
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFunctionalised heterogeneous catalysts for sustainable biomass valorisation
Putla
Sudarsanam
 *a,
Ruyi
Zhong
bc,
Sander
Van den Bosch
a,
Simona M.
Coman
d,
Vasile I.
Parvulescu
*d and
Bert F.
Sels
*a
*a,
Ruyi
Zhong
bc,
Sander
Van den Bosch
a,
Simona M.
Coman
d,
Vasile I.
Parvulescu
*d and
Bert F.
Sels
*a
aCentre for Surface Chemistry and Catalysis, Faculty of Bioscience Engineering, KU Leuven, Celestijnenlaan 200f, B-3001, Heverlee, Belgium. E-mail: sudarsanam.putla@kuleuven.be; putla2007@gmail.com; bert.sels@biw.kuleuven.be; Fax: +32 16 321 998; Tel: +32 16 321 610
bDepartment of Chemistry, Southern University of Science and Technology, Shenzhen, 518055, China
cDalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China
dUniversity of Bucharest, Department of Organic Chemistry, Biochemistry and Catalysis, B-dul Regina Elisabeta 4-12, Bucharest 030016, Romania
First published on 18th September 2018
Abstract
Efficient transformation of biomass to value-added chemicals and high-energy density fuels is pivotal for a more sustainable economy and carbon-neutral society. In this framework, developing potential cascade chemical processes using functionalised heterogeneous catalysts is essential because of their versatile roles towards viable biomass valorisation. Advances in materials science and catalysis have provided several innovative strategies for the design of new appealing catalytic materials with well-defined structures and special characteristics. Promising catalytic materials that have paved the way for exciting scientific breakthroughs in biomass upgrading are carbon materials, metal–organic frameworks, solid phase ionic liquids, and magnetic iron oxides. These fascinating catalysts offer unique possibilities to accommodate adequate amounts of acid–base and redox functional species, hence enabling various biomass conversion reactions in a one-pot way. This review therefore aims to provide a comprehensive account of the most significant advances in the development of functionalised heterogeneous catalysts for efficient biomass upgrading. In addition, this review highlights important progress ensued in tailoring the immobilisation of desirable functional groups on particular sites of the above-listed materials, while critically discussing the role of consequent properties on cascade reactions as well as on other vital processes within the bio-refinery. Current challenges and future opportunities towards a rational design of novel functionalised heterogeneous catalysts for sustainable biomass valorisation are also emphasized.
 Putla Sudarsanam | Dr Putla Sudarsanam (1986), currently Marie Curie Postdoctoral Researcher at KU Leuven (Belgium), obtained his PhD degree in Chemistry (2015) from CSIR-Indian Institute of Chemical Technology (India) under the guidance of Dr B. Mahipal Reddy. His research mainly focuses on developing novel heterogeneous solid catalysts for biomass valorisation, sustainable organic transformations, and automotive exhaust gas purification. He authored about 46 peer-reviewed journal articles with an h-index of 24 and more than 1360 citations. He has been received several awards/fellowships, such as Marie Skłodowska-Curie Fellowship-2016 (Belgium), Young Scientist Award-2017 to participate in the 67th Nobel Laureate Meeting (Germany), Leibniz-DAAD Postdoc Fellowship-2016 (Germany), Best PhD Thesis Award-2015 (Catalysis Society of India), EuropaCat PhD Student Award-2013 (XIth European Congress on Catalysis, France) and Endeavour Research Fellowship-2013 (Australia). |
 Ruyi Zhong | Ruyi Zhong (1987), currently a joint postdoc of Southern University of Science and Technology and Dalian Institute of Chemical Physics, Chinese Academy of Sciences, obtained her PhD degree in 2017 at the Centre for Surface Chemistry and Catalysis under the guidance of Prof. Bert Sels. Her research focuses on the synthesis and functionalization of mesostructured silica–carbon nanocomposites and their application in biomass conversion. Her expertise includes solid acid catalysis in biomass-related reactions, templated synthesis of mesostructures, tuning surface properties of composite catalysts and various characterization methods. |
 Sander Van den Bosch | Sander Van den Bosch (1990), currently postdoctoral researcher at KU Leuven, obtained his PhD degree in 2017 at the Centre for Surface Chemistry and Catalysis under the guidance of Prof. Bert Sels. His research focuses on the improved valorisation of lignin for future biorefining, using innovative lignin-first strategies. His expertise includes heterogeneous catalysis for biomass conversions, lignin chemistry, and biomass characterisation. |
 Simona M. Coman | Simona M. Coman (1969), obtained her PhD degree in 2001 in the field of heterogeneous hydrogenation catalysis. After several research stages in KU Leuven, Belgium, a post-doctoral stage in UC Louvain-la-Neuve, Belgium and a research fellowship of AvH Foundation, in HU Berlin, Germany, she became, in 2008, full professor at University of Bucharest. In 2012, she was awarded the “Gheorghe Spacu” award in the field of Chemical Sciences by the Romanian Academy. Her areas of research interest include heterogeneous catalysis for fine chemical and pharmaceutical intermediates and the biomass conversion. She authored about 90 peer-reviewed papers with an h index of 23. |
 Vasile I. Parvulescu | Vasile I. Parvulescu received his PhD from the Polytechnic University of Bucharest in 1986 with a thesis investigating the selectivity of bi- and multi-metal catalysts in hydrogenation of aromatic hydrocarbons. After several years as high-signor researcher at the Institute of Inorganic and Rare Metals, in 1992 he joined the University of Bucharest, where he becomes full professor in 1999. He is currently director of the Department of Organic Chemistry, Biochemistry and Catalysis. His scientific interest concerns the study of heterogeneous catalysts for green and fine chemistry and environmental protection. He authored more than 370 papers, 27 patents, and 7 books. He was awarded by the Romanian Academy (“Nicolae Teclu” Award, 1990), Romanian President (Knight for Merits, 2008) and The National Grand Lodge of Romania (“Grigore Moisil“ award for exact sciences, 2015). |
 Bert F. Sels | Bert F. Sels (1972), currently full professor at KU Leuven, obtained his PhD degree in 2000 in the field of heterogeneous oxidation catalysis. He was awarded the DSM Chemistry Award in 2000, the Incentive Award by the Belgian Chemical Society in 2005, and the Green Chemistry Award in 2015. He is currently director of the Centre for Surface Chemistry and Catalysis, designing heterogeneous solid catalysts for future challenges in industrial organic and environmental catalysis. His expertise includes heterogeneous catalysis in bio-refineries, design of hierarchical zeolites and carbons and the spectroscopic and kinetic study of active sites for small-molecule activation. He authored about 300 peer-reviewed journal articles with an h index of 70 and more than 16 |
1. Introduction
Rapid population growth and vast economic developments elicited an unforeseen increase in global energy demand that is estimated to double between 2000 and 2035.1 Fossil fuel sources, such as crude oil, coal, and natural gas currently hold the major share of energy supply. However, fossil fuels are non-renewable and global petroleum production is predicted to peak by 2020 due to increasing demand for chemical industries, before decaying.2 The first oil crisis in 1973 raised an awareness about the limited availability of traditional oil and gas sources.3 Another major concern of the 21st century is the increasing levels of greenhouse gas emissions (e.g., CO2) caused by the enormous consumption of fossil fuels.4,5 Global CO2 emissions reached an all-time record of 41.5 ± 4.4 billion tonnes in 2017, contributing to an atmospheric CO2 concentration of 408 ppm, the highest since the beginning of the industrial revolution.6–8 If this situation continues, global average temperatures are estimated to increase drastically in the range of 2.5–5.4 °C above pre-industrial levels by 2050.2 Global warming is a major threat to humankind as well as to the biosphere, as witnessed by the increased rate and intensity of many climate catastrophes in recent times. In order to control global warming, several countries have issued stringent regulations to reduce the utilisation of fossil fuels, moving towards a carbon-neutral society. In December 2015, the 21st Conference of the Parties to the United Nations Framework Convention on Climate Change (COP21/CMP11, Paris) led to an agreement between 195 countries to reduce CO2 emissions, with a target to limit global temperatures to below 1.5 °C above pre-industrial levels.9 The European Union also set the exacting ecological targets, for example a 20% reduction in CO2 emissions by 2020 compared to 1990 levels.10 Part of the European Union goal is also to expand the renewable energy market share to 20% of the total energy supply by 2020. In view of these concerns and challenges, there has been a growing research interest towards the use of clean, sustainable resources for the production of fuels and chemicals.Various renewable resources, such as wind, geothermal, solar, hydropower, and biomass are available for the production of energy and/or chemicals (Fig. 1). As a highly abundant, natural carbon source, biomass is considered as a promising renewable alternative to fossil fuels that can be transformed into a wide range of value-added chemicals, clean solvents, and high-energy density fuels.1,11–16 Global annual production of biomass is estimated to be greater than 10 billion tons (dry basis), equating to more than 10% of global energy supply.17–19 Untapped biomass sources could generate a huge amount of energy, equivalent to more than 2 billion tons of standard coal.19 The National Renewable Energy Action Plans estimated that biomass would provide around 42% of the total renewable energy needed for electricity, heating, and cooling by 2020.20 Several biomass resources, such as animal waste, agricultural crops, wood, and aquatic plants are available for the production of sustainable chemicals.21 These biomass feedstocks can be harnessed within a relatively short period, boosting environmental credentials.
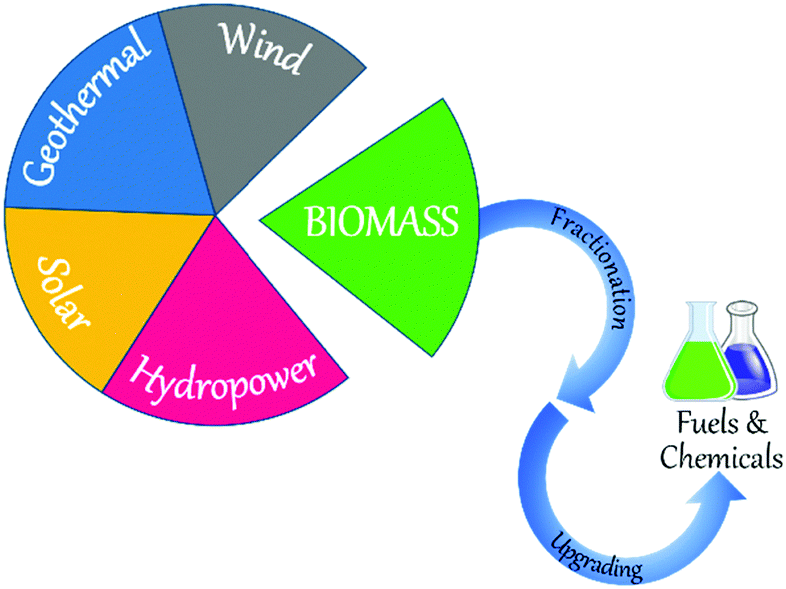 | ||
| Fig. 1 Available renewable energy sources and valorising biomass into fuels and chemicals. | ||
A large number of technologies based on biological, thermal, and chemical processes have been developed for biomass valorisation (Fig. 2).22,23 Among those, chemical processing of biomass is of paramount research interest as the resulting products can exhibit relatively equating characteristics to petro-based products. Various kinds of chemical processes, such as fast pyrolysis, hydro-processing, oxidation, dehydration, hydrolysis, (trans)esterification, isomerisation, and many others have been reported, in which the application of a catalyst is crucial to enhance reaction rates and to obtain high yields of desirable products in a short time period. Indeed, catalysis is a key technology in modern chemical industry and plays an essential role in the production of a vast majority of bulk and commodity chemicals.3 Catalysis greatly contributes to the development of new, greener, and potential chemical processes, offering feasible alternatives to stoichiometric reactions, thus acting as a driving force towards a more sustainable chemical industry.24–26 Homogeneous and heterogeneous catalysts are both used in petrochemical industry as well as in biomass upgrading. Homogeneous catalysts, where the active sites are in the same phase as the reactants, can interact efficiently with the reaction substrates, typically resulting in higher turnover frequency (TOF) rates compared to heterogeneous catalysts. However, homogeneous catalysts are often associated with high toxicity, corrosivity, energy-intensive separation and purification procedures, and inefficient reusability. Stringent government regulations have therefore directed chemical industries to search for alternative catalytic materials. In this respect, heterogeneous catalysis, where the catalyst exists in a different phase (typically solids) as the reactants (mostly liquids or gasses), could offer tremendous potentials for several energy- and environmental-related applications including biomass upgrading.27–30 Availability of facile preparation methods, low production costs, remarkable robustness, high resistance to common reaction conditions (moisture, air, pressure, and temperature) and durable lifetime are some of the primary advantages of heterogeneous solid catalysts. More importantly, solid catalysts can be efficiently recovered from reaction mixtures and can be readily reused in multiple catalytic cycles, making the process cost-effective and more sustainable.
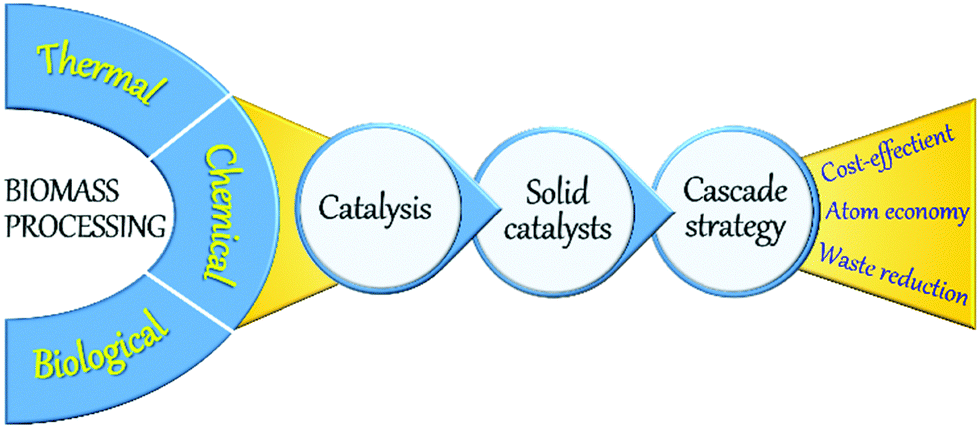 | ||
| Fig. 2 Concept of sustainable biomass processing. | ||
One-pot cascade catalytic reactions play a pivotal role towards sustainable biomass valorisation (Fig. 2).31–34 Cascade reactions can improve the atom economy or the E-factor (kgwaste/kgproduct) by limiting the number of energy-intensive steps needed for the isolation and purification of intermediate products as well as for the catalysts recovery. In this context, the versatile application of multifunctional solid catalysts with adequate amounts of acid, base or redox active sites is essential. Advances in the fields of chemistry, materials science, spectroscopy, and catalysis enabled us to get a profound understanding of key steps involved in the rational design of new, multifunctional solid catalysts as well as in the heterogeneous catalytic reactions. Important aspects are fine-tuning of catalyst properties, efficient functionalisation of solid catalysts, behaviour of reactants/intermediates/products with the solid surface, and the modifications in catalyst properties during and after the reaction. This in-depth understanding has provided several innovative strategies, paving the way for the development of state-of-the-art solid phase catalysts, which are highly valuable not only for petrochemicals production and environmental pollution control, but also for sustainable biomass valorisation.
Over the last few years, a variety of functionalised heterogeneous catalysts has been developed for biomass upgrading, including zeolites, carbon materials, mesoporous silica, metal–organic frameworks, metal oxide supported metals, solid phase ionic liquids, magnetic iron oxides, and organic polymers.21,30,35 In particular, carbon materials, metal–organic frameworks, solid phase ionic liquids, and magnetic iron oxides have received great attention because of exciting scientific breakthroughs and their extensive catalytic applications in biomass valorisation (Fig. 3). Accommodating ample amounts of acid–base and redox functional species is one of the unique features of these materials, making them capable of catalyzing various biomass conversion reactions in a one-pot way. For instance, carbon materials (e.g., activated carbon, graphene, carbon nanotubes, carbon nanofibers, and mesoporous carbons) can play a vital role in biomass valorisation because of their fascinating characteristics associated with tailorable porosity, rich surface chemistry, high specific surface area, and remarkable hydrophobicity.35 In addition, new functional carbon-based materials can be synthesised through pyrolysis of renewable biomass molecules (cellulose, sucrose, glucose, etc.) and their subsequent application as catalyst support or catalytically active phase in the biomass valorisation will be an added advantage in the context of sustainable biomass upgrading.
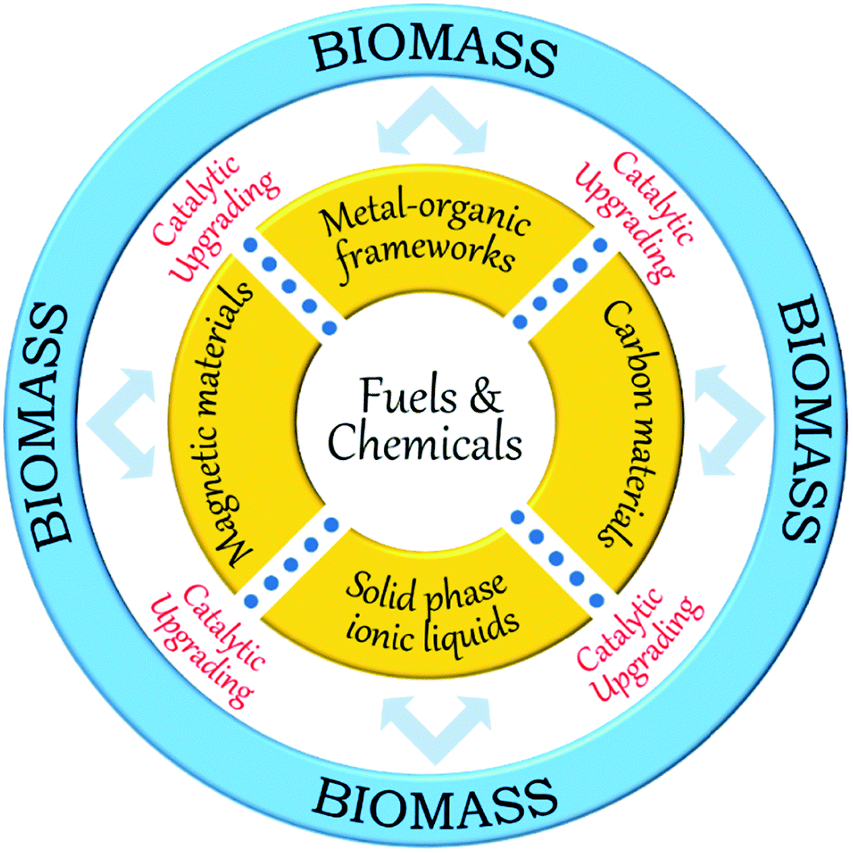 | ||
| Fig. 3 Schematic overview of this review. | ||
Metal–organic frameworks (MOFs), an extraordinary family of crystalline porous materials, are of particular research interest for biomass upgrading and considered as a promising alternative to typical porous zeolite and silica catalysts.36–38 MOFs are composed of metal nodes and organic ligands, which virtually contain considerable amounts of acid–base and redox species. In addition, the distinctive features of MOFs allow to immobilise desired functional acid–base groups (–SO3H, –NH2, etc.) or active metal nanoparticles (Ru, Pd, Cu, etc.) on particular sites of the MOFs. The properties of resulting functionalised MOFs could be significantly different from the pristine analogues, due to the existence of synergistic host-dopant interactions.
Solid phase ionic liquids (SILs), such as supported ILs and polymerised ILs have drawn great attention for both fundamental research and practical applications including catalytic biomass upgrading.39–42 They not only retain the individual properties of ILs and supports/polymers, but also endow novel functional active sites due to the cooperative effect of ILs with supports/polymers. Solid phase ILs can also overcome negative aspects of pristine ILs, such as high preparation costs, active phase leaching, and poor recovery/recycling. Particular interactions between the anionic and cationic parts of ILs can show a conceivable constructive effect in stabilising acid–base and active metal centers, which could be beneficial for cascade biomass upgrading.43
Biomass valorisation often entails the processing of larger molecules in viscous or solid reaction mixtures. The separation of solid catalysts from such type of reaction mixtures requires energy-intensive steps, making the process more complex and expensive. To overcome these concerns, the use of magnetic solid catalysts in biomass conversions is a potential approach. Magnetic catalysts can be efficiently separated from the reaction mixture using an external magnetic field, without affecting their catalytic properties.30,44,45 Particularly, core–shell Fe3O4@SiO2 magnetic nanomaterials have received a great deal of interest in biomass valorisation.46–48 In addition to the benefits of catalyst separation, the existence of silanol groups in Fe3O4@SiO2 can offer facile routes for selective anchoring of acid, base or metal active species that may assist to achieve promising results in biomass conversion reactions.
1.1 Scope of the review
Over the last few years, numerous research efforts have been undertaken towards developing a variety of heterogeneous catalysts for biomass upgrading reactions. Accordingly, several review articles have been reported on this topic. For instance, Zhang et al.49 provided a review on the importance of liquid state ionic liquids for the processing of renewable energy into fuels and chemicals. At the same time, Herbst et al.36 summarised several catalytic applications of MOFs for biomass conversions. In 2016, Liu et al.50 provided a review on the catalytic applications of magnetic materials for biomass upgrading. In another review, Lam and Luong have discussed the role of carbon materials as catalyst supports and as active phase catalysts for the transformation of biomass to fuels and chemicals.35 Li et al.51 reported a critical review, focusing on the valorisation of lignocellulose to biofuels using solid catalysts. Recently, Trombettoni et al.52 reviewed various catalytic applications of sulfonated resin-based solid acids for biomass upgrading. Although these reviews provide useful implications and insights in the respective fields, there seems to remain an unexplored potential in the field of functionalised heterogeneous catalysts for the cascade processing of biomass valorisation. At the same time, there are concerns regarding the added-value of functionalised heterogeneous catalysts for biorefinery industry with respect to pristine analogues. Based on the above background, this review aims to revise and critically evaluate the most significant findings and advances related to the applications of carbon materials, MOFs, solid phase ionic liquids, and magnetic iron oxides for biomass conversions. Throughout the review, special attention has been drawn to facile approaches developed for the immobilisation of functional active sites on the above-listed materials as well as to understand the role of ensuing properties on chemical reactions and mechanisms in biomass valorisation. In addition to one-pot cascade reactions, also other types of vital reactions that normally take place in bio-refineries will be covered. Finally, a summarised overview will highlight the challenges and opportunities of future research towards rational design of promising functionalised heterogeneous catalysts in the context of sustainable biomass upgrading. Considering the significance of tailored catalytic functionalities for efficient biomass upgrading, materials characterization techniques (spectroscopy, microscopy, and sorption) are vital to identify structure–property relationships. This elaborate research field is beyond the scope of the present review, though several recent articles and reviews have already discussed the theory and the application of relevant characterization tools, applicable for the functionalised catalysts reported in this review.53–582. Carbon-based catalysts
Over the last three decades, carbon materials have been intensely investigated in the fields of materials science, nanotechnology, and catalysis, which can be attributed to their fascinating characteristics associated with textural, conductivity, stability, and hydrophobicity.59–62 They have found versatile applications, for example as catalyst support and as active phase in catalysis, as electrode in energy storage devices, in water purification, in gas separation, and as soil additive. Carbon can exhibit three states of hybridisation, namely sp, sp2, and sp3, which drive the formation of well-defined pentagonal, hexagonal or heptagonal structures.60 These ‘magic’ configurations allow the development of a variety of advanced carbon materials, such as fullerenes, carbon nanotubes, graphene, and mesoporous carbon. Although these materials solely consist of carbon atoms, they possess unique properties due to the extraordinary arrangement of carbon atoms with a flexible geometry. The scientific community has recognised the importance of carbon materials with two prestigious Nobel prizes: (1) fullerenes – 1996 (Chemistry, Prof. Robert F. Curl Jr., Prof. Harold W. Kroto and Prof. Richard E. Smalley) and (2) graphene – 2010 (Physics, Prof. Andre Geim and Prof. Konstantin Novoselov).60,612.1 Factors affecting the catalytic activity of carbon materials in biomass upgrading
Carbon materials have emerged as promising catalyst supports as well as metal-free active phase catalysts for various biomass transformation reactions.35,63,64 They exhibit a broad spectrum of crucial catalytic properties: (i) large specific surface area, (ii) tailorable porous structures and surface chemistry, (iii) excellent chemical stability in acid or base media, (iv) remarkable hydrothermal stability, and (v) efficient functionalisation. Many types of conventional carbon materials, such as activated carbon, carbon black, glassy carbon, pyrolytic carbon, and polymer-derived carbon have been employed for stabilizing catalytic active phases. Owing to high specific surface area and rich surface chemistry, these carbon materials allow the formation of highly dispersed metal particles (Pd, Ru, Ni, Cu, Ag, Fe, etc.) throughout the catalyst matrix, resulting in enhanced resistance to sintering even at higher metal loadings and elevated temperature conditions.Advances in materials science and nanotechnology have provided several innovative strategies for the development of new carbon materials, such as carbon nanotubes, graphene, and mesoporous carbons that can be used as catalyst supports or active catalysts.35,63,64 Carbon nanotubes (CNTs) are characterized by a hexagonal arrangement of sp2 carbons with well-controlled cavity geometries. CNTs can be classified as single-walled and multi-walled CNTs based on the number of carbon layers present in the tubular wall. Single-walled CNTs are semiconductive with diameters of around 0.4–2 nm, whereas multi-walled CNTs are metallic.65 Interestingly, the cavities of CNTs can prevent the aggregation of active metal NPs during catalyst synthesis or catalytic reactions.66,67 Graphene is a two-dimensional material consisting of a single layer of carbon atoms in hexagonal sp2 hybridisation.64 Two types of C–C bonds, namely in-plane σ-bond and out of-plane π bond are typically found in graphene. The electronic properties of graphene, controlled by out of-plane π-bonds, play a crucial role in enhancing the interactions of graphene with metallic NPs, and consequently the catalytic performance of graphene in biomass conversions. Mesoporous carbons, mesostructured carbon-based composites, and carbon nanofibers have also received great attention for catalytic biomass upgrading.68–70 Owing to unique shape- and porosity-controlled properties, these carbon materials could offer strong active phase-support interactions, which can lead to unusual catalytic activities and selectivities in biomass upgrading.
Defect sites and surface chemistry are the key parameters that determine the catalytic efficiency of carbon materials in biomass conversions. For example, defect sites incorporated into the sp2 framework of CNTs, graphene or activated carbon can strongly influence surface properties and catalytic functionalities.71 Heteroatom doping is an appealing strategy, which exploits defect structures in carbon materials.72–74 Various elements, including nitrogen, phosphorus, boron, and sulphur have been successfully doped into carbon materials. Interestingly, structural and electronic properties of doped carbon materials can be considerably different from the pristine analogues, due to the synergistic host–dopant interactions. For instance, doping with electron rich nitrogen can adjust the spin density and the charge distribution of carbon atoms. This will lead to enhanced π-binding ability and improved basicity on the carbon surface.72 In addition, N-doping can assist in the formation of new surface nucleation sites and multiple active centres around the N-rich sites.73 In contrast, boron doping generates acid sites in carbon materials. The presence of such functional sites on the carbon surface not only contributes to improved interactions between the carbon support and the dispersed metal NPs, but also enables the use of carbon materials as promising metal-free catalysts.
In order to further improve the catalytic activity and selectivity of pristine or doped carbon materials, the immobilisation of various acid (nitro, sulphate, or phosphate) or base functionalities (–SO3Na, –COONa, –ONa, or –NH2) on the carbon surface has been carried out using appropriate synthesis methods.35 Anchoring of acid functional groups on the carbon surface can occur through strong C–S, C–C, C–O, or C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds.35,75 Hence, these functional carbon materials can act as stable solid Brønsted acids for various important biomass conversion reactions, such as esterification, transesterification, etherification, hydrolysis, and dehydration. For instance, acid-bearing carbon catalysts showed good activity for the dehydration of 2-propanol to propylene in the presence of water vapour, a vital reaction for upgrading biomass-derived oxygenates.76 Base functionalised carbons have also been found excellent catalytic efficiencies, for example in biodiesel synthesis.77 However, these functionalised carbons suffer from several drawbacks, especially in the case of acid groups. For example, acid-bearing carbon materials are usually prepared by sulfonation of partially carbonized organic molecules using highly concentrated hazardous liquid acids, such as H2SO4 or HNO3.78 These protocols show adverse effects on the structure as well as on the stability of carbon materials, which in turn could lead to inefficient recovery of the catalyst from the reaction mixture. Besides, oxygen-containing groups present on the carbon surface may promote side reactions during biomass valorisation (Fig. 4).35 This will lead to a low selectivity of desired products and can even irreversibly poison the catalyst. In order to circumvent these problems, several promising strategies have been suggested, such as selective deposition of –SO3H groups on carbon edges, or deprotonation–carbometalation, followed by base functionalisation, etc.77,78
O bonds.35,75 Hence, these functional carbon materials can act as stable solid Brønsted acids for various important biomass conversion reactions, such as esterification, transesterification, etherification, hydrolysis, and dehydration. For instance, acid-bearing carbon catalysts showed good activity for the dehydration of 2-propanol to propylene in the presence of water vapour, a vital reaction for upgrading biomass-derived oxygenates.76 Base functionalised carbons have also been found excellent catalytic efficiencies, for example in biodiesel synthesis.77 However, these functionalised carbons suffer from several drawbacks, especially in the case of acid groups. For example, acid-bearing carbon materials are usually prepared by sulfonation of partially carbonized organic molecules using highly concentrated hazardous liquid acids, such as H2SO4 or HNO3.78 These protocols show adverse effects on the structure as well as on the stability of carbon materials, which in turn could lead to inefficient recovery of the catalyst from the reaction mixture. Besides, oxygen-containing groups present on the carbon surface may promote side reactions during biomass valorisation (Fig. 4).35 This will lead to a low selectivity of desired products and can even irreversibly poison the catalyst. In order to circumvent these problems, several promising strategies have been suggested, such as selective deposition of –SO3H groups on carbon edges, or deprotonation–carbometalation, followed by base functionalisation, etc.77,78
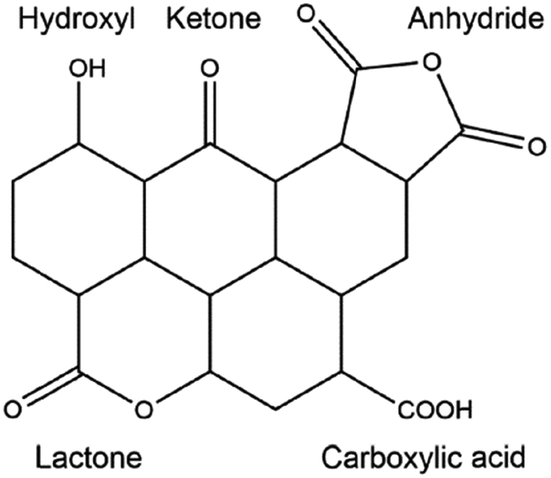 | ||
| Fig. 4 Presence of various oxygen-containing groups in an activated carbon material. Reproduced with permission from ref. 35. Copyright (2014) American Chemical Society. | ||
The simultaneous functionalisation of carbon materials with acid or base species as well as with active metal particles will lead to the design of novel bifunctional catalysts. Bi-functional catalysts have the advantages of integrating several catalytic processes in a one-pot way for sustainable biomass upgrading. For instance, both metallic and acid properties are essential for the direct conversion of levulinic acid to γ-valerolactone. Compared to metal oxides, carbon supports show a stronger ability to stabilise both acid and metal sites because of their high hydrothermal stability and rich surface chemistry. Wang et al.79 developed a facile synthesis procedure for developing a graphene-supported Ru/SO3H bi-functional catalyst. In this process, Ru NPs are initially deposited on graphene, followed by its functionalisation with benzenesulfonic acid groups (Fig. 5). This bifunctional catalyst showed a high selectivity to γ-valerolactone via the Ru catalysed hydrogenation of levulinic acid to 4-hydroxyvaleric acid and the subsequent acid catalysed dehydration to γ-valerolactone. The presence of tailorable porous structures is also crucial for efficient functionalisation of carbon materials, which was well-documented in the case of mesoporous carbon-based composites.80 For example, a porous graphite-like carbon network incorporated in a Sn(IV)-grafted MCM-41 was found to show a good catalytic performance for the conversion of sugars into ethyl lactate in ethanol, which was attributed to synergistic effect of Lewis and weak Brønsted acid sites.81
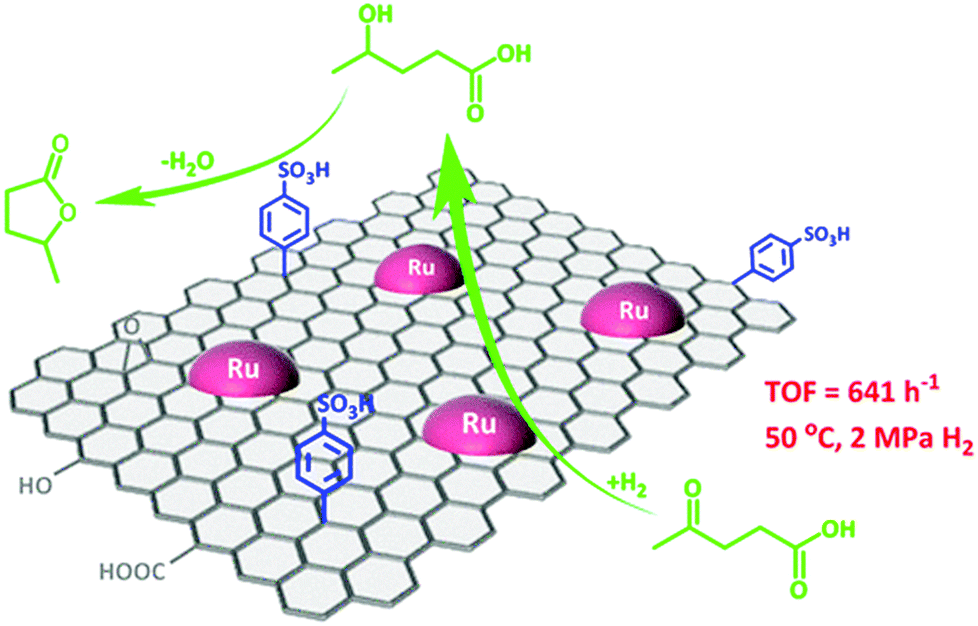 | ||
| Fig. 5 Graphene-based metal/acid bi-functional catalyst for one-pot conversion of levulinic acid to γ-valerolactone. Reproduced with permission from ref. 79. Copyright (2017) American Chemical Society. | ||
In general, high production costs and low yields are the major drawbacks in the synthesis of carbon materials. Especially, in the case of CNTs and graphene, high synthesis costs strongly limit their practical applications in biomass conversions. It is therefore indispensable to use low-cost, abundant, and sustainable substrates for the preparation of carbon-based catalysts. Noticeably, recent studies have provided several facile routes for the transformation of bio-based molecules into new carbon materials with prolific functional sites.82–84 A key advantage of such synthesis processes is that the raw material (biomass) is abundant and renewable. To enhance the activity of biomass-derived carbon catalysts for biomass conversions, various acid–base functional molecules can be immobilised by either ex situ or in situ methods. Devi et al.85 developed a highly stable carbon catalyst derived from glycerol (a major by-product in biodiesel synthesis) incorporating various basic functionalities. The synthesis procedure involves an in situ carbonisation and sulfonation of glycerol to obtain a SO3H–carbon catalyst, followed by a controlled treatment with aqueous NaOH solution. The resulting solid base catalyst showed good performance in the transesterification of sunflower oil with methanol to produce biodiesel, in which glycerol is formed as a by-product that can be used again for the synthesis of carbon-based catalysts, hereby greatly improving process economy and sustainability.
The strong hydrophobic nature of carbon materials is another key driving force that boosts their catalytic applications for biomass upgrading processes. Owing to high resistance capacity towards water, which is the preferred solvent in many bio-refinery processes, carbon surfaces can efficiently prevent the leaching of active phase during catalytic reactions, thus improving the catalyst recyclability, a critical benchmark for industrial applications.76 Moreover, high hydrothermal stability of carbon materials is another significant advantage over other catalytic materials, for example zeolites, which suffer from irreversible deactivation due to deterioration of their crystalline structure in the presence of water. Interestingly, the hydrophilic character of carbon materials can be improved by employing an oxidation treatment in order to facilitate immobilisation of desired functional groups on the carbon surface.86 The catalytic efficiency of several classes of carbon-based materials tested in biomass valorisation was critically discussed in the following paragraphs. In addition, an overview of the most significant catalytic results achieved in carbon catalysed biomass conversions is provided in Table 1.
| Entry | Catalyst | Reaction | Product | Reaction conditions | Catalytic activity | Recycling results | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Conv. % | Yield % | |||||||
| a Delignification efficiency. b RT – room temperature. c HMF – 5-hydroxymethylfurfural. d DMF – 2,5-dimethylfuran. e DMTHF – 2,5-dimethyltetrahydrofuran. f 30 min reaction time. g 200 min reaction time. h GO – graphene oxide. i r-GO – reduced graphene oxide. j OBMF – 5,5′-oxy(bis-methylene)-2-furaldehyde. k CNT – carbon nanotube. l FDCA – 2,5-furandicarboxylic acid. | ||||||||
| 1 | 5 wt% Ru/C | Reductive fractionation of birch wood sawdust | Lignin phenolic monomers | 2 g birch sawdust, 0.3 g Ru/C, 40 mL MeOH, 250 °C, 12 MPa H2 at 250 °C, 3 h | 93a | 50 | Negligible decrease in phenolic monomers yield (48%) after a recycling test | 91 |
| 2 | Ru/activated carbon | Continuous-flow hydrogenation of cellulose oligomers | C6 sugar alcohols | 25 vol% of 2-propanol in water, 180 °C, pH = 2.7, 5 h, liquid hourly space velocity of 4.7 h−1, 60 bar Ar at RTb | — | 36.4% | Stable catalytic activity during 12 h time-on-stream | 103 |
| 3 | Carbon encapsulated Fe NPs | Hydrogenation of levulinic acid | γ-Valerolactone | 5 g levulinic acid, 0.2 g catalyst, 45 mL H2O, 170 °C, 5 bar H2 at RT, 3 h | 99.5 | 99 | 5 (stable catalytic activity) | 104 |
| 4 | Ni–Fe0.5/activated carbon | Hydrogenation of ethyl levulinate | γ-Valerolactone | 100 mg ethyl levulinate, 40 mg catalyst, 10 mL H2O, 100 °C, 4 MPa H2 at RT, 6 h | 99.3 | 99 | 5 (catalyst deactivation, due to leaching of active sites) | 106 |
| 5 | N-Doped carbon decorated Cu/MgAlO | Hydrogenolysis of HMFc | DMFd | 0.5 g HMF, 0.1 g catalyst, 10 mL cyclohexanol, 220 °C, N2 atm | 100 | 96f | 5 (stable catalytic activity) | 110 |
| DMTHFe | 94.6g | |||||||
| 6 | Fe/N-doped carbon | Hydrogenation of furfural | Furfuryl alcohol | 0.5 mmol furfural, 50 mg catalyst, 3 mL of 2-butanol, 160 °C, 15 h | 91.6 | 76 | 5 (catalyst deactivation, due to changes in structure of active phases and pores blocking) | 111 |
| 7 | MoCx NPs/hollow carbon spheres | Hydrodeoxygenation of phenol | Benzene | 3 mmol phenol, 30 mg catalyst, 5 mL n-octane, 350 °C, 80 bar H2 at 350 °C, 4 h | 87 | 74 | Negligible catalytic loss after a recycling test | 119 |
| 8 | Sulfonated carbon | Transesterification of purified palm oil | Biodiesel | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of methanol to oil, 0.5 wt% catalyst based on oil, 270 °C, 30 min :1 molar ratio of methanol to oil, 0.5 wt% catalyst based on oil, 270 °C, 30 min |
— | 95 | 3 (significant decrease in biodiesel yields from 95% to 45%, due to leaching of acidic species) | 123 |
| 9 | Pt9Sn1/C | Oxidation of glycerol | Glyceric acid | 5 mmol glycerol, 100 mg catalyst, 10 mL water, 60 °C, O2 (15 mL min−1), 8 h | 91 | 50 | 4 (stable catalytic activity after treating the catalyst at 400 °C in H2 atmosphere) | 126 |
| 10 | GO–SO3Hh | Decomposition of glucose | Levulinic acid | 30 g glucose, 0.5 g catalyst, 200 mL water, 200 °C, 2 h | 89 | 78 | 5 (catalyst deactivation, due to char formation during reaction) | 132 |
| 11 | WO3/r-GOi | Dehydration of fructose | HMF | 1 mmol fructose, 10 mg catalyst, 2 g ionic liquid, 120 °C, 2 h | 100 | 84.2 | 5 (negligible decrease in catalytic activity) | 133 |
| 12 | Sulfonated Ru/r-GO | Hydrogenolysis of levulinic acid | γ-Valerolactone | 200 mg substrate, 0.5 mg of Ru, 10 mL H2O, 50 °C, 2 MPa H2 at 50 °C, 40 min | 100 | 82 | 8 (significant decrease in product yields, due to sintering of Ru NPs, weight loss of the catalyst, and blockage of active Ru sites) | 79 |
| 13 | SO3H@GO monolith | Esterification of levulinic acid | Benzyl levulinate | 50 mmol substrate, 75 mmol benzyl alcohol, 0.008 g cm−3 catalyst loading, toluene, 110 °C, 90 min | 97 | — | 5 (stable catalytic activity for 5 min reaction in each cycle) | 137 |
| 14 | GO | Etherification of HMF | OBMFj | 63 mg HMF, 20 mg catalyst, 0.5 mL CH2Cl2, 100 °C, 8 h | 98 | 86 | 6 (catalytic activity of GO can be regenerated by treating it under Hummers' conditions) | 138 |
| 15 | Cu–Pd/r-GO | Glycerol hydrogenolysis | Lactic acid | 1:1 molar ratio of NaOH/glycerol, 15 mL H2O, 140 °C, 1.4 N2 MPa at 140 °C, 16 h |
56.2 | 49.5 | 3 (considerable decrease in product yields from 49.5% to 40%) | 139 |
| 16 | VO2–polyaniline/CNTk | Oxidation of HMF | 2,5-Diformylfuran | 1 mmol HMF, 100 mg catalyst, 9.5 wt% of vanadium, 2 mL dimethyl sulfoxide, 120 °C, 1 MPa O2 at RT, 11 h | >99 | 96 | 5 (significant decrease in product yields from 96% to 75%) | 144 |
| 17 | Pt/CNT | Oxidation of HMF | FDCAl | 0.5 mmol HMF, 100:1 molar ratio of HMF/Pt, 20 mL H2O, 95 °C, 0.5 MPa O2 at RT, 14 h |
100 | 98 | 7 (stable catalytic activity) | 146 |
| 18 | Mesoporous carbon-based solid acid | Dehydration of fructose | HMF | 0.5 g fructose, 0.25 g catalyst, 7 mL isopropyl alcohol, 130 °C, 140 min | 97.9 | 87.8 | 5 (about 10% decrease in HMF yields) | 150 |
| 19 | Ordered mesoporous carbon (CMK-5) | Methylation of volatile fatty acids (VFA) | Fatty acid methyl esters | 0.5 vol ratio of VFA/methanol, 65 mg catalyst, 360 °C, 1 atm O2 | — | 98 | 6 (stable catalytic activity) | 152 |
| 20 | Sulfonated silica/carbon nanocomposite | Hydrolysis of cellulose | Glucose | 50 mg catalyst, 5 mL water, 150 °C, 24 h | 60.7 | 50.4 | 3 (small decrease in catalytic activity, due to leaching of acid groups) | 160 |
| 21 | Sulfonated sucrose-derived carbon | Dehydration of xylose | Furfural | 0.4 g xylose, 0.2 g catalyst, 16.5 mL of γ-valerolactone, 170 °C, 30 min | — | 78.5 | 5 (stable catalytic activity) | 169 |
| 22 | Biochar supported SnO2–Co3O4 | Hydrolysis of corncob | Furfural | 0.3 g catalyst, 120 mL water, 180 °C, 200 min | 62.7 | 30 | 6 (significant decrease in product yields from 30% to 15%, due to leaching of acidic species) | 170 |
| 23 | Willow-derived carbon solid acid | Cellulose hydrolysis | Glucose | 50 mg cellulose, 50 mg catalyst, 5 mL water, 160 °C, 8 h | 78 | 65 | 3 (small decrease in glucose yields, due to mass loss of the catalyst during recovery/purification | 174 |
| 24 | Sulfonated bamboo-derived carbon | Microwave-assisted hydrolysis of hemicellulose | Xylo-oligosaccharides | 1:200 mass ratio of catalyst to water, 150 °C, 45 min, microwave reactor |
— | 54.7 | 3 (catalyst deactivation, due to leaching of acidic species) | 179 |
| 25 | Biomass-derived carbon cryogel | Esterification of levulinic acid | Ethyl levulinate | 15:1 molar ratio of ethanol to levulinic acid, 150 °C, 4 h |
87.2 | 86.5 | 5 (catalyst deactivation, due to leaching of active species) | 187 |
| 26 | CoNx@N-doped carbon | Hydrodeoxygenation of eugenol | Propylcyclohexanol | 1 mmol eugenol, 76 mg catalyst, 10 mL n-dodecane, 200 °C, 2 MPa H2 at RT, 2 h | 100 | >99.9 | 4 (catalyst deactivation, due to leaching of active sites) | 193 |
| 27 | Co@N-doped carbon | Hydrodeoxygenation of vanillin | 2-Methoxy-4-methylphenol | 0.5 mmol vanillin, 10 mL water, 180 °C, 0.5 MPa N2 at RT, 4 h, formic acid (0.2 g) as hydrogen source | 95.7 | 95.7 | 5 (stable catalytic activity) | 72 |
| 28 | SO3Na-functionalised glycerol-derived carbon | Transesterification of oil | Biodiesel | 1:5 (wt/vol) ratio of oil to methanol, catalyst amount (20 wt% of oil), 65 °C, 2 h |
100 | 99 | 8 (stable catalytic activity) | 85 |
2.2 Conventional carbon based catalysts
Carbon black and activated carbon have been widely used as supporting materials for the synthesis of promising heterogeneous catalysts because of their low preparation cost and high specific surface area.35,86 Carbon black is an amorphous solid with planar layers of sp2-hybridised carbon. Pyrolysis of hydrocarbon precursors or organic polymers at elevated temperatures is typically used for the synthesis of carbon black. Activated carbon is also an amorphous solid, though with a characteristic twisted geometry of planar-layered structures, which are cross-linked by aliphatic bridging groups that provide a high density of defect sites. Both physical and chemical activation processes are used for the preparation of activated carbons. The raw material is initially pyrolysed at 600–900 °C in the physical activation, and then exposed to an oxidising atmosphere (oxygen or steam) at desirable temperatures. In contrast, chemical activation involves impregnation of carbon precursors with an acid, a strong base, or a salt, followed by a carbonisation step in the range of 450–900 °C. Chemical activation is the preferred process due to the mild conditions (i.e., lower temperatures and shorter times) required for the synthesis of activated carbon. | ||
| Fig. 6 Composition of lignocellulosic biomass with major constituents: cellulose, hemicellulose, and lignin. | ||
Carbon encapsulated Fe NPs were prepared using an ultrasonic method (Fig. 7).104 The effect of the annealing temperature on textural and phase properties was studied. Results revealed a superior catalytic efficiency of carbon-encapsulated Fe NPs for the hydrogenation of levulinic acid to γ-valerolactone (99% yield). The shell structure of carbon prevents leaching of active Fe sites during reaction, resulting in excellent reusability for at least five times (entry 3, Table 1). Carbon-supported bimetallic Ru–Sn catalysts containing different amounts of Sn were studied for the hydrogenation of levulinic acid to γ-valerolactone.105 The catalyst containing equal amounts of Ru and Sn showed a stable activity with a 100% selectivity to γ-valerolactone, while the activity of monometallic Ru/C catalyst was progressively decreased with reaction time. In contrast, the Ru1Sn4/C catalyst was found to be inactive due to the formation of an additional phase (β-Sn), which leaches and sinters during reaction. In another work, γ-valerolactone is produced via hydrogenation of ethyl levulinate using a series of activated carbon supported bimetallic Ni–Fe (Ni–Fe/AC) catalysts.106 Among them, the Ni–Fe0.5/AC catalyst exhibited the highest activity and achieved 99.3% conversion of ethyl levulinate with 99% yield of γ-valerolactone at optimised reaction conditions (entry 4, Table 1). The existence of uniform dispersed Ni–Fe alloy structure in combination with FeOx NPs is essential for high catalytic hydrogenation activity. However, significant catalyst deactivation (around 20% loss in γ-valerolactone yield) was observed after four recycles, due to leaching of active species. A simple calcination step was used to regenerate the catalytic activity of Ni–Fe0.5/AC towards γ-valerolactone production.
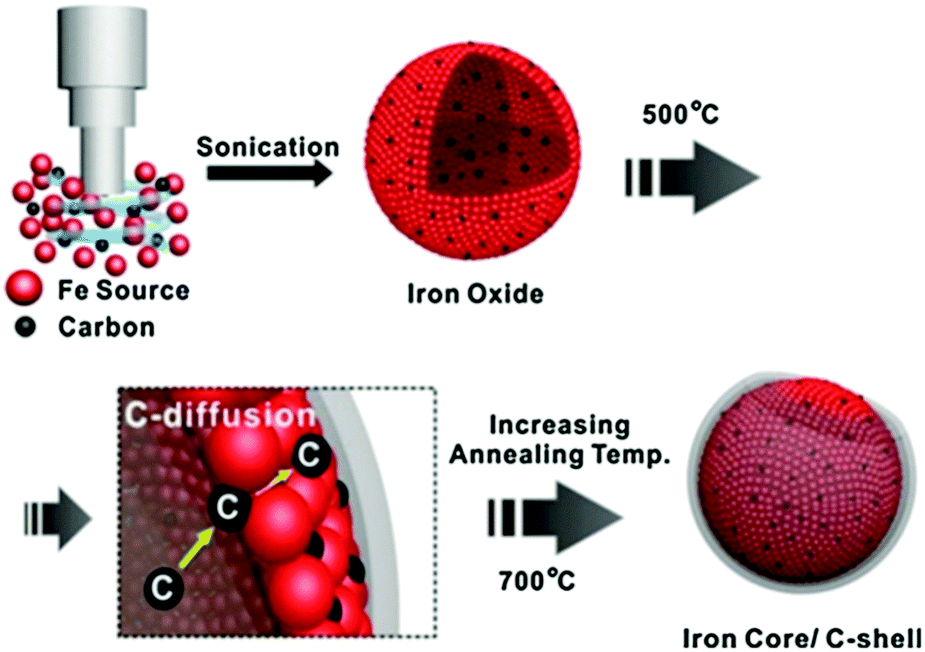 | ||
| Fig. 7 Schematic illustration for the preparation of carbon-encapsulated Fe NPs. Reproduced with permission from ref. 104. Copyright (2014) Springer Nature. | ||
Dehydration of various carbohydrates, such as xylose, glucose, and fructose was studied using a commercial activated carbon (Norit, ROX 0.8).107 This carbon contains several types of functional groups, including carboxylic acids, –SO3H, carbonyls, phenols, thiophenols, quinones, and anhydride sites, which can act as acidic or basic sites. Results reveal that this activated carbon is a highly suitable catalyst for the dehydration of sugars due to its strong hydrophobic character. In addition, the carbon catalyst efficiently suppresses side reactions that produce acids and humins. Several activated carbon supported monometallic Ru and bimetallic RuRe catalysts were studied for sorbitol hydrogenolysis reaction.108 Among them, bimetallic catalysts in combination with Ca(OH)2 as a base promoter exhibited higher activity and selectivity towards 1,2-propanediol, lactic acid, ethylene glycol, and linear alcohols. The aqueous-phase hydrodeoxygenation of sorbitol was also investigated using a Pt–ReOx/C catalyst in a continuous-flow reactor.109 The Pt–ReOx/C catalyst exhibited a 34 times higher activity compared to Pt/Zr–P catalyst (based on a Pt mass), which was attributed to a higher hydrothermal stability of Pt–ReOx/C catalyst related to the carbon support. A catalytic transfer hydrogenolysis of HMF was successfully performed to produce 2,5-dimethylfuran (DMF) and 2,5-dimethyltetrahydrofuran (DMTHF) biofuels using a N-doped carbon-decorated Cu/MgAlO catalyst with cyclohexanol as the hydrogen source.110 About 96% and 94.6% yields of DMF and DMTHF, respectively, were obtained at optimised reaction conditions (entry 5, Table 1). Moreover, the catalyst exhibited a stable activity for at least five cycles without any considerable loss in catalytic performance. The existence of suitable basic sites on the catalyst surface plays a beneficial role in the activation of alcohol hydroxyl moieties in cyclohexanol to release active hydrogen species. Highly dispersed Cu0 NPs and electrophilic Cu+ species catalyse the hydrogen transfer and the subsequent hydrogenolysis of HMF, respectively. Nitrogen-doped carbon supported iron catalysts were found to show a good performance in catalytic transfer hydrogenation of furfural to furfuryl alcohol.111 The effects of the metal center, nitrogen source, pyrolysis temperature, and the support were studied. A 91.6% conversion of furfural with about 76% yield to furfuryl alcohol were achieved, attributed to the coordination of iron cations by pyridinic nitrogen functionalities on the carbon support. A considerable decrease in catalytic efficiency was noticed after five recycles of the catalyst, due to changes in structure of active phases and blockage of the catalyst pores (entry 6, Table 1).
The effect of solvent mixtures in the hydrogenolysis of xylitol to glycols and glycerol was investigated using an activated carbon supported bifunctional Ru–MnO catalyst.112 The product distribution obtained in the case of 90:10 vol% water/1,4-dioxane and water/2-PrOH solutions is very similar to the one obtained in pure water. Interestingly, the overall selectivity to glycols and glycerol is greatly enhanced (up to 70%) in the case of 90:10 vol% water/ROH mixtures (ROH with R = Me, Et, n-Pr, and n-Bu). Hydrodeoxygenation of α-angelica lactone-derived di/trimers to gasoline was studied using carbon supported Pd and Pt catalysts.113 α-Angelica lactone can be produced from levulinic acid. The effect of H2 pressure, reaction temperature, and reaction time on the product selectivities were thoroughly investigated. Considerable amounts of bio-liquids suitable for gasoline application were obtained in this work.
:1) catalyst. In the case of birch wood sawdust, the total yield of monomers even reached up to 39.5 wt% (88% selectivity to propylguaiacol and propylsyringol) over the Ni1–Fe1/AC catalyst, attributed to the synergistic effect of Ni–Fe alloys. A series of bimetallic M–W/AC (M = Ru, Pt, and Pd) catalysts were also studied for hydrogenolysis of lignin and its model compounds.115 Electronic effects of bimetallic catalysts play a pivotal role in stabilising the reaction intermediates. The nature of solvent is also imperative in hydrogenolysis of lignin: higher yields of desired aromatics were obtained with dipolar aprotic solvents, such as n-hexane. Bimetallic catalysts not only cleaved the α-O-4 bonds, but also catalysed hydrogenolysis of other major linkages in lignin, consequently resulting in high yields of liquid oil. Zeng et al.116 developed an efficient Pd(OH)2/C catalyst for cross-coupling of lignin 4-O-5 linkage models with amines via an in situ generated phenol intermediate. The obtained amine derivatives are valuable chemicals for the production of fine chemicals, pharmaceuticals, and electronic materials. Based on the experimental results, a probable mechanism for the production of amine derivatives from lignin model compounds is proposed.
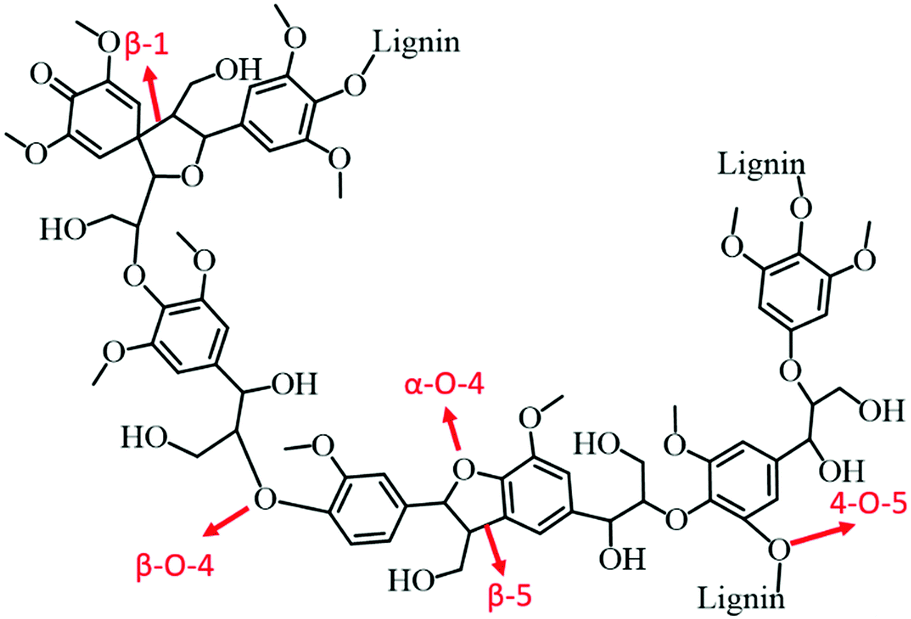 | ||
| Fig. 8 Various important C–C and C–O–C linkages in lignin polymer. | ||
A multi-functional cobalt NP-embedded carbon catalyst (Co@C) was developed via carbonisation of a three-dimensional cobalt coordination polymer.117 This Co@C catalyst showed excellent efficiency in the oxidation of vanillyl alcohol (a lignin model compound) to vanillin with 99% and 100% selectivities using H2O2 and air as oxidants, respectively (Fig. 9). The Co@C catalyst also showed a significantly improved performance compared to other cobalt catalysts and was efficiently re-used without significant loss in catalytic activity. Thorough kinetics and mechanistic studies were performed for the catalytic hydrodeoxygenation and hydrogenation of eugenol, a lignin model compound, using a Ru/C catalyst.118 Results reveal that hydrogenation of the allyl group and benzene ring of eugenol were favored over dehydroxylation and demethoxylation of oxygen groups present on the aromatic ring of eugenol. The mechanism of ketone group formation as well as the factors responsible for cyclohexane-ring transformation to substituted 5-membered ring compounds were also proposed in this work. A detrimental effect of water is observed in the hydrodeoxygenation of phenol in the presence of MoCx NPs supported on hollow carbon spheres (HCS).119 About 87% conversion of phenol with a 74% yield to benzene was obtained over MoCx/HCS catalyst at optimised reaction conditions. A negligible decrease in product yields was noticed after a recycling test (entry 7, Table 1).
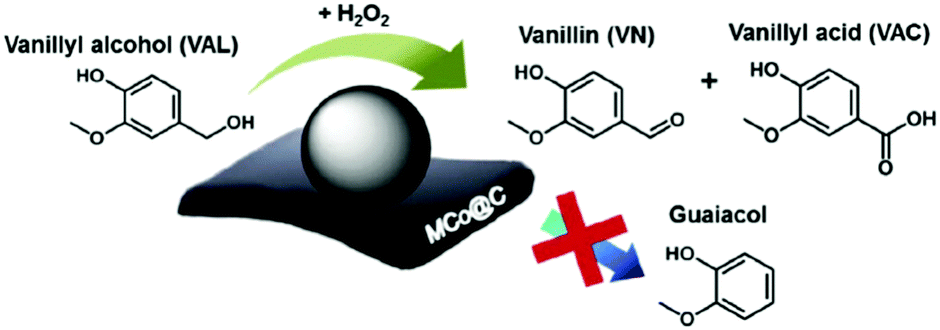 | ||
| Fig. 9 Oxidation of vanillyl alcohol using Co NP-embedded carbon nanocomposite (Co@C) catalyst. Reproduced with permission from ref. 117. Copyright (2018) Elsevier. | ||
Biodiesel (fatty acid methyl esters) is a biodegradable and nontoxic fuel, and hence, considered as a promising alternative to petro-diesel fuel.15,120,121 Several carbon-based catalysts have shown good to excellent efficiencies in the production of biodiesel via transesterification of oils and fats (Fig. 10). An incomplete hydrothermal carbonisation of β-cyclodextrin into polycyclic aromatic carbon sheets, followed by its sulfonation using H2SO4 was performed to develop a novel carbon-based solid acid catalyst.122 The resulting solid acid simultaneously catalysed esterification and transesterification reactions to produce biodiesel from high free fatty acid containing oils, hereby outperforming a conventional solid acid (Amberlyst-15). In another work, an incomplete carbonization of naphthalene in H2SO4 was performed to obtain a carbon-based solid acid catalyst for the production of biodiesel via transesterification of purified palm oil.123 About 95% yield of biodiesel was obtained, which however decreases to 45% after three recycles of the catalyst, due to leaching of acidic species (entry 8, Table 1). Glycerol, an interesting platform chemical, can be largely obtained as a by-product during biodiesel synthesis (Fig. 10).124,125 A series of bimetallic PtM/C (M = Mn, Fe, Co, Ni, Cu, Zn, and Au) catalysts were developed for the oxidation of glycerol.126 Among them, Pt9Sn1/C catalyst showed the highest activity with a 91% of glycerol conversion and a 50% yield of glyceric acid. Modified Pt NPs after the addition of Sn significantly activate oxygen molecules and/or deprotonation of hydroxyl group in glycerol, resulting in improved catalytic performance of Pt9Sn1/C material as well as stable catalytic activity for at least four recycles after treating the catalyst at 400 °C in H2 atmosphere (entry 9, Table 1).
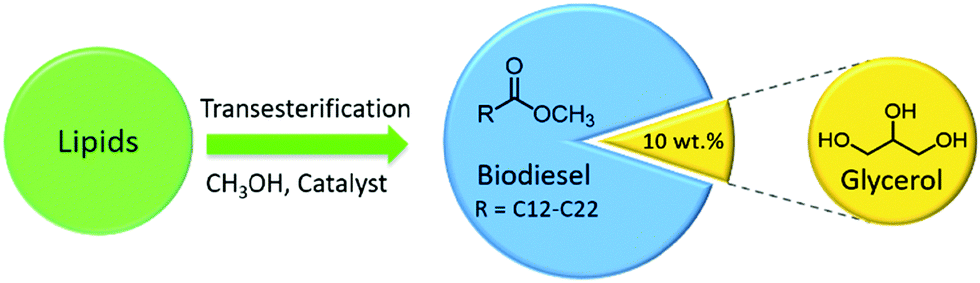 | ||
| Fig. 10 Transesterification of lipids (oils & fats) with alcohols (e.g., methanol) to produce biodiesel with glycerol as a by-product. | ||
2.3 Graphene based catalysts
Graphene and its related materials, such as graphene oxide and reduced graphene oxide represents a fascinating class of materials and continues to provide a fertile ground for various applications including catalysis. Graphene-based materials are ideal supports for anchoring a variety of metal NPs and acid–base functionalities. For the first time, in 2004, Geim et al. developed single layer graphene nanosheets.127,128 Afterwards, several promising synthesis procedures, including graphitisation, physical exfoliation, chemical vapour deposition, and chemical oxidative cleavage have been developed to synthesise well-defined graphene-based materials.129 Graphene oxide (GO) is obtained by the oxidation of graphite, followed by the exfoliation of monolayer sheets in solution. Reduced graphene oxide (r-GO) is simply produced by reduction of graphene oxide and the resulting material partially regains the aromaticity of pristine graphene. The defects and residual O-containing groups present in graphene play an essential role in the synthesis of highly stable and active catalyst systems.128,130,131In an efficient two-step strategy, Ru NPs were first supported on r-GO after which a subsequent functionalisation with benzenesulfonic acid groups was carried out (Fig. 11).79 The obtained catalyst showed high performance in the hydrogenation–dehydration of levulinic acid with a 82% yield of γ-valerolactone. In contrast, 4-hydroxyvaleric acid was the major product over a non-functionalised Ru/r-GO catalyst. The combination of strong acid sites and ample amounts of active metal centers in the sulfonated Ru/r-GO catalyst is the key reason for achieving higher yields of γ-valerolactone. Sintering of Ru NPs, weight loss of the catalyst, and blockage of active Ru sites were noticed after eight recycles of the catalyst, hence a significant decrease in product yields from 82% to 53% (entry 12, Table 1). A few-layer graphene (FLG) supported Ru NPs catalyst was also studied for the conversion of levulinic acid to γ-valerolactone.135 The activity of Ru/FLG is 2–4 times higher than that of Ru particles deposited on activated carbon and other traditional support materials. This was attributed to the higher abundancy of stabilised metallic Ru species in Ru/FLG catalyst. Alternatively, maximal yields of γ-valerolactone (99%) can also be produced from ethyl levulinate in the presence of a r-GO supported polymeric Ru porphyrin catalyst.136 Various graphene oxide-based catalysts were studied for the esterification of levulinic acid with benzyl alcohol as well as the etherification of benzyl alcohol with butanol.137 Among them, a graphene oxide monolith functionalised with chlorosulfonic acid showed excellent catalytic performance in both reactions. This catalyst exhibited a stable activity up to five recycles for 5 min reaction times in esterification of levulinic acid (entry 13, Table 1). Graphene oxide, prepared by a modified Hummers' method, was proven to be an efficient catalyst for the etherification of HMF to produce a polymer building block, 5,5′-oxy(bis-methylene)-2-furaldehyde.138 The solvent plays an important role in this reaction, whereas in situ generated water showed a negative effect on the catalytic performance of graphene oxide. About 86% product yield was obtained, attributed to the presence of abundant oxygen containing functional groups on the graphene oxide catalyst. Reusability studies revealed that the catalytic activity of GO can be regenerated by treating it under Hummers' conditions (entry 14, Table 1).
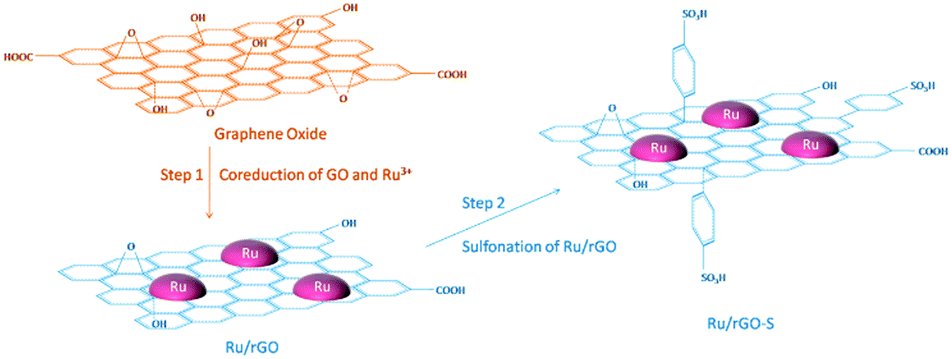 | ||
| Fig. 11 Schematic illustration of the preparation strategy for a bi-functionalised graphene oxide based catalyst. Reproduced with permission from ref. 79. Copyright (2017) American Chemical Society. | ||
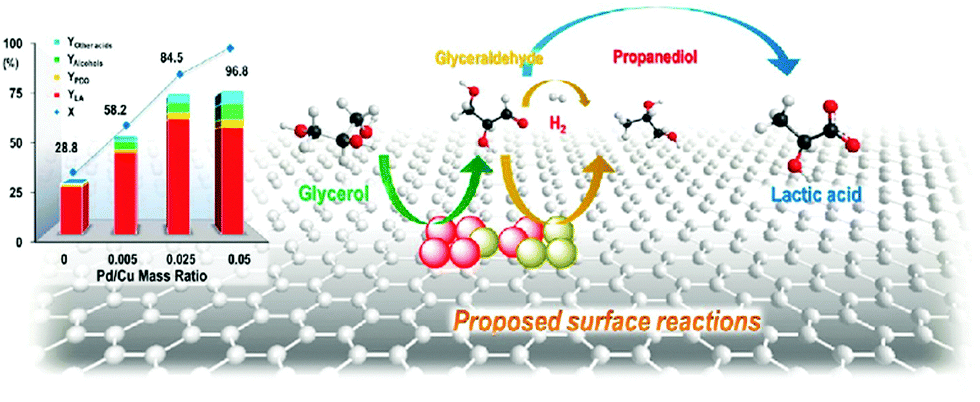 | ||
| Fig. 12 Glycerol conversion and product yields as well as the proposed reaction pathways in glycerol hydrogenolysis over Cu–Pd/r-GO catalysts. Reproduced with permission from ref. 139. Copyright (2013) American Chemical Society. | ||
2.4 Carbon nanotubes based catalysts
Carbon nanotubes (CNTs) offer potential possibilities for stabilising metal NPs, metal oxides, acid–base functional molecules, and even complex hierarchical hybrids. CNTs exhibit defective sp2 carbon surfaces and improved electron transport, which facilitates the interaction of active phases with the CNTs.61,141–143 In addition to preventing particle aggregation, the nanoscale confinement within CNTs can also control the diffusion of reactive species and their interactions with the active phases. These fascinating characteristics have been exploited in the field of biomass upgrading, in order to achieve improved conversion rates with high yields of desired products. Arc discharge, laser vaporisation and recently, chemical vapor deposition have been widely used techniques for the preparation of CNTs.142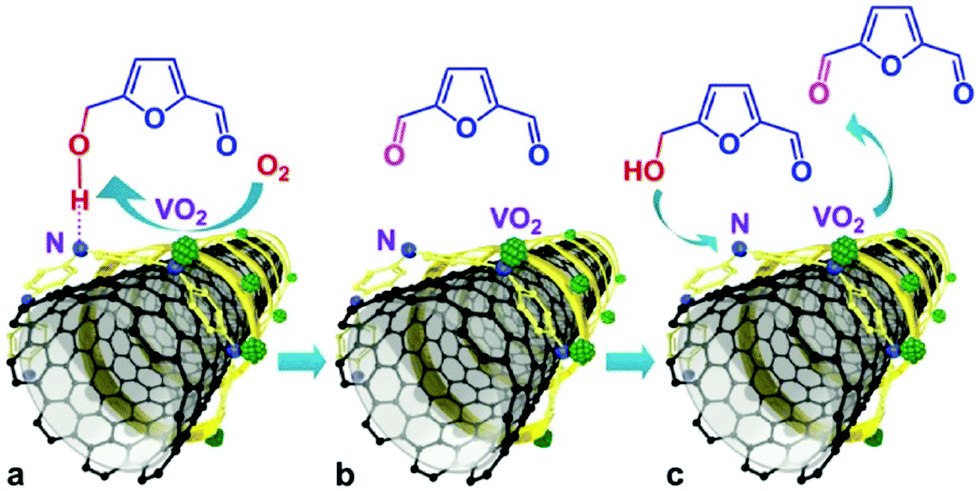 | ||
| Fig. 13 Selective aerobic oxidation of HMF to 2,5-diformylfuran over VO2–PANI/CNT catalyst. Reproduced with permission from ref. 144. Copyright (2015) John Wiley and Sons. | ||
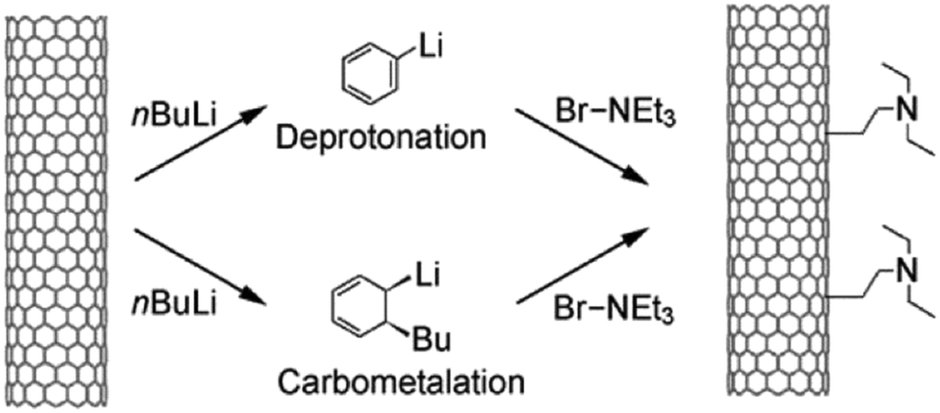 | ||
| Fig. 14 Functionalisation of multi-walled CNTs by deprotonation–carbometalation and subsequent electrophilic attack of bromotriethylamine. Reproduced with permission from ref. 77. Copyright (2009) John Wiley and Sons. | ||
2.5 Mesoporous carbon-based catalysts
Mesoporous carbon materials have gained paramount attention for applications in biomass conversion due to their controllable porous properties, therefore significantly differing from traditional carbon materials.147–149 In addition, they exhibit high specific surface areas and tunable pore channels, fascinating interfacial features, unprecedented control over their morphology and a rich composition. Template-assisted carbonisation methods are widely used for the synthesis of mesoporous carbons as they provide an efficient control over pore arrangement, structure, and mesoporosity. Another potential benefit of mesoporous carbons in biomass conversions is the enhanced interaction between reactive species and the catalytic active phases. For example, cellulose is not soluble in polar solvents. It is therefore difficult to convert completely the lignocellulose to value-added chemicals or biofuels. Owing to large sized uniform pores and abundant functional groups, mesoporous carbons can enhance the affinity between the non-soluble biomass molecules including cellulose and the catalyst, and consequently improved conversion rates in biomass valorisation.147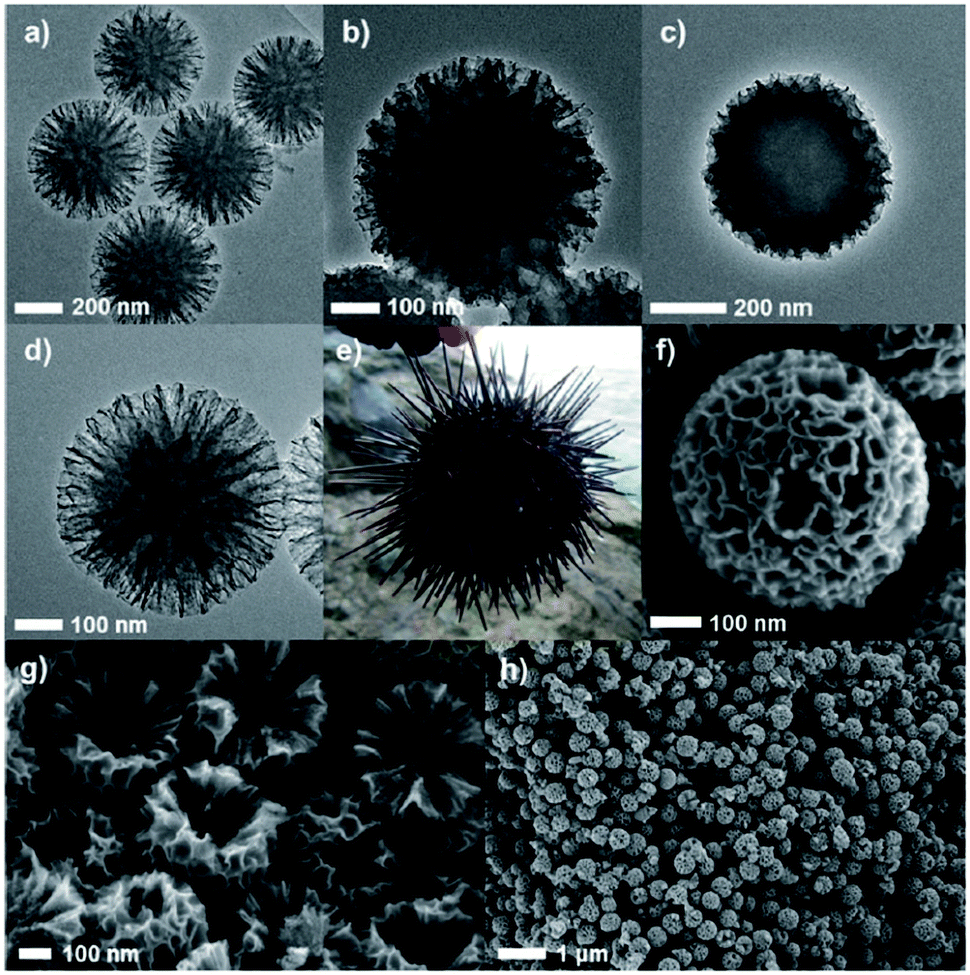 | ||
| Fig. 15 High resolution TEM images of (a) Echinometra mathae-shaped silica (ESS) template, (b) ESS–carbon composite after drying, (c) ESS–carbon composite after aging, (d) carbon nano-Echinometra mathae (CNE), and (e) image of actual Echinometra mathae. SEM images of (f) a particle of CNE, (g) cross-section of CNE, and (h) prepared CNE particles in a large-scale view. Reproduced with permission from ref. 147. Copyright (2013) John Wiley and Sons. | ||
A combined catalytic and biotechnological process was developed for the production of HMF and bioethanol from cellulose.149 In the first step, hydrolysis/dehydration of mechanically activated microcrystalline cellulose was carried out using a mesoporous carbon-based catalyst. Moderate yields of glucose and HMF were obtained. The extracted HMF from the reaction mixture was then subjected to ethanol fermentation. Acid-functionalised periodic mesoporous carbons (CMK-3 and CMK-5) were tested for the continuous production of HMF from fructose in the presence of water.75 Both CMK-3 and CMK-5 showed good catalytic stability compared with acid-functionalised SBA-15 and commercial Nafion SAC-13 catalysts. Zhang et al.150 developed an efficient mesoporous carbon-based solid acid using ethylene tar and concentrated H2SO4 with magnesium acetate as the template. A 87.8% yield of HMF was obtained via fructose dehydration, attributed to superior specific surface area and higher concentration of sulfonic acid groups in the developed mesoporous carbon catalyst. Reusability studies revealed a 10% decrease in HMF yields after five recycles of the catalyst (entry 18, Table 1). Hollow mesoporous carbon spheres (Fig. 16) were used as a support for anchoring ArSO3H groups with controlled loadings (1.8 and 3.2 wt%).151 Owing to strong Brønsted acidity and hollow nanospherical morphology with thin mesoporous shell, the developed catalysts showed excellent activity and stability for the synthesis of ethyl levulinate from levulinic acid or furfuryl alcohol. Tentative reaction mechanisms were proposed in this study (Fig. 16).
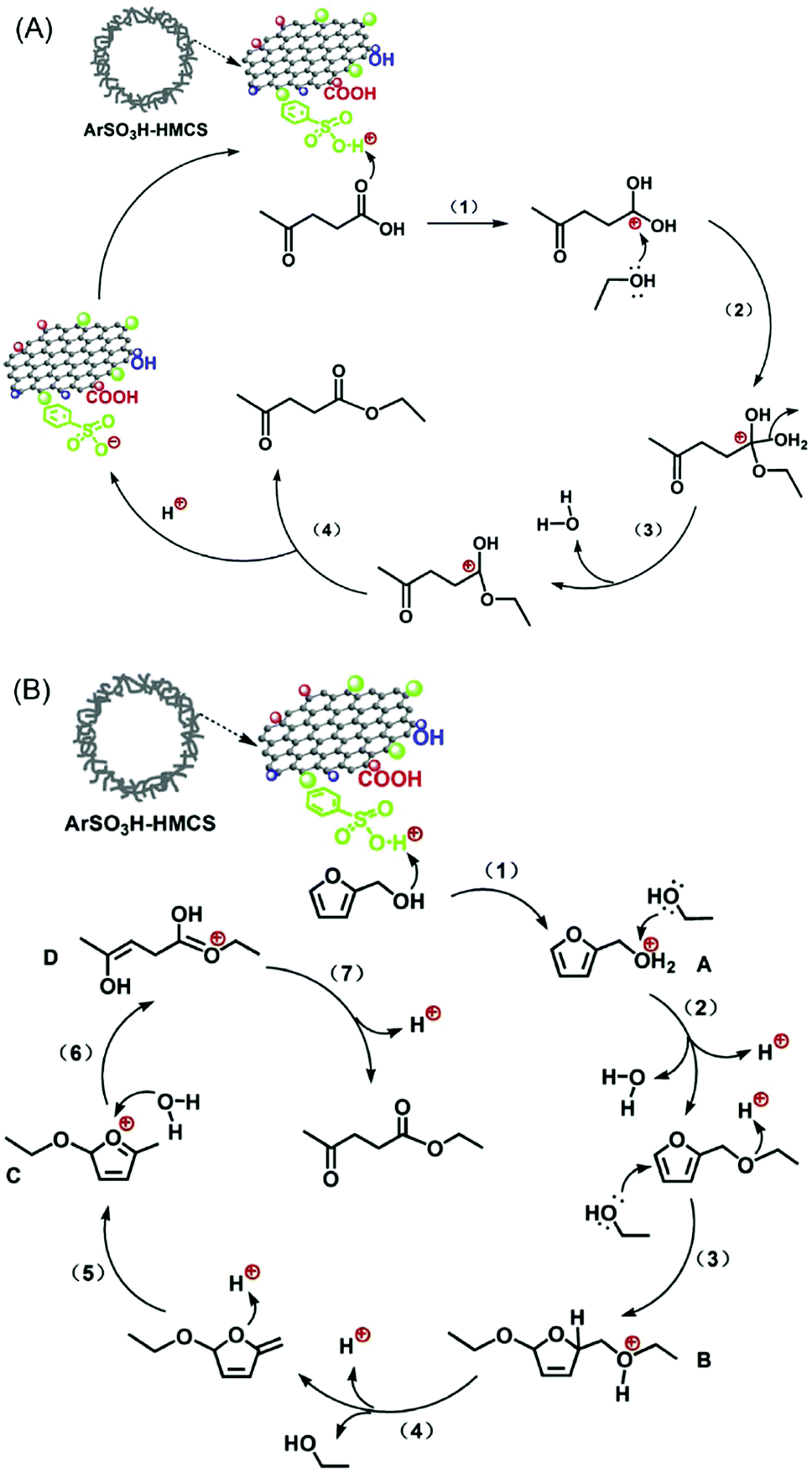 | ||
| Fig. 16 Possible mechanisms for the synthesis of ethyl levulinate (A) from esterification of levulinic acid and (B) from ethanolysis of furfuryl alcohol over ArSO3H-functionalised hollow mesoporous carbon spheres (HMCSs). Reproduced with permission from ref. 151. Copyright (2015) Elsevier. | ||
2.6 Mesostructured carbon-containing composites
The development of mesostructured composites, wherein carbon phase is well-dispersed, is of high research interest, not only for the reason of stabilising catalytically active carbon phase, but also to achieve synergistic properties in the resulting catalysts.80 Over the last few years, a lot of research has been performed towards the design of various mesostructured carbon based composites. In particular, periodic mesoporous organosilicas (PMOs) have attracted significant attention because of their versatile properties towards promising biomass valorisation.80,154 Post-synthetic grafting and co-condensation of organosilanes with silica precursors are often used for the synthesis of PMOs. Nevertheless, the utilization of organosilane precursors is practically unfavorable because of their high expensive nature. Tremendous research efforts have therefore been made to develop PMOs-based analogies, starting with relatively cheap carbon precursors, such as phenolic resin,155,156 2,3-dihydroxynaphthalene,157p-toluenesulfonic acid,158 furfuryl alcohol,81,159 sucrose,160–163 and followed by incorporation of carbon precursors into pre-formed mesoporous hard templates (e.g., ordered mesoporous silica),81,157–159,163 or one-pot formation of mesostructured nanocomposites in the presence of soft template (e.g., tri-block pluronic co-polymer).155,156,160–162 The advantages of mesostructured carbon-containing composites for catalytic biomass valorisation are comparable or superior, depending on the reaction conditions used, to those of mesoporous carbon-based catalysts as discussed in Section 2.5. Interestingly, the presence of robust silica framework in mesoporous silica–carbon composites provides value-added benefits, such as strong thermal stability against elevated temperature/pressure reaction conditions as well as high hydrophilicity for the immobilization of relative functional groups on the catalyst surface.160:C = 33:66) carbonized at 550 °C exhibited high catalytic performance in hydrolysis of cellulose to glucose (up to 50.4% glucose yield).160 This catalytic performance was attributed to the presence of strong and accessible Brønsted acid sites as well as to the hybrid surface structure, which promotes adsorption of reaction substrates on the catalyst surface. A small decrease in catalytic activity of sulfonated silica–carbon nanocomposite was found for cellulose hydrolysis after three recycles, due to leaching of acidic species (entry 20, Table 1). These catalysts were also active for the ethanolysis of fructose to levulinic esters and furanic ethers with the optimized combined yields of ca. 80%, as shown in Fig. 17.162 The developed composites contain a hierarchical pore architecture, of which mesoporosity facilitates fast mass transport, whereas microporosity is beneficial to attain fast sugar catalysis. The observed catalyst deactivation was due to fouling of the acid sites instead of leaching, however, the catalytic activity could be fully regenerated using a simple re-sulfonation step. A modified EISA method was developed for scalable synthesis of acidic mesostructured silica–carbon nanocomposite catalysts.164 The resultant sulfonated nanocomposites displayed good catalytic activity in fructose ethanolysis and in condensation of 2-methylfuran with furfural. This catalytic performance was attributed to several parameters, such as well-ordered mesostructure, tunable hydrophilicity/hydrophobicity and high concentration of strong acidic SO3H sites.
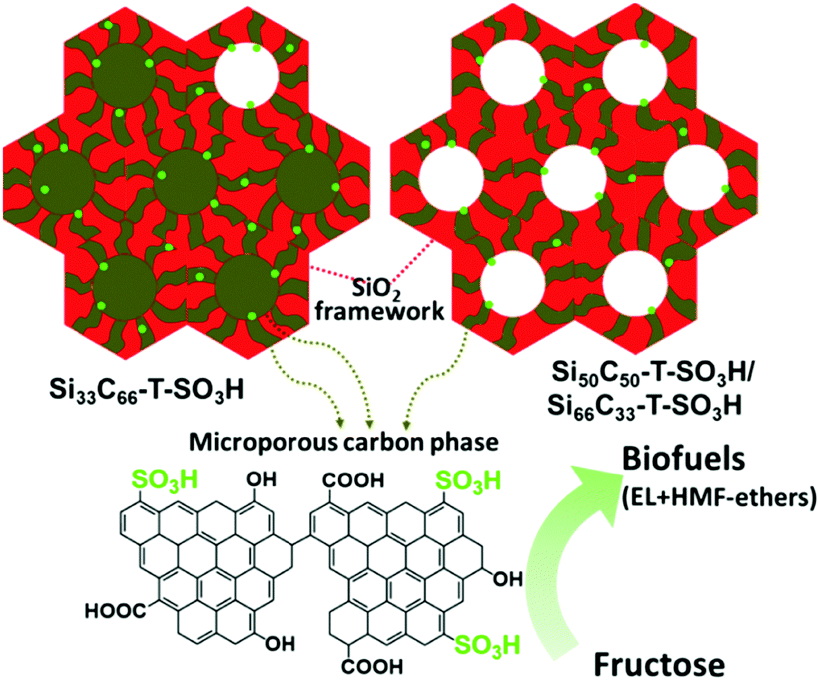 | ||
| Fig. 17 Structural scheme of sulfonated silica–carbon nanocomposites. Red color entity represents silica framework, brown color entity represents microporous carbon phase, green color dots represent SO3H groups, and EL represents ethyl levulinate. Reproduced with permission from ref. 162. Copyright (2017) Elsevier. | ||
EISA method is also applied for the synthesis of ordered mesoporous carbon–alumina nanocomposites, which was then used as a support for loading Pt NPs.156 The resulting nanocomposites showed high catalytic performance in one-pot transformation of cellulose into hexitols, which was attributed to tunable hydrophobicity/hydrophilicity, adjustable metal–support interaction, and strong hydrothermal stability. Mesoporous silica–carbon solid acid catalysts obtained by a hard-template synthesis method were also tested in conversion of fructose, HMF, and furfuryl alcohol with ethanol into HMF ethers and levulinic esters.158 Results reveal that higher yields of desirable bio-products were obtained over the silica–carbon catalysts, containing higher acid sites (1.9–2.3 mmol g−1) and acid strengths.
2.7 Carbon nanofiber based catalysts
Carbon nanofibers (CNFs) have emerged as an interesting class of one-dimensional nanostructured carbon materials, which can exhibit comparable or superior properties to other carbonaceous materials in terms of thermal stability, mechanical strength, and exposed active sites on the outer surface of CNFs.165 In addition, CNFs exhibit strong hydrothermal stability, and thus, often preferred as efficient catalyst supporting materials over metal oxide supports. | ||
| Fig. 18 SEM images of (a) carbon nanofibers grown over Ni/γ-Al2O3 catalyst and (b) microcrystalline cellulose. TEM images of (c) carbon nanofibers and (d) a pear-shaped Ni particle at the tip of a carbon nanofiber. Reproduced with permission from ref. 70. Copyright (2010) John Wiley and Sons. | ||
2.8 Biomass-derived carbon catalysts
Many types of biomass substrates have been evaluated to prepare carbon-based catalysts (e.g. wood, bamboo, oilseed cake, oil palm trunk, sugarcane bagasse, vegetable oil asphalt, and carbohydrates). For this, carbonisation methods are normally used, including pyrolysis, gasification, hydrothermal, and flash carbonisation as these strategies allow to obtain well-controlled micro-/nanostructures of carbon materials with tunable surface chemistries and abundant functional sites.78,82–84 Moreover, applying renewable organic substrates instead of inorganic substrates for catalyst synthesis also contributes to an improved process sustainability.The optimisation of hydrothermal carbonisation conditions, such as temperature and time, was undertaken to prepare a glucose-derived carbon, which was then functionalised with –SO3H species.172 The resulting catalyst showed good activity in cellulose hydrolysis and fructose dehydration reactions with glucose and HMF yields of 44 wt% and 20 wt%, respectively. A one-pot synthesis of –SO3H and COOH-functionalised carbon solid acids was successfully performed by hydrothermal carbonisation of glucose in the presence of sulfosalicylic acid and acrylic acid, respectively.173 The functionalisation led to a change in the particles shape and resulted in good catalytic activity for the hydrolysis of cellulose. Importantly, the HSO3-functionalised carbon solid acid showed excellent stability up to five recycles without leaching of –SO3H species. A sulfonated willow-derived carbon catalyst was successfully prepared through sulfonation of a pencil refill.174 This catalyst, containing –SO3H, –COOH, and –OH groups, is highly suitable for the transformation of cellulose into glucose, reaching 78% conversion and 65% glucose yield, due to its strong acidic strength and well-defined macroporous structure. A minor loss in glucose yields was found after three recycles of the catalyst, due to catalyst mass loss during recovery/purification steps (entry 23, Table 1). Numerous carbon-based solid acids were prepared using different biomass sources, such as glucose, cellulose, bamboo, and rice husk.175 The developed solid acids, containing –SO3H and –COOH groups, showed good catalytic performances for the hydrolysis of cellulose with a maximum 81.8% yield of total reducing sugar for the glucose-derived acid catalyst. An amorphous carbon-based acid catalyst, prepared by sulfonation of bio-char obtained from fast pyrolysis of biomass, was also studied for the hydrolysis of cellulose.176 The complete conversion of cellulose with about 90% yields of α,β-methyl glucosides in methanol was obtained. Sulfonation of carbonised de-oiled seed waste cake, a solid waste from biodiesel production, was carried out to obtain multifunctional carbon-based solid acids.177 The catalysts contain various –OH, –COOH, and –SO3H groups as well as several nitrogen species (pyridinic, pyrrolic, etc.). Improved structure–activity properties were obtained with 4-benzenediazoniumsulfonate as the sulfonating agent, compared to H2SO4. The catalysts showed good activity in cellulose saccharification (glucose yield is in the range of 35–53%) as well as in fatty acid esterification (conversion up to 97%), hereby outperforming a conventional liquid acid (H2SO4) and other solid acids (zeolites, ion-exchange resins, etc.). A biomass residue obtained from the extraction of palm oil was used for the synthesis of a sulfonated carbon solid acid catalyst.178 A positive correlation between the concentration of sulfonic acid groups and the catalytic activity was noticed in the hydrolysis of cellobiose. Moreover, the sulfonated carbon showed higher catalytic activity than commercial acid resins. A sulfonated bamboo-derived carbon catalyst was studied for microwave-assisted conversion of bamboo-derived hemicellulose to xylo-oligosaccharides.179 The catalyst contains various acid groups, such as –SO3H, –COOH, and phenolic –OH. A 54.7 wt% yield of xylo-oligosaccharides based on xylan content was obtained at optimised reaction conditions. Reusability studies revealed a significant decrease in catalytic activity of sulfonated bamboo-derived carbon after three recycles, due to severe leaching of acidic species (entry 24, Table 1).
A novel sulfonated polymer impregnated carbon solid acid catalyst was studied for the dehydration of xylose to furfural. The detailed synthesis procedure is shown in Fig. 19.180 The catalyst exhibited excellent activity and provided almost quantitative conversion of xylose with selective synthesis of furfural. A carbon-based solid acid, prepared using glucose and an aqueous solution of H2SO4 as carbon and sulfonation agent, respectively, was studied for fructose dehydration.181 About 90% yield of HMF was achieved with dimethylsulfoxide as the solvent. The catalyst can be reused for several times without significant loss in catalytic activity. The selective dehydration of fructose to HMF was also investigated over various sulfonated carbon solid acids prepared using fructose as the carbon source.182 About 87% yield of HMF at nearly complete conversion of fructose was achieved. The advantage of microwave-assisted heating over conventional heating was demonstrated, allowing shorter reaction times for the dehydration of fructose.182 Hydrothermal carbonisation of various biomass substrates, such as lignin, cellulose, wood meal, and xylose, followed by sulfonation, was attempted to prepare amorphous carbon-based solid acid catalysts.183 All catalysts possess aromatic structures, as well as hydroxyl and carboxyl functional groups. The catalysts exhibited a good activity in the production of HMF (yields of 47–65%) from inulin, hereby outperforming various traditional solid acid catalysts. A series of metal chloride-modified sulfonated carbon catalysts, synthesised from cellulose, were tested for the selective conversion of carbohydrates to ethyl levulinate in an ethanol medium.183 Various metal chlorides, such as NaCl, KCl, MgCl2, AlCl3, FeCl3, CrCl3, and SnCl4 were used in this study. Among them, the FeCl3 modified carbon catalyst showed the highest performance in the conversion of fructose with a 58 mol% yield of ethyl levulinate. On the other hand, small amounts of water in the reaction mixture favoured the formation of HMF and levulinic acid.
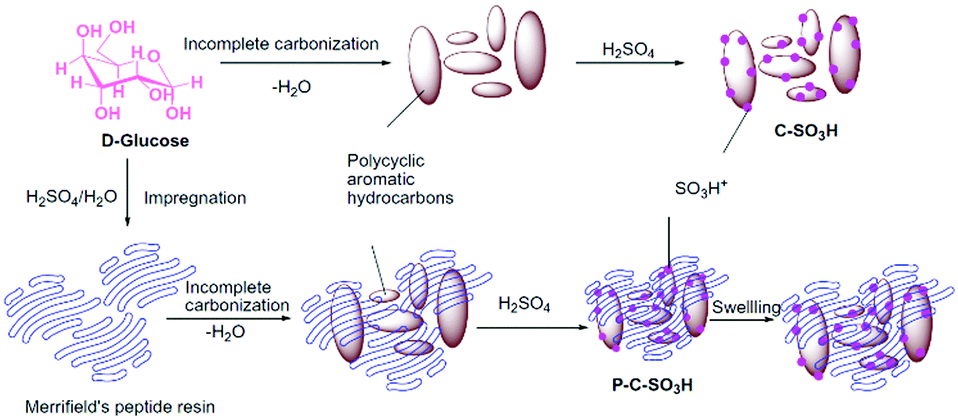 | ||
| Fig. 19 Synthesis procedure for sulfonated polymer impregnated carbon solid acid catalyst. Reproduced with permission from ref. 180. Copyright (2015) Elsevier. | ||
A porous carbon material obtained from bamboo shoots was used as an efficient support for depositing Pt NPs.184 The resulting catalysts possessed a high specific surface area, hierarchical porous structure, uniform dispersion of Pt NPs, large amounts of functional nitrogen and oxygen groups, and excellent water dispersibility. As a result, the carbon-supported Pt catalysts showed good activity for furfural hydrogenation with a controlled selectivity to furfuryl alcohol and cyclopentanone in water. Cyclopentanone is a promising building block for the production of low cost fragrances, fungicides, and perfume ingredients.185 The effect of carbonisation temperature of support, reaction temperature, and reaction pressure were also studied on the products distribution.184 A tandem aldol condensation–hydrogenation of furfural with acetone was carried out using a bi-functional catalyst composed of ultrafine dispersed Pd NPs on a CN@MgO hybrid support (CN = nitrogen doped carbon).186 The hydrophilic N-doped carbon enhanced the water dispersibility, catalyst stability, and the dispersion of Pd NPs (2.2 nm in average size) in the Pd/CN@MgO catalyst. Almost 99% furfural conversion with 95% selectivity to saturated ketones were obtained over the Pd/CN@MgO catalyst. Effects of carbonisation and calcination conditions on the textural and acid properties of carbon cryogel, obtained from a facile sol–gel polycondensation of acidic lignin-furfural mixtures, were studied.187 Calcination of the carbon cryogel led to higher thermal stability and stronger acid sites, and consequently superior catalytic performance in the esterification of levulinic acid, as shown by the 87.2% conversion and 86.5% ethyl levulinate yield at optimised reaction conditions (entry 25, Table 1). Reusability studies revealed that levulinic acid conversion decreases from 87.2% to 70.6% after third cycle, and then, a stable catalytic activity up to 5th cycle. Leaching of active sites is a key reason for the observed catalyst deactivation. Various functionalised carbon based catalysts, prepared by the chemical activation of olive stone waste, were tested for isopropanol decomposition.188 Among the catalysts tested, the H3PO4-activated carbon showed a steady state conversion with propylene as the major product in the absence of oxygen. In the case of base-activated carbon catalysts, variable amounts of acetone and propylene were obtained.
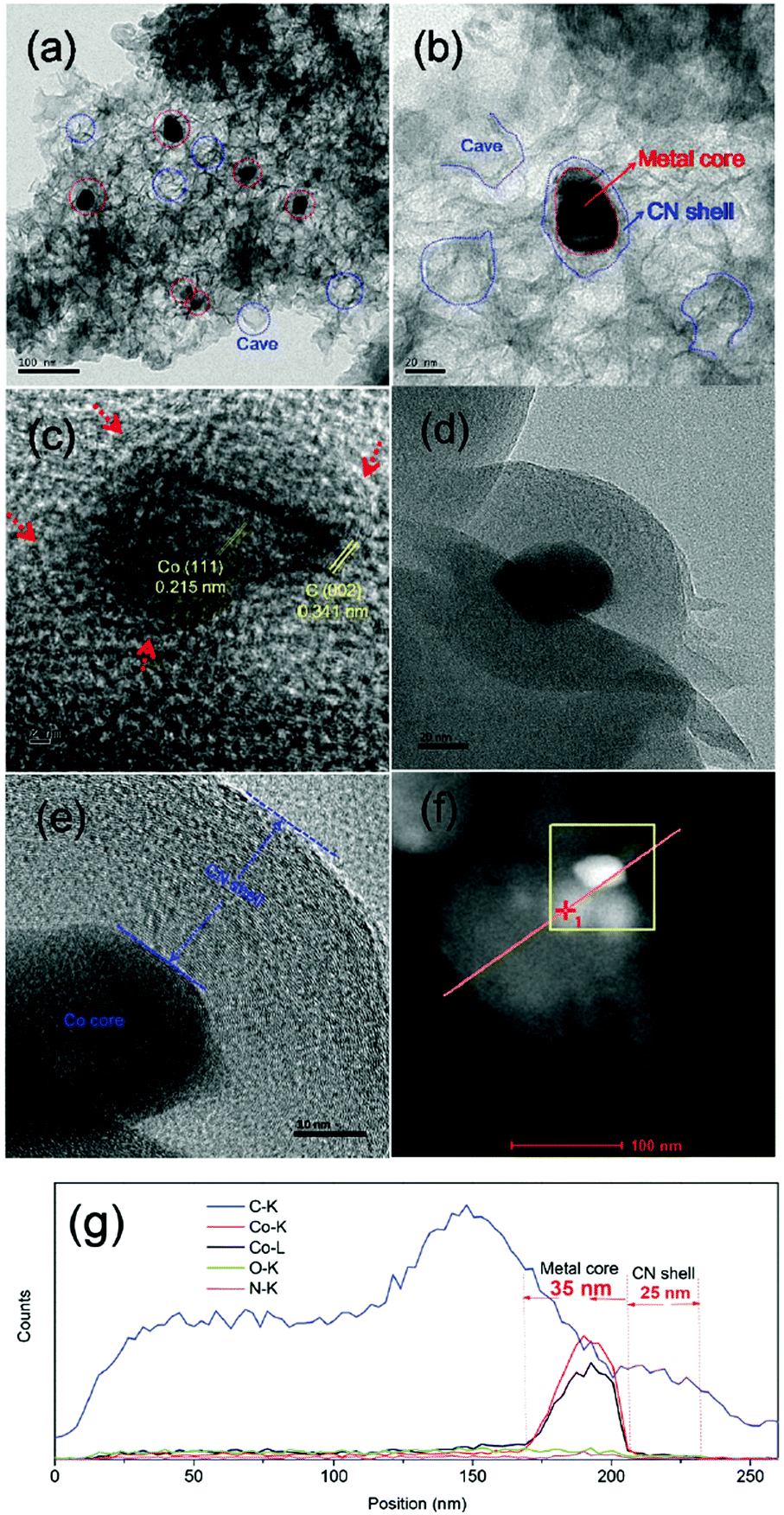 | ||
| Fig. 20 TEM images (a–e), STEM image (f) and line scanning EDS elemental analysis (g) of Co@NC-700 catalyst. Reproduced with permission from ref. 72. Copyright (2017) Royal Society of Chemistry. | ||
Esterification of plant oil with methanol to produce biodiesel was investigated over a series of bagasse-derived carbon solid acids.194 The catalyst, prepared with a dilute acid hydrothermal treatment, showed the best performance towards biodiesel production. This catalyst also demonstrates a more durable activity compared to conventional homogeneous catalysts. Sulfonation of biochar and wood-derived carbon was carried out using H2SO4 and SO3 for the esterification of free fatty acids with methanol.195 Wood-derived carbon solid acids showed higher activity compared to biochar-derived catalysts, which was attributed to the highest density of acid (SO3H) species present in wood-derived carbon solid acids. The catalyst reusability is thoroughly studied and appropriate strategies were suggested to regenerate the catalyst activity after reaction. A facile one-step hydrothermal carbonisation of water hyacinth was performed in the presence of p-toluenesulfonic acid to obtain promising carbon-based solid acids with variable acid density.84 Catalysts with higher amounts of acid sites showed better performance in oleic acid methanolysis as well as in xylose dehydration. Higher hydrothermal carbonisation temperatures (220–240 °C) enhanced the catalyst reusability due to improved graphitisation and hydrophobicity of the carbon surface. Esterification of free fatty acids in waste cooking oil with methanol was studied to produce biodiesel using a carbonaceous solid acid prepared by a one-step mild hydrothermal carbonisation of glucose, followed by sulfonation using hydroxyethylsulfonic acid.196 Catalytic results revealed a strong decrease in the content of free fatty acids (up to 93.4%) in waste cooking oil. Catalyst stability was demonstrated with only a 7% decrease in free fatty acids conversion after five recycles. A bagasse-derived carbon solid acid showed excellent catalytic activity with a 95% yield of methyl oleate from esterification of oleic acid with methanol, hereby outperforming both niobic acid and Amberlyst-15 catalysts.197 The obtained results are comparable or superior to those of starch- and glucose-derived carbon catalysts, respectively. Furthermore, the bagasse-derived carbon solid acid efficiently catalysed the conversion of waste cooking oils (39 wt% free fatty acid content), obtaining a 93.8% biodiesel yield. An incomplete carbonisation of coffee residue followed by sulfonation was carried out to prepare sulfonated carbon catalysts for the esterification of caprylic acid.198 The sulfonation temperature played a crucial role in optimising the –SO3H density on the catalytic surface. The catalyst carbonised at 600 °C and sulfonated at 200 °C showed the highest caprylic acid conversion (71.5%). A robust sulfonated carbon catalyst derived from oil-cake waste was tested for biodiesel production.199 The catalyst exhibited superior acid density, large porosity, high specific surface area, and remarkable thermal stability. As a result, the catalyst showed excellent activity and good reusability in the esterification of free fatty acids into corresponding methyl esters, thus outperforming a homogeneous H2SO4 catalyst. A sulfonated carbon solid acid, obtained after a one-pot carbonisation–sulfonation of cellulose using concentrated H2SO4, was investigated for the esterification of rapeseed oil fatty acids.200 The activity of the carbon solid acid was similar to Amberlyst-15 and led to a 96.5% esterification activity.
Zhang et al.201 investigated the production of Jatropha biodiesel and hydrogen using carbonaceous acid and base catalysts. Two carbon supports were prepared from Jatropha-hull hydrolysate (JHC) and hydrolysis residue (JRC), followed by the deposition of active species to obtain carbonaceous solid acid (C-SO3H@Fe/JHC) and solid base (Na2SiO3@Ni/JRC) catalysts, respectively. The C-SO3H@Fe/JHC catalyst was used for the esterification of free fatty acids in crude Jatropha oil. Subsequent transesterification of remaining triglycerides catalysed by Na2SiO3@Ni/JRC ultimately afforded a high biodiesel yield of 96.7%. Interestingly, the solid base catalyst obtained after three recycles showed a high performance in the hydrothermal gasification of glycerol with 81% yield of H2. In another work, de-oiled Jatropha curcas seed cake waste was used to prepare a carbon-based solid acid catalyst, which was then tested in the esterification of Jatropha curcas in order to reduce free fatty acid content for biodiesel production.202 The conversion of free fatty acids reached 99% under optimised reaction conditions, due to an improved porosity of the carbon framework. Moreover, the biomass-derived catalyst outperformed a conventional sulfuric acid catalyst. A carbon-based solid base catalyst was prepared by treating a SO3H–carbon catalyst obtained from glycerol with an aqueous alkali solution.85 The solid base catalyst contains polycyclic aromatic carbon sheets attached with versatile –SO3Na, –COONa, and –ONa functionalities. The resulting catalyst showed a remarkable catalytic activity in transesterification of sunflower oil with high yields of biodiesel (99%) at atmospheric pressure. In addition, no considerable loss in biodiesel yield was found even after eight recycles of the catalyst (entry 28, Table 1).
The production of biodiesel from oil, obtained by solvent extraction of Calophyllum inophyllum seeds, was studied over a biomass-derived carbon catalyst.203 The catalyst is itself synthesised via carbonisation of the residual cake of Calophyllum inophyllum, followed by sulfonation in concentrated H2SO4. The resulting solid acid efficiently catalysed simultaneous esterification and transesterification of free fatty acids and triglycerides present in Calophyllum inophyllum oil, respectively. Sulfonated carbon-based solid acid catalysts, prepared using a simultaneous carbonisation and sulfonation of glucose with conc. H2SO4, were tested for two reactions, namely transesterification of tributyrin with methanol and dehydration of fructose to HMF.204 The catalyst prepared at 150 °C showed excellent activity in both reactions with 97.2% and 93% yields of methyl butyrate and HMF, respectively, which was attributed to its high density of –SO3H sites.
Glycerol esterification with acetic acid or acetic anhydride is an important reaction that gives three valuable products, namely monoacetin, diacetin, and triacetin.124,205–207 These acetins have great commercial significance as transportation fuel additives and antiknock additives. Moreover, the addition of small amounts of triacetin improves the properties of biodiesel. Hence, this reaction acts as a key driving force to enhance the profitability of biodiesel production. Several sulfonated carbon catalysts were developed from biomass waste at different synthesis conditions.208 Due to their strong acidity and higher thermal stability, excellent activities were obtained for glycerol esterification to a mixture of monoacetin, diacetin, and triacetin. In addition, the strong hydrophobic character of these catalysts resulted in an excellent reusability for glycerol esterification. Sulfonated glucose-derived carbon catalysts showed higher activity for the hydrolysis of alginate, a major constituent of algal biomass, compared with that of H2SO4 and other commercial solid acid catalysts, such as carbon black, activated carbon, Amberlyst-15, and zeolites.209 This performance was ascribed to the high concentration of strong Brønsted acid sites (–SO3H) in the glucose-derived carbon catalysts. Biochar, obtained after fast pyrolysis of pinewood, was used for the encapsulation of Fe NPs.210 The catalyst has a core–shell structure resulting from in situ encapsulation of iron within a graphitic shell. Catalytic experiments demonstrated an efficient conversion of bio-syngas with a good selectivity to liquid hydrocarbons. The catalytic dehydration of 2-butanol was studied using a carbon-based solid acid prepared by chemical activation of olive stone with H3PO4.76 The catalyst shows high surface acidity due to the presence of residual phosphorous species on the catalyst surface, resulting in improved catalytic activities. cis-2-Butene and trans-2-butene are the major products with lower amounts of 1-butene via E2-mechanism (one-step mechanism) of 2-butanol dehydration, which is a rate-determining step.
2.9 Conclusions of carbon-based catalysts
Based on the examples discussed in this chapter, it is clear that the large specific surface area and strong hydrothermal stability of carbon-based catalysts play a favorable role in biomass upgrading. On one hand, immobilization/dispersion of functional species (SO3H, metals, etc.) is facilitated on high surface area carbon supports. This leads to a higher number of surface-exposed active sites available for biomass valorisation. On the other hand, the strong hydrophobic nature of carbon materials prevents leaching of active species during biomass processing reactions, allowing a more efficient catalyst reusability, as evidenced by Table 1. In addition, the unique properties of nanostructured carbon materials, especially graphene, not only play a key role in stabilising acid–base and/or redox functional species (entries 10–13, Table 1), but also enable them to perform as metal-free catalysts in biomass upgrading (entry 14, Table 1). Unfortunately, despite all of these attractive characteristics and potential catalytic applications, carbon materials, especially graphene and CNTs, still suffer from several drawbacks, such as lack of large-scale synthesis methods, high production costs, low bulk density, and mass diffusion constraints, limiting their catalytic applications at the industrial scale.211,212 In this framework, using biomass substrates (Section 2.8) for one-pot synthesis of functionalised carbon-based catalysts would be a promising solution in terms of catalyst costs, functionalisation, and scalability. In parallel, the application of mesostructured carbon catalysts (Sections 5 and 6) in biomass upgrading would be an ideal strategy to overcome the current mass transfer limitations of microporous carbon materials. However, more efforts are still needed to optimise structure–activity features of biomass-derived carbon and mesoporous carbon catalysts to obtain breakthrough catalytic results in biomass valorisation.3. Metal–organic frameworks
Metal–organic frameworks (MOFs) belong to an extraordinary family of crystalline porous materials with unique characteristics and versatile functionalities.36,213,214 They have found plentiful applications in various fields, including catalysis, sensing, luminescence, separation, gas adsorption, H2 storage, and optical devices. MOFs contain two key building blocks: (i) metal ions or metal–oxo clusters and (ii) multidentate organic moieties. The unique nature of metal ions or clusters as lattice nodes and organic moieties as linkers allow the formation of robust metal–ligand coordination frameworks with well-defined porous structures quite similar to those found in zeolites. The capacity to fine-tune pore geometries up to a mesoporous size range is a notable advantage of MOFs over microporous zeolites.36 This pore modification is feasible by selecting a suitable combination of metal ions and organic linkers as well as by tailoring MOFs synthesis parameters. Mesoporous structures can facilitate efficient diffusion of (bulky) biomass substrates towards unsaturated active metal centres in MOFs, thus overcoming mass transport limitations often encountered with microporous zeolites. To meet the requirements of a specific target application, properties of MOFs can be further tuned by anchoring desired active phases. Overall, MOFs are highly tunable materials, which led to more than 20000 synthesised MOFs, described in literature up to date.36 In addition, the high crystallinity of MOFs assists to carry out thorough structure–activity relationship investigations as well as molecular simulations that can provide valuable implications for the rational design of new, high performing MOF materials.
3.1 Factors affecting the catalytic activity of MOFs in biomass upgrading
Over the past few years, MOFs have been in the forefront of research in catalytic biomass valorisation. The diversity of both metal nodes and organic ligands as well as their synergistic interactions enables MOFs to perform as active phase catalysts, catalyst supports or even catalyst synthesis precursors. Owing to durable metal–ligand coordination networks, MOFs can exhibit large pore volumes and superior specific surface areas. Therefore, the individual properties of inorganic and organic components are usually well accessible and uniformly distributed throughout the MOFs, a fundamental advantage over conventional heterogeneous solid catalysts.215 The broad spectrum of starting materials as well as efficient synthesis strategies offer unlimited options for fine-tuning textural properties of MOFs. This is indispensable because biomass feedstocks usually contain larger molecules, hence their diffusion towards catalytic active sites can be improved by carefully adjusting pore dimensions and morphologies. For instance, Li et al.216 reported that the morphology of Fe-MIL-101-NH2 could be modified by simply varying the synthesis times of MOF, without effecting its crystal structure. The resulting MOFs showed good catalytic performances in glycerol oxidation reaction.The individual attributes of metal nodes and organic ligands can offer abundant acid–base properties.215,217 The unsaturated metal sites in MOFs obviously exhibit Lewis acidity. They also show adequate redox and basic properties, depending on the nature of metal–oxygen connection modes of MOFs. Owing to higher Lewis acidity, MOFs can be used as solid acid catalysts for isomerisation of glucose to fructose.218 Notice that Brønsted acid sites are essential in many biomass-upgrading reactions. Nevertheless, MOFs exhibit negligible Brønsted acidity. Therefore, several viable strategies have been developed to attach Brønsted acidic groups onto a particular site of the MOFs.216,219 Organic ligands provide facile approaches to introduce such acid functionalities.220 Mainly, sulfonated-functionalised MOFs have received paramount attention in catalysis because of their superacidity, which is stronger than 100% H2SO4.221 Since functionalised MOFs can combine the advantages of both MOFs and incorporated acid–base functional sites, they are particularly suitable to catalyse cascade chemical reactions for efficient biomass upgrading. For instance, the SO3H-functionalised UiO-66 MOF showed an exceptional catalytic activity in the conversion of levulinic acid and its esters to produce γ-valerolactone (Fig. 21).222 Results indicated that Zr4+–O2− acid–base pair sites, uniformly imbedded in the UiO-66 framework, enhance catalytic transfer hydrogenation of levulinate esters, while –SO3H groups attached to organic linkers catalyse successive intramolecular dealcoholisation to produce high yields of γ-valerolactone.222
 | ||
| Fig. 21 SO3H-Functionalised UiO-66 MOF for catalytic transfer hydrogenation (CTH) of levulinate esters to γ-valerolactone. Note: Zr and O atoms are shown in green and brown, respectively. Reproduced with permission from ref. 222. Copyright (2017) American Chemical Society. | ||
The confinement properties of MOFs stimulated by the existence of rigid coordination frameworks is a key driving force for their application as catalyst supports, but concurrently as protective agents for the development of highly stable heterogeneous catalysts. Metal NPs are likely to undergo sintering at elevated temperature conditions, leading to the formation of larger sized particles with lower specific surface areas, resulting in decreased catalytic activities. The versatile and tailorable porous channels of MOFs offer potential approaches for the encapsulation of active phase metal NPs in order to prevent their agglomeration, hence improving their catalytic performance.223,224 Guo et al.225 demonstrated a beneficial effect of MIL-101(Cr) MOF in stabilising highly dispersed Ru nanoclusters (2.4 nm) (Fig. 22). The resulting Ru/MIL-101(Cr) catalyst showed good activity in the conversion of levulinic acid to γ-valerolactone, hereby outperforming a zeolite supported Ru catalyst.
 | ||
| Fig. 22 Selective hydrogenation of levulinic acid into γ-valerolactone over Ru/MIL-101(Cr) catalyst. Reproduced with permission from ref. 225. Copyright (2016) Springer Nature. | ||
MOFs have also been employed as practicable catalyst precursors as well as the sacrificial templates for the preparation of carbon supported metal catalysts.226–228 Pyrolysis of MOFs at suitable temperatures gives porous carbon materials derived from the organic ligands. The formed carbon matrix in turn serves as a high surface area support, which stabilises the in situ generated metal NPs, originating from the metal nodes. In this manner, highly stable catalyst systems, consisting of uniformly dispersed metallic NPs on a porous carbon matrix, with multifunctional properties can be obtained. Carbon-supported bimetallic catalysts can also be synthesised by tailoring the MOF-templated strategy as reported by Guomin Xiao and his co-workers (Fig. 23).227 They developed an efficient bimetallic CuCo catalyst supported on a porous carbon matrix via thermolysis of a Co-doped Cu–BTC MOF under N2 atmosphere. The effect of pyrolysis temperature from 500 to 800 °C was studied on the size and shape properties of CuCo/C catalysts. The catalytic efficiency of various MOFs-based materials in biomass valorisation are discussed in the following paragraphs. In addition, an overview of the most vital catalytic results obtained in MOFs catalysed biomass upgrading is presented in Table 2.
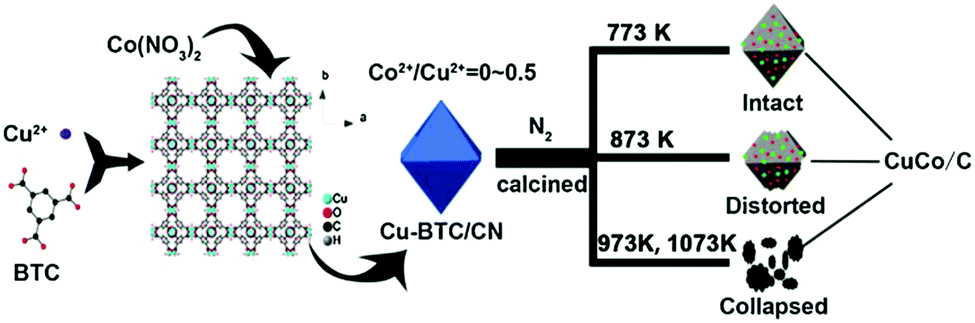 | ||
| Fig. 23 Schematic illustration for the synthesis of CuCo/C catalyst. Reproduced with permission from ref. 227. Copyright (2017) Elsevier. | ||
| Entry | Catalyst | Reaction | Product | Reaction conditions | Catalytic efficiency | Recycling results | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Conv. % | Yield % | |||||||
| a HMF – 5-hydroxymethylfurfural. b TRS – total reducing sugar. c RT – room temperature. d Untreated cellulose with mean particle diameter of 25 μm. e DHMTHF – 2,5-dihydroxymethyl-tetrahydrofuran. f HDO – hydrodeoxygenation. g DFF – 2,5-diformylfuran. h THFOL – tetrahydrofurfuryl alcohol. | ||||||||
| 1 | MIL-53(Al) | Hydrolysis of carboxymethyl cellulose | HMFa | 100 mg substrate, 75 mg catalyst, 80 mL H2O, 200 °C, 4 h | — | 40.3 | 3 (small decrease in product yields, due to catalyst mass loss during recovery/purification and blockage of active sites by humins products) | 230 |
| TRSb | 54.2 | |||||||
| 2 | Zn-ZIF-8 | Sucrose upgrading | Methyl lactate | 225 mg sucrose, 500 mg catalyst, 8 g methanol, 160 °C, 24 h | >99 | 42 | 4 (catalyst deactivation, due to blockage of active sites and structural changes in the catalyst) | 232 |
| 3 | MIL-100(Fe) | Fructose upgrading | Lactic acid | 50 mg fructose, 50 mg catalyst, 10 mL water, 190 °C, 2 h | 87 | 32 | 4 (catalyst deactivation, due to blockage of pores/active sites and leaching of active species. A simple method was used for the regeneration of the catalytic activity) | 233 |
| 4 | Composite of MIL-101(Cr) and activated fly ash | Dehydration of xylose | Furfural | 300 mg xylose, mixture of water and toluene, 150 °C, 3 h | 88.8 | 71 | 10 (stable catalytic activity) | 235 |
| 5 | 1 wt% Pt@MOF-5 | Oxidative condensation of furfural | Furan-2-acrolein | 1 mmol furfural, 25 mg catalyst, 25 mg K2CO3, 15 mL EtOH, 150 °C, 4 h, 0.3 MPa O2 at RTc | 84 | 75.7 | 5 (stable catalytic activity) | 224 |
| 6 | Ru/UiO-66 | Hydrogenation of furfural | Furfuryl alcohol | 0.1 mL furfural in 9.9 mL H2O, 100 mg catalyst, 20 °C, 5 bar H2 at 20 °C, 4 h | — | 94.9 | 5 (negligible decrease in catalytic activity) | 243 |
| 7 | 5 wt% Ru/MIL-101(Cr) | Hydrogenation of levulinic acid | γ-Valerolactone | 1 mmol substrate, 6 mg catalyst, 10 mL H2O, 5 h 70 °C, 1 MPa H2 at RT | 100 | 99 | 4 (catalyst deactivation, due to leaching of active Ru species) | 225 |
| 8 | MIL-101(Cr)–SO3H | Dehydration of glucose | HMF | 1.24 mmol glucose, 5.22 × 10−5 mol catalyst, THF & H2O, 130 °C, 24 h | — | 29 | 4 (catalyst deactivation, due to fouling of the catalyst by humins) | 37 |
| 9 | MIL-101(Cr)–SO3H | Dehydration of fructose | HMF | 500 mg fructose, 300 mg catalyst, 5 mL DMSO, 120 °C, 1 h | >99 | 90 | 5 (small decrease in HMF yields from 90% to 85%, due to accumulation of oligomeric products on the catalyst surface) | 220 |
| 10 | MIL-101(Cr)–SO3H | Ethanolysis of furfuryl alcohol | Ethyl levulinate | 1.15 mmol substrate, 100 mg catalyst, 4 mL ethanol, 140 °C, 2 h | 100 | 79.2 | 5 (small decrease in product yields, due to accumulation of oligomeric products on the catalyst surface) | 249 |
| 11 | 2.4 wt% Ru/PTA-HKUST-1 | Hydrogenolysis of cellulosed | Ethylene glycol | 50 mg substrate, 50 mg catalyst, 8 mL H2O, 245 °C, 4 MPa H2 at RT, 4 h | 96.2 | 50.2 | Catalyst deactivation was found after a recycling test, due to partial decomposition of MOF composite | 253 |
| 12 | 5 wt% Ru/SO3H–UiO-66 | Hydrogenation–cyclisation of methyl levulinate | γ-Valerolactone | 500 mg substrate, 50 mg catalyst, 80 °C, 0.5 MPa H2 at 80 °C, 15 mL H2O, 4 h | 100 | 81 | 5 (stable catalytic activity) | 254 |
| 13 | 5 wt% Pd/MIL-101(Cr)–SO3H | γ-Valerolactone hydrogenation | Ethyl valerate | 10 mmol substrate, 100 mg catalyst, 5.8 mL ethanol, 250 °C, 3 MPa H2 at RT, 10 h | 98 | 83 | 3 (drastic catalyst deactivation, due to aggregation of Pd NPs at elevated reaction temperatures in acidic solution) | 255 |
| 14 | 3 wt% Pd/MIL-101(Al)–NH2 | Hydrogenation of HMF | DHMTHFe | 1 mmol HMF, 20 mg catalyst, 8 mL H2O, 30 °C, 1 MPa H2 at 30 °C | 100 | 96 | 5 (stable catalytic activity was found for 1 h reactions, but product selectivity was significantly decreased from 96% to 80% for 12 h reactions, due to changes in structural and textural properties of the catalyst) | 257 |
| 15 | 2 wt% Pd/MIL-101(Cr)–SO3H | HDO of vanillinf | 2-Methoxy-4-methylphenol | 2 mmol vanillin, 50 mg catalyst, 1 h, 20 mL H2O, 90 °C, 1 MPa H2 at 90 °C | 100 | 98.4 | 7 (stable catalytic activity) | 258 |
| 16 | Fe/C–S | Dehydration-oxidation of fructose | DFFg | 1.2 mmol substrate, catalyst (20 mol% metal), 2 mL EtOH, 100 °C, N2 (1 bar) for 2 h and then O2 (1 bar) for 3 h at RT, 3 h | >99 | >99 | 6 (stable catalytic activity) | 263 |
| 17 | FeCo/C | Oxidation of HMF | DFF | 1 mmol HMF, catalyst (20 mol% metal), 100 °C, 1 MPa O2 at RT, 1 mmol Na2CO3, 2 mL toluene, 6 h | >99 | >99 | 6 (stable catalytic activity) | 264 |
| 18 | Ni/C | Hydrogenation of furfural | THFOLh | 30 mg furfural, 30 mg catalyst, 5 mL of 2-propanol, 120 °C, 1 MPa H2 at RT, 2 h | 100 | 100 | 5 (stable catalytic activity) | 266 |
| 19 | CuNi0.5@C | Hydrogenation of furfural | Cyclopentanone | 5 g furfural, 2 wt% of catalyst based on substrate, 95 mL H2O, 130 °C, 5 MPa H2 at RT, 5 h | 99.3 | 97 | 4 (very small decrease in product yields, due to blockage of active sites by coke formed during reaction) | 228 |
3.2 Pristine MOFs and their composites
In the 1990s, the first catalytic application of MOFs was reported for the cyanosilylation of aldehydes.229 With the development of more advanced MOFs, their catalytic applications in biomass valorisation have been dramatically increased in recent years.Two types of zeolitic imidazolate frameworks (ZIFs), such as Zn-ZIF-8 and Co-ZIF-67 (Fig. 24) were tested for the catalytic conversion of sugars (sucrose, glucose and fructose) to methyl lactate.232 ZIFs are a subclass of MOFs with zeolite-type topology, characterised by a 3D-porous isotropic framework. About 42% yield of methyl lactate was achieved over Zn-ZIF-8 due to its catalytically favourable properties, such as smaller crystal size and higher amounts of acid sites, especially Lewis acid sites. Reusability studies revealed a considerable decrease in product yields from 34.8% to 27.2% after four recycles of ZIF-8 (160 mg catalyst), due to blockage of active sites and structural changes in the catalyst (entry 2, Table 2). The synthesis of lactic acid from carbohydrates is an important step in biomass upgrading.233 Lactic acid is a valuable chemical for the bioplastic industry and functions as a platform molecule towards production of various attractive chemicals, such as acetaldehyde, pyruvic acid, acrylic acid, propylene glycol, 2,3-pentanedione, 1,2-propanediol, and lactic acid esters. Huang et al.233 developed an efficient one-pot transformation of hexose sugars to lactic acid over a Fe-based MIL-100 catalyst. About 32% yield of lactic acid was obtained from fructose, attributed to the unique morphology and textural properties of MIL-100(Fe) MOF. In contrast, only 18% and 20% yields of lactic acid were obtained over Cu–BTC and Cr-based MIL-100, respectively. The MIL-100(Fe) MOF was also found to be effective for the production of lactic acid from glucose, sucrose, inulin, and cellobiose. Owing to blockage of pores/active sites by the reaction substrates and leaching of active species, the catalytic activity of MIL-100(Fe) is significantly decreased after four recycles (entry 3, Table 2). A potential procedure for regeneration of the catalytic activity was used after four recycles, giving about 28% yield of lactic acid from fructose. This regeneration method involves the dissolution of recovered catalyst in HF/HNO3 solution and the subsequent addition of H3BTC ligand. The obtained mixture was oven dried at 150 °C for 12 h, followed by washing with water–ethanol and again dried overnight in air to obtain a regenerated MOF catalyst. In another work, the effect of the metal node on the performance of M-MOF-74 (M = Co, Ni, Mg, and Zn) catalysts was studied for the conversion of sugars to methyl lactate.234 Among them, Mg-MOF-74 exhibited the best catalytic activity with 35% and 47% yields of methyl lactate from glucose and sucrose, respectively. A probable mechanism for the production of methyl lactate from glucose was reported. A composite of MIL-101(Cr) and activated fly ash was found to be effective for the dehydration of xylose to furfural.235 About 71% yield of furfural is obtained at optimised reaction conditions, which is maintained even after ten recycles of the catalyst (entry 4, Table 2). These results were attributed to a remarkable hydrothermal stability and the synergistic effect between Cr species (Lewis acid sites) in MIL-101(Cr) and hydroxyl groups (Brønsted acid sites) on activated fly ash.
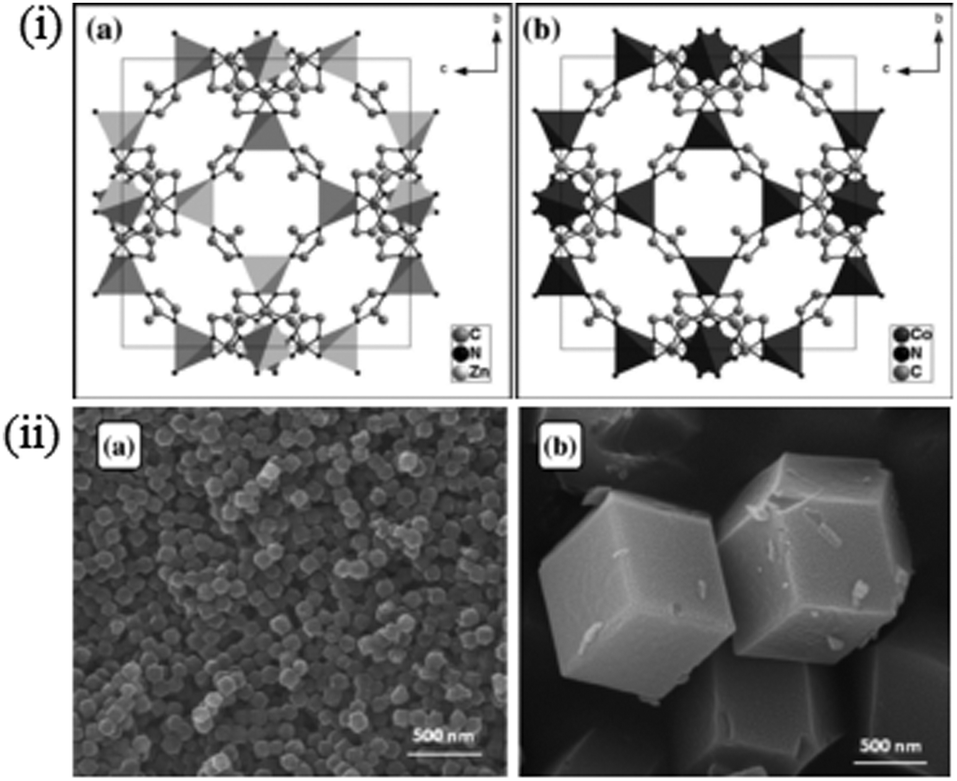 | ||
| Fig. 24 (i) Structures of (a) ZIF-8 and (b) ZIF-67 frameworks and (ii) SEM images of (a) ZIF-8 and (b) ZIF-67. Reproduced with permission from ref. 232. Copyright (2016) Elsevier. | ||
The oxidation of HMF gives a set of highly valuable building blocks for polymer and fine chemicals industries. These include 2,5-diformylfuran, 5-hydroxymethylfuran-2-carboxylic acid, 5-formyl-2-carboxylic acid, and 2,5-furandicarboxylic acid.236 The selectivity of these chemicals can be tuned by varying catalyst compositions and reaction parameters. Lucarelli et al.236 developed a feasible base-free oxidation of HMF to DFF using three types of square planar Ni-based MOFs, i.e. Ni(BDP), Ni(BPEB) and Ni3(BTP)2. Among them, Ni3(BTP)2 showed the best catalytic performance, giving a 27% yield of DFF under mild reaction conditions. The catalytic difference between these three Ni-based MOFs relates to a variable accessibility of the metal centres. An efficient MOF catalysed one-pot process was developed to produce furilydenepropanenitrile derivatives, which are key polymer building blocks.237 The first step i.e. oxidation of HMF was studied using Fe-containing MOFs (MIL-100IJFe and Fe(BTC)) and a Cu containing MOF (Cu3IJBTC)2). MIL-100IJFe post-treated with NH4F showed an exceptional catalytic performance, affording a 100% yield of 2,5-diformylfuran (DFF) from HMF, which was attributed to the presence of abundant MOF-stabilised Fe(III) active species. The second step i.e., Knoevenagel condensation of DFF with malononitrile or ethyl cyanoacetate was performed by exploiting the basicity of the reaction medium. Kikhtyanin et al.238 compared the catalytic efficiency of several MOFs for aldol condensation of furfural with acetone. Results indicated that aldol condensation occurs on the acidic sites of MOFs rather than on basic sites. Particularly, catalysts possessing ample amounts of Brønsted acid sites, which are generated by the interaction of metal cations with surrounding water molecules, showed the best performance. Aerobic oxidative condensation of furfural with alcohols was also studied over various Co-based MOF catalysts.239 Two different strategies were applied for the generation of catalytic active species in these MOFs: (1) removal of coordinated water molecules at 300 °C, resulting in preferentially exposed metal active centres and (2) pyrolysis at 700 °C, generating multi-element carbon–matrix nanocomposites. Interestingly, both MOFs showed good catalytic performances (84.9% conversion and 99% selectivity) in the condensation of furfural with alcohols. Zr-Containing MOFs with terephthalate (UiO-66) or 2-aminoterephthalate ligands (UiO-66-NH2), were studied for the esterification of levulinic acid with simple alcohols as well as biomass-derived complex alcohols.240 The obtained catalytic activities are comparable (and in some cases higher) to those of previously reported solid acid catalysts. Results reveal that the active sites are located at the defect sites associated with ligand deficiencies in these MOFs.
3.3 MOF-supported metal NPs
Transition-metal NPs with small sized particles (usually below 5 nm) show distinctive physicochemical and redox properties as well as unusual catalytic activities that differ considerably from their bulk analogues.216 Owing to strong, well-defined porous frameworks, MOFs can be used as efficient catalyst supports for the stabilisation of metal NPs with controlled size distributions. Compared to conventional porous inorganic materials, such as zeolites and mesoporous silica, MOFs can exhibit synergetic interactions, depending on their nature and composition, with the metal NPs via coordination and π–π forces.2433.4 Acid functionalised MOFs
The catalytic Lewis acidity strength of the MOFs (originating from isolated metal sites in their structure) was investigated, first time, for the cyanosilylation of aldehydes.245 In contrast to the Lewis acidity, understanding the Brønsted acidity of MOFs is challenging and remains less explored. Many biomass conversion reactions, including dehydration, hydrolysis, depolymerisation, hydrodeoxygenation, and esterification/transesterification require abundant Brønsted acid sites. Several practicable strategies have therefore been developed to introduce Brønsted acid functionalities (sulphate, phosphate, nitric acid, etc.) into MOFs. These include encapsulation of Brønsted acid molecules into the framework cavities of MOFs (Fig. 25),219 (post)synthetic functionalisation of organic linkers with acidic groups, or ligation of Brønsted acid moieties to isolated metal nodes.37 Hence, a handful of studies dealing with the conversion of biomass using acid functionalised MOFs have been reported in recent years.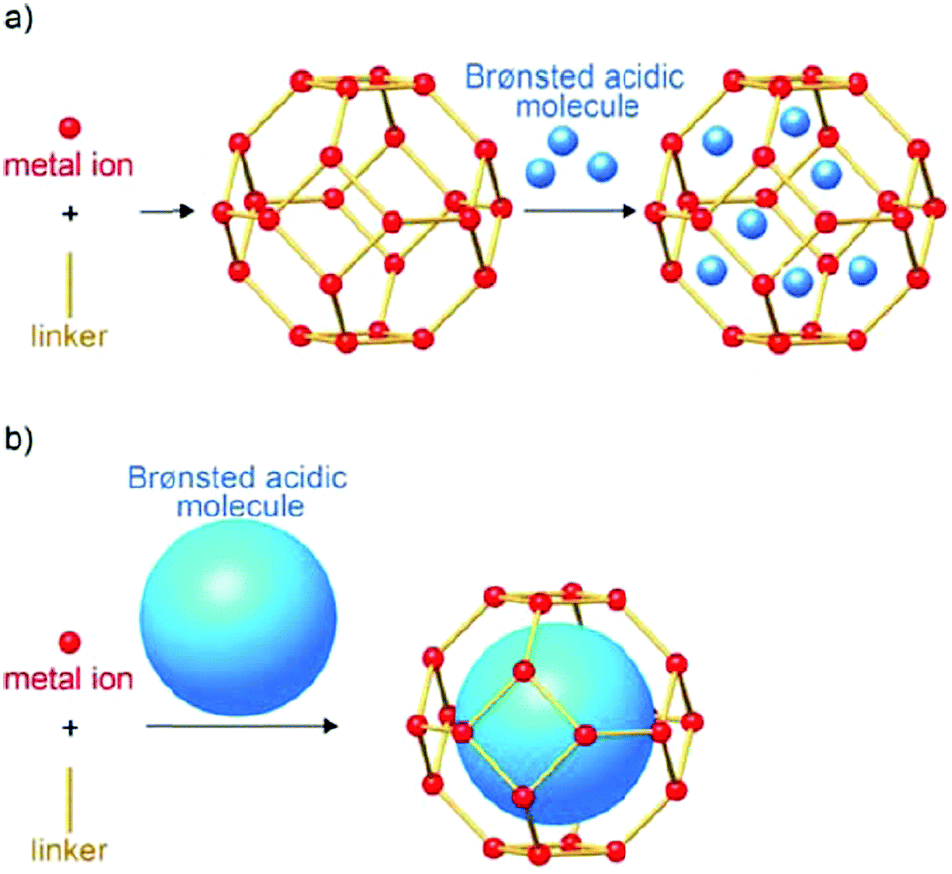 | ||
| Fig. 25 Schematic representation of encapsulation of Brønsted acidic molecules within MOF: (a) two-step method and (b) one-step method. Reproduced with permission from ref. 219. Copyright (2015) American Chemical Society. | ||
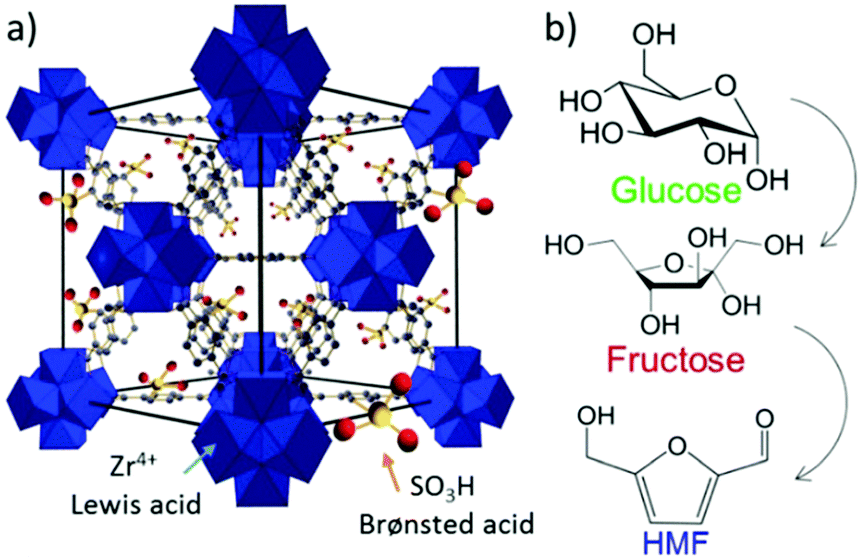 | ||
| Fig. 26 (a) Schematic representation of an acidic UiO-66 framework. (b) Conversion of glucose to HMF via fructose isomerisation. Reproduced with permission from ref. 246. Copyright (2018) John Wiley and Sons. | ||
Fructose, bearing a five-membered ring, is the preferred precursor for the production of HMF. Glucose, on the other hand, has a more stable six-membered ring. Hence, several studies have been undertaken for fructose-to-HMF transformation using MOFs based acid catalysts. For instance, a series of SO3H-functionalised MOFs, synthesised by post-synthetic modification of organic linkers with chlorosulfonic acid, were studied for the dehydration of fructose to HMF.220 The grafting rate of –SO3H determines the Brønsted acid concentration and its strength in the MOF. About 90% HMF yield was achieved at nearly complete conversion of fructose over an optimised MIL-101(Cr)–SO3H catalyst. A minor loss in product yields (from 90% to 85%) was observed after five recycles of the catalyst, due to accumulation of oligomeric products on the catalyst surface (entry 9, Table 2). Hu et al.245 developed two types of hydrothermally stable, sulfonated NUS-6 MOFs, composed of Zr and Hf with high BET surface areas of 550 and 530 m2 g−1, respectively. Both MOFs possess a hierarchical porous structure of coexisting micropores (∼0.5, ∼0.7, and ∼1.4 nm) and mesopores (∼4.0 nm) with strongly connected –SO3H groups. The NUS-6(Hf) MOF showed a higher catalytic performance in the dehydration of fructose with a 98% yield of HMF, which was attributed to its strong Brønsted acidity as well as its more suitable pore size that inhibited side reactions. A multicomponent MOF-based solid acid catalyst, composed of polyvinylpyrrolidone modified halloysite nanotubes encapsulated in a SO3H functionalised UiO-66 MOF, showed good performance for fructose dehydration with a 92.4% HMF yield.248 The concentration of acid sites in the functionalised MOF can be adjusted by simply varying the ratio of support and SO3H species. This MOF catalyst can be efficiently recycled for at least five times without much decrease in its activity and selectivity.
Kuwahara et al.222 synthesised a SO3H-functionalised Zr-MOF (UiO-66) for catalytic transfer hydrogenation of levulinic acid and its esters to γ-valerolactone. Results revealed the formation of strongly connected –SO3H groups in the UiO-66 framework, without affecting the structure of Zr lattice nodes. However, functionalisation resulted in a decreased crystallinity and BET surface area. The optimised MOF, containing a 60 mol% fraction of –SO3H while retaining a high BET surface area, exhibited the best catalytic performance, attributed to synergistic effect of Lewis basic Zr6O4(OH)4 and Brønsted acidic (–SO3H) centers. The transformation of furfuryl alcohol to ethyl levulinate in ethanol was examined using a SO3H functionalised MIL-101(Cr) MOF.249 The as-prepared catalyst showed higher BET surface area, excellent hydrothermal/chemical stability, and enhanced accessibility of Brønsted acid sites. Owing to these fascinating properties, the MIL-101(Cr)–SO3H catalyst showed good activity with a 79.2% yield of ethyl levulinate at full conversion of furfuryl alcohol. Reusability test revealed a minor loss in product yields after five recycles of the catalyst, attributed to adsorption of oligomeric products on the catalyst surface (entry 10, Table 2).
3.5 Bifunctionalised MOFs
Bifunctionality is a synergistic concept stimulated for developing versatile materials that can facilitate catalytic cascade chemical reactions in biomass valorisation. The properties and activities of bifunctional catalysts could be significantly different from the corresponding mono-functional catalysts. Especially, acid and redox properties are vital for efficient biomass valorisation. On the one hand, acid sites (e.g., –SO3H) can fractionate/depolymerise the complex biomass molecules into the model compounds. On the other hand, redox active sites (e.g., metal NPs) can catalyse the subsequent upgrading of model compounds to value-added chemicals. MOFs have the ability to accommodate both acid–base and metal active sites. Tailoring these bifunctional properties is essential for achieving improved catalytic results in biomass upgrading.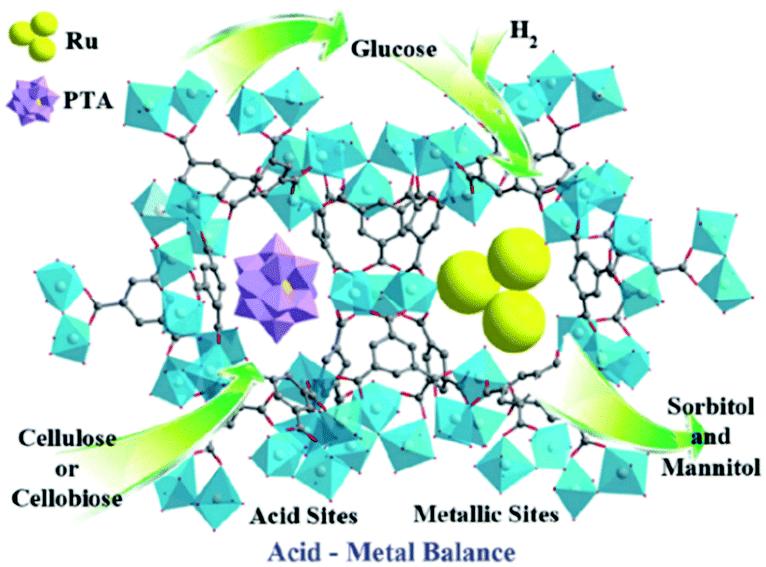 | ||
| Fig. 27 Metal–acid bi-functional Ru–PTA/MIL-100(Cr) catalyst for transformation of cellulose or cellobiose into sugar alcohols. Reproduced with permission from ref. 252. Copyright (2013) John Wiley and Sons. | ||
Although algae-derived bio-oil is regarded as an efficient renewable source of transportation fuel, the existence of large concentrations of oxygen-containing C16–C18 fatty acids leads to several problems, such as polymerisation, strong corrosion, poor stability, and low calorific value. Hence, algae bio-oil cannot be used as transportation fuel without removing these fatty acids. For this, Fang et al.262 developed a facile one-pot strategy to convert palmitic acid into hexadecane over a bifunctional MOF catalyst, consisting of encapsulated phosphotungstic acid inside the MOF structure of PdCu@FeIII-MOF-5. The developed catalyst shows an improved acidity and completely converts palmitic acid with a high selectivity to hexadecane. This superior catalytic performance was attributed to its unique yolk–shell MOF nanostructure, enhanced acidity, and the supercritical fluid medium.
3.6 MOFs-derived catalysts
Highly stable carbon-based metal or metal oxide catalysts can be developed using MOFs as sacrificial templates. A typical concern of MOFs, regarding their relatively low hydrothermal/chemical stability, can thus be turned into an advantage, by employing thermolysis under inert conditions for in situ generation of homogenously dispersed metal NPs on the carbon support.226 The resulting composite catalysts can contain ample amounts of functional sites, hence achieving notable catalytic activities in biomass conversion reactions.227,228:Cu molar ratio of 0.4 and a precursor calcination at 600 °C. Results revealed that doping with Co improved the metal dispersion, while the pyrolysis temperature significantly influences the particle size and chemical state. A one-pot cascade conversion of furfural to cyclopentanone was successfully carried out using bimetallic CuNi NPs embedded in a carbon matrix (CuNi@C).228 Compared with conventional precipitation methods, the MOF-templated procedure gave a higher specific surface area (91.6 m2 g−1) and well-dispersed CuNi NPs (about 15 nm) with a Ni:Cu molar ratio of 0.5. As a result, the CuNi0.5@C catalyst showed the best performance with a 97% yield of cyclopentanone at optimised reaction conditions. Reusability studies revealed a minor loss in product yields after four recycles of the catalyst, due to blockage of active sites by coke formed during reaction (entry 19, Table 2).
 | ||
| Fig. 28 MOF-templated strategy for the design of Fe/C–S catalysts. Reproduced with permission from ref. 263. Copyright (2017) Royal Society of Chemistry. | ||
 | ||
| Fig. 29 Preparation of a Cu/ZnO catalyst from Cu(Zn)-HKUST-1. Reproduced with permission from ref. 268. Copyright (2017) Elsevier. | ||
3.7 Conclusions of MOFs-based catalysts
In conclusion, a large number of research articles have reported on the topic of MOFs-catalysed biomass valorisation. This interest has been mainly stimulated by the unique porosity properties of MOFs compared to various other catalytic materials, such as microporous zeolites, mesoporous silica, and microporous carbon materials. Overall, the tunable porous channels of MOFs seem to play three important roles: (i) minimising mass diffusion limitation, (ii) improving surface area, and (iii) allowing a more efficient immobilisation of functional species. In addition, the building blocks of MOFs, namely metal ions or metal-oxo clusters and organic ligands, exhibit adequate amounts of acid–base and redox properties, which can be tailored in function of specific biomass upgrading reactions. However, several challenges still exist towards fertile catalytic applications of MOF-based materials in biomass valorisation. (1) Developing viable routes for the synthesis of cost-effective MOF-based materials. For this, more efforts should be directed towards one-pot synthesis of functionalised MOFs using relatively cheap precursors. (2) Improving the hydrothermal stability of MOFs and minimizing the leaching of active species,270 for which both strong coordination between ligands and metal centers233 as well as MOFs composites (e.g., with activated fly ash)235 could play a crucial role. (3) In-depth analysis of structure–activity relationships of MOFs during biomass conversions, thus providing useful implications to tune their catalytic activity and selectivity.4. Solid phase ionic liquids based catalysts
Ionic liquids (ILs) have recently emerged as novel clean solvents as well as promising functional catalysts for the development of new frontier catalytic technologies. ILs, popularly known as low temperature or room temperature molten salts, are composed of organic cations and (in)organic anions.271 ILs typically contain bulky and asymmetric ions that will limit their crystallisation, resulting in melting points below 100 °C and occasionally even below room temperature.40 Ethylammonium nitrate with a melting point of 12 °C was the first representative low temperature molten salt, and was synthesised in 1914.272 The development of moisture-stable ILs in 1992 has boosted the applicability of ILs in many fields, including catalysis. The extraordinary properties of ILs mark them as highly appealing “green” alternative to traditional organic solvents for various catalytic processes. Interesting properties include low vapor pressure, non-flammability, wide range of working temperatures, and high ionic conductivity.271,273 Theory estimates a total of 1018 types of ILs can be developed by tailoring the nature and the composition of cation/anion components. However, only about 1000 types of ILs have been synthesised up to date.40,2744.1 Factors affecting the catalytic activity of solid phase ILs in biomass upgrading
ILs have found versatile applications in catalytic biomass processing due to the presence of abundant amounts of both Lewis and Brønsted acid sites.275,276 Important examples are depolymerisation of lignocellulose biomass and its components (cellulose, hemicellulose, and lignin), dehydration of biomass-derived carbohydrates and biodiesel synthesis. As both the anionic and the cationic parts of ILs are easily varied, the properties of ILs can be tuned to fulfil a specific catalytic purpose. This extreme flexibility provides fertile opportunities to design or optimise the most suitable IL systems for achieving enhanced reaction rates in biomass conversions. Owing to the combination of polar and non-polar properties, ILs may also induce a nano-segregation of solvents and substrates, which could potentially improve the chemical reactivity.277 However, several challenges need yet to be solved in order to improve the catalytic applicability of ILs at the industrial scale. For instance, many catalytic processes require significant amounts of ILs, which is economically undesirable because of the high cost of ILs.39–41 The handling of ILs can also be difficult because of their high viscosity, in turn imposing mass transport limitations in catalytic reactions. Moreover, severe leaching of ILs into the reaction mixture, associated with the concerns of their separation and recycling, strongly limits their practical applications in biomass refinery processes.41,278 To overcome these issues, an appealing strategy i.e. combining the advantages of both ILs and solid materials has received great attention that has led to development of a new class of functionalised heterogeneous catalysts, namely ‘solid state ILs’.39–42 Immobilisation of ILs on solid materials by either covalent or non-covalent bonds can be achieved using various feasible synthesis methods, including physisorption, self-assembly, grafting, sol–gel, ionic exchange, or polymerisation. As an example, Fig. 30 illustrates of supported-mono-ILs.39 | ||
| Fig. 30 Representation of supported-mono-ILs. Reproduced with permission from ref. 39. Copyright (2014) Royal Society of Chemistry. | ||
Supported ionic liquids (SILs) offer several advantages for catalytic biomass conversions over the corresponding pristine ILs.40,41,279,280 SILs not only combine the original properties of ILs and supports, but also endows new functional sites and robust stabilisation, due to synergistic IL-support interactions. It is also possible that deposition of ILs on a high specific surface area material can provide increased number of accessible active sites at the catalyst surface, while consuming small amounts of ILs. More importantly, the IL film formed on the support surface provides a homogeneous environment for catalytic reactions, while appearing as a dry solid, allowing facile separation of SILs from the reaction mixture. Overall, these advantageous features of SILs can assist the development of practicable, sustainable catalytic routes for large-scale bio-refinery processes.
The catalytic properties of SILs depend on many factors. Surely, the choice of the anionic and cationic parts will be a dominant factor that governs the catalytic strength of SILs.40 Each IL has a unique set of anionic and cationic parts, resulting in a very specific interaction. As an example, the different types of interactions in a typical imidazolium-based IL are illustrated in Fig. 31.280 Tailorable SILs with desired functional sites can be developed by simply varying the nature and the composition of anionic and cationic parts. The nature of the support is another key factor influencing the catalytic efficiency of SILs. For example, compared with traditional porous silica and polymer supports, the use of oxidised multi-walled carbon nanotubes as a supporting material was found to significantly improve the catalytic performance of SILs.281 Moreover, the particle size of the support as well as the synthesis routes of the SILs can also affect the catalytic activity.279 Particularly, grafting of ILs, a widely used method, can considerably alter the activity of SILs in various ways.282 The advantages of grafting compared to impregnation are confirmed by CO2 diffusivity measurements. Grafted silica exhibits faster CO2 diffusion than impregnated silica, indicating that ILs incorporated into the inner pores of solid materials during impregnation hamper the diffusion of reactive species. In addition, grafting of ILs shows a negligible effect on the blockage of porous structure of solid materials, hence grafted solid surface allows for strong immobilisation of functional groups compared to impregnated surface. Finally, the IL loading can also exert a significant role in tuning the surface properties of SILs to achieve better catalytic activity.279 The loading of ILs can be adjusted by controlling the particle size of the support and the preparation method.
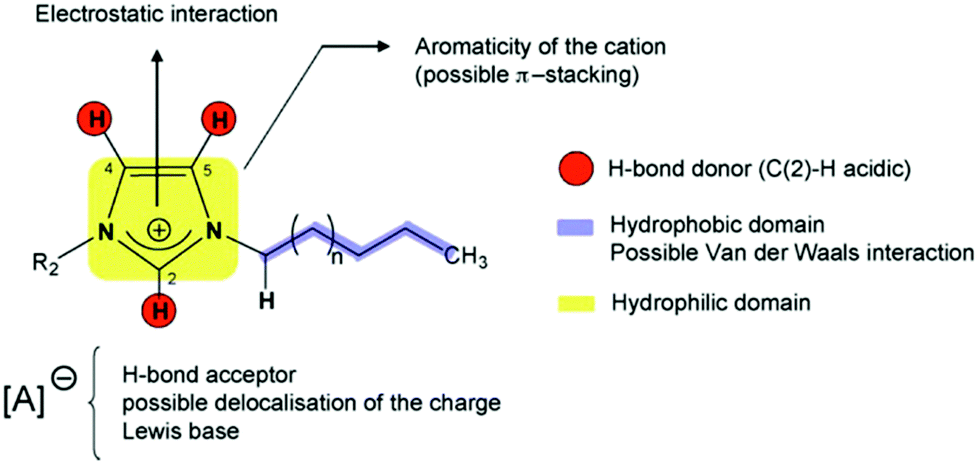 | ||
| Fig. 31 Schematic representation of different types of interactions present in a typical imidazolium-based ionic liquid. Reproduced with permission from ref. 280. Copyright (2010) Elsevier. | ||
Both acid and base functionalised SILs can be developed by simply immobilising acid or base functionalised ILs on the surface of solid supports, respectively.40,41,280,283 ILs with protic ammonium, pyrrolidinium, or imidazolium ions exhibit adequate amounts of acid sites. Strong Brønsted acidic ILs can also be prepared by anchoring –SO3H, carboxylic acids or alkane sulfonic acids as side chains on the cations (Fig. 32).284 On the other hand, a number of ILs with considerable amounts of basic sites exist, including ILs with lactate, formate, acetate, dicyanamide, etc. Interestingly, strong interactions between functionalised ILs and a solid support can prevent leaching of functional sites during biomass transformation reactions. Furthermore, incorporation of metal NPs into functionalised SILs can provide novel, bifunctionalised heterogeneous catalysts (Fig. 32).284 These bifunctionalised SILs possess abundant acid or base and redox properties, which could enable efficient catalysis of cascade biomass conversions in a one-pot way. The existence of various kinds of interactions may play an essential role in stabilising these functional sites and active metal centers (Fig. 31).
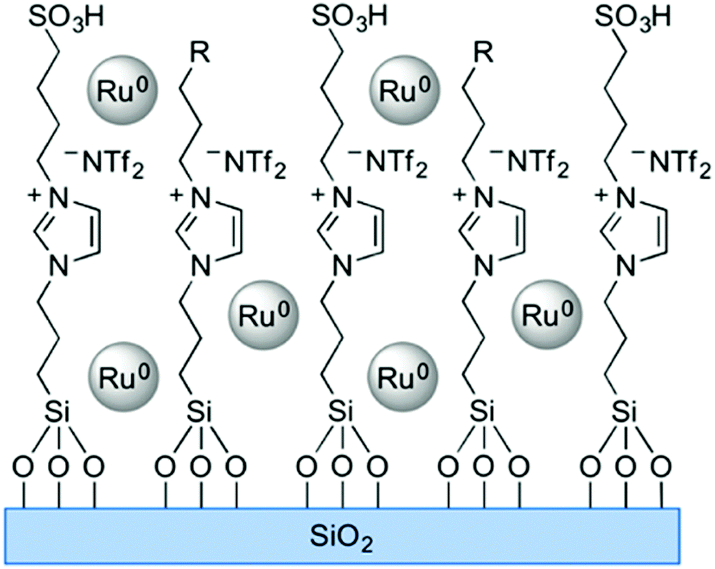 | ||
| Fig. 32 Schematic representation of a bifunctional catalyst composed of an acid-functionalised SIL and Ru NPs. Reproduced with permission from ref. 284. Copyright (2016) American Chemical Society. | ||
Solid polymeric ILs (PILs), a new class of recyclable IL based catalysts, are also perceived as fascinating catalytic alternatives for efficient biomass upgrading.285,286 The incorporation of IL moieties into a polymer chain can combine some of the unique properties of ILs with the common features of polymers. High thermal stability, tunable shaping, and corrosion resistance are some of the added benefits of PILs. PILs can be used as catalyst support or as catalytic active phase, depending on their nature and composition. However, conventional PILs typically exhibit low specific surface areas, resulting in negligible amounts of surface active sites available for catalytic reactions. To overcome this problem, novel porous PILs with well-defined structures and controlled geometries have been developed using appropriate synthesis methods.42,286 In addition to an increased active surface area, porous PILs may also improve mass transport due to tunable porous dimensions. Hence, higher reaction rates in biomass upgrading could be expected. Furthermore, the surface wettability of porous PILs can be efficiently tuned at a molecular level by carefully selecting the synthesis precursors.42 This is vital because surface wettability plays an essential role in tuning the interaction of solid catalysts with reaction substrates. For instance, hydrophobic solid surface favour the adsorption of hydrophobic reactant species. Therefore, an improved hydrophobicity of the catalyst surface can lead to higher activities and excellent selectivities in related reactions. In conclusion, Fig. 33 summarises the key advantages of solid phase ionic liquids. In the following sections, various catalytic biomass conversions were reviewed, partitioned in two categories: (1) supported ILs based catalysts and (2) polymerised ILs based catalysts. In addition, a summary of the most important catalytic results obtained in solid-state ILs catalysed biomass upgrading is presented in Table 3.
 | ||
| Fig. 33 Key advantages of solid phase ionic liquids. | ||
| Entry | Catalyst | IL | Reaction | Product | Reaction conditions | Conv. % (yield %) | Recycling results | Ref. |
|---|---|---|---|---|---|---|---|---|
| a HMF – 5-hydroxymethylfurfural. b DMSO – dimethyl sulfoxide. c FPIL – functional polymeric IL. d [BVIM]Cl – 3-butyl-1-vinylimidazolium chloride. e MPIL – mesoporous poly(IL). f FDCA – 2,5-furandicarboxylic acid. g MMA – methyl methacrylate. h PAL – nanofiber-like palygorskite. i PW – phosphotungstic acid. | ||||||||
| 1 | Acidic IL–SiO2 | 1-(1-Butylsulfonic)-3-methylimidazolium chloride | Cellulose hydrolysis | Reducing sugar | 30 mg cellulose, 2 mL water, 190 °C, 3 h | — (48) | 4 (small decrease in product yields) | 291 |
| 2 | SO3H-IL–Zn/biochar | 1-Trimethoxy-silylpropyl-3-methylimidazolium chloride | Microwave-assisted cellulose hydrolysis | Reducing sugar | 200 mg cellulose, 100 mg catalyst, 1.5 mL H2O, 90 °C, 2 h | — (58.7) | 3 (stable catalytic activity) | 292 |
| 3 | SiO2 supported IL–SO3H–HSO4 | 3-Sulfobutyl-1-(3-propyltriethoxysilane) imidazolium hydrogen sulfate | Fructose dehydration | HMFa | 4:1 mass ratio of fructose to catalyst, 1.8 g DMSOb, 110 °C, 2 h |
>99 (97.5) | 5 (small decrease in HMF yield from 97.5% to 92.7%) | 293 |
| 4 | SiO2 supported IL–HSO4 | 1-(Triethoxy-silyl-propyl)-3-methylimidazolium hydrogen sulfate | Fructose dehydration | HMF | 50 mg fructose, 40 mg catalyst, 0.5 mL DMSO, 130 °C, 30 min | 99.9 (63) | 7 (stable catalytic activity) | 294 |
| 5 | Polypropylene fiber–IL | 3-Sulfopropylpyridinium hydrogen sulfate | Fructose dehydration | HMF | 0.5 g fructose, 7.5 mol% catalyst based on fructose, 10 mL DMSO, 100 °C, 30 min | — (86.2) | 10 (stable catalytic activity) | 295 |
| 6 | Fe25Ru75@SILP | [1-Butyl-3-(3-triethoxy silyl propyl)-imidazolium]NTf2 | Hydrogenation of furfural-acetone | Aromatic alcohol | 0.4 mmol substrate, 40 mg catalyst, 0.5 mL mesitylene, 100 °C, 20 bar H2 at 100 °C, 18 h | >99 (94) | 2 (considerable decrease in product yields from 94% to 83%) | 297 |
| 7 | CrCl2-Im-SBA-15 | 1-(Tri-ethoxy-silyl)-propyl-3-methylimidazolium chloride | Glucose dehydration | HMF | 10 wt% glucose in the aqueous phase, 10 mg catalyst, 150 °C, 3 h | 50 (35) | 2 (catalyst deactivation, due to leaching of active species) | 299 |
| 8 | Cr(salen)-IM-HSO4-MCM-41 | [CPTES-IM-SO3H][HSO4]/[Cl] | Glucose dehydration | HMF | 100 mg substrate, 50 mg catalyst, 2 mL DMSO, 140 °C, 4 h | 99 (43.5) | 5 (minor loss in catalytic activity, due to leaching of active species and adsorption of reaction substrates on the catalyst surface) | 278 |
| 9 | IL/Mg–Al–La | 1-Butyl-3-methylimidazolium hydroxide | Transesterification of fatty oil | Biodiesel | 12:1 molar ratio of methanol to oil, 3 wt% catalyst amount based on oil, 65 °C, 6 h |
— (98.7) | 6 (considerable decrease in biodiesel yield from 98.7% to 85.4%, due to leaching of active basic species) | 304 |
| 10 | Acidic IL/silica | — | Transesterification of oil | Biodiesel | 25:1 molar ratio of methanol to oil, 5 wt% catalyst amount based on oil, 60 °C, 20 h |
87.6 (87.6) | 5 (considerable decrease in biodiesel yield from 87.6% to 70.2%, due to leaching of IL) | 305 |
| 11 | IL-Fe3O4@NH2-MIL-88B(Fe) | 1,4-Butanediyl-3,3′-bis(3-sulfopropyl) imidazolium dihydrogensulfate | Esterification of oleic acid | Biodiesel | 10.5:1 molar ratio of alcohol to acid, 8.5 wt% catalyst based on oleic acid, 90 °C, 4.5 h |
93.2 (93.2) | 6 (noticeable decrease in biodiesel yields, due to catalyst mass loss during recovery/purification) | 306 |
| 12 | CrCl3·6H2O-FPILc | 1-Vinyl-3-propane sulfonate imidazolium | Fructose dehydration | HMF | 100 mg substrate, 30 mg catalyst, 1 mL DMSO, 120 °C, 1 h | 98.7 (90.6) | 5 (stable catalytic activity) | 285 |
| 13 | Poly(IL)–CrCl2 | [BVIM]Cld | Glucose dehydration | HMF | 40 mg substrate, 10 mol% catalyst based on glucose, 1 mL DMF, 120 °C, 3 h | — (65.8) | Significant loss in HMF yields after six recycles | 316 |
| Poly(IL)–Et2AlCl | [BVIM]Cl | — (49) | Minor loss in HMF yields after six recycles | |||||
| 14 | Poly(IL)-H3PW12O40 | 1-Vinyl-3-propane sulfonate imidazolium | Fructose dehydration | HMF | 50 mg fructose, 30 mg catalyst, 0.5 mL DMSO, 130 °C, 1 h | 97.9 (83) | 5 (considerable loss in HMF yields, due to leaching of active species) | 317 |
| 15 | Au1–Pd1@MPILe | 3-Cyanopropyl-1-vinyl imidazolium chloride | HMF oxidation | FDCAf | 0.1 mmol HMF, HMF/metal molar ratio (100), 2 mL water, 90 °C, O2 (10 mL min−1), 12 h | 99.7 (99) | 5 (stable catalytic activity) | 42 |
| 16 | Acidic poly(IL) | — | Esterification of methacrylic acid | MMAg | 1:1.2 molar ratio of substrate/methanol, 5 wt% catalyst based on substrate, 95 °C, 3 h |
100 (100) | 5 (stable catalytic activity) | 319 |
| 17 | Macroporous poly(IL) | 1-Vinyl-3-(3-sulfopropyl) imidazolium hydrogen sulfate | Esterification of oleic acid | Biodiesel | 12:1 molar ratio of alcohol/acid, 8.5 wt% catalyst based on oleic acid, 80 °C, 4.5 h |
92.6 (92.6) | 6 (small decrease in acid conversion from 92.6% to 89.3%, due to catalyst mass loss during recovery/washing steps) | 286 |
| 18 | PAL-poly(IL)h | 1-Butysulfonate-3-vinylimidazole hydrogen sulfate | Esterification of oleic acid | Biodiesel | 12:1 molar ratio of alcohol/acid, 7 wt% catalyst based on oleic acid, 75 °C, 5 h, |
69 (69) | 6 (significant decrease in biodiesel yield from 69% to 22%, due to leaching of IL, decreased surface area, and mass loss of the catalyst) | 320 |
| 19 | PW/poly(IL)i | 1-Butyl-3-vinylimizolium bromide | Isomerisation of α-terpineol | 1,8-Cineole | 1.16 g of α-terpineol, 1.12 wt% catalyst based on substrate, 50 mL cyclohexane, 50 °C, 8 h | 100 (51.7) | 7 (stable catalytic activity) | 321 |
4.2 Supported ILs based catalysts
Supported IL catalysts as a concept was initially introduced in the 1990s.287 Depending on the composition and configuration of ILs immobilised on solid supports, SILs can be classified as supported IL phase, supported IL catalyst, and solid catalyst with an IL layer.288–290 Many solid supports, such as mixed oxides (Mg–Al–O), bio-char, mesoporous silica (SBA-15 and MCM-41), carbon nanotubes, MOFs, and polystyrene have been used for the immobilisation of ILs.Eucalyptol, a major component of eucalyptus oil (∼90%), is as an attractive platform molecule for the production of valuable C10-chemicals, such as menthanes, menthenes, menthadienes, and cymene.284 Several bifunctionalised catalysts consisting of metallic Ru NPs immobilised on acid-functionalised SILs (RuNPs@SILs) were applied in the hydrodeoxygenation of eucalyptol under both batch and continuous-flow conditions.284 The RuNPs@SILs showed excellent catalytic activity and selectivity towards a deoxygenation product, p-menthane, which was attributed to well-balanced acid and metal functionalities. Kinetic experiments under continuous-flow conditions identified the acid catalysed deoxygenation of eucalyptol as the rate-limiting step. The partial substitution of Ru by a non-noble metal (Fe) in RuNPs@SIL led to tailored catalytic activities and selectivities in the hydrogenation of various aromatic substrates including furfural and furfuralacetone.297 A facile organometallic synthesis method was used for the synthesis of FeRuNPs@SIL catalysts. This preparation method allowed a controlled formation of finely dispersed bimetallic FeRu NPs. Among several tested catalysts, bimetallic Fe20Ru80NP@SIL and Fe25Ru75NP@SIL showed the best performance with more than 94% yield to the aromatic alcohol at nearly complete conversion of furfuralacetone. Reusability studies, carried out under shaken conditions rather than magnetically stirred conditions in order to prevent mechanical degradation of the catalyst, revealed a decrease in product yield (83%) after two recycles of Fe25Ru75NP@SIL catalyst (entry 6, Table 3). The effect of the catalyst support, i.e. conventional silica, mesoporous SBA-15, and polystyrene on the catalytic performance of acid functionalised SILs was studied.298 The obtained catalysts were tested for the production of ethylene glycol diacetate via esterification of ethylene glycol with acetic acid. Ethylene glycol diacetate is a useful solvent for paints, coatings, plastics, and wood stains. Interestingly, the conventional silica supported IL showed the best catalytic performance for the synthesis of ethylene glycol diacetate and a pseudo-homogeneous second order kinetic model was proposed, based on thorough kinetic experiments.
Compared to conventional solid supports, the confinement of ILs into the porous materials (mesoporous silica, MOFs, porous bio-char, etc.) can reduce the extent of IL leaching during catalytic reactions.40 Interestingly, confinement effects and interactions of ILs with the pore walls may result in physicochemical properties that significantly differ from conventional SILs. Moreover, these properties may be strongly dependent on the configuration of ILs inside the pores, e.g., as grafted monolayers or physically confined multilayers (Fig. 34).40 A metal chloride (CrCl2) immobilised on a thin layer of IL (1-(triethoxysilyl-propyl)-3-methylimidazolium chloride, (EtO)3–Si–PMImCl) grafted on mesoporous silica (SBA-15) was studied for glucose dehydration in an aqueous medium.299 The coordination of CrCl2 to PMIm+Cl− creates loosely bound, catalytically active Cr2+ species. The high mobility of these complexes was shown to be beneficial for the selective synthesis of HMF from glucose. However, severe catalyst deactivation was noticed after two recycles of the catalyst, due to leaching of large amounts of active species (entry 7, Table 3). The effect of metal chlorides (e.g., CrCl2, AlCl3, CuCl2, and FeCl3) on the structure–activity properties of an immobilised IL/SBA-15 catalyst was also studied for the dehydration of glucose to HMF.300 Results revealed that the ordered mesoporous structure is retained after grafting of the IL, though the specific surface area of SBA-15 was slightly decreased. Higher HMF yields were obtained over the developed SILs compared to various homogeneous metal chloride catalysts. Multifunctional catalysts composed of chromium(III) Schiff base complexes and acidic ILs, both immobilised onto the surface of MCM-41 were investigated for the dehydration of various carbohydrates, such as glucose, fructose, and inulin.278 The reaction temperature, reaction time, and the solvent were optimised for HMF production. About 43.5%, 83.5% and 80.2% yields of HMF were obtained from glucose, fructose, and inulin, respectively. A minor loss in catalytic activity for glucose dehydration was noticed after five recycles of the catalyst, due to leaching of active species and adsorption of reaction substrates on the silica surface (entry 8, Table 3). A series of SBA-15 supported acidic IL catalysts were tested for biphasic alkylation of 2-methylfuran with formalin.301 The developed SIL catalysts showed higher activities and a superior selectivity (90%) towards bis(5-methylfuran-2-yl)methane, compared to commercial sulfonic acid resin catalysts. In addition, the SBA-15 supported acidic IL catalysts were also found to be active for the alkylation of 2-methylfuran with various biomass-derived substrates, such as furfural, glycolaldehyde, glyceraldehyde, and 3,4-dimethoxybenzaldehyde.
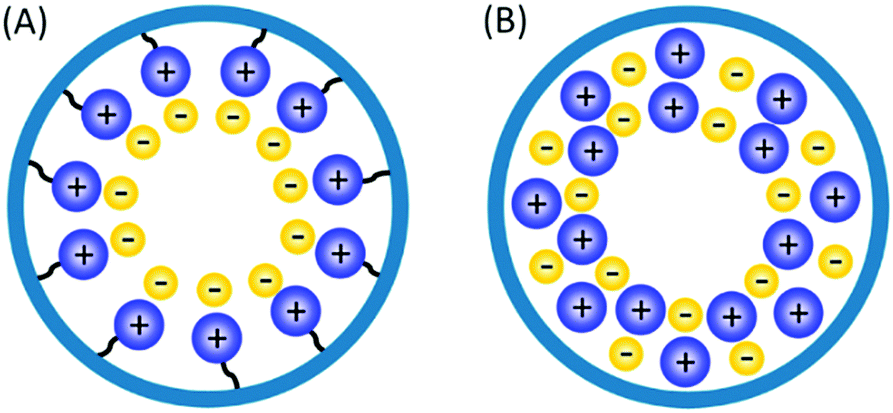 | ||
| Fig. 34 Incorporation of ILs into porous materials by two different strategies: (A) covalent grafting of monolayer IL on the pore wall and (B) physical confinement of multilayers of ILs into the nanopores. Reproduced with permission from ref. 40. Copyright (2017) American Chemical Society. | ||
Multifunctional ILs supported on a porous bio-char were prepared by pyrolysis of cellulose and IL and subsequently tested for the production of levoglucosenone (LGO) and dihydrolevoglucosenone (DLGO) via catalytic reforming of cellulose pyrolysis volatiles (Fig. 35).302 DLGO is an attractive bio-based solvent alternative to dipolar aprotic solvents and can be further transformed to 1,6-hexanediol, a commodity chemical with widespread industrial applications. A 31.6% yield of LGO was obtained on a cellulose carbon basis (24.6 wt%). Moreover, the addition of hydrogen gas and a hydrogenation catalyst in the catalytic bed enabled the production of DLGO. Hollow nanosphere structured N-doped carbon supported sulfonic acid-functionalised ILs were developed for the selective conversion of fructose to promising fuel blends, such as 5-ethoxymethylfurfural and ethyl levulinate.303 Results revealed that strong interactions between functionalised IL groups and the N-doped carbon framework play a favourable role in limiting acid site leaching. Hence, the developed SIL catalyst exhibited large amounts of strong Brønsted acid sites, which efficiently catalyse the ethanolysis of fructose, thus avoiding self-polymerisation of intermediates, leading to higher total yields of 5-ethoxymethylfurfural and ethyl levulinate.
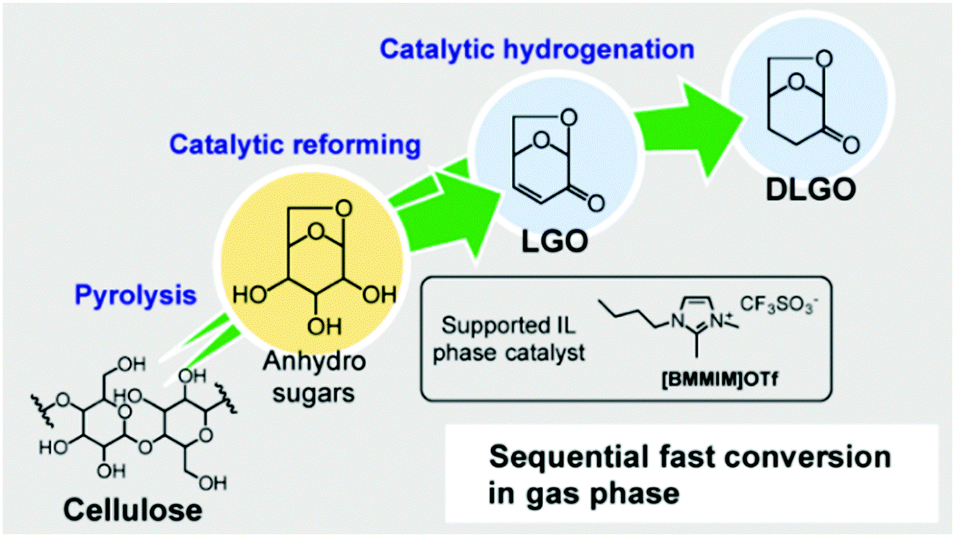 | ||
| Fig. 35 Production of levoglucosenone (LGO) and dihydrolevoglucosenone (DLGO) by catalytic reforming of volatiles obtained from cellulose pyrolysis using supported IL catalysts. Reproduced with permission from ref. 302. Copyright (2017) American Chemical Society. | ||
Various metals incorporated SILs were investigated in the alkylation and acylation of lignin model compounds.309 Results revealed that the nature of the cation controls both catalytic behaviour and active phase leaching. More specifically, silica-supported Fe- and Sn-ILs showed a drastic leaching compared to the Al-ILs. However, the Fe-based IL was found to be more active, which might be due to the homogeneous catalytic reaction because of the Fe leaching into the reaction medium. Leaching issues were also observed when using other supports, such as metal oxides (Nb2O5, TiO2, Na-Y, and Al2O3) or activated charcoal. The formation of highly stable complexes between metal halide anions and aromatic lignin model compounds is the key reason for leaching of active phases. Compared to alkylation, the SILs showed better catalytic results for acylation of lignin model compounds. The effect of metal chlorides (e.g., SnCl2, FeCl3 or CrCl3) immobilised on activated carbon supported IL catalysts was studied for the isomerisation of α-pinene oxide to campholenic aldehyde.310 α-Pinene oxide can be produced from renewable terpenes. Among them, the SnCl2 immobilised SIL showed the best catalytic performance with a 65% yield of campholenic aldehyde and reasonable good reusability. These results were confirmed by mechanistic kinetic models.311 In another work, the isomerisation of β-pinene oxide to myrtanal was investigated over similar types of metal chloride immobilised activated carbon supported IL catalysts.312 Myrtanal is a vital intermediate for fragrance and pharmaceutical industries. The catalytic activity and product distribution were highly dependent on the nature of the ionic liquid. Short reaction times and high reaction temperatures favoured the formation of myrtanal. About 68% yield of myrtanal was obtained over a ZnCl2 immobilised SIL catalyst.
4.3 Polymeric ILs based catalysts
Polymeric ILs (PILs) are solid materials that can be prepared using a variety of strategies, such as incorporation of ILs into the polymeric substrates,313 homo-polymerisation of ILs, copolymerization of ILs with other monomers, or hard templating methods.42,286 Acid or base functionalisation and incorporation of metal NPs have also been carried out to obtain multifunctional PILs.314 In this case, polymerised ILs could fulfil several important roles, like facilitating the stabilisation of metal NPs, controlling the accessibility of reagents/substrates to the active sites, and providing specific environments for an efficient and selective adsorption of the reagents.315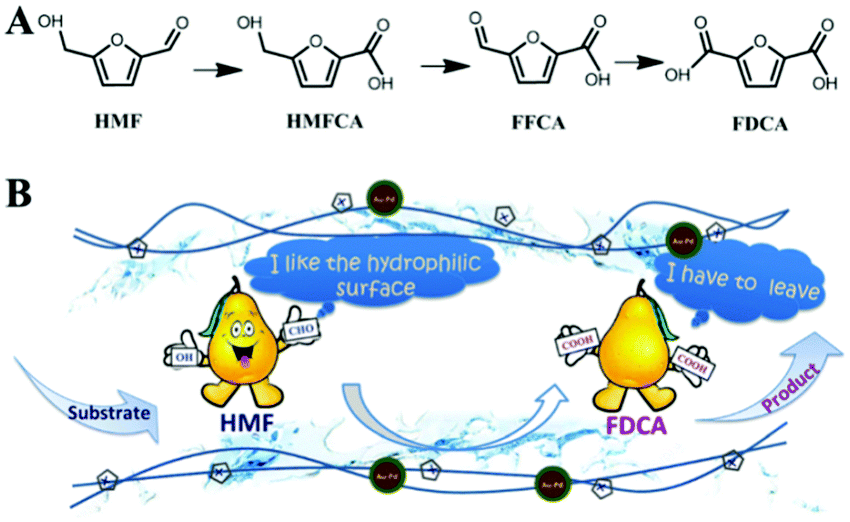 | ||
| Fig. 36 Proposed reaction pathway for the oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA) catalysed by Au1–Pd1@PIL. Reproduced with permission from ref. 42. Copyright (2017) Royal Society of Chemistry. | ||
4.4 Conclusions of solid-state ILs-based catalysts
As discussed in this section, both supported ILs and polymeric ILs possess numerous advantages over the corresponding pristine ILs in terms of hydrothermal stability, recyclability, and the amount of IL needed for a catalytic reaction (Fig. 33 and Table 3). In addition, SILs exhibit new active sites, generated by synergistic effects between the IL and the supporting material, which are vital for achieving improved reaction rates in biomass valorisation. In order to enhance the applicability of SILs based catalysts in biomass upgrading, particularly at the industrial scale, several problems have yet to be solved.40,322 (1) Understanding the role of impurities (e.g., water, halides, unreacted organic or inorganic salts), originating during SILs synthesis, in biomass conversions. This is vital because even trace amounts of impurities could dramatically affect the rate and mechanism of a reaction. (2) Developing eco-friendly procedures for the synthesis of ILs because most of the existed methods use large amounts of hazardous precursors and organic solvents. (3) In-depth structural exploration of ILs in SILs, including intrinsic nature (electrostatic field, ionic strength, etc.), acid–base strength, and ILs/support interaction, which is key to tailor the catalytic activity/selectivity of SILs for specific biomass conversions.5. Magnetic iron oxide based catalysts
Nanoscale metals, metal oxides, or combinations of both display excellent catalytic activities for a broad spectrum of chemical reactions.53,323–325 Tailoring the particle size, morphology, and composition of metallic materials generates changes in their electronic and geometric arrangements, which could result in an improved catalytic activity. Hence, the properties of nanoscale catalysts can be dramatically different from the corresponding macroscopic bulk materials, potentially leading to unusual catalytic results.326–329 However, the separation and recovery of nanoscale catalysts from liquid-phase reaction mixtures for subsequent recycling remains a great challenge. Although filtration or centrifugation are typically used for the recovery of solid catalysts, these post-reaction steps add cost to the entire production, especially in the case of nano-sized catalysts. A promising solution to tackle this issue is the application of magnetic catalytic nanomaterials.330,331 Owing to outstanding paramagnetic properties and inherent insolubility, magnetic catalysts can be efficiently separated from complex reaction mixtures using external magnets without affecting their activity and selectivity (Fig. 37).332 Iron oxide based nanomaterials (mainly Fe3O4) are widely used magnetic catalysts in biomass conversions due to their low cost, facile preparation, and strong magnetic properties.30,50,333 Related magnetic materials, such as γ-Fe2O3, spinel ferrites with a general formula M2+Fe3+2O4 (M = Co, Ni or Mn), Fe-based alloys (FeB, FeNiB, etc.), and iron oxide supported materials also show potential for catalytic biomass conversions.309–311 Overall, magnetically recoverable catalysts have demonstrated high efficiency in a wide range of “one-pot, multi-step” catalytic reactions, including oxidation, epoxidation, hydrogenation, hydroformylation, olefin metathesis, polymerisation, photocatalysis, and C–C bond formation reactions.331,334–338 Moreover, novel applications in asymmetric synthesis, organocatalysis, Knoevenagel condensation, or CO2 cycloaddition reaction have been developed. In this section, the focus lies on the applicability of iron oxide based magnetic catalysts for biomass conversions. | ||
| Fig. 37 Magnetic separation of a finely dispersed Fe3O4 based catalyst used for cellulose hydrolysis. Reproduced with permission from ref. 332. Copyright (2012) Springer Nature. | ||
5.1 Factors affecting the catalytic activity of iron oxides in biomass valorisation
In some applications, the use of magnetic catalysts is particularly interesting because of the difficulties involved with the use of conventional catalyst separation steps, such as filtration or centrifugation. For example, a solid mixture containing hemi(cellulose) pulp and solid catalyst (e.g., Ru/C) is obtained after reductive catalytic fractionation of lignocellulose (Fig. 38).91 Another example is related to the formation of humins (carbonaceous, polymeric by-products), obtained as supplementary solid residues during carbohydrate dehydration towards HMF production.331 These insoluble humins can subsequently hamper the recovery and reusability of the catalyst. Since the separation of a solid catalyst from solid reaction products is highly challenging, the application of catalytic materials having strong magnetic properties could be a valid solution.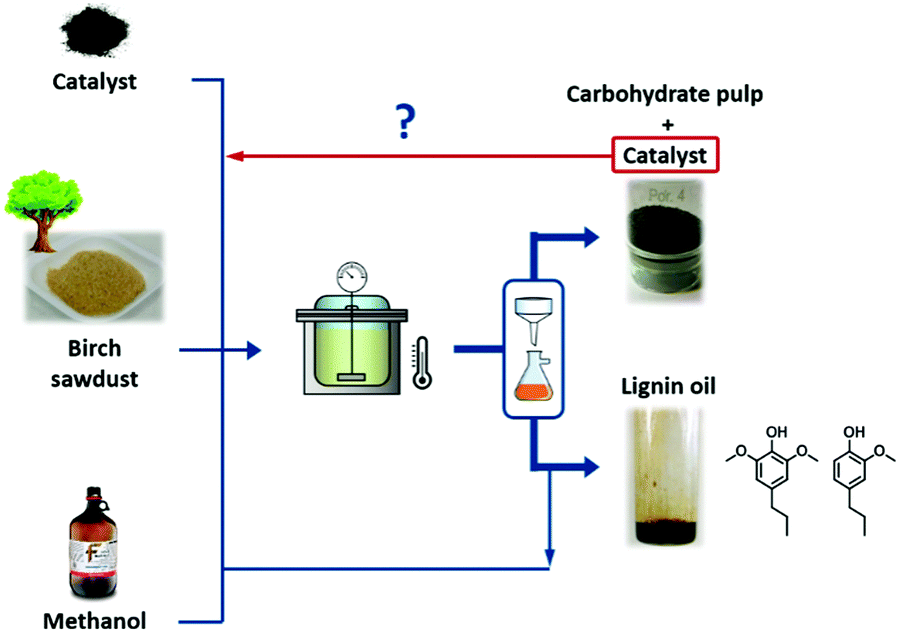 | ||
| Fig. 38 A schematic representation for the catalytic reductive fractionation of lignocellulose sawdust using Ru/C catalyst in methanol under H2 pressure. | ||
Pure iron oxide magnetic NPs suffer from several drawbacks, such as self-interactions which induces particle aggregation, rapid exothermic reactions in the presence of oxygen, and a high sensitivity to air, all resulting in a reduced applicability for magnetic catalytic purposes.47,339,340 In order to preserve the magnetic properties of iron oxide NPs, while improving their catalytic activity, several encapsulating strategies have been developed. Fig. 39 shows some key benefits of encapsulated iron oxide magnetic catalysts. For instance, protection of the Fe3O4 surface with a coating layer has been frequently studied. In this way, highly stable core–shell Fe3O4@M catalysts can be obtained, wherein Fe3O4 constitutes the core and M (coating layer) is the shell. SiO2 is commonly used coating material for Fe3O4 because it is cheap, inert, non-toxic, and besides, it has a high specific surface area.46,47,341 An additional benefit of SiO2 is the promising surface functionality of core–shell Fe3O4@SiO2 magnetic catalysts due to the presence of abundant surface silanol groups (–SiOH). These silanol species offer facile approaches for selective anchoring of acid (–SO3H) or base (–NH2) functional species upon Fe3O4@SiO2 materials.
 | ||
| Fig. 39 Key benefits of encapsulated iron oxide magnetic catalysts. | ||
The stabilisation of magnetic iron oxide NPs can also be achieved using various polymers (e.g., polyphenylquinoxaline, polypyridylphenylene, and hyperbranched polyethylenimine) as the protective agents.342 These polymers are thermally stable up to 400–500 °C, enabling the resulting catalysts for wide-temperature-range reactions in biomass valorisation. The presence of nitrogen-containing species in the repeating units of these polymers offers potential routes for the immobilisation of catalytic active metals (Cu, Co, and Pd) as illustrated in Fig. 40.342 The encapsulation of iron oxide NPs by hydroxyapatite (HAP) is another promising strategy, which not only limits particle aggregation, but also paves the way for the introduction of new catalytic active sites. For example, Ca2+ in the framework of hydroxyapatite can be exchanged with other transition metal cations (Pd2+, Ru2+, etc.).343 Results revealed that a Pd-exchanged HAP encapsulated γ-Fe2O3 (γ-Fe2O3@HAP-Pd) efficiently catalyses the selective oxidation of HMF to FDCA at mild reaction conditions. The exchanged magnetic catalyst synthesis typically involves the cation exchange of Ca2+ in γ-Fe2O3@HAP with Pd2+, followed by a reduction step using NaBH4.
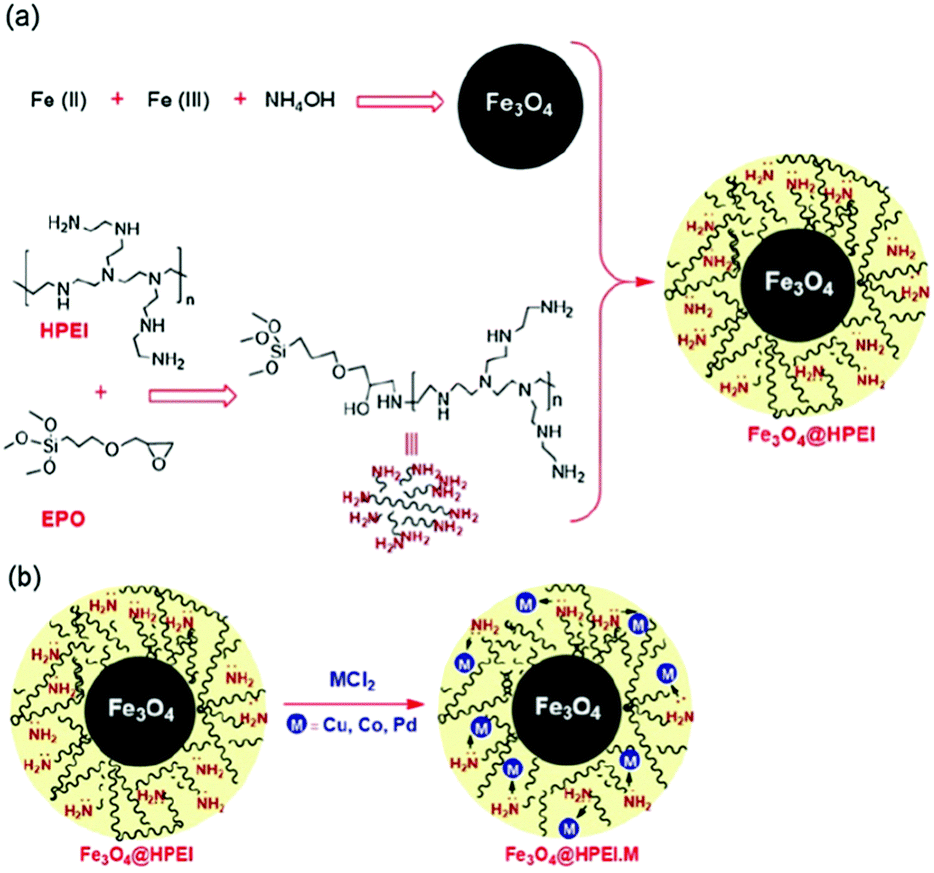 | ||
| Fig. 40 (a) Preparation of hyperbranched polyethylenimine (HPEI) encapsulated magnetic Fe3O4 nanoparticles (Fe3O4@HPEI) and (b) chelation of metal species (Pd, Co and Cu) with nitrogenous ligands in Fe3O4@HPEI. Reproduced with permission from ref. 342. Copyright (2017) John Wiley and Sons. | ||
Spinel-type MFe2O4 ferrites (M = Mn, Ni, Zn, or Co in divalent state and Fe in +3 oxidation state) also exhibit strong magnetic properties with excellent structural and thermal stability, which are highly dependent on the microstructure and cation distribution. To improve the magnetism as well as the activity of ferrites, the partial replacement of Fe3+ in MFe2O4 with transition metal cations (e.g., Ru3+) was successfully attempted without destroying the spinel structure.344 The resulting doped ferrite catalysts were successfully applied for biomass upgrading reactions. Furthermore, Fe-based nanoalloys and Fe3O4 supported catalysts have found considerable applicability in biomass valorisation due to synergistic metal–metal and metal–support interactions, along with improved magnetic properties.344–346 In the following sections, the catalytic applications of magnetic catalysts studied for biomass valorisation are thoroughly discussed, based on two categories, namely core–shell and non-core–shell iron oxide based catalysts. In addition, a summary of the interesting catalytic results obtained in magnetic iron oxides catalysed biomass conversions is presented in Table 4.
| Entry | Catalyst | Reaction | Product | Reaction conditions | Catalytic activity | Recycling results | Ref. | |
|---|---|---|---|---|---|---|---|---|
| Conv. (%) | Yield (%) | |||||||
| a HMF – 5-hydroxymethylfurfural. b FDCA – 2,5-furandicarboxylic acid. c PMO – sulfoacid-containing mesoporous organosilica. d PHA – phosphotungstic acid. e HPW – phosphotungstic acid. f EMF – 5-ethoxymethylfurfural. g BAIL-Al – bifunctional acidic ionic liquid of metal Al substituted. h PPP – poly(pyridylphenylene). i HAP – hydroxyapatite. j DFF – 2,5-diformylfuran. k TEMPO – 2,2,6,6-tetramethylpiperidine-N-oxide. l EG – ethylene glycol and PG – propylene glycol. m Amorphous cellulose. | ||||||||
| 1 | Fe3O4@carbon–SO3H | Hydrolysis of cellulose | Glucose | 50 mg cellulose, 150 mg catalyst, 10 mL H2O, 140 °C, 12 h | 48.6 | 25.3 | 3 (small decrease in cellulose conversion from 48.6% to 45%, due to leaching of acidic species) | 332 |
| 2 | Fe3O4@SiO2–SO3H | Hydrolysis of cellulose | Reducing sugar | 160 mg cellulose, 100 mg catalyst, 2 g ionic liquid & 27 mg H2O, 130 °C, 8 h | — | 73.2 | 6 (small decrease in product yields from 73.2% to 69.4%) | 350 |
| 3 | Fe3O4@SiO2–SO3H | Glucose dehydration | HMFa | 1:4 vol ratio of water/methyl isobutyl ketone, 140 °C, 24 h |
98 | 70.5 | 5 (noticeable decrease in HMF yields from 70.5% to 60.4%, due to accumulation of oligomeric products on the active sites and leaching of acid species) | 340 |
| 4 | Fe3O4@Nb2O5–SiO2 | Hydrolysis of cellulose | Lactic acid | 50 mg cellulose, 50 mg catalyst, 15 mL water, 180 °C, 24 h | 95 | 43.7 | 3 (small decrease in lactic acid yields, due to accumulation of reaction substrates on the catalytic active sites) | 351 |
| 5 | Pd/Fe3O4@carbon | Oxidation of HMF | FDCAb | 0.4 mmol HMF, 40 mg catalyst, 0.2 mmol K2CO3, 8 mL water, 80 °C, O2 (30 mL min−1), 6 h | 98.4 | 86.7 | 5 (very small decrease in FDCA yields from 86.7% to 83.7%) | 352 |
| 6 | Ru(III)/Fe3O4@SiO2–NH2 | Cellulose upgrading | Glycerol | 140 mg cellulose, 60 mg catalyst, 5 mL water, 180 °C, 2 h | 67.3 | 44.4 | 2 (stable catalytic activity) | 353 |
| 7 | Ru@Fe3O4@void@PMO–SO3Hc | Hydrogenolysis of cellulose | Isosorbide | 60 mg cellulose, 60 mg catalyst, 30 mL H2O, 6 MPa H2, 220 °C, 2 h | 98 | 58 | 4 (considerable decrease in isosorbide yields) | 357 |
| 8 | Fe3O4@SiO2@PHAd | Dehydration of glucose | HMF | 0.5 g glucose, 2 g catalyst, 15 mL dimethylformamide, 130 °C, 1 MPa N2 at 130 °C, 3 h | 59.8 | 30.4 | 5 (considerable decrease in HMF yields, due to leaching of acid species) | 359 |
| 9 | Fe3O4@SiO2–HPWe | Etherification of HMF | EMFf | 1 mmol HMF, 150 mg catalyst, 5 mL ethanol, 100 °C, 11 h, N2 atm | 97.9 | 83.2 | 6 (small decrease in EMF yields from 83.2% to 78.8%) | 46 |
| 10 | Fe3O4@Al2O3–BAIL–Alg | Dehydration of xylose | Furfural | 100 mg xylose, 40 mg catalyst, 3 mL dimethyl sulfoxide, 140 °C, 3 h | 97.3 | 67.5 | 5 (noticeable decrease in furfural yields from 67.5% to 58.7%, due to accumulation of reaction substrates on the catalytic active sites) | 360 |
| 11 | 0.3% Pd–Fe3O4–PPPh | Hydrogenation of furfural | Furfuryl alcohol | 2 mL furfural, 48 mL i-propanol, 120 °C, 60 bar H2, 450 min | 98.8 | 98 | 5 (stable catalytic activity) | 361 |
| 12 | γ-Fe2O3@HAPi | Hydrogenation of furfural | Furfuryl alcohol | 1 mmol furfural, 40 mg catalyst, 15 mL of i-propanol, 180 °C, 10 bar N2, 10 h | 96.2 | 91.7 | 6 (stable catalytic activity) | 362 |
| 13 | γ-Fe2O3@HAP–Pd(0) | HMF oxidation | FDCA | 0.4 mmol HMF, 40 mg catalyst, 0.2 mmol K2CO3, 8 mL water, 100 °C, O2 (30 mL min−1), 6 h | 97 | 92.9 | 5 (minor loss of FDCA yields from 92.9% to 90.7%, due to mass loss of the catalyst during recovery/purification steps) | 343 |
| 14 | Fe3O4@SiO2–NH2–Ru(III) | HMF oxidation | DFFj | 100 mg HMF, 150 mg catalyst, 7 mL toluene, 110 °C, O2 (20 mL min−1), 4 h | 99.3 | 86.4 | 6 (small decrease in DFF yields from 86.4% to 80.8%) | 363 |
| 15 | Fe3O4@SiO2–NH2–VO2+ & Fe3O4@SiO2–NH2–Cu2+ as cocatalyst | HMF oxidation | DFF | 0.8 mmol HMF, 100 mg Fe3O4@SiO2–NH2–VO2+, 30 mg Fe3O4@SiO2–NH2–Cu2+, 40 mL toluene, 110 °C, 2.8 bar O2, 1 h | 98.7 | 85.5 | 4 (small decrease in DFF yields from 85.5% to 81.2%) | 48 |
| 16 | Fe3O4@SiO2–TEMPOk | HMF oxidation | DFF | 1 mmol HMF, 2 mol% catalyst, 5 mol% tert-butyl nitrite, 75 mg acetic acid, 2 mL toluene, 50 °C, 18 h, 1 atm O2 | — | >99 | 5 (stable catalytic activity up to four recycles and then a rapid decrease in DFF yield (77%) after 5th recycle) | 364 |
| 17 | WO3HO–VO(salten)–SiO2@Fe3O4 | Dehydration-oxidation of fructose | DFF | (i) 1 mmol fructose, 100 mg catalyst, 80 °C, iso-propanol, 1 h | — | 71 | 5 (minor loss in DFF yields) | 365 |
| (ii) 1 mmol H2O2, 60 °C, 15 h | ||||||||
| 18 | Fe3O4@Nb2O5@Co@Re | Lignin fragmentation | Lignin fragments | 10 mg lignin, 20 mg catalyst, 2.5 mL H2O, 180 °C, 10 bar H2 at RT, 6 h | — | 85 | 6 (stable catalytic activity) | 347 |
| 19 | Ru–Fe3O4–SiO2 (5 wt% Ru) | Hydrogenolysis of cellulose | (EG and PG)l | 300 mg cellulose, 70 mg catalyst, 30 mL water, 255 °C, 60 bar H2 at RT, 50 min | 100 | 19 (EG) | 3 (stable catalytic activity) | 371 |
| 20 (PG) | ||||||||
| 20 | Fe3O4@SBA–SO3H | Hydrolysis of cellulosem | Glucose | 1 g cellulose, 1.5 g catalyst, 15 mL H2O, 150 °C, 3 h | — | 50 | 3 (stable catalytic activity) | 373 |
| 21 | Fe3O4/Mn3O4 | Oxidation of HMF | DFF | 1 mmol HMF, 160 mg catalyst, 7 mL dimethylformamide, 120 °C, O2 (20 mL min−1), 4 h | 99.8 | 82.1 | 6 (small decrease in DFF yields from 82.1% to 78.9%, due to mass loss of the catalyst during recovery/purification steps) | 375 |
| 22 | Fe3O4–CoOx | Oxidation of HMF | FDCA | 70 mg HMF, 100 mg catalyst, 4 mL dimethyl sulfoxide, 0.5 mL of 70% aqueous t-butyl hydroperoxide, 80 °C, 12 h | 97.2 | 68.6 | 2 (stable catalytic activity) | 330 |
| 23 | MnFe2O4 | Oxidation of HMF | FDCA | 1 mmol HMF, 0.015 g cm−3 catalyst, 100 °C, 9 mmol t-butyl hydroperoxide, 5 h | 100 | 85 | 4 (stable catalytic activity) | 376 |
| 24 | NiFe2O4 | Hydrogenation of furfural | Furfuryl alcohol | 2 mmol furfural, 60 mg catalyst, 10 mL of 2-propanol (hydrogen source), 180 °C, 6 h | 99 | 94 | 5 (stable catalytic activity) | 345 |
5.2 Core–shell Fe3O4 based magnetic catalysts
Various coating materials, such as SiO2, γ-Al2O3, Nb2O5–SiO2, organosilica, carbon, polymers, and hydroxyapatite have been used for the preparation of core–shell iron oxide based magnetic catalysts.347,348 Primary applications of these catalysts in biomass valorisation refer to cellulose transformation, lignin fragmentation, and further upgrading of hereby obtained depolymerised (model) compounds.O and/or Nb–O–H), which could be controlled by the niobium content and the applied preparation route. A facile green process was developed for the immobilisation of Pd NPs on core–shell magnetic Fe3O4@carbon microspheres without using any reductant and capping reagents.352 The resulting catalyst showed a high activity and good stability for the oxidation of HMF into FDCA. Several reaction variables, such as temperature, solvent, and base amount were optimised to achieve a 86.7% yield of FDCA, which slightly decreased to 83.7% after five recycles of the catalyst (entry 5, Table 4).
Functionalised Ru-based magnetic catalysts were developed by combining propylamine functionalised SiO2 coated magnetic Fe3O4 NPs with grafted cationic Ru.353 The developed magnetic catalysts are able to convert cellulose into various important platform molecules, including glycerol. The catalyst exhibited 67.3% and 44.4% of cellulose conversion and glycerol yield, respectively, with a stable catalytic performance for at least two recycles (entry 6, Table 4). This is a key advantage over Nb@AlF3, a typical diluted magnetic oxide, which also exhibited good performances in cellulose valorisation.354 A similar type of Ru-based magnetic catalysts also demonstrated high efficiencies for the oxidation of levulinic acid to succinic acid (conversion = 59–79%, selectivity = 96–98%)355 as well as for the cascade conversion of glucose to succinic acid (87.5% yield of succinic acid at nearly complete conversion of glucose).356
A novel “inside-to-outside” synthesis strategy was developed for the synthesis of bifunctional magnetic Ru–SO3H nanoreactors.357 Ru NPs were stabilised on yolk–shell nanoarchitectures composed of a Fe3O4 core and a sulfoacid-containing mesoporous organosilica (PMO) shell (Fig. 41a). The obtained magnetic nanoreactors exhibited various attractive properties, including a highly porous structure with uniform mesopores (∼3.8 nm), superior BET surface areas (>350 m2 g−1), high dispersion of Ru NPs, and enhanced magnetisation. The participation of both Ru NPs and SO3H moieties in catalytic reactions was confirmed by a one-pot conversion of cellulose to isosorbide (Fig. 41b). About 58% yield of isosorbide was achieved at optimised reaction conditions, which however considerably decreased with the repeated use of the catalyst (entry 7, Table 4). Yang et al.358 developed several magnetic base catalysts, composed of functionalised SiO2-coated magnetic Fe3O4 NPs and various organic bases. The resulting magnetic catalysts effectively catalysed the isomerization of glucose to fructose with reasonable good yields in water medium. A 1,2-enediol intermediate mechanism was proposed, which involves the removal of a proton at the C-2 position of glucose by the magnetic base catalyst. The catalytic performance of phosphotungstic acid (PHA) immobilised on a core–shell Fe3O4@SiO2 magnetic catalyst was investigated for the direct synthesis of HMF from glucose.359 The obtained catalytic activity of Fe3O4@SiO2@PHA was comparable to that of a homogeneous PHA catalyst. About 30.4% yield of HMF was obtained at optimised reaction conditions. However, a considerable decrease in HMF yields was noticed after five recycles of the catalyst, due to leaching of acidic species (entry 8, Table 4). Etherification of fructose and HMF to 5-ethoxymethylfurfural (EMF) was investigated using a phosphotungstic acid immobilised core–shell Fe3O4@SiO2 catalyst.46 The resulting catalyst showed excellent activity with 83.2% and 54.8% yields of EMF from etherification of HMF and fructose, respectively. Only a small decrease in EMF yields from 83.2% to 78.8% was found after six recycles of the catalyst (entry 9, Table 4).
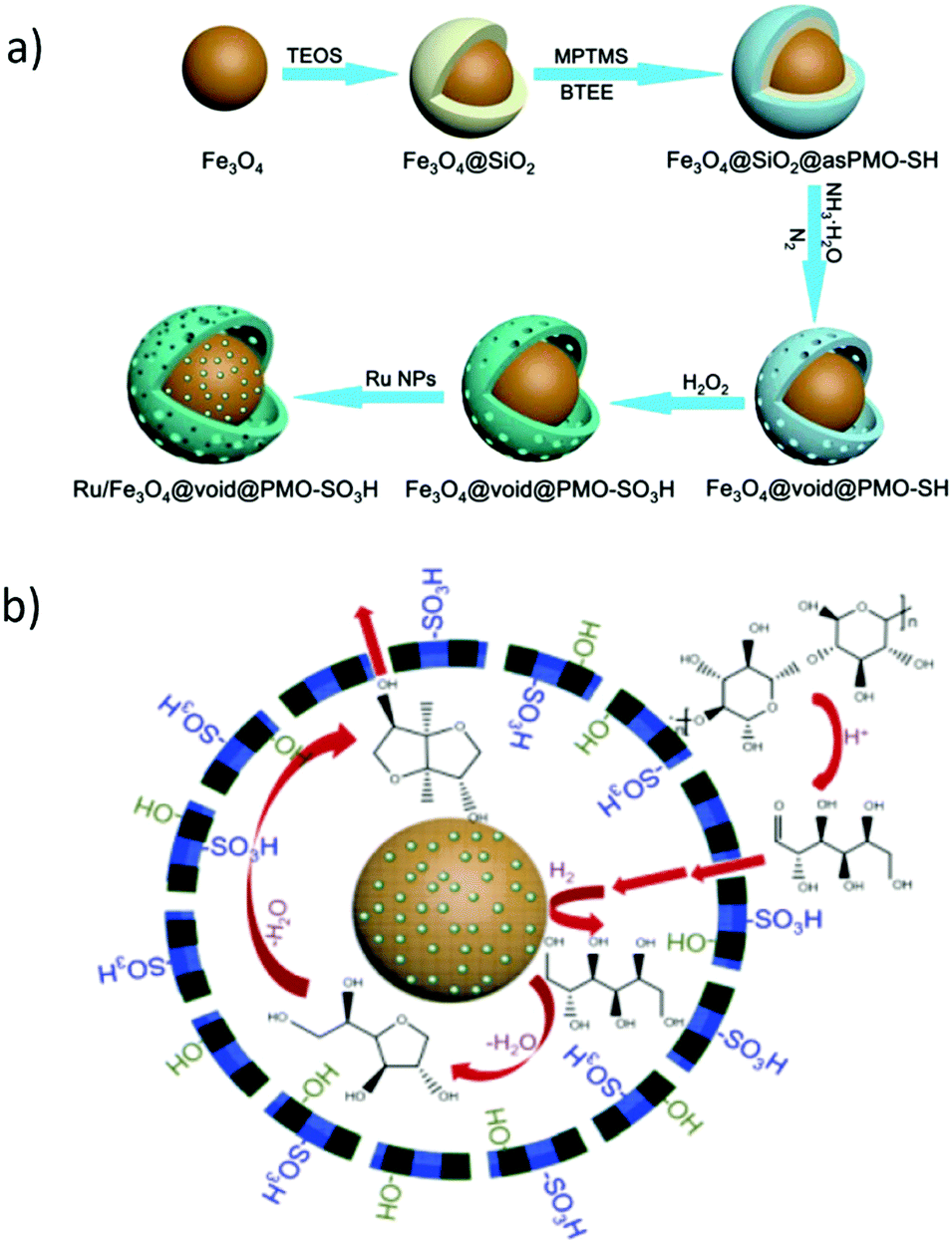 | ||
| Fig. 41 (a) Detailed synthesis strategy for magnetic Ru–SO3H nanoreactor and (b) its application in one-pot conversion of cellulose to isosorbide. Reproduced with permission from ref. 357. Copyright (2018) Royal Society of Chemistry. | ||
The selective dehydration of xylose to furfural was studied using a bifunctional magnetic catalyst composed of an immobilised acid ionic liquid of metal Al substituted onto the surface of Fe3O4@γ-Al2O3.360 A 67.5% yield of furfural with 97.3% xylose conversion was obtained, attributed to abundant amounts of both Brønsted and Lewis acid sites in the developed catalyst. Reusability studies revealed a considerable decrease in furfural yields from 67.5% to 58.7% after five recycles of the catalyst, due to accumulation of reaction substrates on the catalytic active sites (entry 10, Table 4). The selective hydrogenation of furfural to furfuryl alcohol was studied using a magnetic Fe(NiFe)O4@SiO2 catalyst, synthesised by a facile co-precipitation method.2 Good catalytic results were obtained with a more than 93% yield of furfuryl alcohol. A tentative mechanism via a non-hydrogen spillover route was proposed in this work. Highly dispersed nickel species played a pivotal role in the dissociation of H2 into a proton and a hydride, hence enabling the superior catalytic performance of Fe(NiFe)O4@SiO2 for furfural hydrogenation. Polymer stabilised Pt- and Pd-containing Fe3O4 NPs were also studied for the selective hydrogenation of furfural to furfuryl alcohol.361 Two types of polymers, namely polyphenylquinoxaline (PPQ) and polypyridylphenylene (PPP) were used in this work. A higher catalytic activity was observed in the case of hyperbranched PPP-based catalyst compared to that obtained with linear PPQ, which was ascribed to the improved accessibility of catalytic species in PPP-based catalyst. In other words, the longer distance between Pd and Fe3O4 NPs in the case of the PPP-stabilised catalyst endows a higher yield to furfuryl alcohol (98%), with a stable catalytic performance even after five recycles of the 0.3% Pd–Fe3O4–PPP catalyst (entry 11, Table 4). Alternatively, the catalytic transfer hydrogenation of furfural into furfuryl alcohol with alcohols as hydrogen donor was investigated using a hydroxyapatite-encapsulated magnetic γ-Fe2O3 (γ-Fe2O3@HAP) catalyst.362 Results indicated iso-propanol as the best hydrogen donor for the transfer hydrogenation of furfural. A 91.7% yield of furfuryl alcohol was obtained at optimised reaction conditions, which was maintained even after six recycles of the γ-Fe2O3@HAP catalyst (entry 12, Table 4).
A Pd(0) immobilised γ-Fe2O3@HAP catalyst was prepared by the exchange of Pd2+ with Ca2+ in γ-Fe2O3@HAP, followed by the reduction of Pd2+ to Pd(0) using NaBH4.343 The synthesised catalyst was tested for the selective aerobic oxidation of HMF into FDCA. The effect of solvent, base, molar ratio of base to HMF, oxidising agent, catalyst loading, and the reaction temperature was studied for this reaction. About 97% HMF conversion, with a 92.9% FDCA yield was obtained in water after 6 h reaction time at 100 °C. Reusability studies showed a very small decrease in FDCA yields from 92.9% to 90.7% after five recycles of the catalyst, due to mass loss of the catalyst during recovery/purification steps (entry 13, Table 4). Similarly, a Ru exchanged γ-Fe2O3@HAP catalyst was developed for the aerobic oxidation of HMF to DFF.339 A high yield of DFF (89%) at nearly complete conversion of HMF was obtained in this work. In addition, the direct conversion of fructose to DFF was performed, which follows two consecutive steps. The first step involves the dehydration of fructose to HMF (90% yield) over a magnetic acid catalyst (Fe3O4@SiO2–SO3H). Subsequently, the oxidation of HMF to DFF (79% yield based on fructose) is carried out using γ-Fe2O3@HAP-Ru, after removal of Fe3O4@SiO2–SO3H from the reaction mixture with a magnet. Wang et al.363 also studied the aerobic oxidation of HMF to DFF using a Ru(III) catalyst immobilised on a core–shell Fe3O4@SiO2–NH2 catalyst. The developed catalyst showed a good performance with 99.3% conversion of HMF and 86.4% yield of DFF after 4 h. The magnetically recovered catalyst can be reused for at least six times, with a small decrease in DFF yields from 86.4% to 80.8% (entry 14, Table 4). In contrast, Liao et al.48 used non-noble metals i.e., Cu2+ and VO2+ immobilised on amino modified Fe3O4@SiO2 (Fe3O4@SiO2–NH2–Cu2+ and Fe3O4@SiO2–NH2–VO2+) for the production of DFF via HMF oxidation. The reaction is highly dependent on the nature of the solvent. A 98.7% conversion of HMF with 85.5% yield of DFF was obtained over the combination of both catalysts. Only a small decrease in DFF yields from 85.5% to 81.2% was found after four recycles of the catalysts (entry 15, Table 4). In another work, 2,2,6,6-tetramethylpiperidine-N-oxide (TEMPO) immobilised on Fe3O4@SiO2 was studied for the oxidation of HMF into DFF.364 Herein, tert-butyl nitrite and acetic acid were used as additive and co-catalyst, respectively. Quantitative yields of DFF were obtained with excellent chemoselectivity (>99%) and a tentative reaction mechanism was proposed as shown in Fig. 42. Interestingly, a stable catalytic activity was observed up to four recycles of the catalyst, with a rapid decrease in DFF yield (77%) after the 5th recycle (entry 16, Table 4). A bifunctional magnetic catalyst (WO3HO-VO(salten)–SiO2@Fe3O4) was developed for the one-pot conversion of fructose to DFF.365 Two types of functional sites i.e., (i) tungstic acid for fructose dehydration to HMF and (ii) an oxovanadium complex for HMF oxidation to DFF were covalently bonded to the core–shell Fe3O4@SiO2 NPs. The resulting catalyst demonstrated good activity in the conversion of fructose to DFF (71% yield), with a minor loss in catalytic activity after five recycles (entry 17, Table 4).
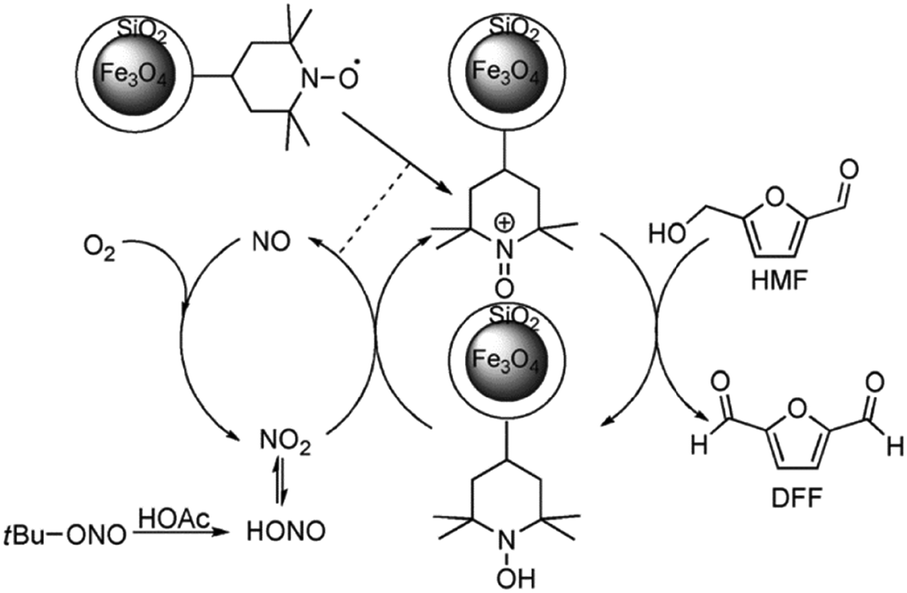 | ||
| Fig. 42 Proposed reaction pathway for the aerobic oxidation of 5-hydroxymethylfurfural (HMF) into 2,5-diformylfuran (DFF) using a Fe3O4@SiO2-TEMPO catalyst. Reproduced with permission from ref. 364. Copyright (2014) John Wiley and Sons. | ||
5.3 Non-core–shell iron-based magnetic catalysts
In this section, applications of non-core–shell iron-based magnetic catalysts, such as Fe3O4 supported metals, mixed oxides, spinel ferrites, and nanoalloys are discussed within the context of biomass conversion.An efficient catalytic protocol for the aerobic oxidation of HMF to DFF was developed using a magnetic Fe3O4 supported Mn3O4 catalyst.375 A 82.1% yield of DFF at nearly complete conversion of HMF was obtained at optimised reaction conditions. The magnetic catalyst can be reused for at least six times and only a small decrease in DFF yields from 82.1% to 78.9% was found, due to inevitable mass loss of the catalyst during recovery/purification steps (entry 21, Table 4). A nanosized magnetic Fe3O4–CoOx catalyst also showed a good activity for the oxidation of HMF, but here FDCA is the major product with t-BuOOH as the oxidant.330 Several reaction parameters were optimised and a 68.6% yield of FDCA was obtained after 12 h reaction, which can be maintained for two recycles of the catalyst (entry 22, Table 4). In another work, a spinel structured magnetic MnFe2O4 catalyst was tested for the oxidation of HMF to FDCA using t-BuOOH as the oxidant.376 About 85% yield of FDCA was obtained at 100 °C in 5 h over the spinel MnFe2O4 catalyst, which was much higher than that obtained with Fe3O4–CoOx.330 A remarkable stable catalytic activity was observed even after four recycles of MnFe2O4 catalyst in HMF oxidation (entry 23, Table 4). This high catalytic performance was ascribed to the presence of variable Mn oxidation states in the spinel MnFe2O4 catalyst. Interestingly, a multicomponent ZnFe1.65Ru0.35O4 magnetic catalyst, prepared via a simple alkaline co-precipitation method, was able to selectively produce either DFF or FDCA from aerobic oxidation of HMF under variable reaction conditions.344 Excellent yields of DFF (93.5%) and FDCA (91.2%) were obtained at different optimised reaction conditions. In addition, the one-pot cascade conversion of fructose to DFF or FDCA was also performed. A functionalised reduced graphene oxide (r-GO–SO3H) based magnetic catalyst was tested for the dehydration of fructose to HMF. The catalyst was then recovered from the reaction mixture using an external magnet.344 Subsequent oxidation of the produced HMF towards DFF or FDCA was then performed in the presence of a ZnFe1.65Ru0.35O4 catalyst. He et al.345 studied the catalytic transfer hydrogenation of furfural over a nickel ferrite (NiFe2O4) catalyst using 2-propanol as both the hydrogen source and the solvent. An excellent yield of furfuryl alcohol (94%) was obtained at 180 °C after 6 h reaction and the catalyst was reused for five recycles without loss of catalytic performance (entry 24, Table 4).
Several heterogeneous magnetic biocatalysts (enzyme immobilised magnetic NPs) were found to exhibit good catalytic activities for the production of glycerol carbonate from glycerol and dimethyl carbonate.380,381 The lipase enzyme was covalently attached to magnetic NPs via 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide or glutaraldehyde.382 The developed magnetic biocatalysts showed about 3-fold higher catalytic efficiency (3.52 × 105 h−1 TOF) than the free lipase (1.16 × 105 h−1 TOF), which was explained as a beneficial effect of the lipase immobilisation and the resulting lipase–Fe3O4 interactions.382,383 Similar catalytic performances were also achieved when starting from “crude” glycerol feed extracted during biodiesel synthesis.384 The efficiency of these magnetic biocatalysts has also been proven for the transesterification of oils extracted from soybean, sunflower, rape, corn, olive, and palm.385
5.4 Conclusions of magnetic iron oxide-based catalysts
The reported examples in this section clearly reveal that the facile recovery of functionalised magnetic nanocatalysts using an external magnetic field is a notable advantage over conventional catalysts. This prevents the use of further steps to remove the catalyst from complex reaction mixture, allowing the efficient recyclability in successive cycles. Notwithstanding, magnetic iron oxide based catalysts still suffer from several drawbacks in terms of synthesis and stability.50,372,386,387 For instance, developing a facile one-pot method for large-scale production of functionalised Fe3O4 based materials, especially core–shell catalysts, is a very challenging task. This is because each constituent material contains disparate properties, hence requires different reaction conditions as well as multi-steps for the synthesis of functionalised Fe3O4 catalysts. In addition, the instability/dissolution of magnetic NPs during longer time catalytic reactions as well as the leaching of functional sites under harsh reaction conditions remain the major problems in several biomass upgrading reactions. Although core–shell Fe3O4 based catalysts exhibit good stability, concerns associated with the necessity of complex synthesis processes and high production costs limit their applications in several catalytic applications including biomass upgrading. Alternatively, non-core–shell Fe3O4 catalysts (Fe3O4 supported metals, spinel ferrites, etc.) are easy to prepare and also show good catalytic stability in biomass conversions. Therefore, more research efforts should be undertaken towards developing novel non-core–shell Fe3O4 catalysts with the required catalytic properties, which is of great significance from both scientific and practical viewpoints.6. Conclusions and future prospects
In view of global warming impact on humankind as well as on the biosphere, the use of biomass alternative to fossil fuels for renewable chemicals production is a hot research topic of the 21st century. Biomass is a promising renewable energy that can be derived from various sustainable sources, including lignocellulose, lipids, starch, and aquatic plants. A large number of value-added chemicals and high-energy density fuels can be produced from biomass using appropriate technologies based on biological, thermal, and chemical processes. Among them, chemical processing of biomass (e.g., hydrolysis, fast pyrolysis, hydroprocessing, oxidation, (trans)esterification, amination, dehydration, isomerisation, etc.) has attracted tremendous attention as the resulting products can exhibit relatively equating characteristics to petroleum-based products, driving towards a more sustainable economy and carbon-neutral society.Catalysis plays a central role in bio-refinery chemical processes. Compared to homogenous catalysts, heterogeneous catalysts can provide promising greener routes for efficient biomass upgrading. Handiness of various feasible synthesis methods, non-toxicity, high stability, and efficient recovery/reusability are some of the key benefits of heterogeneous solid catalysts. The development of multifunctional heterogeneous catalysts with ample amounts of acid, base or redox active sites is vital for achieving higher reaction rates, especially in cascade biomass processes. Recent advances in materials science and catalysis have provided several novel strategies for the development of new appealing catalytic materials, such as carbon materials, metal–organic frameworks, solid phase ionic liquids, and magnetic iron oxides. A distinctive feature of these catalytic materials is to accommodate abundant acid–base as well as redox functional species, hence enabling biomass conversion reactions in a one-pot cascade approach.
In this review, the unique catalytic properties of carbon materials, metal–organic frameworks, solid phase ionic liquids, and magnetic iron oxides were highlighted towards catalytic biomass upgrading. Several potential routes developed for the immobilisation of acid–base and redox functional species on particular sites of above-listed catalysts were discussed in detail. Finally, the subsequent effects of functionalisation on one-pot cascade processes as well as on other types of vital reactions that normally take place in bio-refineries were thoroughly discussed. It can be concluded that the distinctive properties of catalysts play an essential role for robust functionalisation, hence improved catalytic activities in biomass valorisation. For instance, both ex situ and in situ functionalisation are possible in the case of carbon materials. The presence of abundant surface defects and residual O-containing groups, for example in activated carbon, graphene, and carbon nanotubes, allows efficient ex situ functionalisation with acid–base and/or redox active phases.35,79,130 On the other hand, the use of biomass-derived molecules (e.g., glycerol and glucose) as catalyst precursors facilitates in situ functionalisation.85,204 In the case of MOFs, organic ligands offer facile routes to introduce acid–base functional molecules,219 while robust porous cavities afford ample room for the encapsulation of metal NPs.225 ILs fundamentally contain various kinds of synergistic interactions, resulting from organic cations and (in)organic anions, which play a crucial role in stabilising immobilised functional species.284 In addition to the benefits of facile recovery/recyclability, the core–shell or polymer encapsulated magnetic iron oxides allow efficient ionic exchange with the active transition metals, attributed to the presence of abundant surface exchangeable species (e.g., –OH and Ca2+).47,339,343 It must be noted here that most of the biomass upgrading reactions reported in this review are performed on the laboratory scale. This does not necessarily imply industrial relevance, but will provide useful information towards developing large-scale biorefineries in terms of catalyst selection, reaction conditions needed, tailoring activity/selectivity, and potentials to regenerate the catalyst activity.
Although tremendous progress has been achieved towards development of efficient functionalised heterogeneous catalysts for biomass valorisation, further advances are still necessary in view of sustainable bio-refinery industry. Hence, to guide future research in the respective fields we would like to propose an interesting ‘PYSSVR’ concept, which means P – production cost, Y – yield, S – stability, S – selectivity, V – versatility, and R – reusability (Fig. 43).
 | ||
| Fig. 43 Promising features of functionalised heterogeneous catalysts for a more sustainable biomass valorisation. | ||
(1) Production cost: efforts should be made to minimise catalyst production costs. Especially, high synthesis costs of CNTs, graphene, MOFs, and ILs limits their practical applications in biomass upgrading. A promising solution to tackle this problem is the use of low-cost and renewable precursors. For instance, a variety of economical carbon-based catalysts with abundant functional sites can be developed via carbonisation of biomass substrates as discussed in Section 2.8.82–84 The subsequent use of these catalysts in biomass upgrading can greatly improve process economy and sustainability. Moreover, one-pot synthesis of functionalised heterogeneous catalysts via in situ functionalisation as well as the utilisation of non-noble metals can help to reduce catalyst production costs.
(2) Yield: it is also crucial to improve production yields of functionalised heterogeneous catalysts, while maintaining their unique properties. In this context, the selected preparation method must be simple, cost-effective, eco-friendly (i.e. avoiding hazardous organic reagents/solvents) and able to scaled-up. Compared with gas- and solid-phase synthesis methods, efforts needs to be strengthened towards developing wet chemical preparation routes as they can be efficiently modified for economical mass production of solid catalysts.
(3) Stability: hydrothermal stability of functionalised catalysts under harsh aqueous conditions is very important, because water is the preferred reaction medium for many bio-refinery processes. This review emphasised several potential strategies to improve hydrothermal stability of the catalysts, including doping of heteroatoms (e.g., nitrogen) in carbon,186 developing carbon-silica composites,160–162 selective encapsulation/anchoring of active phases in MOFs,245,246 incorporation of ILs into the polymeric substrates,313 and designing robust core–shell Fe3O4@SiO2 catalysts.47
(4) Selectivity: tailoring the selectivity of functionalised catalysts towards a particular product in biomass upgrading is essential to control the formation of unwanted by-products as well as to minimise energy-intensive steps required for the separation/purification of reaction substrates. This can be achieved by selectively anchoring functional species on particular sites of heterogeneous catalysts, which may direct the specificity of interaction with the biomass molecules, hence preferred reaction pathway and formation of desirable products.
(5) Versatility: it is also vital to develop functionalised heterogeneous catalysts, with versatile applications in biomass upgrading. Several examples of this kind are highlighted in this review, including sulfonated silica–carbon nanocomposites (cellulose hydrolysis160 and fructose ethanolysis162), SO3H functionalised MIL-101(Cr) MOF (fructose-to-HMF220 and furfuryl alcohol-to-ethyl levulinate249), and Fe3O4@SiO2–SO3H (glucose-to-HMF340 and cellulose-to-reducing sugar350). For an improved bio-based economy, developing a more number of versatile catalysts with multiple active sites is an urgent task.
(6) Reusability: above all, efficient reusability of the catalysts is of paramount importance in order to avoid post-reaction steps, minimise waste disposal and energy consumption, as well as maximise the productivity. Owing to excellent paramagnetic properties, magnetic iron oxide based catalysts can be efficiently recovered from reaction mixtures using a magnet without affecting their physicochemical properties. Hence, efforts should be directed towards developing composite catalysts consisting of magnetic iron oxides with functionalised carbon materials, MOFs, or ILs. This appealing approach may combine the unique catalytic properties of the respective components, with the magnetic properties of iron oxides, hence avoiding recycling concerns associated with the conventional catalysts. This route may further lead to significantly improved catalytic activities in biomass upgrading, due to availability of abundant functional active sites.
More stimulating research efforts are therefore needed to develop state-of-the-art functionalised heterogeneous catalysts, with necessary functional active sites and various key features as listed in Fig. 43 for a more sustainable biomass valorisation. In this context, the present comprehensive review may provide a systematic background for further advances towards economic mass production of versatile functionalised heterogeneous catalysts for a viable bio-refinery industry.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement number-747968 (project acronym: CATLIGCAR). SVDB acknowledges the internal funds of KU Leuven for a postdoctoral mandate (PDM). VIP kindly acknowledge UEFISCDI for financial support (projects PN-III-P4-ID-PCE-2016-0146 and 32PCCDI-2018). BFS would like to thank BIOFACT Excellence of Science project as well as SPICEY (VLAIO SBO Flemish Government) project.References
- L. T. Mika, E. Cséfalvay and Á. Németh, Chem. Rev., 2018, 118, 505–613 CrossRef PubMed
.
- A. Halilu, T. H. Ali, A. Y. Atta, P. Sudarsanam, S. K. Bhargava and S. B. A. Hamid, Energy Fuels, 2016, 30, 2216–2226 CrossRef
- A. Wittstock and M. Bäumer, Acc. Chem. Res., 2014, 47, 731–739 CrossRef PubMed
- W. Schutyser, T. Renders, S. Van den Bosch, S. F. Koelewijn, G. T. Beckham and B. F. Sels, Chem. Soc. Rev., 2018, 47, 852–908 RSC
- B. Mallesham, P. Sudarsanam, G. Raju and B. M. Reddy, Green Chem., 2013, 15, 478–489 RSC
- K. Sordakis, C. Tang, L. K. Vogt, H. Junge, P. J. Dyson, M. Beller and G. Laurenczy, Chem. Rev., 2018, 118, 372–433 CrossRef PubMed
- G. P. Peters, C. Le Quéré, R. M. Andrew, J. G. Canadell, P. Friedlingstein, T. Ilyina, R. B. Jackson, F. Joos, J. I. Korsbakken, G. A. McKinley, S. Sitch and P. Tans, Nat. Clim. Change, 2017, 7, 848–850 CrossRef
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC
- http://www.cop21.gouv.fr/ .
- https://ec.europa.eu/clima/policies/international/paris_protocol/energy_en .
- S. Van de Vyver, J. Geboers, P. A. Jacobs and B. F. Sels, ChemCatChem, 2011, 3, 82–94 CrossRef
- M. Dusselier, M. Mascal and B. F. Sels, Top. Curr. Chem., 2014, 353, 1–40 CrossRef PubMed
- J. A. Geboers, S. Van de Vyver, R. Ooms, B. Op De Beeck, P. A. Jacobs and B. F. Sels, Catal. Sci. Technol., 2011, 1, 714–726 RSC
- D. M. Alonso, J. Q. Bond and J. A. Dumesic, Green Chem., 2010, 12, 1493–1513 RSC
- P. S. Reddy, P. Sudarsanam, B. Mallesham, G. Raju and B. M. Reddy, J. Ind. Eng. Chem., 2011, 17, 377–381 CrossRef
- S. Santoro, F. Ferlin, L. Luciani, L. Ackermann and L. Vaccaro, Green Chem., 2017, 19, 1601–1612 RSC
- X. Zhang, K. Rajagopalan, H. Lei, R. Ruan and B. K. Sharma, Sustainable Energy Fuels, 2017, 1, 1664–1699 RSC
- W. Y. Hernandez, J. Lauwaert, P. Van Der Voort and A. Verberckmoes, Green Chem., 2017, 19, 5269–5302 RSC
- W. Liu, W. Li, H. Jiang and H. Yu, Chem. Rev., 2017, 117, 6367–6398 CrossRef PubMed
- P. Lamers, R. Hoefnagels, M. Junginger, C. Hamelinck and A. Faaij, GCB Bioenergy, 2015, 7, 618–634 CrossRef
- X. Lian, Y. Xue, Z. Zhao, G. Xu, S. Han and H. Yu, Int. J. Energy Res., 2017, 41, 1798–1816 CrossRef
- T. Ennaert, J. Van Aelst, J. Dijkmans, R. De Clercq, W. Schutyser, M. Dusselier, D. Verboekend and B. F. Sels, Chem. Soc. Rev., 2016, 45, 584–611 RSC
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef PubMed
- A. Aronne, M. Di Serio, R. Vitiello, N. J. Clayden, L. Minieri, C. Imparato, A. Piccolo, P. Pernice, P. Carniti and A. Gervasini, J. Phys. Chem. C, 2017, 121, 17378–17389 CrossRef
- T. Kitanosono, K. Masuda, P. Xu and S. Kobayashi, Chem. Rev., 2018, 118, 679–746 CrossRef PubMed
- F. Yang, D. Deng, X. Pan, Q. Fu and X. Bao, Natl. Sci. Rev., 2015, 2, 183–201 CrossRef
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef PubMed
- A. Kokel, C. Schäfer and B. Török, Green Chem., 2017, 19, 3729–3751 RSC
- Y. Lin and G. W. Huber, Energy Environ. Sci., 2009, 2, 68–80 RSC
- B. Karimi, F. Mansouri and H. M. Mirzaei, ChemCatChem, 2015, 7, 1736–1789 CrossRef
- M. J. Climent, A. Corma, S. Iborra and M. J. Sabater, ACS Catal., 2014, 4, 870–891 CrossRef
- A. Corma, S. B. A. Hamid, S. Iborra and A. Velty, ChemSusChem, 2008, 1, 85–90 CrossRef PubMed
- M. Tao, D. Zhang, H. Guan, G. Huang and X. Wang, Sci. Rep., 2016, 6, 29840 CrossRef PubMed
- S. Song, L. Di, G. Wu, W. Dai, N. Guan and L. Li, Appl. Catal., B, 2017, 205, 393–403 CrossRef
- E. Lam and J. H. T. Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef
- A. Herbst and C. Janiak, CrystEngComm, 2017, 19, 4092–4117 RSC
- A. Herbst and C. Janiak, New J. Chem., 2016, 40, 7958–7967 RSC
- A. A. Ibrahim, A. Lin, F. Zhang, K. M. AbouZeid and M. S. El-Shall, ChemCatChem, 2017, 9, 469–480 CrossRef
- B. Xin and J. Hao, Chem. Soc. Rev., 2014, 43, 7171–7187 RSC
- S. Zhang, J. Zhang, Y. Zhang and Y. Deng, Chem. Rev., 2017, 117, 6755–6833 CrossRef PubMed
- H. Li, P. S. Bhadury, B. Song and S. Yang, RSC Adv., 2012, 2, 12525–12551 RSC
- Q. Wang, W. Hou, S. Li, J. Xie, J. Li, Y. Zhou and J. Wang, Green Chem., 2017, 19, 3820–3830 RSC
- K. L. Luska, P. Migowski and W. Leitner, Green Chem., 2015, 17, 3195–3206 RSC
- P. Ferrini and R. Rinaldi, Angew. Chem., Int. Ed., 2014, 53, 8634–8639 CrossRef PubMed
- Q. Song, F. Wang, J. Cai, Y. Wang, J. Zhang, W. Yu and J. Xu, Energy Environ. Sci., 2013, 6, 994–1007 RSC
- S. Wang, Z. Zhang, B. Liu and J. Li, Catal. Sci. Technol., 2013, 2104–2112 RSC
- Y. Chiang, S. Dutta, C. Chen, Y. Huang, K. Lin, J. C. S. Wu, N. Suzuki, Y. Yamauchi and K. C. W. Wu, ChemSusChem, 2015, 8, 789–794 CrossRef PubMed
- L. Liao, Y. Liu, Z. Li, J. Zhuang, Y. Zhou and S. Chen, RSC Adv., 2016, 6, 94976–94988 RSC
- Z. Zhang, J. Song and B. Han, Chem. Rev., 2017, 117, 6834–6880 CrossRef PubMed
- B. Liu and Z. Zhang, ACS Catal., 2016, 6, 326–338 CrossRef
- H. Li, Z. Fang, R. L. Smith Jr. and S. Yang, Prog. Energy Combust. Sci., 2016, 55, 98–194 CrossRef
- V. Trombettoni, D. Lanari, P. Prinsen, R. Luque, A. Marrocchi and L. Vaccaro, Prog. Energy Combust. Sci., 2018, 65, 136–162 CrossRef
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef PubMed
- A. Villa, N. Dimitratos, C. E. Chan-Thaw, C. Hammond, G. M. Veith, D. Wang, M. Manzoli, L. Prati and G. J. Hutchings, Chem. Soc. Rev., 2016, 45, 4953–4994 RSC
- D. N. Durgasri, T. Vinodkumar, P. Sudarsanam and B. M. Reddy, Catal. Lett., 2014, 144, 971–979 CrossRef
- M. H. Amin, S. Putla, S. Bee Abd Hamid and S. K. Bhargava, Appl. Catal., A, 2015, 492, 160–168 CrossRef
- B. Ren, P. Sudarsanam, A. E. Kandjani, B. Hillary, M. H. Amin, S. K. Bhargava and L. A. Jones, Electroanalysis, 2018, 30, 928–936 CrossRef
- J. C. Yang, M. W. Small, R. V. Grieshaber and R. G. Nuzzo, Chem. Soc. Rev., 2012, 41, 8179–8194 RSC
- Q. Wu, L. Yang, X. Wang and Z. Hu, Acc. Chem. Res., 2017, 50, 435–444 CrossRef PubMed
- A. Lu, G. Hao, Q. Sun, X. Zhang and W. Li, Macromol. Chem. Phys., 2012, 213, 1107–1131 CrossRef
- M. Titirici, R. J. White, N. Brun, V. L. Budarin, D. S. Su, F. del Monte, J. H. Clark and M. J. MacLachlan, Chem. Soc. Rev., 2015, 44, 250–290 RSC
- W. Xin and Y. Song, RSC Adv., 2015, 5, 83239–83285 RSC
- E. Pérez-Mayoral, V. Calvino-Casilda and E. Soriano, Catal. Sci. Technol., 2016, 6, 1265–1291 RSC
- S. Zhu, J. Wang and W. Fan, Catal. Sci. Technol., 2015, 5, 3845–3858 RSC
- V. Georgakilas, J. A. Perman, J. Tucek and R. Zboril, Chem. Rev., 2015, 115, 4744–4822 CrossRef PubMed
- P. Zhang, H. Zhu and S. Dai, ChemCatChem, 2015, 7, 2788–2805 CrossRef
- D. Yu, E. Nagelli, F. Du and L. Dai, J. Phys. Chem. Lett., 2010, 1, 2165–2173 CrossRef
- J. L. Figueiredo, J. Mater. Chem. A, 2013, 1, 9351–9364 RSC
- R. Zhong and B. F. Sels, Appl. Catal., B, 2018, 236, 518–545 CrossRef
- S. Van de Vyver, J. Geboers, M. Dusselier, H. Schepers, T. Vosch, L. Zhang, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, ChemSusChem, 2010, 3, 698–701 CrossRef PubMed
- L. Liu, Y. Zhu, M. Su and Z. Yuan, ChemCatChem, 2015, 7, 2765–2787 CrossRef
- H. Yang, R. Nie, W. Xia, X. Yu, D. Jin, X. Lu, D. Zhou and Q. Xia, Green Chem., 2017, 19, 5714–5722 RSC
- P. Chen, F. Yang, A. Kostka and W. Xia, ACS Catal., 2014, 4, 1478–1486 CrossRef
- M. A. Patel, F. Luo, M. R. Khoshi, E. Rabie, Q. Zhang, C. R. Flach, R. Mendelsohn, E. Garfunkel, M. Szostak and H. He, ACS Nano, 2016, 10, 2305–2315 CrossRef PubMed
- J. M. R. Gallo, R. Alamillo and J. A. Dumesic, J. Mol. Catal. A: Chem., 2016, 422, 13–17 CrossRef
- J. Bedia, J. M. Rosas, J. Márquez, J. Rodríguez-Mirasol and T. Cordero, Carbon, 2009, 47, 286–294 CrossRef
- J. Tessonnier, A. Villa, O. Majoulet, D. S. Su and R. Schlögl, Angew. Chem., Int. Ed., 2009, 48, 6543–6546 CrossRef PubMed
- H. Liu, J. Chen, L. Chen, Y. Xu, X. Guo and D. Fang, ACS Sustainable Chem. Eng., 2016, 4, 3140–3150 CrossRef
- Y. Wang, Z. Rong, Y. Wang, T. Wang, Q. Du, Y. Wang and J. Qu, ACS Sustainable Chem. Eng., 2017, 5, 1538–1548 CrossRef
- F. de Clippel, M. Dusselier, S. Van de Vyver, L. Peng, P. A. Jacobs and B. F. Sels, Green Chem., 2013, 15, 1398–1430 RSC
- F. de Clippel, M. Dusselier, R. Van Rompaey, P. Vanelderen, J. Dijkmans, E. Makshina, L. Giebeler, S. Oswald, G. V. Baron, J. F. M. Denayer, P. P. Pescarmona, P. A. Jacobs and B. F. Sels, J. Am. Chem. Soc., 2012, 134, 10089–10101 CrossRef PubMed
- Q. Ma, Y. Yu, M. Sindoro, A. G. Fane, R. Wang and H. Zhang, Adv. Mater., 2017, 29, 1605361 CrossRef PubMed
- S. Kang, J. Ye, Y. Zhang and J. Chang, RSC Adv., 2013, 3, 7360–7366 RSC
- J. Laohapornchaiphan, C. B. Smith and S. M. Smith, Chem. – Asian J., 2017, 12, 3178–3186 CrossRef PubMed
- B. L. A. Prabhavathi Devi, K. V. Lakshmi, K. N. Gangadhar, R. B. N. Prasad, P. S. Sai Prasad, B. Jagannadh, P. P. Kundu, G. Kumari and C. Narayana, ChemistrySelect, 2017, 2, 1925–1931 CrossRef
-
P. Serp and B. Machado, Nanostructured Carbon Materials for Catalysis, RSC Catalysis Series No. 23, The Royal Society of Chemistry, UK, 2015, ch. 1, pp. 1–45 Search PubMed
- B. M. Upton and A. M. Kasko, Chem. Rev., 2016, 116, 2275–2306 CrossRef PubMed
- M. S. Singhvi, S. Chaudhari and D. V. Gokhale, RSC Adv., 2014, 4, 8271–8277 RSC
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef PubMed
- R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx and B. M. Weckhuysen, Angew. Chem., Int. Ed., 2016, 55, 8164–8215 CrossRef PubMed
- S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S. F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjan and B. F. Sels, Energy Environ. Sci., 2015, 8, 1748–1763 RSC
- W. Schutyser, S. Van den Bosch, T. Renders, T. De Boe, S. F. Koelewijn, A. Dewaele, T. Ennaert, O. Verkinderen, B. Goderis, C. M. Courtin and B. F. Sels, Green Chem., 2015, 17, 5035–5045 RSC
- T. Renders, S. Van den Bosch, T. Vangeel, T. Ennaert, S. Koelewijn, G. Van den Bossche, C. M. Courtin, W. Schutyser and B. F. Sels, ACS Sustainable Chem. Eng., 2016, 4, 6894–6904 CrossRef
- T. Renders, W. Schutyser, S. Van den Bosch, S. Koelewijn, T. Vangeel, C. M. Courtin and B. F. Sels, ACS Catal., 2016, 6, 2055–2066 CrossRef
- S. Van den Bosch, W. Schutyser, S. F. Koelewijn, T. Renders, C. M. Courtin and B. F. Sels, Chem. Commun., 2015, 51, 13158–13161 RSC
- E. M. Anderson, M. L. Stone, R. Katahira, M. Reed, G. T. Beckham and Y. Román-Leshkov, Joule, 2017, 1, 613–622 CrossRef
- I. Kumaniaev, E. Subbotina, J. Sävmarker, M. Larhed, M. V. Galkin and J. S. M. Samec, Green Chem., 2017, 19, 5767–5771 RSC
- B. Op de Beeck, M. Dusselier, J. Geboers, J. Holsbeek, E. Morré, S. Oswald, L. Giebeler and B. F. Sels, Energy Environ. Sci., 2015, 8, 230–240 RSC
- A. Deneyer, T. Ennaert, G. Cavents, J. Dijkmans, J. Vanneste, C. M. Courtin, M. Dusselier and B. F. Sels, Green Chem., 2016, 18, 5594–5606 RSC
- Y. Luo, J. Yi, D. Tong and C. Hu, Green Chem., 2016, 18, 848–857 RSC
- W. Daengprasert, P. Boonnoun, N. Laosiripojana, M. Goto and A. Shotipruk, Ind. Eng. Chem. Res., 2011, 50, 7903–7910 CrossRef
- S. Shen, C. Wang, B. Cai, H. Li, Y. Han, T. Wang and H. Qin, Fuel, 2013, 113, 644–649 CrossRef
- A. Shrotri, H. Kobayashi, A. Tanksale, A. Fukuoka and J. Beltramini, ChemCatChem, 2014, 6, 1349–1356 Search PubMed
- J. Y. Park, M. A. Kim, S. J. Lee, J. Jung, H. M. Jang, P. P. Upare, Y. K. Hwang, J. Chang and J. K. Park, J. Mater. Sci., 2015, 50, 334–343 CrossRef
- S. G. Wettstein, J. Q. Bond, D. M. Alonso, H. N. Pham, A. K. Datye and J. A. Dumesic, Appl. Catal., B, 2012, 117–118, 321–329 CrossRef
- C. Li, G. Xu, Y. Zhai, X. Liu, Y. Ma and Y. Zhang, Fuel, 2017, 203, 23–31 CrossRef
- E. Sairanen, R. Karinen and J. Lehtonen, Catal. Lett., 2014, 144, 1839–1850 CrossRef
- X. Jin, P. S. Thapa, B. Subramaniam and R. V. Chaudhari, ACS Sustainable Chem. Eng., 2016, 4, 6037–6047 CrossRef
- Y. T. Kim, J. A. Dumesic and G. W. Huber, J. Catal., 2013, 304, 72–85 CrossRef
- Z. Gao, C. Li, G. Fan, L. Yang and F. Li, Appl. Catal., B, 2018, 226, 523–533 CrossRef
- J. Li, J. Liu, H. Zhou and Y. Fu, ChemSusChem, 2016, 9, 1339–1347 CrossRef PubMed
- M. Rivière, N. Perret, D. Delcroix, A. Cabiac, C. Pinel and M. Besson, ACS Sustainable Chem. Eng., 2018, 6, 4076–4085 CrossRef
- O. O. Ayodele, F. A. Dawodu, D. Yan, H. Dong, J. Xin and S. Zhang, ChemistrySelect, 2017, 2, 4219–4225 CrossRef
- Y. Zhai, C. Li, G. Xu, Y. Ma, X. Liu and Y. Zhang, Green Chem., 2017, 19, 1895–1903 RSC
- J. Ji, H. Guo, C. Li, Z. Qi, B. Zhang, T. Dai, M. Jiang, C. Ren, A. Wang and T. Zhang, ChemCatChem, 2018, 10, 415–421 CrossRef
- H. Zeng, D. Cao, Z. Qiu and C. J. Li, Angew. Chem., Int. Ed., 2018, 57, 3752–3757 CrossRef PubMed
- H. Lai, Y. Chou, M. Lee and K. A. Lin, Chem. Eng. J., 2018, 332, 717–726 CrossRef
- A. Bjelić, M. Grilc and B. Likozar, Chem. Eng. J., 2018, 333, 240–259 CrossRef
- J. Engelhardt, P. Lyu, P. Nachtigall, F. Schüth and Á. M. García, ChemCatChem, 2017, 9, 1985–1991 CrossRef
- B. Mallesham, P. Sudarsanam and B. M. Reddy, Catal. Sci. Technol., 2014, 4, 803–813 RSC
- P. Sudarsanam, B. Mallesham, A. N. Prasad, P. S. Reddy and B. M. Reddy, Fuel Process. Technol., 2013, 106, 539–545 CrossRef
- X. Fu, J. Chen, X. Song, Y. Zhang, Y. Zhu, J. Yang and C. Zhang, J. Am. Oil Chem. Soc., 2015, 92, 495–502 CrossRef
- P. Chanchaochai, P. Boonnoun, N. Laosiripojana, M. Goto, B. Jongsomjit, J. Panpranot, O. Mekasuwandumrong and A. Shotipruk, Chem. Eng. Commun., 2013, 200, 1542–1552 CrossRef
- B. Mallesham, P. Sudarsanam and B. M. Reddy, Ind. Eng. Chem. Res., 2014, 53, 18775–18785 CrossRef
- S. B. A. Hamid, N. A. Daud, D. D. Suppiah, W. A. Yehya, P. Sudarsanam and S. K. Bhargava, Polyhedron, 2016, 120, 154–161 CrossRef
- J. Dou, B. Zhang, H. Liu, J. Hong, S. Yin, Y. Huang and R. Xu, Appl. Catal., B, 2016, 180, 78–85 CrossRef
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef PubMed
- E. Furimsky, Ind. Eng. Chem. Res., 2017, 56, 11359–11371 CrossRef
- B. Qiu, M. Xing and J. Zhang, Chem. Soc. Rev., 2018, 47, 2165–2216 RSC
- C. Su and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275–2285 CrossRef PubMed
- V. K. Das, Z. B. Shifrina and L. M. Bronstein, J. Mater. Chem. A, 2017, 5, 25131–25143 RSC
- P. P. Upare, J. Yoon, M. Y. Kim, H. Kang, D. W. Hwang, Y. K. Hwang, H. H. Kung and J. Chang, Green Chem., 2013, 15, 2935–2943 RSC
- H. Han, H. Zhao, Y. Liu, Z. Li, J. Song, W. Chu and Z. Sun, RSC Adv., 2017, 7, 3790–3795 RSC
- P. A. Russo, S. Lima, V. Rebuttini, M. Pillinger, M. Willinger, N. Pinna and A. A. Valente, RSC Adv., 2013, 3, 2595–2603 RSC
- C. Xiao, T. Goh, Z. Qi, S. Goes, K. Brashler, C. Perez and W. Huang, ACS Catal., 2016, 6, 593–599 CrossRef
- T. Zhang, Y. Ge, X. Wang, J. Chen, X. Huang and Y. Liao, ACS Omega, 2017, 2, 3228–3240 CrossRef
- A. V. Nakhate and G. D. Yadav, ACS Sustainable Chem. Eng., 2016, 4, 1963–1973 CrossRef
- H. Wang, Y. Wang, T. Deng, C. Chen, Y. Zhu and X. Hou, Catal. Commun., 2015, 59, 127–130 CrossRef
- X. Jin, L. Dang, J. Lohrman, B. Subramaniam, S. Ren and R. V. Chaudhari, ACS Nano, 2013, 7, 1309–1316 CrossRef PubMed
- J. Cheng, Y. Qiu, J. Zhang, R. Huang, W. Yang and Z. Fan, Bioresour. Technol., 2017, 244, 569–574 CrossRef PubMed
- X. Guo, J. Guan, B. Li, X. Wang, X. Mu and H. Liu, Sci. Rep., 2015, 5, 16451 CrossRef PubMed
- M. Melchionna, S. Marchesan, M. Prato and P. Fornasiero, Catal. Sci. Technol., 2015, 5, 3859–3875 RSC
- S. A. Miners, G. A. Rance and A. N. Khlobystov, Chem. Soc. Rev., 2016, 45, 4727–4746 RSC
- Y. Guo and J. Chen, ChemPlusChem, 2016, 80, 1760–1768 CrossRef
- J. Chen, J. Zhong, Y. Guo and L. Chen, RSC Adv., 2015, 5, 5933–5940 RSC
- C. Zhou, W. Deng, X. Wan, Q. Zhang, Y. Yang and Y. Wang, ChemCatChem, 2015, 7, 2853–2863 CrossRef
- D. S. Park, D. Yun, T. Y. Kim, J. Baek, Y. S. Yun and J. Yi, ChemSusChem, 2013, 6, 2281–2289 CrossRef PubMed
- L. P. P. Sukma and E. W. Qian, Environ. Prog. Sustainable Energy, 2018, 37, 850–860 CrossRef
- K. N. Sorokina, O. P. Taran, T. B. Medvedeva, Y. V. Samoylova, A. V. Piligaev and V. N. Parmon, ChemSusChem, 2017, 10, 562–574 CrossRef PubMed
- S. Zhang, Z. Zheng, C. Zhao and L. Zhang, ACS Omega, 2017, 2, 6123–6130 CrossRef
- D. Song, S. An, B. Lu, Y. Guo and J. Leng, Appl. Catal., B, 2015, 179, 445–457 CrossRef
- J. Lee, J. Jung, H. J. Kim, T. Kim, K. Kim and E. E. Kwon, ACS Sustainable Chem. Eng., 2017, 5, 7433–7438 CrossRef
- L. Peng, A. Philippaerts, X. Ke, J. Van Noyen, F. De Clippel, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, Catal. Today, 2010, 150, 140–146 CrossRef
- U. Díaz, D. Brunel and A. Corma, Chem. Soc. Rev., 2013, 42, 4083–4097 RSC
- R. Liu, Y. Shi, Y. Wan, Y. Meng, F. Zhang, D. Gu, Z. Chen, B. Tu and D. Zhao, J. Am. Chem. Soc., 2006, 128, 11652–11662 CrossRef PubMed
- J. Xu, A. Wang, X. Wang, D. Su and T. Zhang, Nano Res., 2011, 4, 50–60 CrossRef
- P. Valle-Vigón, M. Sevilla and A. B. Fuertes, Appl. Surf. Sci., 2012, 261, 574–583 CrossRef
- P. A. Russo, M. M. Antunes, P. Neves, P. V. Wiper, E. Fazio, F. Neri, F. Barreca, L. Mafra, M. Pillinger, N. Pinna and A. A. Valente, Green Chem., 2014, 16, 4292–4305 RSC
- L. Fang, K. Zhang, L. Chen and P. Wu, Chin. J. Catal., 2013, 34, 932–941 CrossRef
- S. Van de Vyver, L. Peng, J. Geboers, H. Schepers, F. de Clippel, C. J. Gommes, B. Goderis, P. A. Jacobs and B. F. Sels, Green Chem., 2010, 12, 1560–1563 RSC
- R. Zhong, L. Peng, F. de Clippel, C. Gommes, B. Goderis, X. Ke, G. Van Tendeloo, P. A. Jacobs and B. F. Sels, ChemCatChem, 2015, 7, 3047–3058 CrossRef
- R. Zhong, F. Yu, W. Schutyser, Y. Liao, F. de Clippel, L. Peng and B. F. Sels, Appl. Catal., B, 2017, 206, 74–88 CrossRef
- L. Fang, K. Zhang, X. H. Li, H. H. Wu and P. Wu, Chin. J. Catal., 2012, 33, 114–122 CrossRef
- R. Zhong, L. Peng, R. I. Iacobescu, Y. Pontikes, R. Shu, L. Ma and B. F. Sels, ChemCatChem, 2017, 9, 65–69 CrossRef
- W. Song, Z. Chen, W. Lai, I. Rodríguez-Ramos, X. Yi, W. Weng and W. Fang, Appl. Catal., A, 2017, 540, 21–30 CrossRef
- S. Van de Vyver, J. Geboers, W. Schutyser, M. Dusselier, P. Eloy, E. Dornez, J. W. Seo, C. M. Courtin, E. M. Gaigneaux, P. A. Jacobs and B. F. Sels, ChemSusChem, 2012, 5, 1549–1558 CrossRef PubMed
- T. van Haasterecht, C. C. I. Ludding, K. P. de Jong and J. H. Bitter, J. Catal., 2014, 319, 27–35 CrossRef
- Y. Xu, P. Jiang and Q. Li, Acta Phys. -Chim. Sin., 2013, 29, 1041–1047 Search PubMed
- T. Zhang, W. Li, Z. Xu, Q. Liu, Q. Ma, H. Jameel, H. Chang and L. Ma, Bioresour. Technol., 2016, 209, 108–114 CrossRef PubMed
- Q. Liu, F. Yang, Z. Liu and G. Li, J. Ind. Eng. Chem., 2015, 26, 46–54 CrossRef
- Y. Wang, F. Delbecq, W. Kwapinski and C. Len, Mol. Catal., 2017, 438, 167–172 CrossRef
- P. Wataniyakul, P. Boonnoun, A. T. Quitain, M. Sasaki, T. Kida, N. Laosiripojana and A. Shotipruk, Catal. Commun., 2018, 104, 41–47 CrossRef
- X. Qi, Y. Lian, L. Yan and R. L. Smith, Catal. Commun., 2014, 57, 50–54 CrossRef
- Z. Sun, M. Tao, Q. Zhao, H. Guang, T. Shi and X. Wang, Cellulose, 2015, 22, 675–682 CrossRef
- Y. Bai, L. Xiao and R. Sun, Cellulose, 2014, 21, 2327–2336 CrossRef
- S. Dora, T. Bhaskar, R. Singh, D. V. Naik and D. K. Adhikari, Bioresour. Technol., 2012, 120, 318–321 CrossRef PubMed
- L. J. Konwar, P. Mäki-Arvela, E. Salminen, N. Kumar, A. J. Thakur, J. Mikkola and D. Deka, Appl. Catal., B, 2015, 176–177, 20–35 CrossRef
- A. D. C. Fraga, C. P. B. Quitete, V. L. Ximenes, E. F. Sousa-Aguiar, I. M. Fonseca and A. M. B. Rego, J. Mol. Catal. A: Chem., 2016, 422, 248–257 CrossRef
- Y. Bai, L. Xiao and R. Sun, Biomass Bioenergy, 2015, 75, 245–253 CrossRef
- P. K. Khatri, N. Karanwal, S. Kaul and S. L. Jain, Tetrahedron Lett., 2015, 56, 1203–1206 CrossRef
- J. Zhao, C. Zhou, C. He, Y. Dai, X. Jia and Y. Yang, Catal. Today, 2016, 264, 123–130 CrossRef
- Q. Wang, J. Hao and Z. Zhao, Aust. J. Chem., 2018, 24–31 CrossRef
- J. Zhang and J. Chen, J. Energy Chem., 2016, 25, 747–753 CrossRef
- X. Liu, B. Zhang, B. Fei, X. Chen, J. Zhang and X. Mu, Faraday Discuss., 2017, 202, 79–98 RSC
- P. Sudarsanam, L. Katta, G. Thrimurthulu and B. M. Reddy, J. Ind. Eng. Chem., 2013, 19, 1517–1524 CrossRef
- M. Li, X. Xu, Y. Gong, Z. Wei, Z. Hou, H. Li and Y. Wang, Green Chem., 2014, 16, 4371–4377 RSC
- M. M. Zainol, N. A. S. Amin and M. Asmadi, Fuel Process. Technol., 2017, 167, 431–441 CrossRef
- J. Bedia, J. M. Rosas, D. Vera, J. Rodríguez-Mirasol and T. Cordero, Catal. Today, 2010, 158, 89–96 CrossRef
- S. B. Abd Hamid, M. M. Ambursa, P. Sudarsanam, L. H. Voon and S. K. Bhargava, Catal. Commun., 2017, 94, 18–22 CrossRef
- M. M. Ambursa, T. H. Ali, H. V. Lee, P. Sudarsanam, S. K. Bhargava and S. B. A. Hamid, Fuel, 2016, 180, 767–776 CrossRef
- M. M. Ambursa, P. Sudarsanam, L. H. Voon, S. B. A. Hamid and S. K. Bhargava, Fuel Process. Technol., 2017, 162, 87–97 CrossRef
- S. Gbadamasi, T. H. Ali, L. H. Voon, A. Y. Atta, P. Sudarsanam, S. K. Bhargava and S. B. Abd Hamid, RSC Adv., 2016, 6, 25992–26002 RSC
- X. Liu, L. Xu, G. Xu, W. Jia, Y. Ma and Y. Zhang, ACS Catal., 2016, 6, 7611–7620 CrossRef
- M. Zhang, A. Sun, Y. Meng, L. Wang, H. Jiang and G. Li, Catal. Surv. Asia, 2015, 19, 61–67 CrossRef
- J. R. Kastner, J. Miller, D. P. Geller, J. Locklin, L. H. Keith and T. Johnson, Catal. Today, 2012, 190, 122–132 CrossRef
- I. F. Nata, M. D. Putra, C. Irawan and C. Lee, J. Environ. Chem. Eng., 2017, 5, 2171–2175 CrossRef
- W. Lou, Q. Guo, W. Chen, M. Zong, H. Wu and T. J. Smith, ChemSusChem, 2012, 5, 1533–1541 CrossRef PubMed
- K. Ngaosuwan, J. G. Goodwin and P. Prasertdham, Renewable Energy, 2016, 86, 262–269 CrossRef
- L. J. Konwar, R. Das, A. J. Thakur, E. Salminen, P. Mäki-Arvela, N. Kumar, J. Mikkola and D. Deka, J. Mol. Catal. A: Chem., 2014, 388–389, 167–176 CrossRef
- K. Malins, J. Brinks, V. Kampars and I. Malina, Appl. Catal., A, 2016, 519, 99–106 CrossRef
- F. Zhang, X. Tian, Z. Fang, M. Shah, Y. Wang, W. Jiang and M. Yao, Energy Convers. Manage., 2017, 142, 107–116 CrossRef
- H. H. Mardhiah, H. C. Ong, H. H. Masjuki, S. Lim and Y. L. Pang, Energy Convers. Manage., 2017, 144, 10–17 CrossRef
- F. A. Dawodu, O.
O. Ayodele, J. Xin and S. Zhang, J. Chem. Technol. Biotechnol., 2014, 89, 1898–1909 CrossRef
- C. Wang, F. Yuan, L. Liu, X. Niu and Y. Zhu, ChemPlusChem, 2015, 80, 1657–1665 CrossRef
- P. S. Reddy, P. Sudarsanam, G. Raju and B. M. Reddy, Catal. Commun., 2010, 11, 1224–1228 CrossRef
- P. S. Reddy, P. Sudarsanam, G. Raju and B. M. Reddy, J. Ind. Eng. Chem., 2012, 18, 648–654 CrossRef
- P. Sudarsanam, B. Mallesham, P. S. Reddy and B. M. Reddy, J. Chem. Sci. Technol., 2013, 2, 161–168 Search PubMed
- M. Tao, H. Guan, X. Wang, Y. Liu and R. Louh, Fuel Process. Technol., 2015, 138, 355–360 CrossRef
- C. Ban, W. Jeon, G. Park, H. C. Woo and D. H. Kim, ChemCatChem, 2017, 9, 329–337 CrossRef
- Q. Yan, C. Wan, J. Liu, J. Gao, F. Yu, J. Zhang and Z. Cai, Green Chem., 2013, 15, 1631–1640 RSC
- B. F. Machado and P. Serp, Catal. Sci. Technol., 2012, 2, 54–75 RSC
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 RSC
- Q. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468–5512 RSC
- O. Fleker, A. Borenstein, R. Lavi, L. Benisvy, S. Ruthstein and D. Aurbach, Langmuir, 2016, 32, 4935–4944 CrossRef PubMed
- Y. Zhou, Y. Chen, Y. Hu, G. Huang, S. Yu and H. Jiang, Chem. – Eur. J., 2014, 20, 14976–14980 CrossRef PubMed
- X. Li, A. K. Tjiptoputro, J. Ding, J. M. Xue and Y. Zhu, Catal. Today, 2017, 279, 77–83 CrossRef
- L. Zhu, X. Liu, H. Jiang and L. Sun, Chem. Rev., 2017, 117, 8129–8176 CrossRef PubMed
- M. Yabushita, P. Li, T. Islamoglu, H. Kobayashi, A. Fukuoka, O. K. Farha and A. Katz, Ind. Eng. Chem. Res., 2017, 56, 7141–7148 CrossRef
- J. Jiang and O. M. Yaghi, Chem. Rev., 2015, 115, 6966–6997 CrossRef PubMed
- J. Chen, K. Li, L. Chen, R. Liu, X. Huang and D. Ye, Green Chem., 2014, 16, 2490–2499 RSC
- J. Jiang, F. Gándara, Y. Zhang, K. Na, O. M. Yaghi and W. G. Klemperer, J. Am. Chem. Soc., 2014, 136, 12844–12847 CrossRef PubMed
- Y. Kuwahara, H. Kango and H. Yamashita, ACS Sustainable Chem. Eng., 2017, 5, 1141–1152 CrossRef
- V. Pascanu, A. Bermejo Gómez, C. Ayats, A. E. Platero-Prats, F. Carson, J. Su, Q. Yao, M. À. Pericàs, X. Zou and B. Martín-Matute, ACS Catal., 2015, 5, 472–479 CrossRef
- L. Ning, S. Liao, H. Cui, L. Yu and X. Tong, ACS Sustainable Chem. Eng., 2018, 6, 135–142 CrossRef
- Y. Guo, Y. Li, J. Chen and L. Chen, Catal. Lett., 2016, 146, 2041–2052 CrossRef
- T. B. Čelič, M. Grilc, B. Likozar and N. N. Tušar, ChemSusChem, 2015, 8, 1703–1710 CrossRef PubMed
- Y. Wang, Y. Miao, S. Li, L. Gao and G. Xiao, Mol. Catal., 2017, 436, 128–137 CrossRef
- Y. Wang, S. Sang, W. Zhu, L. Gao and G. Xiao, Chem. Eng. J., 2016, 299, 104–111 CrossRef
- P. Hester, S. Xu, W. Liang, N. Al-Janabi, R. Vakili, P. Hill, C. A. Muryn, X. Chen, P. A. Martin and X. Fan, J. Catal., 2016, 340, 85–94 CrossRef
- G. Zi, Z. Yan, Y. Wang, Y. Chen, Y. Guo, F. Yuan, W. Gao, Y. Wang and J. Wang, Carbohydr. Polym., 2015, 115, 146–151 CrossRef PubMed
- P. Huang and L. Yan, Chin. J. Chem. Phys., 2016, 29, 742–748 CrossRef
- B. Murillo, B. Zornoza, O. de la Iglesia, C. Téllez and J. Coronas, J. Catal., 2016, 334, 60–67 CrossRef
- S. Huang, K. Yang, X. Liu, H. Pan, H. Zhang and S. Yang, RSC Adv., 2017, 7, 5621–5627 RSC
- X. Lu, L. Wang and X. Lu, Catal. Commun., 2018, 110, 23–27 CrossRef
- A. Chatterjee, X. Hu and F. L. Lam, Catal. Today, 2018, 314, 137–146 CrossRef
- C. Lucarelli, S. Galli, A. Maspero, A. Cimino, C. Bandinelli, A. Lolli, J. Velasquez Ochoa, A. Vaccari, F. Cavani and S. Albonetti, J. Phys. Chem. C, 2016, 120, 15310–15321 CrossRef
- A. Rapeyko, K. S. Arias, M. J. Climent, A. Corma and S. Iborra, Catal. Sci. Technol., 2017, 7, 3008–3016 RSC
- O. Kikhtyanin, D. Kubička and J. Čejka, Catal. Today, 2015, 243, 158–162 CrossRef
- L. Ning, S. Liao, X. Liu, L. Yu, X. Zhuang and X. Tong, J. Catal., 2017, 352, 480–490 CrossRef
- F. G. Cirujano, A. Corma and F. X. Llabrés i Xamena, Chem. Eng. Sci., 2015, 124, 52–60 CrossRef
- I. Larasati, D. Winarni, F. R. Putri, Q. A. Hanif and W. W. Lestari, IOP Conf. Ser.: Mater. Sci. Eng., 2017, 214, 012006 Search PubMed
- F. G. Cirujano, A. Corma and F. X. Llabrés i Xamena, Catal. Today, 2015, 257, 213–220 CrossRef
- Q. Yuan, D. Zhang, L. V. Haandel, F. Ye, T. Xue, E. J. M. Hensen and Y. Guan, J. Mol. Catal. A: Chem., 2015, 406, 58–64 CrossRef
- S. B. A. Hamid, N. Basiron, W. A. Yehye, P. Sudarsanam and S. K. Bhargava, Polyhedron, 2016, 120, 124–133 CrossRef
- Z. Hu, Y. Peng, Y. Gao, Y. Qian, S. Ying, D. Yuan, S. Horike, N. Ogiwara, R. Babarao, Y. Wang, N. Yan and D. Zhao, Chem. Mater., 2016, 28, 2659–2667 CrossRef
- R. Oozeerally, D. L. Burnett, T. W. Chamberlain, R. I. Walton and V. Degirmenci, ChemCatChem, 2018, 10, 706–709 CrossRef PubMed
- Y. Su, G. Chang, Z. Zhang, H. Xing, B. Su, Q. Yang, Q. Ren, Y. Yang and Z. Bao, AIChE J., 2016, 62, 4403–4417 CrossRef
- M. Liu, Y. Zhang, E. Zhu, P. Jin, K. Wang, J. Zhao, C. Li and Y. Yan, ChemistrySelect, 2017, 2, 10413–10419 CrossRef
- X. Liu, H. Li, H. Zhang, H. Pan, S. Huang, K. Yang and S. Yang, RSC Adv., 2016, 6, 90232–90238 RSC
- Z. Hasan, J. W. Jun and S. H. Jhung, Chem. Eng. J., 2015, 278, 265–271 CrossRef
- W. Xie, X. Yang and P. Hu, Catal. Lett., 2017, 147, 2772–2782 CrossRef
- J. Chen, S. Wang, J. Huang, L. Chen, L. Ma and X. Huang, ChemSusChem, 2013, 6, 1545–1555 CrossRef PubMed
- S. Wang, J. Chen and L. Chen, Catal. Lett., 2014, 144, 1728–1734 CrossRef
- Z. Lin, X. Cai, Y. Fu, W. Zhu and F. Zhang, RSC Adv., 2017, 7, 44082–44088 RSC
- D. Zhang, F. Ye, Y. Guan, Y. Wang and E. J. M. Hensen, RSC Adv., 2014, 4, 39558–39564 RSC
- R. Insyani, D. Verma, S. M. Kim and J. Kim, Green Chem., 2017, 19, 2482–2490 RSC
- J. Chen, R. Liu, Y. Guo, L. Chen and H. Gao, ACS Catal., 2015, 5, 722–733 CrossRef
- F. Zhang, Y. Jin, Y. Fu, Y. Zhong, W. Zhu, A. A. Ibrahim and M. S. El-Shall, J. Mater. Chem. A, 2015, 3, 17008–17015 RSC
- H. Yu, Z. Guo, Y. Zang, Y. Jin, T. Shi, G. Tu, C. Xu, Y. Fu, F. Zhang, J. Xie and W. Zhu, Adv. Sci. Lett., 2017, 23, 5821–5823 CrossRef
- F. Zhang, S. Zheng, Q. Xiao, Y. Zhong, W. Zhu, A. Lin and M. Samy El-Shall, Green Chem., 2016, 18, 2900–2908 RSC
- Q. Guan, B. Wang, X. Chai, J. Liu, J. Gu and P. Ning, Fuel, 2017, 205, 130–141 CrossRef
- X. Fang, Y. Shi, K. Wu, J. Liang, Y. Wu and M. Yang, RSC Adv., 2017, 7, 40581–40590 RSC
- R. Fang, R. Luque and Y. Li, Green Chem., 2017, 19, 647–655 RSC
- R. Fang, R. Luque and Y. Li, Green Chem., 2016, 18, 3152–3157 RSC
- P. Jin, Y. Zhang, Y. Chen, J. Pan, X. Dai, M. Liu, Y. Yan and C. Li, J. Taiwan Inst. Chem. Eng., 2017, 75, 59–69 CrossRef
- Y. Su, C. Chen, X. Zhu, Y. Zhang, W. Gong, H. Zhang, H. Zhao and G. Wang, Dalton Trans., 2017, 46, 6358–6365 RSC
- B. Mallesham, P. Sudarsanam, B. V. S. Reddy and B. M. Reddy, Appl. Catal., B, 2016, 181, 47–57 CrossRef
- L. Zheng, X. Li, W. Du, D. Shi, W. Ning, X. Lu and Z. Hou, Appl. Catal., B, 2017, 203, 146–153 CrossRef
- A. Li, K. Shen, J. Chen, Z. Li and Y. Li, Chem. Eng. Sci., 2017, 166, 66–76 CrossRef
- H. Zhang, J. Nai, L. Yu and X. W. D. Lou, Joule, 2017, 1, 77–107 CrossRef
- T. Torimoto, T. Tsuda, K. Okazaki and S. Kuwabata, Adv. Mater., 2010, 22, 1196–1221 CrossRef PubMed
- M. Koel, Crit. Rev. Anal. Chem., 2005, 35, 177–192 CrossRef
- V. I. Pârvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615–2665 CrossRef PubMed
- Q. Zhang, S. Zhang and Y. Deng, Green Chem., 2011, 13, 2619–2637 RSC
- A. M. Da Costa Lopes and R. Bogel-Lukasik, ChemSusChem, 2015, 8, 947–965 CrossRef PubMed
- A. S. Amarasekara, Chem. Rev., 2016, 116, 6133–6183 CrossRef PubMed
- C. C. Weber, A. F. Masters and T. Maschmeyer, Org. Biomol. Chem., 2013, 11, 2534–2542 RSC
- Y. Wang, Z. Gu, W. Liu, Y. Yao, H. Wang, X. Xia and W. Li, RSC Adv., 2015, 5, 60736–60744 RSC
- H. Zhou, L. Yang, W. Li, F. Wang, W. Li, J. Zhao, X. Liang and H. Liu, Ind. Eng. Chem. Res., 2012, 51, 13173–13181 CrossRef
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef
- L. Han, H. Li, S. Choi, M. Park, S. Lee, Y. Kim and D. Park, Appl. Catal., A, 2012, 429–430, 67–72 CrossRef
- J. Zhu, B. He, J. Huang, C. Li and T. Ren, Microporous Mesoporous Mater., 2018, 260, 190–200 CrossRef
- D. R. MacFarlane, J. M. Pringle, K. M. Johansson, S. A. Forsyth and M. Forsyth, Chem. Commun., 2006, 1905–1917 RSC
- K. L. Luska, P. Migowski, S. El Sayed and W. Leitner, ACS Sustainable Chem. Eng., 2016, 4, 6186–6192 CrossRef
- H. Li, Q. Zhang, X. Liu, F. Chang, Y. Zhang, W. Xue and S. Yang, Bioresour. Technol., 2013, 144, 21–27 CrossRef PubMed
- Z. Wu, C. Chen, Q. Guo, B. Li, Y. Que, L. Wang, H. Wan and G. Guan, Fuel, 2016, 184, 128–135 CrossRef
-
E. Benazzi, A. Hirschauer, J. F. Joly, H. Olivier and J. Y. Berhard, EP0553009, 1993
- C. P. Mehnert, R. A. Cook, N. C. Dispenziere and M. Afeworki, J. Am. Chem. Soc., 2002, 124, 12932–12933 CrossRef PubMed
- T. Selvam, A. Machoke and W. Schwieger, Appl. Catal., A, 2012, 445–446, 92–101 CrossRef
- M. Haumann and A. Riisager, Chem. Rev., 2008, 108, 1474–1497 CrossRef PubMed
- B. Wiredu and A. S. Amarasekara, Catal. Commun., 2014, 48, 41–44 CrossRef
- C. Zhang, Z. Fu, B. Dai, S. Zen, Y. Liu, Q. Xu, S. R. Kirk and D. Yin, Cellulose, 2014, 21, 1227–1237 CrossRef
- H. Xu, H. Zhao, H. Song, Z. Miao, J. Yang, J. Zhao, N. Liang and L. Chou, J. Mol. Catal. A: Chem., 2015, 410, 235–241 CrossRef
- K. B. Sidhpuria, A. L. Daniel-da-Silva, T. Trindade and J. A. P. Coutinho, Green Chem., 2011, 13, 340–349 RSC
- X. Shi, M. Zhang, Y. Li and W. Zhang, Green Chem., 2013, 15, 3438–3445 RSC
- J. Miao, H. Wan and G. Guan, Catal. Commun., 2011, 12, 353–356 CrossRef
- K. L. Luska, A. Bordet, S. Tricard, I. Sinev, W. Grünert, B. Chaudret and W. Leitner, ACS Catal., 2016, 6, 3719–3726 CrossRef
- J. Yang, L. Zhou, X. Guo, L. Li, P. Zhang, R. Hong and T. Qiu, Chem. Eng. J., 2015, 280, 147–157 CrossRef
- V. Degirmenci, E. A. Pidko, P. C. M. M. Magusin and E. J. M. Hensen, ChemCatChem, 2011, 3, 969–972 CrossRef
- V. Degirmenci and E. J. M. Hensen, Environ. Prog. Sustainable Energy, 2014, 33, 657–662 CrossRef
- H. Li, S. Saravanamurugan, S. Yang and A. Riisager, ACS Sustainable Chem. Eng., 2015, 3, 3274–3280 CrossRef
- S. Kudo, N. Goto, J. Sperry, K. Norinaga and J. Hayashi, ACS Sustainable Chem. Eng., 2017, 5, 1132–1140 CrossRef
- Y. Sun, Q. Zhang, P. Zhang, D. Song and Y. Guo, ACS Sustainable Chem. Eng., 2018, 6, 6771–6782 CrossRef
- J. Sun, J. Yang, S. Li and X. Xu, Catal. Commun., 2016, 83, 35–38 CrossRef
- Y. Cao, H. Zhou and J. Li, Renewable Sustainable Energy Rev., 2016, 58, 871–875 CrossRef
- Z. Wu, C. Chen, H. Wan, L. Wang, Z. Li, B. Li, Q. Guo and G. Guan, Energy Fuels, 2016, 30, 10739–10746 CrossRef
- H. Zhang, H. Li, H. Pan, X. Liu, K. Yang, S. Huang and S. Yang, Energy Convers. Manage., 2017, 138, 45–53 CrossRef
- M. K. Munshi, S. T. Lomate, R. M. Deshpande, V. H. Rane and A. A. Kelkar, J. Chem. Technol. Biotechnol., 2010, 85, 1319–1324 CrossRef
- M. H. Valkenberg, C. DeCastro and W. F. Holderich, Green Chem., 2002, 4, 88–93 RSC
- E. Salminen, P. Mäki-Arvela, P. Virtanen, T. Salmi and J. Mikkola, Top. Catal., 2014, 57, 1533–1538 CrossRef
- E. Salminen, P. Mäki-Arvela, P. Virtanen, T. Salmi, J. Wärnå and J. Mikkola, Ind. Eng. Chem. Res., 2014, 53, 20107–20115 CrossRef
- E. Salminen, L. Rujana, P. Mäki-Arvela, P. Virtanen, T. Salmi and J. Mikkola, Catal. Today, 2015, 257, 318–321 CrossRef
- A. Eftekhari and T. Saito, Eur. Polym. J., 2017, 90, 245–272 CrossRef
- Y. Wang, J. Liu and C. Xia, Tetrahedron Lett., 2011, 52, 1587–1591 CrossRef
- J. Restrepo, R. Porcar, P. Lozano, M. I. Burguete, E. García-Verdugo and S. V. Luis, ACS Catal., 2015, 5, 4743–4750 CrossRef
- D. D. J. Liu and E. Y. X. Chen, Biomass Bioenergy, 2013, 48, 181–190 CrossRef
- H. Li, X. He, Q. Zhang, F. Chang, W. Xue, Y. Zhang and S. Yang, Energy Technol., 2013, 1, 151–156 CrossRef
- H. Zhou, J. Song, X. Kang, J. Hu, Y. Yang, H. Fan, Q. Meng and B. Han, RSC Adv., 2015, 5, 15267–15273 RSC
- J. Zhang, S. Zhang, J. Han, Y. Hu and R. Yan, Chem. Eng. J., 2015, 271, 269–275 CrossRef
- W. Zhang, M. Li, J. Wang, Y. Zhao, S. Zhou and W. Xing, Appl. Clay Sci., 2017, 146, 167–175 CrossRef
- R. Wu, L. Song, G. Ma, Y. Su, Y. Hong, H. Wang and J. Li, Catal. Lett., 2017, 147, 2736–2744 CrossRef
- C. D. Hubbard, P. Illner and R. van Eldik, Chem. Soc. Rev., 2011, 40, 272–290 RSC
- P. Sudarsanam, B. Hillary, M. H. Amin, S. B. A. Hamid and S. K. Bhargava, Appl. Catal., B, 2016, 185, 213–224 CrossRef
- Y. Li and W. Shen, Chem. Soc. Rev., 2014, 43, 1543–1574 RSC
- B. Hillary, P. Sudarsanam, M. H. Amin and S. K. Bhargava, Langmuir, 2017, 33, 1743–1750 CrossRef PubMed
-
D. Astruc, Nanoparticles and Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed
- P. Sudarsanam, B. Hillary, B. Mallesham, B. G. Rao, M. H. Amin, A. Nafady, A. M. Alsalme, B.
M. Reddy and S. K. Bhargava, Langmuir, 2016, 32, 2208–2215 CrossRef PubMed
- S. Putla, M. H. Amin, B. M. Reddy, A. Nafady, K. A. Al Farhan and S. K. Bhargava, ACS Appl. Mater. Interfaces, 2015, 7, 16525–16535 CrossRef PubMed
- P. Sudarsanam, B. Hillary, D. K. Deepa, M. H. Amin, B. Mallesham, B. M. Reddy and S. K. Bhargava, Catal. Sci. Technol., 2015, 5, 3496–3500 RSC
- S. Wang, Z. Zhang and B. Liu, ACS Sustainable Chem. Eng., 2015, 3, 406–412 CrossRef
- S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428–3459 CrossRef PubMed
- C. Zhang, H. Wang, F. Liu, L. Wang and H. He, Cellulose, 2013, 20, 127–134 CrossRef
- N. Mei and B. Liu, Int. J. Hydrogen Energy, 2016, 41, 17960–17966 CrossRef
- V. Polshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J. Basset, Chem. Rev., 2011, 111, 3036–3075 CrossRef PubMed
- A. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef PubMed
- J. Lee, Y. Lee, J. K. Youn, H. B. Na, T. Yu, H. Kim, S. Lee, Y. Koo, J. H. Kwak, H. G. Park, H. N. Chang, M. Hwang, J. Park, J. Kim and T. Hyeon, Small, 2008, 4, 143–152 CrossRef PubMed
- V. Polshettiwar and R. S. Varma, Green Chem., 2010, 12, 743–754 RSC
- N. Candu, C. Rizescu, I. Podolean, M. Tudorache, V. I. Parvulescu and S. M. Coman, Catal. Sci. Technol., 2015, 5, 729–737 RSC
- Z. Zhang, Z. Yuan, D. Tang, Y. Ren, K. Lv and B. Liu, ChemSusChem, 2014, 7, 3496–3504 CrossRef PubMed
- I. Elsayed, M. Mashaly, F. Eltaweel, M. A. Jackson and E. B. Hassan, Fuel, 2018, 221, 407–416 CrossRef
- K. Rajkumari, J. Kalita, D. Das and L. Rokhum, RSC Adv., 2017, 7, 56559–56565 RSC
- A. Ramazani, M. Khoobi, F. Sadri, R. Tarasi, A. Shafiee, H. Aghahosseini and S. W. Joo, Appl. Organomet. Chem., 2018, 32, e3908 CrossRef
- Z. Zhang, J. Zhen, B. Liu, K. Lv and K. Deng, Green Chem., 2015, 17, 1308–1317 RSC
- Z. Yang, W. Qi, R. Su and Z. He, Energy Fuels, 2017, 31, 533–541 CrossRef
- J. He, S. Yang and A. Riisager, Catal. Sci. Technol., 2018, 8, 790–797 RSC
- X. Long, P. Sun, Z. Li, R. Lang, C. Xia and F. Li, Chin. J. Catal., 2015, 36, 1512–1518 CrossRef
- C. Opris, B. Cojocaru, N. Gheorghe, M. Tudorache, S. M. Coman, V. I. Parvulescu, B. Duraki, F. Krumeich and J. A. van Bokhoven, ACS Catal., 2017, 7, 3257–3267 CrossRef
- C. Opris, B. Cojocaru, N. Gheorghe, M. Tudorache, S. M. Coman, V. I. Parvulescu, B. Duraki, F. Krumeich and J. A. van Bokhoven, J. Catal., 2016, 339, 209–227 CrossRef
- A. Negoi, I. T. Trotus, O. Mamula Steiner, M. Tudorache, V. Kuncser, D. Macovei, V. I. Parvulescu and S. M. Coman, ChemSusChem, 2013, 6, 2090–2094 CrossRef PubMed
- Y. Xiong, Z. Zhang, X. Wang, B. Liu and J. Lin, Chem. Eng. J., 2014, 235, 349–355 CrossRef
- N. Candu, F. Anita, I. Podolean, B. Cojocaru, V. I. Parvulescu and S. M. Coman, Green Process Synth., 2017, 6, 255–264 Search PubMed
- B. Liu, Y. Ren and Z. Zhang, Green Chem., 2015, 17, 1610–1617 RSC
- I. Podolean, A. Negoi, N. Candu, M. Tudorache, V. I. Parvulescu and S. M. Coman, Top. Catal., 2014, 57, 1463–1469 CrossRef
- V. Kuncser, S. M. Coman, E. Kemnitz and V. I. Parvulescu, J. Appl. Phys., 2015, 117, 17D724 CrossRef
- I. Podolean, V. Kuncser, N. Gheorghe, D. Macovei, V. I. Parvulescu and S. M. Coman, Green Chem., 2013, 15, 3077–3082 RSC
- I. Podolean, C. Rizescu, C. Bala, L. Rotariu, V. I. Parvulescu, S. M. Coman and H. Garcia, ChemSusChem, 2016, 9, 2307–2311 CrossRef PubMed
- Y. Yang, W. Zhang, F. Yang, B. Zhou, D. Zeng, N. Zhang, G. Zhao, S. Hao and X. Zhang, Nanoscale, 2018, 10, 2199–2206 RSC
- Q. Yang, S. Zhou and T. Runge, J. Catal., 2015, 330, 474–484 CrossRef
- Y. Wang, Z. Hu, G. Fan, J. Yan, G. Song and J. Li, Waste Biomass Valorization, 2018 DOI:10.1007/s12649-018-0242-9
- C. Wu, W. Yuan, Y. Huang, Y. Xia, H. Yang, H. Wang and X. Liu, Catal. Lett., 2017, 147, 953–963 CrossRef
- K. Alibegovic, D. G. Morgan, Y. Losovyj, M. Pink, B. D. Stein, N. V. Kuchkina, E. S. Serkova, K. E. Salnikova, Z. B. Shifrina, V. G. Matveeva, E. M. Sulman and L. M. Bronstein, ChemistrySelect, 2017, 2, 5485–5491 CrossRef
- F. Wang and Z. Zhang, ACS Sustainable Chem. Eng., 2017, 5, 942–947 CrossRef
- S. Wang, Z. Zhang, B. Liu and J. Li, Ind. Eng. Chem. Res., 2014, 53, 5820–5827 CrossRef
- B. Karimi, H. M. Mirzaei and E. Farhangi, ChemCatChem, 2014, 6, 758–762 CrossRef
- N. Mittal, G. M. Nisola, L. B. Malihan, J. G. Seo, H. Kim, S. Lee and W. Chung, RSC Adv., 2016, 6, 25678–25688 RSC
- M. Raita, J. Arnthong, V. Champreda and N. Laosiripojana, Fuel Process. Technol., 2015, 134, 189–197 CrossRef
- M. R. Mehrasbi, J. Mohammadi, M. Peyda and M. Mohammadi, Renewable Energy, 2017, 101, 593–602 CrossRef
- B. Thangaraj, Z. Jia, L. Dai, D. Liu and W. Du, Arabian J. Chem., 2016 DOI:10.1016/j.arabjc.2016.09.004
- Q. Zhang, Z. Zheng, C. Liu, C. Liu and T. Tan, Colloids Surf., B, 2016, 140, 446–451 CrossRef PubMed
- M. Kalantari, M. Kazemeini and A. Arpanaei, Biochem. Eng. J., 2013, 79, 267–273 CrossRef
- O. V. Manaenkov, J. J. Mann, O. V. Kislitza, Y. Losovyj, B. D. Stein, D. G. Morgan, M. Pink, O. L. Lependina, Z. B. Shifrina, V. G. Matveeva, E. M. Sulman and L. M. Bronstein, ACS Appl. Mater. Interfaces, 2016, 8, 21285–21293 CrossRef PubMed
- N. Cherkasov, V. Jadvani, J. Mann, Y. B. Losovyj, Z. B. Shifrina, L. M. Bronstein and E. V. Rebrov, Fuel Process. Technol., 2017, 167, 738–746 CrossRef
- D. Lai, L. Deng, J. Li, B. Liao, Q. Guo and Y. Fu, ChemSusChem, 2011, 4, 55–58 CrossRef PubMed
- T. Komanoya, H. Kobayashi, K. Hara, W. Chun and A. Fukuoka, Appl. Catal., A, 2011, 407, 188–194 CrossRef
- B. Liu, Z. Zhang, K. Lv, K. Deng and H. Duan, Appl. Catal., A, 2014, 472, 64–71 CrossRef
- A. B. Gawade, A. V. Nakhate and G. D. Yadav, Catal. Today, 2018, 309, 119–125 CrossRef
- Y. N. Regmi, J. K. Mann, J. R. McBride, J. Tao, C. E. Barnes, N. Labbé and S. C. Chmely, Catal. Today, 2018, 302, 190–195 CrossRef
- K. Liu, R. Wang and M. Yu, RSC Adv., 2017, 7, 51814–51821 RSC
- L. Lin, S. Vittayapadung, X. Li, W. Jiang and X. Shen, Environ. Prog. Sustainable Energy, 2013, 32, 1255–1261 CrossRef
- M. Tudorache, L. Protesescu, S. Coman and V. I. Parvulescu, Green Chem., 2012, 14, 478–482 RSC
- M. Tudorache, G. Ghemes, A. Nae, E. Matei, I. Mercioniu, E. Kemnitz, B. Ritter, S. Coman and V. I. Parvulescu, Cent. Eur. J. Chem., 2014, 12, 1262–1270 CrossRef
- M. Tudorache, L. Protesescu, A. Negoi and V. I. Parvulescu, Appl. Catal., A, 2012, 437–438, 90–95 CrossRef
- M. Tudorache, A. Negoi, L. Protesescu and V. I. Parvulescu, Appl. Catal., B, 2014, 145, 120–125 CrossRef
- M. Tudorache, A. Nae, S. Coman and V. I. Parvulescu, RSC Adv., 2013, 3, 4052–4058 RSC
- M. Tudorache, A. Negoi, B. Tudora and V. I. Parvulescu, Appl. Catal., B, 2014, 146, 274–278 CrossRef
- D. Wang and D. Astruc, Chem. Rev., 2014, 114, 6949–6985 CrossRef PubMed
- M. Shokouhimehr, Catalysts, 2015, 5, 534–560 CrossRef
| This journal is © The Royal Society of Chemistry 2018 |