
Clinical applications of infrared and Raman spectroscopy: state of play and future challenges
Matthew J.
Baker
 a,
Hugh J.
Byrne
*b,
John
Chalmers
c,
Peter
Gardner
d,
Royston
Goodacre
d,
Alex
Henderson
d,
Sergei G.
Kazarian
e,
Francis L.
Martin
f,
Julian
Moger
g,
Nick
Stone
g and
Josep
Sulé-Suso
h
a,
Hugh J.
Byrne
*b,
John
Chalmers
c,
Peter
Gardner
d,
Royston
Goodacre
d,
Alex
Henderson
d,
Sergei G.
Kazarian
e,
Francis L.
Martin
f,
Julian
Moger
g,
Nick
Stone
g and
Josep
Sulé-Suso
h
aWestCHEM, Technology and Innovation Centre, Department of Pure and Applied Chemistry, University of Strathclyde, 99 George Street, Glasgow G1 1RD, UK
bFOCAS Research Institute, Dublin Institute of Technology, Kevin Street, Dublin 8, Ireland. E-mail: Hugh.Byrne@dit.ie
cVS Consulting, Tame Bridge, Stokesley, TS9 5NW, UK
dManchester Institute of Biotechnology (MIB), University of Manchester, 131 Princess Street, Manchester, M1 7DN, UK
eDepartment of Chemical Engineering, Imperial College London, South Kensington Campus, London, SW7 2AZ, UK
fSchool of Pharmacy and Biomedical Sciences, University of Central Lancashire, Preston PR1 2HE, UK
gBiomedical Physics, School of Physics and Astronomy, University of Exeter, Exeter EX4 4QL, UK
hInstitute for Science and Technology in Medicine, Keele University, Guy Hilton Research Centre, Thornburrow Drive, Stoke on Trent ST4 7QB, UK
First published on 27th February 2018
Abstract
Vibrational spectroscopies, based on infrared absorption and/or Raman scattering provide a detailed fingerprint of a material, based on the chemical content. Diagnostic and prognostic tools based on these technologies have the potential to revolutionise our clinical systems leading to improved patient outcome, more efficient public services and significant economic savings. However, despite these strong drivers, there are many fundamental scientific and technological challenges which have limited the implementation of this technology in the clinical arena, although recent years have seen significant progress in addressing these challenges. This review examines (i) the state of the art of clinical applications of infrared absorption and Raman spectroscopy, and (ii) the outstanding challenges, and progress towards translation, highlighting specific examples in the areas of in vivo, ex vivo and in vitro applications. In addition, the requirements of instrumentation suitable for use in the clinic, strategies for pre-processing and statistical analysis in clinical spectroscopy and data sharing protocols, will be discussed. Emerging consensus recommendations are presented, and the future perspectives of the field are assessed, particularly in the context of national and international collaborative research initiatives, such as the UK EPSRC Clinical Infrared and Raman Spectroscopy Network, the EU COST Action Raman4Clinics, and the International Society for Clinical Spectroscopy.
Matthew J. Baker is a Senior Lecturer in Chemistry at the University of Strathclyde. He obtained his MChem (UMIST 2004), PhD (University of Manchester 2007) and EPSRC LSI Fellowship (2007–2010). He then worked at the DSTL before returning to academia. He is a Director of the International Society for Clinical Spectroscopy (CLIRSPEC). |
Hugh J. Byrne is Head of the FOCAS Research Institute, DIT. He received his B.A. (mod) (1985), PhD (1989) in Physics, from Trinity College Dublin. He worked as a postdoctoral fellow in TCD (1989–91), Research Scientist at the Max-Planck-Institut für Festkörperforschung (1991–95), visiting scientist in NIMCR, Japan (1995–96), and joined DIT in 1996. He is a Director of CLIRSPEC. |
John M. Chalmers, now retired, was an industrial vibrational spectroscopist with ICI plc for 34 years. After this, he operated as a self-employed consultant for a number of years (2000–2016) during which time he also held a part-time position (2000–2010) at the School of Chemistry at the University of Nottingham. He is an Honorary Member of CLIRSPEC. |
Peter Gardner is Professor of Analytical and Biomedical Spectroscopy, School of Chemical Engineering and Analytical Science, University of Manchester. He obtained his BSc in Chemistry (1984) and PhD in Physical Chemistry (1988) from the University of East Anglia. He worked as a postdoctoral research fellow at the Fritz-Haber Institute, Berlin (1988–1992), and University of Cambridge (1992–1994), before moving to Manchester in 1994. He is a Director of CLIRSPEC. |
Roy Goodacre (FRSC, FLSW) is Professor of Biological Chemistry at the University of Manchester. His research interests are in Raman spectroscopy, SERS and MS-based metabolomics for the characterisation and understanding of biological systems; he develops novel machine learning approaches to achieve this. |
Alex Henderson is a Senior Research Data Analyst at the University of Manchester. His research interests include chemometric and cheminformatic treatment of hyperspectral image data from infrared, Raman, SIMS and MALDI experiments. He is director of both CLIRSPEC and SurfaceSpectra Ltd. |
Sergei G. Kazarian is Professor of Physical Chemistry at the Department of Chemical Engineering of Imperial College London. Most of his research is focused on the development of FTIR spectroscopic imaging and its applications for characterisation of polymeric, pharmaceutical, forensic and biomedical systems. His research also encompasses confocal Raman microscopy and tip-enhanced Raman scattering for materials characterisation. |
Francis L. Martin FIBMS is Professor of Biosciences and Biosciences theme lead at University of Central Lancashire. He has a strong interest in the application of spectrochemical methodologies in clinical and biological settings with a following interest in the computational algorithms that might be used to achieve such novel insights. |
Julian Moger is Associate Professor in Biophotonics at the University of Exeter, UK. His research is in the field of Biophotonics – the interface between Biology and Photonics that deals with the interaction between light and biological systems. Julian currently focuses on the development and application of cutting-edge optical technologies to tackle healthcare and global sustainability research challenges. |
Nick Stone holds the position of Professor of Biomedical Imaging and Biosensing and Head of Physics and Astronomy at the University of Exeter. He is a founder Director of CLIRSPEC. Nick has worked to pioneer the field of novel optical diagnostics within the clinical environment. He has published over 150 papers and book chapters. |
Josep Sulé-Suso is Professor of Oncology at the University Hospital of North Midlands and Keele University, UK. He obtained his MB ChB in Barcelona (1987) and PhD in Medicine from Keele University (2003). He was a Research Fellow at the National Cancer Institute, Milan, Italy (1993–1995). He is a founding member of the CLIRSPEC. |
Introduction
The potential of vibrational spectroscopy for biomedical applications has been well established through many proof of concept studies over the past decades.1–4 Due to its unique fingerprinting capability, vibrational spectroscopy can play a significant role in histopathology, cytology, biopsy targeting, surgical targets, treatment, monitoring and drug studies. However, translation into the clinical environment has been slow, and a recent discussion paper has outlined the challenges facing progress to the realisation of applications potential.5 At present, the field of spectral diagnosis is in a dynamic evolutionary phase, and a number of key issues remain to be addressed; there has been very little progress towards standardisation of either sample preparation, measurement protocols or reporting and very few studies on the scale of clinical trials have been undertaken. Although this is an exciting time, the lack of consistency in the field may deter investors, hamper product development and delay translation.Progress towards the objectives of translation requires close collaboration across the range of disciplines involved, and a clear identification and understanding of what the un-met clinical needs are, and the limitations of current “gold standard” techniques. These techniques are based on subjective clinical evaluation of morphological changes in cells and/or tissue, and therefore consultation with clinicians is imperative, such that improved performance of spectroscopic approaches can be compared to consensus standards.
Understanding the complexities of light interaction with the physically and chemically complex milieu of biological samples is, in itself, a challenge. Although vibrational spectroscopy has the potential to provide characteristic chemical signatures of disease, the chemical and physical inhomogeneity of the samples can confound these signatures, and, although significant progress has been made towards understanding scattering processes and dealing with them during data pre-processing routines,6–10 there remain questions surrounding interference effects in some measurement modalities.11–14
Standardisation of sample preparation and measurement protocols is also critical, and although collective efforts have been made to establish consensus in a research context,15–17 the development of protocols for the preparation of cells, tissue and biofluids for clinical spectroscopy should be primarily guided first and foremost by the demands of current clinical practice. Validation of the robustness of the techniques through inter-laboratory “round robin” or ring trials is also required.
Data pre-processing and analysis techniques remain the subject of ongoing debate and development, in an academic context.18 However, it has become increasingly important that a consensus on optimal data pre-treatment is reached and adopted such that the limits and scope of the data analysis can be established and demonstrated in a clinical setting. Equally, the requirement to establish sharing protocols for the increasingly complex datasets is rapidly emerging.
The field of clinical application of vibrational spectroscopy is also enriched by the emergence of novel techniques and methodologies which potentially add analytical capabilities and capacity, but also introduce new challenges associated with technical aspects as well as with integration into a clinical workflow.
From fundamental research to clinical translation, the field is broadly multi- and interdisciplinary, involving as it does fundamental and applied research, chemometrics, biomedical applications and ultimately clinical translation. There is, therefore, a critical need for concerted effort which integrates all stakeholders, through consortia such as the UK EPSRC Clinical Infrared and Raman Spectroscopy Network, the EU COST Action Raman4Clinics, and CLIRSPEC – the International Society for Clinical Spectroscopy. This paper reviews the state of play of efforts to realise the translation of vibrational spectroscopic techniques into the clinical setting, while addressing the challenges identified, and projecting the future perspectives of the field.
State of the art
In vivo-intraoperative characterisation of tumour resection margins
Perhaps one of the most striking recent demonstrations of the potential for clinical translation of vibrational spectroscopy has been the deployment of fibre based Raman spectroscopy for intra-operative guidance of brain surgery.19 Identification of tumour margins is critical to minimise the potential for recurrence, and this is particularly difficult for Gliomas, which have poorly defined margins. The benefit of minimising the amount of tissue removed is obvious, particularly in the case of brain surgery, and therefore the additional guidance of a spectroscopic probe to detect invasive brain cancer in situ in real time in patients is potentially invaluable. Jerym et al. demonstrated the use of a handheld contact fiber optic Raman spectroscopy probe to distinguish brain cancer (glioma) from normal brain in situ. Using state-of-the-art neuro-navigation techniques, MRI was used for visualisation and for estimating the location of each Raman measurement on the preoperative images. The system was able to measure in vivo spectra and when the data was used to build a boosted tree machine learning classification model, using the gold-standard for training, it was possible to show that the Raman signals could accurately differentiate normal brain from dense cancer and normal brain invaded by cancer cells, with a sensitivity of 93% and a specificity of 91%. The probe enabled identification of the diffusely invasive brain cancer cells at cellular resolution in patients with grade 2 to 4 gliomas. In a further study, the impact of the ambient environment of the operating theatre on the performance of the probe system was characterised.20 In a study of 35 patients, Vaqas et al. similarly deployed Raman spectroscopy to analyse tissue at the brain surface, en route to the tumour, in superficial and deep tumour zones and resection margins in vivo during surgery, demonstrating that, in comparison to matched core biopsy samples verified by routine histopathology, meningiomas and metastases as well as low and high grade gliomas could be identified with high accuracy.21 These and other recent developments of applications of Raman based spectroscopy for improving the accuracy of brain tumour surgery have been reviewed by Hollon et al.22 In the field of oral cancer, Barroso et al.23 have demonstrated discrimination between oral cancer and healthy tissue based on water content of freshly excised tongue specimens, determined by Raman spectroscopy in the high wavenumber region. This dramatic signature difference can be used to determine the location of the tumor border in oral cancer surgery.24In vivo-endoscopic probes for disease detection
Fibre based in vivo applications have also been explored for endoscopic gastrointestinal probes. Early work by Shim et al. showed the potential25 but insufficient signal to noise was obtained for early diagnosis of malignancies. Huang et al. have been exploring different probe configurations to provide diagnostic signals from the lining of hollow organs such as the oesophagus.26 These multifibre probes have been shown to be efficient in terms of light collection, and ongoing work has enabled more specific sampling of diagnostically relevant surface signals. The collaborative works of Stone et al., have been devoted to developing and testing a confocal probe able to measure signals from only the surface 100–200 μm of the oesphageal mucosa,27,28 shown to be the optimum depth for identification of early dysplastic changes leading to cancers.29 This system is currently undergoing in vivo trials, funded by UK NIHR.A sequence of early feasibility studies have shown that Raman spectroscopy can accurately identify metastatic invasion in lymph nodes from the axilla, mediastinum, and head and neck (H&N) using Raman microscopic mapping of the cut surface of the nodes.30–32 Foundation studies in head and neck malignancies using NIR Raman have enabled the demonstration that Raman can identify the pathology of swollen lymph nodes. Raman spectra can clearly be separated into reactive nodes (those swollen from reaction to infection), primary malignancies (lymphomas) and secondary malignancies (metastatic squamous cell carcinomas and adenocarcinomas) (90% sensitivity/86% specificity33).
This performance has been reproduced in the operating theatre, using a low-cost commercially available, portable probe-based Raman system to measure the molecular fingerprint of the excised sentinel lymph nodes (SLN). Greater that 85% sensitivity and 96% specificity was achieved for identification of metastatic nodes. The performance of Raman in the breast SLN feasibility study versus other intra-operative results showed Raman measurements to be equivalent to the molecular assay approaches, but they require no tissue destruction (allowing for follow up histopathology). Furthermore, Raman provided more rapid results and could be applied in vivo.34
A smart Raman needle probe has been developed and tested for potential in vivo and ex vivo use, capable of measuring Raman molecular tissue signals in <1–2 seconds down a hypodermic needle.35 The hand-held device containing the key optical components is coupled to disposable needle probe tips.36 Initial ex vivo feasibility testing of the technique was performed on excised head and neck lymph nodes from 62 patients undergoing surgery. A Principal Components/Linear Discriminant Analysis classification model tested using leave-one-patient-out cross-validation showed sensitivities and specificities of this first feasibility test exceeding 80%.37
For in vivo surgical applications, an interesting study by Ashton et al. explored the translation of Raman spectroscopic signatures of diseased tissue in to audio signals to provide real-time guidance to surgeons without disturbing the visual focus on the patient.38
The field of Raman Spectroscopy for cancer detection and cancer surgery guidance has recently been reviewed by Santos et al., demonstrating real progress towards clinical translation of these techniques and adoption by the medical community, as well as highlighting challenges.44
In contrast, there have been few demonstrations of in vivo clinical applications of mid – IR spectroscopy. However, initial studies of the skin have demonstrated that high peak power Quantum Cascade Laser (QCL) light sources may allow the measurement of signals below the stratum corneum in the skin.45 Indeed, the goal of the EU MINERVA project is to achieve in vivo MIR molecular vibrational spectroscopy,46,47 promising further developments in this aspect of the field.
Ex vivo-spectroscopic histopathology
Ex vivo applications of vibrational spectroscopy include analyses of samples taken directly from the body for diagnostic purposes, namely tissue biopsies (histological samples), cell biopsies (cytological samples) and liquid biopsies (bodily fluids such as blood plasma/serum, saliva, etc.).In general, adoption by the medical community is regarded as a key stage of development, and, in this context, minimal disruption to the normal workflow is recommended. The adoption of appropriate clinical sample preparation and presentation for spectroscopic analytical procedures is therefore critical. Ideally, spectroscopic diagnostic techniques should add technical/medical value without compromising cost and/or efficiency. In some cases, however, it may be argued that significant improvements over an established gold standard may warrant significant changes to currently accepted protocols. Such disruptive innovations may not be the most strategic targets for translation of the technology into the clinic, however, and so adaptation of the state of the art spectroscopic capabilities to the clinical environment should be prioritised, rather than vice versa. The development of protocols for the preparation of cells, tissue and biofluids for clinical spectroscopy should therefore be guided first and foremost by current clinical practice.
In the context of surgical procedures, an important application of spectroscopic monitoring has been identified as “near patient” screening of tissue biopsies, taken intra-operatively to guide the surgeon in identifying tumour resection margins. Ideally, analysis should be performed on fresh tissue, and screening time, including data processing, should be kept to a minimum.
The incorporation of Focal Plane Array (FPA) detectors in FTIR microscopes has greatly improved data acquisition and processing speeds,48 enabling the acquisition of several thousand spectra simultaneously. Nevertheless, images from a large area of tissue (cm2) or from a large tissue micro array (TMA), often take several hours to acquire. Data analysis and management of these burgeoning data sets is also a major problem. Reducing the spectral resolution or reducing the number of co-added scans can reduce acquisition times but ultimately deteriorates the quality of the data. Recently, it has been suggested that the full spectral range is not required for accurate disease diagnosis using spectral histopathology.49,50 These studies suggest that, once spectral biomarkers are known, it may be possible to diagnose disease based on a limited number of discrete spectral features. In this context, recent developed high intensity infrared sources, including free electron lasers (FEL), optical parametric oscillators (OPO) super continuum and QCLs, have indicated a route towards significant further advances in the field.
QCLs51 are high intensity, tuneable semiconductor lasers which can emit across the mid infrared region of the electromagnetic spectrum. Availability of these high intensity tuneable sources has renewed interest in unmultiplexed, scanning infrared spectrometers utilising discrete frequencies.52 In a recent study,53 a QCL imaging system was used to image a large TMA consisting of 207 breast core biopsies. In addition to rapid imaging at a pixel size of 4.25 × 4.25 μm2, the high magnification optical configuration with a pixel size of 1.36 × 1.36 μm2 can measure high definition images providing a greater level of detail of finer structures not visible using a conventional desktop FTIR imaging system that has a pixel size of 5.5 × 5.5 m2 (Fig. 1). Using the QCL microscope to collect a chemical image of the amide I band, the full TMA composed of a 10 × 12 array of infrared tiles was collected in just 9 minutes. Measurements on the same sample using a state of the art FTIR FPA system required approximately 19 hours. Although the direct comparison is unfair, in that the FTIR collects the full spectral range and the QCL only collected one discrete frequency, the potential speed advantage over FTIR is undeniable, enabling the possibility of high throughput imaging of tissue biopsies from a large numbers of patients. One consideration is the speed of collection for each infrared tile when multiple frequencies are being collected. FTIR spectroscopy collects all wavelengths simultaneous, and sufficient scans are co-added to ensure adequate signal to noise. QCLs are not continuous sources, requiring the source to be tuned to the desired wavelength, and data points need to be recorded stepwise for each frequency of interest.54 Despite this, once spectral biomarkers have been identified, the instrument could be assigned to collect a limited number of key features, enabling high throughput imaging. Recent work has shown that excellent classification of cancerous sample can be achieved with as few as 25 frequencies, although classification accuracy starts to drop off rapidly as the number of frequencies is reduced.55
 | ||
| Fig. 1 (a) Expanded view of TMA core (b) FTIR chemical image of amide I, pixel size = 5.5 × 5.5 μm2. (c) QCL chemical image of amide I, pixel size = 1.36 × 1.36 μm2.53 | ||
Although the speed advantage of QCL discrete frequency imaging is clear, the main focus of research has been on throughput rather than accurate disease diagnosis. There are still significant obstacles that need to be overcome before such technology can be used routinely. For example, it is still not clear how to deal with baseline correction when measuring highly scattering samples such as glandular prostate tissue. Acceptance of this new technology will depend on whether high quality infrared spectra can be rapidly acquired and still maintain the diagnostic accuracy demonstrated using FTIR spectroscopy. Furthermore, given that imaging with a QCL system presents a quantum leap in current technology, it needs to be demonstrated that infrared spectra similar to those using a conventional FTIR spectroscopic system can be acquired. Despite these exciting developments, large scale trials are now required to correctly assess the future of the technology, and its potential in the clinical setting.
For screening of large sample areas such as tissue biopsies, the required pixel dwell times of the order of 0.1 to 10 seconds limits the application of high-resolution Raman microspectroscopy in many areas of clinical practice, particularly histological screening. Coherent Raman scattering (CRS)56 provides enhancement of signals by several orders of magnitude and, when applied in a microscopy format, benefits from the non-linear nature of the process which confines the signal to a sub-micron focus that can be scanned in space, allowing rapid 3D mapping of bio-molecules with sub-micron resolution.57 CRS microscopy can be implemented by recording the coherent anti-Stokes Raman scattering (CARS) or stimulated Raman scattering (SRS) signal. While the former has gained popularity for rapid screening of large areas, its nonlinear dependence on the number of scatterers means that the spectral profiles are not trivially interpreted, and so SRS is favoured for analytical applications.58
While other reviews have provided a more comprehensive summary of the technical implementation and applications of CRS,59–61 this report aims to highlight the current state-of-the-art in the clinical translation of CRS which is paving the way towards its integration into widespread clinical use. The applications of CRS microscopies to clinical and biological studies have also been reviewed by Schie et al.62Fig. 2 shows an example of the use of CARS for the rapid subcellular analysis of Hep2 liver cells.62 CRS techniques also have significant potential to overcome the time limitations of conventional histology. Ji et al.63 demonstrated that SRS microscopy enables rapid, in situ label-free acquisition of H&E like images based on the ratio of Raman signals at 2930 and 2845 cm−1, reflecting the different lipid and protein contents. Using this technique, they were able to differentiate tumour from non-neoplastic tissue in a human glioblastoma xenograft mouse model and demonstrated excellent correlation between SRS and conventional H&E microscopy for detection of glioma infiltration. Orringer et al.64 have demonstrated the clinical translation of SRS microscopy for intraoperative tumour margin detection, a significant innovation being the application of a balanced-detection scheme that allowed SRS to be performed using compact portable fibre lasers and an image-processing method (stimulated Raman histology, SRH) to convert the SRS signals into virtual H&E slides so the existing clinical expertise could be applied to make on the spot decisions, in this application, an SRH mosaic of ∼0.6 × 1.0 mm was recorded in ∼2.5 minutes. SRH was shown to have remarkable agreement with conventional histology, showing an accuracy >92%. Most recently, Francis et al.65 introduced an improvement to narrowband SRS pathology through spectral slicing of broadband (femtosecond) pulses. Drawing upon the higher peak power of femtosecond pulses while preserving the spectral resolution of picosecond pulses, they demonstrated the same chemical specificity as narrowband SRS while achieving a higher signal-to-noise ratio (SNR).
 | ||
| Fig. 2 (a) CARS image of cellular lipid droplet accumulations in HepG2 cells after the exposure to 250 μM of oleic acid (OA) and 250 μM palmitic acid (PA). Raman spectra were taken from the indicated cellular lipid droplets. The pie charts show the relative amount of oleic and palmitic acid in the individual lipid droplets determined from the Raman spectra. The percentile values are plotted below. The GC values indicate the amount for OA and PA determined from lipid droplets of millions of cells. It can be seen that the values based on Raman spectroscopy and GC are in good agreement. (b) Multiphoton image of HepG2 cells. The CARS signal from cellular lipid droplets is in green, and TPEF signal of green fluorescence protein expressed in peroxisome in blue. The unnormalized Raman spectra for the indicated point are shown below. The peak intensity of the CH2 stretch vibration at 2854 cm−1 is 9.8 times lower from peroxisome [ref. 62, reproduced with permission]. | ||
The examples above rely on narrowband (picosecond) excitation, in which the excitation bandwidth is confined to the line width of the Raman mode used for contrast. While this scheme provides efficient excitation of single Raman bands, spectral mapping requires sequential acquisition of multiple Raman modes to acquire morphochemical contrast. However, for spectral imaging, they suffer from limitations in motion artefacts between acquisition of images at sequential Raman shifts and the speed of laser tuning. Broadband CRS techniques overcome this limitation by using broadband (femtosecond) pulses to simultaneously excite multiple Raman bands which are then separated using spectroscopic detection. There are many examples of biomedical applications of broadband CRS, and, notably Camp et al.66 recently presented a technique for efficient hyperspectral CRS image acquisition over the full spectral range (500–3000 cm−1) using intrapulse three-colour excitation to achieve high-resolution microspectroscopy with pixel dwell times of a few microseconds.
Ex vivo-spectroscopic cytology
Cytological screening is commonly employed as a routine preventative measure for early stage disease detection, notably for cervical and oral disease.67,68 In the case of cervical, screening programmes are well established in the “first world” and so sample throughput is very high. This puts increased demands on the cost effectiveness of alternatives or adjuncts to currently employed clinical practice.The use of vibrational spectroscopy as an adjunct or alternative to currently employed cytological screening methods may be a viable strategic target objective. Particularly in the case of automated liquid based cytology methods, it can conceivably be easily integrated into the workflow. Using Raman spectroscopy to characterise Thin Prep exfoliated cervical cytological samples, Ramos et al. reported sensitivities and specificities of 94% and 95% for low grade squamous intraepithelial lesion and 84 and 100% high grade squamous intraepithelial lesion.69 When calculated according to the histological grading system of cervical intraepithelial neoplasia, the sensitivities and specificities improved further. Sensitivity to HPV infection has also been reported promising an integrated screening for early stage disease.70
A limited number of infrared absorption spectroscopy studies have demonstrated the feasibility of using oral exfoliated cells. Papamarkakis et al.71 demonstrated that oral cells could be classified according to anatomical region through principal components analysis (PCA). In addition, samples from patients with reactive atypical changes or malignancy associated changes were seen to be spectrally similar to a sample from a patient with squamous cell carcinoma, rather than samples from healthy volunteers. Furthermore, spectral changes were identified in oral cells infected with the herpes simplex virus. Further studies from the same group72,73 showed that spectra from exfoliated cells from the tongues of healthy volunteers could be discriminated from exfoliated cells from the tongues of patients with oral dysplasia and cancer. Although there have been few studies of exfoliated oral cells, the protocol for cervical spectrocytology should be relatively easily translated, as demonstrated recently by Behl et al.74
Ex vivo-liquid biopsies
Screening of bodily fluids (“liquid biopsy”) is a further ex vivo application which is currently attracting increasing attention and may represent a strategic, achievable target for clinical translation.75 Biofluids offer an ideal diagnostic medium due to their routine use, ease of collection, acceptance by patients and the fact that they offer the ability to provide a snapshot of human health (e.g. via systemic bodily fluids such as blood (serum, plasma etc.)) and specific tissue function assessment via specific biofluids (e.g. bile, sputum etc.)76 Suspended or dissolved analytes are present in rather low concentrations, however, and many studies to date have been performed on dried samples.77,78Pioneering work by Petrich et al. demonstrated the use of serum spectroscopic diagnostics to be able to aid in the triage of patients with acute chest pain discriminating between acute myocardial infarction and chest pain of other origin to sensitivities and specificities of 88.5% and 85.1% respectively.79 Recent work on serum spectroscopic diagnostics has shown the ability of ATR-FTIR spectroscopy to distinguish between a brain cancer serum set of 433 patients (3987 spectra) demonstrating the ability to distinguish between cancer vs. non-cancer, metastatic cancer vs. organ confined, brain cancer severity and organ of metastatic disease with optimum sensitivities and specificities between 80.0 and 100.0% depending upon the question being asked.80 In addition, Raman spectroscopy has recently been shown to be sensitive to disease signature for bodily fluid analysis by enabling the discrimination of cirrhotic patients with hepatocellular carcinoma (HCC) and without to overall rates of accuracy of 84.5% to 90.2% for dried serum drops and 86% and 91.5% for freeze-dried serum drops.81 Importantly this study was conducted with a benign inflammatory disease background showing the capability to distinguish the malignant disease.
Translational work by the group from Monash University, based on the use of ATR-FTIR spectroscopy for the determination of malaria parasitemia in whole blood samples,82 has been recently demonstrated in field trials in austere environments proving the robustness and capability of serum biofluid diagnostics.
In vitro-drug screening and companion diagnostics
In vitro studies of cell cultures are plentiful in the literature and are invaluable for the development and validation of new measurement technologies, measurement and data processing and analysis protocols, and ultimately the exploration of the limits of the techniques in identifying and screening biomarkers associated with biological function and dysfunction. The techniques may also provide valuable information on, for example, radiation and chemotherapeutic resistance, and present opportunities in their own right for potential applications in screening for drug delivery mechanisms and efficacy, radiation damage and toxicology,83–88 given the drive for a reduction in the use of animal models for evaluating toxicity, due to regulatory developments in both the EU and US (EU Directive-2010/63/EU and US Public Law 106–545, 2010, 106th Congress) generally based on the principle of the 3 R's, to replace, reduce and refine the use of animals used for scientific purposes.In this context, the development of 3D cell culture models which better mimic the in vivo environment is critical. As an example, multicellular tumour spheroids are commonly used to provide a better model of tumour structure,89,90 in particular to model redox and pH gradients, which can be probed by functionalised surface enhanced Raman spectroscopy probes.91
The ability of vibrational spectroscopy to characterise responses of resistant and sensitive cell types to drugs opens up potential clinical applications as a Companion Diagnostics (CD) tool, and ultimately personalised medicine approaches.92,93 Companion diagnostics is a fast-emerging area of in vitro diagnostics that seeks to identify a patient's response to a specific therapy and thus guide disease intervention. Currently, Companion Diagnostic techniques rely on single biomarker detection, but, with increasing understanding of disease pathophysiology, advanced techniques need to be developed, based on patient profiling and multiplex platforms.
Farhane et al.94,95 demonstrated that Raman spectroscopy could be employed to differentiate the respective responses of two different lung cancer lines (A549 and Calu I) to doxorubicin (DOX) exposure, indicating that Raman can be employed to help understand cellular resistance pathways. Cellular resistance pathways have also been targeted by Yosef et al.96 and El-Mashtoly et al.,97 who used Raman spectral imaging to investigate the oncogenic mutation resistance to epidermal growth factor receptor targeting therapy. Specifically, in related studies, colon cancer cells with and without oncogenic mutations such as KRAS and BRAF mutations were treated with erlotinib and pantinolib, inhibitors of epidermal growth factor receptor, in order to detect the impact of these mutations on Raman spectra of the cells as markers of cell resistance. Rutter et al.98 utilised a cell cloning technique to specifically isolate sensitive and resistant cells from a mixed cell population, and investigated the difference in response of gemcitabine-sensitive and gemcitabine-resistant CALU-1 epidermoid lung cancer cells to the commercial drug gemcitabine, using IR spectroscopy. Furthermore, Siddique et al. showed that it was also possible to identify differences of nilotinib-sensitive and nilotinib resistant K562 (a chronic myelogenous leukaemia cell line) cloned cells, using both FTIR and Raman microspectroscopies.99
Technological challenges
In vivo-fibre probes
The major technological challenges of in vivo use of vibrational spectroscopies lie in the limited penetration depth of mid-IR light in water rich tissues and the relatively weak signals from Raman scattering, which can be compromised by background signals from measurement systems, scattering and/or fluorescence. Use of near-infrared laser illumination can minimise tissue fluorescence contributions, but signals from the silica fibres can dominate Raman spectra without recourse to careful optical design and filtration; these signals can be subtracted but the accompanying shot noise can not. This may be sufficient to impact on the relatively small Raman signals obtained in the ideally short time scales. In vivo measurements need relatively low intensity illumination to ensure no thermal damage ensues and short acquisition times (a few seconds or less) to minimise movement and patient discomfort. A recent review provides more detail on the strategies to optimise in vivo Raman measurements with Raman probes.100Beyond accessing disease specific molecular signals using fibre probes, the rapidly developing technique of deep Raman spectroscopy is emerging as a powerful in vivo tool. Early studies, in pioneering the field of deep Raman spectroscopy for biomedical applications, have established the basic feasibility of recovering Raman signals (both native and exogenous) from depths of several centimetres beneath the surface of animal tissue. This is, by around two orders of magnitude, deeper than achievable with conventional approaches such as confocal Raman microscopy. Deep Raman sampling involves the use of either transmission (TRS) illumination and collection geometries, or spatially offset Raman spectroscopy (SORS), whereby illumination and collection points are spatially separated on the outer surface of the tissue.39–41 Initial work has involved the study of signals from substances such as those found in calcified tissues, whereby the signal is distinct from that found in soft tissues. Clinically relevant concentrations of calcifications have been detected to depths of up to 40 mm in animal tissue phantoms (i.e. at near clinically relevant depths; X-ray mammographic screening compression ranges from 1.9 to 5 cm thick) demonstrating the basic viability of scanning human breasts.42 This penetration depth was accomplished by dramatically increasing the Raman photon gathering rates (by around 100 times) when compared with the first TRS Raman system used.43 This indicates the feasibility of using TRS for non-invasive analysis of breast tissues in vivo.
One of the most significant requirements of in vivo Raman systems relates to the specific sampling volume of the clinical application and sampling method. NIR light is highly scattered and minimally absorbed in tissue, and therefore optical designs that do not restrict light collection to the surface scattered photons can result in deep signal collection, which may confound results. An example of this is when no beam steering or focussing is used in hollow organ endoscopic fibre probes. The relevant diagnostic signal may originate from the surface 100–200 μm in organs such as the oesophagus or bladder, and any deeper signals may contain contributions from, for example, normal cell division and adipose tissue on the outer organ surface.101 As an illustration, Fig. 3 compares spectra obtained using a confocal Raman microscope of bladder biopsy surfaces versus that obtained with a simple in vivo probe design of six collection fibres around one illumination fibre.
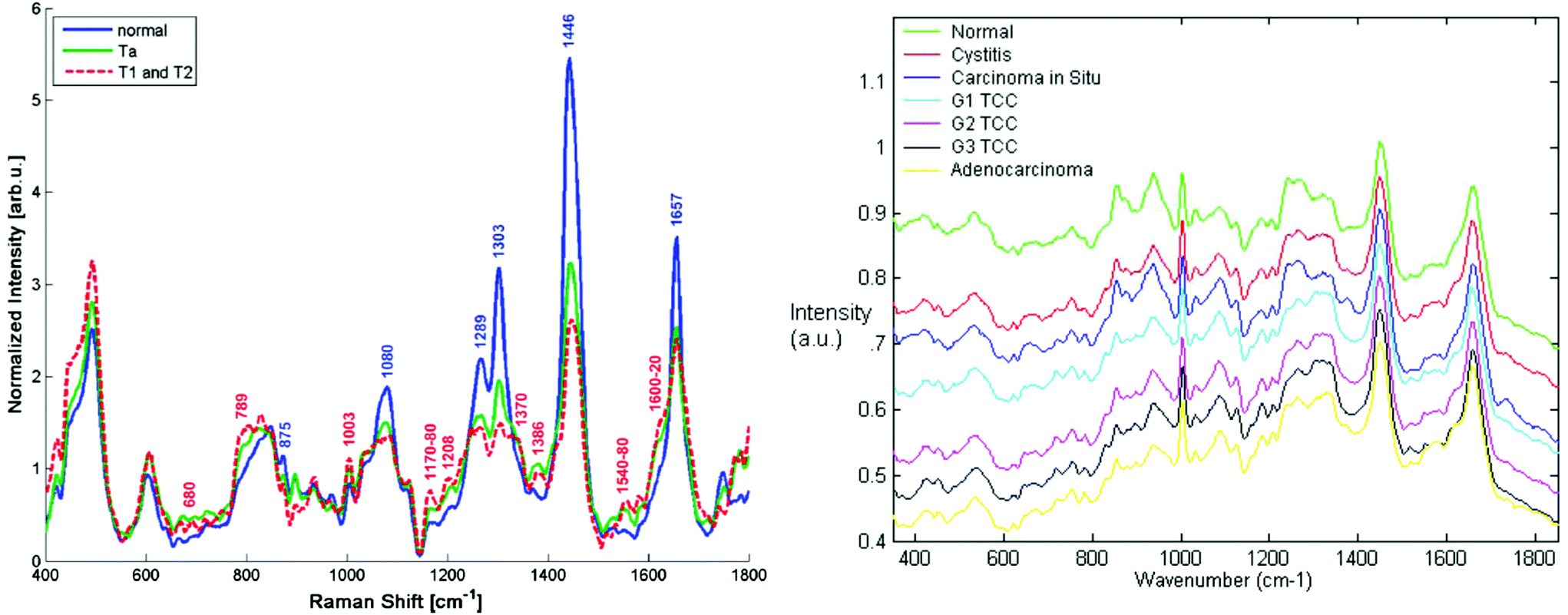 | ||
| Fig. 3 Comparison of Raman spectra from the bladder surface (left) measured using a six collection fibres around one illumination fibre probe with no beam steering/focussing at 785 nm (ref. 101 reproduced with permission) and (right) Raman spectra measured from bladder biopsy surface using a semi-confocal, 830 nm Raman microspectrometer (ref. 102 reproduced with permission). The right hand spectra are the ‘true’ surface spectra, most demonstrating early malignant changes, whereas the in vivo spectra show spectra combining the spectral signals from all layers in the bladder wall including the adipose tissue surrounding the outside of the bladder wall. This is clear from the highly lipid-rich spectra and highlights the importance of designing probes to optimally sample tissue regions/volumes of interest. | ||
Ex vivo-spectroscopic histopathology
The long standing problem of optical aberrations in FTIR microspectroscopy have been systematically addressed,103–108 by introducing a novel, practical, yet elegant, approach to remove scattering artefacts, whereby the tissue biopsy is inverted such that the CaF2 window sits between the biopsy and objective. Then, an added lens, of the same material as the window, is placed on top of the window, centred to the optical axis, to form a pseudo hemisphere. In this way, the rays of light from the sample to the objective pass normal to the CaF2 surface and do not refract. The scattering is also reduced or removed as the refractive index of the biopsy more closely matches that of the CaF2 than it does air. Both these advantages make this added lens approach ideal for measuring biopsies.106,108 The originality and simplicity of this approach has surprised many working in the field, as it was thought that optical aberrations are unavoidable when infrared windows were used as a substrate.109 The other important advantages to this added lens approach are the increase in magnification and the removal of chromatic aberration, the latter of which would otherwise occur when measuring samples through a window, for example imaging of live cells in microfluidic devices.108,110,111 As such, this method is a state-of-the-art method to acquire high-quality spectra without chromatic aberration and scattering.High spatial resolution in spectroscopic imaging is vital for revealing histological domains or structures on the micrometre scale within biological samples. Until recently, biomedical applications of FTIR spectroscopic imaging have mainly been performed in transmission mode but it has been demonstrated that FTIR imaging in ATR mode can provide images with much higher spatial resolution. In particular, a stringent test for the assessment of the spatial resolution achieved in FTIR spectroscopic imaging in both ATR and transmission modes has been presented and it has been demonstrated that the true spatial resolution is much lower than what is expected from the projected pixel size, which is often used, albeit incorrectly, as a value for spatial resolution.103,105,108 Recently micro ATR-FTIR spectroscopic images of living cells have been analysed with disrelation mapping which allowed differentiating molecular states of water as well as studying hydration of proteins within living cells (Fig. 4).112 he most recent developments in the area of FTIR spectroscopic imaging have been reviewed, where the current state of the art is summarised in detail.106,108,113–116 Notably, Kimber and Kazarian specifically highlight improvements in spatial resolution and spectral quality through the use of novel added lenses and computational algorithms, as well as quantum cascade laser imaging systems, which offer advantages over traditional Fourier transform infrared systems with respect to the speed of acquisition and field of view.117
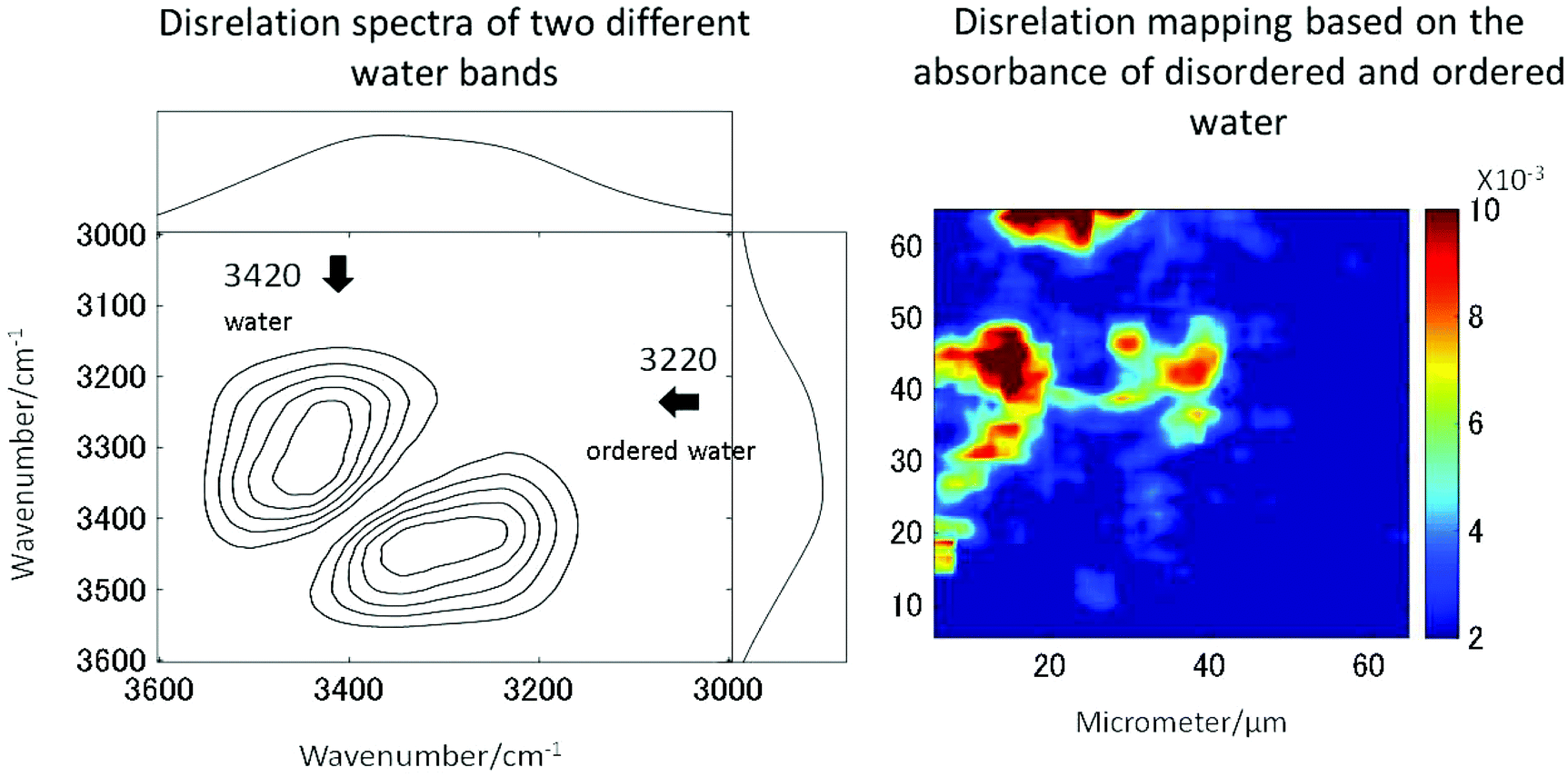 | ||
| Fig. 4 Two molecular states of water in HEK cell. The disrelation map emphasises the regions where water molecules interact with proteins (i.e. hydration). Reproduced from ref. 112. | ||
In the case of infrared spectral pathology (using either FTIR or QCL microscopes), there are technological challenges associated with the most appropriate substrate to use. The obvious choice, from the spectroscopists point of view, is to use an infrared transparent substrate such as CaF2 or BaF2, both of which also happen to be transparent in the visible, and contribute minimally to Raman spectra in the regions of interest for biological samples, and are therefore ideal for microscopy applications. The low frequency cut-off for these materials is ∼900 cm−1 and 700 cm−1, respectively, meaning that most of the important biological spectral features can be recorded. The use of such slides for thin tissue sections, measured in standard transmission mode, results in clean spectra with very little spectral distortion other than scattering from the sample itself. However, these slides are brittle and less robust than glass meaning that there is a significant risk of fracturing in the automated sample preparation equipment commonly used in pathology labs.
An alternative solution is to use infrared reflective microscope slides. These can be stainless steel, aluminium or more commonly a glass slide with a reflective coating, such as commercially available low e-slides,103 whereupon the measurement is performed in reflection mode (more commonly known as transflection mode). The transflection mode effectively doubles the path-length through the sample giving better signal to noise ratio for the same thickness and they are relatively inexpensive. The cost is a significantly less than that of CaF2 or BaF2 although, at ∼€1 each, they are still slightly more expensive than conventional glass. The drawback of the transflection mode is that the spectra often display frequency dependent non-Beer–Lambert absorption behaviour as a function of thickness. Brooke et al.104 first reported this phenomenon explicitly when investigating multilayer polymer spheres and found that absorption band intensities did not scale linearly with the thickness of the films. They suggested that the variation could be approximated by a squared sinusoidal function and proposed that the effect was caused by the electric field standing wave (EFSW), a view supported by Filik et al.13 The EFSW effect occurs due the interference of the incident and reflected wave. An oscillating electric field in the plane of the reflecting surface is very effectively screened such that it forces a node at the surface. This means that the strength of the electric field just above the surface, within a wavelength or so, is frequency dependent, and therefore different thicknesses of tissue will give slightly different intensity spectra even when normalised. The work of Bassan et al.11,12 conclusively demonstrated the existence of the physical phenomenon and highlighted the implications this could have on the analysis of tissue. They showed that the classification accuracy was significantly influenced by sample thickness, which was not the case for the same samples measured in transmission. This work led to a flurry of papers on the subject, making the case either for or against the use of transflection. In the case of the former, several authors suggested that the use of the second derivative spectrum essentially negates the non-Beer–Lambert absorption behaviour, while others suggested that all tissue studies should be performed in transmission. Recently Lee105 has cast doubt on the EFSW being responsible for the phenomenon, suggesting instead that it is due to a simple interference effect between the reflecting surface and the surface of the sample layer. Regardless, it is important to note that the non-linear absorption as a function of thickness is not in dispute, and that therefore measurements in the transflection mode may compromise diagnostic accuracy.
A key question in terms of sample preparation for Clinical Applications, both histological and cytological, is, can we afford not to work with glass microscope slides? In the research environment, many of the proof of concept studies have been conducted with optimised experimental conditions, including choice of substrate, to minimise spectroscopic interference in the case of Raman,118 or maximise signal by transmission or transflection in the case of IR absorption.16 However, routine clinical practice, notably, for example, the high throughput ThinPrep procedure for routine cervical screening, uses standard microscope slides and the cost implications of a change in this practice must be considered.
Based on estimates from the Royal Preston Hospital, Royal Stoke University Hospital and the Coombe Women & Infants University Hospital, Dublin, for a population base of 65 million (UK, 2015), a total of 2.6 million tissue biopsies + 10 million cytological smears are carried out annually. The current cost of Raman grade CaF2 substrates, in orders of 100+, is ∼£25, implying an additional annual cost of ∼£65 million for analysis of tissue biopsies and ∼£250 million for routine cytological screening programmes, based on spectroscopy. The requirement for sample archiving implies the substrates are not reusable, and although it may be argued that widespread adoption of the techniques will, in the future, drive down the cost of the spectroscopically optimal substrates and potentially eradicate the need for associated consumables such as cellular and tissue stains, any substantially increased running cost such as this will not aid in the drive for clinical translation. The implications for adaptation of the spectroscopic protocols should therefore be considered.
Bassan et al. suggested that, for a simple case of distinguishing malignant from non-malignant tissue using FTIR transmission, a simple glass microscope slide could be used as substrate.119 Although the low wavenumber cut-off for such a side is 2500 cm−1, implying no access to any of the fingerprint region, there is still enough information in the remaining spectrum to get a reasonable classification. This work was followed up by Pilling et al. who used standard coverslipped H&E stained sections and TMA cores, and again demonstrated that tissue pathology and malignant classification could be obtained.120Fig. 5 shows the mean spectra of the different tissue regions 182 H&E stained prostate tissue cores measured in the glass transmission window, while Fig. 6 shows the false colour spectroscopic images of the classified prostate tissue cores. The ability to perform spectro-histolopathology on glass substrates is important, since this would mean no disruption to the sample preparation workflow, thus removing a significant barrier to clinical adoption. It remains to be seen, however, if more detailed diagnostic information can be obtained using such a limited portion of the spectrum.
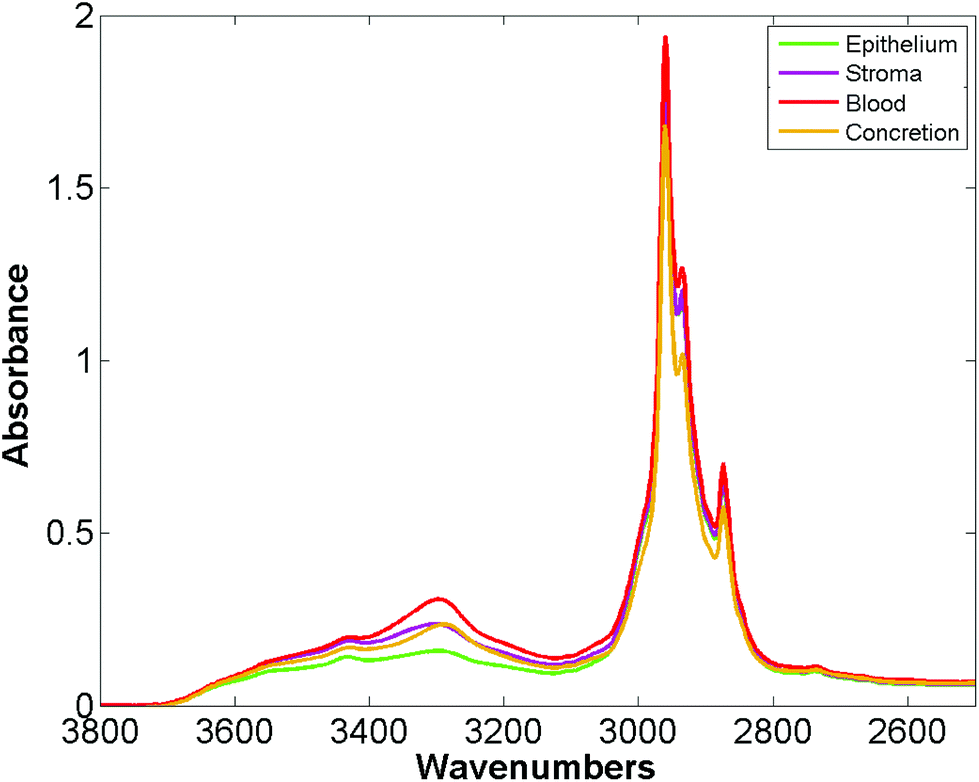 | ||
| Fig. 5 Mean spectra of epithelium, stroma, blood and concretion in the glass transmission window obtained for 182 H&E stained prostate tissue cores.120 | ||
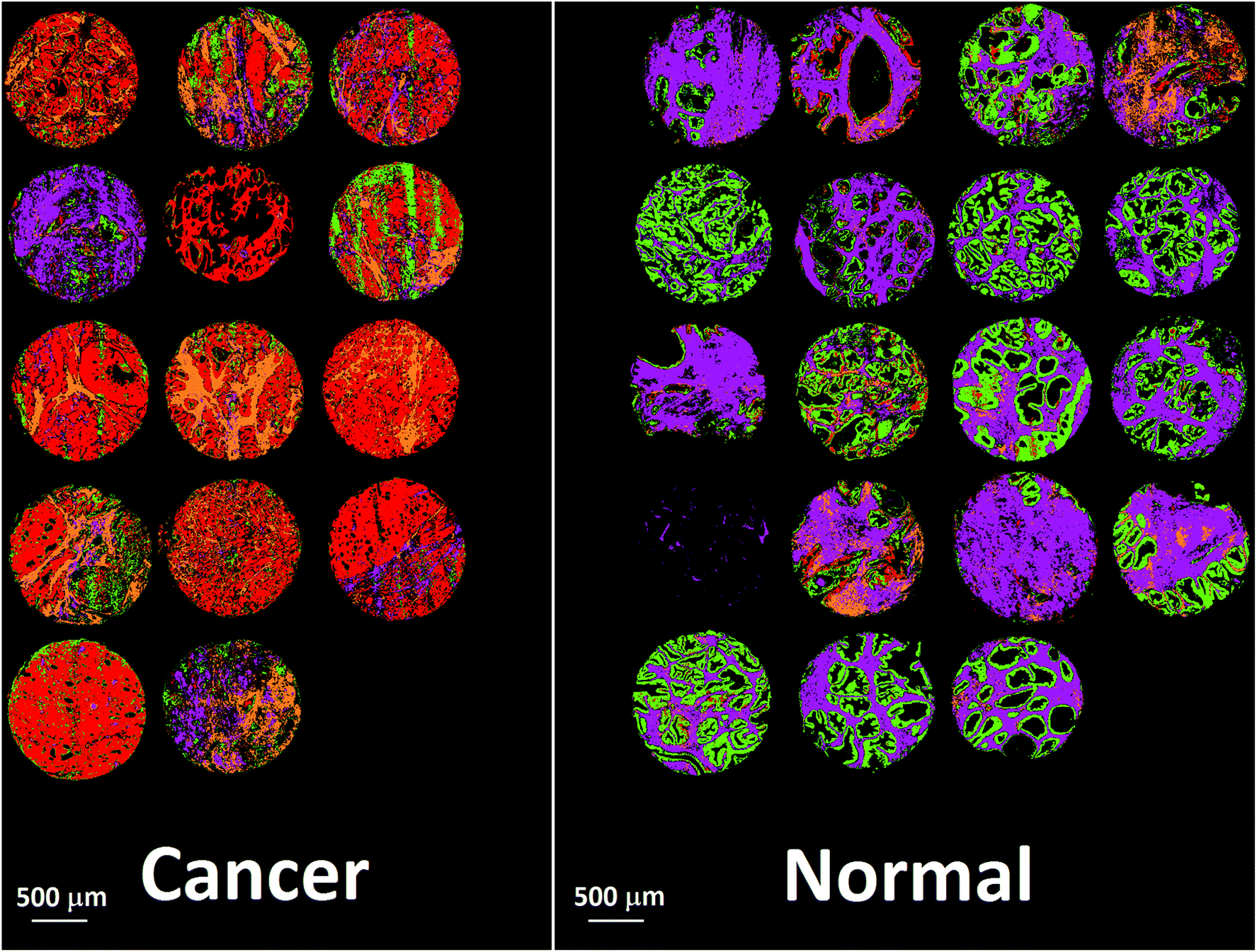 | ||
| Fig. 6 False colour image of the classified prostate tissue cores: red = malignant epithelium, orange = cancer associated stroma, green = normal epithelium, purple = normal stroma.120 | ||
The effect of substrate choice for spectral histopathology by Raman microspectroscopy, has been studied by Fullwood et al., highlighting the difficulties of operating with 785 nm on glass substrates.121 However, Raman microspectroscopy may be favoured for cytological samples because the smaller spotsize/higher spatial resolution enables specific targeting of the nucleus or even nucleoli to achieve better classification.122,123 In development of Raman protocols, choice of wavelength is intimately linked with choice of substrate, and the recent comprehensive study of substrates for cytological samples and their dependence on commonly used Raman sources by Kerr et al.118 demonstrated that, although not the optimum for spectral quality, the optimal wavelength for glass microscope slides is 532 nm. At this wavelength, the contribution of the substrate spectrum is minimum, and can be routinely subtracted as an independent component by automated routines such as Non-negatively Constrained Least Squares Analysis, as previously demonstrated for the so-called digital dewaxing procedure,8,124 or EMSC, as demonstrated by Kerr et al.125
Recently, it has been demonstrated that readily available and inexpensive aluminium (Al) foil can be used as a versatile and suitable substrate for preparing diverse cytology and histology specimens for ATR-FTIR, transflection FTIR or Raman spectroscopic measurements.126 The low and almost featureless background spectra of Al foil enables the acquisition of high-quality IR and Raman spectra without substrate interference and sacrificing important fingerprint biochemical information of biomedical specimen. Furthermore, low-cost stainless steel slides have been reported as potential candidate substrates in a routine pathology laboratory setting.127 These potential new substrates would need to be incorporated into a robust protocol acceptable to a diagnostic setting and clinically trialled.
The form of tissue presentation for measurement obviously depends on the clinical applications and sample availability. Fresh (frozen) sections are available for near patient intraoperative screening and are closest to in vivo tissue architecture and biochemistry. However, fixed sections are preferred for histopathology,128 and, for research purposes, analyses of archived tissue libraries may add much to understanding disease progression and patient prognosis. Standardised protocols for spectroscopic analysis of Formalin Fixed Paraffin Processed (FFPP) are therefore important.
Notably, it has been demonstrated that it is not necessary to remove the paraffin to obtain usable spectral information.124,129 Leaving the paraffin in place reduces scattering artefacts in FTIR and background scattering in Raman spectroscopy. Recognising the increased variability of the signal in Raman microspectroscopy due to the scattering of the polarised source by microcrystalline wax residues, improved “digital dewaxing” protocols have recently been demonstrated by Ibrahim et al., for the case or oral tissue.124 However, the debate over whether to dewax or not to dewax continues. It may be argued that greater consistency of spectral information is achieved when sections are deparaffinised, and deparaffinising enables histological staining of the sections. However, it has been noted and emphasised that the efficiency of the deparaffinisation process, and therefore staining procedures, can be very variable and even depend on the tissue pathology.130 Measurement of dewaxed tissue samples increases issues of scattering in both FTIR and Raman spectroscopy, although water immersion has been shown to substantially reduce scattering background in Raman spectra of tissue samples, due to the optical phase matching effects of the aqueous environment,131 while also substantially reducing any photothermal damage, due to the heat sinking effect.132 These effects are less problematic in cytological samples, however.
Ex vivo-spectroscopic cytology
Sample processing protocols for routine clinical cytological screening are well established for the case of cervical screening and have been shown to be compatible with spectroscopic analysis, in the case of Raman spectroscopy.69 Similar sample collection and ThinPrep preparation protocols have been adapted for analysis of exfoliated oral cells.133 In liquid based cytology applications, operation at 532 nm may imply the need for additional washing protocols. In cytology laboratories, cell suspensions prepared for ThinPrep or smear samples are annotated according to a blood scale, whereby 0 indicates a clear solution and 3 indicates a bloody sample. Samples presenting a grade 2 or 3 on the blood scale are either rejected as being unsuitable or treated using Cytolyte or various solutions to wash the cells before preparing the samples. A study by Bonnier et al., demonstrated that even samples graded as 0 or 1 can present significant influences of trace amounts of blood in their Raman profiles using 532 nm as source, resulting in a significant variability in spectral profiles of normal samples.134 Haemoglobin is resonant at 532 nm and so the Raman contribution is significantly enhanced compared to the cellular signal, resulting in the enhanced sensitivity to even trace amounts of blood in the sample. The employment of an additional hydrogen peroxide (H2O2) treatment of the ThinPrep slides was demonstrated to eradicate the impact of the blood contributions. The additional washing process was seen to have no impact on the cellular classification and could easily be introduced into the automated sample processing protocol.Operation at such wavelengths raises questions in relation to sample degradation, and an early study by Puppels et al., demonstrated that, in live cells, whereas at 633 nm negligible photodamage is observable, this is not the case at 532 nm.135 Photodamage associated with a photon energy dependence is normally associated with photoxidation, however, and the reduced oxygen content in fixed cytological samples significantly reduces the impact of photodegradation. In unstained, fixed cytological samples, 532 nm causes negligible observable damage to the samples over the measurement period. However, photodamage due to absorption and/or large fluorescent backgrounds due to clinical stains presents a significant problem for the use of Raman spectroscopy as an adjunct to cytological screening, although not an issue for IR absorption studies.136 Conventionally stained samples cannot be simultaneously optically and spectroscopically screened, suggesting that a fully automated procedure for unstained cell recognition, spectral analysis and assessment may be required. In such a procedure, however, registration of cell identification for transfer to the pathologist would be required. As an alternative, new stains are required which, while rendering the cell morphology visible under white light, do not absorb at the Raman source wavelength of 532 nm.
Ex vivo-liquid biopsies
Biofluids potentially offer the ideal option for a spectroscopic clinical trial due to the advantages mentioned previously. However, the analysis of such samples can suffer from problems associated with relatively low concentrations of the analyte of interest. Concentration of samples using centrifugal filtration devices has been shown to offer an alternative which allows measurement of the analytes in the native aqueous environment. Although Raman appears most promising in this context, due to the relatively lower contribution of the water, sufficient concentration of the sample also allows analysis of the fingerprint region by FTIR spectroscopy.137 Centrifugal filtration also allows fractionation according to molecular weight of the constituent analytes, potentially allowing targeting of molecular biomarkers of disease.138Alternatively, the samples can be dried for analysis. However, this introduces chemical and physical inhomogeneity into the sample during the drying process, due to the so-called coffee ring effect, cracking and gelation patterns, reducing reproducibility and sensitivities.139,140 Compositions and concentrations vary significantly across the dried droplet, due to different molecular weights and relative molecular solubilities, and can cause band saturation in transmission FTIR26 but Raman signals are highly reproducible within the ring. Lovergne et al. performed an infrared microspectroscopic imaging study on pure, 2 fold, 3 fold and 4 fold diluted serum (serum diluted with deionised water).141 The images (Fig. 7) highlight the spectral and visible differences between the different sample states. It can be seen that the total signal intensity varies within a drop and the highest intensity occurs at the drop edge, confirming a gradient of concentration of molecules from the centre to the periphery as a result of the drying process, resulting in this “coffee ring effect”, which in the case of human serum is more appropriately known as the Vroman effect.142 This relationship is repeated for the diluted samples with concurrent decrease in overall intensity. It can also be seen that major molecular components (proteins-amide I and II 1648 cm−1 and 1542 cm−1, lipids 1452 cm−1 and nucleic acids 1078 cm−1) all follow this same trend. This trend is accentuated in the diluted samples. The variability of the composition requires mapping of the droplet profile, increasing significantly the screening time, particularly for Raman analysis, although a quicker and simpler regime of measuring 5–10 spectra in the ring has been shown to provide reproducible signals. The Vroman effect is an unavoidable result of sample preparation but current efforts to reduce this effect have focused on the analysis of minute volumes, and the spraying of the biofluid onto a surface. Bulk ATR-FTIR spectroscopic measurements have been shown to reduce such effects,143 in effect providing an average spectrum of the dried droplet. Furthermore, fractionation of human serum by centrifugal filtration has been demonstrated to improve the quantitative analysis of low molecular weight biomarkers using ATR-IR spectroscopy, for the case of the variation of glucose levels in patient samples, such that it is comparable to currently employed clinical analysis techniques144 and quantification of glycine to a level 50 times lower than when using whole human serum. Raman spectroscopy has similarly been demonstrated to accurately predict Hepatitis C viral loads in dried patient plasma samples,145 although the inherently higher spatial resolution of Raman microscopy renders it less favourable for integrating measurements over an inhomogeneous, dried deposit. Ultimately, in order to enable clinical translation there needs to be a focus on large population studies and procedural and instrumental standardisation to enable regulatory requirements to be met.146
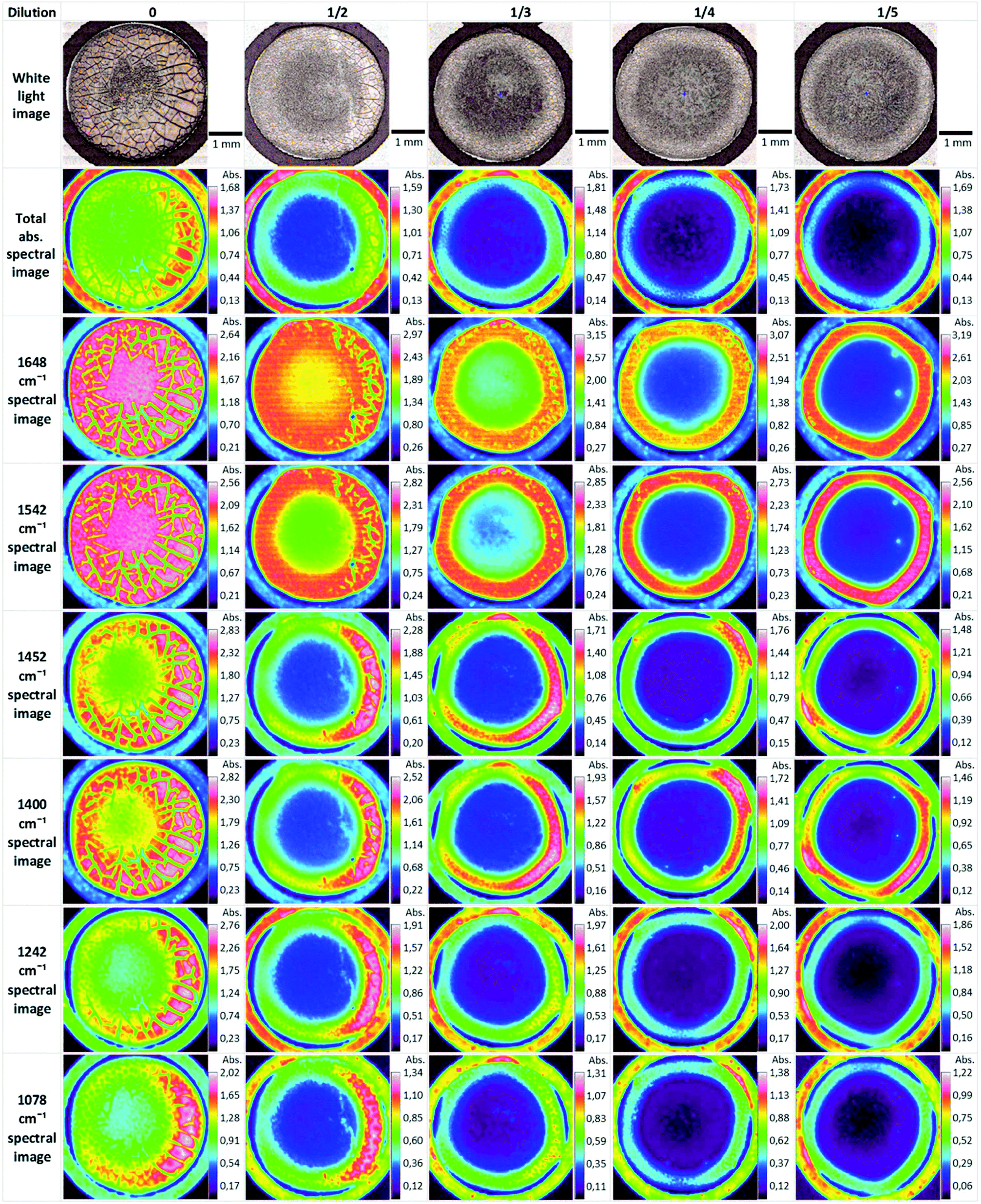 | ||
| Fig. 7 ATR-FTIR serum dilution study of dried drops. Original spectral images were atmospheric corrected. Other spectral images were constructed using a single wavenumber. Reproduced from ref. 141. | ||
In vitro
For in vitro cytological studies, formalin fixation has been demonstrated to best reproduce the characteristics of live cells.147 Protein coated substrates148 and 3-D matrices have also been explored to improve measurement protocols.149,150 Bonnier et al.,151 and Casey et al.152 have demonstrated that caution must be taken when applying conventional 2D cytotoxicity assay and drug dosing protocols to cells in a 3D environment, due to differing bioavailabilities of the assay or drug. Nevertheless, it is generally accepted that such 3D environments better mimic the in vivo extracellular matrix. Addressing the depth of a 3D environment by optical methods is, however, challenging. Recent studies have used functionalised nanoparticles to probe physicochemical gradients of potential clinical relevance,153 as well as the effects of radiation therapies in 3D models154 based on the SERS technique. Such multicellular tumour spheroids are relatively small in diameter (∼500 μm), however, and thicker tissue mimicking samples may require similar tissue processing techniques for analysis.155“Biological characteristics that are objectively measured and evaluated as indicators of normal biological processes, pathological processes or pharmacologic responses to a therapeutic intervention”.156
In terms of infrared and Raman spectroscopies, these will be multivariate signatures that allow one, for example, to differentiate between patients with disease and healthy controls or in longitudinal studies investigate the effect of radiotherapeutic, drug or nutritional intervention on a cohort of individuals.
In the biomarker discovery process, samples are taken from individuals (e.g. a body fluid), and are then measured in an unbiased way using spectroscopy. Data preprocessing is applied, and analyses are used to elucidate which are the discriminatory peaks or patterns within the data. However, before pursuing this path, one needs to consider the intrinsic variations in the experiment. These may be at the biological level, the way in which the samples have been prepared and the spectra measured, and of course could be related to the instrument itself. Spectral signatures of disease are often subtle perturbations to the norm, and therefore any unwanted experimental variability needs to be considered before any spectroscopic analyses are performed, as experimental design is key to avoiding false discovery. Indeed, there is a significant problem with many clinical studies in which the aim is to discover new biomarkers, in part due to poor experimental design and a lack of reproducibility,157–160 leading to the current crisis in biomarker discovery.161–164
To affect good design of experiments, is recommended to enlist the help of a (bio-) statistician at the planning stage of a project, to ensure that the appropriate individuals are selected so that intrinsic factors in human biology are accounted for. These include, but are not limited to: age, gender, genotype, reproductive cycle or other diurnal patterns, as well as body mass index.165 If this is done correctly, in case-control studies, the only non-random factor is that which is being testing for and the judicious experimental design will have resulted in the correct patients with disease (the cases) being carefully matched with healthy individuals (the controls).
An important consideration in the application of multivariate methods to spectral data analysis is the requisite size of dataset. Statistical significance is a critical consideration, and when patient diagnosis is the outcome, misclassification has serious consequences. Beleites et al., have carried out a study examining the effects of sample size on multivariate classifier models for clinical biospectroscopy.166 It was demonstrated that, while learning curves for dataset sizes common to small scale academic studies (2–25) can indicate acceptable performance, the model testing is itself limited by the dataset size and that datasets of 75–100 samples are required to produce “a good but not perfect classifier”.
Once the spectra data has been collected, the next stage entails the data analysis and this, and the above processes, are shown in a flow diagram in Fig. 8. The approaches and processes within the ‘Chemometrics Zoo’ are immense and this zoo contains many data pre-processing, data pre-treatment and data analysis (processing) algorithms. Several reviews of spectroscopic data processing and analysis protocols have been published, for guidance.18,167 Many different algorithms are used commonly used and these may have to be optimized. In the chemometrics community, the adage ‘no free lunch’ is often used and this is because, if one finds the optimum data analysis method for one scenario, then it is not guaranteed to translate and work on the next problem. The biological question of interest should drive the selection of the most appropriate type of multivariate analyses (MVA) to be used and exemplars of these are briefly detailed in Table 1. The computational code for these and links to other resources are available to the clinical spectroscopy community here: clirspec.org/resources/.
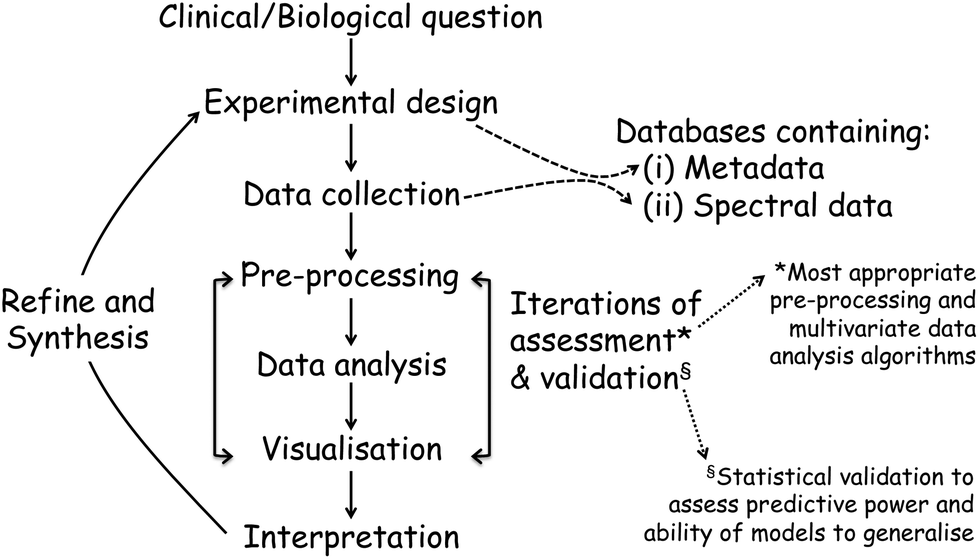 | ||
| Fig. 8 The experimental pipeline in a typical spectroscopic experiment. In all cases this pipeline starts with a clinical or biological question and ends up with interpretation of the system, which may then be feedback to refine the experiment and to generate (synthesise) new knowledge of the biological system. | ||
| Data analysis approach | Research question being asked | Algorithm features | Chemometric MVA solutions |
|---|---|---|---|
| Exploratory data analysis | • What can we find out about the relationship between these samples? | • No prior information is required | • Principal component analysis (PCA) |
| • e.g., to investigate gender or age | • Unsupervised method is used | • Dendrograms | |
| • Self-organising Map (SOM) | |||
| Classification of samples | • Can I classify samples into different groups? | • Prediction of unseen samples into groups | • Linear discriminant analysis (LDA) |
| • e.g., case-control studies | • Supervised learning is used | • Partial least squares-DA (PLS-DA) | |
| • Should include validation | • Support vector machines (SVM) | ||
| • Random forests (RFs) | |||
| • Neural networks | |||
| Quantitative analysis | • Can spectra be correlated with a continuous variable | • Allows quantitative prediction of unseen samples to some continuous variable | • PLS regression |
| • e.g., time, age, disease status, drug dose | • Principal component regression (PCR) | ||
| • Support vector regression (SVR) | |||
| Explanatory analysis | • What are the differences in chemical or physical state between samples | • Highlights spectral changes as function of group membership | • Many algorithms can be used: |
| • e.g., can I find vibrational features that relate to group separation or a continuous feature | • Need to know which group each spectrum belongs to | • Based on loadings vectors; e.g., LDA, PLS-DA, PLSR, PCR | |
| • Feature selection in RFs; use of Gini | |||
| • Genetic algorithms (GAs) as input selectors for MVA |
Critically, during the data analysis phase, statistical validation should be used to assess how robust the supervised learning methods (Table 1) have been. This is because, as the name suggests, the analysis is being supervised with training pairs of inputs (X-data from infrared or Raman) and outputs (Y-data representing the prediction; either categorical for classification or quantitative for continuous outputs). Thus, some data not used in model construction should be used to assess how well the model has been calibrated. Validation is usually achieved by resampling the data and this is performed in several different ways, such as Leave-one-out validation, K-fold validation, bootstrapping validation, external validation, for which new samples are obtained to generate an independent data set to test the models, and may be from a different cohort of individuals from a different geographical location and can be blind encoded to the investigated for additional rigour. This validation step is critically important, and may also be prone to error.168
In a clinical context, it is imperative that spectroscopic data is validated according to clinical “gold standards”, including appropriate controls. The subjectivity and therefore variability of these standards themselves is often quoted as a driving force for the development of objective technologies such as spectroscopy, and therefore the use of consensus is strongly, where possible, and, when not, the study should highlight the standard agreement of pathologists for the clinical condition in question. Classification models must be validated, and it is insufficient to provide training results only. Furthermore, when reporting performance against the gold standard, one should consider using the same measures used to compare two or more observers, such as Kappa statistics, and to provide the performance demonstrated in the literature for the gold standard discrimination of the pathologies under consideration.
Visualisation of the results of data analysis is essential, as multivariate analyses are used to reduce and simplify the data, and it is recommended that any figures are presented as objectively as is possible. If scores plots of latent variables are generated from, for example, principal component analysis or some discriminant analysis, then biplots should be used of latent variable 1 (LV1) versus latent variable 2 (LV2) or LV1 versus LV3, and so on. 2-Dimensional projections of a 3-D plot (LV1 v. LV2 v. LV3) with colour coding showing the groups to be differentiated could be biased, as these scatter ordination plots may have been rotated to show what the researcher wants to see, rather than the results being displayed in a completely objective manner.
Chemical image analysis is also highly visual and can also be performed using infrared or Raman spectroscopy, whereby peak areas or LV scores are used to construct images using false-shading from some arbitrary low to arbitrary high value. In a nice article by Ashton et al.169 it was demonstrated that different shading approaches can guide the eye to the wrong conclusion. For example, in rainbow scaling from red to violet, the average value on this scale is yellow and yet this is a very bright and dominant colour. In this scenario, the eye can be falsely drawn into thinking this is significant, when clearly it is not. A much better approach would be to use a more linear colour gradient from, for example, blue for low values to yellow for high values of each pixel.
The IR data acquired during this experiment could also be used in later experiments, for example to see whether different pathologists identify slightly different regions of the tissue, thereby changing the model produced, or used in the identification of tissue architecture. All pathologists will follow the relevant guidelines, but classification of cancer is not a straightforward issue. Therefore, it may be interesting to reinterpret existing data in the light of evolving pathology guidelines.
The clinical translation of vibrational spectroscopies will result in a range of non-ideal environmental conditions such as temperature, humidity, sample thickness, sample storage conditions, substrate quality and variation, magnification etc. The requirement for high throughput will also impact signal-to-noise characteristics. Having archival data with a range of provenance will aid identification of robust algorithms.
An increasing number of funders require data, produced as a result of their financial input, to be made openly available. On a more prosaic level, a field able to indicate the extent and usefulness of its data can help to justify the financial and resource outlay of the past, secure continuing investment and foster a growing community.
Data sharing has some practical considerations; what to share and where to share. Along with many other fields, the community of clinical IR and Raman spectroscopists currently has no domain-specific repository. Most journals offer a small amount of space in which to store information supplementary to an article, but only subscribers to the journal can access this data. To cater for the long tail of science, general purpose data repositories have emerged, examples of which include figshare,170 Mendeley Data171 and Zenodo.172 Some journals now offer to host large volumes of data, relating to Open Access publications173 and many universities and research institutes are establishing their own repositories to host data from their researchers. All these repositories will allocate a unique digital object identifier (DOI)174 to the data set, enabling a permanent indicator to the location of the data, ‘permanent’ typically implying in excess of 10 years.
In order to fill the gap between a domain-specific repository and widely scattered data stores, a Community has been created on the Zenodo platform for clinical IR and Raman data.175 The underlying Zenodo repository allows for an unlimited number of data sets per user, each up to 50 GB in size, larger data sets being accommodated on request. There is no fee for contributing data, or downloading it. Embargo periods are available on this, and most other platforms, to enable the data owner time to publish additional papers before the data is released openly.
In determining what to share, it can be helpful to divide the available data into broad categories: spectra, including hyperspectral images; metadata, which is the information describing the sample, experiment and the workflow executed; and computer code used to process the spectra. These categories will be taken in turn.
Spectra and hyperspectral image files should be stored in their raw form in the proprietary format of the instrument vendor. These files contain the most information that it is possible to collect regarding the experiment performed. In addition, to cater for those without the software able to access these files, a number of de facto standards can be used. Open standards available to the IR and Raman community include JCAMP-DX,176,177 SPC and HDF5,178 and some vendors’ software will export data to files in these formats directly. The reason for sharing in both proprietary and open formats is to maximise the information content of the resource. If there is only capacity for one type, then the open version is preferred. If data size becomes an issue, file compression can be used.
The spectra and hyperspectral images are what one might usually consider to be ‘data’; however, on their own they lack the context to be useful. Metadata, sometimes described as data about data, supplies that context and describes all parameters involved in the experiment lifecycle. The various stages that the experiment comprises can include the following:
• Patient information such as gender, age, the organ of interest, biobank identifiers and other ethically restricted information;
• Sample type; tissue, bodily fluid, bone;
• Sampling method; in vivo, ex vivo, orientation of sample in original organ
• Time and state of sampling; before/after drug treatment
• Sample preparation; storage, filtration, fixation and embedding processes;
• Experiment performed; instrumental parameters and acquisition conditions;
• Experiment repetition; biological replicates, or technical replicates;
• Data treatment; algorithms and parameterisation of quality tests and other operations.
Some of this information will be captured in the research paper, but the level of detail can be important179 and therefore as much information as practically possible should be included. Some information will be captured in standard operating procedures (SOP) which should be stored separately, in an open manner, and referred to via a DOI. Deviations from a recorded SOP should be documented and released as a new SOP, referencing the original to highlight the modifications. The separation of the SOP(s) from a given experiment allows for re-use of the protocol in a different context. SOPs for clinical IR and Raman can be uploaded to the CLIRSPEC Zenodo Community repository mentioned above.
Where patient data is restricted for ethical reasons, it may be possible to store identifiers such that other researchers, granted access to the controlled information, will be able to match the same patients to their spectroscopic data. Care should be taken to abide by any data protection guidelines in force.
The methods employed for data preprocessing and analysis should be recorded, together with all relevant parameters such as the window size for differentiation, the number of principal components for PCA denoising, or the discrimination levels for quality testing. The computer code used to perform these steps should be identified so that the end-user can repeat the steps on this, or other, data, or in a different order. The version number of software should be recorded. This is particularly important for regularly updated, open source computer code; for example ref. 180–185. In-house computer codes should be stored in version controlled source code repositories such as GitHub186 or Bitbucket.187 These resources record versions of the code as it evolves. Users of the GitHub platform can automatically generate a DOI for a software release on the Zenodo platform,188 allowing for a historical snapshot with a unique identifier. Similar to the case for SOPs, software should be stored separately from the data, with unique identifiers supplied relating the two entities.
When re-using, data it is important to recognise any restrictions imposed by the original owner. These are listed in the form of licence conditions attached during deposition. For spectroscopic data, metadata and SOPs the Creative Commons CC-BY licence is recommended.189 This allows the user to adapt and redistribute the data, but requires that appropriate credit is given to the original owner, and any modifications identified. For software, a permissive licence such as the GNU Lesser General Public License (LGPL)190 is preferred. This allows for re-use for any purpose, but requires acknowledgement of the original author, and that any modified versions of the code be released under an equally liberal licence.191 In order for a researcher to re-use data, they must first find it. It is recommended that any papers making use of the data, including its original creation, cite the DOI of the storage point. This is a requirement if published under a CC-BY licence. The repository should also list the DOI of any papers making use of the data. This cross-linking will give the data as much publicity, and therefore potential for re-use, as possible.
The (meta)data generated has the potential to assist in the growth and understanding of the clinical IR and Raman field, and the sharing of data, simply another form of sharing expertise and knowledge. It loses value, however, if the appropriate information about patient cohort, instrument calibration, pre- and post processing parameters etc. are provided. Journal editors, reviewers and indeed readers should strive to enforce the requirement for the provision and deposition of all such data, since, without it, the knowledge content of the research is greatly limited.
Outlook: clinical translation
Most spectroscopic analysis and imaging systems are state-of-the-art research grade instruments designed for the research laboratory and often come with “bells and whistles” appropriate for a range of different analytical applications. They are analytical instruments rather than purpose built medical diagnostics instruments and their designs are not necessarily compatible with a clinical environment. It is clear that many of the potential functions are simply not required for many clinical applications.As they address many potential analytical applications, the market for such instrumental technologies is broad, ranging from academic research, to food science, the pharma industry, and forensic science, to name but a few application areas. Instrument manufacturers are therefore, by and large, reluctant to dedicate instrument design to the specific area of clinical/medical diagnostics, in which the techniques have not yet been firmly established.
It is worth noting that, currently, commercially available spectroscopic instrumentation has been, by and large, optimised for sophisticated analysis of chemically pure materials. Perhaps a significant step towards clinical translation may be the design of instrumentation which is specifically tailored to accommodate the chemical and physical inhomogeneities and complexities of clinical samples. An example of such an approach could be the inclusion of integrating collection optics as default, to minimise scattering losses.192 In the case of Raman spectroscopy for in vivo applications, the situation is somewhat further advanced. RiverD International B.V. in the Netherlands has developed Raman instrumentation specifically for in vivo skin measurement, although applications for the study penetration and transdermal delivery of topically applied materials are predominantly promoted.193 Furthermore, Raman probes have been developed specifically for in vivo clinical applications, although each research group has their own design.
It is important to ask “what is the minimum quality of instrumentation and the minimum number of functions/flexibility that is required for a clinical spectroscopy system/microscope”. This has not yet been evaluated, since it very much depends on the specimen under study. The requirements will be different for applications involving tissue, cells, biofluids, whether the instrument is to be situated in an operating theatre, a pathology laboratory, or indeed employed for remote field trials, whether the samples will be frozen or not etc.
The question of “How good is good enough?”, is one that has been extensively discussed by the community over the past decade. Some of the deliberations are summarised in, for example the editorials of the special issues of Analyst, Optical Diagnostics, derived largely from panel or group discussions of the SPEC 2014 and SPEC 2016 conferences.5,194 The Faraday Discussions (2016) on Advanced Vibrational Spectroscopy for Biomedical Applications includes aspects of clinical translation, but also many of the other more technical consideration.115 It is clear that there is no single answer to the question, and that required performance depends on the application, and critically clinical demand. In applications such as evaluation of resection margins in neurosurgery, there is no alternative comparator and the value to the patient is in estimable. In routine screening applications such as cervical cytological screening, current protocols are well established, and improved performance must be more closely scrutinised. Justification of improved performance must also consider, for example, clinical workflow and health economics. Indeed, an indication of some progress in the field is the evolution of the language used by the spectroscopy community, which demonstrated an increased understanding of clinical requirements. The patient perspective is of course of critical importance.195
It is also important that the robustness of the techniques is evaluated, in terms of reproducibility of the diagnostic performance of individual instruments and comparison of different instruments, and operators, sites, etc. In recent years, a limited amount of attention has been paid to such considerations, and notably, Isabelle et al. conducted a UK based multi-centre trial of Raman spectral mapping of oesophageal cancer tissues.196 They developed a classification model to discriminate between adenocarcinoma (AC) and non-cancerous intraepithelial metaplasia (IM) oesophageal tissue samples, measured on three different Raman instruments in three different locations. Spectra were corrected using system transfer spectral correction algorithms including wavenumber shift (offset) correction, instrument response correction and baseline removal which minimized the instrument and sample quality variations within and between the instrument sites. Notably, strict calibration protocols were also required (around 0.2 cm−1 peak reproducibility). The EU COST Action Raman4clinics is currently (sept 2016–sept 2018) undertaking a series of more extensive ring trials which will encompass laboratories across Europe.197
It is clear that significant advances towards demonstration of the potential of vibrational spectroscopy for clinical applications continue to be made. Many of the technical challenges have been and/or are being addressed, and emerging technologies such as QCL based IR microscopy and nonlinear Raman techniques are poised to dramatically improve the acquisition timescales. Much of the proof of concept research is performed on within an academic context, and such research has addressed many of the fundamental challenges of dealing with the interaction of light with complex media, analysis of the complex data sets, and many advances in the associated technologies have been made. However, such academic research, by its nature, requires innovation on the timescale of postgraduate qualifications (3–4 years). Within this context, standardisation of protocols and methodologies is difficult to implement, and, in general, translation requires a significantly longer timescale to affect.198 Increasingly, however, the academic community has been working ever closer with clinical and instrument manufacturer partners, towards a better understanding of the clinical needs and identification of realistic strategic target applications. In this context, the importance of collaborative frameworks such as the UK EPSRC Clinical Infrared and Raman Spectroscopy for Medical Diagnosis Network, and the EU COST Raman4Clinics Action are invaluable. Much of the current EU wide community was brought together through the EU FP6 SSA DASIM, which led to significant advances in understanding and addressing issues of resonant scattering in IR spectroscopy.6–8 It is hoped that the formalism of the International Society for Clinical Spectroscopy (http://www.clirspec.org) will continue to facilitate progress in a similar fashion, promoting consensus of sample presentation and measurement as well as data processing protocols, towards the translation of spectroscopy into the clinical environment, for the general benefit of patients, to improve patient diagnosis and prognosis.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support of the Clinical Infrared and Raman Spectroscopy for Medical Diagnosis Network, EPSRC grant number EP/L012952/1, as well as the kind permission of Iwan Schie, Christophe Krafft and Juergen Popp to reproduce Fig. 2.Notes and references
- M. Diem, A. Mazur, K. Lenau, J. Schubert, B. Bird, M. Miljković, C. Krafft and J. Popp, J. Biophotonics, 2013, 6, 855–886 CrossRef CAS PubMed.
- D. I. Ellis, D. P. Cowcher, L. Ashton, S. O'Hagan and R. Goodacre, Analyst, 2013, 138, 3871–3884 RSC.
- C. Kendall, M. Isabelle, F. Bazant-Hegemark, J. Hutchings, L. Orr, J. Babrah, R. Baker and N. Stone, Analyst, 2009, 134, 1029–1045 RSC.
- A. Nijssen, S. Koljenović, T. C. B. Schut, P. J. Caspers and G. J. Puppels, J. Biophotonics, 2009, 2, 29–36 CrossRef CAS PubMed.
- H. J. Byrne, M. Baranska, G. J. Puppels, N. Stone, B. Wood, K. M. Gough, P. Lasch, P. Heraud, J. Sulé-Suso and G. D. Sockalingum, Analyst, 2015, 140, 2066–2073 RSC.
- P. Bassan, H. J. Byrne, J. Lee, F. Bonnier, C. Clarke, P. Dumas, E. Gazi, M. D. Brown, N. W. Clarke and P. Gardner, Analyst, 2009, 134, 1171–1175 RSC.
- P. Bassan, H. J. Byrne, F. Bonnier, J. Lee, P. Dumas and P. Gardner, Analyst, 2009, 134, 1586–1593 RSC.
- P. Bassan, A. Kohler, H. Martens, J. Lee, H. J. Byrne, P. Dumas, E. Gazi, M. Brown, N. Clarke and P. Gardner, Analyst, 2010, 135, 268–277 RSC.
- T. Konevskikh, R. Lukacs, R. Blumel, A. Ponossov and A. Kohler, Faraday Discuss., 2016, 187, 235–257 RSC.
- F. Bonnier, A. Mehmood, P. Knief, A. Meade, W. Hornebeck, H. Lambkin, K. Flynn, V. McDonagh, C. Healy, T. C. Lee, F. M. Lyng and H. J. Byrne, J. Raman Spectrosc., 2011, 42, 888–896 CrossRef CAS.
- P. Bassan, J. Lee, A. Sachdeva, J. Pissardini, K. M. Dorling, J. S. Fletcher, A. Henderson and P. Gardner, Analyst, 2013, 138, 144–157 RSC.
- P. Bassan, A. Sachdeva, J. Lee and P. Gardner, Analyst, 2013, 138, 4139–4146 RSC.
- J. Filik, M. D. Frogley, J. K. Pijanka, K. Wehbe and G. Cinque, Analyst, 2012, 137, 853–861 RSC.
- T. P. Wrobel, B. Wajnchold, H. J. Byrne and M. Baranska, Vib. Spectrosc., 2013, 69, 84–92 CrossRef CAS.
- Biomedical Applications of Synchrotron Infrared Microspectroscopy: A Practical Approach, ed. D. Moss, RSC Analytical Spectroscopy Monographs No. 11, 2011, ISBN: 978-0-85404-154-1 Search PubMed.
- M. J. Baker, J. Trevisan, P. Bassan, R. Bhargava, H. J. Butler, K. M. Dorling, P. R. Fielden, S. W. Fogarty, N. J. Fullwood, K. Heys, C. Hughes, P. Lasch, P. L. Martin-Hirsch, B. Obinaju, G. D. Sockalingum, J. Sulé-Suso, R. J. Strong, M. J. Walsh, B. R. Wood, P. Gardner and F. L. Martin, Nat. Protoc., 2014, 9, 1771–1791 CrossRef CAS PubMed.
- H. J. Butler, L. Ashton, B. Bird, G. Cinque, K. Curtis, J. Dorney, K. Esmonde-White, N. J. Fullwood, B. Gardner, P. L. Martin-Hirsch, M. J. Walsh, M. R. McAinsh, N. Stone and F. L. Martin, Nat. Protoc., 2016, 11, 664–687 CrossRef CAS PubMed.
- H. J. Byrne, P. Knief, M. E. Keating and F. Bonnier, Chem. Soc. Rev., 2016, 45, 1865–1878 RSC.
- M. Jermyn, K. Mok, J. Mercier, J. Desroches, J. Pichette, K. Saint-Arnaud, L. Bernstein, M.-C. Guiot, K. Petrecca and F. Leblond, Sci. Transl. Med., 2015, 7, 274ra19 CrossRef CAS PubMed.
- J. Desroches, M. Jermyn, K. Mok, C. Lemieux-Leduc, J. Mercier, K. St-Arnaud, K. Urmey, M.-C. Guiot, E. Marple, K. Petrecca and F. Leblond, Biomed. Opt. Express, 2015, 6, 2381 Search PubMed.
- B. Vaqas, K. O'Neill, M. Short, H. Zeng, I. Patel and U. Faiz, Neuro-Oncology, 2016, 18, vi194–vi195 CrossRef.
- T. Hollon, S. Lewis, C. W. Freudiger, S. Xie and D. A. Orringer, Neurosurg. Focus, 2016, 40, E9 CrossRef PubMed.
- E. M. Barroso, R. W. Smits, T. C. Bakker Schut, I. Ten Hove, J. A. Hardillo, E. B. Wolvius, R. J. Baatenburg de Jong, S. Koljenović and G. J. Puppels, Anal. Chem., 2015, 17, 2419–2426 CrossRef PubMed.
- R. W. Barroso, R. W. Smits, C. G. van Lanschot, P. J. Caspers, I. Ten Hove, H. Mast, A. Sewnaik, J. A. Hardillo, C. A. Meeuwis, R. Verdijk, V. Noordhoek Hegt, R. J. Baatenburg de Jong, E. B. Wolvius, T. C. Bakker Schut, S. Koljenović and G. J. Puppels, Cancer Res., 2016, 76, 5945–5953 CrossRef PubMed.
- M. G. Shim, L. M. Song, N. E. Marcon and B. C. Wilson, Photochem. Photobiol., 2000, 72, 146–150 CAS.
- J. Wang, K. Lin, W. Zheng, K. Y. Ho, M. Teh, K. G. Yeoh and Z. Huang, Anal. Bioanal. Chem., 2015, 407, 8303–8310 CrossRef CAS PubMed.
- L. M. Almond, J. Hutchings, G. Lloyd, H. Barr, N. Shepherd, J. Day, O. Stevens, S. Sanders, M. Wadley, N. Stone and C. Kendall, Gastrointest Endosc., 2014, 79, 37–45 CrossRef PubMed.
- J. C. C. Day, R. Bennett, B. Smith, C. Kendall, J. Hutchings, G. M. Meaden, C. Born, S. Yu and N. Stone, Phys. Med. Biol., 2009, 54, 7077–7087 CrossRef CAS PubMed.
- G. Shetty, C. Kendall, N. Shepherd, N. Stone and H. Barr, Br. J. Cancer, 2006, 94, 1460–1464 CrossRef CAS PubMed.
- J. Smith, C. Kendall, A. Sammon, J. Christie-Brown and N. Stone, Technol. Cancer Res. Treat., 2003, 2, 4 Search PubMed.
- J. D. Horsnell, J. A. Smith, M. Sattlecker, A. Sammon, J. Christie-Brown, C. Kendall and N. Stone, Surgeon, 2012, 10, 123–127 CrossRef PubMed.
- M. Isabelle, K. Rogers and N. Stone, J. Biomed. Opt., 2010, 15, 026030 CrossRef PubMed.
- G. R. Lloyd, L. E. Orr, J. Christie-Brown, K. McCarthy, S. Rose, M. Thomas and N. Stone, Analyst, 2013, 138, 3900–3908 RSC.
- J. Horsnell, P. Stonelake, J. Christie-Brown, G. Shetty, J. Hutchings, C. Kendall and N. Stone, Analyst, 2010, 135, 3042–3047 RSC.
- J. Day and N. Stone, Appl. Spectrosc., 2013, 67, 349–354 CrossRef CAS PubMed.
- E. Ingeborg, I. Petterson, J. C. C. Day, L. M. Fullwood, B. Gardner and N. Stone, Anal. Bioanal. Chem., 2015, 407, 8311–8320 CrossRef PubMed.
- A. P. Dudgeon, L. M. Fullwood, G. R. Lloyd, C. A. Kendall, S. Hopcrofte, O. Stevens, C. Hall, J. C. C. Day and N. Stone, Br. J. Cancer Search PubMed , submitted Oct (2018).
- K. M. Ashton, A. Brodbelt, C. Davis, T. P. Dawson, R. W. Lea, C. Walker, R. Stables, D. Vicinanza, M. D. Jenkinson, G. Clemens and M. J. Baker, Neuro-Oncology, 2014, 16(suppl_6), vi21 CrossRef.
- P. Matousek and N. Stone, Chem. Soc. Rev., 2015, 45, 1794–1802 RSC.
- N. Stone and P. Matousek, Cancer Res., 2008, 68, 4424–4430 CrossRef CAS PubMed.
- N. Stone, R. Baker, K. Rogers, A. W. Parker and P. Matousek, Analyst, 2007, 132, 899–905 RSC.
- A. Ghita, P. Matousek and N. Stone, J. Biophotonics, 2018, 11, e201600260 CrossRef PubMed.
- P. Matousek and N. Stone, J. Biomed. Opt., 2007, 12, 024008 CrossRef PubMed.
- I. P. Santos, E. M. Barroso, T. C. Bakker Schut, P. J. Caspers, C. G. F. van Lanschot, D.-H. Choi, M. F. van der Kamp, R. W. H. Smits, R. van Doorn, R. M. Verdijk, V. Noordhoek Hegt, J. H. von der Thüsen, C. H. M. van Deurzen, L. B. Kaoppert, G. J. L. H. van Leenders, P. C. Ewing-Graham, H. C. van Doorn, C. M. F. Dirven, M. B. Busstra, J. Hardillo, A. Sewnaik, I. ten Hove, H. Mast, D. A. Monserez, C. Meeuwis, T. Nijsten, E. B. Wolvius, R. J. Baatenburg de Jong, G. J. Puppels and S. Koljenović, Analyst, 2017, 142, 3025–3047 RSC.
- A. P. M. Michel, S. Liakat, K. Bors and C. F. Gmachl, Biomed. Opt. Express, 2013, 4, 520–530 CrossRef PubMed.
- http://www.bioopticsworld.com/articles/print/volume-9/issue-2/biooptics-features/mid-infrared-spectroscopy-bioimaging-moving-toward-mir-optical-biopsy.html .
- EU ICT-8-3.5 – Core and disruptive photonic technologies, Project Reference 317803: http://minerva-project.eu/.
- P. Bassan, A. Sachdeva, J. H. Shanks, M. D. Brown, N. W. Clarke and P. Gardner, Proc. SPIE 9041, Med. Imaging: Digital Pathol., 2014, 90410D Search PubMed.
- P. Bassan, A. Sachdeva, J. H. Shanks, M. D. Brown, N. W. Clarke and P. Gardner, Proc. SPIE, Prog. Biomed. Optics Imaging, 2014, 9041 Search PubMed.
- L. S. Leslie, A. Kadjacsy-Balla and R. Bhargava, Proc. SPIE, Med. Imaging 2015: Digital Pathol., 2015, 9420, 94200I–994208 Search PubMed.
- J. Faist, F. Capasso, D. L. Sivco, C. Sirtori, A. L. Hutchinson and A. Y. Cho, Science, 1994, 264, 553–556 CAS.
- H. Sreedhar, V. Varma, B. Bird, G. Guzman and M. J. Walsh, Proc. SPIE 10140, Med. Imaging: Digital Pathol., 2017, 1014004 Search PubMed.
- P. Bassan, M. J. Weida, J. Rowlette and P. Gardner, Analyst, 2014, 139, 3856–3859 RSC.
- M. J. Pilling, A. Henderson and P. Gardner, Anal. Chem., 2017, 89, 7348–7355 CrossRef CAS PubMed.
- M. J. Pilling, A. Henderson, B. Bird, M. D. Brown and P. Gardner, Faraday Discuss., 2016, 187, 135–154 RSC.
- P. D. Maker and R. W. Terhune, Phys. Rev., 1965, 137, A801 CrossRef.
- A. Zumbusch, G. R. Holtom and X. S. Xie, Phys. Rev. Lett., 1999, 82, 20 CrossRef.
- C. W. Freudiger, W. Min, B. G. Saar, S. Lu, G. R. Holtom, C. He, J. C. Tsai, J. X. Kang and X. S. Xie, Science, 2008, 322 Search PubMed.
- C. L. Evans and X. S. Xie, Annu. Rev. Anal. Chem., 2008, 1, 883–909 CrossRef PubMed.
- W. Min, Annu. Rev. Phys. Chem., 2011, 62, 507–530 CrossRef CAS PubMed.
- C. Zhang, Annu. Rev. Biomed. Eng., 2015, 17, 415–445 CrossRef CAS PubMed.
- I. W. Schie, C. Krafft and J. Popp, Analyst, 2015, 140, 3897–3909 RSC.
- M. Ji, D. A. Orringer, C. W. Freudiger, S. Ramkissoon, X. Liu, D. Lau, A. J. Golby, I. Norton, M. Hayashi, N. Y. R. Agar, G. S. Young, C. Spino, S. Santagata, S. Camelo-Piragua, K. L. Ligon, O. Sagher and X. S. Xie, Sci. Transl. Med., 2013, 5, 201 Search PubMed.
- D. A. Orringer, B. Pandian, Y. S. Niknafs, T. C. Hollon, J. Boyle, S. Lewis, M. Garrard, S. L. Hervey-Jumper, H. J. L. Garton, C. O. Maher, J. A. Heth, O. Sagher, D. A. Wilkinson, M. Snuderl, S. Venneti, S. H. Ramkissoon, K. A. McFadden, A. Fisher-Hubbard, A. P. Lieberman, T. D. Johnson, X. S. Xie, J. K. Trautman, C. W. Freudiger and S. Camelo-Piragua, Nat. Biomed. Eng., 2017, 1, 0027 CrossRef PubMed.
- A. Francis, K. Berry, Y. Chen, B. Figueroa and D. Fu, PLoS One, 2017, 12, e0178750 Search PubMed.
- C. H. Camp Jr., Y. J. Lee, J. M. Heddleston, Ch. M. Hartshorn, A. R. Hight Walker, J. N. Rich, J. D. Lathia and M. T. Cicerone, Nat. Photonics, 2014, 8, 627–634 CrossRef CAS PubMed.
- F. M. Lyng, D. Traynor, I. R. M. Ramos, F. Bonnier and H. J. Byrne, Anal. Bioanal. Chem., 2015, 407, 8279–8289 CrossRef CAS PubMed.
- S. P. Singh, O. Ibrahim, H. J. Byrne, J. W. Mikkonen, A. P. Koistinen, A. M. Kullaa and F. M. Lyng, Head Neck, 2016, 38, E2403–E2411 CrossRef PubMed.
- I. R. Ramos, A. D. Meade, O. Ibrahim, H. J. Byrne, M. McMenamin, M. McKenna, A. Malkin and F. M. Lyng, Faraday Discuss., 2016, 187, 187–198 RSC.
- K. M. Ostrowska, A. Malkin, A. Meade, J. O'Leary, C. Martin, C. Spillane, H. J. Byrne and F. M. Lyng, Analyst, 2010, 135, 3087–3093 RSC.
- K. Papamarkakis, B. Bird, J. M. Schubert, M. Miljkovic, R. Wein, K. Bedrossian, N. Laver and M. Diem, Lab. Invest., 2010, 90, 589–598 CrossRef PubMed.
- M. Diem, A. Mazur, K. Lenau, J. Schubert, B. Bird, M. Milikovic, C. Krafft and J. Popp, J. Biophotonics, 2013, 6, 855–886 CrossRef CAS PubMed.
- M. Miljkovíc, B. Bird, K. Lenau, A. I. Mazur and M. Diem, Analyst, 2013, 138, 3975–3982 RSC.
- I. Behl, G. Calado, O. Ibrahim, A. Malkin, S. Flint, H. J. Byrne and F. M. Lyng, Anal. Methods, 2017, 9, 937 RSC.
- See for example: J. Biophotonics, special issue: photonic biofluid diagnostics, 2014, 7, 145–275 Search PubMed.
- M. J. Baker, S. R. Hussain, L. Lovergne, V. Untereiner, C. Hughes, R. A. Lukaszewski, G. Thiéfinbg and G. D. Sockalingum, Chem. Soc. Rev., 2016, 45, 1803–1818 RSC.
- D. Rohleder, G. Kocherscheidt, K. Gerber, W. Kiefer, W. Kohler, J. Mocks and W. Petrich, J. Biomed. Opt., 2005, 10, 031108 CrossRef CAS PubMed.
- K. W. Poon, F. M. Lyng, P. Knief, O. Howe, A. D. Meade, J. F. Curtin, H. J. Byrne and J. Vaughan, Analyst, 2013, 137, 1807 RSC.
- W. Petrich, K. B. Lewandrowski, J. B. Muhlestein, M. E. Hammond, J. L. Januzzi, E. L. Lewandrowski, R. R. Pearson, B. Dolenko, J. Früh, M. Haass, M. M. Hirschl, W. Köhler, R. Mischler, J. Möcks, J. Ordóñez-Llanos, O. Quarder, R. Somorjai, A. Staib, C. Sylvén, G. Werner and R. Zerback, Analyst, 2009, 134, 1092–1098 RSC.
- J. R. Hands, G. Clemens, R. Stables, K. Ashton, A. Brodbelt, C. Davis, T. P. Dawson, M. D. Jenkinson, R. W. Lea, C. Walker and M. J. Baker, J. Neuro-Oncol., 2016, 127, 463–472 CrossRef PubMed.
- I. Taleb, G. Thiéfin, C. Gobinet, V. Untereiner, B. Bernard-Chabert, A. Heurgué, C. Truntzer, P. Hillon, M. Manfait, P. Ducoroy and G. D. Sockalingum, Analyst, 2013, 138, 4006–4014 RSC.
- S. Roy, D. Perez-Guaita, D. W. Andrew, J. S. Richards, D. McNaughton, P. Heraud and B. R. Wood, Anal. Chem., 2017, 89, 5238–5245 CrossRef CAS PubMed.
- Z. Farhane, F. Bonnier and H. J. Byrne, Anal. Bioanal. Chem., 2017, 409, 1333–1346 CrossRef CAS PubMed.
- A. Derenne, V. Van Hemelryck, D. Lamoral-Theys, R. Kiss and E. Goormaghtigh, Biochim. Biophys. Acta, Mol. Basis Dis., 2013, 1832, 46–56 CrossRef CAS PubMed.
- C. Hughes, G. Clemens and M. J. Baker, Trends Biotechnol., 2015, 33, 429–430 CrossRef CAS PubMed.
- L. E. Jamieson and H. J. Byrne, Vib. Spectrosc., 2017, 91, 16–30 CrossRef CAS.
- A. D. Meade, O. Howe, V. Unterreiner, G. D. Sockalingum, H. J. Byrne and F. M. Lyng, Faraday Discuss., 2016, 187, 213–234 RSC.
- E. Efeoglu, A. Casey and H. J. Byrne, Analyst, 2016, 141, 5417–5431 RSC.
- L. E. Jamieson, D. J. Harrison and C. J. Campbell, Analyst, 2015, 140, 3910–3920 RSC.
- I. Vega-Naredo, R. Loureiro, K. A. Mesquita, I. A. Barbosa, L. C. Tavares, A. F. Branco, J. R. Erickson, J. Holy, E. L. Perkins, R. A. Carvalho and P. J. Oliveira, Cell Death Differ., 2014, 21, 1560–1574 CrossRef CAS PubMed.
- L. E. Jamieson, V. L. Camus, P. O. Bagnaninchi, K. M. Fisher, G. D. Stewart, W. H. Nailon, D. B. McLaren, D. J. Harrison and C. J. Campbell, Nanoscale, 2016, 8, 16710 RSC.
- A. Agarwal, D. Ressler and G. Snyder, Pharmgenomics Pers. Med., 2015, 8, 99–110 Search PubMed.
- S. Naylor and T. Cole, Drug Discovery World, 2010,(Spring), 67–69 Search PubMed.
- Z. Farhane, F. Bonnier, M. A. Maher, J. Bryant, A. Casey and H. J. Byrne, J. Biophotonics, 2017, 10, 151–165 CrossRef CAS PubMed.
- Z. Farhane, F. Bonnier, O. Howe, A. Casey and H. J. Byrne, J. Biophotonics, 2018, 11, e201700060 CrossRef PubMed.
- H. K. Yosef, L. Mavarani, A. Maghnouj, S. Hahn, S. F. El-Mashtoly and K. Gerwert, Anal. Bioanal. Chem., 2015, 407, 8321–8331 CrossRef CAS PubMed.
- S. F. El-Mashtoly, H. K. Yosef, D. Petersen, L. Mavarani, A. Maghnouj, S. Hahn, C. Kötting and K. Gerwert, Anal. Chem., 2015, 87, 7297–7304 CrossRef CAS PubMed.
- A. V. Rutter, M. R. Siddique, J. Filik, C. Sandt, P. Dumas, G. Cinque, G. D. Sockalingum, Y. Yang and J. Sulé-Suso, Cytometry, Part A, 2014, 85, 688–697 CrossRef PubMed.
- M. R. Siddique, A. V. Rutter, K. Wehbe, G. Cinque, G. Bellisola and J. Sulé-Suso, Analyst, 2017, 142, 1299–1307 RSC.
- O. Stevens, E. Ingeborg, I. Petterson, J. Day and N. Stone, Chem. Soc. Rev., 2015, 45, 1919–1934 RSC.
- R. O. P. Draga, M. C. M. Grimbergen, P. L. M. Vijverberg, C. F. P. van Swol, T. G. N. Jonges, J. A. Kummer and J. L. H. R. Bosch, Anal. Chem., 2010, 82, 5993–5999 CrossRef CAS PubMed.
- P. Crow, J. S. Uff, J. A. Farmer, M. P. Wright and N. Stone, BJU Int., 2004, 93, 1232–1236 CrossRef CAS PubMed.
- J. A. Cohen and C. Virnelson, US Pat, 5160826, 1991 Search PubMed.
- H. Brooke, D. L. Perkins, B. Setlow, P. Setlow, B. V. Bronk and M. L. Myrick, Appl. Spectrosc., 2008, 62, 881–888 CrossRef CAS PubMed.
- J. Lee, Vib. Spectrosc., 2017, 90, 104–111 CrossRef CAS.
- K. L. A. Chan and S. G. Kazarian, Anal. Chem., 2013, 85, 1029–1036 CrossRef CAS PubMed.
- A. Sroka-Bartnicka, J. A. Kimber, L. Borkowski, M. Pawlowska, I. Polkowska, G. Kalisz, A. Belcarz, K. Jozwiak, G. Ginalska and S. G. Kazarian, Anal. Bioanal. Chem., 2015, 407, 775–7785 CrossRef PubMed.
- J. A. Kimber, L. Foreman, B. Turner, P. Rich and S. G. Kazarian, Faraday Discuss., 2016, 187, 69–85 RSC.
- G. L. Carr, Rev. Sci. Instrum., 2001, 72, 1613–1619 CrossRef CAS.
- K. L. A. Chan and S. G. Kazarian, Analyst, 2013, 138, 4040–4047 RSC.
- K. L. A. Chan and S. G. Kazarian, Chem. Soc. Rev., 2016, 45, 1850–1864 RSC.
- H. Shinzawa, B. Turner, J. Mizukado and S. G. Kazarian, Analyst, 2017, 142, 2475–2483 RSC.
- S. G. Kazarian and K. L. A. Chan, Appl. Spectrosc., 2010, 64, 135A–152A CrossRef CAS PubMed.
- S. G. Kazarian and K. L. A. Chan, Analyst, 2013, 138, 1940–1951 RSC.
- Advanced Vibrational Spectroscopy for Biomedical Applications, Faraday Discussions, The Royal Society of Chemistry, 2016, vol. 187 Search PubMed.
- M. Pilling and P. Gardner, Chem. Soc. Rev., 2016, 45, 1935–1957 RSC.
- J. A. Kimber and S. G. Kazarian, Anal. Bioanal. Chem., 2017, 409, 5813–5820 CrossRef CAS PubMed.
- L. T. Kerr, H. J. Byrne and B. M. Hennelly, Anal. Methods, 2015, 7, 5041–5052 RSC.
- P. Bassan, J. Mellor, J. Shapiro, K. J. Williams, M. Lisanti and P. Gardner, Anal. Chem., 2014, 86, 1648–1653 CrossRef CAS PubMed.
- M. J. Pilling, A. Henderson, J. H. Shanks, M. D. Brown, N. W. Clarke and P. Gardner, Analyst, 2017, 142, 1258–1268 RSC.
- L. M. Fullwood, D. Griffiths, K. Ashton, T. Dawson, R. W. Lea, C. Davis, F. Bonnier, H. J. Byrne and M. J. Baker, Analyst, 2014, 139, 446–454 RSC.
- J. K. Pijanka, N. Stone, A. V. Rutter, N. Forsyth, G. D. Sockalingum, Y. Yang and J. Sule-Suso, Analyst, 2013, 138, 5052–5058 RSC.
- Z. Farhane, F. Bonnier, A. Casey, A. Maguire, L. O'Neill and H. J. Byrne, Analyst, 2015, 140, 5908–5919 RSC.
- O. Ibrahima, A. Maguire, A. D. Meade, S. Flint, M. Toner, H. J. Byrne and F. M. Lyng, Anal. Methods, 2017, 9, 4709–4717 RSC.
- L. T. Kerr and B. M. Hennelly, Chemom. Intell. Lab. Syst., 2016, 158, 61–68 CrossRef CAS.
- L. Cui, H. J. Butler, P. L. Martin-Hirsch and F. L. Martin, Anal. Methods, 2016, 8, 481–487 RSC.
- A. T. Lewis, R. Gaifulina, M. Isabelle, J. Dorney, M. L. Woods, G. R. Lloyd, K. Lau, M. Rodriguez-Justo, C. Kendall, N. Stone and G. M. Thomas, J. Raman Spectrosc., 2017, 48, 119–125 CrossRef CAS PubMed.
- J. D. Bancroft and M. Gamble, Theory and Practice of Histological Techniques, Churchill Livingstone, London, 2002 Search PubMed.
- A. Tfayli, C. Gobinet, V. Vrabie, R. Huez, M. Manfait and O. Piot, Appl. Spectrosc., 2009, 63, 564–570 CrossRef CAS PubMed.
- L. M. Fullwood, D. Griffiths, K. Ashton, T. Dawson, R. W. Lea, C. Davis, F. Bonnier, H. J. Byrne and M. J. Baker, Analyst, 2014, 139, 446–454 RSC.
- F. Bonnier, A. Mehmood, P. Knief, A. Meade, W. Hornebeck, H. Lambkin, K. Flynn, V. McDonagh, C. Healy, T. C. Lee, F. M. Lyng and H. J. Byrne, J. Raman Spectrosc., 2011, 42, 888–896 CrossRef CAS.
- F. Bonnier, S. M. Ali, P. Knief, H. Lambkin, K. Flynn, V. McDonagh, C. Healy, T. C. Lee, F. M. Lyng and H. J. Byrne, Vib. Spectrosc., 2012, 61, 124–132 CrossRef CAS.
- I. Behl, G. Calado, O. Ibrahim, A. Malkin, S. Flint, H. J. Byrne and F. M. Lyng, Anal. Methods, 2017, 9, 937 RSC.
- F. Bonnier, D. Traynor, P. Kearney, C. Clarke, P. Knief, C. Martin, J. J. O'Leary, H. J. Byrne and F. Lyng, Anal. Methods, 2014, 6, 7831–7841 RSC.
- G. J. Puppels, J. H. F. Olminkhof, G. M. J. Segers-Nolten, C. Otto, F. F. M. de Mul and J. Greve, Exp. Cell Res., 1991, 195, 361 CrossRef CAS PubMed.
- J. Pijanka, G. D. Sockalingum, A. Kohler, Y. Yang, F. Draux, P. Dumas, C. Sandt, G. Parkes, D. G. van Pittius, G. Douce, V. Untereiner and J. Sulé-Suso, Lab. Invest., 2010, 90, 797–807 CrossRef CAS PubMed.
- F. Bonnier, F. Petitjean, M. J. Baker and H. J. Byrne, J. Biophotonics, 2014, 7, 167–179 CrossRef CAS PubMed.
- J. Filik and N. Stone, J. Raman Spectrosc., 2009, 40, 218–224 CrossRef CAS.
- J. Filik and N. Stone, Analyst, 2007, 132, 544–550 RSC.
- J. Filik and N. Stone, Anal. Chim. Acta, 2008, 616, 177–184 CrossRef CAS PubMed.
- L. Lovergne, G. Clemens, V. Untereiner, R. A. Lukaszweski, G. D. Sockalingum and M. J. Baker, Anal. Methods, 2015, 7, 7140 RSC.
- L. Vroman, A. L. Adams, G. C. Fischer and P. C. Munoz, Blood, 1980, 55, 156 CAS.
- K. M. Dorling and M. J. Baker, Trends Biotechnol., 2013, 31, 327–328 CrossRef CAS PubMed.
- F. Bonnier, H. Blasco, C. Wasselet, G. Brachet, R. Respaud, L. F. C. S. Carvalho, D. Bertrand, M. J. Baker, H. J. Byrne and I. Chourpa, Analyst, 2017, 142, 1285–1298 RSC.
- H. Nawaz, N. Rashid, M. Saleem, I. Amin, M. Iqbal, M. Rahman, O. Ibrahim, S. M. Baig, M. Ahmed, F. Bonnier and H. J. Byrne, J. Raman Spectrosc., 2017, 48, 623–777 CrossRef.
- K. Spalding, R. Board, T. Dawson, M. D. Jenkinson and M. J. Baker, Brain Behav., 2016, 8, e00502 CrossRef PubMed.
- A. D. Meade, C. Clarke, F. Draux, G. D. Sockalingum, M. Manfait, F. M. Lyng and H. J. Byrne, Anal. Bioanal. Chem., 2010, 396, 1781–1791 CrossRef CAS PubMed.
- A. D. Meade, F. M. Lyng, P. Knief and H. J. Byrne, Anal. Bioanal. Chem., 2007, 387, 1717–1728 CrossRef CAS PubMed.
- F. Bonnier, A. D. Meade, S. Merzha, P. Knief, K. Bhattacharya, F. M. Lyng and H. J. Byrne, Analyst, 2010, 135, 1697–1703 RSC.
- F. Bonnier, P. Knief, B. Lim, A. D. Meade, J. Dorney, K. Bhattacharya, F. M. Lyng and H. J. Byrne, Analyst, 2010, 135, 3169–3177 RSC.
- F. Bonnier, M. E. Keating, T. P. Wróbel, K. Majzner, M. Baranska, A. Garcia-Munoz, A. Blanco and H. J. Byrne, Toxicol. in Vitro, 2015, 29, 124–131 CrossRef CAS PubMed.
- A. Casey, M. Gargotti, F. Bonnier and H. J. Byrne, Toxicol. in Vitro, 2016, 33, 99–104 CrossRef CAS PubMed.
- L. E. Jamieson, V. L. Camus, P. O. Bagnaninchi, K. M. Fisher, G. D. Stewart, W. H. Nailon, D. B. McLaren, D. J. Harrisong and C. J. Campbell, Nanoscale, 2016, 8, 16710 RSC.
- V. L. Camus, G. Stewart, W. H. Nailon, D. B. McLarend and C. J. Campbell, Analyst, 2016, 141, 5056 RSC.
- S. M. Ali, F. Bonnier, K. Ptasinski, H. Lambkin, K. Flynn, F. M. Lyng and H. J. Byrne, Analyst, 2013, 138, 3946–3956 RSC.
- Biomarkers Definitions Working Group; Biomarkers and surrogate endpoints: preferred definitions and conceptual framework, Clin. Pharmacol. Ther., 2001, 69, 89–95 Search PubMed.
- G. Poste, Nature, 2011, 469, 156–157 CrossRef CAS PubMed.
- M. R. Munafò, B. A. Nosek, D. V. M. Bishop, K. S. Button, C. D. Chambers, N. P. du Sert, U. Simonsohn, E.-J. Wagenmakers, J. J. Ware and J. P. A. Ioannidis, Nat. Hum. Behav., 2017, 1, 0021 CrossRef.
- R. Bouça-Machado, M. Rosário, J. Alarcão, L. Correia-Guedes, D. Abreu and J. J. Ferreira, BMC Palliative Care, 2017, 16, 10 CrossRef PubMed.
- H. C. Kraemer, Am. J. Psychiatry, 2016, 173, 672–679 CrossRef PubMed.
- D. Broadhurst and D. B. Kell, Metabolomics, 2006, 2, 171–196 CrossRef CAS.
- M. Mamas, W. B. Dunn, L. Neyses and R. Goodacre, Arch. Toxicol., 2011, 85, 5–17 CrossRef CAS PubMed.
- J. P. A. Ioannidis and P. M. M. Bossuyt, Clin. Chem., 2017, 63, 963–972 CAS.
- A. Mordente, E. Meucci, G. E. Martorana and A. Silvestrini, Adv. Exp. Med. Biol., 2015, 867, 9–26 CrossRef CAS PubMed.
- D. K. Trivedi, K. A. Hollywood and R. Goodacre, New Horiz. Transl. Med., 2017, 3, 6 Search PubMed.
- C. Beleites, U. Neugebauer, T. Bocklitz, C. Krafft and J. Popp, Anal. Chim. Acta, 2013, 760, 25–33 CrossRef CAS PubMed.
- P. Lasch, Chemom. Intell. Lab. Syst., 2012, 117, 100–114 CrossRef CAS.
- S. Guo, T. Bocklitz, U. Neugebauer and J. Popp, Anal. Methods, 2017, 9, 4410–4417 RSC.
- L. Ashton, K. A. Hollywood and R. Goodacre, Analyst, 2015, 140, 1852–1858 RSC.
- https://figshare.com/ , (accessed August 2017).
- https://data.mendeley.com/ , (accessed August 2017).
- https://zenodo.org/ , (accessed August 2017).
- https://www.nature.com/sdata/ , (accessed August 2017).
- https://www.doi.org/ , (accessed August 2017).
- https://zenodo.org/communities/clirspec/ .
- R. S. McDonald and P. A. Wilks Jr., Appl. Spectrosc., 1988, 42, 151–162 CrossRef CAS.
- P. Lampen, J. Lambert, R. J. Lancashire, R. S. McDonald, P. S. McIntyre, D. N. Rutledge, T. Fröhlich and A. N. Davies, Pure Appl. Chem., 1999, 71, 1549–1556 CrossRef CAS.
- https://support.hdfgroup.org/HDF5/ , (accessed August 2017).
- W. C. Hines, Y. Su, I. Kuhn, K. Polyak and M. J. Bissell, Cell Rep., 2014, 6, 779–781 CrossRef CAS PubMed.
- http://hyperspec.r-forge.r-project.org/ , (accessed August 2017).
- J. Trevisan, P. P. Angelov, A. D. Scott, P. L. Carmichael and F. L. Martin, Bioinformatics, 2013, 29, 1095–1097 CrossRef CAS PubMed.
- Irootlab. An open-source toolbox for biospectroscopy in MATLAB, http://trevisanj.github.io/irootlab/, (accessed August 2017).
- ChiToolbox, https://bitbucket.org/AlexHenderson/chitoolbox, (accessed August 2017).
- M. Toplak, G. Birarda, S. Read, C. Sandt, S. M. Rosendahl, L. Vaccari, J. Demšar and F. Borondics, Synchrotron Radiat. News, 2017, 30(4), 40–45 CrossRef.
- Orange for spectral data, https://github.com/markotoplak/orange-infrared, (accessed August 2017).
- GitHub, https://github.com/, (accessed August 2017).
- Atlassian Bitbucket, https://bitbucket.org, (accessed August 2017).
- Making your code citable, https://guides.github.com/activities/citable-code/, (accessed August 2017).
- Creative Commons Attribution 4.0 International (CC BY 4.0), https://creativecommons.org/licenses/by/4.0/, (accessed August 2017).
- GNU Lesser General Public License, https://www.gnu.org/licenses/lgpl-3.0.en.html, (accessed August 2017).
- A. Morin, J. Urban and P. Sliz, PLoS Comput. Biol., 2012, 8, e1002598 CAS.
- A. Dazzi, A. Deniset-Besseaua and P. Lasch, Analyst, 2013, 138, 4191 RSC.
- http://www.riverd.com/skin-analysis .
- K. M. Gougha and F. Leblond, Analyst, 2017, 142, 1177–1178 RSC.
- C. Hughes and M. J. Baker, Analyst, 2016, 141, 467–475 RSC.
- M. Isabelle, J. Dorney, A. Lewis, G. R. Lloyd, O. Old, N. Shepherd, M. Rodriguez-Justo, H. Barr, K. Lau, I. Bell, S. Ohrel, G. Thomas, N. Stone and C. Kendall, Faraday Discuss., 2016, 187, 87–103 RSC.
- https://www.raman4clinics.eu/save-the-date-symposium-on-the-raman-round-robin-experiment/ .
- Z. Slote Morris, S. Wooding and J. Grant, J. R. Soc. Med., 2011, 104, 510–520 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |