
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multi-element ion-exchange chromatography and high-precision MC-ICP-MS isotope analysis of Mg and Ti from sub-mm-sized meteorite inclusions†
K. K.
Larsen
 *,
D.
Wielandt
and
M.
Bizzarro
*,
D.
Wielandt
and
M.
Bizzarro
Centre for Star and Planet Formation, Natural History Museum of Denmark, University of Copenhagen, DK-1350, Denmark. E-mail: kirstenl@snm.ku.dk
First published on 15th March 2018
Abstract
The analytical improvement of new generation mass spectrometers has reached a level required for high-precision isotope analysis of very small and unique natural samples. The multi-element isotopic signatures of meteorite inclusions can potentially provide detailed insight into the origin of our solar system. As such, in-line separation and isotope analysis of multiple elements from such unique samples is highly desirable, but rarely accommodated by current chromatographic purification procedures necessary for accurate isotope analysis using multiple collector inductively coupled plasma source mass spectrometry (MC-ICP-MS). Here, we report a new multi-element ion chromatographic purification procedure, specifically developed for the separation of very small amounts of Mg (∼5 μg) and Ti (0.7–1 μg) from individual samples. Highly isotopically anomalous sub-mm sized materials, such as Ca–Al-rich inclusions in primitive meteorites, can be routinely analysed for their mass-independent μ26Mg*, μ46Ti*, μ48Ti*, μ49Ti* and μ50Ti* isotope compositions with an estimated external reproducibility of 4.1, 11, 8.8, 18 and 12 ppm (2sd), respectively. These procedures consume significantly less material than previous methods and as such represents a 6- to 10-fold improvement in precision. The method ensures >99.9% Mg and >97% Ti recovery, thereby allowing for the accurate determination of mass-dependent compositions with a precision better than ∼60 ppm amu−1 for both Mg and Ti. It can further be adapted to accommodate the in-line separation of e.g. Ca, Cr, Fe, Ni, W, Mo, Ru, V, Zr and Hf from the same sample matrices. Doping tests on a synthetic Ti standard demonstrate that isobaric interference can be adequately corrected for provided that atomic ratios are Ca/Ti < 0.07, V/Ti < 0.04 and Cr/Ti < 0.005 and Zr2+, Mo2+ and Ru2+ have no influence on Ti measurements when atomic ratios are Zr/Ti < 0.002, Mo/Ti < 0.01 and Ru/Ti < 0.001, which is ensured by the chromatographic procedures. Magnesium and titanium isotope data for terrestrial geostandards and isotopically anomalous chondrite meteorites (OC, CR, CV, and CI) and two CV CAIs are in excellent agreement with literature values, demonstrating the accuracy of our methods. The two CAIs plot on an extension of the mass-independent correlation line between μ46Ti* and μ50Ti* defined by inner solar system materials, suggesting residual nucleosynthetic effects in CAI precursors.
1. Introduction
Magnesium and titanium are both important elements in natural environments on Earth and throughout the solar system. While Mg is an important ingredient of terrestrial planets,1 Ti is a major refractory element, which are enriched in the solar system's first solids, Ca–Al-rich inclusions (CAIs), found in primitive meteorites.2 Studying their isotope composition potentially provides constraints on, for example, the timing and efficiency of planetary differentiation processes,3–6 mixing and transport of primitive components in the solar protoplanetary disk,7–9 thermal processing of presolar dust,10 nucleosynthetic sources that contributed to the astrophysical birth environment of our young Sun,11 biogeochemical processes on Earth12 and evaporation and condensation processes.13,14Given that Mg is a moderately volatile element and Ti a highly refractory one, potential differences in the degree of stable isotope fractionation may discern the condensation and/or evaporation history of CAIs. Also, the identification of planetary-scale mass-independent heterogeneity in both Mg (most likely associated with heterogeneities in the short-lived radioactive nuclide 26Al15–17) and Ti isotopes10 calls for an evaluation of the large-scale mixing/unmixing processes of pre-solar components associated with the establishment of the protoplanetary disk and the formation of the first planetesimals. The coupled isotope systematics of various elements (including Mg and Ti) from unique solar system components, including disk solids such as CAIs, AOAs (Amoeboid Olivine Aggregates) and chondrules from primitive meteorites, may aid in such a quest.
Ion chromatographic purification required for high-precision isotope analysis using Multi-Collector Inductively Coupled Plasma Mass Spectrometry (MC-ICP-MS) and Thermal Ionisation Mass Spectrometry (TIMS) is often accomplished using chromatographic separation protocols tailored for specific elements involving a combination of various ion exchangers.18–20 The combined analysis of the isotope composition of multiple elements extracted from the exact same sample matrices may, however, be very valuable for a critical evaluation of their prehistory. Such multi-element isotope analyses are especially advantageous when handling small and unique samples, such as sub-mm meteoritic inclusions.
With the advent of new generation high-precision mass spectrometers, such as the Thermo Scientific Neptune Plus MC-ICP-MS and its Jet/X cone interface, the precision of isotope measurements has increased significantly. This has unlocked the possibility of precisely analysing very small amounts of analytes for the isotope composition of sub-mm inclusions in primitive meteorites. However, such an increase in analytical precision further tightens the demands on ion chromatographic purification protocols required to diminish the number of matrix elements generating instrumental interference and thus inaccurate measurements. It also puts constraints on the amount of impurities, known as ‘blanks’, introduced through chemical purification of the analyte. The scarcity of material available in small silicate samples calls for effective purification procedures that allow for the separation and high-precision isotope analysis of many elements from the same samples, while at the same time minimizing impurities introduced into the sample through chemical processing.
Given these requirements, we present a novel multi-element chromatographic separation protocol that allows for high-precision isotope analysis of Mg and Ti from the same digestion aliquots of small silicate samples. We show that sub-mm meteoritic inclusions, such as highly isotopically anomalous CAIs, can be accurately and precisely measured for their Mg and Ti isotope compositions with a resolution significantly exceeding that required to resolve mass-dependent and mass-independent variability amongst most inner solar system materials.
2. Sample preparation and ion exchange chromatography
2.1 Materials, reagents and sample digestion
Experimental and analytical procedures were carried out in ultra-clean laboratory environments with HEPA-filtered laminar flow hoods. All reagents were either purchased as ultrapure chemicals (HF, H2O2, and NH4OH) or purified through distillation (HNO3, HCl, acetone, and Milli-Q water). These reagents were all blank tested to ensure a sufficiently low level of impurities prior to chromatographic purification. CAIs were microdrilled using tungsten-carbide drill bits on a computer-assisted New Wave Micromill at the Centre for Star and Planet Formation, University of Copenhagen. Whole-rock powders, crushed silicate rock specimens and microdrilled samples (0.1–2 mg) were digested in Savillex PFA beakers in a mixture of concentrated HF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 (3:1) (∼0.5 ml) for 2 days at 120 °C and evaporated to dryness. To ensure complete dissolution of all samples, this step was followed by digestion in concentrated aqua regia (HCl:HNO3 = 3:1) (∼0.5 ml) at 120 °C for another 2 days, thereby also destroying any fluorides formed during HF–HNO3 digestion. The aqua regia treatment was repeated until there were no remaining solids. Prior to sample digestion all PFA beakers were pre-cleaned using the same mixture of acids as that used for sample digestion. Following decomposition, all samples were converted to NO3 salts through two evaporation cycles with 7 M HNO3 in preparation for chromatographic purification.
:HNO3 (3:1) (∼0.5 ml) for 2 days at 120 °C and evaporated to dryness. To ensure complete dissolution of all samples, this step was followed by digestion in concentrated aqua regia (HCl:HNO3 = 3:1) (∼0.5 ml) at 120 °C for another 2 days, thereby also destroying any fluorides formed during HF–HNO3 digestion. The aqua regia treatment was repeated until there were no remaining solids. Prior to sample digestion all PFA beakers were pre-cleaned using the same mixture of acids as that used for sample digestion. Following decomposition, all samples were converted to NO3 salts through two evaporation cycles with 7 M HNO3 in preparation for chromatographic purification.
2.2 In-line chromatographic separation and purification of Mg and Ti
To ensure a minimum impurity contribution from resin and reagents, we developed a new low-level chromatographic separation protocol for in-line separation and purification of multiple elements from small amounts of silicate samples. This ensures a sample load of <5% of the full cation exchange capacity (CEC) of the 1st chromatographic column, thereby preventing peak fronting during elution and thus ensuring the availability of sufficient ion exchange sites.21 For the current setup of the exchanger columns, this corresponds to a typical sample load of approximately 1–2 mg (from which Si has been removed prior to chromatographic separation by volatilization through HF–HNO3 digestion) depending on cation valences in individual sample matrices. The capacity of the exchanger columns can be scaled up to accommodate larger amounts of samples. This may be of relevance for the separation of elements, such as W, Mo, Ru, V, Zr and Hf, given their common presence in trace amounts in most types of sample matrices.The ion chromatographic separation protocol involves a series of discrete ion chromatographic separation steps, as summarized in Fig. 1. It takes advantage of novel sample pre-treatment procedures to ensure a high Cr recovery (>95%)22 and is further refined from a Mg purification protocol for larger samples (∼100 μg Mg), known to provide high-purity Mg for high-precision isotope analysis by MC-ICP-MS.23 Although applicable to most types of inorganic sample matrices, the purification method described here is specifically tailored for samples with highly anomalous isotope compositions whose measured isotope composition may be compromised by minute analyte impurity contributions. The protocol is well suited for sample matrices with particularly high amounts of Mg, Al, Ca and Fe, such as CAIs and chondrules.
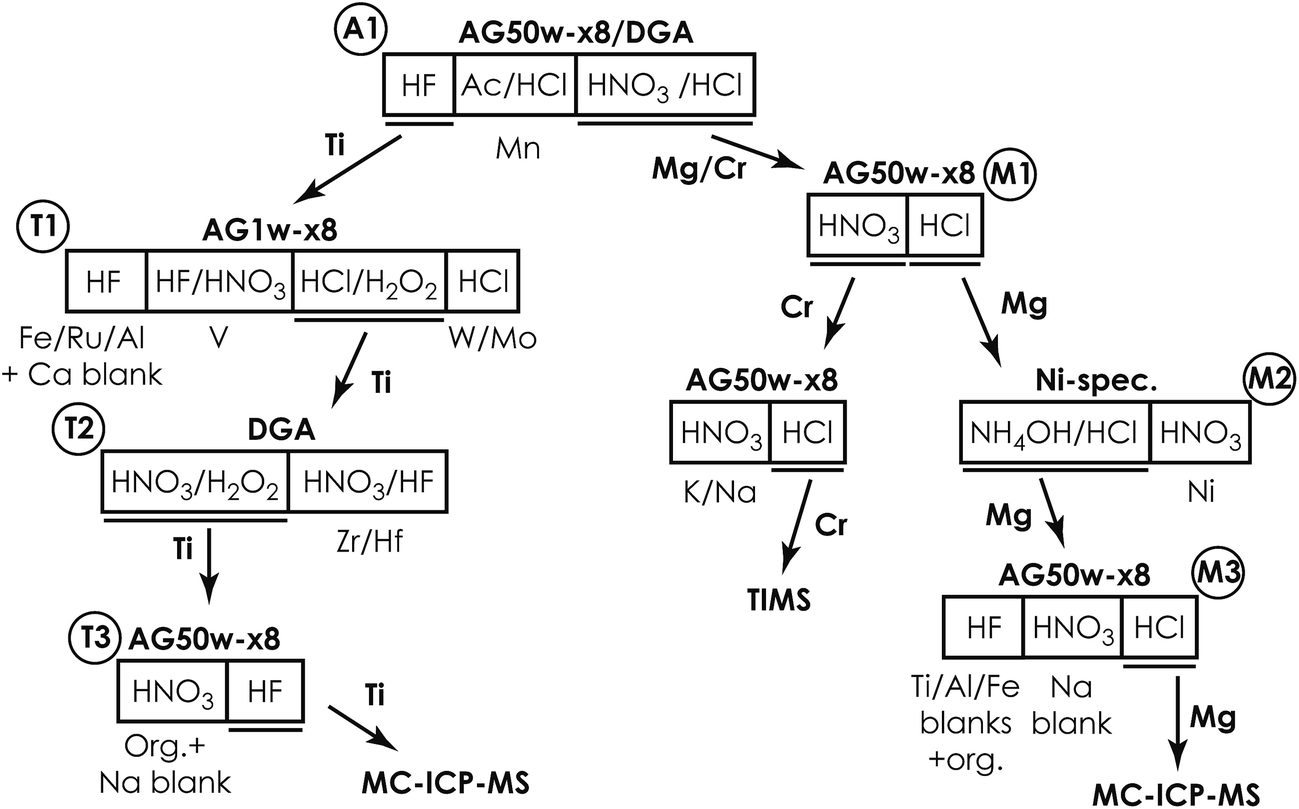 | ||
| Fig. 1 Schematic diagram of the multi-element ion chromatographic separation protocol. | ||
|
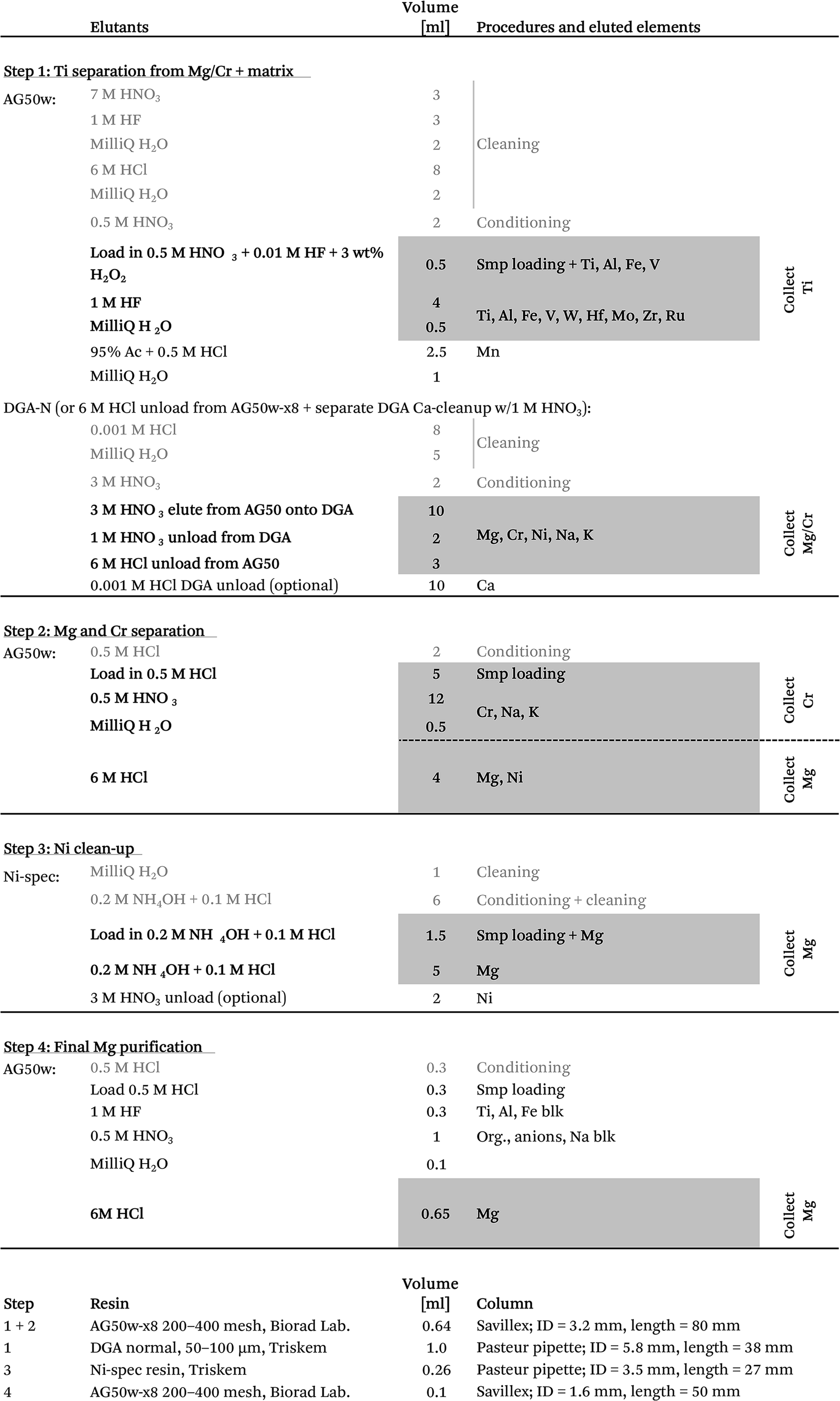
 | ||
| Fig. 2 Ion chromatographic elution profiles of elements separated on (a) the 1st separation column (A1) using a cation exchanger (AG50w-x8 200–400 mesh, ID = 3.2 mm, L = 80 mm) for eluting Al, Ti, Fe and Mn, followed by Mg, Cr, Ni, Na, K and Ca elution onto a faster-flowing DGA-N (50–100 μm) resin-filled anion exchange column, which retains Ca while eluting the other elements and (b) the 2nd Mg/Cr separation column (M1), which separates Cr (together with Na and K) from Mg and Ni by reusing the cation exchange column in A1. | ||
After the 5–7 day equilibration procedure described above, the sample was loaded directly onto the column in 0.5 M HNO3 + 3 wt% H2O2 + 0.01 M HF. Titanium was subsequently collected together with Fe, Al, V, Re, W and Mo by elution in 4 ml of 1 M HF (Table 1). This was followed by Mn elution using a 95% acetone + 0.5 M HCl mixture.24 Calcium was finally separated from the residual sample matrix by unloading Mg, Ca, Ni, Cr, Na and K from the cation exchanger using 10 ml of 3 M HNO3 and eluting directly onto a pre-cleaned faster-flowing Bio-Rad column containing 0.7 ml DGA-N (TrisKem) resin. The high affinity of Ca for the DGA resin in 3 M HNO3 (ref. 25) ensures its efficient separation from Mg, Cr, Ni, Na and K, which is collected for further purification. The pore-volumes of DGA and AG50w-x8 resin were subsequently emptied into the collected aliquot using 2 ml of 1 M HNO3 and 3 ml of 6 M HCl, respectively. Calcium can be unloaded from the DGA column using a few column volumes of 0.001 M HCl. Alternatively, Ca can be cleaned up in a separate step by eluting Mg, Na, K, Cr and Ni together with Ca from the cation exchanger in 4 ml of 6 M HCl (instead of 3 M HNO3 elution directly onto DGA) and later eluting Mg, Na, K, Cr and Ni through a 0.5 ml DGA column in 5 ml of 1 M HNO3, while efficiently retaining Ca. Chromatographic recoveries from this 1st column, as asserted on elution aliquots around the collected Ti and Mg/Cr cuts for different types of sample matrices (including BHVO-2, BIR-1, BCR-2, chondrites (CCs & OCs) and CAIs), were >99% for Ti and Cr and >99.95% for Mg.
:1) on a hotplate at 130 °C and subsequently converted to the Cl form using ∼6 M HCl in preparation for the final Mg purification step. The resin-bound Ni–DMG complex can be destroyed and Ni can be eluted with a few column volumes of an acidic solution, such as 2 M HNO3. The chromatographic Mg recoveries from this column were >99.99%.
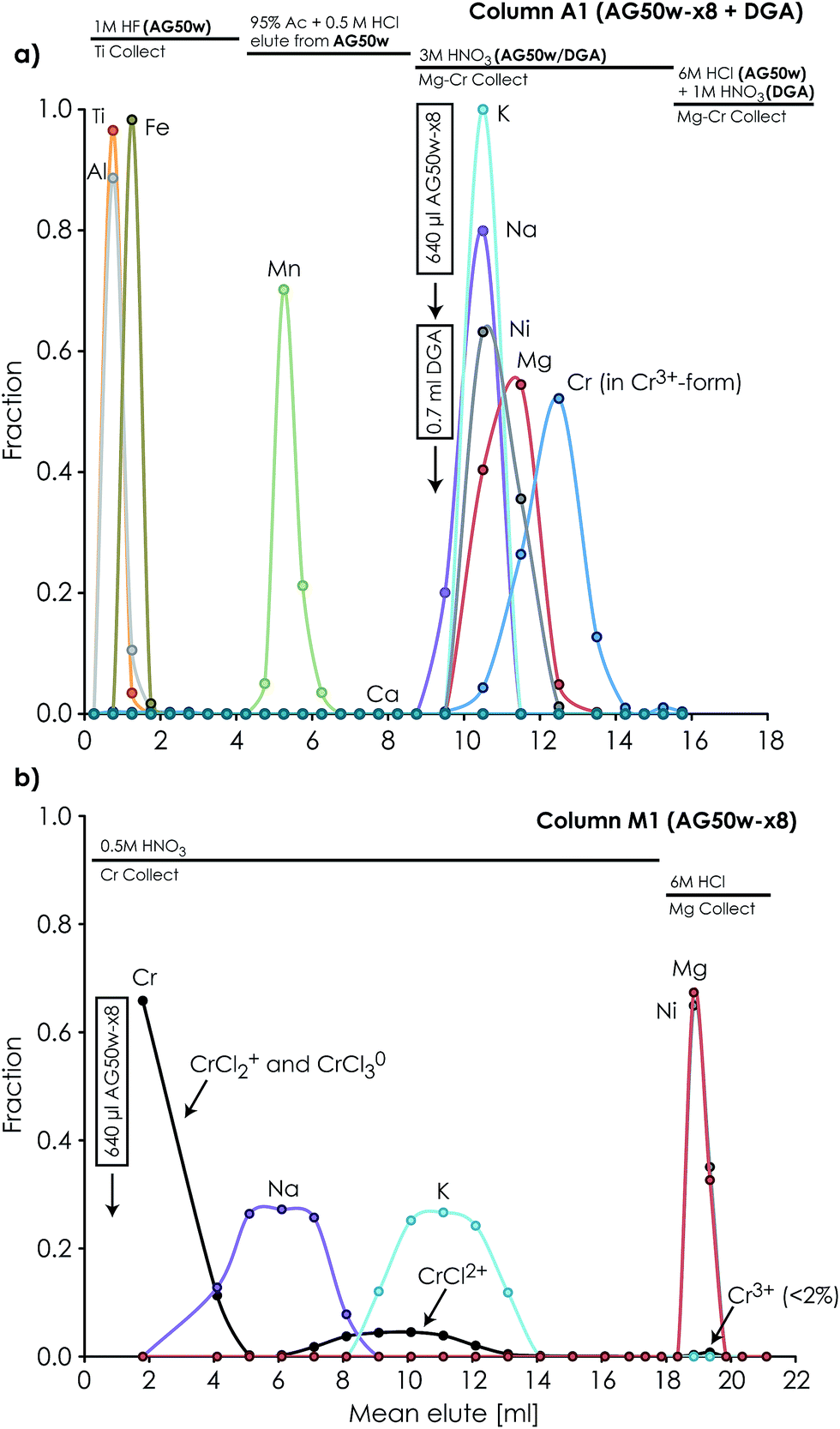
 | ||
| Fig. 3 Diagram showing the effect that impurity contribution (known as ‘blanks’ with isotopically normal composition) from chemical purification of an analyte has on the maximum isotope anomaly, which can be accurately determined within the stated uncertainty. For example, a procedural Ti ‘blank’ contribution of 0.8 ng introduced into a 1 μg Ti sample through chemical purification provides an anomaly/uncertainty contrast of 1250. With a stated external reproducibility of 12 ppm for μ50Ti*, this allows for the accurate analysis of materials with anomalies in μ50Ti* of up to 15000 ppm. | ||

|
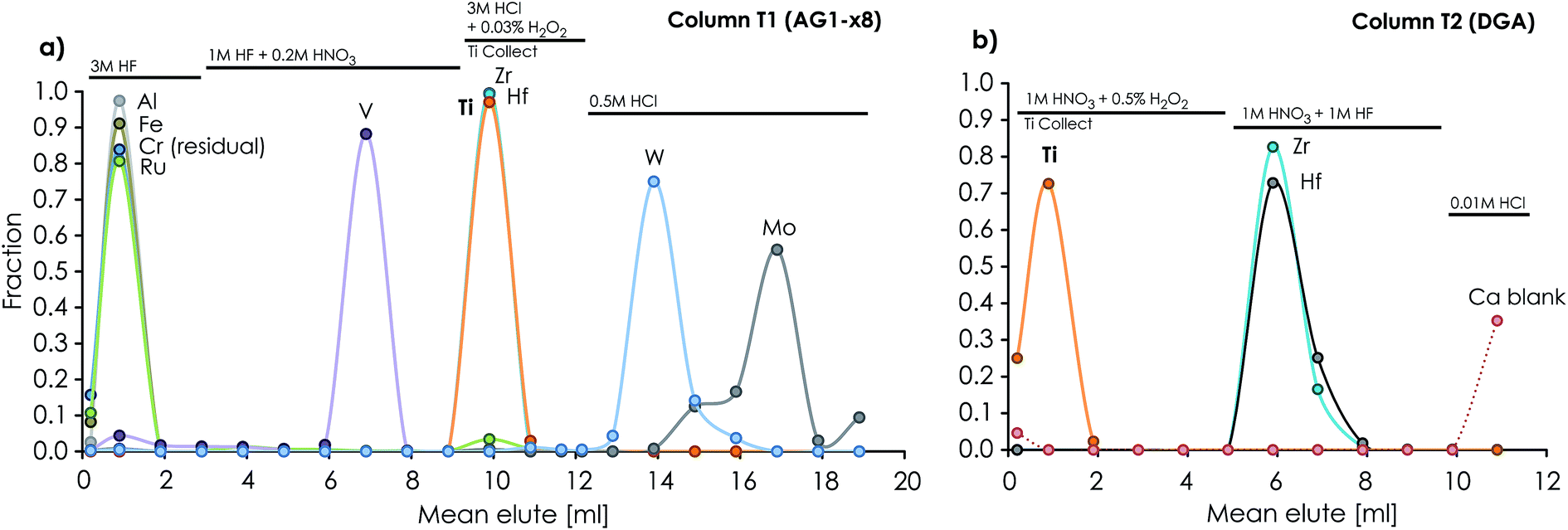 | ||
| Fig. 4 Ion chromatographic elution profiles of elements separated on (a) the anion exchanger (600 μl AG1-x8 200–400 mesh) column T1 (ID = 3.5 mm, L = 62 mm), which separates Ti (together with Zr and Hf) from Al, Fe, Ru, V, W and Mo, and (b) the 3rd Ti purification column (T2, ID = 3.5 mm, L = 27 mm) packed with 250 μl of DGA-N 50–100 μm resin. | ||
000 ppm (Fig. 3), i.e. well within the range of most solar system materials, including FUN-type CAIs.
3. Mass spectrometry
3.1 Mg isotope analysis using MC-ICP-MS
High-precision Mg isotope analysis was performed using the Thermo-Finnigan Neptune Plus MC-ICP-MS located at the Centre for Star and Planet Formation (Natural History Museum of Denmark, University of Copenhagen), using procedures based on Bizzarro et al. 2011.23 In brief, purified Mg was introduced into the plasma source by means of an Apex IR desolvating nebuliser in a 2% HNO3 run solution at 50 μl min−1 using a Thermo Fisher Jet sample cone and skimmer X-cone interface in high-resolution mode. The typical sensitivity of the instrument was ∼200 V ppm−1 of 24Mg and samples were typically analysed at ∼50 V. Magnesium isotope data were acquired in static mode using three Faraday collectors connected to amplifiers with 1011 ohm feedback resistors. Samples and standards were analyzed 10 times with each analysis consisting of 100 cycles of 16.78 second integrations separated by a total of 822 seconds of on-peak zero blank measurements in clean 2% HNO3 solution.Corrections for instrumental mass bias were obtained by sample–standard bracketing with sample/standard peak intensities matching to better than 5%. Stable Mg isotope ratios are reported in parts per million (ppm) deviation relative to the composition of the Mg reference standard DTS-2B27 according to: μiMg = [(iMg/24Mg)sample/(iMg/24Mg)DTS-2B − 1] × 106, where i = 25 or 26. The mass-independent μ26Mg* (radiogenic ingrowth and nucleosynthetic effects) is calculated as the deviations of the corrected 26Mg/24Mg from the reference standard, normalized to 25Mg/24Mg = 0.126896 (ref. 23), assuming that instrumental mass discrimination follows the exponential mass-fractionation law. Data reduction was conducted offline using the freeware software package Iolite.28
3.2 Ti isotope analysis using MC-ICP-MS
High-precision Ti isotope analysis was performed by standard–sample–standard bracketing using the Thermo-Finnigan Neptune Plus MC-ICP-MS located at the Centre for Star and Planet Formation (Natural History Museum of Denmark, University of Copenhagen). Following Ti purification, the samples were dissolved in 0.3 M HNO3 + 0.001 M HF and introduced into the plasma source by means of an Apex HF desolvating nebuliser (with Ar + N2) at a sample uptake rate of 30–45 μl min−1. To enhance ion transmission and sensitivity, we used a Thermo Fisher Jet sample cone and skimmer X-cone interface. The measurements were performed on the low mass side of peak shoulders. We used high mass resolution mode, defined as mass resolving power > 8000(m/(m0.95 − m0.05)), measured in the center cup. These conditions are sufficient to efficiently filter out slightly heavier polyatomic interference from e.g.36Ar14N+ on 50Ti, 14N216O+ or 22Ne+ on 44Ca, and 40Ar12C+ or 36Ar16O+ (+38Ar14N+) on 52Cr (shown in Fig. 5 for a 0.1 ppb solution of Ti, V and Cr); see Section 4.4 for detailed discussion on the measurement conditions. For a 250 ppb run solution, this resulted in a typical sensitivity of ∼12 V of 48Ti. Standard and sample intensities were matched to within 5% and tune settings were optimized before each analytical session to maximize the sensitivity and signal stability.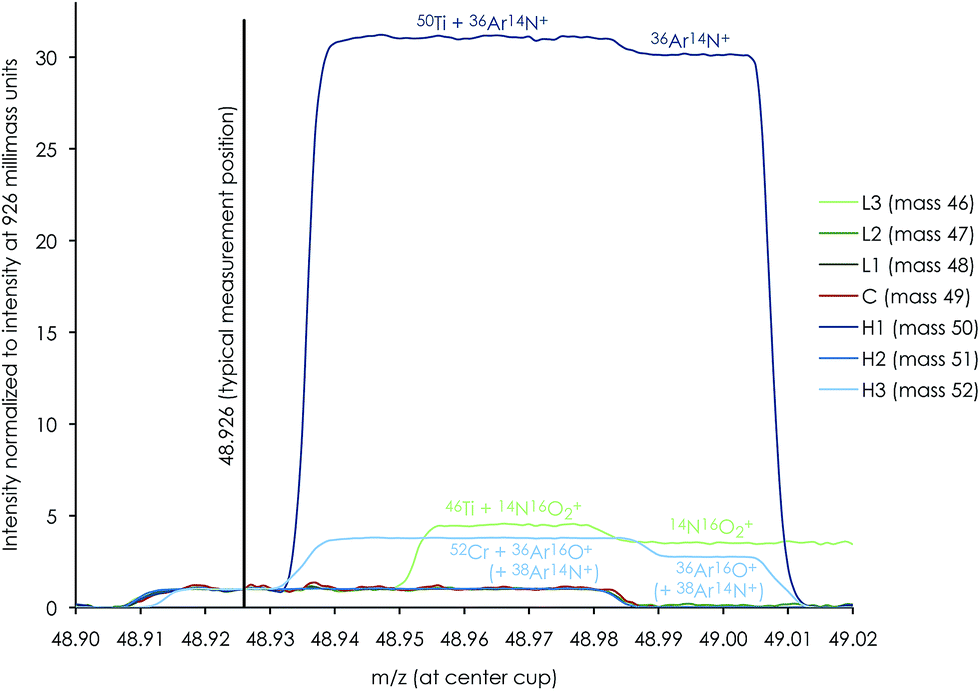 | ||
| Fig. 5 Mass peak scan profile of a dilute (0.1 ppb) mixture of Ti, V and Cr demonstrating that gas-based molecular interference from 36Ar14N+ on 50Ti, 14N16O2+ on 46Ti and 36Ar16O+ (+38Ar14N+) on 52Cr is clearly resolved from the typical measurement position at X.926 in high-resolution mode. The abundances of other potential molecular interference on the other titanium isotopes (47Ti, 48Ti and 49Ti) and 51V are negligible. | ||
Ti isotope data were acquired in dynamic mode by peak jumping using Faraday collectors connected to amplifiers with 1011 ohm feedback resistors for 46,47,48,49,50Ti, 51V, 52Cr and 44Ca and a 1013 ohm feedback resistor to monitor doubly charged 91Zr2+ at half-mass 45.5. Each sequence consisted of a number of repeated measurements on a sample bracketed by standards and blanks, each consisting of one cycle of 33.6 seconds of integration for 44Ca in line 1 followed by 200 cycles of 2.1 seconds of integration for Ti isotopes, 51V and 52Cr in line 2 (Table 3). A peak centering procedure was automatically performed prior to each standard analysis. With a sample uptake time of 100 seconds, this corresponds to a total effective analysis time of 8.9 hours for 10 repeats.

|
After correction for isobaric interference, stable Ti isotope ratios were calculated as parts per million (ppm) deviation from the composition of the Origins Laboratory Ti reference standard (OL-Ti)29 according to: μiTi = [(iTi/47Ti)sample/(iTi/47Ti)OL-Ti − 1] × 106, where i = 46, 48, 49 or 50. The mass-independent μ46Ti*, μ48Ti*, μ49Ti* and μ50Ti* components represent deviations in part per million (ppm) from the bracketing standard, OL-Ti, after correction for mass-dependent fractionation by internal normalization to either 49Ti/47Ti = 0.749766 or 48Ti/47Ti = 10.070565 using the exponential mass-fractionation law.10,18,30,31 Data reduction was conducted offline using the freeware software package Iolite.28
4. Results and discussion
4.1 Minimizing procedural blanks and optimizing the performance of chromatographic procedures
Due to potential matrix effects and isobaric interferences, isotope analysis by MC-ICP-MS requires sufficient ion exchange chromatographic purification of analytes to ensure measurement accuracy.32 The need for such chemical processing of samples prior to data acquisition, however, unavoidably results in small impurity contributions, known as ‘blanks’, introduced either through (1) release from the ion exchange resins, (2) the chemical reagents used for sample digestion and during chromatographic purification, or (3) other exogenous contributions introduced during sample handling. Blanks introduced through chromatographic purification can potentially dilute the true isotopic signature of the ‘purified’ element and thus generate false results. This delicate balance between chemical purification and blank contributions becomes increasingly important when handling samples that are highly isotopically anomalous. This is because of the large isotopic contrast between the sample and typical ‘Earth’-like blanks. Amongst some of the most isotopically anomalous samples are the first solids known to have formed in our solar system, namely Ca–Al-rich inclusions (CAIs) in primitive chondritic meteorites. These inclusions commonly exhibit large mass-dependent and/or mass-independent anomalies in the permil range in a wide range of elements and thus contain important isotopic fingerprints of the very earliest evolution of our solar system.10,15,33–37 As such, the chemical and analytical procedures described here were optimized to obtain accurate and precise isotope data from isotopically anomalous materials.Taking advantage of the increased precision of modern mass spectrometric techniques and thus the small amount of sample that can be precisely analysed simultaneously requires sufficient reduction of procedural blanks introduced through chemical processing. Minimizing procedural blanks demands a sufficient reduction in the amount of resins and acid used through chromatographic purification. Thus, in order to ensure effective separation of most of the sample matrix during the first purification step, the first column ensures a removal of >75% of matrix elements (Fe, Al, Ti, Ca, V and Mn) from Mg and Cr for a variety of rock samples (Table S1†). Ti is effectively separated from the residual matrix in its 2nd purification step, requiring subsequent removal of only Zr. This effectively minimizes the amount of resins and reagents required in the subsequent purification steps, thereby reducing major blank contributions.
The separation of Mg from elements that may generate doubly charged interference (e.g.48Ca2+, 48Ti2+, 50Ti2+, 50V2+, 50Cr2+ and 52Cr2+) and/or affect the instrumental mass bias of the sample as compared to the bracketing standard during analysis (e.g. Al, Fe, Na, Ni, Mn and Ca) is critical for reliable and accurate Mg isotope measurements using MC-ICP-MS.23,38 Ca is specifically known to be problematic for isotope analysis of meteoritic materials, as it may induce significant isobaric interference on mass 24 from doubly charged 48Ca and can result in apparent μ26Mg* anomalies.32,38 Thus, in handling sample matrices with high Ca levels compared to Mg, such as CAIs, efficient separation of Ca from Mg is important. Cation to Mg ratios were monitored in purified samples processed through the chromatographic separation protocol and found to be <∼0.001 (examples are given in Table S1†).
Accurate and precise mass spectrometric analysis of Ti is further compromised by direct isobaric interference from Ca (on masses 46 (46Ca ∼ 0.004%) and 48 (48Ca ∼ 0.187%)), V (on mass 50 (50V ∼ 0.2497%)) and Cr (on mass 50 (50Cr ∼ 4.3452%)) and potential doubly charged interference from Zr (on masses 46, 47 and 48), Mo (on all Ti isotopes) and Ru (on masses 48, 49 and 50) (Table 3). Therefore, the chromatographic purification procedure was designed to ensure sufficient separation of these elements from Ti. However, Ca blanks may be especially problematic, since Ca can be introduced through general sample handling from gloves and pipette tips. Thus, all chromatographic columns used in the purification of Ti were designed to separate potential Ca blanks introduced on the sample from Ti. The last column further ensures the effective separation of potential Ca and Cr blanks from the purified Ti fraction. Moreover, all reagents used in the chromatographic chemistry, as well as analytical solutions, were blank tested in order to ensure a sufficiently low Ca blank introduced on the samples. Finally, to eliminate potential interference from chlorides (35Cl14N+ on 49Ti, 35Cl15N+ and 1H14N35Cl+ on 50Ti, 36Cl15N+ and 35Cl16O+ on 51V and 35Cl16O1H+ and 35Cl17O+ on 52Cr), HCl-based reagents were avoided on the last two columns.
4.2 Efficiency of the multi-element ion chromatographic separation protocol
Our protocol allows for the efficient separation of multiple elements from the same sample aliquots (Fig. 1). Fig. 2 and 4 show the high resolution achieved for individual elution peaks as exemplified by a synthetic CAI composition. The elution profiles of other types of sample matrices (including a dunite (DTS-2B), basalt (BIR-1), lherzolite (J11), ordinary chondrite (Tennasilm), a CAI, and synthetic mixtures) were equally well resolved, demonstrating the reproducibility and efficiency of the chromatographic procedures in separating multiple elements from various types of sample matrices. The efficiency of the chromatographic protocol in separating elements other than Mg, Ti and Cr was evaluated by measuring K, Ca, V, Fe, Ni, Zr, Mo, Ru, Hf, and W eluted from the respective columns. Their recoveries were found to be near 100% for K, Ca, V, Fe, Ni, Zr, Mo, Hf and W, and >92% for Ru in their respective collection aliquots (Tables 1 and 2). The lower recovery of Ru most likely results from complications associated with chemical speciation and redox conditions in the eluting acid, thereby distributing Ru species with variable selectivity over the column. Prior to future isotope work on these elements, their purity in the collected aliquots needs to be addressed and further purification for some is required.4.3 Accuracy and reproducibility of Mg isotope measurements
The procedures described here for Mg purification and analysis are optimized to handle considerably smaller samples than previous high-precision Mg isotope protocols,23i.e. 20 times less. This is attained by minimizing the amount of resin and reagents used in the chemical processing of the sample and optimizing the chromatographic separation scheme, as well as by taking advantage of the Jet and X cone interface. Our results show that isotopically anomalous samples, such as CAIs, can be routinely analysed to an external long-term reproducibility for their stable Mg isotope composition of ±61 ppm (2sd) on μ25Mg and ±4.1 ppm for the mass-independent μ26Mg* component (Table 5). This is comparable to, although less precise, the protocols designed for high-precision Mg isotope analysis of larger samples.23 The typical internal standard error in each repeated measurement was on the order of 5–15 ppm for μ25Mg and 4–6 ppm for μ26Mg*, whereas the external precision (2se) for 10 repeated measurements on the same aliquot was on the order of 3–10 ppm for μ25Mg and 2–5 ppm for μ26Mg*. This level of precision is adequate to resolve the often very large isotope differences (a few ‰) between refractory inclusions, such as AOAs and CAIs, in primitive meteorites.15,33,34 5 μg Mg typically consumed during a full analysis corresponds to the amount of Mg typically found in a spherical CAI of about 350 μm in diameter. This allows for the measurement of the Mg isotope composition of inclusions in non-CV carbonaceous chondrites, such as CR, CO and CM chondrites, which have previously been inaccessible to high-precision isotope analysis by MC-ICP-MS due to their small sizes.An external reproducibility of 4.1 ppm on μ26Mg* restricts the Mg blank contribution (with μ26Mg* ∼ 0) allowed on a sample with e.g. a very extreme anomaly in μ26Mg* of 5000 ppm (such as a canonical CV CAI fragment with an anomalously high 27Al/24Mg ratio of 13.5) to <1‰. Thus, for typical 5 μg Mg required for high-precision isotope analysis following the present analytical procedures, this corresponds to a maximum blank level of 5 ng Mg (Fig. 3), which is fulfilled by our ion chromatographic procedures.
In addition to the reproducibility test, our analysis shows that three terrestrial rock standards (J11, BIR-1 and BCR-2) are all within the error of the bracketing standard DTS-2B (Table 5). Similarly, measurements on a range of chondritic meteorites (CI, CV, CR and OC) and another CAI (31E) are within the error of other high-precision μ25Mg and μ26Mg* data previously reported in ref. 3, 8, 15 and 39. This demonstrates the accuracy of our methods.
4.4 Optimization of Ti mass-spectrometric analytical protocols
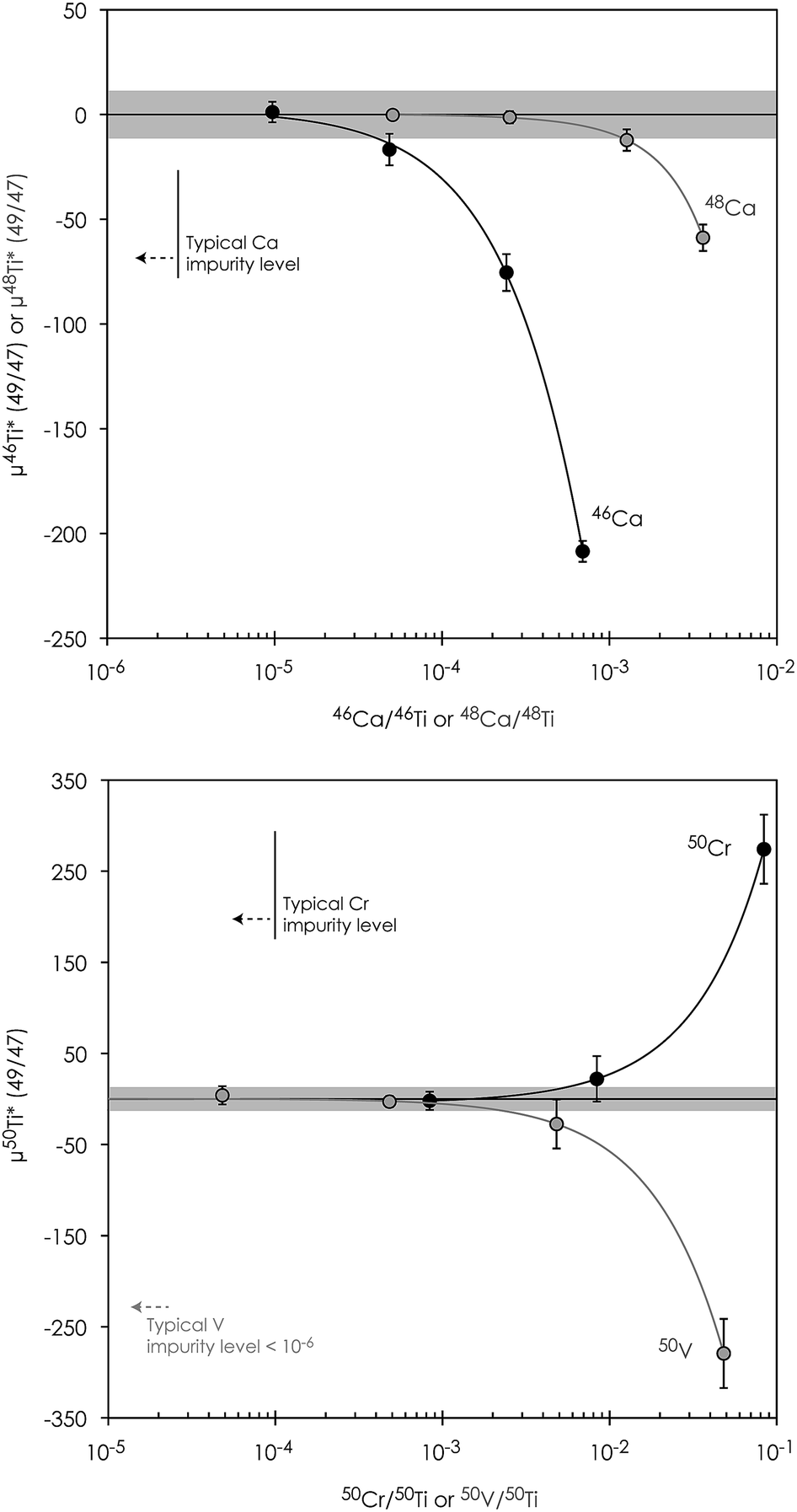 | ||
| Fig. 6 Titanium isotope composition of a Ca-, V- and Cr-doped Alfa Aesar Ti standard solution after interference correction showing their impact on the accuracy of Ti isotope measurements. Grey shaded regions represent the external reproducibility (2sd) on μ46Ti*, μ46Ti* and μ46Ti* as asserted on repeated measurements on the Allende CV CAI ‘A3’. Error bars represent the 2se external error in individual runs. | ||
| Atomic ratio | μ49Ti | μ46Ti* (49/47) | μ48Ti* (49/47) | μ49Ti* (48/47) | μ50Ti* (49/47) | Na |
|---|---|---|---|---|---|---|
| a Number of repeated measurements. | ||||||
| Zr/Ti = 0.002 | 7 ± 63 | 1.3 ± 8.1 | −0.6 ± 3.5 | 1.3 ± 7.0 | −5.8 ± 4.3 | 3 |
| Zr/Ti = 0.0005 | 11 ± 23 | 10.0 ± 10.0 | −1.8 ± 3.3 | 3.6 ± 6.5 | −14.0 ± 11.0 | 7 |
| Mo/Ti = 0.01 | 29 ± 33 | 19.0 ± 21.0 | −0.2 ± 5.7 | 0.0 ± 11.0 | −12.6 ± 8.2 | 3 |
| Mo/Ti = 0.001 | 1 ± 19 | −5.8 ± 6.6 | −0.6 ± 5.3 | 1.0 ± 10.0 | −0.1 ± 8.4 | 10 |
| Ru/Ti = 0.001 | 2 ± 16 | −4.8 ± 9.0 | −1.1 ± 3.0 | 2.2 ± 5.9 | −5.5 ± 7.5 | 10 |
| Sample | Type of material | μ25Mg | μ26Mg* | Na |
|---|---|---|---|---|
| a Number of repeated measurements. | ||||
| Terrestrial rock standards and samples | ||||
| J11(1) | Olivine separated from spinel lherzolite | −13 ± 4 | −1.1 ± 5.5 | 10 |
| J11(2) | Olivine separated from spinel lherzolite | −19 ± 19 | −4.3 ± 4.2 | 10 |
| BIR-1 | Icelandic basalt, USGS rock standard | 33 ± 4 | −0.4 ± 2.4 | 10 |
| BCR-2 | Basalt, Columbia River, USGS rock standard | 0 ± 3 | −1.3 ± 1.3 | 10 |
|
||||
| Meteoritic samples | ||||
| Ivuna | CI chondrite | 12 ± 5 | 4.2 ± 2.1 | 10 |
| Allende | CV3ox chondrite | 1 ± 8 | 10.4 ± 3.1 | 10 |
| Alfianello | Ordinary chondrite (L6) | 25 ± 3 | −4.0 ± 2.6 | 10 |
| Tennasilm | Ordinary chondrite (L4) | 4 ± 26 | −4.3 ± 1.3 | 10 |
| NWA 6043 | CR2 chondrite | 14 ± 4 | −4.6 ± 2.2 | 10 |
| 31E | CAI from Efremovka (CV3red) | 6050 ± 7 | 1166.2 ± 4.2 | 10 |
|
||||
| Reproducibility test | ||||
| A3 (1) | CAI from Allende (CV3ox) | −1486 ± 8 | 1321.2 ± 4.9 | 10 |
| A3 (2) | −1512 ± 10 | 1323.5 ± 2.7 | 10 | |
| A3 (3) | −1501 ± 10 | 1321.0 ± 3.8 | 10 | |
| A3 (4) | −1442 ± 3 | 1318.5 ± 4.6 | 10 | |
| Average and 2sd | −1485 ± 61 | 1321.1 ± 4.1 | ||
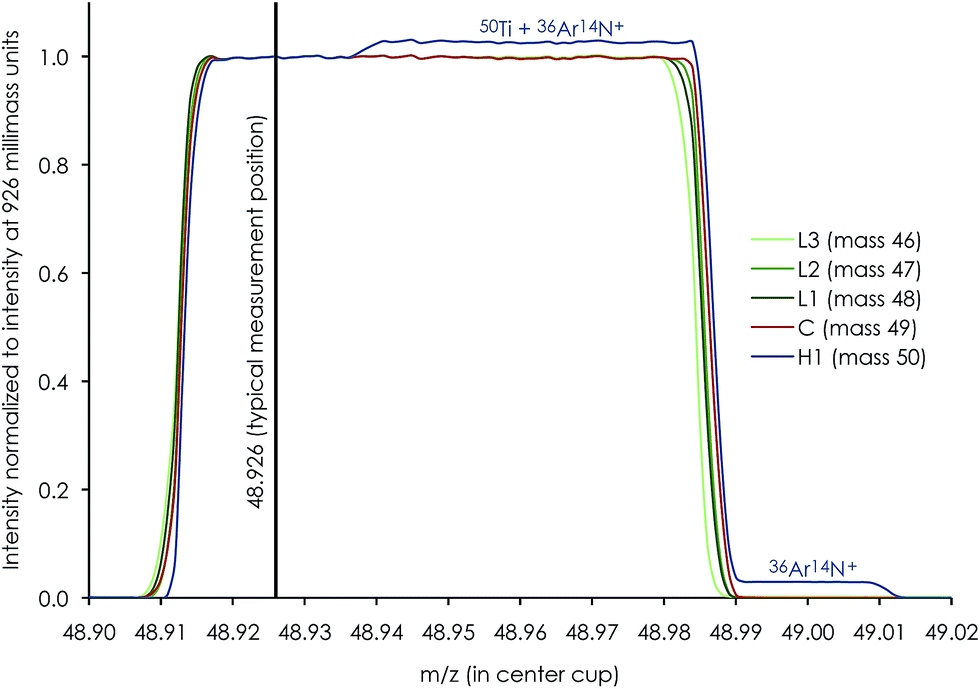 | ||
| Fig. 7 Mass peak scan profile of a 250 ppb Ti run solution (0.3 M HNO3 + 0.001 M HF) showing the alignment of individual Ti cups and a sufficiently flat low-mass shoulder plateau of >15 millimass units between peak edges and interference from 36Ar14N+ on 50Ti in high-resolution mode. The typical measurement position at X.926 is positioned in the middle of this plateau. | ||
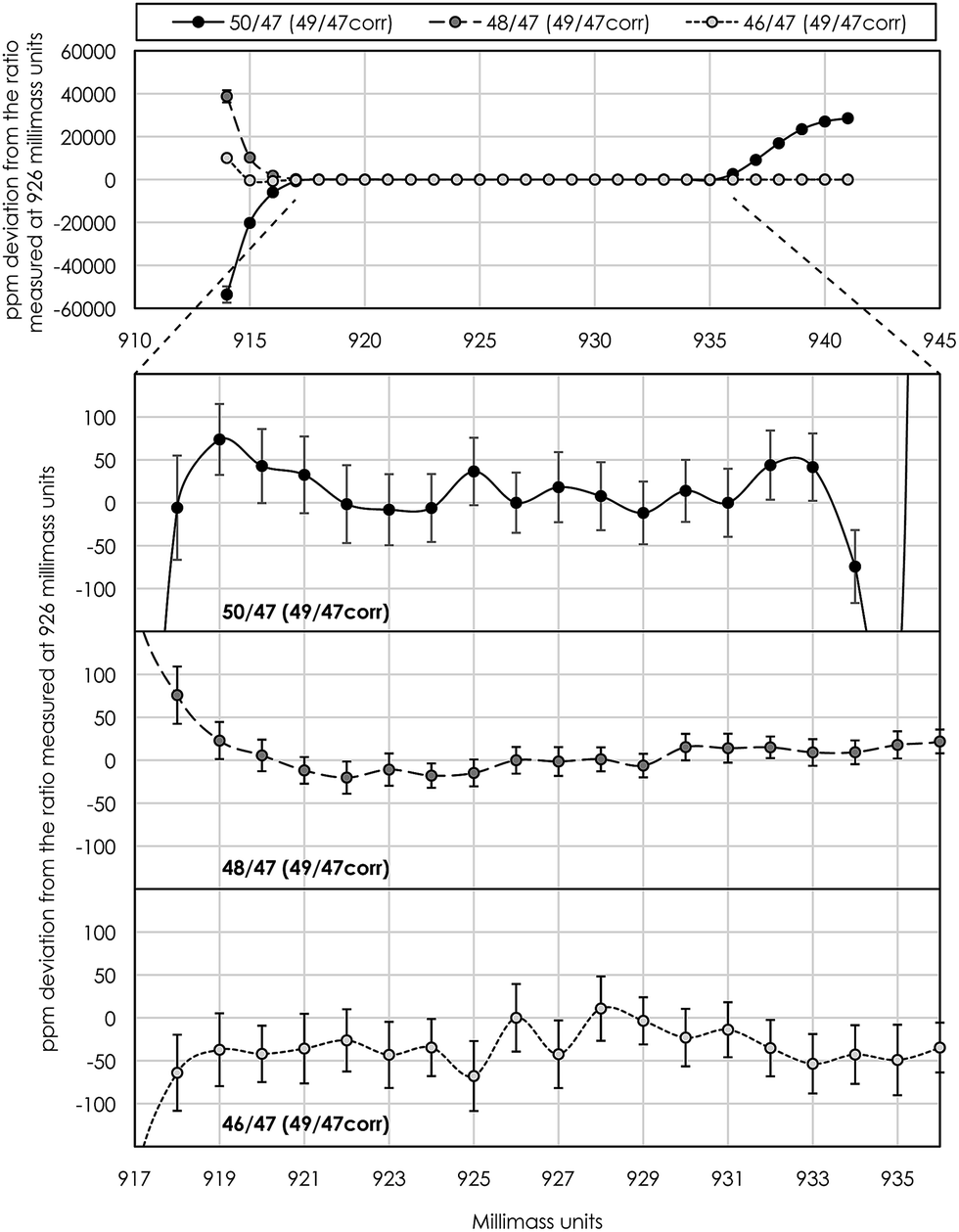 | ||
| Fig. 8 An example of a ‘peak-shoulder-flatness test’ used for asserting the mass range on the low-mass side of peaks where the internally 49Ti/47Ti normalized mass-fractionation corrected isotope ratios approach constant values, i.e. a flat plateau. The typical measurement position at 926 millimass units is in the middle of this flat shoulder plateau, which was acquired by repeated scans over peaks at 8.4 seconds of integration for 1 millimass unit increments from 914 to 941 for a duration of ∼15 minutes. Top panel shows the internally normalized mass-fractionation corrected Ti isotope ratios measured over the full mass range. Ratios diverge to extreme values at low-mass peak edges due to small misalignments of individual cups of a few millimass units. The 50Ti/47Ti ratio becomes anomalously high at >935 millimass units because of gas-based interference from 36Ar14N+ on 50Ti. The three lower panels show the finer details of the flat plateau from 917 to 936 millimass units. This routine was performed prior to each analytical session and the measurement position was adjusted if necessary, thereby ensuring the accuracy, precision and reproducibility of our isotope analysis. Error bars represent the 2se at each mass position. | ||
Instrumental drift due to magnet instabilities between two peak centers (prior to each standard measurement) was typically less than 1 millimass unit, which is much less than the peak-shoulder-flat plateau of >15 millimass units on the low-mass shoulder. With a typical measurement position in the middle of the shoulder plateau at X.926 mass units, this ensures a flat peak plateau of >7 millimass units on either side of the shoulder measurement position, i.e. sufficient to neglect measurement inaccuracy associated with magnet instabilities during an analytical session.
Another obstacle towards optimizing measurement precision during Ti isotope analysis in dynamic peak jumping mode is the idle times (prior to actual measurement) needed for amplifiers and magnets to settle when jumping between peaks in two cup configuration lines. Conventional analytical protocols for Ti isotope analysis utilize consecutive peak-jumping between two cup configuration lines for a given number of cycles,10,18 thereby measuring each line an equal number of times. This approach, however, uses a relatively large volume of sample solution because of the idle time necessary for magnet stabilization between two peak jumps. Our analysis shows that an idle time of at least 3 seconds is required for magnet and amplifier stabilization (Fig. 9). For 200 cycles at 2.1 second integration with 10 repeated measurements at 250 ppb Ti, this corresponds to 3–4 μg of Ti consumed during a full analysis. Thus, in order to minimize sample consumption during analysis, without compromising the time spend on the actual Ti isotope measurements (as well as V and Cr) in line 2, we measured the Ca interference in line 1 only once at the beginning of each repeated measurement at 33.6 seconds of integration. This limits the amount of Ti consumed per 10 repeats to 0.75–1 μg, i.e. 4 times less than that by the conventional mode. Hence, the precision of the actual Ti isotope measurements stays the same, because all Ti isotopes are measured in line 2. Although compromising the effectiveness of the Ca interference correction routine, considerably less material is being consumed. This is highly desirable when dealing with very small and unique samples, such as sub-mm-sized CAIs.
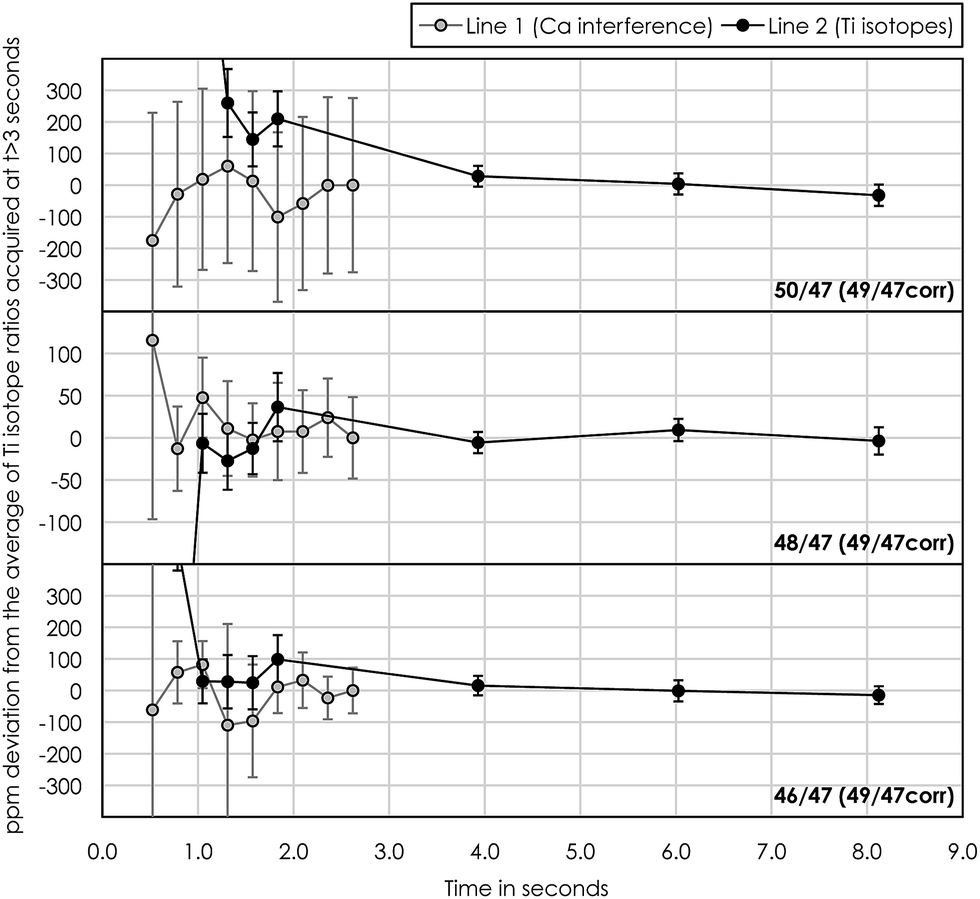 | ||
| Fig. 9 Test showing the influence of the idle time necessary for the magnet and amplifiers to settle prior to the actual isotope measurements when jumping between peaks in lines 1 (Ca interference) and 2 (Ti isotopes). Internally 49Ti/47Ti normalized mass-fractionation corrected Ti isotope ratios approach constant values after an idle time of ∼3 seconds. | ||
000 ppm accuracy limitation on μ50Ti* of our Ti procedures (Fig. 3) fully allows the accurate determination of minor inner solar system isotope signatures, as well as the extreme isotopic signatures in FUN-type CAIs and most hibonite crystals found in carbonaceous chondrites.7,40,41 Finally, both stable and mass-independent Ti isotope data for terrestrial geostandards (BIR-1, BCR-2 and BHVO-2) are normal within error, demonstrating the accuracy of our methods.
| Sample | Type of material | μ49Ti | μ46Ti* (49/47) | μ48Ti* (49/47) | μ49Ti* (48/47) | μ50Ti* (49/47) | N a |
|---|---|---|---|---|---|---|---|
| a Number of repeated measurements. | |||||||
| Terrestrial rock standards and samples | |||||||
| Alfa Aesar Ti | Pure Ti ICP standard from Alfa Aesar | 89 ± 16 | −9.5 ± 7.5 | 2.0 ± 5.6 | −5.0 ± 10.0 | −3.6 ± 8.6 | 10 |
| BIR-1 (1) | Icelandic basalt, USGS rock standard | −24 ± 12 | −11.9 ± 7.6 | 0.3 ± 4.0 | −0.6 ± 8.0 | −2.3 ± 5.9 | 10 |
| BIR-1 (2) | Icelandic basalt, USGS rock standard | −40 ± 19 | −3.7 ± 8.4 | −1.3 ± 5.3 | 3.0 ± 10.0 | −3.0 ± 9.2 | 10 |
| BCR-2 | Basalt, Columbia River, USGS rock standard | −38 ± 30 | −8.3 ± 5.0 | 2.9 ± 2.6 | −6.1 ± 5.2 | −3.8 ± 7.7 | 10 |
| BHVO-2 (1) | Basalt, Hawaiian Volcanic Observatory, USGS rock standard | 10 ± 37 | 4.5 ± 6.6 | 1.4 ± 2.9 | −2.7 ± 5.7 | −2.9 ± 7.0 | 10 |
| BHVO-2 (2) | Basalt, Hawaiian Volcanic Observatory, USGS rock standard | 56 ± 11 | −2.5 ± 5.3 | 0.9 ± 3.6 | −1.9 ± 7.2 | −7.6 ± 5.2 | 10 |
|
|||||||
| Meteoritic samples | |||||||
| Alfianello | Ordinary chondrite (L6) | 180 ± 21 | −15.3 ± 8.5 | −0.2 ± 5.9 | 0.0 ± 12.0 | −63.8 ± 9.5 | 8 |
| NWA 6043 | CR2 chondrite | 1 ± 42 | 29.9 ± 9.6 | −14.0 ± 2.7 | 27.7 ± 5.4 | 228.0 ± 10.0 | 10 |
| 31E | CAI from Efremovka (CV3red) | 270 ± 67 | 169.5 ± 4.6 | 34.4 ± 3.5 | −68.2 ± 7.0 | 947.0 ± 12.0 | 10 |
|
|||||||
| Reproducibility test | |||||||
| A3 (1) | CAI from Allende (CV3ox) | 479 ± 26 | 223.0 ± 10.0 | 8.7 ± 1.7 | −17.2 ± 3.4 | 1459.5 ± 4.3 | 10 |
| A3 (2) | 406 ± 23 | 232.6 ± 7.9 | 8.5 ± 2.9 | −16.9 ± 5.8 | 1460.0 ± 10.0 | 10 | |
| A3 (3) | 522 ± 32 | 224.0 ± 13.0 | 1.6 ± 8.4 | −3.0 ± 17.0 | 1469.8 ± 9.4 | 10 | |
| A3 (4) | 397 ± 44 | 223.0 ± 11.0 | 12.1 ± 2.7 | −23.9 ± 5.4 | 1470.0 ± 11.0 | 10 | |
| Average and 2sd | 451 ± 120 | 228.2 ± 10.8 | 7.7 ± 8.8 | −15.3 ± 17.6 | 1464.8 ± 11.7 | ||
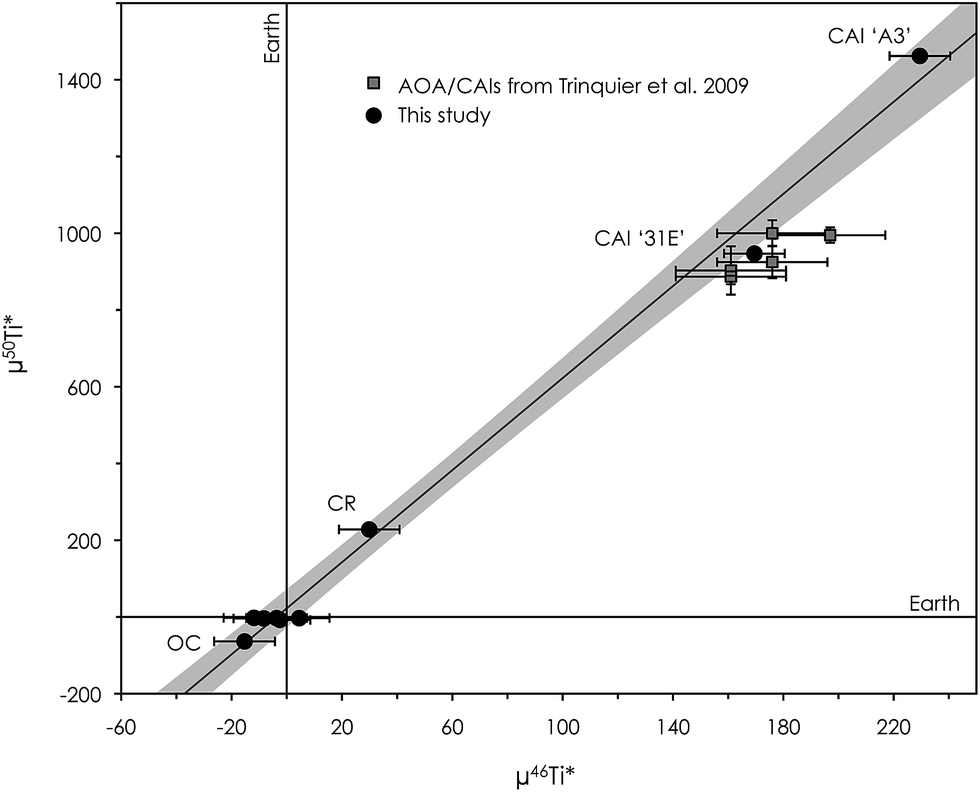 | ||
| Fig. 10 μ50Ti* versus μ46Ti* diagram showing the correlation between inner solar system materials measured in this study (Earth, one ordinary chondrite (Alfianello) and one CR chondrite (NWA 6043)), which is in accordance with the observations of Trinquier et al. 2009 and Zhang et al. 2011. The correlation extends to include Ca–Al-rich inclusions (CAIs) with divergent anomalies. The CAI, ‘31E’, from the Efremovka CV chondrite has a Ti isotope composition similar to that reported for CAIs and AOAs in Trinquier et al. 2009, while the Allende CV CAI, ‘A3’, shows significantly larger anomalies in both μ50Ti* and μ46Ti*. Error bars are the internal 2se precision or external reproducibility (2sd), whichever is larger. | ||
Conclusions
We have developed a new multi-element ion chromatographic separation protocol, specifically tailored to handle small amounts of isotopically anomalous material, such as Ca–Al-rich inclusions in primitive meteorites, for which impurity contributions must be minimized. We present refined analytical procedures for high-precision Mg and Ti isotope analysis using MC-ICP-MS. The high-precision isotope analysis results of a range of isotopically anomalous chondritic meteorites and refractory inclusions are identical to data previously reported for the same types of materials, demonstrating the accuracy of our methods. Data for terrestrial geostandards are within the error of the OL-Ti standard used for standard–sample bracketing. We show that the protocols allow for the accurate determination of the extreme mass-dependent and mass-independent Mg and Ti isotopic signatures found in both normal CAIs and FUN-type CAIs, as well as some anomalous hibonite crystals in CM chondrites (μ26Mg* up to ±4000 ppm and μ50Ti* up to 15000 ppm). Finally, our results support the observation of correlated anomalies in μ46Ti* and μ50Ti* for inner solar system materials, which extend to include two CAIs (31E and A3) with highly divergent mass-independent Ti isotope compositions. Such effects cannot be explained by mass-dependent stable isotope fractionation and must invoke variable nucleosynthetic effects in CAIs.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Marc-Alban Millet and Christophe Cloquet for providing the OL-Ti standard. Funding for this project was provided through grants from the Danish National Research Foundation (grant number DNRF97) and from the European Research Council (ERC Consolidator grant agreement 616027-STARDUST2ASTEROIDS) to M. B.Notes and references
- F.-Z. Teng, Rev. Mineral. Geochem., 2017, 82, 219–287 CrossRef.
- J. N. Connelly, M. Bizzarro, A. N. Krot, Å. Nordlund, D. Wielandt and M. A. Ivanova, Science, 2012, 338, 651–655 CrossRef CAS PubMed.
- K. K. Larsen, M. Schiller and M. Bizzarro, Geochim. Cosmochim. Acta, 2016, 176, 295–315 CrossRef CAS PubMed.
- M.-A. Millet, N. Dauphas, N. D. Greber, K. W. Burton, C. W. Dale, B. Debret, C. G. Macpherson, G. M. Nowell and H. M. Williams, Earth Planet. Sci. Lett., 2016, 449, 197–205 CrossRef CAS.
- N. D. Greber, N. Dauphas, A. Bekker, M. P. Ptáček, I. N. Bindeman and A. Hofmann, Science, 2017, 357, 1271–1274 CrossRef CAS PubMed.
- M. Schiller, J. A. Dallas, J. Creech, M. Bizzarro and J. A. Baker, Geochemical Perspectives Letters, 2017, 22–31 CrossRef.
- M.-C. Liu, K. D. McKeegan, J. N. Goswami, K. K. Marhas, S. Sahijpal, T. R. Ireland and A. M. Davis, Geochim. Cosmochim. Acta, 2009, 73, 5051–5079 CrossRef CAS.
- E. M. M. E. Van Kooten, D. Wielandt, M. Schiller, K. Nagashima, A. Thomen, K. K. Larsen, M. B. Olsen, Å. Nordlund, A. N. Krot and M. Bizzarro, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 2011–2016 CrossRef CAS PubMed.
- S. Gerber, C. Burkhardt, G. Budde, K. Metzler and T. Kleine, Astrophys. J., 2017, 841, L17 CrossRef.
- A. Trinquier, T. Elliott, D. Ulfbeck, C. Coath, A. N. Krot and M. Bizzarro, Science, 2009, 324, 374–376 CrossRef CAS PubMed.
- F. R. Niederer, D. A. Papanastassiou and G. J. Wasserburg, Astrophys. J., 1980, 240, L73–L77 CrossRef CAS.
- V. Mavromatis, A. Immenhauser, D. Buhl, B. Purgstaller, A. Baldermann and M. Dietzel, Geochim. Cosmochim. Acta, 2017, 207, 139–153 CrossRef CAS.
- A. M. Davis and F. M. Richter, in Treatise on Geochemistry, Elsevier Ltd., 2nd edn, 2014, vol. 1, pp. 335–360 Search PubMed.
- J. Zhang, S. Huang, A. M. Davis, N. Dauphas, A. Hashimoto and S. B. Jacobsen, Geochim. Cosmochim. Acta, 2014, 1–47 Search PubMed.
- K. K. Larsen, A. Trinquier, C. Paton, M. Schiller, D. Wielandt, M. A. Ivanova, J. N. Connelly, Å. Nordlund, A. N. Krot and M. Bizzarro, Astrophys. J., Lett., 2011, 735, L37 CrossRef.
- M. Schiller, J. N. Connelly, A. C. Glad, T. Mikouchi and M. Bizzarro, Earth Planet. Sci. Lett., 2015, 420, 45–54 CrossRef CAS PubMed.
- J. Bollard, J. N. Connelly, M. J. Whitehouse, E. A. Pringle, L. Bonal, J. K. Jørgensen, Å. Nordlund, F. Moynier and M. Bizzarro, Sci. Adv., 2017, 3, e1700407 CrossRef PubMed.
- J. Zhang, N. Dauphas, A. M. Davis and A. Pourmand, J. Anal. At. Spectrom., 2011, 26, 2197 RSC.
- D. Wielandt and M. Bizzarro, J. Anal. At. Spectrom., 2011, 26, 366 RSC.
- N. S. Saji, D. Wielandt, C. Paton and M. Bizzarro, J. Anal. At. Spectrom., 2016, 1–20 Search PubMed.
- F. Nelson, T. Murase and K. A. Kraus, J. Chromatogr., 1964, 13, 503–535 CrossRef CAS PubMed.
- K. K. Larsen, D. Wielandt, M. Schiller and M. Bizzarro, J. Chromatogr. A, 2016, 1443, 162–174 CrossRef CAS PubMed.
- M. Bizzarro, C. Paton, K. K. Larsen, M. Schiller, A. Trinquier and D. Ulfbeck, J. Anal. At. Spectrom., 2011, 26, 565 RSC.
- F. W. E. Strelow, A. H. Victor, C. R. Van Zyl and C. Eloff, Anal. Chem., 1971, 43, 870–876 CrossRef CAS.
- A. Pourmand and N. Dauphas, Talanta, 2010, 81, 741–753 CrossRef CAS PubMed.
- A. Makishima, X.-K. Zhu, N. S. Belshaw and R. K. O'Nions, J. Anal. At. Spectrom., 2002, 17, 1290–1294 RSC.
- M. B. Olsen, M. Schiller, A. N. Krot and M. Bizzarro, Astrophys. J., 2013, 776, L1 CrossRef.
- C. Paton, J. Hellstrom, B. Paul, J. Woodhead and J. Hergt, J. Anal. At. Spectrom., 2011, 26, 2508 RSC.
- M.-A. Millet and N. Dauphas, J. Anal. At. Spectrom., 2014, 29, 1444–1458 RSC.
- I. Leya, M. Schönbächler, U. Wiechert, U. Krähenbühl and A. N. Halliday, Int. J. Mass Spectrom., 2007, 262, 247–255 CrossRef CAS.
- A. M. Davis, J. Zhang, N. D. Greber, J. Hu, F. L. H. Tissot and N. Dauphas, Geochim. Cosmochim. Acta, 2017, 275–295 Search PubMed.
- A. Galy, N. S. Belshaw, L. Halicz and R. K. O'Nions, Int. J. Mass Spectrom., 2001, 208, 89–98 CrossRef CAS.
- T. Lee, D. A. Papanastassiou and G. J. Wasserburg, Astrophys. J., Lett., 1977, 211, L107–L110 CrossRef CAS.
- M. Bizzarro, J. A. Baker and H. Haack, Nature, 2004, 431, 275–278 CrossRef CAS PubMed.
- D. Wielandt, K. Nagashima, A. N. Krot, G. R. Huss, M. A. Ivanova and M. Bizzarro, Astrophys. J., 2012, 748, L25 CrossRef.
- J. Holst, M. B. Olsen, C. Paton, K. Nagashima, M. Schiller, D. Wielandt, K. K. Larsen, J. N. Connelly, J. K. Jørgensen, A. N. Krot, Å. Nordlund and M. Bizzarro, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8819–8823 CrossRef CAS PubMed.
- M. Schiller, C. Paton and M. Bizzarro, Geochim. Cosmochim. Acta, 2015, 149, 88–102 CrossRef CAS PubMed.
- M. Schiller, J. A. Baker and M. Bizzarro, Geochim. Cosmochim. Acta, 2010, 74, 4844–4864 CrossRef CAS.
- R. C. Hin, C. D. Coath, P. J. Carter, F. Nimmo, Y.-J. Lai, P. A. E. P. von Strandmann, M. Willbold, Z. M. Leinhardt, M. J. Walter and T. Elliott, Nature, 2017, 549, 511–515 CrossRef PubMed.
- T. R. Ireland, Geochim. Cosmochim. Acta, 1990, 54, 3219–3237 CrossRef CAS.
- L. Kööp, D. Nakashima, P. R. Heck, N. T. Kita, T. J. Tenner, A. N. Krot, K. Nagashima, C. Park and A. M. Davis, Geochim. Cosmochim. Acta, 2017, 1–44 Search PubMed.
- J. Zhang, N. Dauphas, A. M. Daivs, I. Leya and A. Fedkin, Nat. Geosci., 2012, 5, 251–255 CrossRef CAS.
- C. D. Williams, P. E. Janney, R. R. Hines and M. Wadhwa, Chem. Geol., 2016, 436, 1–10 CrossRef CAS.
- J. I. Simon, M. K. Jordan, M. J. Tappa, E. A. Schauble, I. E. Kohl and E. D. Young, Earth Planet. Sci. Lett., 2017, 472, 277–288 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7ja00392g |
| This journal is © The Royal Society of Chemistry 2018 |