
An air-stable N-heterocyclic carbene iminoxyl borate radical zwitterion†
Youngsuk
Kim
 ab and
Eunsung
Lee
*abc
ab and
Eunsung
Lee
*abc
aCenter for Self–assembly and Complexity, Institute for Basic Science (IBS), Pohang, 37673, Republic of Korea
bDepartment of Chemistry, Pohang University of Science and Technology, Pohang, 37673, Republic of Korea. E-mail: eslee@postech.ac.kr
cDivision of Advanced Materials Science, Pohang University of Science and Technology, Pohang, 37673, Republic of Korea
First published on 27th March 2018
Abstract
A remarkably stable radical zwitterion derived from N-heterocyclic carbene nitric oxide and B(C6F5)3 is reported. The presented radical was generated by steric and electronic protection of the nitric oxide moiety using B(C6F5)3, which secured its stability toward air and moisture. An analogous yet less stable radical derived from C(C6H5)3+ is also synthesized and characterized.
Stable organic radicals are of great interest to chemists since they offer fundamental understanding of reactive intermediates, as well as they have numerous applications as functional materials.1 While the majority of organic radicals are thermodynamically and kinetically unstable, there are several kinds of stable organic radicals, namely triarylmethyl, nitroxyl, and hydrazyl radicals, for example.2 In the past decade, N-heterocyclic carbenes (NHCs)3 have been introduced to stabilise various organic radicals and radical ions,4 as a variety of previously inaccessible organic radicals have been successfully prepared and characterised with the aid of NHCs.5 To date, NHC-stabilised carbonyl (A and B),6 propargyl (C),7 aminyl (D and E),8 phosphinyl (F),9 and many other organic radicals have been isolated or spectroscopically characterised by the groups of Bertrand, Roesky, Curran, and many others (Scheme 1).10 These radicals were successfully stabilised due to the π-accepting properties of NHCs that delocalise the spin density,11 as well as the steric protection of the bulky NHC substituents.12
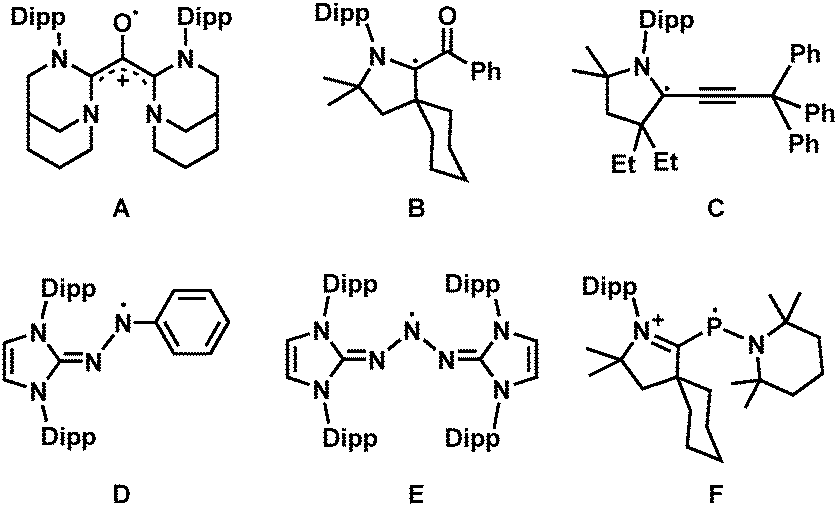 | ||
| Scheme 1 Selected examples of organic radicals stabilised by NHC (Dipp = 2,6-diisopropylphenyl). | ||
In this context, our group recently reported the synthesis of persistent iminoxyl radicals derived from NHCs with nitric oxide.10a,13 The iminoxyl radical 1 was quite stable in the solid state; however, in the solution phase, it slowly decomposed even under an inert atmosphere of nitrogen. Here we report the synthesis and characterisation of a remarkably stable radical zwitterion 2 obtained from the steric protection of 1 using tris(pentafluorophenyl)borane (B(C6F5)3) (Scheme 2).
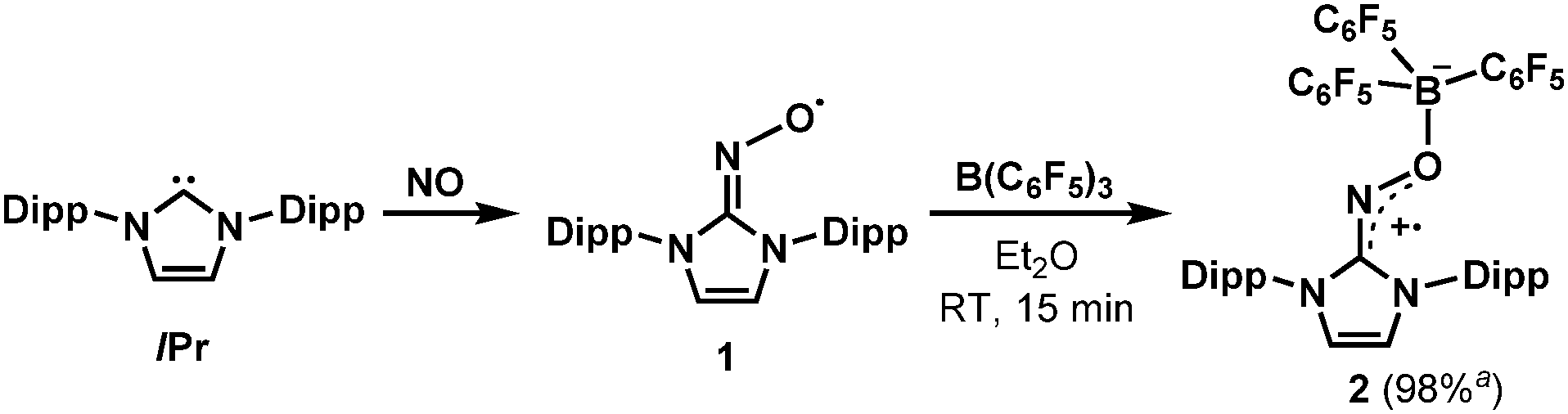 | ||
| Scheme 2 Synthesis of the radical zwitterion 2 from IPr (1,3-bis-(2,6-diisopropylphenyl)imidazol-2-ylidene) and NO (nitric oxide). a Isolated yield. | ||
The radical 2 can be synthesised in high yield by mixing equimolar amounts of 1 and B(C6F5)3 in diethyl ether solution under a N2 atmosphere. The reaction mixture turned dark brown immediately, and after 15 minutes, volatiles were removed under vacuum. After washing the resulting solid using n-pentane, 2 was isolated as a dark brown solid in 98% yield. It is notable that a similar reactivity was recently reported for the (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) radical with B(C6F5)3.14 The molecular structure of 2 was unambiguously determined using single crystal X-ray crystallography (Fig. 1). The bond lengths of C1–N3 (1.339(2) Å) and N3–O1 (1.344(2) Å) indicate a bond order of 1.5. The (imidazole ring)–N3–O1–B1 group is planar, which also suggests delocalisation of the radical through π-conjugation. The structural parameters of 2 were well reproduced using density functional theory (DFT) calculations at the B3LYP/6-31G(d,p) level of theory. The Wiberg bond orders were calculated for the C1–N3 (1.49), N3–O1 (1.44), and O1–B1 (0.86) bonds, which were consistent with the structure obtained from single crystal X-ray analysis. The experimental electron paramagnetic resonance (EPR) spectrum shows a complex splitting pattern (Fig. 2), which was well reproduced in the simulated spectra. DFT calculations suggest that the N3 atom has the largest spin density (44%), consistent with the largest hyperfine coupling constant on N3 (23.4 MHz). The calculation also shows that the singly occupied molecular orbital (SOMO) of 2 is delocalised over the molecular plane (Fig. S1, ESI†). The UV-vis absorption spectrum of 2 in benzene at room temperature shows a peak at λmax = 448 nm (Fig. S8, ESI†). The cyclic voltammogram of 2 reveals one reversible redox peak at E1/2 = −0.022 V (versus saturated Ag/AgCl electrode), showing that 2 is a weak oxidant (Fig. S13, ESI†).
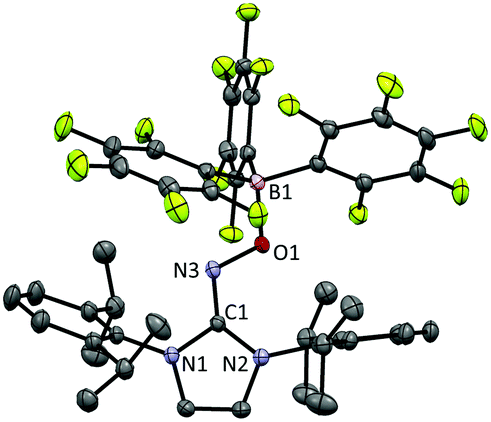 | ||
| Fig. 1 Molecular structure of 2 from X-ray crystallography. The thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms and solvent molecules (n-pentane) were omitted for clarity. | ||
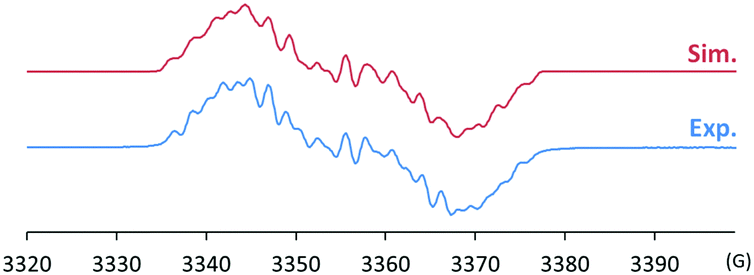 | ||
| Fig. 2 Experimental (bottom) and simulated (top) EPR spectra of 2 (g = 2.0107; hyperfine coupling constants: a(14N) = 23.4, 9.1, 6.9 MHz, a(11B) = 6.7 MHz, a(1H) = 5.3, 3.6 MHz, a(19F) = 2.6, 1.7 MHz). | ||
It is notable that 2 shows remarkable stability toward air and moisture. For example, a solution of 2 in wet technical-grade benzene was monitored using UV-vis, which showed no detectable decomposition over 12 days (Fig. 3). In addition, 2 was stable even under silica chromatographic conditions as the benzene solution of 2 was still EPR active even after the filtration through silica gel under air (Fig. S6, ESI†).
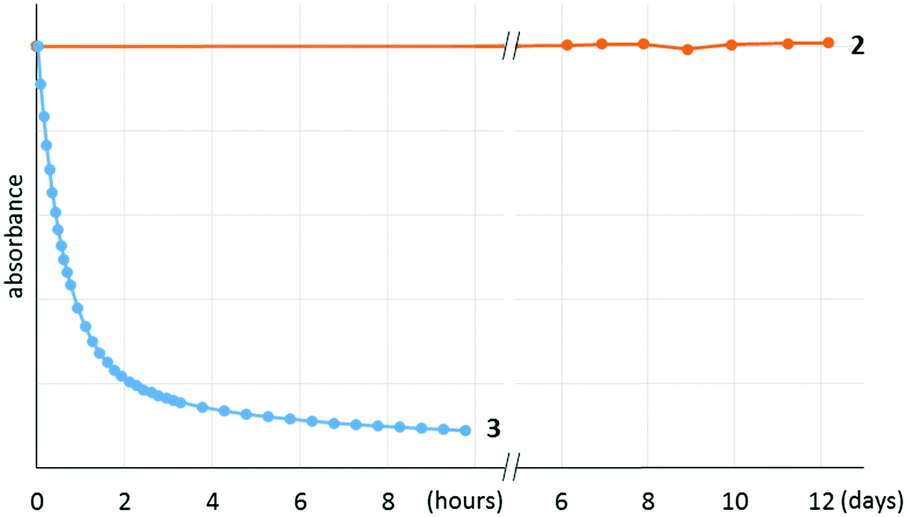 | ||
| Fig. 3 Decay of the radicals 2 and 3 in wet benzene solution under air; monitored by UV-vis. | ||
After discovering the remarkable stability of the radical 2, we were curious whether the radical cation 3 with almost equivalent steric bulk is also stable or not. When 1 was treated with a stoichiometric amount of trityl tetrakis(pentafluorophenyl)borate (Ph3C+B(C6F5)4−) in diethyl ether solution under a N2 atmosphere, the color of the reaction mixture changed immediately to dark brown (Scheme 3). After 5 minutes, the product was precipitated by the addition of n-pentane, and subsequently washed to yield 3 as a dark brown solid (96%). Single crystal X-ray crystallographic analysis revealed the molecular structure of 3 with the B(C6F5)4− counteranion (Fig. 4). The planar molecular structure and bond lengths of C1–N3 (1.339(1) Å) and N3–O1 (1.361(1) Å) of 3 were almost identical with the structure of 2. The calculated Wiberg bond lengths of C1–N3 (1.50), N3–O1 (1.38), and O1–C2 (0.87) bonds of 3 were also similar with those of 2. On the other hand, the experimental and simulated EPR spectra of 3 were very different from those of 2 due to the absence of nearby boron and fluorine atoms (Fig. 5). The UV-vis spectrum of 3 in benzene was recorded at room temperature, which showed a peak at λmax = 458 nm (Fig. S9, ESI†). In contrast to 2, 3 is a quite strong oxidant as its cyclic voltammogram shows one reversible redox peak at E1/2 = 0.582 V versus sat. Ag/AgCl (Fig. S14, ESI†). One-electron reduction of 3 using decamethylferrocene also resulted in the neutral oxime compound (see the ESI†). Interestingly, 3 was much more sensitive toward air and moisture than 2 as the half-life of 3 in wet technical-grade benzene was approximately 35 minutes (Fig. 3).
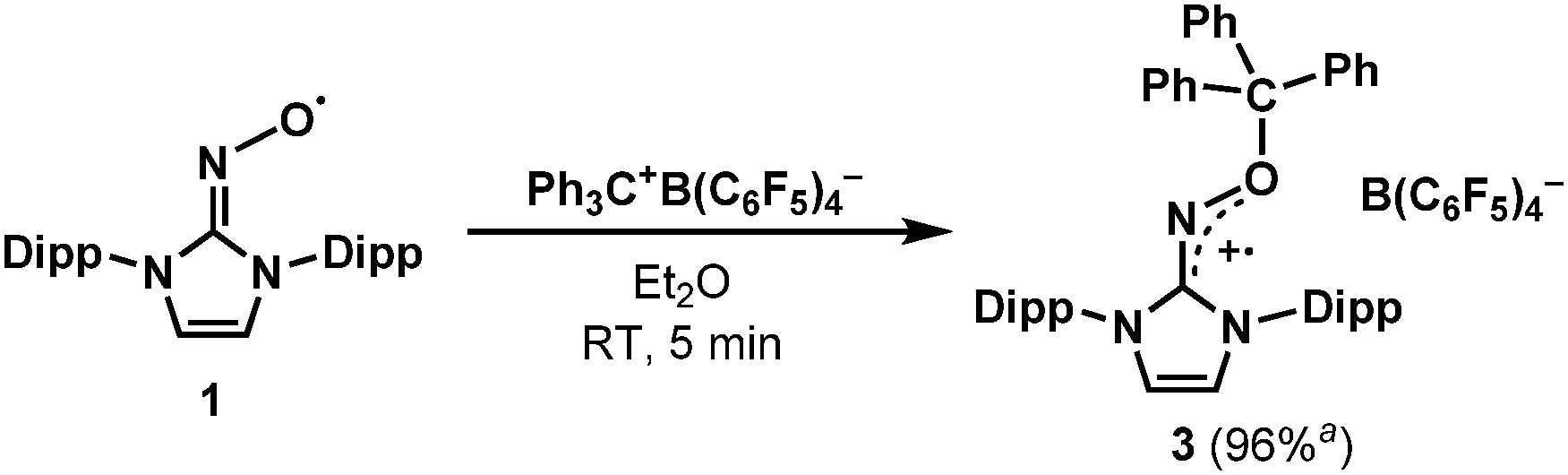 | ||
| Scheme 3 Synthesis of the radical cation 3. a Isolated yield. | ||
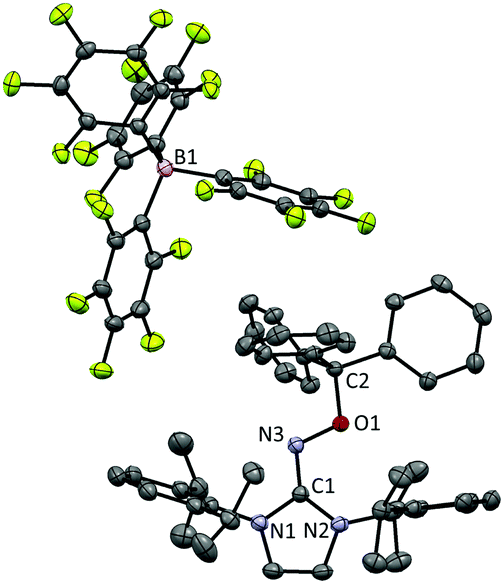 | ||
| Fig. 4 Molecular structure of 3 from X-ray crystallography. The thermal ellipsoids are shown at the 50% probability level. Hydrogen atoms were omitted for clarity. | ||
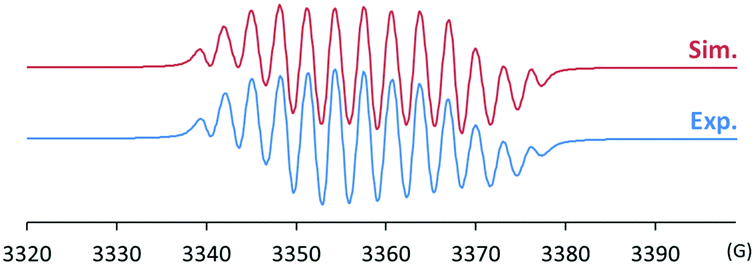 | ||
| Fig. 5 Experimental (bottom) and simulated (top) EPR spectra of 3 (g = 2.0098; hyperfine coupling constants: a(14N) = 26.6, 9.3, 7.5 MHz, a(1H) = 10.2, 6.6 MHz). | ||
Therefore, simply protecting the radical center using a bulky substituent did not guarantee the stability of the radical. DFT calculations at the B3LYP/6-31G(d,p) level using the SMD solvation model showed that the trityl group is not binding strongly enough to the oxygen atom of 1 compared to the B(C6F5)3 group. The standard free energy of the dissociation of the B(C6F5)3 group from 2 is energetically uphill by 12.6 kcal mol−1 in benzene solution, while the trityl group of 3 required only 1.4 kcal mol−1 for dissociation (Fig. S2, ESI†). Additional calculations using BPh3 and C(C6F5)3+ groups also suggested that the introduction of electron-withdrawing fluorine atoms makes huge difference in the dissociation energies, as perfluorinated substituents have much higher electrophilicity (see the ESI†). The dissociation of the trityl group from 3 was also evidenced by the crossover experiment: adding 2 equivalents of B(C6F5)3 to a solution of 3 successfully generated 2 along with the trityl group as observed by EPR (Fig. S7, ESI†) and UV-vis (Fig. S12, ESI†).
In summary, two different radicals 2 and 3 were synthesised from 1 and B(C6F5)3 or Ph3C+B(C6F5)4−, respectively. The radical zwitterion 2 showed remarkable stability toward air, moisture, and even toward silica. On the other hand, 3 showed limited stability upon exposure to air and moisture, showing a half-life of about 35 minutes in wet benzene solution. The difference of stability is mainly because the trityl moiety of 3 binds weaker than the B(C6F5)3 group of 2 to the oxygen, as analysed via DFT calculations. With the help from the novel properties of NHCs, this work clearly shows a successful example of designing a stable radical. Possible applications of the stable radical 2 are currently under active investigation.
This work was supported by Institute for Basic Science (IBS) [IBS-R007-D1]. The X-ray diffraction experiment with synchrotron radiation was performed at the Pohang Accelerator Laboratory (Beamline 2D).
Conflicts of interest
There are no conflicts to declare.Notes and references
- I. Ratera and J. Veciana, Chem. Soc. Rev., 2012, 41, 303–349 RSC.
- R. G. Hicks, Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds, John Wiley & Sons, Ltd, 2010 Search PubMed.
- (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (b) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed; (c) A. Igau, H. Grutzmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS; (d) A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS; (e) V. Lavallo, Y. Canac, C. Prasang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed; (f) T. W. Hudnall and C. W. Bielawski, J. Am. Chem. Soc., 2009, 131, 16039–16041 CrossRef CAS PubMed; (g) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed.
- (a) C. D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2013, 4, 3020–3030 RSC; (b) D. P. Curran, A. Solovyev, M. Makhlouf Brahmi, L. Fensterbank, M. Malacria and E. Lacote, Angew. Chem., Int. Ed., 2011, 50, 10294–10317 CrossRef CAS PubMed; (c) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed; (d) J. P. Moerdyk, D. Schilter and C. W. Bielawski, Acc. Chem. Res., 2016, 49, 1458–1468 CrossRef CAS PubMed; (e) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS PubMed.
- (a) O. Back, M. A. Celik, G. Frenking, M. Melaimi, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2010, 132, 10262–10263 CrossRef CAS PubMed; (b) O. Back, B. Donnadieu, P. Parameswaran, G. Frenking and G. Bertrand, Nat. Chem., 2010, 2, 369–373 CrossRef CAS PubMed; (c) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930–5933 CrossRef CAS PubMed; (d) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610–613 CrossRef CAS PubMed; (e) H. Tanaka, M. Ichinohe and A. Sekiguchi, J. Am. Chem. Soc., 2012, 134, 5540–5543 CrossRef CAS PubMed; (f) K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, I. Tkach, H. Wolf, D. Kratzert, R. Herbst-Irmer, B. Niepotter and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 1801–1805 CrossRef CAS PubMed.
- (a) J. K. Mahoney, D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2013, 135, 18766–18769 CrossRef CAS PubMed; (b) D. Martin, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7014–7017 CrossRef CAS PubMed; (c) J. K. Mahoney, D. Martin, F. Thomas, C. E. Moore, A. L. Rheingold and G. Bertrand, J. Am. Chem. Soc., 2015, 137, 7519–7525 CrossRef CAS PubMed; (d) C. L. Deardorff, R. Eric Sikma, C. P. Rhodes and T. W. Hudnall, Chem. Commun., 2016, 52, 9024–9027 RSC; (e) V. Regnier, F. Molton, C. Philouze and D. Martin, Chem. Commun., 2016, 52, 11422–11425 RSC; (f) J. K. Mahoney, R. Jazzar, G. Royal, D. Martin and G. Bertrand, Chem. – Eur. J., 2017, 23, 6206–6212 CrossRef CAS PubMed.
- (a) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2017, 139, 15620–15623 CrossRef CAS PubMed; (b) Y. Li, K. C. Mondal, P. P. Samuel, H. Zhu, C. M. Orben, S. Panneerselvam, B. Dittrich, B. Schwederski, W. Kaim, T. Mondal, D. Koley and H. W. Roesky, Angew. Chem., Int. Ed., 2014, 53, 4168–4172 CrossRef CAS PubMed; (c) L. Jin, M. Melaimi, L. Liu and G. Bertrand, Org. Chem. Front., 2014, 1, 351–354 RSC; (d) M. M. Hansmann, M. Melaimi and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2206–2213 CrossRef CAS PubMed; (e) M. M. Hansmann, M. Melaimi, D. Munz and G. Bertrand, J. Am. Chem. Soc., 2018, 140, 2546–2554 CrossRef CAS PubMed.
- (a) L. Y. Eymann, A. G. Tskhovrebov, A. Sienkiewicz, J. L. Bila, I. Zivkovic, H. M. Ronnow, M. D. Wodrich, L. Vannay, C. Corminboeuf, P. Pattison, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2016, 138, 15126–15129 CrossRef CAS PubMed; (b) J. Back, J. Park, Y. Kim, H. Kang, Y. Kim, M. J. Park, K. Kim and E. Lee, J. Am. Chem. Soc., 2017, 139, 15300–15303 CrossRef CAS PubMed.
- O. Back, B. Donnadieu, M. von Hopffgarten, S. Klein, R. Tonner, G. Frenking and G. Bertrand, Chem. Sci., 2011, 2, 858 RSC.
- (a) J. Park, H. Song, Y. Kim, B. Eun, Y. Kim, D. Y. Bae, S. Park, Y. M. Rhee, W. J. Kim, K. Kim and E. Lee, J. Am. Chem. Soc., 2015, 137, 4642–4645 CrossRef CAS PubMed; (b) S. Roy, A. C. Stuckl, S. Demeshko, B. Dittrich, J. Meyer, B. Maity, D. Koley, B. Schwederski, W. Kaim and H. W. Roesky, J. Am. Chem. Soc., 2015, 137, 4670–4673 CrossRef CAS PubMed; (c) S. Styra, M. Melaimi, C. E. Moore, A. L. Rheingold, T. Augenstein, F. Breher and G. Bertrand, Chem. – Eur. J., 2015, 21, 8441–8446 CrossRef CAS PubMed; (d) A. D. Ledet and T. W. Hudnall, Dalton Trans., 2016, 45, 9820–9826 RSC; (e) S. Kundu, P. P. Samuel, S. Sinhababu, A. V. Luebben, B. Dittrich, D. M. Andrada, G. Frenking, A. C. Stuckl, B. Schwederski, A. Paretzki, W. Kaim and H. W. Roesky, J. Am. Chem. Soc., 2017, 139, 11028–11031 CrossRef CAS PubMed; (f) S. Kundu, S. Sinhababu, S. Dutta, T. Mondal, D. Koley, B. Dittrich, B. Schwederski, W. Kaim, A. C. Stuckl and H. W. Roesky, Chem. Commun., 2017, 53, 10516–10519 RSC; (g) B. Li, S. Kundu, A. C. Stuckl, H. Zhu, H. Keil, R. Herbst-Irmer, D. Stalke, B. Schwederski, W. Kaim, D. M. Andrada, G. Frenking and H. W. Roesky, Angew. Chem., Int. Ed., 2017, 56, 397–400 CrossRef CAS PubMed; (h) M. F. Silva Valverde, P. Schweyen, D. Gisinger, T. Bannenberg, M. Freytag, C. Kleeberg and M. Tamm, Angew. Chem., Int. Ed., 2017, 56, 1135–1140 CrossRef CAS PubMed; (i) Y. Kim, K. Kim and E. Lee, Angew. Chem., Int. Ed., 2018, 57, 262–265 CrossRef CAS PubMed.
- (a) D. Martin, N. Lassauque, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2012, 51, 6172–6175 CrossRef CAS PubMed; (b) J. P. Moerdyk, G. A. Blake, D. T. Chase and C. W. Bielawski, J. Am. Chem. Soc., 2013, 135, 18798–18801 CrossRef CAS PubMed; (c) H. Song, Y. Kim, J. Park, K. Kim and E. Lee, Synlett, 2016, 477–485 CAS; (d) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- (a) S. Würtz and F. Glorius, Acc. Chem. Res., 2008, 41, 1523–1533 CrossRef PubMed; (b) C. Valente, S. Calimsiz, K. H. Hoi, D. Mallik, M. Sayah and M. G. Organ, Angew. Chem., Int. Ed., 2012, 51, 3314–3332 CrossRef CAS PubMed; (c) Y. Kim, Y. Kim, M. Y. Hur and E. Lee, J. Organomet. Chem., 2016, 820, 1–7 CrossRef CAS.
- (a) M. Sajid, A. Stute, A. J. Cardenas, B. J. Culotta, J. A. Hepperle, T. H. Warren, B. Schirmer, S. Grimme, A. Studer, C. G. Daniliuc, R. Frohlich, J. L. Petersen, G. Kehr and G. Erker, J. Am. Chem. Soc., 2012, 134, 10156–10168 CrossRef CAS PubMed; (b) J. C. Pereira, M. Sajid, G. Kehr, A. M. Wright, B. Schirmer, Z. W. Qu, S. Grimme, G. Erker and P. C. Ford, J. Am. Chem. Soc., 2014, 136, 513–519 CrossRef CAS PubMed.
- X. Tao, G. Kehr, X. Wang, C. G. Daniliuc, S. Grimme and G. Erker, Chem. – Eur. J., 2016, 22, 9504–9507 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, EPR spectra, X-ray crystal data, DFT calculation results. CCDC 1823243 and 1823244. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8cc01399c |
| This journal is © The Royal Society of Chemistry 2018 |