
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntroducing unactivated acyclic internal aliphatic olefins into a cobalt catalyzed allylic selective dehydrogenative Heck reaction†
Soham
Maity
,
Pravas
Dolui
,
Rajesh
Kancherla
and
Debabrata
Maiti
 *
*
Department of Chemistry, IIT Bombay, Powai, Mumbai-400076, India. E-mail: dmaiti@chem.iitb.ac.in
First published on 16th May 2017
Abstract
Unactivated acyclic internal aliphatic olefins are often found to be unreactive in conventional alkenylation reactions. To address this problem, a cobalt catalyzed allylic selective dehydrogenative Heck reaction with internal aliphatic olefins has been developed. The method is highly regio- and stereoselective, the conditions are mild and a wide variety of functional groups can be tolerated. Remarkably, both internal and terminal aliphatic olefins can be employed, thereby significantly expanding the scope of alkenylation chemistry with aliphatic olefins.
Introduction
The selective incorporation of unactivated aliphatic olefins into arenes is a highly sought-after transformation in modern Heck chemistry.1 The ubiquity and synthetic potential of aliphatic olefins and α-olefins have inspired significant developments in recent times.2 An existing problem of this methodology, however, is the inertness of internal aliphatic olefins. This could be attributed to a number of factors, such as inappropriate coordination or sensitivity towards the enhanced steric environment in the carbometalation step.3 To solve this problem, the discovery and development of new catalysts seems essential. Here we report a method that overcomes these limitations and successfully introduces internal aliphatic olefins in a highly regio- and stereoselective manner.Our recent studies suggested that an inexpensive and readily available cobalt catalyst could facilitate an allylic selective Heck-type reaction with a variety of terminal aliphatic olefins.4 The unorthodox allylic selectivity was attributed to a flexible framework consisting of a bis-chelating directing scaffold, which placed the metal suitably to generate the allylic product. However, an attempt to extend this reaction to internal aliphatic olefins remained unsuccessful, as competitive reductive elimination predominated.5 We hypothesized that a structural modification of the 5,7-membered fused metallacycle intermediate (such as IIb) could suppress the annulation pathway. As a consequence, unbiased internal aliphatic olefins could be selectively included (Scheme 1B).
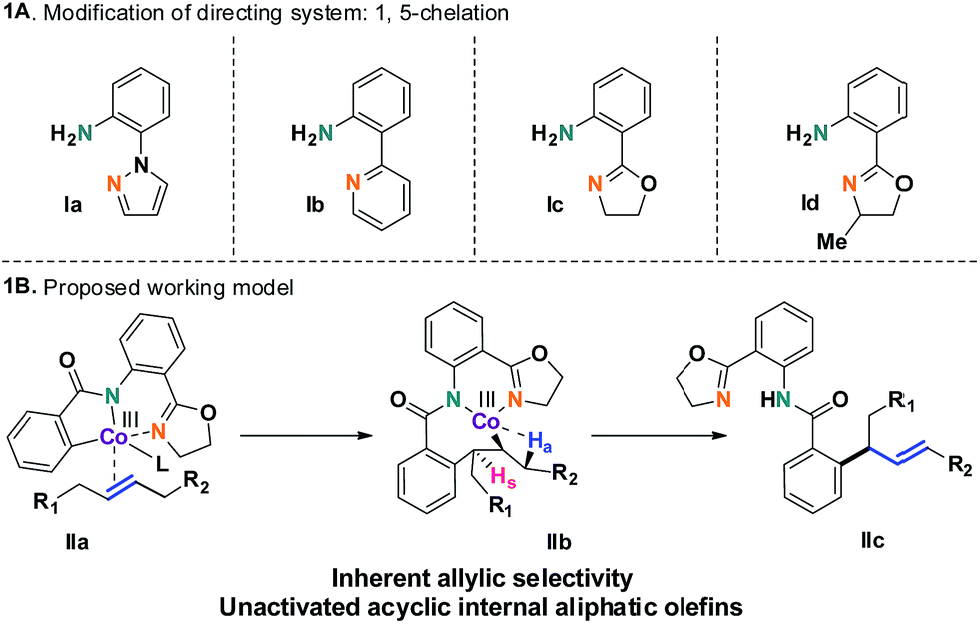 | ||
| Scheme 1 Evaluation of directing groups with internal aliphatic olefins. | ||
This hypothesis led to the synthesis and evaluation of several 1,5-bis-chelating systems with attached pyrazole, pyridine and dihydrooxazole moieties. Out of this selection of directing groups, 2-(4,5-dihydrooxazol-2-yl)aniline (Ic, Scheme 1A), first developed by the Yu group, showed promising reactivity with commercially available cobalt catalysts.6 Optimization of the reaction parameters and inclusion of aliphatic acid additives considerably increased the efficiency of the method (Table 1). Under the optimized conditions, completely allylic selective alkenylated products could be obtained in high synthetic yields with very high diastereomeric ratios (Scheme 2).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Oxidant | Solvent | Yield |
| a With 1 equiv. isobutyric acid additive and 3 equiv. NaHCO3 base. | ||||
| 1 | Co(OAc)2·4H2O | Ag2SO4 | DCE | 19% |
| 2 | Co(OAc)2·4H2O | Ag2SO4 | 1,4-Dioxane | 38% |
| 3 | Co(OAc)2·4H2O | Ag2CO3 | 1,4-Dioxane | 35% |
| 4 | Co(OAc)2·4H2O | PhI(OAc)2 | 1,4-Dioxane | 19% |
| 5 | Co(OAc)2·4H2O | Ag2SO4 | 1,4-Dioxane | 90%a |
| 6 | Co(acac)2 | Ag2SO4 | 1,4-Dioxane | 51% |
| 7 | CoBr2 | Ag2SO4 | 1,4-Dioxane | 56% |

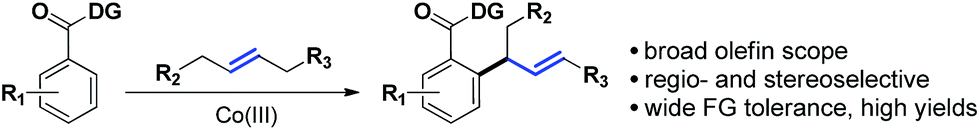 | ||
| Scheme 2 Introducing acyclic internal aliphatic olefins in dehydrogenative Heck chemistry. | ||
Results and discussion
We began to study the scope of the reaction with trans-4-octene as a model olefin. These olefins have historically proven to be difficult substrates for any Heck-type transformations and Fujiwara–Moritani reactions.7 In the absence of electronic bias, the migratory insertion step is less facile and the methods often suffer from poor reaction rates. Even if the metal could bind with the olefin effectively, the carbometalation intermediate (IIb) might lead to a deleterious mixture of styrenyl/allylic products.8 In the present system, however, the affiliation of the 1,5-chelating directing scaffold and the cobalt catalyst proved to be inherently allylic selective.9,10 Although the origin of this selectivity is yet to be fully established, given the thermodynamic non-preference of allylic isomers (vs. styrenyl), this observation demanded a more detailed evaluation of the scope of the reaction.Therefore, we tested different benzoic acid derivatives under standard conditions (Scheme 3). It was observed that the reaction was quite general, as both electron donating as well as electron withdrawing substituents could be tolerated at the ortho-, meta- or para-positions of the arene ring. Even multiple fluoro groups did not affect the efficiency of the method (3b–3e and 3q). Among the synthesized compounds, entry 3c was characterized through X-ray crystallography, thereby unambiguously assigning the trans-geometry to the α-branched internal allylarene product. Different functional groups such as –CN, –Cl and –OMe were well suited (3n, 3p and 3q; 75–80%). When both the ortho-positions were unsubstituted, some amount of di-allylation (7–10%) was observed, which could be easily purified through column chromatography. For substrates with moderate yields (3i and 3l), the unreacted starting materials were recovered in considerable amounts. Heteroaromatic benzoic acids such as thiophene (3g, 70%) and benzothiophene (3j, 60%) carboxylic acids were also found to be useful substrates. Interestingly, a chiral center was generated in all these cases; thus future development of an asymmetric variant of the method could be envisaged.
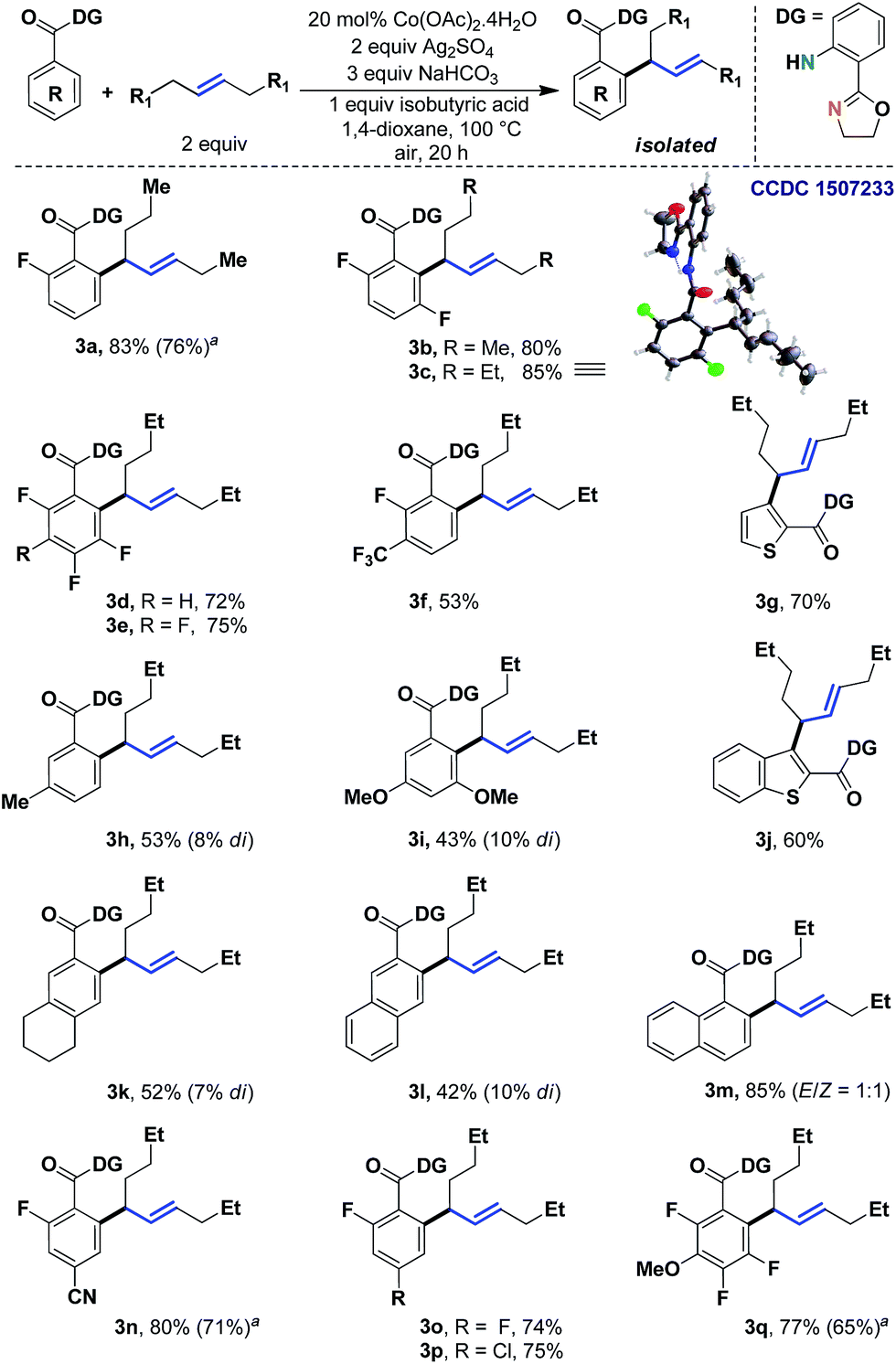 | ||
| Scheme 3 Allylic selective alkenylation with unactivated acyclic internal aliphatic olefins. aYields with 10 mol% Co. | ||
The scope of the method was evaluated further with different internal aliphatic olefins (Scheme 4). As expected, a higher homologue, trans-5-decene, was found to be equally reactive (4a, 89%). Although we observed a slight erosion of efficiency with trans-3-hexene (4b), this might be attributed to the volatility of the olefin substrate. A free alcohol containing olefin could be successfully employed without oxidation of the alcohol functional group (4g, 65%). Other unsymmetrical internal olefins were also useful in this reaction (entries 4g–4l). An internal allyl ester was particularly interesting, as regioselective allylation was observed in an 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (4e, 60%). Although in principle two allyl isomers are possible, the conjugation with the carbonyl group determined the geometry of the major allyl isomer. A similar reactivity pattern was observed with internal olefins derived from naturally occurring citronellol (4k) and nerol (4l), which had additional tri-substituted double bonds. Unfortunately, despite our best efforts, cis-olefins proved to be difficult substrates (4c, 48%). Even a prolonged reaction time failed to improve the synthetic yield to a considerable extent. Nevertheless, considering the documented difficulties with internal aliphatic olefins, these studies seemed very encouraging.
:1 ratio (4e, 60%). Although in principle two allyl isomers are possible, the conjugation with the carbonyl group determined the geometry of the major allyl isomer. A similar reactivity pattern was observed with internal olefins derived from naturally occurring citronellol (4k) and nerol (4l), which had additional tri-substituted double bonds. Unfortunately, despite our best efforts, cis-olefins proved to be difficult substrates (4c, 48%). Even a prolonged reaction time failed to improve the synthetic yield to a considerable extent. Nevertheless, considering the documented difficulties with internal aliphatic olefins, these studies seemed very encouraging.
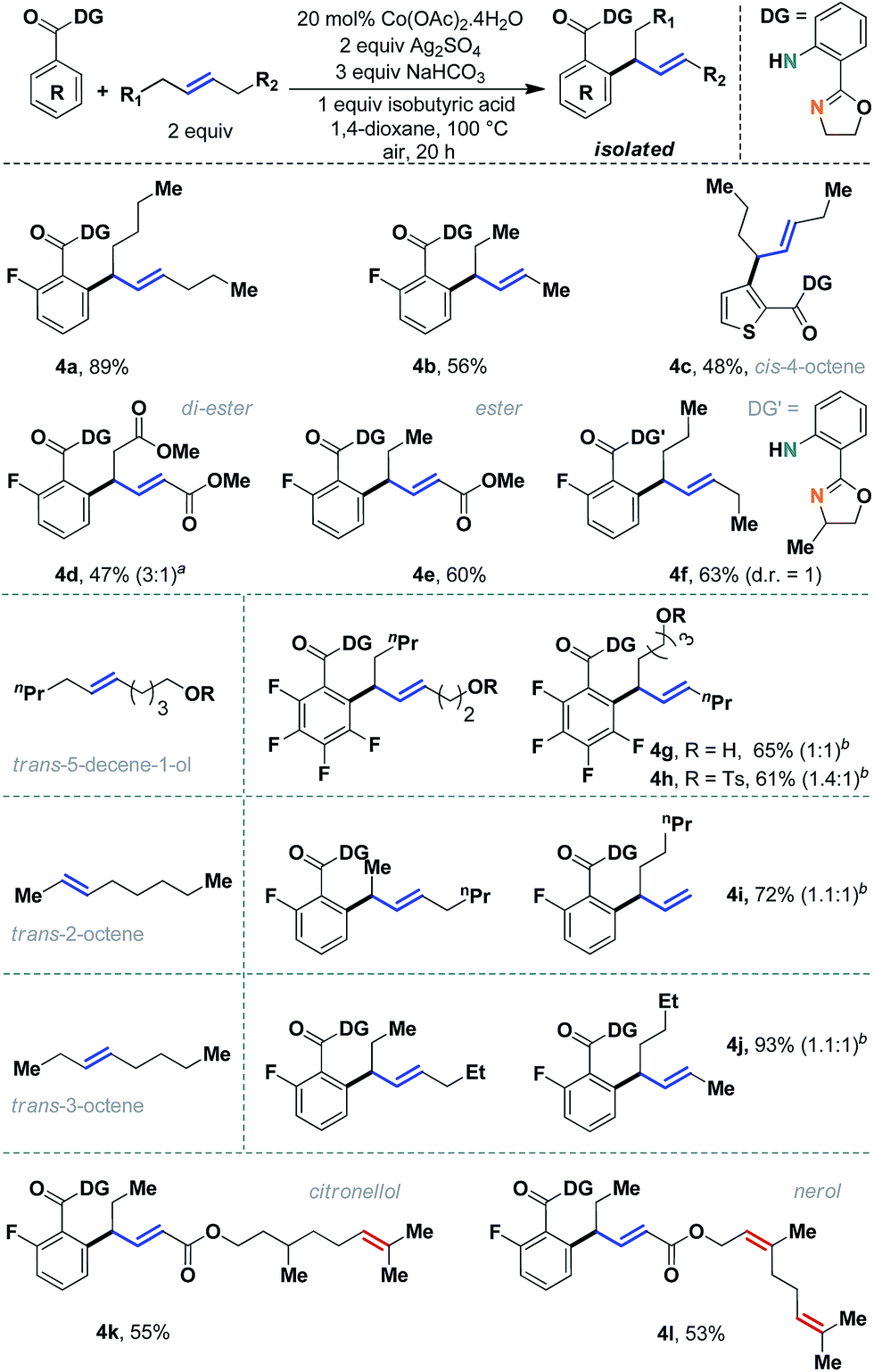 | ||
| Scheme 4 Evaluation of unactivated acyclic internal aliphatic olefins. aAnother isomer can be detected in minor amounts; bregioisomeric ratios are mentioned in parentheses. | ||
To gain additional insight as to whether this reactivity was internally specific, we decided to assess terminal aliphatic olefins as well (Scheme 5). We were pleased to observe that a wide variety of terminal alkenes could be incorporated with excellent yields and complete allylic selectivity. Simple benzoic acids as well as heteroaromatic acids, without any bias, were useful substrates, thereby significantly expanding the scope of allylation with aliphatic olefins (5a–5i). Additionally, sequential allylation could be performed with equal ease (5s–5u).11
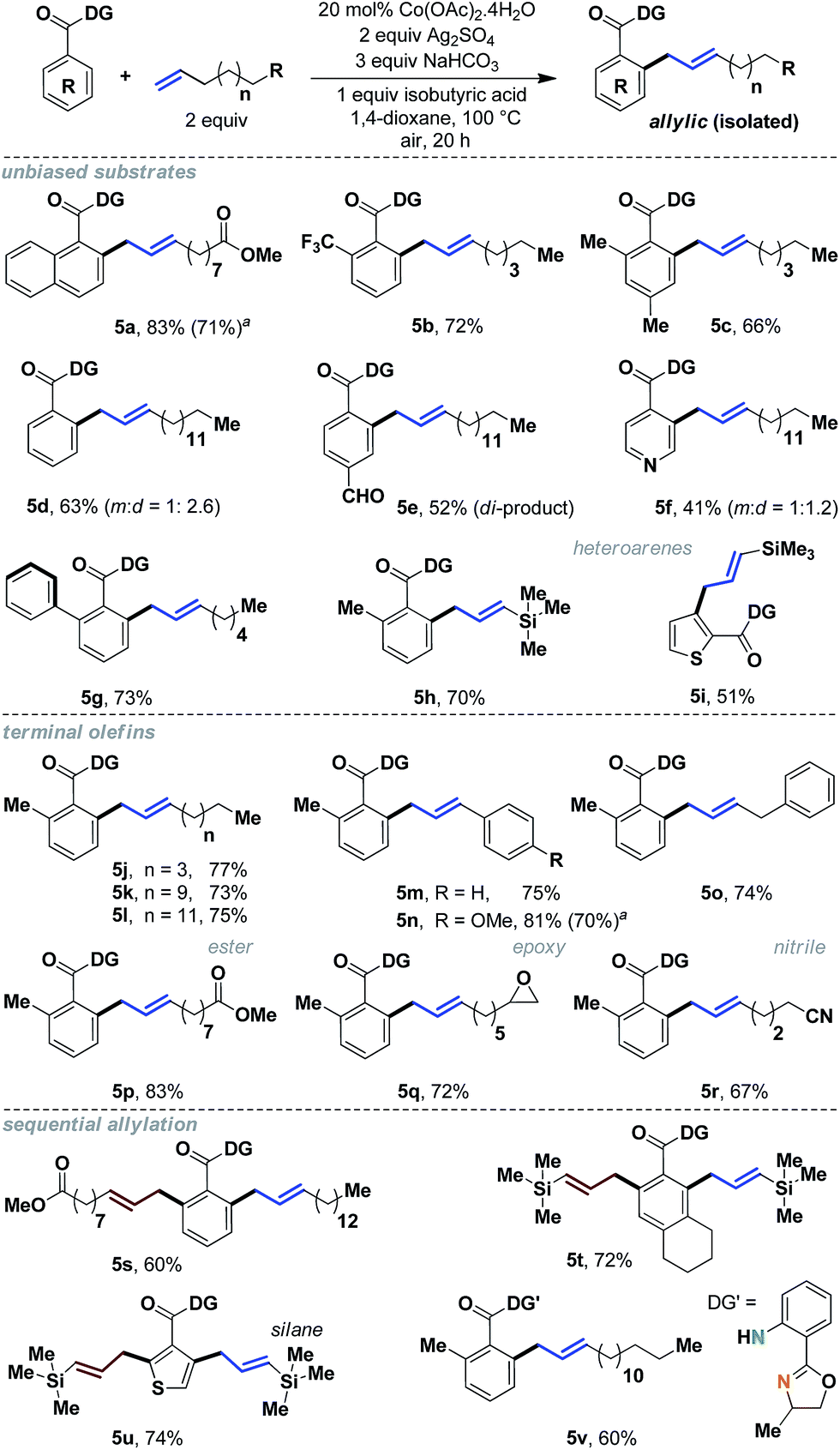 | ||
| Scheme 5 Allylic selectivity with terminal aliphatic olefins. aYields with 10 mol% Co. | ||
We next decided to study the comparative reactivity of different aliphatic olefins (Scheme 6). An intermolecular competition experiment revealed that 1-octene could react preferentially in a 14:1 ratio over trans-4-octene (6a, 60%). Similarly, a substrate with both terminal and internal olefins reacted exclusively at the terminal site, while the internal olefin remained completely unreacted (6b). Kinetic studies further showed that a trans-olefin reacts 2.5 times faster than the cis-isomer, which partially explained the apparent lower reactivity of cis-olefins under the standard conditions.
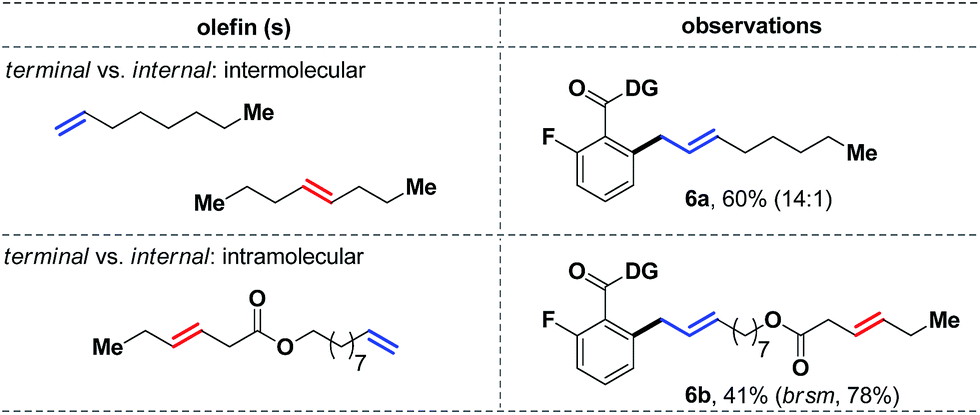 | ||
| Scheme 6 Competition experiments with aliphatic olefins. | ||
A plausible mechanism has been depicted in Scheme 7. The reaction was thought to be initiated by an in situ generated Co(III) species under aerobic conditions. C–H activation (C), olefin coordination (D), and subsequent migratory insertion led to a unique seven-membered intermediate (E), the stability of which was crucial for the progress of the reaction. In all probability, the geometry of this particular intermediate determined the regioselective outcome of the reaction.
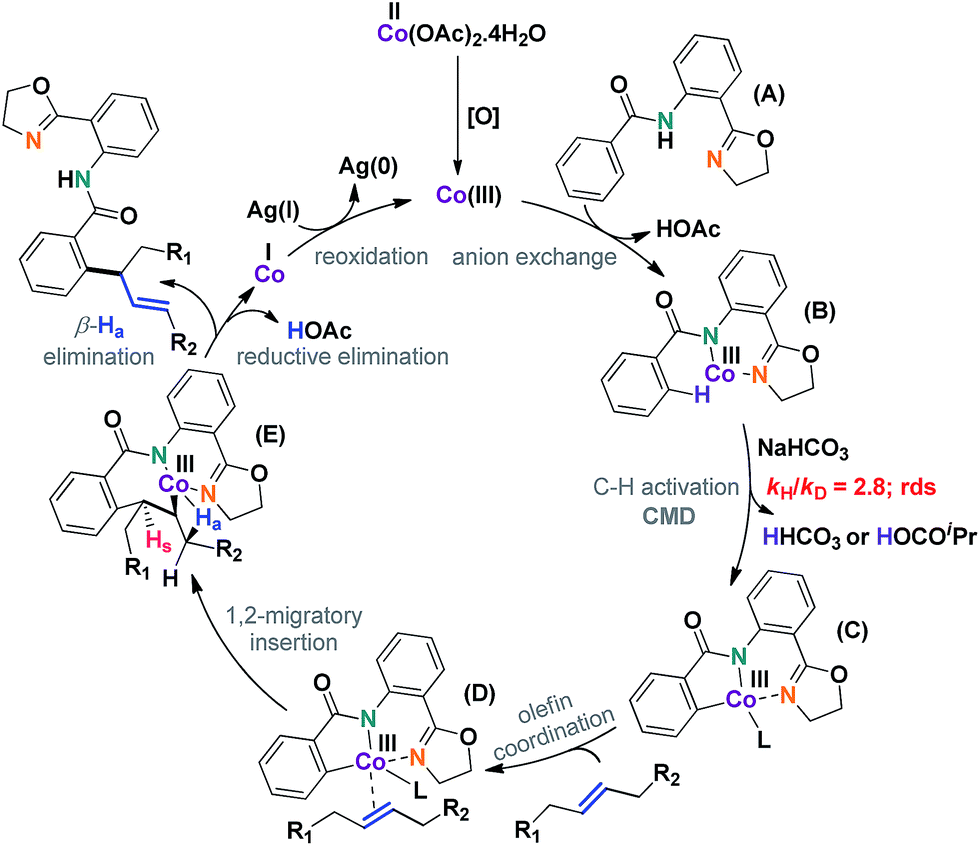 | ||
| Scheme 7 Plausible mechanism for allylic selective alkenylation with internal aliphatic olefins. | ||
It was found that the aliphatic acid additives had a profound influence on the rate enhancement. In sharp contrast, omitting NaHCO3 slightly altered the rate of the reaction (Scheme 8). This could be justified by considering the superiority of the aliphatic carboxylate (iPrCO2Na) as the proton abstracting agent in the C–H activation step (B), thus clearly indicating that a concerted metalation–deprotonation or CMD-type pathway was operative.
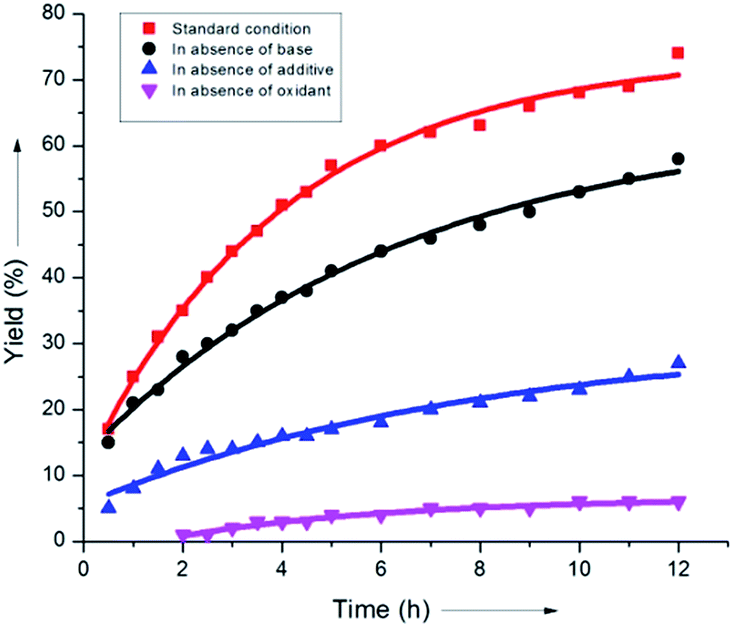 | ||
| Scheme 8 Kinetic investigation of the reaction components. | ||
Interestingly, labeling studies revealed a kH/kD value of 2.8, thereby suggesting C–H activation as the rate determining step (r.d.s.) of the overall transformation (Scheme 9). This observation was supported further by the fact that both internal and terminal olefins could react at nearly the same rate in parallel experiments (see ESI†).
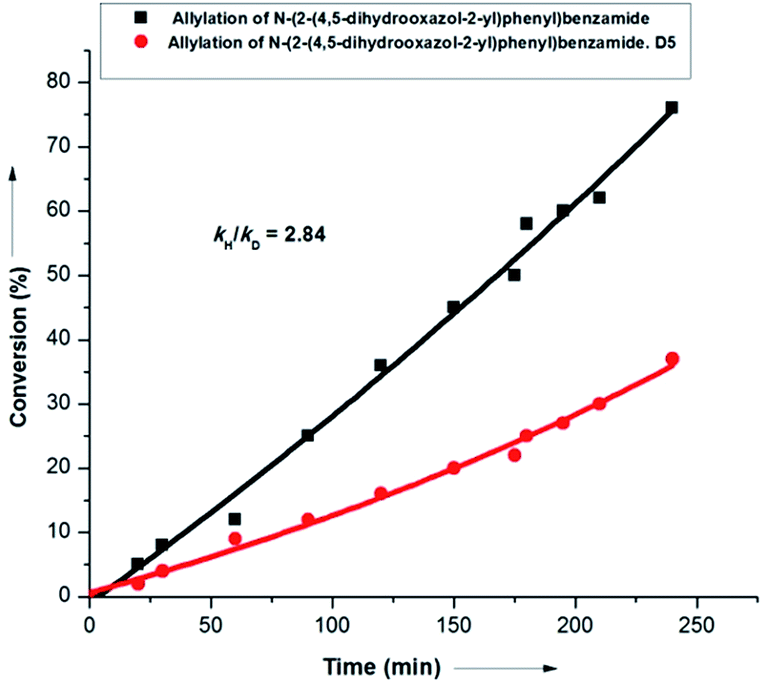 | ||
| Scheme 9 Determination of the kinetic isotope effect. | ||
Conclusions
In summary, we have developed a cobalt catalyzed allylic selective alkenylation with unactivated internal aliphatic olefins. The excellent regio- and stereoselectivity, broad scope, and high synthetic yields are some of the noteworthy features of the present method. Remarkably, a variety of acyclic internal aliphatic olefins were successfully incorporated. The development of a related asymmetric transformation and detailed mechanistic investigations are presently underway in our laboratory.Acknowledgements
This activity is supported by DST, India {SR/NM/NS-1065/2015 (G)}. Fellowships from CSIR-New Delhi and UGC India (for S. M. and P. D., respectively) are gratefully acknowledged.References
- For selected references of Heck reactions with aliphatic olefins, see: (a) J. H. Delcamp, A. P. Brucks and M. C. White, J. Am. Chem. Soc., 2008, 130, 11270 CrossRef CAS PubMed; (b) E. W. Werner and M. S. Sigman, J. Am. Chem. Soc., 2010, 132, 13981 CrossRef CAS PubMed; (c) E. W. Werner and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 9692 CrossRef CAS PubMed; (d) A. Deb and D. Maiti, Eur. J. Org. Chem., 2017, 1239 CrossRef CAS.
- For selective alkenylation with aliphatic olefins, see: (a) Y. Lu, D.-H. Wang, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 5916 CrossRef CAS PubMed; (b) A. S. Tsai, M. Brasse, R. G. Bergman and J. A. Ellman, Org. Lett., 2011, 13, 540 CrossRef CAS PubMed; (c) X. Li, X. Gong, M. Zhao, G. Song, J. Deng and X. Li, Org. Lett., 2011, 13, 5808 CrossRef CAS PubMed; (d) R. Matsubara, A. C. Gutierrez and T. F. Jamison, J. Am. Chem. Soc., 2011, 133, 19020 CrossRef CAS PubMed; (e) L. Qin, X. Ren, Y. Lu, Y. Li and J. Zhou, Angew. Chem., Int. Ed., 2012, 51, 5915 CrossRef CAS PubMed; (f) C. Zheng, D. Wang and S. S. Stahl, J. Am. Chem. Soc., 2012, 134, 16496 CrossRef CAS PubMed; (g) C. S. Sevov and J. F. Hartwig, J. Am. Chem. Soc., 2014, 136, 10625 CrossRef CAS PubMed; (h) A. Deb, S. Bag, R. Kancherla and D. Maiti, J. Am. Chem. Soc., 2014, 136, 13602 CrossRef CAS PubMed; (i) X. Xue, J. Xu, L. Zhang, C. Xu, Y. Pan, L. Xu, H. Li and W. Zhang, Adv. Synth. Catal., 2016, 358, 573 CrossRef CAS.
- For significant developments of Heck chemistry with aliphatic alkenols and cyclic olefins, see: (a) E. W. Werner, T.-S. Mei, A. J. Burckle and M. S. Sigman, Science, 2012, 338, 1455 CrossRef CAS PubMed; (b) T.-S. Mei, E. W. Werner, A. J. Burckle and M. S. Sigman, J. Am. Chem. Soc., 2013, 135, 6830 CrossRef CAS PubMed; (c) L. Xu, M. J. Hilton, X. Zhang, P.-O. Norrby, Y.-D. Wu, M. S. Sigman and O. Wiest, J. Am. Chem. Soc., 2014, 136, 1960 CrossRef CAS PubMed; (d) T.-S. Mei, H. H. Patel and M. S. Sigman, Nature, 2014, 508, 340 CrossRef CAS PubMed; (e) H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 3462 CrossRef CAS PubMed; (f) C. Zhang, C. B. Santiago, J. M. Crawford and M. S. Sigman, J. Am. Chem. Soc., 2015, 137, 15668 CrossRef CAS PubMed; (g) H. H. Patel and M. S. Sigman, J. Am. Chem. Soc., 2016, 138, 14226 CrossRef CAS PubMed; (h) F. Ozawa, A. Kubo and T. Hayashi, J. Am. Chem. Soc., 1991, 113, 1417 CrossRef CAS; (i) Z. Yang and J. Zhou, J. Am. Chem. Soc., 2012, 134, 11833 CrossRef CAS PubMed; (j) C. Wu and J. Zhou, J. Am. Chem. Soc., 2014, 136, 650 CrossRef CAS PubMed; (k) G. Yue, K. Lei, H. Hirao and J. Zhou, Angew. Chem., Int. Ed., 2015, 54, 6531 CrossRef CAS PubMed; (l) A. Faulkner and J. F. Bower, Angew. Chem., Int. Ed., 2012, 51, 1675 CrossRef CAS PubMed; (m) A. Faulkner, N. J. Race, J. S. Scott and J. F. Bower, Chem. Sci., 2014, 5, 2416 RSC; (n) I. R. Hazelden, X. F. Ma, T. Langer and J. F. Bower, Angew. Chem., Int. Ed., 2016, 55, 11198 CrossRef CAS PubMed.
- S. Maity, R. Kancherla, U. Dhawa, E. Hoque, S. Pimparkar and D. Maiti, ACS Catal., 2016, 6, 5493 CrossRef CAS.
- (a) L. Grigorjeva and O. Daugulis, Org. Lett., 2014, 16, 4684 CrossRef CAS PubMed. For similar reactivity with alkynes, see: (b) L. Grigorjeva and O. Daugulis, Angew. Chem., Int. Ed., 2014, 53, 10209 CrossRef CAS PubMed; (c) R. Mei, H. Wang, S. Warratz, S. A. Macgregor and L. Ackermann, Chem.–Eur. J., 2016, 22, 6759 CrossRef CAS PubMed.
- The Yu group developed and utilized this directing scaffold in Pd- and Cu-catalyzed reactions: (a) R. Giri, N. Maugel, B. M. Foxman and J.-Q. Yu, Organometallics, 2008, 27, 1667 CrossRef CAS; (b) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, Org. Lett., 2014, 16, 5666 CrossRef CAS PubMed; (c) M. Shang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 3354 CrossRef CAS PubMed; (d) M. Shang, S.-Z. Sun, H.-L. Wang, B. N. Laforteza, H.-X. Dai and J.-Q. Yu, Angew. Chem., Int. Ed., 2014, 53, 10439 CrossRef CAS PubMed; (e) M. Shang, H.-L. Wang, S.-Z. Sun, H.-X. Dai and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 11590 CrossRef CAS PubMed; (f) S.-Z. Sun, M. Shang, H.-L. Wang, H.-X. Lin, H.-X. Dai and J.-Q. Yu, J. Org. Chem., 2015, 80, 8843 CrossRef CAS PubMed.
- (a) R. F. Heck, Acc. Chem. Res., 1979, 12, 146 CrossRef CAS; (b) I. Moritani and Y. Fujiwara, Tetrahedron Lett., 1967, 8, 1119 CrossRef.
- (a) R. F. Heck, J. Am. Chem. Soc., 1971, 93, 6896 CrossRef CAS; (b) K. Kikukawa and T. Matsuda, Chem. Lett., 1977, 159 CrossRef CAS; (c) K. Kikukawa, K. Nagira, F. Wada and T. Matsuda, Tetrahedron, 1981, 37, 31 CrossRef CAS; (d) J. G. Taylor, A. V. Moro and C. R. D. Correia, Eur. J. Org. Chem., 2011, 1403 CrossRef CAS; (e) Y. Takahama, Y. Shibata and K. Tanaka, Chem.–Eur. J., 2015, 21, 9053 CrossRef CAS PubMed.
- For previous instances of allylic selectivity with aliphatic olefins, see: (a) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315 CrossRef CAS PubMed; (b) K. M. Engle, D.-H. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2010, 132, 14137 CrossRef CAS PubMed; (c) H. Li, Y. Li, X.-S. Zhang, K. Chen, X. Wang and Z.-J. Shi, J. Am. Chem. Soc., 2011, 133, 15244 CrossRef CAS PubMed; (d) B. Liu, Y. Fan, Y. Gao, C. Sun, C. Xu and J. Zhu, J. Am. Chem. Soc., 2013, 135, 468 CrossRef CAS PubMed; (e) Y. Takahama, Y. Shibata and K. Tanaka, Org. Lett., 2016, 18, 2934 CrossRef CAS PubMed; (f) R. Manoharan, G. Sivakumar and M. Jeganmohan, Chem. Commun., 2016, 52, 10533 RSC; (g) T. Yamaguchi, Y. Kommagalla, Y. Aihara and N. Chatani, Chem. Commun., 2016, 52, 10129 RSC.
- For allylation with activated olefins, see: (a) D. Leow, G. Li, T.-S. Mei and J.-Q. Yu, Nature, 2012, 486, 518 CrossRef CAS PubMed; (b) S. Bag, T. Patra, A. Modak, A. Deb, S. Maity, U. Dutta, A. Dey, R. Kancherla, A. Maji, A. Hazra, M. Bera and D. Maiti, J. Am. Chem. Soc., 2015, 137, 11888 CrossRef CAS PubMed.
- For selected references on important allylation chemistry, see: (a) M. R. Luzung, C. A. Lewis and P. S. Baran, Angew. Chem., Int. Ed., 2009, 48, 7025 CrossRef CAS PubMed; (b) T. Yao, K. Hirano, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2011, 50, 2990 CrossRef CAS PubMed; (c) Y. Makida, H. Ohmiya and M. Sawamura, Angew. Chem., Int. Ed., 2012, 51, 4122 CrossRef CAS PubMed; (d) S. Asako, L. Ilies and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 17755 CrossRef CAS PubMed; (e) H. Wang, N. Schroeder and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 5386 CrossRef CAS PubMed; (f) X. Cong, Y. Li, Y. Wei and X. Zeng, Org. Lett., 2014, 16, 3926 CrossRef CAS PubMed; (g) H. Jiang, W. Yang, H. Chen, J. Li and W. Wu, Chem. Commun., 2014, 50, 7202 RSC; (h) T. Gensch, S. Vasquez-Cespedes, D.-G. Yu and F. Glorius, Org. Lett., 2015, 17, 3714 CrossRef CAS PubMed; (i) Y. Suzuki, B. Sun, K. Sakata, T. Yoshino, S. Matsunaga and M. Kanai, Angew. Chem., Int. Ed., 2015, 54, 9944 CrossRef CAS PubMed; (j) H.-P. Deng, D. Wang and K. J. Szabó, J. Org. Chem., 2015, 80, 3343 CrossRef CAS PubMed; (k) G. Cera, T. Haven and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 1484 CrossRef CAS PubMed; (l) D. Zell, Q. Bu, M. Feldt and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7408 CrossRef CAS PubMed; (m) C. C. Pattillo, I. I. Strambeanu, P. Calleja, N. A. Vermeulen, T. Mizuno and M. C. White, J. Am. Chem. Soc., 2016, 138, 1265 CrossRef CAS PubMed; (n) H. Zhang, R.-B. Hu, N. Liu, S.-X. Li and S.-D. Yang, Org. Lett., 2016, 18, 28 CrossRef CAS PubMed; (o) S. E. Korkis, D. J. Burns and H. W. Lam, J. Am. Chem. Soc., 2016, 138, 12252 CrossRef CAS PubMed; (p) R. Y. Liu, Y. Yang and S. L. Buchwald, Angew. Chem., Int. Ed., 2016, 55, 14077 CrossRef CAS PubMed; (q) W. Liu, S. C. Richter, Y. Zhang and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 7747 CrossRef CAS PubMed; (r) S. Y. Lee and J. F. Hartwig, J. Am. Chem. Soc., 2016, 138, 15278 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1507233. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7sc01204g |
| This journal is © The Royal Society of Chemistry 2017 |