
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Layered gadolinium hydroxides for simultaneous drug delivery and imaging†
Yadong
Xu
a,
Alvaro
Goyanes
a,
Yuwei
Wang
a,
Andrew J.
Weston
a,
Po-Wah
So
 b,
Carlos F. G. C.
Geraldes
c,
Andrew M.
Fogg
*d,
Abdul W.
Basit
a and
Gareth R.
Williams
*a
b,
Carlos F. G. C.
Geraldes
c,
Andrew M.
Fogg
*d,
Abdul W.
Basit
a and
Gareth R.
Williams
*a
aUCL School of Pharmacy, University College London, 29-39 Brunswick Square, London, WC1N 1AX, UK. E-mail: g.williams@ucl.ac.uk; Tel: +44(0) 207 753 5868
bDepartment of Neuroimaging, Institute of Psychiatry, Psychology and Neuroscience, King's College London, Maurice Wohl Clinical Neuroscience Institute, 5 Cutcombe Road, London, SE5 9RX, UK
cDepartment of Life Sciences and Coimbra Chemistry Center, Faculty of Science and Technology, University of Coimbra, Coimbra, Portugal
dDepartment of Chemical Engineering, University of Chester, Thornton Science Park, Cheshire, CH2 4NU, UK. E-mail: a.fogg@chester.ac.uk; Tel: +44 (0)1244 512516
First published on 11th January 2018
Abstract
The potential of the layered gadolinium hydroxide (LGdH) [Gd2(OH)5]Cl·yH2O (LGdH-Cl) for simultaneous drug delivery and magnetic resonance imaging was explored in this work. Three non-steroidal anti-inflammatory drugs (diclofenac [dic], ibuprofen [ibu], and naproxen [nap]) were intercalated into LGdH-Cl for the first time, using three different routes (ion exchange intercalation, coprecipitation, and exfoliation-self-assembly). X-ray diffraction, elemental microanalysis and IR spectroscopy confirmed successful incorporation of the drug into the interlayer spaces of the LGdH in all cases. From a comparison of the guest anion sizes and interlayer spacings, the active ingredients are believed to adopt intertwined bilayer configurations between the LGdH layers. The materials prepared by coprecipitation in general have noticeably higher drug loadings than those produced by ion exchange or self-assembly, as a result of the incorporation of some neutral drug into the composites. The LGdH-drug intercalates are stable at neutral pH, but rapidly degrade in acidic conditions to free Gd3+ into solution. While LGdH-nap releases its drug loading into solution very rapidly (within ca. 1.5 h) at pH 7.4, LGdH-dic shows sustained release over 4 h, and LGdH-ibu extends this to 24 h. The latter composites therefore can be incorporated into enteric-coated tablets to provide sustained release in the small intestine. The drug intercalates are highly biocompatible and retain the proton relaxivity properties of the parent LGdH-Cl, with the materials most promising for use as negative contrast agents in MRI. Overall, the LGdH-drug intercalation compounds appear to have great potential for use in theranostic applications.
Introduction
Layered materials have attracted a great deal of attention in the literature. Such systems typically contain two-dimensional networks of metal cations surrounded by anions, and a three-dimensional lamellar structure is formed by the stacking of these layers.1 This structure endows layered materials with a rich interlayer chemistry, and the ability to be exfoliated into individual layers. If the layers carry an overall charge, ions are located between them to balance this. The latter are frequently exchangeable, and such materials can be divided into two broad classes: cation-exchange and anion-exchange systems. In comparison to the wealth of cation-exchangeable layered materials (e.g. smectite clays,2 metal phosphates and phosphonates,1 aluminophosphates3), anion-exchangeable systems are much less common.In anion-exchangeable layered systems, atoms in the host layers interact with each other through covalent bonding to form sheets bearing an overall positive charge. This is balanced by anions located between the layers, and electrostatic interactions with these hold the layers together in a three-dimensional stack.4 The most commonly explored class of such materials are the layered double hydroxides (LDHs). These contain positively charged mixed-metal hydroxide layers and can be described with the generic formula [M2+1−xM3+x(OH)2][An−]x/n·yH2O, where usually M2+ = Ca2+, Mg2+, Zn2+, Ni2+ and M3+ = Al3+, Ga3+, Fe3+, or Mn3+. An− is a charge-compensating inorganic or organic anion (e.g. CO32−, Cl−, SO42− and NO3−), and x is normally between 0.2–0.33.5 LDHs have been very widely explored as as flame retardants,6,7 catalysts and catalyst precursors,8 CO2 adsorbents,9–13 cement additives,14 and drug delivery systems.15–18
Drug anions have been intercalated into the interlayer gallery of LDHs on a number of occasions. There are four synthesis routes that have been commonly employed: ion exchange (replacement of an initial interlayer anion such as Cl− or NO3− with the drug),19–21 reconstruction (calcination followed by reacting the calcined LDH with a solution of the drug),22,23 exfoliation-reassembly (separation of the LDH into single layers, and then reassembly in a solution of the drug),24 and coprecipitation (addition of a mixture of metal salts to an alkaline solution of the drug of interest to prepare an intercalate in a single-step).25–28 Beyond LDHs, there exist other layered hydroxides which can anion-exchange and have been explored for drug delivery, such as the hydroxy double salts29,30 or layered rare earth hydroxides (LRHs).
LRHs with general formula [Ln(OH)2]An−1/n·yH2O have been known for decades, but only infrequently studied since the 1970s.31–34 This is likely due to the fact that the anions in these compounds are usually incapable of being replaced, because they are directly coordinated to the lanthanide centers. Anion-exchangeable LRHs were not discovered until 2006, when Monge et al.35 reported a new type of pillared materials enabling anion-exchange in the interlayer gallery. These compounds, Yb4(OH)10[C14H6O2(SO3)2]·4H2O and Y4(OH)10[C10H6(SO3)2]·4H2O, were proposed to be promising in green chemistry because of the active metal centers in the hydroxide layers. Two years later, [Ln2(OH)5]NO3·1.5H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 36 and [Ln2(OH)5]Cl·yH2O37–39 were successfully synthesized. Since this time, the intercalation chemistry of LRHs has received increasing attention.
36 and [Ln2(OH)5]Cl·yH2O37–39 were successfully synthesized. Since this time, the intercalation chemistry of LRHs has received increasing attention.
The combination of a rich intercalation chemistry and the presence of rare earth elements with magnetic and fluorescent properties in the LRH layers can lead to integrated materials with many applications in medical science,40,41 catalysis,42 separation science,43 sensors,44 and luminescence devices.45–55 For instance, Yang et al.48 intercalated organic sensitizers into layered europium hydroxides and observed enhanced red luminescence of Eu3+. As a result of their magnetic properties, layered gadolinium hydroxides (LGdHs) have potential for use as contrast agents in magnetic resonance imaging (MRI).56 This was first reported by Lee et al. in 2009.40 Multimodal contrast agents for MRI and fluorescent imaging can also be prepared via surface modification of the LGdH nanosheets with phospholipids, and intercalation with fluorescein anions.41
The anion exchange ability of LRHs should permit them to act as effective drug delivery systems, similarly to LDHs. However, to date there has been little effort expended to explore this, with only four studies in the literature. In the first, Stefanakis et al. intercalated several pharmaceutical anions including an antibiotic, amino acids, and a fatty acid into the interlayer region of LGdH matrix by ion exchange, but the functional performance of the products was not explored.56 In the second, Roa et al. reported the intercalation of microRNA into the LGdH matrix, and found the resultant material to have promising delivery and MRI properties.57 More recently, Gu and co-workers generated a naproxen-intercalated layered europium hydroxide, which was found to release the incorporated drug over about 200 min, retain the inherent luminescence of Eu3+, and to be highly biocompatible.58 Finally, Ju and Gu have reported an aspirin intercalate of a layered terbium hydroxide formed by ion exchange, and observed enhanced Tb luminescence intensity and sustained drug release over 10 h.59 Therefore, it appears that LRH-drug composites have great promise, but more work is required to explore this in detail.
In this work, we systematically study the intercalation of the common non-steroidal anti-inflammatory drugs ibuprofen, naproxen and diclofenac (Fig. 1) into [Gd2(OH)5]Cl·yH2O (LGdH-Cl). We investigate the effect of the intercalation method used on the nature of the composite formed, probe the drug release profiles, and quantify the biocompatibility and magnetic resonance properties of the composites.
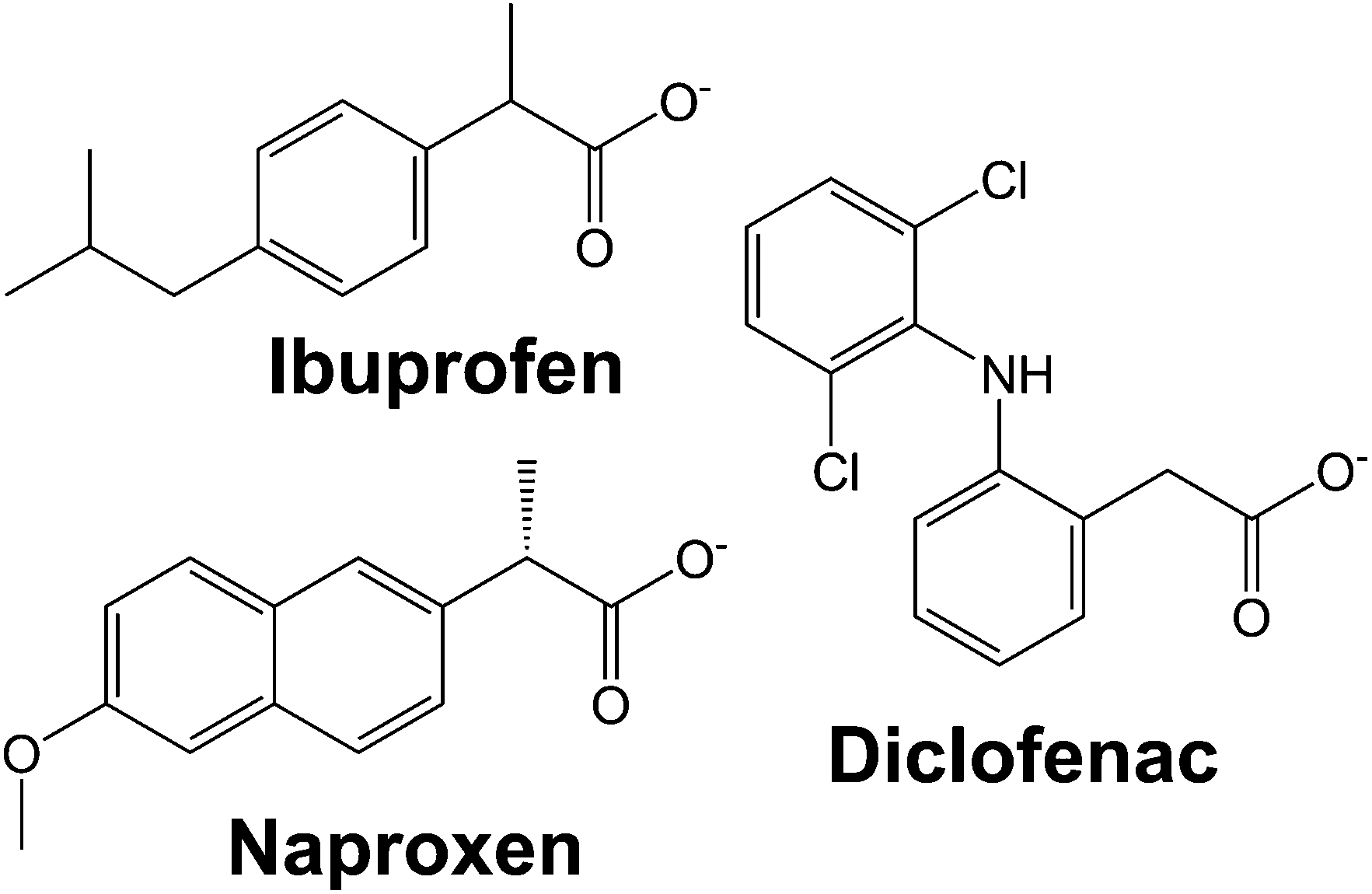 | ||
| Fig. 1 The chemical structures of the anions of diclofenac, ibuprofen, and naproxen. | ||
Experimental
Materials
Gadolinium chloride hexahydrate was supplied by Alfa Aesar. Diclofenac sodium and naproxen sodium were procured from Acros Organics, while ibuprofen sodium was obtained from Santa Cruz Biotechnology. All water was deionized prior to use, and all other chemicals were of analytical grade and used without further purification.Synthesis
Ion exchange was then performed by taking 150 mg of well crystallized LGdH-Cl precursor (ca. 462 g mol−1) and dispersing this in 15 mL of an aqueous solution containing a 3-fold molar excess of the drug ion of interest (310 mg for diclofenac sodium, 222 mg for ibuprofen sodium, and 245 mg for naproxen sodium). Ion exchange reactions were conducted with vigorous stirring at 60 °C for 24 h. The product was collected by filtration, washed with deionized water and ethanol, and dried at 40 °C for 24 h.
Characterisation
Cell culture
Caco-2 cells, a colorectal adenocarcinoma cell line (ATCC HTB-37), were employed for in vitro studies. Cells were cultured at 37 °C under 5% CO2, in Dulbecco's modified Eagle's medium (DMEM-HG; Gibco) supplemented with penicillin–streptomycin (1% v/v) and L-glutamine (1% v/v) solutions (Life Technologies), non-essential amino acid solution (1% v/v, Life Technologies), and 10% v/v heat-inactivated fetal bovine serum (Gibco) (termed “complete DMEM”).For viability tests, Biolite 96 well Multidish clear plates (ThermoFisher) were used. The seeding density was 5.6 × 104 cells per mL, and each well contained 180 μL of cell suspension. 10 mg mL−1 suspensions of selected LGdH-drug formulations were prepared in sterile culture medium. These were then either added to the cells directly, or solubilised with 1% v/v DMSO and filtered through a 0.22 μm filter before being added to the cells. In either case, 5 or 10 μL of the LGdH/culture medium mixture was added to the wells of the plate, giving final concentrations of 526 or 270 μg mL−1. The cells were incubated with the formulations for 24 h. Cell viability was determined with the CellTiter-Glo™ assay (Promega) according to the manufacturer's instructions. After addition of the fluorescent reagent (100 μL per well), the plate was left for 30 min at room temperature before luminescence was recorded using a SpectraMax M2e spectrophotometer (Molecular Devices). The viability of the cells was then calculated as follows:
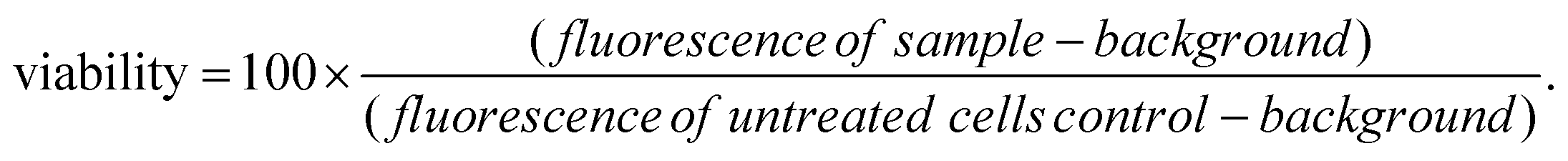
Three independent experiments were performed, with triplicate conditions within each experiment. Data are presented as mean ± S.D.
Results and discussion
Ion exchange intercalation
XRD patterns of the materials obtained from ion exchange intercalation with diclofenac (dic), ibuprofen (ibu) and naproxen (nap) are given in Fig. 2. The pattern for the LGdH-Cl raw material clearly matches well the pattern calculated from the reported structure.37 Strong basal (00l) reflections show the material to have a layered structure, while the large number of non-basal reflections confirms that there is extensive in-layer ordering. After intercalation, the reflections become broader and weaker, indicative of a loss of crystallinity. The (00l) reflections also shift to lower angle, consistent with the layers moving apart to accommodate intercalation of a larger guest. The (001) reflection of LGdH-Cl at 8.45 Å is not visible in any of the drug intercalates, confirming complete reaction. A summary of the interlayer spacings is provided in Table 1.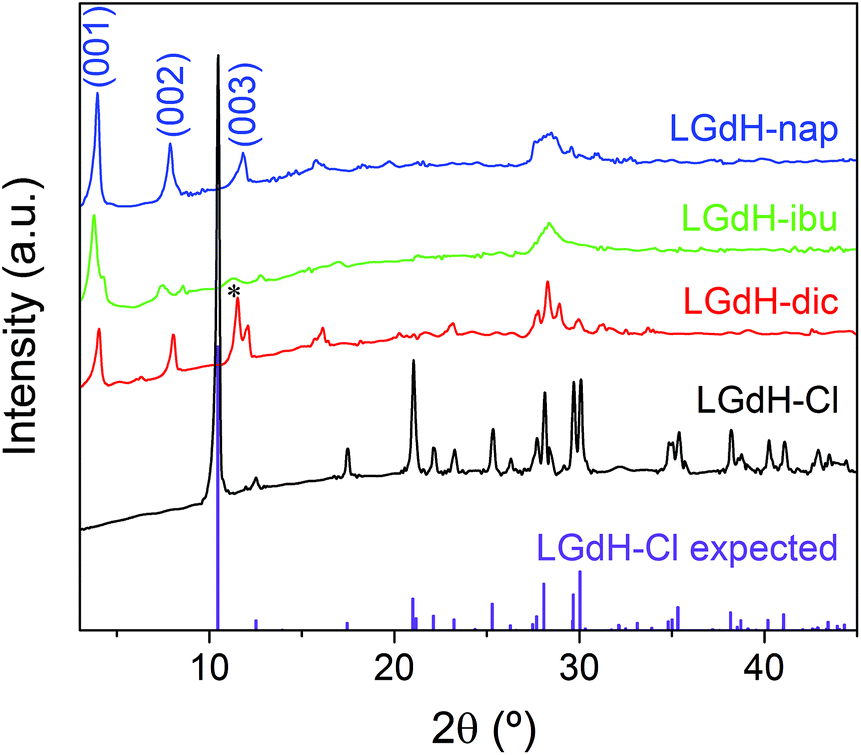 | ||
| Fig. 2 XRD patterns for intercalation compounds of LGdH prepared by ion exchange. *: the (003) basal reflection of LGdH-dic overlaps with the in-plane (101) reflection. | ||
| Material | d 001 (Å) | Chemical formula | Drug loading (%) |
|---|---|---|---|
| LGdH-Cl | 8.45 | [Gd2(OH)5]Cl0.8(CO3)0.1·H2O | 0 |
| LGdH-dic | 21.96 | [Gd2(OH)5](C14H10Cl2NO2)0.8Cl0.2·H2O | 35.7 |
| LGdH-ibu | 23.46, 20.73 | [Gd2(OH)5](C13H17O2)0.9Cl0.1·1.5H2O | 30.0 |
| LGdH-nap | 22.30 | [Gd2(OH)5](C14H13O3)0.82Cl0.18·1.75H2O | 30.1 |
For LGdH-dic and LGdH-nap, there appears to be a single phase present; with LGdH-ibu the (001) reflection is a doublet, suggesting that two intercalates with slightly different interlayer spacings have been formed. The end-to-end lengths of dic, ibu, and nap have previously been estimated as 11.76,29 10.30,61 and 12.8829 Å respectively. The layer thickness of LGdH is approximately 6.5 Å,48 which gives gallery heights for LGdH-dic, LGdH-ibu and LGdH-nap of 15.46, 16.96, and 15.80 Å. These values are around 1.2 to 1.6 times greater than the guest sizes, suggesting that the drug anions form intertwined bilayers in the interlayer space.29 This is illustrated in Fig. 3. The interlayer spacing for the nap intercalate is close to that reported for the analogous layered europium hydroxide, where the presence of two intercalate phases with different interlayer spacings was also noted.58
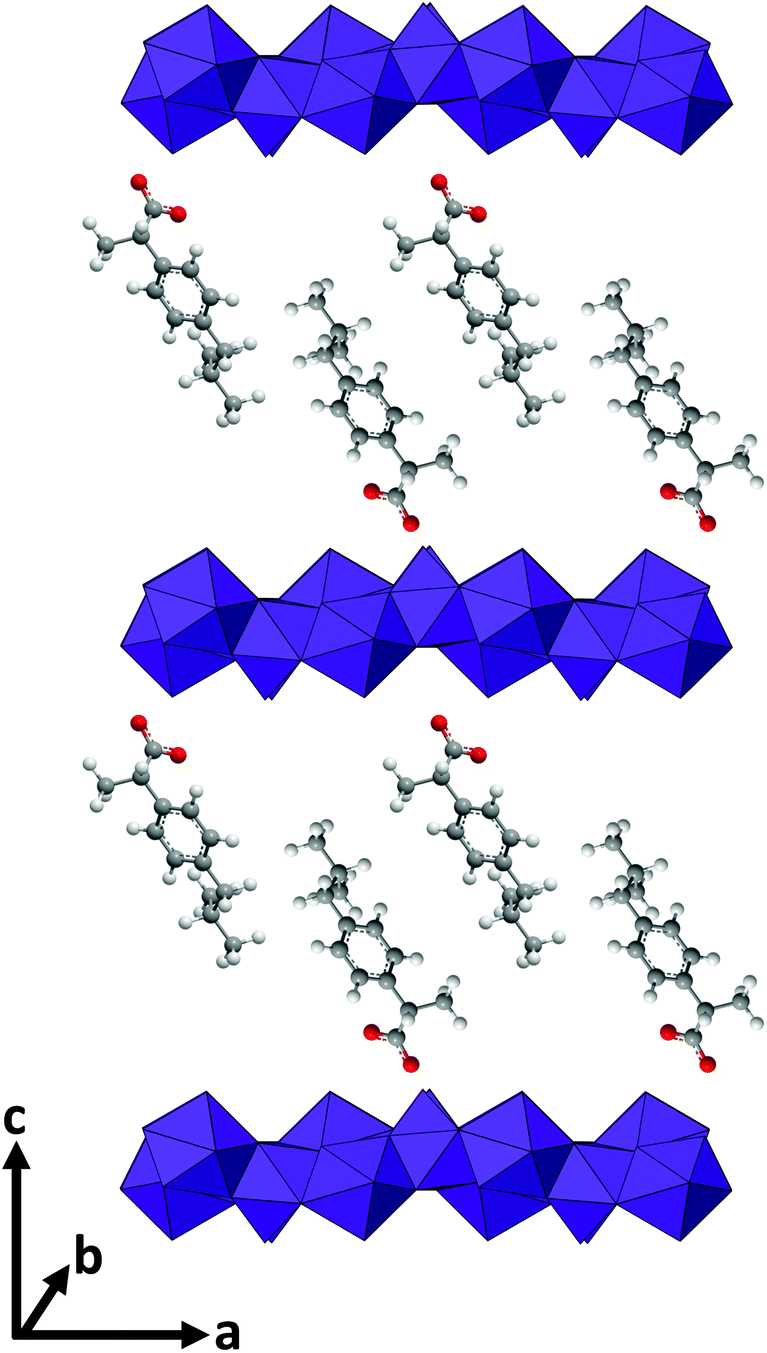 | ||
| Fig. 3 A schematic illustrating the orientation of the anions in LGdH-ibu. | ||
The elemental analysis data (Table 1 and Table S1, ESI†) reveal that there is still some residual Cl− present in the system, with around 80–90% of this having been replaced. These Cl ions are presumably distributed between the same layers as the drug anions, since there is none of the starting LGdH-Cl phase present (see XRD data in Fig. 2).
The successful incorporation of the drug ions was verified by IR spectroscopy, and the spectra are depicted in Fig. 4. LGdH-Cl shows broad bands ranging from 3646 to 3383 cm−1, which can be attributed to stretching vibrations of the hydroxyl groups from both interlayer water and the hydroxide layers. The absorption peak at around 1667 cm−1 corresponds to the δ-bend of water. Considering the IR spectra of the LGdH-drug hybrids, again there is a broad band from OH stretching at around 3500 cm−1. All the intercalates exhibit a range of additional bands, which arise in very similar positions to those in the spectra of the pure drug salts, except that they are often shifted to lower wavenumbers as a result of electrostatic interactions between the drug anions and hydroxide layers.62 The similarity of the spectra of the intercalates and drug salts confirms successful intercalation.
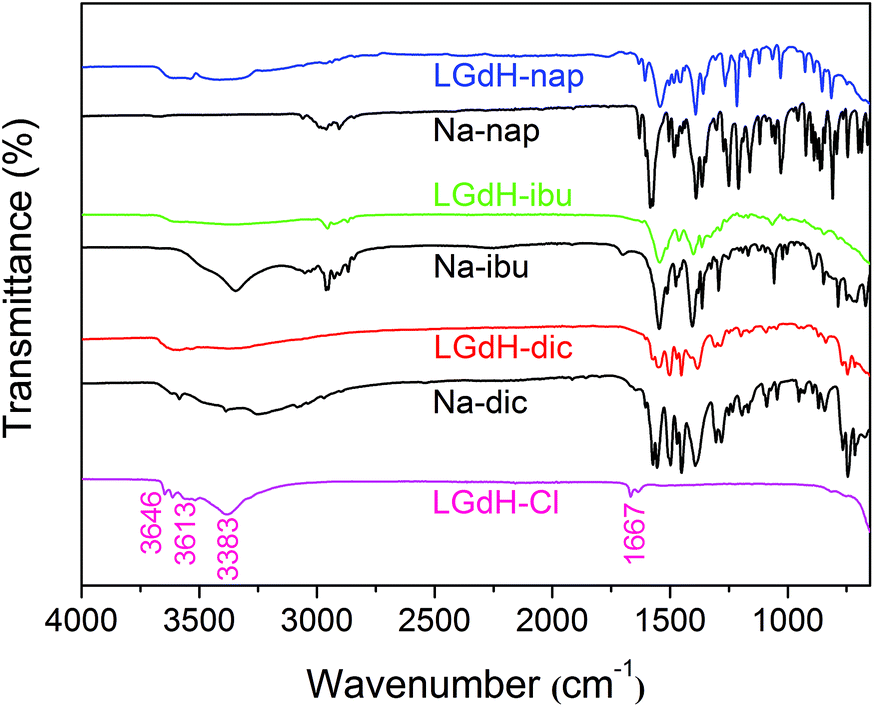 | ||
| Fig. 4 IR spectra for the products of ion exchange reactions and the guest drug species. | ||
Scanning electron microscopy (SEM) images of the LGdH particles are presented in Fig. 5. LGdH-Cl and all the drug intercalates prepared by ion exchange comprise large (5–10 μm) aggregates of rod- and plate like particles with sizes from around 100 nm to 1–2 μm. The particle morphology is less regular and the average size smaller after the ion exchange process.
 | ||
| Fig. 5 SEM images of LGdH-Cl and the products of ion exchange reactions. | ||
Coprecipitation
Coprecipitation intercalation of the drug ions was explored at pH 8, 10, and 12. The XRD patterns of the products obtained (denoted LGdH-drug-c) are presented in the ESI, Fig. S1.† In the case of dic, it proved impossible to stabilise the pH at 8, and thus experiments were undertaken at pH 7.7 and 9.3, as close to 8 as could be obtained. For all three drugs and at all pHs explored, intercalation appeared to be successful, with the patterns exhibiting basal (00l) reflections and no Bragg reflections attributable to LGdH-Cl. However, the products appear more poorly crystalline than those from ion exchange, with broader reflections and in some cases only the (001) reflection visible in the data. Impurities were also observed in some cases. Elemental microanalysis was obtained in selected cases to confirm intercalation, and a summary of these results, plus the interlayer spacings from XRD is provided in Table 2. Full data are given in Table S2†.| Material | pH | d 001 (Å) | Chemical formula | Drug loading (%) |
|---|---|---|---|---|
| LGdH-dic-c | 7.7 | 22.99 | NM | NM |
| 9.3 | 23.36 | NM | NM | |
| 10 | 22.18 | NM | NM | |
| 12 | 21.96 | [Gd2(OH)5](C14H10Cl2NO2)0.9Cl0.1·4H2O | 35.9 | |
| LGdH-ibu-c | 8 | 22.86 | [Gd2(OH)5](C13H17O2)(C13H18O2)·H2O | 49.6 |
| 10 | 23.85 | [Gd2(OH)5](C13H17O2)(C13H18O2)0.8·H2O | 47.0 | |
| 12 | 24.52 | [Gd2(OH)5](C13H17O2)(C13H18O2)0.25·H2O | 38.1 | |
| LGdH-nap-c | 8 | 20.82 | NM | NM |
| 10 | 20.82 | NM | NM | |
| 12 | 23.48 | [Gd2(OH)5](C14H13O3)(C14H14O3)0.4·1.75H2O | 42.7 |
The interlayer spacings obtained by coprecipitation are similar to those from ion exchange. There is no clear relationship between the pH and interlayer spacing, with this tending to decline in the case of dic and increase with ibu and nap. In general, it can be said that the products are better crystallised at higher pH, with more basal reflections visible. In all cases, the distinctive bands of the drug ions can be seen in the IR spectra of the coprecipitation products (Fig. S2, ESI†), confirming successful intercalation. The elemental analysis data indicate that higher drug loadings are obtained from coprecipitation than ion exchange (Table S2 cf. Table S1†). The amounts of drug calculated to be incorporated are greater than those which can be intercalated on the basis of charge balance alone, suggesting that some neutral drug is present, either in the interlayer space or surface adsorbed. It should be noted however that the agreement between observed and calculated values is less close for the coprecipitation products than those from ion exchange.
SEM images for the LGdH-dic-c intercalates (Fig. S3†) reveal them to consist of large aggregates of platelet-shaped particles. In general, the secondary particle size decreases and the primary particle size increases as the pH rises. The LGdH-ibu-c materials (Fig. S4†) also show an increase in the primary particle size with pH; their morphology is similar to the LGdH-dic-c analogues, but with more regular habits. Similar trends can be observed with LGdH-nap-c (Fig. S5†), except that there are also large amorphous-looking particles at pH 8 and 10. Overall, it is clear that the products of coprecipitation have less regular morphologies than the analogous systems prepared by ion exchange.
Self-assembly
Intercalation by self-assembly was also explored. This involves the exfoliation of the LRH into individual layers, followed by restacking around the desired guest. XRD patterns of LGdH-drug intercalates prepared by this route (LGdH-dic-sa, LRH-ibu-sa, and LRH-nap-sa) are shown in Fig. 6.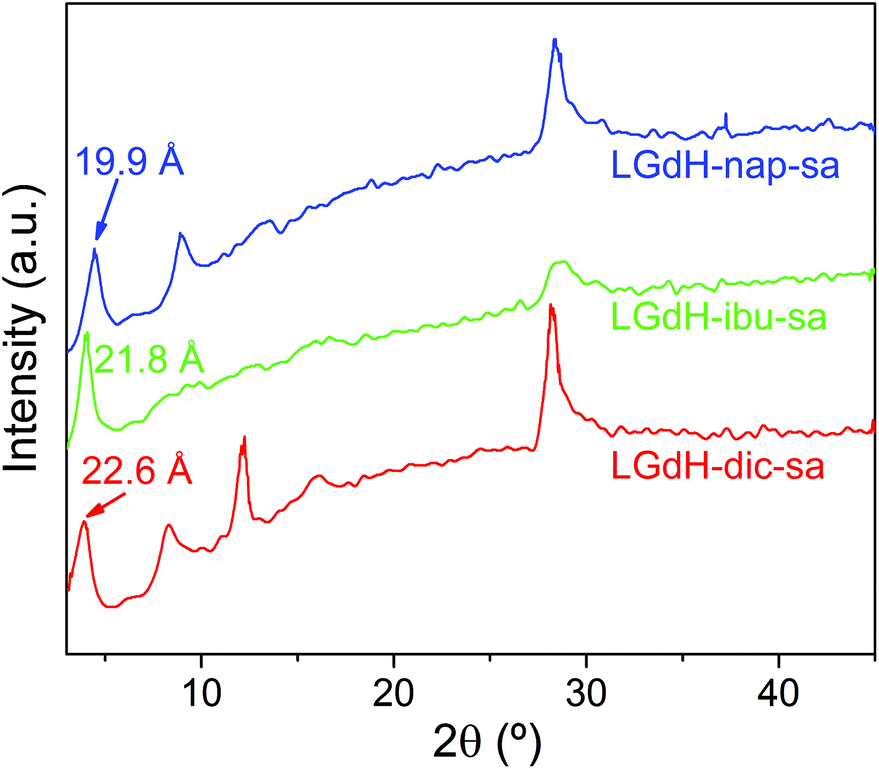 | ||
| Fig. 6 XRD patterns for intercalation compounds of LGdH prepared by self-assembly. | ||
As was observed with the ion exchange and coprecipitation routes, it is possible to incorporate all three drug ions into the LGdH material using self-assembly. However, the reflections in the XRD patterns are broad, indicative of stacking defects and poor crystallinity. While LGdH-dic-sa and LGdH-nap-sa have a series of (00l) reflections, only the (001) can be seen for LGdH-ibu-sa, suggesting a greater degree of disorder for this material. IR spectra (ESI, Fig. S6†) confirm the successful intercalation of the drug anions. The interlayer spacings and chemical formulae are detailed in Table 3 and Table S3.† The former are somewhat lower for nap and ibu compared to the other intercalation methods, possibly as a result of different guest orientations in the interlayer spaces. The formulae and drug loadings are very similar to those obtained by ion exchange, with better agreement between the observed and calculated values than with the coprecipitation products.
| Material | d 001 (Å) | Chemical formula | Drug loading (%) |
|---|---|---|---|
| LGdH-dic-sa | 22.60 | [Gd2(OH)5](C14H10Cl2NO2)0.8 Cl0.2·1.3H2O | 35.4 |
| LGdH-ibu-sa | 21.80 | [Gd2(OH)5](C13H17O2)0.95Cl0.05·2H2O | 30.8 |
| LGdH-nap-sa | 19.90 | [Gd2(OH)5](C14H13O3)0.88Cl0.12·1.5H2O | 31.9 |
As for the materials prepared by ion exchange and coprecipitation, the self-assembled materials comprise multimicron aggregates of plate-like primary particles, with the latter being ca. 100–500 nm in size (Fig. S7†). The morphology is more similar to the coprecipitation products than to those from ion exchange.
Stability
Free Gd3+ is toxic to humans. Therefore, we assessed the stability of the formulations at pH 1.5 (representing the stomach) and pH 7.4 (the general physiological pH), using the Arsenazo III assay.63 Arsenazo III forms strongly-bound 1:1 complexes with Gd3+, the absorbance of which is highly dependent on pH.64 The λmax of Gd3+-Arsenazo complexes is reported to be 650 nm over the pH ranges from 3 to 4 and 6.4 to 8.65–67 The results of these assays are given in Fig. S8† and Fig. 7.
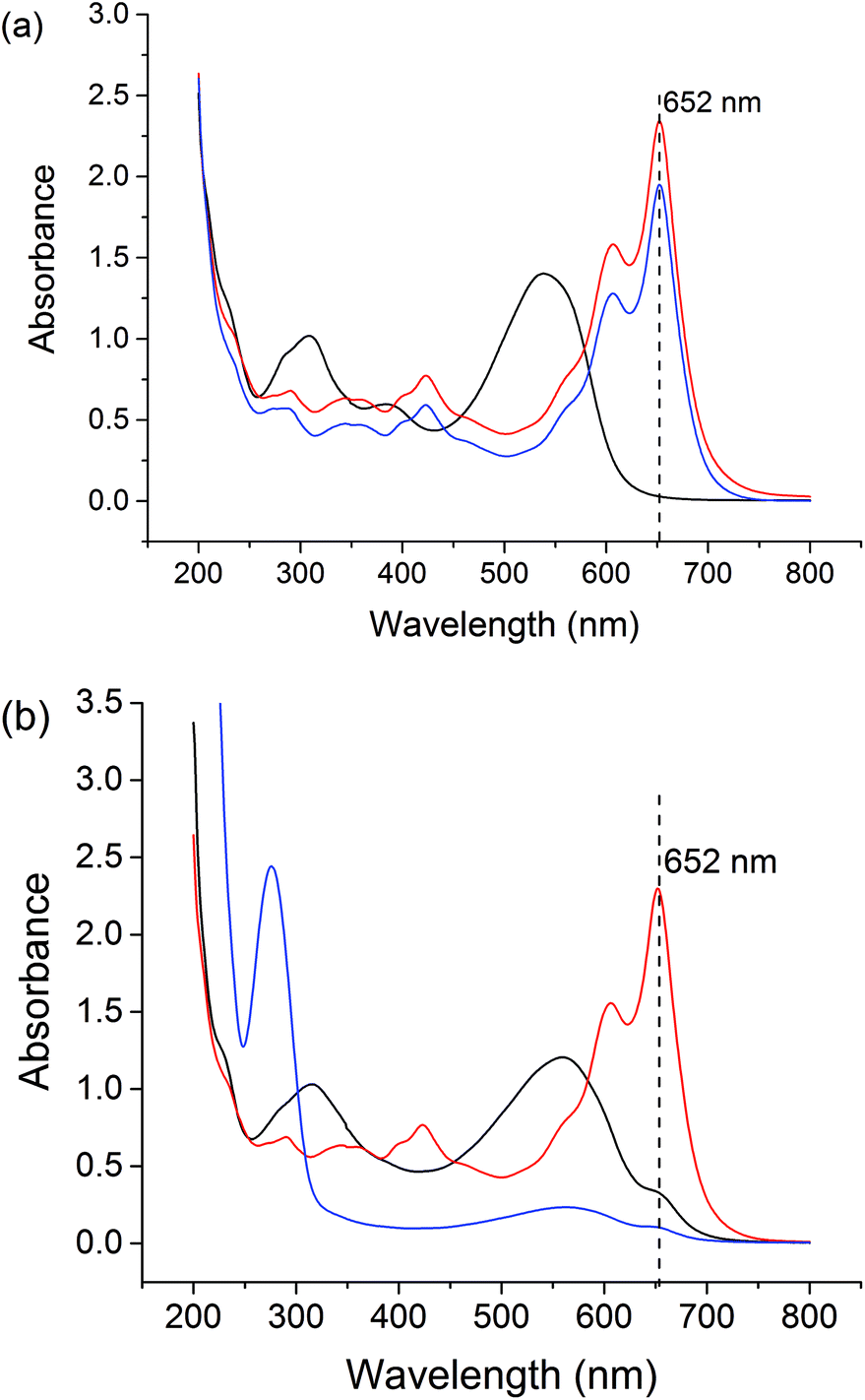 | ||
Fig. 7 Assessment of the stability of LGdH-dic at (a) pH 1.5 for 2 h, and (b) pH 7.4 for 24 h, as measured using the Arsenazo III assay. Data are shown for the release medium (negative control; HCl or PBS; –); GdCl3 (positive control;  ), and LGdH-Cl ( ), and LGdH-Cl ( ). ). | ||
The stabilities of the LGdH-Cl precursor and LGdH-dic were investigated in acidic solution and neutral PBS. Samples were incubated at 37 °C in pH 1.5 HCl solution for 2 h or in pH 7.4 PBS for 24 h. GdCl3 and HCl/PBS incubated with Arsenazo III were used as positive and negative controls respectively. Under both pH conditions, the GdCl3 positive control shows strong absorbance at 652 nm, corresponding to complex formation, while the HCl and PBS solutions show very low absorbance at this wavelength. After the immersion of LGdH-Cl and LGdH-dic in a pH 1.5 medium, the solution has strong absorbance at 652 nm, demonstrating that free Gd3+ leaches from the material at this pH. A calibration curve was constructed (data not shown), and based on this it was calculated that approximately 43% of the Gd3+ from LGdH-Cl and 48% from LGdH-dic was released after 2 h. In contrast, at pH 7.4 the absorbance at 652 nm is similar to the negative control, even after 24 h. Therefore, it can be concluded that the LGdH-drug composites are stable at neutral pH, but not in acidic conditions. In order to be suitable for drug delivery and diagnostic purposes, the LGdH composites must thus be encapsulated inside an enteric coating to protect them from the acidic conditions in the stomach.
Drug release
The release of the incorporated active ingredients was studied for selected systems in Krebs buffer. This is a carbonate-based buffer which more accurately represents the physiological environment of the lower parts of the intestinal tract (where the LGdH-drug particles would first encounter physiological media if administered in an enteric coated formulation as suggested above) than traditional phosphate buffers. The release plots are shown in Fig. 8.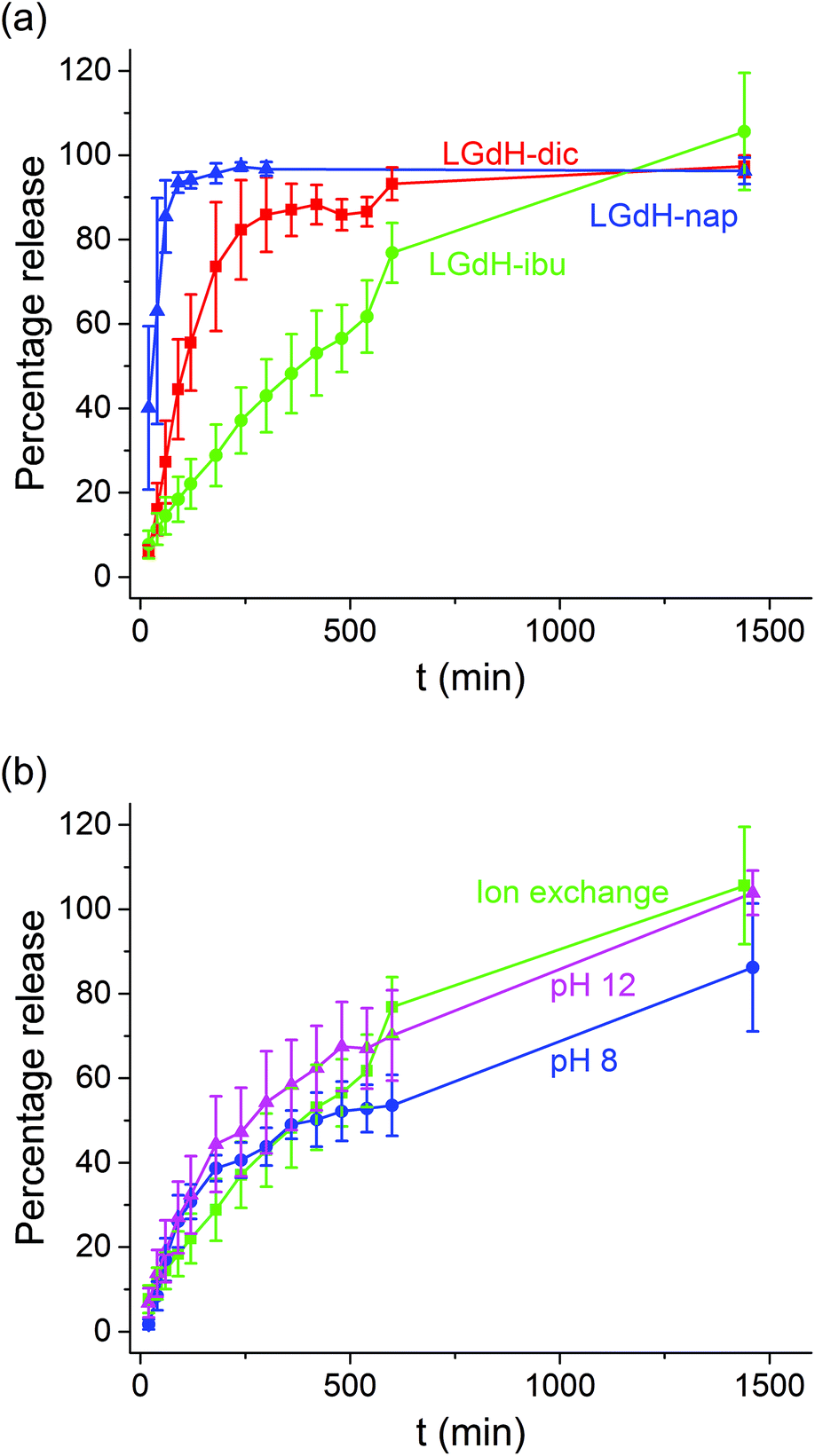 | ||
| Fig. 8 Drug release from (a) the intercalates prepared by ion exchange and (b) the LGdH-ibu composites produced using different synthetic procedures. Data are reported as mean ± S.D. from three independent experiments. | ||
It is clear from Fig. 8(a) that the different active ingredients release at varied rates from their LGdH intercalates. Nap releases very quickly, while dic is slower and ibu releases most slowly. However, the final release percentage is greatest for LGdH-ibu, reaching 105.6 ± 13.9% as compared to 97.3 ± 2.59% for LGdH-dic and 96.3 ± 3.13% for LGdH-nap. The synthesis route used does not appear to have a marked effect on the release profiles (Fig. 8(b)). The material prepared by ion exchange is perhaps freeing its drug cargo fractionally faster than the coprecipitation products, but this effect does not appear to be significant. The final release percentages reached are also similar, with both LGdH-ibu and LGdH-ibu-c prepared at pH 12 reaching 100% release. The material from coprecipitation at pH 8 reaches only 86.2 ± 15.2%, perhaps reflecting the larger ibu loading of this sample. The release profiles observed suggest that the LGdH composites could be used for extended release in the small intestine, if they were packaged into an enteric coated capsule. Release from LGdH-nap is rather fast, but the LGdH-dic system shows sustained release over ca. 4 h, while the LGdH-ibu and LGdH-ibu-c systems extend this to 24 h. The drug loadings are relatively high (30–50% w/w), and thus it would be feasible to use the intercalates to deliver a standard dose of ibuprofen (1200–3200 mg day−1) or diclofenac (150–225 mg day−1).
XRD studies were performed on the residual solid from drug release experiments (ESI, Fig. S9†). These materials are largely amorphous, with some small reflections at around 12.5° corresponding to LGdH-CO3 in the case of dic and ibu. The LGdH-ibu residue also has a small peak corresponding to the drug intercalate, indicating that release was not quite complete. The release kinetics were fitted using the Bhaskar and Avarmi-Erofe'ev models (eqn (1) and (2)).
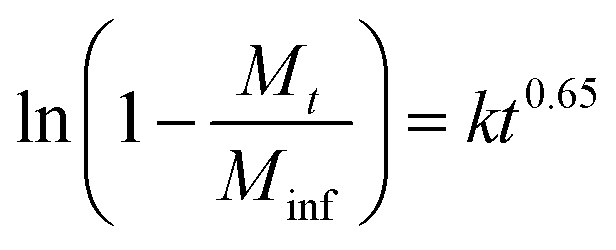 | (1) |
| ln(−ln(1 − α)) = nlnk + nlnt | (2) |
In eqn (1), Mt is the amount of drug released at time t, and Minf is the amount of drug initially present in the carrier. In eqn (2), α is the extent of reaction and can be regarded as equivalent to Mt/Minf for drug release data, while n is an exponent providing information on the reaction mechanism. In both equations, k is a rate constant and t the time.
The Bhaskar model assumes that diffusion through the particle is the rate limiting step to release, making it appropriate for ion exchange process.68,69 The Avrami-Erofe'ev equation allows for a wide range of kinetic processes, with the value of n providing information on the reaction mechanism.70 Fits of both models to the experimental data are given in Fig. S10 (ESI†). In the majority of cases, it can be seen that the models provide a good fit to the data. In the case of LGdH-nap, there are very few data points on the line because of the rapid nature of the release, and so caution must be taken in interpreting the data, but nevertheless the models appear to give a good fit. The close fits obtained with the Bhaskar model suggest that the rate limiting step to release is the movement of drug ions out of the interlayer spaces, and their replacement with carbonate ions from the release medium. In the Avrami-Erofe'ev model, the values of n obtained lie between 0.81 and 1.29. It is not possible to unambiguously determine the reaction mechanism from these values, but they are consistent with deceleratory nucleation followed by diffusion control. This is sensible, since the drug-filled interlayers are the nucleation sites, and all are present at the start of the reaction (hence deceleratory nucleation).
There is one system where neither model provides a good fit to the data, which is LGdH-ibu-c prepared at pH 8. The elemental analysis data for this system indicated the presence of twice as much ibuprofen as is required to charge-balance, suggesting surface adsorption of some excess ibuprofen. Therefore, in this case it may be that the surface adsorbed and intercalated drug are freed through two different mechanisms, resulting in the simple Bhaskar and Avrami-Erofe'ev models not being sufficiently complex to describe this system.
Relaxivity measurements
In order to evaluate the effectiveness of LGdH-Cl and its drug intercalates as MRI contrast agents (CAs), their relaxivities (r1 and r2) in water suspensions were determined from the value of the longitudinal (1/T1) or transverse (1/T2) relaxation rates normalized to the gadolinium concentration in the composites (eqn (3)).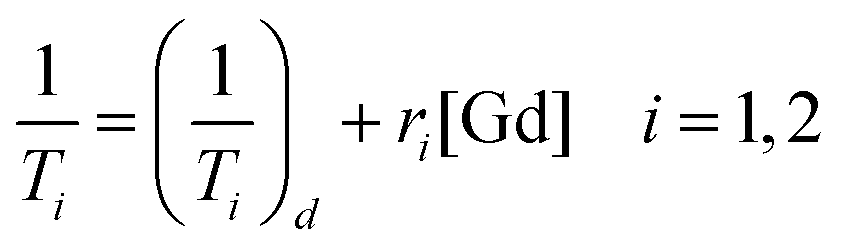 | (3) |
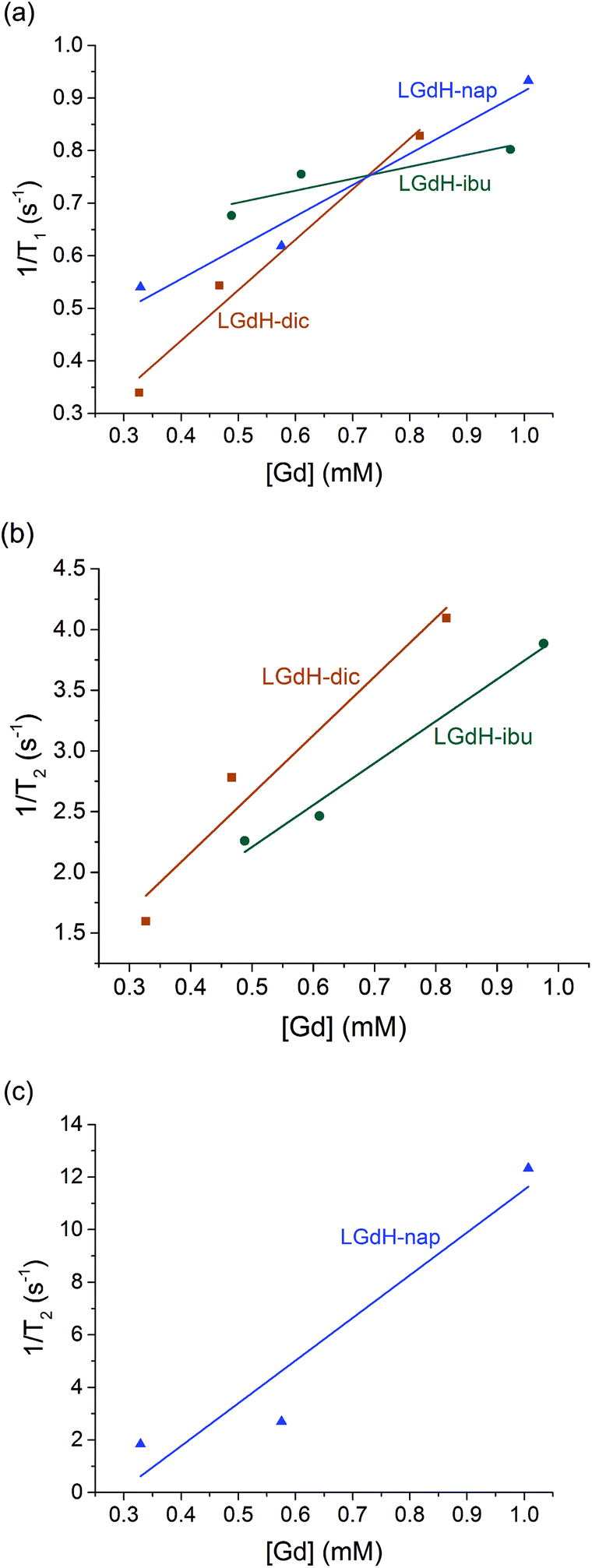 | ||
| Fig. 9 Relaxivity data on the LGdH-drug composites prepared by ion exchange, showing (a) r1, (b) and (c) r2 plots. | ||
The r1 values are determined by the quantity of Gd3+ ions at the surface of the material exposed to solvent water molecules, which facilitate proton exchange of coordinated water molecules with bulk water and thereby accelerate T1 relaxation though an inner-sphere mechanism.71 LGdH-Cl has particle surfaces comprising Gd2(OH)5+ layers, with each Gd3+ coordinated by both OH groups and bound water. The particle surfaces will have many Gd3+ ions exposed to bulk water in the suspension, which might be expected to exchange with Gd-bound water.40 The experimental r1 values are small for all the samples studied in this work, indicating that virtually no exchange with bulk water occurs. This could be due to a very slow water exchange rate, or to the binding of agarose to the surface of the particles, which could hinder the exchange process.72 The contribution of the outer-sphere mechanism to r1 is also rather weak. The incorporation of the drug ions does not seem to significantly change the water accessibility of the surface Gd3+ ions, as would be expected. Although the space between the layers increases after intercalation, which could potentially allow more water molecules to diffuse into the interlayer space, the exchange between interlayer and bulk water will still be hindered, leading to a minimal effect on r1.
T 2 relaxation is dominated by the outer-sphere contribution, created by local magnetic field inhomogeneities induced by the tumbling magnetic nanoparticles. The presence of agarose (applied as an emulsifier) adsorbed at the particle surface during relaxivity measurements is expected to slow down water diffusion near the particle surfaces due to the formation of hydrogen bonds with the agarose chains, affecting the r2 value.73 However, the particle size and shape also determine the field perturbation area experienced by outer-sphere protons. LRHs comprise platelet-shaped particles, and hence their tumbling generates a large area of local field inhomogeneity and therefore larger r2 values. While the intercalation of dic and ibu cause a small r2 decrease compared to the parent LGdH-Cl, nap leads to a more than a two-fold r2 increase. This could result from slower and/or closer water diffusion, or a more asymmetric tumbling of the modified LGdH particles. However, a more complete study of the relaxation mechanisms of these materials is beyond the scope of the present study.
CAs with higher relaxivities give higher contrast and clearer images at lower dosage.74 MRI CAs can be classified into two groups based on the ratio of r2 to r1. T1 CAs, otherwise known as positive contrast agents, have r2 to r1 ratios close to 1 (normally between 1 and 2) and can enhance signal intensity, whereas T2 CAs with higher r2/r1 (>10), have a dominant T2 shortening effect, causing a reduction in signal intensity.74 Those CAs with r2/r1 between 2 and 10 can function either as positive or negative agents.74,75 Thus, LGdH-dic can act as either a positive or negative CA, while LGdH-ibu, LGdH-nap, and LGdH-Cl are better suited as negative CAs (see Table S4†).
The materials prepared using the different methods show some variations in the calculated r1 and r2 values, but there are no obvious trends. What is however clear is that the drug intercalates retain the relaxivity properties of the parent LGdH-Cl, and in some cases enhance them. Their ability to be used in MRI was verified in a preliminary imaging study (Fig. 10). Thus, the new materials prepared in this work have the potential to be used for simultaneous drug delivery and imaging.
 | ||
| Fig. 10 T 1-Weighted MRI images of selected LGdH-drug intercalates. Images are shown for (A) a 1% w/v agarose gel (negative control); suspensions of LGdH-ibu with Gd concentrations of (B) 0.5 and (C) 0.25 mM; and, suspensions of LGdH-dic at Gd concentrations of (D) 0.5 and (E) 0.25 mM. | ||
Biocompatibility
The biocompatibility of selected LGdH-drug composites was assessed using in vitro cell viability studies. The results of performing these with the solid LGdH materials are presented in Fig. 11. At concentrations of 526 μg mL−1, both LGdH-Cl and LGdH-ibu are highly biocompatible, and in fact appear to encourage cell growth. In contract, LGdH-dic causes some cell death, with a mean viability of 71%. At a lower concentration of 270 μg mL−1 all three LGdH materials explored have very good biocompatibility, resulting in cell counts higher than the untreated cells control. Further experiments were undertaken with solutions made from LGdH suspensions (see Fig. S11, ESI†). In all cases here the cell viability was indistinguishable from the untreated cells, thereby confirming the biocompatibility of the LGdH-drug materials.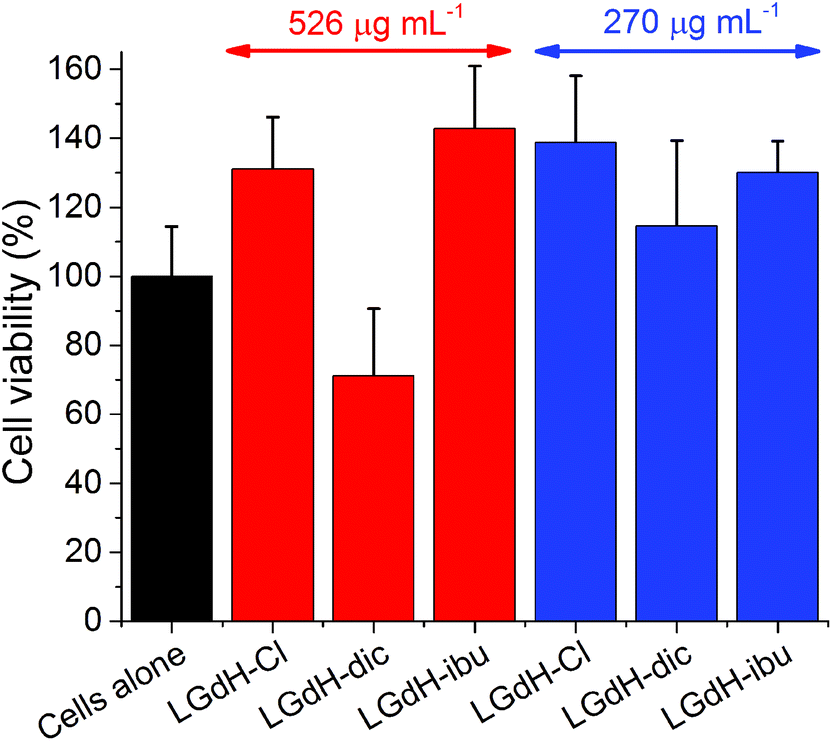 | ||
| Fig. 11 The results of in vitro cell viability studies with selected LGdH materials. Experiments were performed with suspensions of the LGdHs at concentrations of 526 (red bars) or 270 (blue bars) μg mL−1. Results are shown as mean ± S.D. from three independent experiments, each containing three replicates. | ||
Conclusions
Three non-steroidal anti-inflammatory drugs (diclofenac, ibuprofen, and naproxen) were intercalated into the layered gadolinium hydroxide (LGdH) [Gd2(OH)5]Cl·yH2O for the first time. Intercalation could be achieved successfully using ion exchange, coprecipitation or exfoliation-self-assembly approaches, and in all cases X-ray diffraction and IR spectroscopy confirmed the successful incorporation of the drug anions into the interlayer space. An intertwined bilayer arrangement of anions is proposed. The products obtained from the different routes are similar, but those obtained from coprecipitation generally had higher drug loadings. In the latter case, the drug content calculated is in most samples greater than that required to balance the charge of the layers, and thus some neutral drug molecules are also present, either intercalated or surface adsorbed. The LGdH-drug composites are stable at neutral pH, but degrade rapidly in acidic conditions to free Gd3+ into solution. In drug release assays, LGdH-nap freed its drug cargo very quickly at pH 7.4, with 80% release in 60 min. The other materials showed sustained release, over ca. 4 h for LGdH-dic and 24 h for LGdH-ibu. The drug intercalates retain the relaxivity properties of the parent LGdH material, with the materials most promising for use as negative contrast agents in MRI. The LGdH-drug composites are further found to be highly biocompatible. Overall, the new materials prepared in this work can be said to have great potential for theranostic applications, where simultaneous delivery of a drug and imaging are required.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Dr Tom Gregory at the UCL Institute of Archaeology for assistance with EDX measurements. CFGCG thanks the Portuguese Foundation for Science and Technology for grant no. UID/QUI/00313/2013, and the FEDER−European Regional Development Fund for funding through the COMPETE Programme (Operational Programme for Competitiveness; Grant No. PEst-OE/QUI/UI0313/2014). The China Scholarship Council is also thanked for funding YW's work at UCL.Notes and references
- F. Geng, R. Ma and T. Sasaki, Acc. Chem. Res., 2010, 43, 1177–1185 CrossRef CAS PubMed.
- S. P. Newman and W. Jones, J. Solid State Chem., 1999, 148, 26–40 CrossRef CAS.
- T. Kimura, Microporous Mesoporous Mater., 2005, 77, 97–107 CrossRef CAS.
- P. Braterman, Z. Xu and F. Yarberry, Layered Double Hydroxides, in Handbook of Layered Materials, ed. S. M. Auerbach, K. A. Carrado and P. K. Dutta, Marcel Dekker, New York, 2004, p. 373 Search PubMed.
- Q. Wang and D. O'Hare, Chem. Rev., 2012, 12, 4124–4155 CrossRef PubMed.
- C. Nyambo, P. Songtipya, E. Manias, M. M. Jimenez-Gasco and C. A. Wilkie, J. Mater. Chem., 2008, 18, 4827–4838 RSC.
- C. Manzi-Nshuti, J. M. Hossenlopp and C. A. Wilkie, Polym. Degrad. Stab., 2009, 94, 782–788 CrossRef CAS.
- X. Xu, R. Lu, X. Zhao, S. Xu, X. Lei, F. Zhang and D. G. Evans, Appl. Catal., B, 2011, 102, 147–156 CrossRef CAS.
- Q. Wang, J. Luo, Z. Zhong and A. Borgna, Energy Environ. Sci., 2011, 4, 42–55 CAS.
- Q. Wang, H. H. Tay, D. J. W. Ng, L. Chen, Y. Liu, J. Chang, Z. Zhong, J. Luo and A. Borgna, ChemSusChem, 2010, 3, 965–973 CrossRef CAS PubMed.
- Q. Wang, Z. Wu, H. H. Tay, L. Chen, Y. Liu, J. Chang, Z. Zhong, J. Luo and A. Borgna, Catal. Today, 2011, 164, 198–203 CrossRef CAS.
- Q. Wang, H. H. Tay, Z. Guo, L. Chen, Y. Liu, J. Chang, Z. Zhong, J. Luo and A. Borgna, Appl. Clay Sci., 2012, 55, 18–26 CrossRef CAS.
- Q. Song, W. Liu, C. D. Bohn, R. N. Harper, E. Sivaniah, S. A. Scott and J. S. Dennis, Energy Environ. Sci., 2013, 6, 288–298 CAS.
- J. Plank, D. Zhimin, H. Keller, F. V. Hössle and W. Seidl, Cem. Concr. Res., 2010, 40, 45–57 CrossRef CAS.
- A. Alcantara, P. Aranda, M. Darder and E. Ruiz-Hitzky, J. Mater. Chem., 2010, 20, 9495–9504 RSC.
- V. Rives, M. Del Arco and C. Martin, J. Controlled Release, 2013, 169, 28–39 CrossRef CAS PubMed.
- L. Li, W. Gu, J. Chen, W. Chen and Z. P. Xu, Biomater., 2014, 35, 3331–3339 CrossRef CAS PubMed.
- V. Rives, M. del Arco and C. Martín, Appl. Clay Sci., 2014, 88–89, 239–269 CrossRef CAS.
- M. Delarco, A. Fernandez, C. Martin and V. Rives, Appl. Clay Sci., 2009, 42, 538–544 CrossRef CAS.
- L. Perioli, T. Posati, M. Nocchetti, F. Bellezza, U. Costantino and A. Cipiciani, Appl. Clay Sci., 2011, 53, 374–378 CrossRef CAS.
- Z. Wang, R. Ma, L. Yan, X. Chen and G. Zhu, Chem. Commun., 2015, 51, 11587–11590 RSC.
- Z. Wang, E. Wang, L. Gao and L. Xu, J. Solid State Chem., 2005, 178, 736–741 CrossRef CAS.
- D. Pan, H. Zhang, T. Fan, J. Chen and X. Duan, Chem. Commun., 2011, 47, 908–910 RSC.
- B.-I. Lee, S.-Y. Lee and S.-H. Byeon, J. Mater. Chem., 2011, 21, 2916–2923 RSC.
- C. Chen, L. K. Yee, H. Gong, Y. Zhang and R. Xu, Nanoscale, 2013, 5, 4314–4320 RSC.
- J.-M. Oh, S.-J. Choi, G.-E. Lee, S.-H. Han and J.-H. Choy, Adv. Funct. Mater., 2009, 19, 1617–1624 CrossRef CAS.
- M. S. San Román, M. J. Holgado, B. Salinas and V. Rives, Appl. Clay Sci., 2012, 55, 158–163 CrossRef.
- Y. Yang, X. Zhao, Y. Zhu and F. Zhang, Chem. Mater., 2012, 24, 81–87 CrossRef CAS.
- A. Y. Kaassis, S.-M. Xu, S. Guan, D. G. Evans, M. Wei and G. R. Williams, J. Solid State Chem., 2016, 238, 129–138 CrossRef CAS.
- A. Y. A. Kaassis, M. Wei and G. R. Williams, J. Mater. Chem. B, 2016, 4, 5789–5793 RSC.
- R. Klevtsova and P. Klevtsov, J. Struct. Chem., 1967, 7, 524–527 CrossRef.
- J. M. Haschke, Inorg. Chem., 1974, 13, 1812–1818 CrossRef CAS.
- D. Mullica, E. Sappenfield and D. A. Grossie, J. Solid State Chem., 1986, 63, 231–236 CrossRef CAS.
- M. Louett and D. Louer, Eur. J. Solid State Inorg. Chem., 1989, 26, 241 Search PubMed.
- F. Gandara, J. Perles, N. Snejko, M. Iglesias, B. Gomez-Lor, E. Gutierrez-Puebla and M. A. Monge, Angew. Chemie, 2006, 45, 7998–8001 CrossRef CAS PubMed.
- L. J. McIntyre, L. K. Jackson and A. M. Fogg, Chem. Mater., 2008, 20, 335–340 CrossRef CAS.
- F. Geng, Y. Matsushita, R. Ma, H. Xin, M. Tanaka, F. Izumi, N. Iyi and T. Sasaki, J. Am. Chem. Soc., 2008, 130, 16344–16350 CrossRef CAS PubMed.
- F. Geng, H. Xin, Y. Matsushita, R. Ma, M. Tanaka, F. Izumi, N. Iyi and T. Sasaki, Chem. – Eur. J., 2008, 14, 9255–9260 CrossRef CAS PubMed.
- L. Poudret, T. J. Prior, L. J. McIntyre and A. M. Fogg, Chem. Mater., 2008, 20, 7447–7453 CrossRef CAS.
- B. I. Lee, K. S. Lee, J. H. Lee, I. S. Lee and S. H. Byeon, Dalton Trans., 2009, 2490–2495 RSC.
- Y.-S. Yoon, B.-I. Lee, K. S. Lee, G. H. Im, S.-H. Byeon, J. H. Lee and I. S. Lee, Adv. Funct. Mater., 2009, 19, 3375–3380 CrossRef CAS.
- F. Gándara, E. G. Puebla, M. Iglesias, D. M. Proserpio, N. Snejko and M. A. Monge, Chem. Mater., 2009, 21, 655–661 CrossRef.
- W. Li, Q. Gu, F. Su, Y. Sun, G. Sun, S. Ma and X. Yang, Inorg. Chem., 2013, 52, 14010–14017 CrossRef CAS PubMed.
- Y. Xiang, X. F. Yu, D. F. He, Z. Sun, Z. Cao and Q. Q. Wang, Adv. Funct. Mater., 2011, 21, 4388–4396 CrossRef CAS.
- Q. Zhu, J.-G. Li, C. Zhi, X. Li, X. Sun, Y. Sakka, D. Golberg and Y. Bando, Chem. Mater., 2010, 22, 4204–4213 CrossRef CAS.
- B. I. Lee and S. H. Byeon, Chem. Commun., 2011, 47, 4093–4095 RSC.
- Q. Zhu, J. G. Li, C. Zhi, R. Ma, T. Sasaki, J. X. Xu, C. H. Liu, X. D. Li, X. D. Sun and Y. Sakka, J. Mater. Chem., 2011, 21, 6903 RSC.
- N. Chu, Y. Sun, Y. Zhao, X. Li, G. Sun, S. Ma and X. Yang, Dalton Trans., 2012, 41, 7409–7414 RSC.
- Q. Gu, Y. Sun, N. Chu, S. Ma, Z. Jia and X. Yang, Eur. J. Inorg. Chem., 2012, 2012, 4407–4412 CrossRef CAS.
- X. Wu, J. G. Li, Q. Zhu, J. Li, R. Ma, T. Sasaki, X. Li, X. Sun and Y. Sakka, Dalton Trans., 2012, 41, 1854–1861 RSC.
- Q. Gu, N. Chu, G. Pan, G. Sun, S. Ma and X. Yang, Eur. J. Inorg. Chem., 2014, 2014, 559–566 CrossRef CAS.
- Q. Gu, F. Su, L. Ma, S. Ma, G. Sun and X. Yang, J. Mater. Chem. C, 2015, 3, 4742–4750 RSC.
- Q. Gu, F. Su, S. Ma, G. Sun and X. Yang, Chem. Commun., 2015, 51, 2514–2517 RSC.
- H. Kim, B.-I. Lee, H. Jeong and S.-H. Byeon, J. Mater. Chem. C, 2015, 3, 7437–7445 RSC.
- T. Shen, Y. Zhang, W. Liu and Y. Tang, J. Mater. Chem. C, 2015, 3, 1807–1816 RSC.
- D. Stefanakis and D. F. Ghanotakis, J. Nanopart. Res., 2010, 12, 1285–1297 CrossRef CAS.
- S. S. Yoo, R. Razzak, E. Bedard, L. Guo, A. R. Shaw, R. B. Moore and W. H. Roa, Nanotechnology, 2014, 25, 425102 CrossRef PubMed.
- Q. Gu, W. Chen, F. Duan and R. Ju, Dalton Trans., 2016, 45, 12137–12143 RSC.
- R. Ju and Q. Gu, Appl. Organomet. Chem., 2018, 32, e3926 CrossRef.
- A. Goyanes, G. B. Hatton, H. A. Merchant and A. W. Basit, Int. J. Pharm., 2015, 484, 103–108 CrossRef CAS PubMed.
- A. N. Ay, B. Zumreoglu-Karan, A. Temel and V. Rives, Inorg. Chem., 2009, 48, 8871–8877 CrossRef CAS PubMed.
- Z. Gu, A. Wu, L. Li and Z. P. Xu, Pharmaceutics, 2014, 6, 235–248 CrossRef PubMed.
- E. Rowatt and R. Williams, Biochem. J., 1989, 259, 295–298 CrossRef CAS PubMed.
- H. Rohwer and E. Hosten, Anal. Chim. Acta, 1997, 339, 271–277 CrossRef CAS.
- S. Savvin, Talanta, 1961, 8, 673–685 CrossRef CAS.
- R. Borissova and E. Mitropolitska, Talanta, 1979, 26, 543–547 CrossRef CAS PubMed.
- D. Gladilovich, V. Kubáň and L. Sommer, Talanta, 1988, 35, 259–265 CrossRef CAS PubMed.
- R. Bhaskar, R. Murthy, B. Miglani and K. Viswanathan, Int. J. Pharm., 1986, 28, 59–66 CrossRef CAS.
- H. Zhang, D. Pan and X. Duan, J. Phys. Chem. C, 2009, 113, 12140–12148 CAS.
- S. F. Hulbert, J. Br. Ceram. Soc., 1969, 6, 11 CAS.
- J. Y. Park, M. J. Baek, E. S. Choi, S. Woo, J. H. Kim, T. J. Kim, J. C. Jung, K. S. Chae and G. H. Lee, ACS Nano, 2009, 3, 3663–3669 CrossRef CAS PubMed.
- G. A. Pereira, D. Ananias, J. Rocha, V. S. Amaral, R. N. Muller, L. Vanda Elst, É. Tóth, J. A. Peters and C. F. G. C. Geraldes, J. Mater. Chem., 2005, 15, 3832–3837 RSC.
- M. Norek, G. A. Pereira, C. F. G. C. Geraldes, A. Denkova and W. Zhou, J. Phys. Chem. C, 2007, 111, 10240–10246 CAS.
- H. Dong, S. R. Du, X. Y. Zheng, G. M. Lyu, L. D. Sun, L. D. Li, P. Z. Zhang, C. Zhang and C. H. Yan, Chem. Rev., 2015, 115, 10725–10815 CrossRef CAS PubMed.
- D. Stefanakis, I. Seimenis and D. Ghanotakis, J. Nanopart. Res., 2014, 16, 2954 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Elemental analysis data; additional X-ray diffraction, SEM and IR spectroscopy data; results of stability studies; fits of kinetic models to the release data; relaxivity data; and, additional in vitro experimental results. See DOI: 10.1039/c7dt03729e |
| This journal is © The Royal Society of Chemistry 2018 |