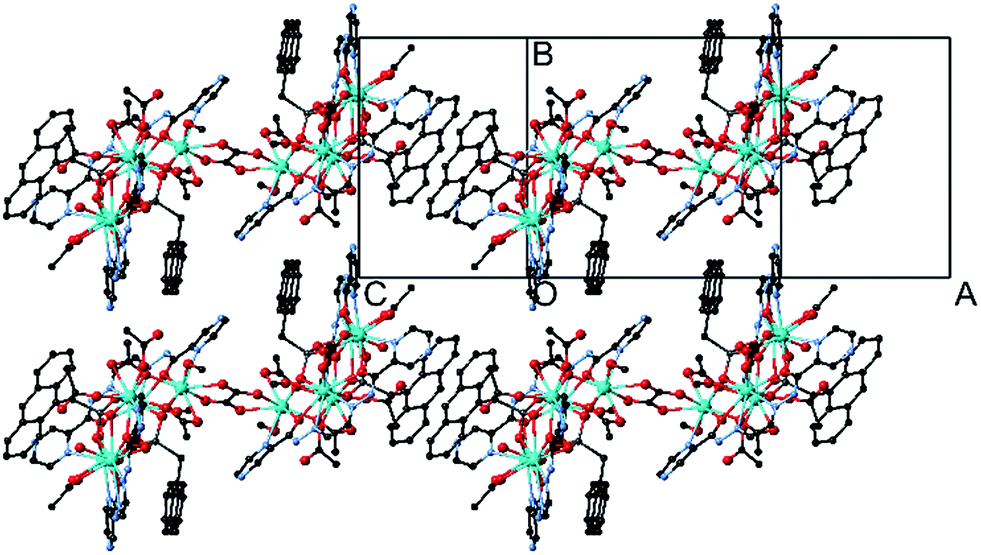 | ||
| Fig. 1 Structure of I showing two parallel chains connected by the supramolecular π–π overlap of the anthracene moieties of adjacent molecules (blue circle highlight) while the other anthracene is face-to-face with the EDDC2− ligand (red circle). | ||
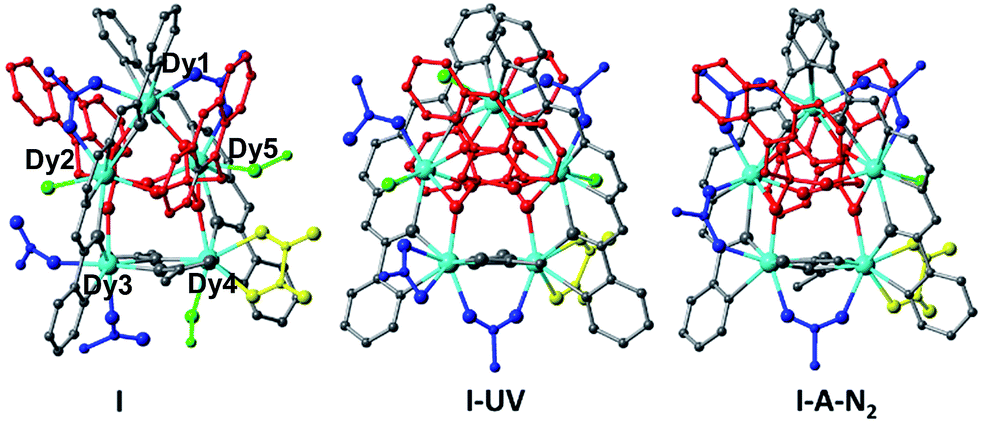 | ||
| Fig. 2 The pentameric units in the structures of I, I-UV and I-A-N2. The common fragments (2 EDDC2− and 2 acetate) are shown in grey, Dy in cyan, other acetate in blue, C15H11PO3 in red, oxalate in yellow, and H2O and CH3OH solvents in green. | ||
II was synthesized as red hexagonal crystals by reacting H2EDDC, Dy(OAc)3·4H2O, triethylamine and C15H11PO3H2 in methanol/chloroform solution at room temperature. It crystallizes in the trigonal chiral space group R32 with Z = 9. The structure contains two kinds of cyclic pentanuclear rings of different compositions {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)3(AcO)2(H2O)2} (II-molecule A) and {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)(μ4-C15H11PO3)(μ2-AcO)4(AcO)2(CH3OH)2} (II-molecule B). It also contains the common basic building block, 5DyIII, 2EDDC2−, 2C15H11PO32−, and 2 acetate forming the star ring, in both independent molecules (Fig. 3 and Table S2†).
 | ||
| Fig. 3 The structures of the two pentamers in II. The colour coding is as used in Fig. 1. | ||
The remaining ligands adopt different coordination modes in all the five different pentamers of compounds I and II. Table 2 lists the modes of coordination of each ligand and the total coordination bonds per pentamer. We find that there are additionally a μ2-bridge (syn–syn mode), chelate and terminal acetate (shown in blue). The two phosphonates (shown in red) per pentamers can be only μ5- or mixed with μ4-C15H11PO32− as in I and II-molecule B. They sit on each face of the pentamers with a P⋯P distance of ca. 3.7 Å. Methanol or water molecules (shown in green) are also present in prepared I which contains two coordinated methanol and one water molecules. When it is exposed to UV light it is transformed to I-UV where the methanol molecules are replaced by two water molecules, presumably from the atmosphere. Upon heating I-UV at 100 °C under a flow of N2 on a diffractometer it loses two of the three molecules of water to form I-A-N2. During the two transformations major reconstructions have taken place as can be seen from Table 2 and in the ESI.† After the subsequent exposure of I-A-N2 in air, it reabsorbs two water molecules to I-A-N2-cool which has almost the same molecular content and structure as I-UV (Tables 1 and S2†). When I-UV is heated at 100 °C under argon in a glovebox it loses its solvents forming I-A-Ar. Upon subsequent exposure to air it quickly absorbs water to give the same structure as I-A-N2-cool and I-UV, confirmed by the crystal structure determination (Tables S1 and S3†).
| I | I-UV | I-A-N2 | II-molecule A | II-molecule B | |
|---|---|---|---|---|---|
| Formula | {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)(μ4-C15H11PO3)(μ2-AcO)2(AcO)2(H2O)(CH3OH)2}2(μ4-C2O4) | {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)2(c-AcO)(t-AcO)(H2O)3}2(μ4-C2O4) | {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)4(H2O)}2(μ4-C2O4) | {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)2(μ2-AcO)3(c-AcO)2(H2O)2} | {Dy5(EDDC)2(μ3-AcO)2(μ5-C15H11PO3)(μ4-C15H11PO3)(μ2-AcO)3(c-AcO)2(CH3OH)2} |
| Acetate | 2 μ3syn–syn–anti (Dy3–Dy4), 2 μ2syn–syn (Dy1–Dy2, Dy1–Dy5), 2 terminal (Dy3) | 2 μ3syn–syn–anti (Dy3–Dy4), 2 μ2syn–syn (Dy1–Dy5, Dy3–Dy4), 1 chelate (Dy3), 1 terminal (Dy2) | 2 μ3syn–syn–anti (Dy3–Dy4), 4 μ2syn–syn (Dy1–Dy2, Dy1–Dy5, Dy2–Dy3, Dy3–Dy4) | 2 μ3syn–syn–anti (Dy4–Dy5), 3 μ2syn–syn (Dy2–Dy3, Dy2–Dy1, Dy4–Dy5), 2 chelate (Dy4, Dy5) | 2 μ3syn–syn–anti (Dy6–Dy7), 3 μ2syn–syn (Dy8–Dy9, Dy9–Dy10, Dy6–Dy7), 2 chelate (Dy6, Dy7) |
| Oxalate | Chelate (Dy4) | Chelate (Dy4) | Chelate (Dy4) | — | — |
| H2O | 1 terminal (Dy2) | 3 terminal (Dy1, Dy2, Dy5) | 1 terminal (Dy5) | 2 terminal (Dy3, Dy5) | 2 terminal (Dy8, Dy10) |
| CH3OH | 2 terminal (Dy4, Dy5) | None | None | None | None |
| PO3 | P1 – 2 μ2 (Dy1–Dy2, Dy1–Dy5), 1 terminal (Dy4), P2 – 1 μ2 (Dy1–Dy2), 2 terminal (Dy4, Dy5) | P1 – 2 μ2 (Dy1–Dy5, Dy4–Dy5), 1 terminal (Dy2), P2 – 2 μ2 (Dy1–Dy2, Dy2–Dy3), 1 terminal (Dy5) | P1 – 2 μ2 (Dy1–Dy5, Dy4–Dy5), 1 terminal (Dy2), P2 – 2 μ2 (Dy1–Dy2, Dy2–Dy3), 1 terminal (Dy5) | P1 – 2 μ2 (Dy2–Dy1, Dy5–Dy1), 1 terminal (Dy3), P2 – 2 μ2 (Dy2–Dy3, Dy3–Dy4), 1 terminal (Dy1) | P3 – 2 μ2 (Dy8–Dy9, Dy7–Dy8), 1 terminal (Dy10), P4 – 1 μ2 (Dy9–Dy10), 2 terminal (Dy6, Dy8) |
| EDDC | 2 N–O chelate (Dy1, Dy3), 1 N–N chelate (Dy2), 2 N–O chelate (Dy1, Dy4), 1 N–N chelate (Dy5), carbonyl bridges (Dy1–Dy2, Dy1–y5, Dy2–Dy3, Dy4–Dy5) | 2 N–O chelate (Dy1, Dy3), 1 N–N chelate (Dy2), 2 N–O chelate (Dy1, Dy4), 1 N–N chelate (Dy5), carbonyl bridges (Dy1–Dy2, Dy1–Dy5, Dy2–Dy3, Dy4–Dy5) | 2 N–O chelate (Dy1, Dy3), 1 N–N chelate (Dy2), 2 N–O chelate (Dy1, Dy4), 1 N–N chelate (Dy5), carbonyl bridges (Dy1–Dy2, Dy1–Dy5, Dy2–Dy3, Dy4–Dy5) | 2 N–O chelate (Dy2, Dy4), 1 N–N chelate (Dy3), 2 N–O chelate (Dy2, Dy5), 1 N–N chelate (Dy1), carbonyl bridges (Dy2–Dy3, Dy2–Dy5, Dy3–Dy5, Dy5–Dy1) | 2 N–O chelate (Dy9, Dy7), 1 N–N chelate (Dy8), 2 N–O chelate (Dy9, Dy6), 1 N–N chelate (Dy10), carbonyl bridges (Dy9–Dy10, Dy10–Dy6, Dy9–Dy8, Dy9–Dy7) |
| Coordination bonds | 42 (3 × 8 + 2 × 9) | 43 (2 × 8 + 3 × 9) | 44 (1 × 8 + 4 × 9) | 42 (3 × 8 + 2 × 9) | 41 (4 × 8 + 1 × 9) |
| Coordination geometries | Dy1 – triangular dodecahedron (D2d), Dy2 – Hula-hoop (C2v), Dy3 – Snub diphenoid J84 (D2d), Dy4 – spherical capped square antiprism (C4v), Dy5 – Hula-hoop (C2v) | Dy1 – triangular dodecahedron (D2d), Dy2 – Hula-hoop (C2v), Dy3 – spherical tricapped trigonal prism (D3h), Dy4 – spherical capped square antiprism (C4v), Dy5 – Hula-hoop (C2v) | Dy1 – square antiprism (D4d), Dy2 – Hula-hoop (C2v), Dy3 – biaugmented trigonal prism (C2v), Dy4 – spherical capped square antiprism (C4v), Dy5 – Hula-hoop (C2v) | Dy1 – spherical tricapped trigonal prism (D3h), Dy2 – Hula-hoop (C2v), Dy3 – square antiprism (D4d) | Dy4 – spherical capped square antiprism (C4v), Dy5 – Hula-hoop (C2v), Dy6 - square antiprism (D4d) |
Because of the changes in the coordination of the ligands the coordination number (CN) per cluster is also varied from 41 to 44. For example, it is 42 for I, meaning that 3 × CN = 8 and 2 × CN = 9, 43 for I-UV (2 × CN = 8 and 3 × CN = 9), 44 for I-A-N2 (1 × CN = 8 and 4 × CN = 9), 42 for II-molecule A (3 × CN = 8 and 2 × CN = 9), and 41 for II-molecule B (4 × CN = 8 and 1 × CN = 9). The different coordination geometries suggested by the Shape analysis program (SChM)40 are listed in Tables 2 and S5–S8.† While the coordination number of the Dy sharing the two EDDC2− is always eight, their geometries are close to either a triangular dodecahedron (D2d) or square antiprism (D4d). The coordination numbers of the other atoms are eight or nine and are random with a wide range of geometries.
The oxalate anion serves as a bis-chelate bridge through its four oxygen atoms between the two pentamers in the different structures of compound I. The Dy⋯Dy distances over the oxalate bridges are 6.32 Å for I, 6.30 Å for I-UV and 6.21 Å for I-A-N2, respectively. It is planar in each case.
The Dy–O and Dy–N bond lengths for the five pentamers are in the range 2.1–2.5 Å and 2.4–2.7 Å, respectively. One of the two anthracene of the coupled pentamers in the structure of I is oriented nearly face-to-face to the aromatic rings of the EDDC2− and the other protrudes out of the long axis of the molecules in such a way to form π–π interaction (plane-to-plane distance of 3.39 Å and centroid-to-centroid distance of 3.68 Å) with neighbouring molecules to form chains (Fig. 1 and S7†). But for II the situation is completely different. The two anthracenes are oriented towards the EDDC2− ligand and no π–π interaction is possible. However, the structure may be viewed as a 2D supramolecular plane with a network arrangement of weakly intermolecular H-bonds between the pentamers (Fig. S15†). The shortest intermolecular Dy⋯Dy distance is 9.1 Å.
Given the short distance of less than 4.2 Å between the peripheral anthracene moieties in I, it may be favourable for solid-state [4π + 4π] photodimerization reaction. With this in mind, UV-irradiation with 365 nm of compound I was performed for 3 days at room temperature. It experiences SC–SC transformation to I-UV (Fig. 2). However, the structure of the UV irradiated crystal suggests that photodimerization does not occur but the methanol molecules of the original compound I were replaced by two water molecules. The replacement is not at the same Dy atom hosting the methanol. There are several concerted steps: (i) the departure of methanol on Dy4 is compensated by the bridging of the terminal acetate of Dy3, (ii) the methanol of Dy5 is replaced by water and (iii) the bridging acetate (Dy1–Dy2) breaks one bond with Dy1 and a water molecule takes its place and (iv) the terminal acetate (Dy3) then becomes chelated to Dy3. The π–π interaction between the anthracene groups in I-UV becomes weaker with a plane-to-plane distance of 3.42 Å and a centroid-to-centroid distance of 4.40 Å. The transformation of I-UV to I-A-N2 has two modifications: (i) the departing water of Dy1 promotes the bridging of the terminal acetate on Dy2 – a reverse of the changes from I to I-UV, and (ii) the chelate acetate of Dy3 then forms a bridge between Dy3 and Dy2.
The lattice solvent molecules in I and II are heavily disordered and thus treated with the PLATON/SQUEEZE program. The solvent accessible volumes per unit cell are 665 Å3 for I, 1420 Å3 for I-UV, 1538 Å3 for I-A-N2 and 8741 Å3 for II, respectively. It is surprising that the volume of I is significantly smaller than that of the other related compounds. After a careful analysis of the structures of I and I-UV, we found that the methyl groups from the coordinated methanol molecules (C72 and C73) and the terminal acetate (C68) in I occupy partial voids which is different from the case of I-UV (Fig. S16 and S17†). This may explain the fact that the solvent accessible void in I is largely reduced compared with that in I-UV. The number of lattice solvent molecules is determined by thermal analyses.
Magnetic properties
The direct-current (dc) magnetic susceptibility was measured as a function of temperature (1.8–300 K) in a field of 1 kOe on polycrystalline samples of I, I-UV, I-A-Ar and II (Fig. S18†). At 300 K, the χMT (cm3 K mol−1) values per Dy10 are 139.9 for I, 137.4 for I-UV, 135.6 for I-A-Ar and 140.4 for II, close to the expected values of 141.7 for ten non-interacting DyIII (S = 5/2, L = 5, 6H15/2, g = 4/3, and C = 14.17 cm3 K mol−1). The χMT of all the compounds behaves similarly showing a decrease on lowering the temperature from 300 to 30 K, which is mainly due to the progressive depopulation of the excited Stark sublevels of the mJ states of the DyIII.41–44 Below 30 K, weak intra-/inter-molecular antiferromagnetic coupling becomes operative and the χMT shows a sharp decline to 88.1 (I), 79.2 (I-UV), 81.5 (I-A-Ar) and 86.7 cm3 K mol−1 (II). This behaviour is typical of DyIII.10,11The isothermal magnetizations up to 70 kOe were measured for each sample at different temperatures (Fig. S19†). The magnetization rises abruptly in a low field then slowly reach the maximum values of 53.1 (I), 51.4 (I-UV) 49.9 (I-A-Ar) and 59.2 μB (II) at 1.8 K. The magnetizations do not saturate at 70 kOe and reach values which are lower than the expected 100 μB for ten DyIII, and are associated with the considerable crystal-field effects.45,46 Furthermore, the non-saturation of the magnetization together with the non-superimposed M vs. H/T curves (Fig. S20†) suggests the presence of significant magnetic anisotropy and/or low-lying excited states that might be populated when a dc field is used.47–49
Alternating-current (ac) susceptibility measurements were carried out on polycrystalline samples in the temperature range 1.8 to 26 K without a dc field in the frequency range 1 to 1000 Hz to investigate the dynamics of the magnetization reversal.21
The behaviours of I, I-UV and I-A-Ar are drastically different from one another. I exhibits a lack of slow dynamics with clear frequency independence of both χ′ (in-phase) and χ′′ (out-of-phase) (Fig. 4a). This is speculatively associated with facile tunnelling allowing demagnetization without overcoming the barrier.
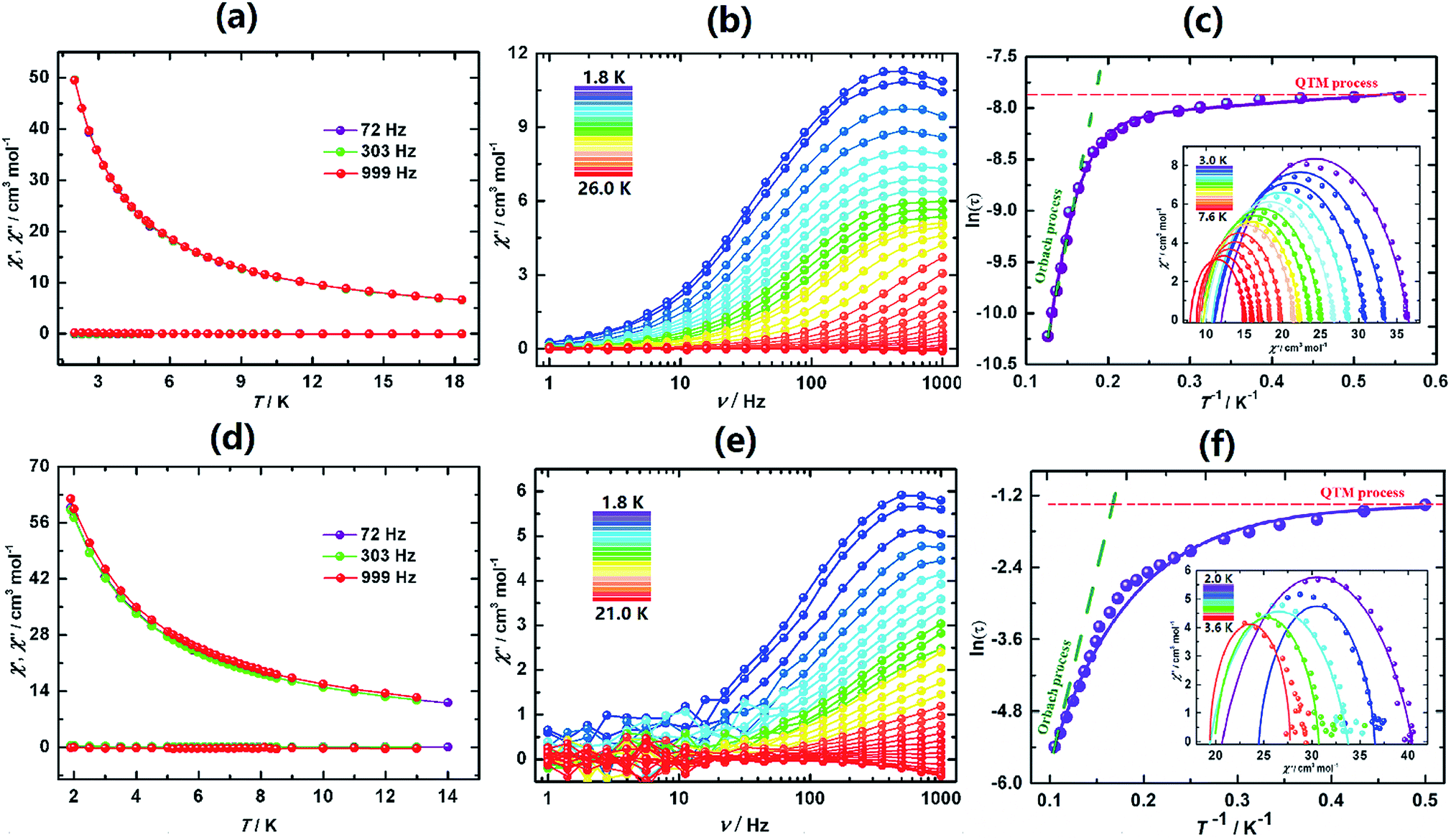 | ||
| Fig. 4 Magnetic properties of I in its different forms. (a) Temperature dependence of the ac-susceptibilities of I for three different frequencies. (b) Frequency dependence of the out-of-phase ac susceptibility of I-UV and (c) Arrhenius plots of the relaxation rate and its fitting (see the text) for I-UV; inset: Cole–Cole plot and fits at different temperatures. (d) Temperature dependence of the ac-susceptibilities of I-A-Ar for three different frequencies. (e) Frequency dependence of the out-of-phase ac susceptibility of I-A-Ar exposed to air and (f) Arrhenius plots of the relaxation rate and its fitting (see the text) for I-A-Ar exposed to air; inset: Cole–Cole plot and fits at different temperatures. | ||
However, upon transforming I to I-UV by exposure to 365 nm light for 3 days strong frequency dependent ac-susceptibilities are now observed indicating that slow relaxation is completely integrated (Fig. 4b, S21 and S22†). The Cole–Cole plots (χ′′ vs. χ′)50 are quasi-semicircles fitting well to a generalized Debye model between 3.0 and 7.6 K (Fig. 4c, inset and Table S9†). To quantify the effective energy barrier to magnetization reversal, the relaxation time was obtained from the fit of the frequency-dependent data between 1.8 and 7.6 K. The plot of lnτ versus 1/T shows three distinct regions based on an Orbach process of high temperatures, a Raman process for intermediate temperatures and a QTM process at lower temperatures.51–54 These data follow the function including the three above-mentioned relaxation processes, τ−1 = AT + B + CTn + τ0−1exp(−Ueff/kT),55 with the following parameters: Ueff = 47.3 K (32.8 cm−1), τo = 1.26 × 10−7 s, A = 3.58 s−1 K−1, B = 0.0036 s−1, C = 0.0011 s−1 K−5.21, n = 5.21 and α ≈ 0.15 (Fig. 4c). The α parameter indicates a narrow distribution of slow relaxation times (τ).56,57 This drastic change of behaviour may be brought about by the different geometrical changes of the coordination spheres of the five Dy atoms of the asymmetric unit.
Upon annealing I-UV under Ar at 100 °C the crystal is transformed to I-A-Ar and its ac-susceptibilities are reverted back to being non-SMM. When I-A-Ar is allowed to stand in air under ambient conditions, it regains its SMM characteristics, albeit with slight modification of its metrics, Ueff/kB = 37.6 K (26.1 cm−1), τo = 1.04 × 10−7 s, A = 2.23 s−1 K−1, B = 0.0031 s−1, C = 0.0009 s−1 K−4.79, n = 4.79 and α ≈ 0.19 (Fig. 4e and f). This means that annealing at 100 °C can be reverted by standing under ambient conditions but I-UV cannot be reverted back to I. This corroborates well with the crystal structures which indicates that annealing by heating at 100 °C removes two water molecules and it takes up the water reversibly upon standing in air.
The ac susceptibilities of II (Fig. 5, S25 and S26†) exhibit progressive frequency dependence characteristics of SMM. Analyses of the data led to characteristic parameters, Ueff = 138 K (95.9 cm−1), τo = 2.79 × 10−7 s, A = 2.62(3) s−1 K−1, B = 0.19(6) s, C = 0.0014 s−1 K−1 and n = 4.83(5). The energy barrier is much higher than that of I-UV.
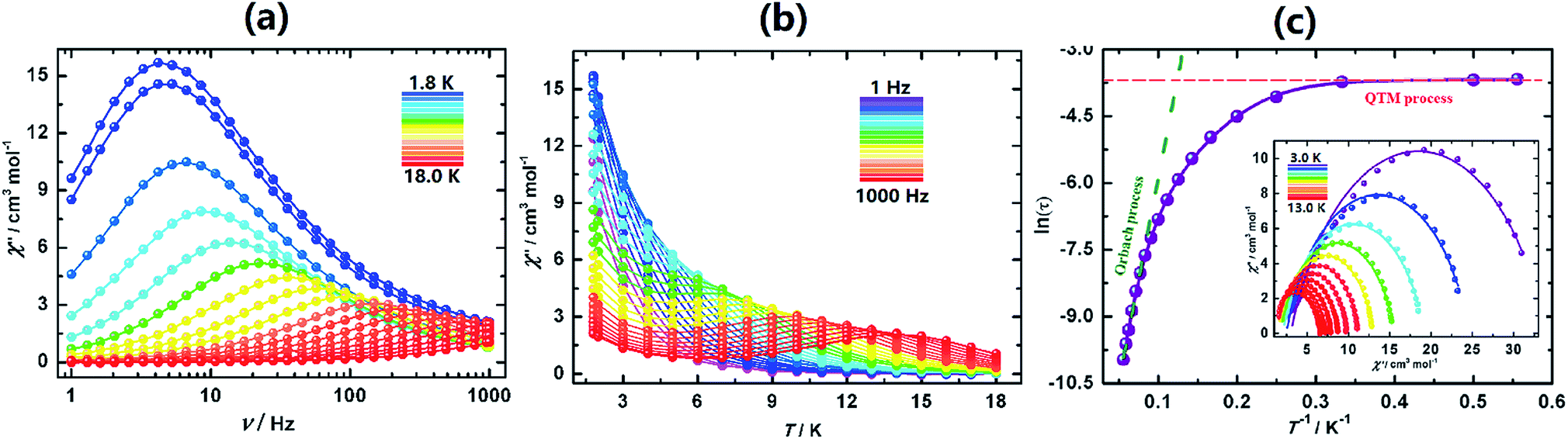 | ||
| Fig. 5 Magnetic properties of II. (a) Frequency and (b) temperature dependence of the out-of-phase ac-susceptibilities. (c) Arrhenius plots of the relaxation rate and its fitting (see the text); inset: Cole–Cole plot and fits at different temperatures. | ||
The contrasting relaxation processes of the different structures of compound I are quite astonishing and have not been seen previously. Although I is a non-SMM, its UV irradiated form I-UV behaves as a SMM and its heat annealed form under N2 or argon I-A is reverted back to a non-SMM and II behaving as a SMM, and they all contain pentameric units which are structurally related. Therefore, the fusion of two pentamers with an oxalate bridge in I, I-UV and I-A cannot be the cause of the switching compared to the isolated pentamers in II which is a SMM.
The fortuitous advantage of having the above process with complete single-crystal transformation to single-crystals allows for a thorough structure–property correlation. The differences in the magnetic dynamics may be correlated with the different crystal field environments with the distinct coordination modes of the peripheral assistant ligands for crystallographically independent DyIII ions. Careful analysis of the three cyclic arrangements reveals important disparities (Fig. 2). One piece of evidence is that the cyclic pentanuclear unit of I is surrounded by six acetate groups adopting three different coordination modes (Fig. 2a), while three of the six acetate groups in I-UV show one kind of crossfade based on the terminal and chelate modes (Fig. 2b). Only one coordination mode (bidentate fashion) can be observed for the four peripheral acetate groups of I-A-N2 (Fig. 2c). On the other hand, the coordination spheres of the central metal ions of I are completed by one water and two methanol molecules, while it has three terminal water molecules in I-UV and only one water molecule in I-A-N2. Obviously, these structural modifications are most likely to influence the emergence of magnetic anisotropy through alteration of some crucial parameters of the structure (such as Dy–O/N bond lengths and Dy–O–Dy angles), and the coordination geometries of DyIII ions (see Table S1† for details), hence causing the drastic difference of magnetization dynamic behaviours.
In order to get an insight into the magneto-structure relationship, theoretical investigation was conducted for all these complexes. Complete-active-space self-consistent field (CASSCF) calculations on individual DyIII fragments for complexes I, I-UV, I-A-N2, II-molecule A and II-molecule B on the basis of X-ray determined geometries have been carried out by the CASSCF/RASSI method with the MOLCAS 8.2 program package58–60 (see the ESI† for details). The Single_Aniso61–63 program was used to obtain the g tensors, energy levels, magnetic axes, etc., based on the above CASSCF/RASSI calculations. The lowest eight spin–orbit energies, the corresponding g tensors and the mJ values of individual DyIII fragments for complexes I, I-UV, I-A-N2, II-molecule A and II-molecule B are shown in Table S12,† where the calculated energy gaps of the lowest two Kramers doublets (KDs) and the ground g tensors of five or three types of individual DyIII fragments for each complex are much different from each other. The tunnelling gaps of individual DyIII fragments for each complex are also different from each other (Fig. S28–S32†). Thus, it is difficult to differentiate them only from the energy gaps of the lowest two KDs and the ground g tensors of individual DyIII fragments for the five complexes. From the energy states and g tensors of the five complexes in Table S12,† however, we can find some differences between them. For I, the energy gap of the lowest two KDs of the Dy5 fragment is only 15.1 cm−1, much smaller than that of the others, and its ground gz value is far from 20. For I-UV, both of the energy gaps of the lowest two KDs of five individual DyIII fragments and the ground gz values are larger than those of I, which can be one of the reasons for its showing SMM behaviour.
As usual, the energy barriers of the polynuclear lanthanide complexes are mainly from the single-ion anisotropy of the DyIII centres. But the Dy–Dy interactions have some influence on the relaxation rate of incoherent quantum tunnelling to give drastically different effective relaxation barriers. For the current systems, it is difficult to fit the Dy–Dy interactions using the Lines model64 since each complex has a large pair of interactions. However, the dipole–dipole interactions between DyIII ions can be calculated exactly according to the ground gz values and the magnetic axes shown in Fig. S33.† Simply, we only gave the nearest neighbouring Dy–Dy dipole–dipole interactions in Table 3 for the five complexes. The parameters J1, J2, J3, J4, and J5 represent Dy1–Dy2, Dy2–Dy3, Dy3–Dy4, Dy4–Dy5, and Dy5–Dy1 dipole–dipole interactions, respectively, for each complex. It is evident that most of the dipole–dipole interactions are antiferromagnetic. But the interactions are too complicated to deduce further interesting conclusions from them.
| Compd | I | I-UV | I-A-N2 | II-molecule A | II-molecule B |
|---|---|---|---|---|---|
| J dip | J dip | J dip | J dip | J dip | |
| J 1 | 1.76 | 0.73 | 2.32 | −0.82 | 2.26 |
| J 2 | −1.56 | −0.56 | −1.21 | 0.34 | −0.13 |
| J 3 | −1.17 | −0.34 | −1.87 | −3.21 | −3.59 |
| J 4 | −0.30 | −1.84 | 2.91 | 1.86 | −0.07 |
| J 5 | −1.38 | −2.49 | −0.56 | 0.93 | −2.20 |
Conclusions
In this paper, we report the first examples of odd-numbered cyclic pentamer rings of lanthanide, e.g. decanuclear clusters of I, I-UV and I-A-N2 containing oxalate bridged pentamers as well as a pentanuclear cluster of II containing two crystallographically distinguished pentamers. The switching ON and OFF of the single-molecule magnetism followed by single-crystal-to-single-crystal structural transformation has been evidenced for the first time in a decanuclear cluster of I upon a simple and modest physical process like irradiation with UV light and annealing at a moderate temperature of 100 °C. The work opens new possibilities to achieve switchable ON–OFF SMM behaviour using dynamic cluster systems.Experimental
Materials and physical measurements
9-Anthrylmethylphosphonic acid (C15H11PO3H2)65 and (N′,N′′E,N′,N′′E-N′,N′′-(ethane-1,2-diylidene)dipyrazine-2-carbohydrazide (H2EDDC)36 were prepared according to the methods reported in the literature. All other reagents were purchased from commercial suppliers and used as received.Structure determinations
Single crystals of I, I-UV and II were attached to glass fibres and mounted on a Bruker D8 system for data collection at 123(2) K, using monochromatic Mo-Kα radiation (λ = 0.71073 Å). The data of I-A-N2 were collected at 373 K by heating a single crystal of Iin situ under a flow of nitrogen on the diffractometer (Bruker D8 system) for half an hour. The data of I-A-N2-cool, obtained by exposing the same single crystal of I-A-N2 to air at 25 °C for a week, were collected at 123 K.The structures were solved by direct methods and refined on F2 by full matrix least squares using SHELXTL.66 All non-hydrogen atoms were refined with anisotropic thermal parameters. All hydrogen atoms were either put in calculated positions or found from the difference Fourier maps and refined isotropically. In the cases of I and II, residual electron densities in the solvent-accessible void due to disordered solvent molecules were treated with the PLATON/SQUEEZE program.67 In each case of I-UV and I-A-N2-cool, 11 lattice water molecules were found from the F-map. Except for O14W (1.0 occupancy), the other water molecules (O4W, O5W, O6W, O7W, O8W, O9W, O10W, O11W, O12W and O13W) are disordered with half occupancy. The hydrogen atoms of disordered water molecules are not placed. In all cases, quite large (3.1–4.9 e A3) residual electron density peaks are all located near the Dy atoms in the corresponding structures and could not be modelled with reasonably disordered atoms. The refinement of I-A-N2-cool gave unsatisfactory R1 and wR2 values of 11.1% and 27.1% due to the poor quality of data. Hence this structure was not further refined and deposited in CCDC.
A summary of the lattice parameters is presented in Tables 1 and S1.† Selected bond lengths and angles are given in Tables S2–S4.† CCDC 1819929–1819933 contain the supplementary crystallographic data for this paper.
Physical measurements
Elemental analyses for C, N and H were performed with a Perkin Elmer 240C elemental analyzer. Infrared spectra were recorded on a Bruker Tensor 27 spectrometer by transmission from KBr pellets containing the compounds in the range of 400–4000 cm−1. Magnetization measurements were performed in the temperature range 1.8–300 K, field of ±70 kOe and frequency range of 1 to 1000 Hz using a vibrating sample magnetometer (VSM) of a Quantum Design MPMS SQUID-VSM system. The diamagnetic contributions of the samples were estimated from Pascal's constant.41Synthesis of I
To a slurry of H2EDDC (59.6 mg, 0.20 mmol) and triethylamine (0.07 mL, 0.5 mmol) in mixed water/methanol (20 mL, 1:3), solid Dy(OAc)3·4H2O (82.4 mg, 0.20 mmol) and sodium oxalate (20.1 mg, 0.15 mmol) were added at room temperature. After stirring for 24 h, C15H11PO3H2 (54.4 mg, 0.2 mmol) was then added to the resulting yellow solution and stirred for another 24 h at room temperature. Subsequently, this mixture was transferred to 25 mL glassware, sealed and kept in a vacuum drying oven at 100 °C. Dark red single crystals, suitable for X-ray diffraction analysis, were formed after 7 days as a single-phase product. Yield: 47 mg (39%, based on Dy). Elemental analysis (%) calcd for C138H132Dy10N32O54P4·6H2O: C, 33.39, H, 2.90, N, 9.03: found C, 32.95, H, 2.52, N, 10.11. IR (KBr, cm−1): 3419(vs), 1655(m), 1606(s), 1582(vs), 1501(s), 1452(m), 1420(s), 1364(m), 1218(w), 1177(m), 1161(m), 1137(m), 1105(m), 1024(s), 975(m), 943(w), 805(m), 776(m), 724(w), 676(m), 651(m), 603(w), 563(m), 06(m), 473(w), 417(w).
I was converted to I-UV by UV-irradiation at 365 nm for 3 days. By annealing at 100 °C under a flow of nitrogen on a diffractometer it was transformed to I-A-N2 and subsequently kept at 25 °C in air for one week (I-A-N2-cool). When I-UV was annealed at 100 °C under argon in a glovebox for 2 d, I-A-Ar was obtained which was loaded into a capsule in a glovebox and carefully immobilised in n-eicosane for magnetic measurements.
Synthesis of II
A mixture of H2EDDC (44.7 mg, 0.15 mmol) and Dy(OAc)3·4H2O (61.8 mg, 0.15 mmol) in methanol (20 mL) was stirred with triethylamine (0.14 mL, 1.0 mmol) at room temperature. After 12 h, C15H11PO3H2 (27.2 mg, 0.1 mmol) was added to the solution and stirred overnight at room temperature. The resulting yellow precipitate was filtered off, dissolved in chloroform (5 mL), and evaporated to 2 mL under vacuum. Subsequently, this solution was transferred to a 5 mL glass tube. Red hexagonal crystals, suitable for X-ray diffraction analysis, were formed as a single-phase product by slow diffusion of n-hexane (2 mL) into the mother liquor after one week. Yield: 34 mg (45% based on Dy). Elemental analysis (%) calcd for C140H132Dy10N32O52P4Cl6: C, 33.26, H, 2.63, N, 8.86; found C, 32.61, H, 2.47, N, 9.24. IR (KBr, cm−1): 3415(s), 1669(m), 1610(s), 1576(vs), 1505(s), 1427(s), 1365(m), 1279(w), 1224(w), 1185(m), 1154(s), 1132(s), 1076(m), 1029(s), 1009(m), 982(m), 935(w), 865(w), 787(m), 732(m), 678(m), 747(m), 601(w), 561(w), 483(m), 420(w).Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support by the National Key R&D Program of China (2017YFA0303203) and the National Natural Science Foundation of China (21731003, 11774178, and U1532110) is acknowledged. MK is funded by the CNRS, France.Notes and references
- (a) O. Sato, Nat. Chem., 2016, 8, 644–656 CrossRef PubMed; (b) X.-Z. Song, S.-Y. Song, S.-N. Zhao, Z.-M. Hao, M. Zhu, X. Meng, L.-L. Wu and H.-J. Zhang, Adv. Funct. Mater., 2014, 24, 4034–4041 CrossRef; (c) M. Zhu, X.-Z. Song, S.-Y. Song, S.-N. Zhao, X. Meng, L.-L. Wu, C. Wang and H.-J. Zhang, Adv. Sci., 2015, 2, 1500012 CrossRef PubMed.
- R. Sessoli, D. Gatteschi, A. Caneschi and M. A. Novak, Nature, 1993, 365, 141–143 CrossRef.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef PubMed.
- F. Troiani and M. Affronte, Chem. Soc. Rev., 2011, 40, 3119–3129 RSC.
- R. Vincent, S. Klyatskaya, M. Ruben, W. Wernsdorfer and F. Balestro, Nature, 2012, 488, 357–360 CrossRef PubMed.
- R. Bagai and G. Christou, Chem. Soc. Rev., 2009, 38, 1011–1026 RSC.
- D. Gatteschi, R. Sessoli and J. Villain, Molecular Nanomagnets, Oxford University Press, 2006 Search PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef PubMed.
- R. A. Layfield and M. Murugesu, Lanthanides and Actinides in Molecular Magnetism, Wiley, 2015 Search PubMed.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef PubMed.
- P. Zhang, Y.-N. Guo and J. Tang, Coord. Chem. Rev., 2013, 257, 1728–1763 CrossRef.
- M. Ren and L.-M. Zheng, Acta Chim. Sin., 2015, 73, 1091–1113 CrossRef.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- Y.-S. Ding, N. F. Chilton, R. E. P. Winpenny and Y.-Z. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef PubMed.
- J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef PubMed.
- C. A. P. Goodwin, F. Ortu, D. Reta, N. F. Chilton and D. P. Mills, Nature, 2017, 548, 439–442 CrossRef PubMed.
- S. Demir, M. I. Gonzalez, L. E. Darago, W. J. Evans and J. R. Long, Nat. Commun., 2017, 9, 2144 CrossRef PubMed.
- D. Tanaka, T. Inose, H. Tanaka, S. Lee, N. Ishikawa and T. Ogawa, Chem. Commun., 2012, 48, 7796–7798 RSC.
- B. S. Dolinar, S. Gómez-Coca, D. I. Alexandropoulos and K. R. Dunbar, Chem. Commun., 2017, 53, 2283–2286 RSC.
- C. M. Dickie, A. L. Laughlin, J. D. Wofford, N. S. Bhuvanesha and M. Nippe, Chem. Sci., 2017, 8, 8039–8049 RSC.
- K. Suzuki, R. Sato and N. Mizuno, Chem. Sci., 2013, 4, 596–600 RSC.
- X. Zhang, V. Vieru, X. Feng, J.-L. Liu, Z. Zhang, B. Na, W. Shi, B.-W. Wang, A. K. Powell, L. F. Chibotaru, S. Gao, P. Cheng and J. R. Long, Angew. Chem., Int. Ed., 2015, 54, 9861–9865 CrossRef PubMed.
- J.-Y. Ge, L. Cui, J. Li, F. Yu, Y. Song, Y.-Q. Zhang, J.-L. Zuo and M. Kurmoo, Inorg. Chem., 2017, 56, 336–343 CrossRef PubMed.
- G. Cosquer, M. Morimoto, M. Irie, A. Fetoh, B. K. Breedlove and M. Yamashita, Dalton Trans., 2015, 44, 5996–6002 RSC.
- L.-F. Wang, J.-Z. Qiu, J.-L. Liu, Y.-C. Chen, J.-H. Jia, J. Jover, E. Ruiz and M.-L. Tong, Chem. Commun., 2015, 51, 15358–15361 RSC.
- M. Ren, S.-S. Bao, N. Hoshino, T. Akutagawa, B. Wang, Y.-C. Ding, S. Wei and L.-M. Zheng, Chem.–Eur. J., 2013, 19, 9619–9628 CrossRef PubMed.
- S. Mohapatra, B. Rajeswaran, A. Chakraborty, A. Sundaresan and T. K. Maji, Chem. Mater., 2013, 25, 1673–1679 CrossRef.
- Q. Zhou, F. Yang, B. Xin, G. Zeng, X. Zhou, K. Liu, D. Ma, G. Li, Z. Shi and S. Feng, Chem. Commun., 2013, 49, 8244–8246 RSC.
- J. Tang, I. Hewitt, N. T. Madhu, G. Chastanet, W. Wernsdorfer, C. E. Anson, C. Benelli, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2006, 45, 1729–1733 CrossRef PubMed.
- A. Soncini and L. F. Chibotaru, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 132403 CrossRef.
- L. Ungur, S.-Y. Lin, J. Tang and L. F. Chibotaru, Chem. Soc. Rev., 2014, 43, 6894–6905 RSC.
- S. K. Langley, B. Moubaraki, C. M. Forsyth, I. A. Gass and K. S. Murray, Dalton Trans., 2010, 39, 1705–1708 RSC.
- V. Chandrasekhar, P. Bag and E. Colacio, Inorg. Chem., 2013, 52, 4562–4570 CrossRef PubMed.
- L. G. Westin, M. Kritikos and A. Caneschi, Chem. Commun., 2003, 1012–1013 RSC.
- H. Tian, S.-S. Bao and L.-M. Zheng, Dalton Trans., 2015, 44, 14208–14212 RSC.
- L. Zhao, S. Xue and J. Tang, Inorg. Chem., 2012, 51, 5994–5996 CrossRef PubMed.
- (a) S. Biswas, S. Das, J. Acharya, V. Kumar, J. van Leusen, P. Kögerler, J. M. Herrera, E. Colacio and V. Chandrasekhar, Chem.–Eur. J., 2017, 23, 5154–5170 CrossRef PubMed; (b) X.-Y. Zheng, J.-B. Peng, X.-J. Kong, L.-S. Long and L.-S. Zheng, Inorg. Chem. Front., 2016, 3, 320–325 RSC; (c) X.-Y. Zheng, J. Xie, X.-J. Kong, L.-S. Long and L.-S. Zheng, Coord. Chem. Rev., 2017 DOI:10.1016/j.ccr.2017.10.023.
- (a) H. Tian, S.-S. Bao and L.-M. Zheng, Chem. Commun., 2016, 52, 2314–2317 RSC; (b) H. Tian, S.-S. Bao and L.-M. Zheng, Eur. J. Inorg. Chem., 2016, 3184–3190 CrossRef.
- M. Pinsky and D. Avnir, Inorg. Chem., 1998, 37, 5575–5582 CrossRef PubMed.
- O. Kahn, Molecular Magnetism, Wiley-VCH, New York, 1993 Search PubMed.
- P. H. Lin, T. J. Burchell, R. Clerac and M. Murugesu, Angew. Chem., Int. Ed., 2008, 47, 8848–8851 CrossRef PubMed.
- L. Ungur, S. K. Langley, T. N. Hooper, B. Moubaraki, E. K. Brechin, K. S. Murray and L. F. Chibotaru, J. Am. Chem. Soc., 2012, 134, 18554–18557 CrossRef PubMed.
- M. L. Kahn, R. Ballou, P. Porcher, O. Kahn and J. P. Sutter, Chem.–Eur. J., 2002, 8, 525–531 CrossRef.
- S. Osa, T. Kido, N. Matsumoto, N. Re, A. Pochaba and J. Mrozinski, J. Am. Chem. Soc., 2004, 126, 420–421 CrossRef PubMed.
- R. J. Blagg, L. Ungur, F. Tuna, J. Speak, P. Comar, D. Collison, W. Wernsdorfer, E. J. L. McInnes, L. F. Chibotaru and R. E. P. Winpenny, Nat. Chem., 2013, 5, 673–678 CrossRef PubMed.
- P. Zhang, M. Perfetti, M. Kern, P. P. Hallmen, L. Ungur, S. Lenz, M. R. Ringenberg, W. Frey, H. Stoll, G. Rauhut and J. van Slageren, Chem. Sci., 2018, 9, 1221–1230 RSC.
- E. Moreno Pineda, N. F. Chilton, R. Marx, M. Dörfel, D. O. Sells, P. Neugebauer, S.-D. Jiang, D. Collison, J. van Slageren, E. J. L. McInnes and R. E. P. Winpenny, Nat. Commun., 2014, 5, 5243–5251 CrossRef PubMed.
- S. G. McAdams, A. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef.
- K. S. Cole and R. H. Cole, J. Chem. Phys., 1941, 9, 341–351 CrossRef.
- I. J. Hewitt, J. Tang, N. T. Madhu, C. E. Anson, Y. Lan, J. Luzon, M. Etienne, R. Sessoli and A. K. Powell, Angew. Chem., Int. Ed., 2010, 49, 6352–6356 CrossRef PubMed.
- J. J. Le Roy, M. Jeletic, S. I. Gorelsky, I. Korobkov, L. Ungur, L. F. Chibotaru and M. Murugesu, J. Am. Chem. Soc., 2013, 135, 3502–3510 CrossRef PubMed.
- K. L. M. Harriman, J. L. Brosmer, L. Ungur, P. L. Diaconescu and M. Murugesu, J. Am. Chem. Soc., 2017, 39, 1420–1423 CrossRef PubMed.
- B. S. Dolinar, D. I. Alexandropoulos, K. R. Vignesh, T. James and K. R. Dunbar, J. Am. Chem. Soc., 2018, 140, 908–911 CrossRef PubMed.
- A. J. Brown, D. Pinkowicz, M. R. Saber and K. R. Dunbar, Angew. Chem., Int. Ed., 2015, 54, 5864–5868 CrossRef PubMed.
- G. Abbas, Y. Lan, G. E. Kostakis, W. Wernsdorfer, C. E. Anson and A. K. Powell, Inorg. Chem., 2010, 49, 8067–8072 CrossRef PubMed.
- Y. Bi, Y.-N. Guo, L. Zhao, Y. Guo, S. -Y. Lin, S. -D. Jiang, J. Tang, B. -W. Wang and S. Gao, Chem.–Eur. J., 2011, 17, 12476–12481 CrossRef PubMed.
- F. Aquilante, L. De Vico, N. Ferré, G. Ghigo, P.-Å. Malmqvist, P. Neogrády, T. B. Pedersen, M. Pitonak, M. Reiher, B. O. Roos, L. Serrano-Andrés, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224 CrossRef PubMed.
- V. Veryazov, P. -O. Widmark, L. Serrano-Andres, R. Lindh and B. O. Roos, Int. J. Quantum Chem., 2004, 100, 626 CrossRef.
- G. Karlström, R. Lindh, P.-Å. Malmqvist, B. O. Roos, U. Ryde, V. Veryazov, P.-O. Widmark, M. Cossi, B. Schimmelpfennig, P. Neogrády and L. Seijo, Comput. Mater. Sci., 2003, 28, 222 CrossRef.
- L. F. Chibotaru, L. Ungur and A. Soncini, Angew. Chem., Int. Ed., 2008, 47, 4126 CrossRef PubMed.
- L. Ungur, W. Van den Heuvel and L. F. Chibotaru, New J. Chem., 2009, 33, 1224 RSC.
- L. F. Chibotaru, L. Ungur, C. Aronica, H. Elmoll, G. Pilet and D. Luneau, J. Am. Chem. Soc., 2008, 130, 12445 CrossRef PubMed.
- M. E. Lines, J. Chem. Phys., 1971, 55, 2977 CrossRef.
- I. P. Beletskaya and M. A. Kazankova, Russ. J. Org. Chem., 2002, 38, 1391–1430 CrossRef.
- SHELXTL (version 5.0), Reference Manual, Siemens Industrial Automation, Analytical Instruments, Madison, WI, 1995 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1819929–1819933. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8sc01228h |
| This journal is © The Royal Society of Chemistry 2018 |